
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Carbonic anhydrases (CAs, EC 4.2.1.1) activators were shown to be involved in memory enhancement and learning in animal models of cognition. Here we investigated the CA activating effects of a large series of histamine based compounds, including histamine receptors (H1R – H4R) agonists, antagonists and other derivatives of this autacoid. CA activators may be thus useful for improving cognition as well as in diverse therapeutic areas (phobias, obsessive-compulsive disorder, generalised anxiety, post-traumatic stress disorders), for which activation of this enzyme was recently shown to be involved.
1. Introduction
CO2 is generated in most metabolic processes, being one of the simplest molecules involved in crucial physiologic processes in all life kingdomsCitation1,Citation2. The carbonic anhydrases (CAs, EC 4.2.1.1) are metalloenzymes which catalyse its interconversion to bicarbonate (EquationEquation (1)(1)
(1) Citation3–6, generating also a proton, and thus a pH disequilibrium, which is used in most biological systems as a readily available buffering systemCitation7.
(1)
(1)
The reaction also occurs without a catalyst, but at physiological pH values it is exceedingly slow for meeting metabolic needs, as CO2 is a poorly water-soluble gas, which can also damage cellular components (e.g. membranes, mitochondria, etc.)Citation6,Citation7. On the other side, its conversion to water-soluble ions (bicarbonate and protons) counteracts this effect, and although interfering with the pH balance, is used to control homeostasis and metabolism, making CAs crucial enzymes in many physiological and pathological conditionsCitation3–7. In fact, in vertebrates at least 16 CA isoforms belonging to the α-CA genetic family are known, whereas in other organisms all over the phylogenetic tree at least seven other CA families were described so far, the β-, γ-, δ-, ζ-, η-, θ- and ι-CAsCitation8–14. In humans 15 CAs are expressed, 12 of which are catalytically active: the cytosolic CA I-III, VII and XIII, the membrane-bound CA IV, the mitochondrial CA VA and VB, the secreted (in saliva and tears) CA VI, and the transmembrane CA IX, XII and XIV (the acatalytic forms are CA VIII, X and XI)Citation4,Citation15–19. many of these enzymes are drug targets, as their inhibitors show pharmacological applications for drugs treating edoema, glaucoma, obesity, epilepsy and tumoursCitation4–6.
The human central nervous system (CNS), as well as the choroid plexus, contains a multitude of CA isoforms, although their particular functions are not yet completely understoodCitation17. We will consider here mainly the CAs present in CNS, as the compounds investigated here for modulating their activity (i.e. the CA activators – CAAs) may also have interesting applications in therapy, which started to be considered only recentlyCitation20–23. The nervous system CA isoforms comprise: the cytosolic CA I (expressed in the motor neurons in the spinal cord), CA II (present in the choroid plexus, oligodendrocytes, myelinated tracts, astrocytes and myelin sheaths); CA III (in the choroid plexus), the membrane-associated CA IV (located on the luminal surface of cerebral capillaries and associated with the blood-brain barrier, being present also in the cortex, hippocampus and thalamus). The mitochondrial CA VA is expressed in astrocytes and in neurons, whereas CA VB seems to be absent in the SNCCitation17. CA VII and VIII are present in high levels throughout the cortex, hippocampus and thalamus, although CA VIII is acatalytic, whereas CA VII shows a good enzymatic activity with CO2/bicarbonate as substratesCitation4. The acatalytic CA X is expressed in the myelin sheath, whereas CA XI (also acatalytic) is present in the neural cell body and astrocytesCitation17. CA IX and CA XII are transmembrane proteins overexpressed in many neurologic cancersCitation18,Citation19, whereas CA XIII seems not to be present in the brain. CA XIV is expressed in nuclei and nerve tracts associated with pontine, medullary and hippocampal functions being also located on the plasma membrane of some neurons and on axons of mammalian brainCitation17.
