
Abstract
Herein, a new wave of bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxaline derivatives have been successfully designed and synthesised. The synthesised derivatives were biologically investigated for their cytotoxic activities against HepG2 and MCF-7. Also, the tested compounds were further examined in vitro for their VEGFR-2 inhibitory activity. The most promising derivative 23j was further investigated for its apoptotic behaviour in HepG2 cell lines using flow cytometric and western-plot analyses. Additional in-silico studies were performed to predict how the synthesised compounds can bind to VEGFR-2 and to determine the drug-likeness profiling of these derivatives. The results revealed that compounds 23a, 23i, 23j, 23l, and 23n displayed the highest antiproliferative activities against the two cell lines with IC50 values ranging from 6.4 to 19.4 µM. Furthermore, compounds 23a, 23d, 23h, 23i, 23j, 23l, 23 m, and 23n showed the highest VEGFR-2 inhibitory activities with IC50 values ranging from 3.7 to 11.8 nM, comparing to sorafenib (IC50 = 3.12 nM). Moreover, compound 23j arrested the HepG2 cell growth at the G2/M phase and induced apoptosis by 40.12% compared to the control cells (7.07%). As well, such compound showed a significant increase in the level of caspase-3 (1.36-fold), caspase-9 (2.80-fold), and BAX (1.65-fold), and exhibited a significant decrease in Bcl-2 level (2.63-fold).
1. Introduction
Cancer is a rebound system represented by unrestricted cell growthCitation1. Cancer originates in the humanoid body from the buildup of genetic and epigenetic variations in the normal cellsCitation2. In spite of the huge efforts directed towards cancer treatment and prevention, cancer remains one of the foremost public health problems all over the worldCitation3. Consequently; increasing interest in the current medicinal chemistry has been dedicated to the design and synthesis of more effective anticancer agents with low side effectsCitation4.
Angiogenesis is a multi-stage process to produce new vessels from quiescent pre-existing onesCitation5–7. It is interrelated to several physiological functions and also, it is a fundamental step in numerous diseases including cancerCitation8. Furthermore, it has a significant role in tumour progression and developmentCitation9,Citation10.
At present, there are main approaches in targeting angiogenesis that has been verified in clinical trials and officially approved in clinical practiceCitation11: i) monoclonal antibodies which bind to vascular endothelial growth factor-A (VEGF-A) e.g. bevacizumabCitation12; ii) VEGF-trap e.g. aflibercept which binds to VEGF-A, VEGF-B, and placental growth factor (PGF)Citation13; iii) monoclonal antibodies targeting VEGF receptors and block the binding of natural VEGFR ligands e.g. ramucirumab, iv) tyrosine kinase inhibitors (TKIs) which are drugs that inhibit the kinase activity of VEGF receptors through binding to the ATP binding site. Hence, TKIs hinder the phosphorylation of the tyrosine residue and subsequent transmission of signalling down the intercellular pathway. Among this class of agents, sorafenib 1 and sunitinib 2 are the prototypesCitation14. There have been numerous known tyrosine kinase receptors such as vascular endothelial growth factor receptors (VEGFRs) and endothelial growth factor receptors (EGFRs)Citation15.
It has been stated that the vascular endothelial growth factor (VEGF) can stimulate the production and movement of vascular endothelial cells and regulate the formation of blood vesselsCitation12,Citation16. There are three main vascular endothelial growth factor receptors (VEGFR-1, VEGFR-2, and VEGFR-3), which are strategic intermediates in angiogenesis and in the construction of new networks of blood vessels required to hoard oxygen and nourishment for cancer growthCitation17.
Vascular endothelial growth factor receptor-2 (VEGFR-2) is the most critical regulator of angiogenic factors that plays a significant role in tumour survival, angiogenesis, and migrationCitation18. Binding of VEGFR-2 to VEGF leads to inspiration of downstream signalling pathway and certain endothelial reactions, such as improved permeability of vascular cells and increased endothelial cell multiplying and propagation, consequently, lead to angiogenesisCitation19. Hence, blockage of VEGF/VEGFR-2 system is considered a favourable approach for anti-angiogenic therapy in retarding cancer growthCitation20,Citation21. Additionally, VEGFR-2 has been confirmed to motivate apoptosis in cancer cells which synergistically enhances the antitumor effectCitation22–25.
Apoptosis has two major pathways; the intrinsic pathway which is controlled by the Bcl-2 family (involve BAX and Bcl-2) and the extrinsic pathway which is controlled by a subgroup of tumour necrosis factor receptors superfamily (TNFR)Citation26. Immediately as the apoptosis process is instigated, caspases, a family of protease enzymes, are activated. Caspases are classified into initiator caspases as caspases 8 and 9, effector caspases as caspases 3, 6, and, 7 and inflammatory caspases as caspases 1, 4, 5, 11, and 12Citation26. Caspase 9 is the initiator of intrinsic apoptosis while caspase 3 plays a central role in the execution phase of apoptosisCitation27. Cancer cells can avoid apoptosis by hindering caspase function, decreasing BAX expression level or increasing the gene expression level of anti-apoptotic Bcl-2Citation27,Citation28.
