
Abstract
Facile syntheses of 3-O-carbamoyl, -sulfamoyl, or -pivaloyl derivatives of 13α-oestrone and its 17-deoxy counterpart have been carried out. Microwave-induced, Ni-catalysed Suzuki–Miyaura couplings of the newly synthesised phenol esters with phenylboronic acid afforded 3-deoxy-3-phenyl-13α-oestrone derivatives. The carbamate and pivalate esters proved to be suitable for regioselective arylations. 2-(4-Substituted) phenyl derivatives were synthesised via Pd-catalysed, microwave-assisted C–H activation reactions. An efficient, one-pot, tandem methodology was elaborated for the introduction of the carbamoyl or pivaloyl group followed by regioselective C-2-arylation and subsequent removal of the directing group. The antiproliferative properties of the novel 13α-oestrone derivatives were evaluated in vitro on five human adherent cancer cell lines of gynaecological origin. 3-Sulfamate derivatives displayed substantial cell growth inhibitory potential against certain cell lines. The newly identified antiproliferative compounds having hormonally inactive core might be promising candidates for the design of more active anticancer agents.
Introduction
At the beginning of the 2000s, transition metals and, in particular, palladium came into focus in regard to the development of carbon–carbon coupling reactions. In principle, palladium is one of the few metals, which has a unique ability to activate various organic compounds. Essentially, two molecules are brought close to each other via forming metal–carbon bonds. The two partners couple together through establishing a new carbon–carbon single bondCitation1. The significance of palladium-catalysed C–C cross couplings was highlighted by the shared Nobel Prize in Chemistry in 2010. Richard F. Heck, Ei-ichi Negishi, and Akira Suzuki received the Prize for “palladium-catalysed cross couplings in organic synthesis”Citation2. These cross-coupling reactions have found remarkable utility in the synthesis of natural products and biologically active compounds. A wide range of their applications in the pharmaceutical industry increased their value even more. These coupling reactions are catalysed by zerovalent palladium, utilising organohalides as electrophilic and organometallic compounds as nucleophilic partners. Suzuki coupling facilitates the synthesis of biaryl compounds by employing organoboron nucleophilesCitation3. Recently, direct arylations have come in the focus of attention. Aromatic C–H activation allows the formation of biaryl derivatives by avoiding the use of an organometallic nucleophilic partner. Palladium-catalysed chelation-directed C–H activations of phenol derivatives utilise “directing groups (DGs)”, which facilitate and direct the substitution in a regioselective manner. Literature describes several nitrogen-containing DGs, such as N-heterocycles, amides, imines, and aminesCitation4–8. In order to broaden the applicability of chelation-directed C–H activations, other types of substrates have been applied as well. Phenol estersCitation9–13 offer a good alternative, owing to their usability under mild conditions (Scheme 1). Carbamates, sulfamates, or pivalates have already been examined as DGs in Pd-catalysed ortho-arylation of phenolsCitation4,Citation10. These DGs are readily prepared, robust, easily removable, and might show important biological properties on their own rightCitation10. It should be emphasised that these phenolic derivatives are generally stable under Pd-catalysed reaction conditionsCitation10–13. Beside the synthetic advantages of aryl carbamates, sulfamates, or pivalates in directed C–H activations, these phenol derivatives are popular coupling partners in cross-coupling reactions (Scheme 1)Citation9,Citation14. The oxygen-based electrophiles provide a greener alternative to commonly used organohalides, since the halide-containing waste is avoided. Concerning the hydrolytic properties of the O-acylated phenol derivatives, the robust pivalate esters appear to be the right choice. Pivalates are attractive candidates considering cost and they are useful in both academic and industrial applicationsCitation15. The pronounced stability of carbamates and sulfamates allows their application in coupling reactions using heterocyclic boronic acidsCitation9. Boronic acids are beneficial coupling partners in Suzuki–Miyaura reactions, owing to their wide availability, low toxicity, high stability, and broad functional group tolerance. Ni- or Fe-catalysed C(sp2)–C(sp2) couplings of the mentioned phenol derivatives with boronic acids or aryl trifluoroborates are known in the literatureCitation9,Citation14,Citation16. The methodology based on Ni-catalysed coupling of halogen-free substrates described recently, applying non-toxic boronic acids in green solvents, might find application even in industryCitation9.
Scheme 1. Formation of biaryl products by catalytic cross coupling or directed C–H activation.