The most investigated CAAs are the amino acids, the biogenic amines (histamine, serotonin, catecholamines and their derivatives), and to some extent also the oligopeptides or small proteins, although these activators were less investigatedCitation20. The CAAs were demonstrated to participate in the catalytic cycle of the enzyme, forming enzyme-activator complexes, as described in EquationEquation (2)(2)
(2) :Citation20
(2)
(2)
The activator molecule forms a complex with the enzyme, binding in an active site region distinct of that of the classical CA inhibitorsCitation24,Citation25, which generally bind to the metal ionCitation4–6. The activator molecule must incorporate proton shuttling moieties, which take part to the rate-determining step of the catalytic cycle, i.e. the transfer of protons from the zinc-coordinated water molecule to the external reaction medium, with formation of the nucleophilic, zinc hydroxide species of the enzymeCitation20. In the wild type enzyme, this proton shuttling is achieved by residue His64 (in many CA isoforms), found within the middle of the active site cleft, and which possess the imidazole moiety able to transfer protons in the pH range of 6–8Citation20–24. His64 was shown to possess two conformations: the in one, orientated towards the bottom of the active site, and the out one, orientated towards the external part of the active site, favouring thus the proton wiringCitation20,Citation24. In such processes, within the enzyme-activator complexes, the proton transfer becomes intramolecular, being more efficient compared to the intermolecular transfer to buffer molecules (which are not bound within the enzyme cavity)Citation20. X-ray crystallography has been performed on several other hCA I/II – activator complexes, among which those with histamine, L- and D-His, L- and D-Phe, D-Trp, L-adrenaline as well as pyridinium derivatives of histamineCitation20,Citation23–26. A schematic representation of the activators bound to CA is shown in .
Figure 1. CA activation mechanisms. Activators bind in the middle of the active site and contain a proton shuttle moiety (PSM) of the amine, imidazole or carboxylate type with an appropriate pKa for the proton transfer processes, usually in the range of 6–8.
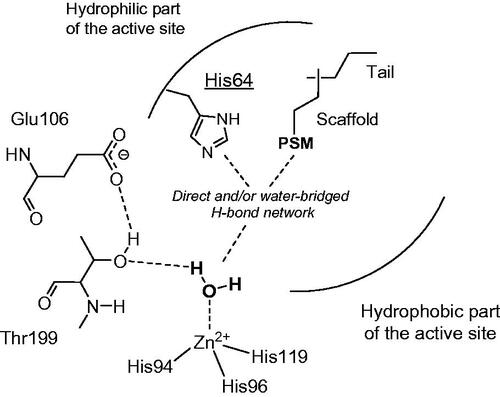
Thus, histamine was the main compound used to obtain new CAAsCitation27, but many of its rather simple derivatives as well as drugs belonging to the histamine receptors (H1R, H2R, H3R and H4R) agonists/antagonists, were not yet been investigated for their potential activating effects. Here we report the first such study, including in our investigations 28 such derivatives which have been assayed as activators of four pharmacologically significant isoforms, hCA I, II and VII (cytosolic isoforms) and hCA IV (membrane-anchored enzyme).
2. Materials and methods
2.1. Chemistry
Histamine 1 and compounds 2–30 were commercially available, highest purity reagents from Sigma-Aldrich, Milan Italy.
2.2. Carbonic anhydrase activation
A stopped-flow methodCitation28 has been used for assaying the CA catalysed CO2 hydration activity with Phenol red as indicator, working at the absorbance maximum of 557 nm, following the initial rates of the CA-catalysed CO2 hydration reaction for 10–100 s. For each activator, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of activator (0.1 mM) were prepared in distilled-deionized water and dilutions up to 0.1 nM were done thereafter with the assay buffer. The activation constant (KA), defined similarly with the inhibition constant (KI), was obtained by considering the classical Michaelis–Menten EquationEquation (3)(3)
(3) , which has been fitted by nonlinear least squares by using PRISM 3:
(3)
(3)
where [A]f is the free concentration of activator.
Working at substrate concentrations considerably lower than KM ([S]≪KM), and considering that [A]f can be represented in the form of the total concentration of the enzyme ([E]t) and activator ([A]t), the obtained competitive steady-state equation for determining the activation constant is given by EquationEquation(4)
(4) Equation (4)
(4)
(4) :
(4)
(4)
where v0 represents the initial velocity of the enzyme-catalysed reaction in the absence of an activatorCitation29–32. Enzyme concentrations in the assay system were in the range of 6.5–12.0 nM.
3. Results and discussion
As mentioned above, histamine 1 () was one of the first CAAs to be investigated in detailCitation24, but except for histidine (L- and D-enantiomers), other histamine derivatives were not yet assayed for their potential CA activating effects.
Figure 2. Histamine 1 and derivatives 2–30 acting as histamine receptors agonists/antagonists (for reviews see refs.Citation33,Citation34) investigated here as CAAs.
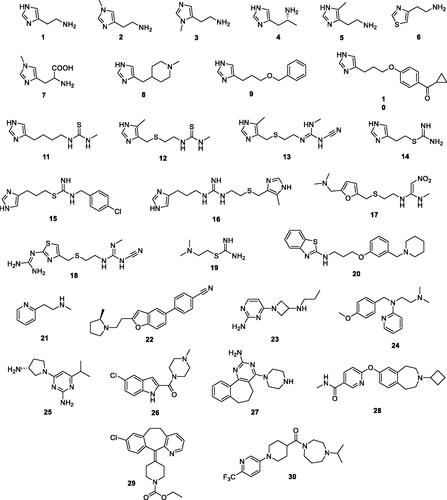
Considering the relatively large number of histamine receptors (H1R-H4R) as well as the huge number of agonists/antagonists developed for the management of various disorders, among which allergies, gastritis and gastric ulcers, narcolepsy, acute unilateral vestibulopathy, and atopic dermatitisCitation33,Citation34, there is a large number of compounds incorporating fragments of the histamine chemotype as well as a wealth of structural modifications which mimic this autacoid.