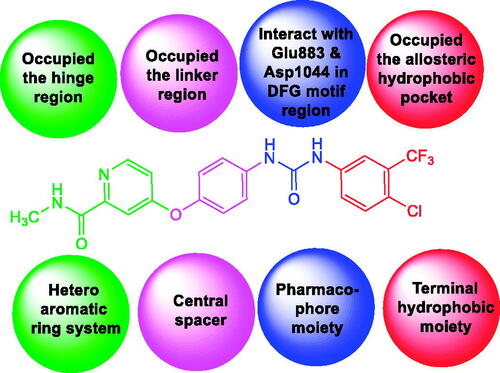
A literature study revealed that VEGFR-2 inhibitors have main pharmacophoric featuresCitation29–31. (i) A flat heteroaromatic ring system that can accommodate the hinge regionCitation29. (ii) A central spacer moiety that can occupy the linker region between the hinge region and DFG domain of the enzymeCitation32. (iii) A pharmacophore moiety consists of an H-bond acceptor (HBA) and an H-bond donor (HBD). Such pharmacophore moiety can interact with the two vital amino acid residues (Asp1044 and Glu883) in the DFG motif regionCitation33. (iv) A terminal hydrophobic moiety that occupies the allosteric hydrophobic pocket via numerous hydrophobic interactionsCitation34 (.
Figure 1. The essential structural requirements of reported VEGFR-2 inhibitors.
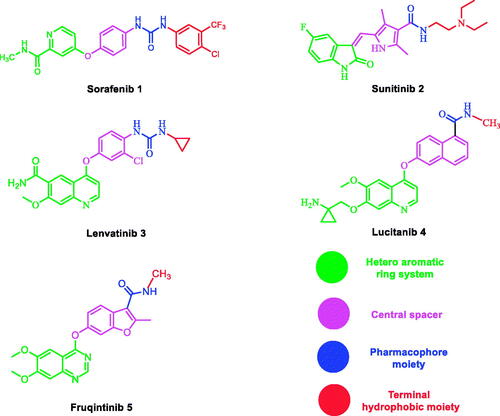
Also, a literature review revealed that many VEGFR-2 inhibitors have been approved as potent anticancer agents such as sorafenib 1Citation35, Sunitinib 2Citation36, lenvatinib 3Citation37,Citation38, lucitanib 4Citation37,Citation38, and fruquinitinib 5Citation39. All of them fulfilled the main pharmacophoric features of VEGFR-2 inhibitors (.
Figure 2. Structures of some representative VEGFR-2 inhibitors.
1.1. Rationale of molecular design
Based on facts mentioned above and in the extension of our efforts to discover novel molecules with potential anticancer activityCitation5,Citation40–48, we design and synthesise a new wave of bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]quinoxaline derivatives having the basic crucial pharmacophoric features of VEGFR-2 inhibitors.
At first, bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]quinoxaline moiety was selected as a heteroaromatic ring system to occupy the hinge region in the ATP binding site. Such moiety was accomplished to attain more rigid structures that could increase binding affinity against the active site. Then, sulfanyl-N-phenylacetamide moiety was utilised as a central spacer to occupy the linker region. Such spacer was expected to increase the flexibility of the designed compounds. Regarding the DFG-motif region, we used two different moieties having the essential HBA/HBD features to play the role of the pharmacophore. The pharmacophores were designed to be amide moiety (compounds 23a–n), or diamide moiety (compounds 24a–c). Finally, the allosteric hydrophobic region can be occupied by different terminal aliphatic moieties (23a–f) or terminal aromatic moieties (23 g–n and 24a–c). These wide varieties of modifications helped us to study the SAR of these compounds as efficient anticancer agents with potential VEGFR-2 inhibitory activities. All modification pathways and molecular design rationale were illustrated and summarised in . As shown in , the target compounds achieved the essential pharmacophoric requirements of VEGFR-2 inhibitors.
Figure 3. Schematic representation showing the designing strategy.
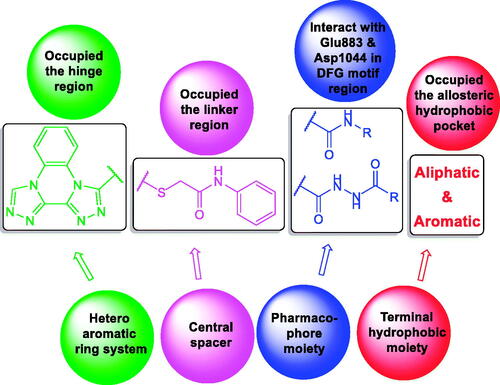
Figure 4. The target compounds fulfilled the pharmacophoric features of VEGFR-2 inhibitors.
To attain our aim of this work, cytotoxicity and VEGFR-2 inhibitory effects of the synthesised compounds were assessed. Besides, flow cytometric analyses were carried out for the most active candidate to estimate its potential against apoptosis induction and cell cycle arrest. Furthermore, such a compound was examined against several apoptotic markers include caspase 3/9 and BAX, and Bcl-2.