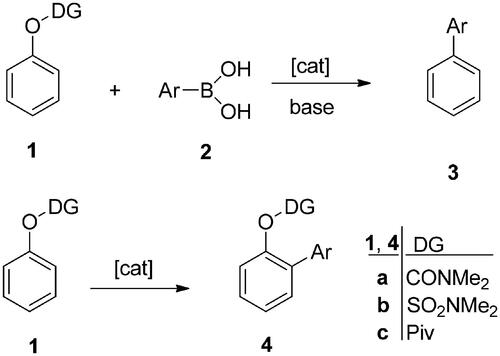
We have recently described Pd-catalysed cross couplings at the C-2 or C-4 position of the 13α-estrane core (Scheme 2)Citation17–20. Starting from steroidal aryl halides (5–7), microwave-assisted C–C, C–N, or C–P couplings were performed. The 2-(substituted 4-phenyl) moiety was attached to the A-ring of the steroid via C(sp2)–C(sp2) or C(sp2)–C(sp) coupling. Suzuki–Miyaura reactions were carried out using substituted phenylboronic acids as reagents leading to biphenyl derivatives (8–10, Y═Ph)Citation17. The indirect introduction of a phenyl group was performed in Sonogashira reactions through a linear C≡C linker, furnishing phenylethynyl derivatives (8–10, Y═PhC≡C)Citation18. The Buchwald–Hartwig aminations of the bromoarene substrates (5–7) with aniline derivatives led to phenylamino compounds (8–10, Y═PhNH)Citation19. The Hirao couplings facilitated the functionalisation of the A-ring with substituents differing in size and polarityCitation20. Steroidal phosphonates and tertiary phosphine oxide derivatives (8–10, Y═P(O)Z2) were synthesised. In addition to positions C-2 or C-4, the 3-OH group was also modified by synthesising 3-O-alkyl and 3-O-aralkyl derivatives. The majority of 13α-oestrone derivatives modified in the A-ring proved to be biologically active. Certain biphenyl derivatives displayed substantial antiproliferative action against human reproductive cancer cell linesCitation17. The 3-hydroxy-2-phenylethynyl compounds exerted marked inhibitory action against 17β-hydroxysteroid dehydrogenase 1 enzyme (17β-HSD1), which catalyses the last step of oestradiol biosynthesisCitation18. C-2 phosphonated derivatives proved to be dual organic anion-transporting polypeptide (OATP2B1) and 17β-HSD1 inhibitorsCitation20. OATP2B1 is a membrane transporter facilitating the cellular uptake of various endogenous compounds, drugs, and hormonal steroidsCitation21–23. Since increased uptake of hormones by OATP2B1 might lead to marked tumour proliferation, inhibition of the transporter might be a powerful antitumoural approachCitation24,Citation25. It should be underlined that the biological activity of 13α-oestrone derivatives modified at the A-ring greatly depends on the substitution pattern of the aromatic ring. 13α-Oestrone is readily available from natural oestrone, and this core-modification results in marked decrease in the oestrogenic effectCitation26,Citation27. The latter suggest that 13α-oestrone is a valuable starting material in the design of bioactive oestrone-based agents lacking oestrogenic side effects.
Scheme 2. Pd-catalysed cross couplings of 13α-estrones as aryl halides.
Encouraged by our recent results, here we report the synthesis of 13α-oestrone carbamates, sulfamates, and pivalates suitable for C–H activation and cross-coupling reactions. Our study included microwave-assisted, Pd-catalysed regioselective ortho-arylations of phenol esters. Phenylation at C-3 was performed via Ni-catalysed Suzuki–Miyaura coupling using phenylboronic acid. Evaluation of in vitro antiproliferative action of the newly synthesised compounds against five human reproductive cancer cell lines was also accomplished.
Materials and methods
Chemical syntheses, characterisation data of the reported compounds, as well as experimental conditions of antiproliferative assays performed are described in the Supporting Information.
Results and discussion
Chemistry
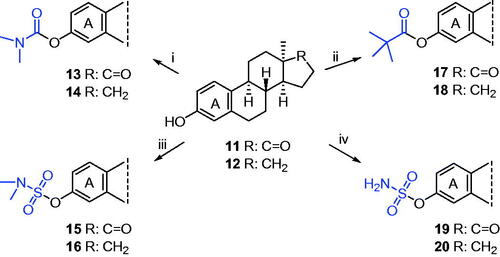
Modification of the phenolic hydroxyl function with DGs was carried out starting from 13α-oestrone (11) or its 17-deoxy counterpart (12, Scheme 3). Reactions of the steroidal substrates with N,N-dimethylcarbamoyl chloride using sodium hydride as a base afforded the desired carbamate esters (13, 14) in high yields. Pivalate and sulfamate derivatives (15–20) were synthesised in a microwave reactor. Esterifications of the substrates (11 or 12) using pivaloyl chloride as a reagent and triethylamine and (4-dimethylamino)pyridine as bases furnished the required pivalates (17, 18) in excellent yields. Microwave irradiation of the starting compounds (11 or 12) with sodium hydride and sulfamoyl or N,N-dimethylsulfamoyl chloride yielded the corresponding sulfamates (15, 16, 19, 20) in moderate to high yields.
Scheme 3. Syntheses of 13α-oestrone carbamates (13, 14), pivalates (17, 18), and sulfamates (15, 16, 19, 20). Reagents and conditions: (i) N,N-dimethylcarbamoyl chloride (1.0 equiv.), NaH (1.3 equiv.), DMF, rt, 30 min; (ii) pivaloyl chloride (1.2 equiv.), DMAP (0.1 equiv.), NEt3 (1.2 equiv.), CH2Cl2, MW, 40 °C, 1 h; (iii) N,N-dimethylsulfamoyl chloride (1.0 equiv.), NaH (1.3 equiv.), toluene, MW, 100 °C, 30 min; (iv) sulfamoyl chloride (1.0 equiv.), NaH (1.3 equiv.), toluene, MW, 75 °C or 100 °C, 30 min.
There is information in the literature about the syntheses of 3-deoxy-3-phenyl derivatives of natural oestrone, starting from its trifluoromethanesulfonyl or triazinyl esterCitation28–31. The cross-coupling reactions of the esters with boronic acid derivatives afforded the desired products. However, certain less common, but more robust phenol derivatives might also be utilised as substrates in cross-coupling reactions. Quasdorf et al. described Suzuki–Miyaura coupling reactions of phenol carbamates, sulfamates, and pivalatesCitation32,Citation33. The activation of a rather inert aryl C–O bond was achieved via NiCl2(PCy3)2-promoted transformations. The couplings with boronic acid reagents were accomplished under heating in the temperature range of 110–150 °C in 20–24 h.