Some of these compounds, possessing structures 2–30 (), were included in our study for investigating their possible CA activating effects against four pharmacologically relevant human isoforms, hCA I, II, IV and VII. The compounds were numbered according to their similarity to the lead histamine 1 and are: the H1R agonist 2-(2-aminoethyl)thiazole 6; the H2R agonists impromidine 16 and nordimaprit 19; the H3R agonists Nπ-methylhistamine 3, α-methylhistamine 4, methimmepip 8, proxyfan 9, imetit 14, VUF16839 23; the H1R antagonists pyrilamine 24, loratadine 29; the H2R antagonists metiamide 12, cimetidine 13, ranitidine 17, tiotidine 18, zolantidine 20; the H3R antagonists ciproxifan 10, clobenpropit 15, ABT239 22, GSK189254A 28, GSK334429B 30; the H4R antagonists JNJ39758979 25, JNJ7777120 26, A940894 27; the mixed modulators of the histaminergic system Nτ-methylhistamine 2, 4-methylhistamine 5, 1-methylhistidine 7, burimamide 11, betahistine 21.
In some of these compounds, such as the methyl-histamines 2–5, the thiazolyl derivative 6 or τ-methyl-His 7, the histamine chemotype is readily observable, whereas the remaining compounds incorporate more drastic changes of the basic structure, but all of them possess moieties which can in principle shuttle protons in the pH range of 6–8 which, as mentioned earlierCitation20, lead to CA activation.
Table 1. hCA I, II, IV and VII activation with compounds 2–30 () by a stopped-flow CO2 hydrase assay.Citation28 Histamine 1 used as standard.
The following structure-activity relationship (SAR) can be worked out from the data reported in for activation of the four isoforms hCA I, II, IV and VII:
Compounds 17, 20, 22, 25–30 did not induce any activation of the tested CA isoforms (KAs >100 µM). Consistently, these derivatives do not possess the histamine chemotype in their structures and/or other moieties that clearly make CA activation possible. As a unique exception, compound 30 reported a 5.06 µM selective activation of hCA VII. It should be stressed that other derivatives, such as 18, 19, 21, 23 and 24, do not directly include imidazole-like scaffolds in their chemical structure, but showed however to possess significant CA activation profiles in a low micromolar range (KAs between 1.36 and 45.5 µM) and are thus included in the SAR discussion. These compounds possess however protonatable moieties of the secondary amine or guanidine type in their molecule, which like the imidazole may shuttle protons and thus act as CAAs.
The cytosolic and ubiquitous hCA isoforms I and II were quite efficiently activated by most active derivatives (that are 2–16, 18, 19, 21, 23 and 24), in a low micromolar to submicrolar range. Intriguingly, derivatives 15, 21 and 23 did not produce any hCA I activation up to 100 µM, whereas 18 did not activates neither hCA I nor hCA II. The methylation of histamine 1 at position Nτ (2), α (4), 4 (5) and the imidazole/thiazole swap (6) increased up to one order of magnitude the hCA I activation profile (from 2.1 to 0.11 µM). The presence of an extra proton transfer group (COOH) as in 7 (Nτ-methyl-histidine) further improved 2-fold the KA (52 vs 110 nM) against hCA I compared to compound 2. The Nπ-methylation of histamine (3, KA of 3.1 µM) slightly decreased the activation efficacy of the molecule, situations also encountered for the inclusion of the aliphatic amine into a cycle (8, KA of 3.16 µM) or the amine/ether swap (as in 9 and 10, KAs of 3.15 and 4.29 µM). Substituting the amine group with N-linked thioureas as in 11 and 12 improved 2-fold the KAs towards hCA I with respect to histamine. Among the remaining derivatives, only the bis-imidazole 16 reported an improved KA over the lead towards this isoform (KA of 0.72 µM). In fact, the presence of S-linked thioureas worsened the activation efficacy 2- (14, KA of 4.25 µM) or 4-fold (13, KA of 8.79 µM) with respect to the lead histamine. Oddly, the S-linked thiourea 19 showed a 2-fold improved KA compared to histamine, although bearing a dimethylamino group in place of the imidazole ring. As an exception, the N-rich compound 24 interestingly activated hCA I just two times less than histamine, in spite of a completely diverse structure. On the contrary, all histamine derivatives here reported showed a superior hCA II activation efficacy with respect to the lead (KA > 100 μM). Among mostly low micromolar CAAs (KAs in the range 2.1–13.5 μM), derivatives 3, 4 and 7 stood out as the most potent hCA II modulators of the study (KAs in the range 82 nM–0.57 μM). In particular, the α-methylation of histamine, as in 4, induced the largest increase of efficacy, up to a KA of 82 nM, when compared to the ineffective (as CAA) lead molecule.