2. Results and discussion
2.1. Chemistry
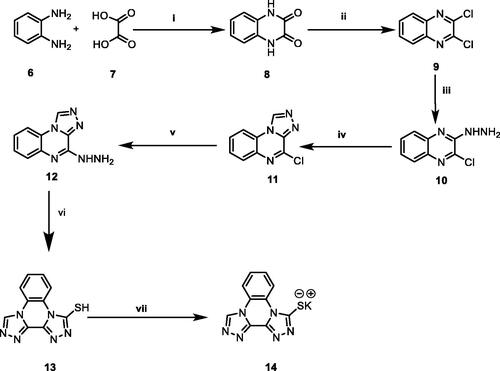
The general synthetic pathways adopted for the synthesis of the designed compounds are illustrated in Schemes 1–3. Scheme 1 depicts the synthesis of potassium bis[1, 2, 4]triazolo[4,3-a]quinoxaline-4-thiolate 14. Initially, o-phenylenediamine 6 was refluxed with oxalic acid 7 in the presence of 4 N HCl to afford 2,3-(1H,4H)-quinoxalinedione 8Citation49. Chlorination of compound 8 was done by refluxing with thionyl chloride yielding 2,3- dichloroquinoxaline 9Citation49. Subsequent treatment of the latter with hydrazine hydrate in absolute ethanol afforded 2-chloro-3-hydrazinylquinoxaline 10Citation50. Heating of compound 10 with triethyl orthoformate gave 4-chloro[1, 2, 4]triazolo[4,3-a]quinoxaline 11Citation50. The obtained compound 11 was heated with hydrazine hydrate to afford 4-hydrazinyl-[1, 2, 4] triazolo[4,3-a]quinoxaline 12Citation51. Moreover, reflux of 12 in an alcoholic mixture of carbon disulphide and potassium hydroxide afforded bis[1, 2, 4]triazolo[4,3-a:3′,4′-c]quinoxaline-3-thiol 13Citation44. Heating compound 13 with an alcoholic solution of potassium hydroxide gave the corresponding potassium salt 14Citation44. (Scheme 1)
Scheme 1. Synthetic pathway for compound 14; Reagents and conditions: (i) 4 N conc. HCl/reflux/6 h, (ii) SOCl2/DCE/reflux/4 h, (iii) NH2NH2.H2O/ethanol/r.t., (iv) triethyl orthoformate/reflux/4 h, (v) NH2NH2.H2O/ethanol/reflux/4 h, (vi) absolute ethanol/KOH/CS2/reflux/6 h, (vii) absolute ethanol/KOH/heating/10 min.
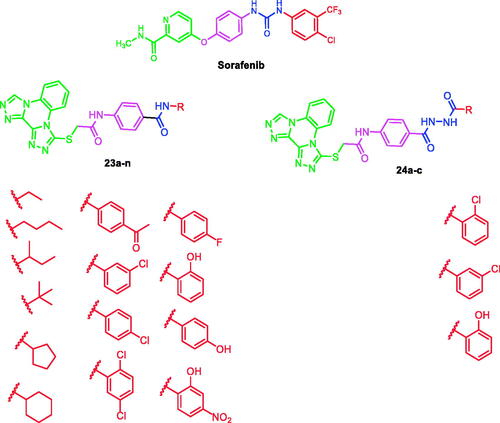
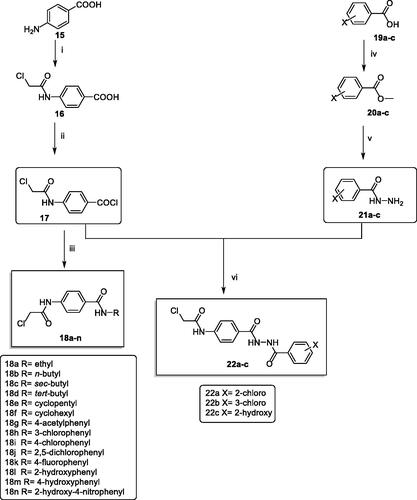
Scheme 2 was carried out to prepare the key intermediates (18a–n and 22a–c). At first, acetylation of p-amino benzoic acid 15 with chloroacetyl chloride in DMF in the presence of NaHCO3 provided the key product 4–(2-chloroacetamido)benzoic acid 16. Compound 16 was acylated using thionyl chloride to produce the benzoyl chloride derivative 17Citation52. Furthermore, stirring of compound 17 with commercially available amines namely, ethylamine, n-butylamine, sec-butylamine, tert- butylamine, cyclopentylamine, cyclohexylamine, 4-aminoacetophenone, 3-chloroaniline, 4-chloroaniline, 2,5-dichloroaniline, 4-fluoroaniline, 2-hydroxyaniline, 4-hydroxyaniline, and 2-hydroxy-4-nitroaniline at room temperature in acetonitrile/TEA mixture afforded the corresponding intermediates 18a–n, respectively. On the other hand, to prepare the ester derivatives 20a–c, appropriate acid derivatives 19a–c namely, 2-chlorobenzoic acid, 3-chlorobenzoic, and 2-hydroxybenzoic acid were refluxed in methanol in the presence of sulphuric acid according to the reported proceduresCitation42,Citation53,Citation54. In addition, reflux of 20a–c with hydrazine hydrate afforded the corresponding acid hydrazides 21a–cCitation42. In the end, in acetonitrile and TEA mixture, compound 17 was stirred with the acid hydrazides 21a–c to produce the corresponding diamide derivatives 22a–c (Scheme 2).
Scheme 2. Synthetic pathway for compounds 18a–n and 22a–c; Reagents and conditions: (i) ClCH2COCl/DMF/NaHCO3/r.t./1 h, (ii) SOCl2/DCE/DMF/reflux/4 h, (iii) RNH2/acetonitrile/TEA/r.t./8 h (iv) methanol/conc.H2SO4/reflux/8 h, (v) NH2NH2.H2O/ethanol/reflux/8 h, (vi) acetonitrile/TEA/r.t./8 h.
The structures of compounds 18a–n and 22a–c were confirmed by spectral and elemental data. IR spectra of such compounds showed strong bands around 3370 − 3100 cm−1 corresponding to NHs. Also, IR spectra showed strong C=O absorption bands at a range of 1770 − 1624 cm−1. Moreover, 1H NMR spectra showed singlet signals around δ 10.50 and 8.35 ppm corresponding to the two amidic NHs. Additionally, CH2 protons appeared at around δ 4.30 ppm. Matching with such results, 13C NMR spectra also confirmed the validity of suggested structures where characteristic peaks were displayed around δ 165.60, 165.05, and 44.00 ppm corresponding to the two C=O and CH2 groups, respectively.