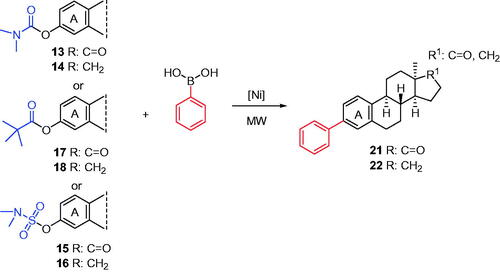
Here, we performed C(sp2)–C(sp2) couplings of 13α-oestrone esters according to the literature methodologyCitation32, but using microwave heating (Scheme 4). The Ni-catalysed couplings of steroidal substrates were carried out with phenylboronic acid as a reagent and K3PO4 as the base in various solvents. The Ni(II) precatalyst NiCl2(PCy3)2 is readily available and does not require glovebox handling. Conversions of carbamates (13, 14) or sulfamates (15, 16) were appreciably higher in toluene; however, reactions of pivalates (17, 18) proceeded more effectively in acetonitrile. Reaction times varied from 30 to 60 min, depending on the nature of the DG. The most significant improvement in reaction time was observed in the case of pivalate (17, 18) couplings. Owing to microwave irradiation, the reaction time could markedly be shortened, compared to those reported earlier applying other methodologies. It should be noted that 3-phenylation of 17-ketones took place with higher conversions than those of their 17-deoxy counterparts. The newly synthesised 3-deoxy-3-phenyl derivatives of 13α-oestrone (21, 22) might be interesting from biological point of view. Biochemical investigations of the derivatives bearing a large, apolar moiety at C-3 might provide valuable structure–activity relationship. The removal of one or both oxygen-containing moieties (17-keto and/or 3-OH) might have great influence on the biological activity of the compounds.
Scheme 4. Syntheses of 3-deoxy-3-phenyl-13α-estrones (21, 22). Reagents and conditions: from carbamates (13 or 14): NiCl2(PCy3)2 (10 mol%), phenylboronic acid (4 equiv.), K3PO4 (7.2 equiv.), toluene, MW, 130 °C, 1 h; from pivalates (17 or 18): NiCl2(PCy3)2 (10 mol%), phenylboronic acid (4 equiv.), K3PO4 (7.2 equiv.), MeCN, MW, 75 °C, 30 min; from sulfamates (15 or 16): NiCl2(PCy3)2 (10 mol%), phenylboronic acid (4 equiv.), K3PO4 (7.2 equiv.), toluene, MW, 130 °C, 1 h.
Phenylations of natural or 13α-oestrone derivatives at C-2 were earlier carried out via Pd-catalysed cross-coupling reactions of steroidal aryl halides with boronic acid reagentsCitation17,Citation34,Citation35. However, this strategy requires multiple steps and prefunctionalisation of the reagents. An alternative methodology, based on C–H activation, might circumvent the inconveniences of cross-coupling reactions, such as halogenation of the substrates and the use of organometallic nucleophilic coupling partners. Regioselective C–H bond arylation of 3-carbamoylestrone with aryl iodides was described by Bedford et al.Citation36 In this transformation, Pd(OAc)2 was used as a catalyst, AgOAc as a base in TFA solvent, and the mixture was stirred at 60 °C for 18 h. Arylation occurred at the C-2 ortho-position owing to the directing ability of the carbamate group. The removal of the DG was achieved in a subsequent step, by treatment with LiAlH4 followed by acidic hydrolysis. Note that not only 3-carbamoylestrone, but its 3-pivaloyl derivative also proved to be suitable for the regioselective arylation via C–H activation. Palladium-catalysed transformation of oestrone pivalate with the appropriate hypervalent iodonium salt gave the 2-tolyl derivativeCitation37. The reaction mixture was stirred at room temperature for 24 h.
With these considerations in mind, here we intended to explore the applicability of the earlier elaborated C–H activation methodologies on the hormonally inactive 13α-oestrone, utilising microwave irradiation (Scheme 5). The newly synthesised 3-carbamoyl (13), -pivaloyl (17), or -sulfamoyl (15) 13α-oestrone derivatives were selected as substrates. The rational of this choice included both the DG ability of the mentioned 3-O-moieties and their important potential biological activities in their own rightCitation10,Citation38,Citation39. First, the transformation of 3-carbamoyl derivative 13 was carried out. As a first attempt, the combination of aryl iodide (4 equiv.) and silver acetate (2 equiv.) was used with Pd(OAc)2 (10 mol%) catalyst in TFA solvent. In earlier studies, this methodology proved to be suitable for ortho-arylation of anilides and O-carbamoylphenolsCitation5,Citation10,Citation36. The reaction of compound 13 with iodobenzene was performed under both conventional heating and in a microwave reactor at different temperatures. Microwave irradiation markedly improved the efficiency of the C–H activations, since conventional heating at 50 °C gave only moderate yields of the desired 23a product. Microwave heating at 50 °C for 1 h, in turn, afforded the best results in site-selective 2-phenylation with both high product yields and high regio- and chemoselectivity. Formation of neither the 4-phenyl regioisomer nor 2,4-bis products was observed. The explanation of exclusive 2-selectivity might be the steric hindrance of the B-ring. Similar yields were obtained under the same reaction conditions (time and temperature) by replacing AgOAc (2 equiv.) with Cs2CO3 (2 equiv.) or K2CO3 (2 equiv.). Considering the price of the mentioned bases, K2CO3 is the best choice.