No submicromolar KA values was measured for 1–30 as hCA IV activators. Indeed, all KAs are in a rather flat low micromolar range (KAs in the range 1.02–13.9 μM), making this membrane-associated isozyme the less activated one by the compounds tested in this work. Interestingly, with the exception of compound 18, all derivatives were more efficient CAAs than the lead histamine, which showed a KA of 25.3 μM. Of note, the imidazole/thiazole swap led to the most effective activation increase with respect to the lead, with a KA of 1.02 μM in case of derivative 6.
The other cytosolic isoform investigated here, hCA VII, was the most effectively activated one by the compounds investigated in this study. Indeed, a wide subset of KAs were detected in a submicromolar range (from 0.10 to 45.5 μM) for some of these derivatives. All of them showed much better activation profile than the reference compound histamine (KA of 37.5 μM) towards hCA VII. Contrariwise to hCA I and II, the most efficient CAAs were not detected among the methylhistamine derivatives 2–5: Nτ-methylhistidine 7, N-methylpiperidine 8 and the aryl ether 10, showed KAs ranging between 110 and 190 nM. The bis-imidazole 16 stood out as the most effective hCA VII activator of the study with a KA of 100 nM. Intriguingly, compound 23 did not activate hCA VII below 100 μM, whereas derivative 30, previously classified among the inactive compounds for the other CA isoforms, weakly activated this CNS-associated CA (KA of 5.06 μM). In fact, this isoform is one of the most widely spread in the brain, probably being involved in crucial metabolic/pH regulation processes, while it is not expressed in other tissues. It is thus relevant that a rather wide set of compounds was detected here (8, 9, 10, 13, 16), which showed a promising isoform selectivity towards hCA VII over the ubiquitous CAs (up to 100-fold over hCA II).
Table 2 also include the literature references regarding the compounds ability to cross the BBB, which presumably should also lead to brain CA activating effects, as well as evidences for their action at central level.
4. Conclusions and future perspectives
In the present study, we investigated the CA activating effects of a series of histamine receptors agonists/antagonists (compounds 2–30 in ) towards four hCA isoforms expressed in human brain, that are CA I, II, IV and VII. Though all derivatives possess moieties which can in principle shuttle protons in the pH range of 6–8, a consistent subset of them (17, 20, 22, 25–30), not having the histamine chemotype in their structure, did not report any activation effect for the tested CA isoforms (KAs >100 µM). hCA I and II were effectively activated by methylhistamine derivatives (2–7), whereas more intriguing SAR were observed for hCA VII with more lipophilic groups (as in 6, 8 or 10) promoting greater and more selective isoform activation. Of note, a subset of selective hCA VII activators was identified, that could serve to drive the identification and optimisation of new brain specific CAAs. We are currently witnessing a second youth period for CAAs, because of innovative pharmacological studies spurring researchers to take into account these lately neglected agents for their potential clinical relevance in the treatment of emotional memory disorders, including the improvement of the clinical efficacy of exposure-based treatments of obsessive-compulsive disorders, phobias, generalised anxiety, and post-traumatic stress disorder. The here gathered data might also provide more insights on the pharmacodynamics of therapeutically used histamine modulators, whose therapeutic action and/or side effects could be related to polypharmacology. Overall, this work might bring new lights on the intricate relationship between CA activation and brain physiology.
Acknowledgements
This paper is dedicated to the 90th anniversary of Professor Alexandru T. Balaban, mentor and friend to some of us.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- (a) Arias-Hidalgo M, Yuan Q, Carta F, et al. CO2 permeability of rat hepatocytes and relation of CO2 permeability to CO2 production. Cell Physiol Biochem 2018;46:1198–208.(b) Arias-Hidalgo M, Hegermann J, Tsiavaliaris G, et al. CO2 and HCO3– permeability of the rat liver mitochondrial membrane. Cell Physiol Biochem 2016;39:2014–24.