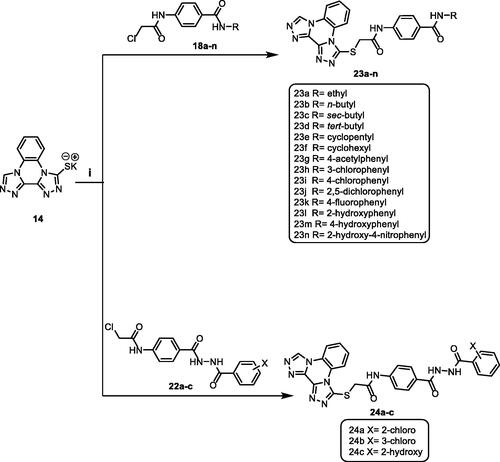
Scheme 3 demonstrated the synthetic pathway of the final target compounds (23a–n and 24a–c). Compound 14 was heated with the formerly synthesised intermediates (18a–n and 22a–c) in dry DMF using KI to furnish the titled compounds 23a–n and 24a–c, respectively.
Scheme 3. Synthetic pathway for compounds 23a–n and 24a–c; Reagents and conditions: (i) DMF/KI/reflux/6 h.
The spectral and elemental data supported the structures of the obtained derivatives, where the 1H NMR spectra of compounds 23a–n and 24a–c displayed characteristic downfield singlet signals around δ 10.75 ppm. The mass spectra were also consistent with the expected structures. Taking compound 23d as a representative example, the IR spectrum demonstrated stretching bands at 2968 and 2929 cm−1 corresponding to aliphatic CH bonds. The 1H NMR spectrum of this compound showed an up-field singlet signal at δ 1.38 ppm corresponding to tertiary butyl moiety. Furthermore, 13C NMR spectrum showed the presence of two peaks at δ 51.16 and 29.11 corresponding to CH and three CH3 of tert-butyl moiety.
2.2. Biological evaluation
2.2.1. In vitro anti-proliferative activity
All newly prepared compounds were assessed for their in vitro cytotoxic efficiencies via standard MTT methodCitation55–57, against breast cancer (MCF-7) and hepatocellular carcinoma (HepG2) cell lines. Sorafenib was applied as a standard anticancer drug. The growth inhibitory concentration (IC50) values were concluded for each final compound and depicted in .
Table 1. In vitro anti-proliferative activities of the tested compounds against MCF-7 and HepG2 cell lines, and in vitro enzymatic inhibitory activities against VEGFR-2.
Overall, HepG2 cells were more sensitive to the tested compounds than MCF-7 except for compound 23k. Among the series, compound 23j showed the highest anti-proliferative activities against MCF-7 and HepG2 cell lines with IC50 values of 10.3 and 6.4 µM, respectively. In addition, compounds 23a, 23i, 23 l, and 23n exhibited good anti-proliferative activities against the tested cell lines with IC50 values ranging from 7.1 to 19.4 µM, comparing to sorafenib (IC50 = 3.51 and 2.17 µM against MCF-7 and HepG2, respectively). Moreover, compounds 23d, 23h, 23m, 24a, 24b, and 24c displayed moderate anti-proliferative activities against the tested cell lines with IC50 values ranging from 15.2 to 48.3 µM. On the other hand, the rest of the compounds displayed weak anti-proliferative activities against the tested cell lines.
2.2.2. In vitro VEGFR-2 enzyme assay inhibition
All the synthesised compounds were subjected to further assay for their ability to inhibit VEGFR-2 using sorafenib as a positive control. The results were stated as growth inhibitory concentration (IC50) values and illuminated in .
Compound 23j was the most potent VEGFR-2 inhibitor with an IC50 value of 3.7 nM, nearly equal to that of sorafenib (IC50 = 3.12 nM). Moreover, compounds 23a, 23d, 23h, 23i, 23l, 23m, and 23n showed promising activities with IC50 values ranging from 5.8 to 11.8 nM. On the other hand, compounds 23c, 23e, 23f, 23k, and 24a–c exhibited moderate to weak activity with IC50 values ranging from 20.7 to 49.6 nM. Finally, compounds 23b and 23g exhibited the lowest anti VEGFR-2 activities with IC50 values of 71.6 and 62.7 nM, respectively.
2.2.3. Structure–activity relationship (SAR)
Inspecting the results of different biological analyses (anti-proliferative activity and VEGFR-2 assay); we concluded a valuable SAR.
At First, the effect of pharmacophore moiety on the activity was explored. It was noticed that the amide derivatives 23h (IC50 = 37.2 and 22.3 µM against MCF-7 and HepG2, respectively & 11.7 nM against VEGFR-2) and 23 l (IC50 = 19.4 and 11.3 µM against MCF-7 and HepG2, respectively & 5.8 nM against VEGFR-2) displayed higher activities than the corresponding diamide derivatives 24b (IC50 = 42.7 and 30.3 µM against MCF-7 and HepG2, respectively & 22.3 nM against VEGFR-2) and 24c (IC50 = 40.7 and 29.8 µM against MCF-7 and HepG2, respectively & 20.7 nM against VEGFR-2).