Scheme 5. Syntheses of 2-(4-substituted phenyl)-13α-oestrone derivatives (23a,b–25a,b). Reagents and conditions: (i) carbamate 13, Pd(OAc)2 (10 mol%), iodobenzene (4 equiv.) or 1-chloro-4-iodobenzene (4 equiv.), K2CO3 (2 equiv.), TFA, MW, 50 °C, 60 min; (ii) pivalate 17, Pd(OAc)2 (10 mol%), iodobenzene (4 equiv.) or 1-chloro-4-iodobenzene (4 equiv.), K2CO3 (2 equiv.), TFA, MW, 50 °C, 60 min; (iii) sulfamate 15, Pd(OAc)2 (10 mol%), iodobenzene (4 equiv.) or 1-chloro-4-iodobenzene (4 equiv.), K2CO3 (2 equiv.), TFA, MW, 50 °C, 60 min; (iv) 2-phenyl carbamate 23a, TFA, MW, 150 °C, 30 min; (v) 2-phenyl pivalate 24a, TFA, MW, 100 °C, 30 min; (vi) 2-phenyl sulfamate 25a, TFA, MW, 150 °C, 30 min; (vii) N,N-dimethylcarbamoyl chloride (1.0 equiv.), NaH (1.3 equiv.), DMF, rt, 30 min; (viii) pivaloyl chloride (1.2 equiv.), DMAP (0.1 equiv.), NEt3 (1.2 equiv.), CH2Cl2, MW, 40 °C, 1 h; (ix) N,N-dimethylsulfamoyl chloride (1.0 equiv.), NaH (1.3 equiv.), toluene, MW, 100 °C, 30 min.
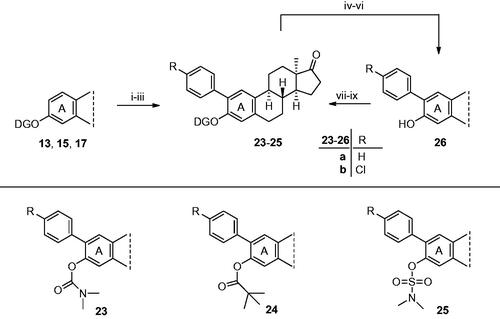
When the reaction mixture submitted to a 1-h irradiation at 50 °C was additionally exposed to a treatment at 150 °C for 1 h, complete removal of the DG occurred. Consequently, our present microwave-assisted methodology allows one-pot ortho-arylation and removal of the DG in a tandem reaction.
Next, we explored the reactions of 3-pivalate 17. Substrate 17 was subjected to C–H activation by utilising the microwave-assisted methodology elaborated here. The desired 2-phenyl derivative (24a) was obtained only in moderate yield. The pivalate ester-directed approach seemed to be less efficient than that of the carbamate. According to the literature, in the reaction of pivalates with hypervalent iodonium salts, acyloxy-directed Pd(II)-insertion into the C–H bond of phenol derivatives might be promoted with the use of TFA as solventCitation4. The acid is capable of tuning the electrophilicity of the transition metal. However, in the case of 13α-oestrone pivalate 17, phenylation with iodobenzene did not meet our expectations concerning product yield. If the reaction mixture was exposed to an additional microwave irradiation at 100 °C for 45 min, the pivaloyl DG could completely be removed. N,N-Dimethylsulfamoyl derivative 15 was also subjected to microwave-assisted C–H activations with Pd(OAc)2 as catalyst; however, no reaction occurred.
Next, we focussed on the synthesis of the 2-phenyl derivatives (23–25) via an alternative approach. The facile, one-pot, microwave-assisted methodology furnishing the 3-deprotected 2-phenyl derivative (26a) allowed the synthesis of the desired 13α-oestrone derivatives (23a–25a) in an indirect manner. 2-Phenyl-13α-oestrone (26a) was synthesised from its carbamate derivative (23a) according to the MW procedure described above, and a pivalate or a sulfamate DG was introduced in the next step. This two-step reaction route provided the 2-phenyl-3-protected derivatives (24a, 25a) in high overall yields. Based on our recent publication, concerning the promising antiproliferative action of 2-(4-chlorophenyl)-13α-oestrone (26b)Citation17, arylations with 1-chloro-4-iodobenzene were also performed. 2-(4-Chlorophenyl) derivatives (23b–25b) were synthesised via the two-step reaction pathway described above. C–H activations with Pd(OAc)2 as the catalyst, starting from carbamate 13 or pivalate 17 and 1-chloro-4-iodobenzene as the reagent, under the above-described microwave conditions, delivered the desired 2-(4-chlorophenyl) derivatives (23b–25b) in high yields. The presence of chlorine at para position relative to iodine seemed to be advantageous concerning the yields of the desired products. The indirect approach for the synthesis of 3-protected 2-substituted phenyl derivatives (23b–25b) included the introduction of the DGs onto the phenolic hydroxy function of compound 26b, synthesised from carbamate 13 (Scheme 5, vi–viii).
Pharmacology
The antiproliferative properties of the newly synthesised compounds (13–25) and their parent derivatives 11 or 12 were investigated in vitro on a panel of human adherent cancer cell lines representing the most relevant cancers of gynaecological origin. The panel included different breast (MCF-7 and MDA-MB-231), cervical (HeLa and SiHa), and ovarian (A2780Citation40,Citation41) cancer cell lines. While MCF-7 is an oestrogen receptor positive cell line, MDA-MB-231 is lacking receptors for oestrogens, progesterone, and HER2 growth factorCitation41–44. HeLa and SiHa differ in their HPV status: they represent HPV-18 and HPV-16 positive cases, respectivelyCitation45,Citation46. The determination of cell growth inhibitory action of test compounds was performed by MTT assayCitation47. The cancer selectivity of the most effective derivatives was additionally determined by the same method using NIH/3T3 mouse fibroblast cells.