- (a) Itel F, Al-Samir S, Öberg F, et al. CO2 permeability of cell membranes is regulated by membrane cholesterol and protein gas channels. Faseb J 2012;26:5182–91. (b) Endeward V, Musa-Aziz R, Cooper GJ, et al. Evidence that aquaporin 1 is a major pathway for CO2 transport across the human erythrocyte membrane. Faseb J 2006;20:1974–81.
- (a) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. (b) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. (c) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (d) Supuran CT. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin Drug Discov 2020;15:671–86.
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.(b) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.(c) De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810.
- (a) Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35. (b) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21. (c) Supuran CT. The management of glaucoma and macular degeneration. Expert Opin Ther Pat 2019;29:745–7. (d) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47.
- (a) Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16.(b) Supuran CT, Altamimi ASA, Carta F. Carbonic anhydrase inhibition and the management of glaucoma: a literature and patent review 2013–2019. Expert Opin Ther Pat 2019;29:781–92.(c) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.(d) Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95.
- (a) Lee SH, McIntyre D, Honess D, et al. Carbonic anhydrase IX is a pH-stat that sets an acidic tumour extracellular pH in vivo. Br J Cancer 2018;119:622–30.(b) Ruusuvuori E, Kaila K. Carbonic anhydrases and brain pH in the control of neuronal excitability. Subcell Biochem 2014;75:271–90. (c) Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends Neurosci 1992;15:396–402.(d) Occhipinti R, Boron WF. Role of carbonic anhydrases and inhibitors in acid-base physiology: insights from mathematical modeling. Int J Mol Sci 2019;20:3841.
- (a) Mishra CB, Tiwari M, Supuran CT. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev 2020;40:2485–565.(b) Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32.(c) Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov 2019;14:1175–97.
- (a) Capasso C, Supuran CT. Inhibition of bacterial carbonic anhydrases as a novel approach to escape drug resistance. Curr Top Med Chem 2017;17:1237–48. (b) Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704. (c) Ozensoy Guler O, Supuran CT, Capasso C. Carbonic anhydrase IX as a novel candidate in liquid biopsy. J Enzyme Inhib Med Chem 2020;35:255–60. (d) Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin Ther Pat 2018;28:745–54.
- (a) Kaur J, Cao X, Abutaleb NS, et al. Optimization of acetazolamide-based scaffold as potent inhibitors of vancomycin-resistant enterococcus. J Med Chem 2020;63:9540–62.(b) Supuran CT, Capasso C. Antibacterial carbonic anhydrase inhibitors: an update on the recent literature. Expert Opin Ther Pat 2020;30:963–82.
- (a) Del Prete S, Bua S, Supuran CT, Capasso C. Escherichia coli γ-carbonic anhydrase: characterisation and effects of simple aromatic/heterocyclic sulphonamide inhibitors. J Enzyme Inhib Med Chem 2020;35:1545–54.(b) Del Prete S, De Luca V, Bua S, et al. The effect of substituted benzene-sulfonamides and clinically licensed drugs on the catalytic activity of CynT2, a carbonic anhydrase crucial for escherichia coli life cycle. Int J Mol Sci 2020;21:4175.(c) Angeli A, Ferraroni M, Pinteala M, et al. Crystal structure of a tetrameric type II β-carbonic anhydrase from the pathogenic Bacterium Burkholderia pseudomallei. Molecules 2020;25:2269.
- (a) Del Prete S, Nocentini A, Supuran CT, Capasso C. Bacterial ι-carbonic anhydrase: a new active class of carbonic anhydrase identified in the genome of the Gram-negative bacterium Burkholderia territorii. J Enzyme Inhib Med Chem 2020;35:1060–8.(b) Angeli A, Pinteala M, Maier SS, et al. Inhibition of α-, β-, γ-, δ-, ζ- and η-class carbonic anhydrases from bacteria, fungi, algae, diatoms and protozoans with famotidine. J Enzyme Inhib Med Chem 2019;34:644–50.
- (a) Angeli A, Etxebeste-Mitxeltorena M, Sanmartín C, et al. Tellurides bearing sulfonamides as novel inhibitors of leishmanial carbonic anhydrase with potent antileishmanial activity. J Med Chem 2020;63:4306–14.(b) Nocentini A, Osman SM, Almeida IA, et al. Appraisal of anti-protozoan activity of nitroaromatic benzenesulfonamides inhibiting carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani. J Enzyme Inhib Med Chem 2019;34:1164–71. (c) da Silva Cardoso V, Vermelho AB, Ricci Junior E, et al. Antileishmanial activity of sulphonamide nanoemulsions targeting the β-carbonic anhydrase from Leishmania species. J Enzyme Inhib Med Chem 2018;33:850–7.