Next, we investigated the effect of the terminal hydrophobic moiety. With respect to the terminal aliphatic hydrophobic moieties, it was found that the VEGFR-2 inhibitory activities decreased in the order of ethyl (23a, IC50 = 7.1 nM) > tert-butyl (23d, IC50 = 11.8 nM) > cyclohexyl (23f, IC50 = 39.5 nM) > cyclopentyl (23e, IC50 = 39.8 nM) > sec-butyl (23c, IC50 = 47.1 nM) > n-butyl (23b, IC50 = 71.6 nM). In addition, the effect of the substitution on the aromatic hydrophobic moieties has been examined. For the amide derivatives, it was found that the VEGFR-2 inhibitory activities decreased in the order of 2,5-dichloro (23j, IC50 = 3.7 nM) >2-hydroxy (23l, IC50 = 5.8 nM) >2-hydroxy −4-nitro (23n, IC50 = 7.4 nM) >4-chloro (23i, IC50 = 9.4 nM) >4-hydroxy (23m, IC50 = 9.7 nM) >4-fluoro (23k, IC50 = 49.6 nM). While the diamide derivatives revealed decrease in VEGFR-2 inhibitory activities in the order of 2- hydroxy (24c, IC50 = 20.7 nM) >3-chloro (24b, IC50 = 22.3 nM) > 2-chloro (24a, IC50 = 23.9 nM).
2.2.4. Correlation of cytotoxicity with VEGFR-2 inhibition
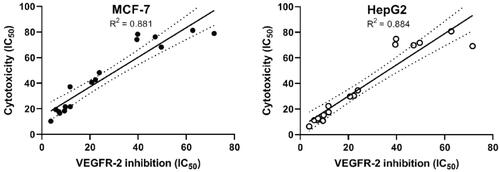
From the aforementioned results, we can conclude that our tested compounds can inhibit VEGFR-2 in the tested cell lines. To prove that inhibition of VEGFR-2 is the major prominent cause of cell mortality, we compared the cytotoxicity results of the synthesised candidates with their corresponding VEGFR-2 inhibitory activities utilising a simple linear regression analysis. The coefficients of determination (R2) were determined. The R2 values for MCF-7 and HepG2 were 0.881 (p values >.0001) and 0.800 (p values > .0001), respectively (. Such high values of R2 indicate the high correlation between the dependent variable (VEGFR-2 inhibition) and the independent one (cytotoxicity).
Figure 5. Correlation of cytotoxicity with VEGFR2 inhibition on two cell line models MCF-7and HepG2. MCF-7 (p value >.0001) & HepG2 (p value >.0001).
2.2.5. Cellular mechanistic study
Compound 23j which demonstrated remarkable cytotoxic potency and significant inhibitory activity against VEGFR-2 was nominated for further cellular mechanistic study. This involved study of its influence on cell cycle progression and induction of apoptosis in HepG2 cells.
2.2.5.1. Effect on cell cycle progression
In this work, HepG2 cell line was treated with compound 23j at a concentration of 6.4 μM (the IC50 value of compound 23j) and incubated for 24 h. Then, the cells were stained with propidium iodide and analysed for cell distribution during the various phases of the cell cycle against untreated HepG2 cells. Flow cytometry results exhibited that the percent of HepG2 cells decreased at the Sub-G1, G1 and S phases. For Sub-G1 phase, it decreased from 1.46% to 1.21%, for G1 phase it decreased from 57.75 to 37.34% while for S phase it decreased from 28.65% to 25.79%. On the other hand, the percentage of HepG2 cells significantly increased at G2/M phase from 12.13% to 35.64%. Such results indicated that compound 23j inhibited proliferation of HepG2 cells via cessation of the growth of the cell cycle at G2/M phase ( and .
Figure 6. Cell cycle analysis of HepG2 cells treated with compound 23j. **p < .01 and ***p < .001.
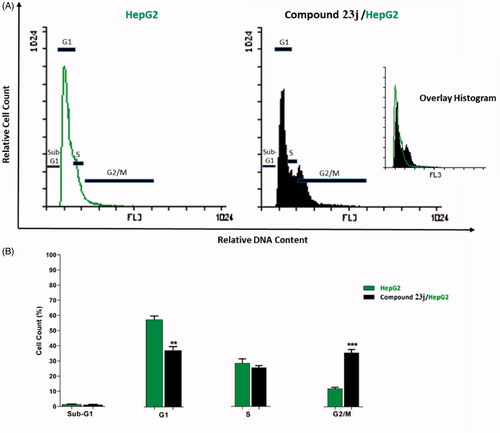
Table 2. Effect of compound 23j on cell cycle progression in HepG2 cells after 24 h treatment.
2.2.5.2. Apoptosis analysis
To confirm the apoptotic ability of compound 23j, a flow cytometry technique was performed. In such technique, HepG2 cells were stained with annexin V/propidium iodide (PI) and incubated for 24 h with compound 23j (6.4 μM). It was revealed that compound 23j triggered more apoptotic cells comparing to untreated control cells. In details, compound 23j induced apoptosis by 40.12% (early apoptosis = 39.97% & late apoptosis = 0.15%), compared to 7.07% in the control cells (early apoptosis = 6.88% & late apoptosis = 0.19%). Such findings revealed that compound 23j could induce apoptosis in HepG2 cells ( & .
Figure 7. Flow cytometric analysis of apoptosis in HepG2 cells exposed to compound 23j. ***p < .001.
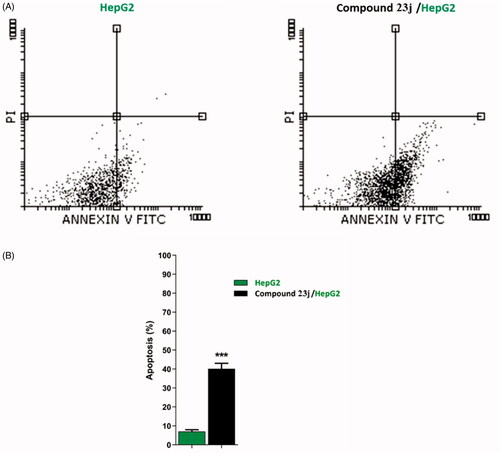
Table 3. apoptotic effect of compound 23j against HepG2 cells.