We have recently reported the synthesis of potent antiproliferative core-modified oestrone derivatives obtained by inversion of the C-13 configuration or opening the D-ringCitation17,Citation48–55. Certain test compounds displayed submicromolar IC50 values, occasionally with high cell line selectivity. The 13α- and/or D-seco compounds possessed modifications mainly at positions C-2, C-4, and/or 3-OH. It was shown that the nature, size, and polarity of the introduced substituents greatly influence the cell growth inhibitory properties of the compounds. 3-Hydroxy derivatives proved to be generally less potent than their 3-ether counterparts. Introduction of a benzyl or benzyltriazolyl group onto 3-OH improved the antiproliferative properties substantiallyCitation51–55. Having found functional groups responsible for the biological effect, we turned our attention to attach all crucial structural elements to the hormonally inactive 13α-oestrone core. As a result, certain derivatives were identified as dually acting agents, possessing both enzyme inhibitory and antiproliferative propertiesCitation20,Citation55. We demonstrated that a few of our potent anticancer compounds exert a direct effect on the tubulin-microtubule system by increasing the rate of tubulin polymerisationCitation50,Citation52,Citation55. These compounds might give an important basis for the design of oestrone-based potent anticancer agents, lacking oestrogenic activity.
Concerning the modification of the phenolic hydroxy function of oestrone derivatives, important structure–activity relationships are described in the literature. Introduction of a sulfamoyl moiety onto 3-OH of oestrogens resulted in compounds possessing remarkable biological activitiesCitation56–59. Their aryl O-sulfamate pharmacophore facilitated steroid sulfatase inhibitory activity, leading to anticancer effect. Nevertheless, certain oestrone sulfamates proved to be active against triple-negative breast cancer owing to their triple effectCitation60. Beside their microtubule disruptor properties, they displayed proapoptotic and anti-angiogenic actionCitation60. Furthermore, carbamoylation of oestrogens at C-3-O afforded N-mustard carbamates as potent cytotoxic agents against prostatic adenocarcinomaCitation61.
In addition to modifications at C-3, introduction of substituents onto C-2 of the estrane core led to potent anticancer agents of high value. 2-Halogenated and 2-phenylated derivatives were identified as efficient inhibitors against enzymes involved in the metabolism or biosynthesis of oestrogensCitation34,Citation62. It was highlighted that 2-phenyl derivatives display potent CYP1B1 inhibitory actionCitation34. CYP1B1 is responsible for the bioactivation of certain procarcinogens, thereby catalysing the synthesis of mutagenic compounds. The combination of a CYP1B1 inhibitor with an anticancer agent might be suitable for the treatment of drug-resistant cancersCitation63,Citation64. We recently described our results with respect to the antiproliferative action of 2- or 4-(substituted phenyl)-13α-estrones and their 3-benzyl ethersCitation17. 2-(4-Chlorophenyl)-13α-oestrone was found to be the most potent compound with low micromolar cell growth inhibitory action against MCF-7 and HeLa cell lines. An important structure–activity relationship was found, since the 3-benzyl ether counterpart proved to be ineffective. A substantial distinction was observed between the two pairs of breast and cervical cancer cell lines, concerning the cell growth inhibitory action. The triple negative breast and the HPV-16 positive cervical cell lines seemed to be less sensitive to the test compound.
Taking into consideration of the above-mentioned promising pharmacological properties of natural and 13α-oestrone derivatives, here we combined the essential structural elements with the aim of getting more potent antiproliferative agents and important structure–activity relationship data. The key pharmacophores identified on the natural estrane core were transferred to the hormonally inactive 13α-estone basic compound. Before testing the newly designed derivatives, the cell growth inhibitory properties of the starting 13α-oestrone (11) and its 17-deoxy counterpart (12) were compared. As it can be seen in , compound 12 lacking the 17-keto group displayed stronger inhibitory actions than 13α-oestrone 11. As a consequence, it seemed rational to test the influence of the modification at the C-3-O group on both 17-keto and 17-deoxy core structures on the antitumoural action. Carbamoylation leading to compounds 13 or 14 did not improve the cell growth inhibitory properties. However, introduction of a pivaloyl moiety seemed to lower the IC50 values of 17-deoxy derivative 18 (against MCF-7 and HeLa). The most potent compound group is represented by the 3-O-sulfamoyl derivatives. Sulfamates bearing NH2-function (19, 20) generally exerted less potent action, than the N,N-dimethyl derivatives (15, 16). The latter structural element, combined with a 17-keto group resulted in a highly potent derivative (15). Phenylations at C-3 via Suzuki–Miyaura coupling afforded 3-deoxy-3-phenyl derivatives (21, 22), possessing weak antiproliferative action. These results indicate that introduction of a large, apolar moiety onto C-3 by the simultaneous removal of the oxygen-containing moiety is rather disadvantageous concerning the cell growth inhibitory action on the tested cell lines. Summarising the results obtained for C-3-O-modified 13α-oestrone derivatives, it can be emphasised, that the determined antiproliferative effect greatly depends on the nature of introduced moieties. This screening suggests that compound 15 is the most promising candidate for further evaluations.
Table 1. Antiproliferative properties of the synthesised compounds.