- (a) Vermelho AB, da Silva Cardoso V, Ricci Junior E, et al. Nanoemulsions of sulfonamide carbonic anhydrase inhibitors strongly inhibit the growth of Trypanosoma cruzi. J Enzyme Inhib Med Chem 2018;33:139–46.(b) Vermelho AB, Capaci GR, Rodrigues IA, et al. Carbonic anhydrases from Trypanosoma and Leishmania as anti-protozoan drug targets. Bioorg Med Chem 2017;25:1543–55.(c) Nocentini A, Cadoni R, Dumy P, et al. Carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani chagasi are inhibited by benzoxaboroles. J Enzyme Inhib Med Chem 2018;33:286–9.
- (a) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. (b) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25. (c) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8.
- (a) Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother 2016;16:961–8. (b) Supuran CT. Acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6.
- (a) Halmi P, Parkkila S, Honkaniemi J. Expression of carbonic anhydrases II, IV, VII, VIII and XII in rat brain after kainic acid induced status epilepticus. Neurochem Int 2006;48:24–30. (b) Thiry A, Dogne JM, Supuran CT, Masereel B. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr Top Med Chem 2007;7:855–64. (c) Patrikainen MS, Tolvanen MEE, Aspatwar A, et al. Identification and characterization of a novel zebrafish (Danio rerio) pentraxin-carbonic anhydrase. PeerJ 2017;5:e4128.
- (a) Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev 2018;38:1799–836. (b) Supuran CT. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs 2018;27:963–70. (c) Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008–2018). Expert Opin Ther Pat 2018;28:729–40.
- (a) Angeli A, Carta F, Nocentini A, et al. Carbonic anhydrase inhibitors targeting metabolism and tumor microenvironment. Metabolites 2020;10:412.(b) McDonald PC, Chia S, Bedard PL, et al. A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am J Clin Oncol 2020;43:484–90.(c) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- (a) Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73.(b) Blandina P, Provensi G, Passsani MB, et al. Carbonic anhydrase modulation of emotional memory. Implications for the treatment of cognitive disorders. J Enzyme Inhib Med Chem 2020;35:1206–14.
- Canto de Souza L, Provensi G, Vullo D, et al. Carbonic anhydrase activation enhances object recognition memory in mice through phosphorylation of the extracellular signal-regulated kinase in the cortex and the hippocampus. Neuropharmacology 2017;118:148–56.
- Schmidt SD, Costa A, Rani B, et al. The role of carbonic anhydrases in extinction of contextual fear memory. Proc Natl Acad Sci USA 2020;117:16000–8.
- (a) Izquierdo I, Furini CR, Myskiw JC. Fear memory. Physiol Rev 2016;96:695–750.(b) Provensi G, Costa A, Izquierdo I, et al. Brain histamine modulates recognition memory: possible implications in major cognitive disorders. Br J Pharmacol 2020;177:539–56.(c) Provensi G, Passani MB, Costa A, et al. Neuronal histamine and the memory of emotionally salient events. Br J Pharmacol 2020;177:557–69.(d) Benetti F, Furini CR, de Carvalho Myskiw J, et al. Histamine in the basolateral amygdala promotes inhibitory avoidance learning independently of hippocampus. Proc Natl Acad Sci USA 2015;112:E2536–42.
- (a) Briganti F, Mangani S, Orioli P, et al. Carbonic anhydrase activators: X-ray crystallographic and spectroscopic investigations for the interaction of isozymes I and II with histamine. Biochemistry 1997;36:10384–92.(b) Temperini C, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase activators. Activation of isozymes I, II, IV, VA, VII, and XIV with l- and d-histidine and crystallographic analysis of their adducts with isoform II: engineering proton-transfer processes within the active site of an enzyme. Chemistry 2006;12:7057–66.(c) Temperini C, Scozzafava A, Supuran CT. Carbonic anhydrase activators: the first X-ray crystallographic study of an adduct of isoform I. Bioorg Med Chem Lett 2006;16:5152–6.
- (a) Temperini C, Scozzafava A, Supuran CT. Carbonic anhydrase activation and the drug design. Curr Pharm Des 2008;14:708–15.(b) Temperini C, Innocenti A, Scozzafava A, et al. Carbonic anhydrase activators: L-Adrenaline plugs the active site entrance of isozyme II, activating better isoforms I, IV, VA, VII, and XIV. Bioorg Med Chem Lett 2007;17:628–35.(c) Temperini C, Innocenti A, Scozzafava A, Supuran CT. Carbonic anhydrase activators: kinetic and X-ray crystallographic study for the interaction of D- and L-tryptophan with the mammalian isoforms I-XIV. Bioorg Med Chem 2008;16:8373–8.(d) Temperini C, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase activators. Activation of isoforms I, II, IV, VA, VII, and XIV with L- and D-phenylalanine and crystallographic analysis of their adducts with isozyme II: stereospecific recognition within the active site of an enzyme and its consequences for the drug design. J Med Chem 2006;49:3019–27.