2.2.5.3. Caspase 3/9 assay
To study the effect of compound 23j, the most promising member, on caspase-3 and caspase-9 levels, western blot technique was carried out. HepG2 cells were treated with 23j (6.4 µM) for 24 h. The results revealed that compound 23j produced a significant increase in the cellular levels of caspase-3 (1.36-fold) and caspase-9 (2.80-fold) compared to the control cells ( and .
Figure 8. The immunoblotting of BAX, Bcl-2, Caspase-9, and Caspase-3 (Normalized to β-actin). *p < .05, **p < .01, ***p < .001.
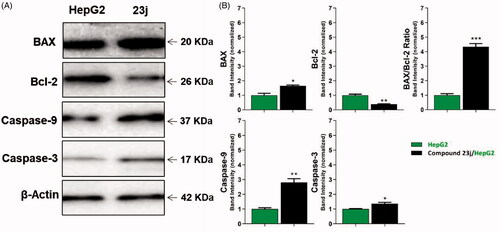
Table 4. Effect of compound 23j on levels of BAX, Bcl-2, active caspase-9, and active caspase-3 proteins expression
2.2.5.4. Evaluation of BAX and Bcl-2 expressions
BAX and Bcl-2 cellular levels were assessed for compound 23j after 24 h of its application on HepG2 cells using western blot technique. The results showed that compound 23j produced an increase of the pro-apoptotic factor BAX by 1.65-fold while anti-apoptotic protein Bcl-2 demonstrated a concentration decreased by 2.63-fold. Moreover, compound 23j increased BAX/Bcl-2 ratio to be 4.34 which indicates the efficiency of compound 23j on apoptosis cascade ( and .
2.3. Molecular modelling study
2.3.1. Docking study
Molecular docking study was accomplished to get further insight into the binding modes and orientations of the newly synthesised compounds into the ATP binding site of VEGFR-2 enzyme. Sorafenib was used as a reference ligand. Validation of the docking procedure was achieved via re-docking of the co-crystallised ligand against the active pocket of VEGFR-2. The results revealed that the RMSD value between the re-docked pose and the co-crystallised one was 1.02. This value revealed the validity of the docking process (. The calculated ΔG (binding free energies) of the synthesised compounds and the reference drug against VEGFR-2 were summarised in .
Figure 9. Alignment of the co-crystallised pose (green) and the redocked pose (Orange) of the same ligand inside the protein.
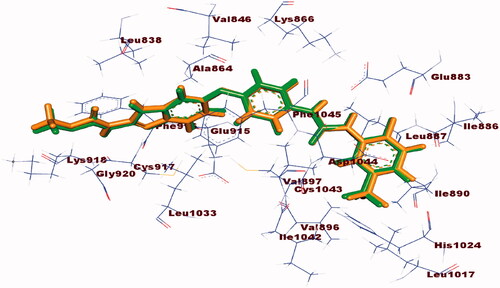
Table 5. The calculated ΔG of the synthesised candidates and sorafenib (ΔG in kcal/mole).
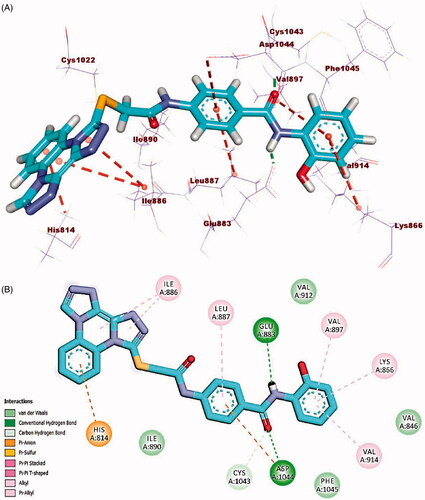
Sorafenib interactions with the amino acids of the active site have been studied and displayed in 2 D and 3 D style in . The proposed binding mode of sorafenib revealed an affinity value of −22.48 kcal/mol. The N-methylpicolinamide moiety formed five hydrophobic interactions with Val846, Ala864, Phe1045, Leu1033, and Leu838. Likewise, it formed one hydrogen bond with Cys917. Moreover, the phenyl moiety was buried in the linker region forming five hydrophobic interactions with Phe1045, Cys1043, Val846, Val914, and Val897. Furthermore, the pharmacophore moiety (urea group) formed three hydrogen bonds with the two crucial amino acids Glu883 and Asp1044. Finally, the 1-chloro-2-(trifluoromethyl)benzene moiety conquered the allosteric hydrophobic region forming many hydrophobic and electrostatic interactions with Ile886, Leu887 Leu1017, His1024, Ile890, and Asp1044Citation34,Citation58.
Figure 10. (A) 3D binding mode of sorafenib into VEGFR-2. (B) 2D binding mode sorafenib into VEGFR-2.
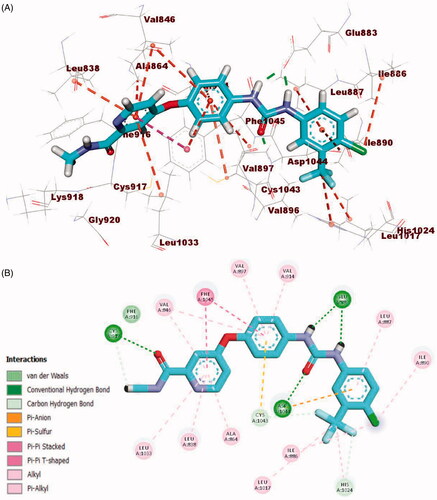
Docking simulation of compound 23i revealed that it has a good fitting into the enzyme active sites with docking score of −23.63 kcal/mol. In DFG region, the amide moiety (pharmacophore) formed two hydrogen bonds with carboxylate moiety of Glu883 (1.50 Å) and NH group of Asp1044 (1.82 Å). Furthermore, the phenyl ring (central spacer) occupied the linker region forming five hydrophobic interactions with Cys1043, Phe1045, Val897, and Val914. Also, the bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]quinoxaline moiety occupied the hinge region forming two hydrophobic and one hydrogen bond interactions with Leu838. Additionally, the terminal hydrophobic (4-chlorophenyl) moiety formed three hydrophobic interactions with Ile886 and Leu887 and one electrostatic interaction with Asp1044 (.