Determination of the antiproliferative activities of the newly synthesised compounds was continued by testing 2-(substituted 4-phenyl)-13α-oestrone derivatives (23a,b–25a,b) bearing different DGs. In the carbamate compound group (13; 23a,b), phenylations did not improve the cytostatic properties. 2-Phenyl pivalate (24a) displayed somewhat higher effect on MCF-7 cell line than that of its 2-H counterpart (17). However, the sulfamate compound set (15; 25a,b) provides interesting correlation results. It is worth comparing the data obtained for the three 17-keto sulfamates, namely, compound 15 with no modification at C-2 and the two 2-phenyl derivatives (25a,b). There was no marked difference in the effects exerted on the MCF-7 cell line: each compound displayed high potency with IC50 values in the low micromolar range. HeLa seemed to be correspondingly sensitive to the sulfamates, but the cell growth inhibitory activity was improved by introducing the 4-chlorophenyl moiety onto C-2. The most significant improvement caused by 4-chlorophenylation was observed on SiHa and A2780 cell lines. The growth of HPV-16 positive cervical cells was substantially inhibited by compound 25b in a very low micromolar range. To the best of our knowledge, this is the first 13α-oestrone derivative with such a high potency against SiHa described in the literature. This test compound, exerting outstanding activity against both HPV-18 and HPV-16 positive cervical cancers, might be of great importance in the design of anticancer agents targeting cervical carcinomas, since the majority of these carcinomas are caused by these two types of HPV. For invasive cervical cancer, HPV-16 is the most prevalent type (approximately 60%), HPV-18 is the second (15%), and HPV-45 is the third most common typeCitation65.
With the aim of getting preliminary results concerning the tumour selectivity of the detected action, certain test compounds were additionally tested against a mouse fibroblast cell line (NIH/3T3). 17-Deoxy-13α-oestrone 12 exerted the most potent antiproliferative action against the NIH/3T3 cell line with an IC50 value of 15.29 µM. However, the rest of the test compounds influenced the growth of the fibroblasts scarcely (less than 51% inhibition even at 30 μM). It can be stated, that the NIH/3T3 cells are more sensitive to reference agent cisplatin than to the 13α-oestrone derivatives tested.
Conclusions
We introduced N- and/or O-containing DGs onto the phenolic 3-OH function of 13α-oestrone and its 17-deoxy counterpart. The resulting carbamate, pivalate, or sulfamate esters proved to be suitable for regioselective ortho-arylations via Pd-catalysed C–H activation. A mild and efficient microwave-assisted methodology was elaborated. Arylation of a carbamate or pivalate and the removal of the DG were achieved via a one-pot, tandem, microwave procedure. The newly synthesised phenol esters were suitable electrophilic substrates in microwave-induced, Ni-catalysed Suzuki–Miyaura couplings with phenylboronic acid as a nucleophilic reagent. Biphenyl derivatives formed by C(sp2)–C(sp2) couplings represent an interesting novel class of 13α-estrane derivatives lacking one or two oxygen-containing functionalities. The antitumoural properties of the newly synthesised 13α-oestrone derivatives were determined in vitro on five human cancer cell lines of gynaecological origin. Certain potent antiproliferative compounds were identified and important structure–activity relationships were established. Sulfamate derivatives seemed to be superior concerning their substantial antiproliferative potential. 2-(4-Chlorophenyl)-13α-oestrone sulfamate 25b displayed outstanding growth inhibitory action against the two cervical cancer cell lines with different HPV-status. The presence of an N,N-dimethylsulfamate pharmacophore together with the 2-(4-chlorophenyl) moiety improved the antitumoural action. Considering that HPV-16 and HPV-18 play a causative role in the majority of cervical cancer cases, newly identified 25b with its hormonally inactive core might be a promising candidate in the design of new anticancer agents acting selectively.
Supplemental Material
Download PDF (327.4 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Molnár Á. Palladium-catalyzed coupling reactions: practical aspects and future developments. Weinheim: Wiley-VCH; 2013.
- Seechurn CCCJ, Kitching MO, Colacot TJ, Snieckus V. Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize. Angew Chem Int Ed Engl 2012;51:5062–85.
- Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev 1995;95:2457–83.
- Xiao B, Fu Y, Xu J, et al. Pd(II)-catalyzed C–H activation/aryl-aryl coupling of phenol esters. J Am Chem Soc 2010;132:468–9.
- Daugulis O, Zaitsev VG. Anilide ortho-arylation by using C–H activation methodology. Angew Chem Int Ed Engl 2005;44:4046–8.
- Shi BF, Maugel N, Zhang YH, Yu JQ. Pd(II)-catalyzed enantioselective activation of C(sp2)-H and C(sp3)-H bonds using monoprotected amino acids as chiral ligands. Angew Chem Int Ed Engl 2008;47:4882–6.
- Desai LV, Hull KL, Sanford MS. Palladium-catalyzed oxygenation of unactivated sp3 C–H bonds. J Am Chem Soc 2004;126:9542–3.
- Li JJ, Mei TS, Yu JQ. Synthesis of indolines and tetrahydroisoquinolines from arylethylamines by Pd(II)-catalyzed C–H activation reactions. Angew Chem Int Ed Engl 2008;47:6452–5.
- Mesganaw T, Garg NK. Ni- and Fe-catalyzed cross-coupling reactions of phenol derivatives. Org Proc Res Dev 2013;17:29–39.
- Bedford RB, Webster RL, Mitchell C. Palladium-catalysed ortho-arylation of carbamate-protected phenols. J Org Biomol Chem 2009;7:4853–7.