- (a) Dave K, Ilies MA, Scozzafava A, et al. An inhibitor-like binding mode of a carbonic anhydrase activator within the active site of isoform II. Bioorg Med Chem Lett 2011;21:2764–8. (b) Bhatt A, Mondal UK, Supuran CT, et al. Crystal structure of carbonic anhydrase II in complex with an activating ligand: implications in neuronal function. Mol Neurobiol 2018;55:7431–7.
- (a) Saada MC, Montero JL, Vullo D, et al. Carbonic anhydrase activators: gold nanoparticles coated with derivatized histamine, histidine, and carnosine show enhanced activatory effects on several mammalian isoforms. J Med Chem 2011;54:1170–7. (b) Scozzafava A, Supuran CT. Carbonic anhydrase activators – part 21. Novel activators of isozymes I, II and IV incorporating carboxamido and ureido histamine moieties. Eur J Med Chem 2000;35:31–9. (c) Scozzafava A, Supuran CT. Carbonic anhydrase activators: high affinity isozymes I, II, and IV activators, incorporating a beta-alanyl-histidine scaffold. J Med Chem 2002;45:284–91. (d) Ilies M, Banciu MD, Ilies MA, et al. Carbonic anhydrase activators: design of high affinity isozymes I, II, and IV activators, incorporating tri-/tetrasubstituted-pyridinium-azole moieties. J Med Chem 2002;45:504–10.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Vullo D, Innocenti A, Nishimori I, et al. Carbonic anhydrase activators: activation of the human isoforms VII (cytosolic) and XIV (transmembrane) with amino acids and amines. Bioorg Med Chem Lett 2007;17:4107–12.
- (a) Pastorekova S, Vullo D, Nishimori I, et al. Carbonic anhydrase activators: activation of the human tumor-associated isozymes IX and XII with amino acids and amines. Bioorg Med Chem 2008;16:3530–6. (b) Vullo D, Nishimori I, Innocenti A, et al. Carbonic anhydrase activators: an activation study of the human mitochondrial isoforms VA and VB with amino acids and amines. Bioorg Med Chem Lett 2007;17:1336–40.
- (a) Saada MC, Vullo D, Montero JL, et al. Mono- and di-halogenated histamine, histidine and carnosine derivatives are potent carbonic anhydrase I, II, VII, XII and XIV activators. Bioorg Med Chem 2014;22:4752–8. (b) Saada MC, Vullo D, Montero JL, et al. Carbonic anhydrase I and II activation with mono- and dihalogenated histamine derivatives. Bioorg Med Chem Lett 2001;21:4884–7.
- (a) Supuran CT, Barboiu M, Luca C, et al. Carbonic anhydrase activators. Part 14. Synthesis of mono- and bis- pyridinium salt derivatives of 2-amino-5-(2-aminoethyl)- and 2-amino-5-(3-aminopropyl)-1,3,4-thiadiazole, and their interaction with isozyme II. Eur J Med Chem 1996;31:597–606. (b) Scozzafava A, Supuran CT. Carbonic anhydrase activators. Part 24. High affinity isozymes I, II and IV activators, derivatives of 4-(4-chlorophenylsulfonylureido-amino acyl)ethyl-1H-imidazole. Eur J Pharm Sci 2000;10:29–41.
- Tiligada E, Ennis M. Histamine pharmacology: from Sir Henry Dale to the 21st century. Br J Pharmacol 2020;177:469–89.
- (a) Passani MB, Benetti F, Blandina P, et al. Histamine regulates memory consolidation. Neurobiol Learn Mem 2017;145:1–6. (b) Provensi G, Costa A, Passani MB, Blandina P. Donepezil, an acetylcholine esterase inhibitor, and ABT-239, a histamine H3 receptor antagonist/inverse agonist, require the integrity of brain histamine system to exert biochemical and procognitive effects in the mouse. Neuropharmacology 2016;109:139–47.
- Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol Neurobiol 2000;20:131–47.
- Kitanaka J, Kitanaka N, Hall SF, et al. Histamine H3 receptor agonists decrease hypothalamic histamine levels and increase stereotypical biting in mice challenged with methamphetamine. Neurochem Res 2011;36:1824–33.
- Giovannini MG, Bartolini L, Bacciottini L, et al. Effects of histamine H3 receptor agonists and antagonists on cognitive performance and scopolamine-induced amnesia. Behav Brain Res 1999;104:147–55.