Figure 11. (A) 3D binding mode of compound 23i into VEGFR-2. (B) 2D binding mode of compound 23i into VEGFR-2.
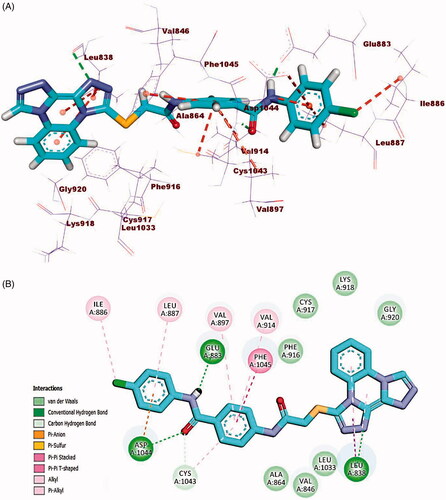
The docking findings of compound 23j revealed affinity value of −23.87 kcal/mol. The amide moiety formed two hydrogen bonds with Glu883 (COO-, 1.75 Å) and Asp1044 (NH, 2.20 Å). Additionally, in the hinge region, the docked compound formed four hydrophobic interactions via its bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]quinoxaline moiety with Leu838 and Phe916. Also, the central phenyl ring moiety formed three hydrophobic interactions with Val914, Val897, and Cys1043. Finally, the 2,5-dichlorophenyl moiety formed electrostatic and hydrophobic interactions with Asp1044, Ile890, and Leu 887 (.
Figure 12. (A) 3D binding mode of compound 23j into VEGFR-2. (B) 2D binding mode of compound 23j into VEGFR-2.
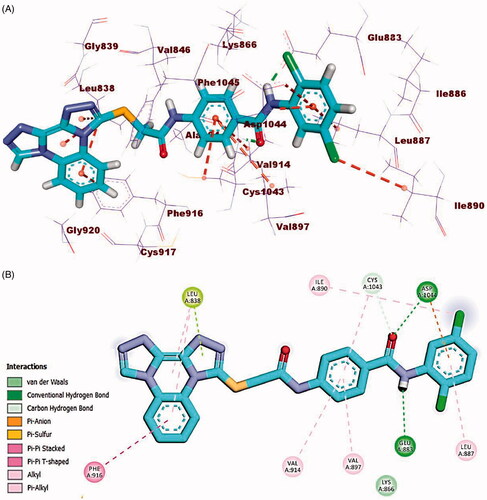
Compound 23l (affinity value of −23.26 kcal/mol) combined with the receptor protein as follows; In DFG domain, the amide moiety formed two hydrogen bonds with Glu883 (distance: 2.28 Å) and Asp1044 (distance: 1.76 Å). Moreover, in the hinge region, bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]quinoxaline moiety formed three hydrophobic interactions with His814 and Ile886. Also, in the linker region, the central phenyl ring moiety formed two hydrophobic interactions with Asp1044 and Leu887. Additionally, the terminal hydrophobic (2-hydroxyphenyl moiety) formed three hydrophobic interactions with Val897, Val914, and Lys866 (.
Figure 13. (A) 3D binding mode of compound 23 l into VEGFR-2. (B) 2D binding mode of compound 23 l into VEGFR-2.
2.3.2. In silico ADME analysis
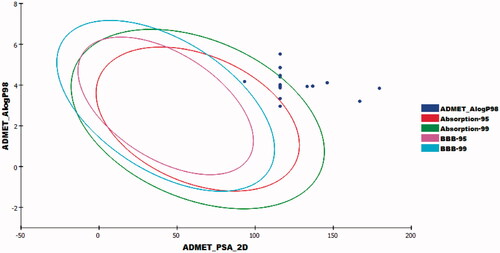
To investigate pharmacokinetics properties of the prepared compounds, computer aided ADME studies were accomplished using Accelrys Discovery Studio 4.0 software. Sorafenib was used as a reference molecule. These studies include the estimation of certain parameters. 1) Blood brain barrier penetration which measures the ability of molecule to diffuse through blood brain barrier. 2) Absorption level which determines human intestinal absorption (HIA) of a chemical after its oral administration. A well-absorbed compound is one that is absorbed at least 90% into the blood streamCitation59,Citation60. iii) Solubility level in which the solubility of a chemical in water was predicted at 25 °C. iv) CYP2D6 binding which analyzes cytochrome P450 2D6 enzyme inhibitionCitation61,Citation62. v) Plasma protein binding which predicts the fraction of drug bound to plasma proteins in the bloodCitation63. The results were predicted and listed in .
Table 6. ADME screening of the designed compounds
The results revealed that the estimated compounds have very low BBB penetration levels.
Accordingly, the CNS side effects of all compounds were expected to be low. Regarding absorption levels, compound 23a demonstrated good absorption level, compounds 23b–e and 23k exhibited moderate absorption levels. On the other hand, compounds 23f–j, 23l–n, and 24a–c showed poor and very poor intestinal absorption. With respect to aqueous solubility, all compounds demonstrated low and very low levels of aqueous solubility. Moreover, the effect on cytochrome P450 2D6 was investigated. The results showed that all examined members were non-inhibitors of CYP2D6. Consequently, liver side effect is not expected upon their administration. The plasma protein binding model exhibited that all compounds were anticipated to bind plasma protein less than 90% ().