- Zhao X, Yeung CS, Dong VM. Palladium-catalyzed ortho-arylation of O-phenylcarbamates with simple arenes and sodium persulfate. J Am Chem Soc 2010;132:5837–44.
- Nishikata T, Abela AR, Huang S, Lipshutz BH. Cationic palladium(II) catalysis: C–H activation/Suzuki–Miyaura couplings at room temperature. J Am Chem Soc 2010;132:4978–9.
- Yamazaki K, Kawamorita S, Ohmiya H, Sawamura M. Directed ortho borylation of phenol derivatives catalyzed by a silica-supported iridium complex. Org Lett 2010;12:3978–81.
- Baghbanzadeh M, Pilger C, Kappe CO. Rapid nickel-catalyzed Suzuki–Miyaura cross-couplings of aryl carbamates and sulfamates utilizing microwave heating. J Org Chem 2011;76:1507–10.
- Guan BT, Wang Y, Li BJ, et al. Biaryl construction via Ni-catalyzed C–O activation of phenolic carboxylates. J Am Chem Soc 2008;130:14468–70.
- Molander GA, Beaumard F. Nickel-catalyzed C–O activation of phenol derivatives with potassium heteroaryltrifluoroborates. Org Lett 2010;12:4022–5.
- Jójárt R, Ali H, Horváth G, et al. Pd-catalyzed Suzuki–Miyaura couplings and evaluation of 13α-estrone derivatives as potential anticancer agents. Steroids 2020;164:108731.
- Bacsa I, Jójárt R, Wölfling J, et al. Synthesis of novel 13α-estrone derivatives by Sonogashira coupling as potential 17β-HSD1 inhibitors. Beilstein J Org Chem 2017;13:1303–9.
- Bacsa I, Szemerédi D, Wölfling J, et al. The first Pd-catalyzed Buchwald–Hartwig aminations at C-2 or C-4 in the estrone series. Beilstein J Org Chem 2018;14:998–1003.
- Jójárt R, Pécsy S, Keglevich G, et al. Pd-catalyzed microwave-assisted synthesis of phosphonated 13α-estrones as potential OATP2B1, 17β-HSD1 and/or STS inhibitors. Beilstein J Org Chem 2018;14:2838–45.
- Hagenbuch B, Stieger B. The SLCO (former SLC21) superfamily of transporters. Mol Aspects Med 2013;34:396–412.
- Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol 2009;158:693–705.
- König J, Cui Y, Nies AT, Keppler D. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am J Physiol Gastrointest Liver Physiol 2000;278:156–64.
- Giacomini KM, Huang SM, Tweedie D. Membrane transporters in drug development. NIH Public Access 1937;9:215–36.
- Kovacsics D, Patik I, Özvegy-Laczka C. The role of organic anion transporting polypeptides in drug absorption, distribution, excretion and drug–drug interactions. Expert Opin Drug Metab Toxicol 2017;13:409–24.
- Schönecker B, Lange C, Kotteritzsch M, et al. Conformational design for 13alpha-steroids. J Org Chem 2000;65:5487–97.
- Ayan D, Roy J, Maltais R, Poirier D. Impact of estradiol structural modifications (18-methyl and/or 17-hydroxy inversion of configuration) on the in vitro and in vivo estrogenic activity. J Steroid Biochem Mol Biol 2011;127:324–30.
- Huang L, Ackerman LKG, Kang K, et al. LiCl-accelerated multimetallic cross-coupling of aryl chlorides with aryl triflates. J Am Chem Soc 2019;141:10978–83.
- Iranpoor N, Panahi F, Jamedi F. Nickel-catalyzed one-pot synthesis of biaryls from phenols and arylboronic acids via C–O activation using TCT reagent. J Organomet Chem 2015;781:6–10.
- Li XJ, Zhang JL, Geng Y, Jin Z. Nickel-catalyzed Suzuki–Miyaura coupling of heteroaryl ethers with arylboronic acids. J Org Chem 2013;78:5078–84.
- Ciattini PG, Morera E, Ortar G. Palladium-catalyzed cross-coupling reactions of vinyl and aryl triflates with tetraarylborates. Tetrahedron Lett 1992;33:4815–8.
- Quasdorf KW, Tian X, Garg NK. Cross-coupling reactions of aryl pivalates with boronic acids. J Am Chem Soc 2008;130:14422–3.
- Quasdorf KW, Antoft-Finch A, Liu P, et al. Suzuki–Miyaura cross-coupling of aryl carbamates and sulfamates: experimental and computational studies. J Am Chem Soc 2011;133:6352–63.
- Dutour R, Roy J, Cortés-Benítez F, et al. Targeting cytochrome P450 (CYP) 1B1 enzyme with four series of A-ring substituted estrane derivatives: design, synthesis, inhibitory activity, and selectivity. J Med Chem 2018;61:9229–45.
- Ivanov A, Ejaz SA, Shah SJA, et al. Synthesis, functionalization and biological activity of arylated derivatives of (+)-estrone. Bioorg Med Chem 2017;25:949–62.
- Bedford RB, Brenner PB, Durrant SJ, et al. Palladium-catalyzed ortho-arylation of carbamate-protected estrogens. J Org Chem 2016;81:3473–8.
- Xiao B, Gong T-J, Liu Z-J, et al. Synthesis of dibenzofurans via palladium-catalyzed phenol-directed C–H activation/C–O cyclization. J Am Chem Soc 2011;133:9250–3.
- Teiber JF, Billecke SS, La Du BN, Draganov DI. Estrogen esters as substrates for human paraoxonases. Arch Biochem Biophys 2007;461:24–9.