- Kitbunnadaj R, Zuiderveld OP, Christophe B, et al. Identification of 4-(1H-Imidazol-4(5)-ylmethyl)pyridine (Immethridine) as a Novel, Potent, and Highly Selective Histamine H3 Receptor Agonist. J Med Chem 2004;47:2414–7.
- Henry MB, Zheng S, Duan C, et al. Hwa antidiabetic properties of the histamine H3 receptor protean agonist proxyfan. Endocrinology 2011;152:828–35.
- Ligneau X, Lin J, Vanni-Mercier G, et al. Neurochemical and behavioral effects of ciproxifan, a potent histamine H3-receptor antagonist. J Pharmacol Exp Ther 1998;287:658–66.
- Hough LB, Nalwalk JW, Barnes WG, et al. A third life for burimamide. Discovery and characterization of a novel class of non-opioid analgesics derived from histamine antagonists. Ann N Y Acad Sci 2000;909:25–40.
- Nowak JZ. Effects of histamine H1- and H2-receptor antagonists on dopamine, noradrenaline and serotonin systems in rat brain. Pol J Pharmacol Pharm 1980;32:451–61.
- Jonsson KA, Eriksson SE, Kagevi I, et al. Cimetidine, but not oxmetidine, penetrates into the cerebrospinal fluid after a single intravenous dose. Br J Clin. Pharmac 1982;14:815–9.
- Clapp RH, Luckman SM. Proxyfan acts as a neutral antagonist of histamine H3 receptors in the feeding-related hypothalamic ventromedial nucleus. Br J Pharmacol 2012;167:1099–110.
- Mochizuki T, Jansen FP, Leurs R, et al. Brain penetration of the histamine H3 receptor antagonists thioperamide and clobenpropit in rat and mouse, determined with ex vivo [125I]iodophenpropit binding. Brain Res 1996;743:178–83.
- Boertje SB, Le Beau D, Ward S. Impromidine-induced changes in the permeability of the blood-brain barrier of normotensive and spontaneously hypertensive rats. Res Commun Chem Pathol Pharmacol 1990;69:249–52.
- Slugg PH, Haug MT, Pippenger CE. Ranitidine pharmacokinetics and adverse central nervous system reactions. Arch Intern Med 1992;152:2325–9.
- Hough LB, Nalwalk JW. Inhibition of morphine antinociception by centrally administered histamine H2 receptor antagonists. Eur J Pharmacol 1992;215:69–74.
- Calcutt CR, Ganellin CR, Griffiths R, et al. Zolantidine (SK&F 95282) is a potent selective brain-penetrating histamine H2-receptor antagonist. Br J Pharmacol 1988;93:69–78.
- Tighilet B, Trottier S, Lacour M. Dose- and duration-dependent effects of betahistine dihydrochloride treatment on histamine turnover in the cat. Eur J Pharmacol 2005;523:54–63.
- Esbenshade TA, Fox GB, Krueger KM, et al. Pharmacological properties of ABT-239 [4-(2-{2-[(2R)-2-methylpyrrolidinyl]ethyl}-benzofuran-5-yl)benzonitrile]: I. Potent and selective histamine H3 receptor antagonist with drug-like properties. J Pharmacol Exp Ther 2005;313:165–75.
- Wágner G, Mocking TAM, Arimont M, et al. 4-(3-Aminoazetidin-1-yl)pyrimidin-2-amines as High-Affinity Non-imidazole Histamine H3 Receptor Agonists with in Vivo Central Nervous System Activity. J Med Chem 2019;62:10848–66.
- Yamazaki M, Terasaki T, Yoshioka K, et al. Carrier-mediated transport of H1-antagonist at the blood-brain barrier: Mepyramine uptake into bovine brain capillary endothelial cells in primary monolayer cultures. Pharm Res 1994;11:975–8.
- Dettori I, Gaviano L, Melani A, et al. A selective histamine h4 receptor antagonist, JNJ7777120, is protective in a rat model of Transient Cerebral Ischemia. Front Pharmacol. 2018;9:e01231.
- Plisson C, Gunn RN, Cunningham VJ, et al. 11C-GSK189254: a selective radioligand for in vivo central nervous system imaging of histamine H3 receptors by PET. J Nucl Med 2009;50:2064–72.
- Nakamura T, Hiraoka K, Harada R, et al. Brain histamine H1 receptor occupancy after oral administration of desloratadine and loratadine. Pharmacol Res Perspect 2019;7:e00499.
- Medhurst AD, Briggs MA, Bruton G, et al. Structurally novel histamine H3 receptor antagonists GSK207040 and GSK334429 improve scopolamine-induced memory impairment and capsaicin-induced secondary allodynia in rats. Biochem Pharmacol 2007;73:1182–94.