- Gieling RG, Babur M, Mamnani L, et al. Antimetastatic effect of sulfamate carbonic anhydrase IX inhibitors in breast carcinoma xenografts. J Med Chem 2012;55:5591–6000.
- Lee YK, Lim J, Yoon SY, et al. Promotion of cell death in cisplatin-resistant ovarian cancer cells through KDM1B-DCLRE1B modulation. Int J Mol Sci 2019;20:2443.
- Schelz Z, Ocsovszk I, Bózsity N, et al. Antiproliferative effects of various furanoacridones isolated from Ruta graveolens on human breast cancer cell lines. Anticancer Res 2016;36:2751–8.
- Chavez KJ, Garimella SV, Lipkowitz S. Triple negative breast cancer cell lines: one tool in the search for better treatment of triple negative breast cancer. Breast Dis 2010;32:35–48.
- Anders CK, Zagar TM, Carey LA. The management of early-stage and metastatic triple-negative breast cancer: a review. Hematol Oncol Clin North Am 2013;27:737–49.
- Suba Z. Triple-negative breast cancer risk in women is defined by the defect of estrogen signaling: preventive and therapeutic implications. Onco Targets Ther 2014;7:147–64.
- Goodman A. HPV testing as a screen for cervical cancer. BMJ 2015;350:h2372.
- Ghittoni R, Accardi R, Chiocca S, Tommasino M. Role of human papillomaviruses in carcinogenesis. Ecancermedicalscience 2015;9:526.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Mernyák E, Kovács I, Minorics R, et al. Synthesis of trans-16-triazolyl-13α-methyl-17-estradiol diastereomers and the effects of structural modifications on their in vitro antiproliferative activities. J Steroid Biochem Mol Biol 2015;150:123–34.
- Szabó J, Pataki Z, Wölfling J, et al. Synthesis and biological evaluation of 13α-estrone derivatives as potential antiproliferative agents. Steroids 2016;113:14–21.
- Bózsity N, Minorics R, Szabó J, et al. Mechanism of antiproliferative action of a new d-secoestrone-triazole derivative in cervical cancer cells and its effect on cancer cell motility. J Steroid Biochem Mol Biol 2017;165:247–57.
- Szabó J, Jerkovics N, Schneider G, et al. Synthesis and in vitro antiproliferative evaluation of C-13 epimers of triazolyl-d-secoestrone alcohols: the first potent 13α-d-secoestrone derivative. Molecules 2016;21:611.
- Mernyák E, Szabó J, Bacsa I, et al. Syntheses and antiproliferative effects of d-homo- and d-secoestrones. Steroids 2014;87:128–36.
- Mernyák E, Fiser G, Szabó J, et al. Synthesis and in vitro antiproliferative evaluation of d-secooxime derivatives of 13β- and 13α-estrone. Steroids 2014;89:47–55.
- Kiss A, Mernyák E, Wölfling J, et al. Stereoselective synthesis of the four 16-hydroxymethyl-3-methoxy- and 16-hydroxymethyl-3-benzyloxy-13α-estra-1,3,5(10)-trien-17-ol isomers and their antiproliferative activities. Steroids 2018;134:67–77.
- Jójárt R, Tahaei SAS, Trungel-Nagy P, et al. Synthesis and evaluation of anticancer activities of 2- or 4-substituted 3-(N-benzyltriazolylmethyl)-13α-oestrone derivatives. J Enzyme Inhib Med Chem 2021;36:58–67.
- Elger W, Schwarz S, Hedden A, et al. Sulfamates of various estrogens are prodrugs with increased systemic and reduced hepatic estrogenicity at oral application. J Steroid Biochem Mol Biol 1995;55:395–403.
- Woo LW, Howarth NM, Purohit A, et al. Steroidal and nonsteroidal sulfamates as potent inhibitors of steroid sulfatase. J Med Chem 1998;41:1068–83.
- Howarth NM, Purohit A, Reed MJ, Potter BV. Estrone sulfamates: potent inhibitors of estrone sulfatase with therapeutic potential. J Med Chem 1994;37:219–21.
- Thomas MP, Potter BV. Discovery and development of the aryl O-sulfamate pharmacophore for oncology and women's health. J Med Chem 2015;58:7634–58.
- Andring JT, Dohle W, Tu C, et al. 3,17β-Bis-sulfamoyloxy-2-methoxyestra-1,3,5(10)-triene and nonsteroidal sulfamate derivatives inhibit carbonic anhydrase IX: structure–activity optimization for isoform selectivity. J Med Chem 2019;62:2202–12.
- Petrow V, Padilla GM. Design of cytotoxic steroids for prostate cancer. Prostate 1986;9:169–82.
- Bacsa I, Herman BE, Jójárt R, et al. Synthesis and structure–activity relationships of 2- and/or 4-halogenated 13β- and 13α-estrone derivatives as enzyme inhibitors of estrogen biosynthesis. J Enzyme Inhib Med Chem 2018;33:1271–82.
- Murray GI, Taylor MC, McFadyen MC, et al. Tumor-specific expression of cytochrome P450 CYP1B1. Cancer Res 1997;57:3026–31.
- Cui J, Li S. Inhibitors and prodrugs targeting CYP1: a novel approach in cancer prevention and therapy. Curr Med Chem 2014;21:519–52.
- Rumfield SC, Roller N, Pellom ST, et al. Therapeutic vaccines for HPV-associated malignancies. Immunotargets Ther 2020;9:167–200.