Figure 14. The ADME plot of the synthesised compounds.
2.3.3. Toxicity studies
Toxicity profile of the prepared candidates was assessed as stated by the validated and constructed models in Discovery studio software version 4.0Citation64,Citation65. These models involve: 1) FDA rodent carcinogenicity. 2) Carcinogenic potency TD50Citation66. 3) Rat maximum tolerated doseCitation67,Citation68. 4) Developmental toxicity potentialCitation69,Citation70. 5) Rat oral LD50Citation71. 6) Rat chronic LOAELCitation72,Citation73.
Regarding FDA rodent carcinogenicity model, all the tested candidates were forecasted to be non-carcinogenic. For carcinogenic potency TD50 rat model, compounds 23a,b, 23 l,m, and 24c showed TD50 values ranging from 33.907 to 44.001 g/kg body weight, which are higher than sorafenib (14.244). With respect to rat maximum tolerated dose model, compounds 23a–c, 23 h–n, and 24a–c demonstrated maximum tolerated dose with a range of 0.099 to 0.391 g/kg body weight which are higher than that of sorafenib (0.089 g/kg body weight). On the other hand, compounds 23d–g showed a maximum tolerated dose lower than that of sorafenib with a range of 0.083 to 0.088 g/kg body weight. Additionally, all compounds were predicted to be non-toxic against developmental toxicity potential model. For rat oral LD50 model, compounds 23a–d, 23g, 23i, 23m, and 24a–c revealed oral LD50 values in a range of 0.864 to 2.477 g/kg body weight which are higher than that of sorafenib (0.823 g/kg body weight). For rat chronic LOAEL model, the tested compounds exhibited LOAEL values ranging from 0.064 to 0.329 g/kg body weight. These values are higher than sorafenib (0.005 g/kg body weight) .
Table 7. Toxicity study of the synthesised compounds.
3. Conclusion
In the present study, a new series of bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]quinoxaline derivatives were synthesised as potential cytotoxic agents and VEGFR-2 inhibitors. The anticancer efficiency of these derivatives was evaluated against MCF-7 and HepG2 cancer cell lines. Four compounds 23a (IC50 = 17.7 and 11.3 µM), 23i (IC50 = 18.3 and 10.8 µM), 23j (IC50 = 10.3 and 6.4 µM), 23l (IC50 = 19.4 and 11.3 µM), and 23n (IC50 =16.5 and 12.7 µM) were the most active antiproliferative members against MCF-7 and HepG2, respectively. The in vitro VEGFR-2 assay revealed that compounds 23a, 23d, 23h, 23i, 23j, 23l, 23m, and 23n exhibited the highest inhibitory activity versus VEGFR-2 with IC50 values of 7.1, 11.8, 11.7, 9.4, 3.7, 5.8, 9.7, and 7.4 nM, respectively. SAR revealed that the amide derivatives (compounds 23h and 23l) exhibited higher activities than the corresponding diamide derivatives (compounds 24b and 24c). Furthermore, compound 23j, the most potent member, arrested the HepG2 cell growth at G2/M phase and induced apoptosis by 40.12% compared to the control cells (7.07%). Moreover, caspase activation assay was performed for 23j in HepG2 cell lines. The results showed significant increase of caspase 3 (1.36-fold) and caspase 9 (2.80-fold). BAX and Bcl-2 concentration levels were also evaluated and showed a titre increase of the pro-apoptotic protein BAX (1.65-fold) and a decrease of Bcl-2 (2.63-fold). Additionally, compound 23j increased BAX/Bcl-2 ratio to be 4.34. Finally, computational physicochemical assessment of the synthesised compounds showed that they have favourable properties with satisfactory drug-like profiles.
4. Experimental
4.1. Chemistry
All the reagents, chemicals, and apparatus were presented in Supplementary data. Compounds 8, 9, 10, 11, 12, 13, 14, 16, 17, 20a–c, and 21a–c were prepared in accordance with the reported proceduresCitation41,Citation42,Citation44,Citation49,Citation50,Citation52.
4.1.1. General procedure for synthesis of compounds 18a–n
A mixture of 17 (2 mmol) and appropriate amines namely, ethylamine, n-butylamine, sec-butylamine, tert-butylamine, cyclopentylamine, cyclohexylamine, 4-aminoacetophenone, 3-chloroaniline, 4-chloroaniline, 2,5-dichloroaniline, 4-flouroaniline, 2-hydroxyaniline, 4-hydroxyaniline, and 2-hydroxy-4-nitroaniline (2 mmol) was stirred in acetonitrile (50 ml) in the presence of trimethylamine (1 ml) for 8 h. The obtained precipitates were filtered and crystallised from ethanol to give the corresponding derivatives, 18a–n, respectively.
4.1.1.1. 4-(2-Chloroacetamido)-N-ethylbenzamide (18a)
Yellow crystal (yield, 80%); m.p. = 190 − 192 °C; FT-IR (νmax, cm−1): 3376, 3273 (NH), 2971(C–H aliphatic), 1709, 1636 (C=O), 1605 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 8.36 (s, 1H), 7.80 (d, J = 9.7 Hz, 2H), 7.63 (d, J = 8.3 Hz, 2H), 4.26 (s, 2H), 3.29 − 3.21 (q, 2H), 1.14 − 1.05 (t, 3H).; 13C NMR (101 MHz, DMSO-d6) δ 165.73 (2C), 165.40 (2C), 141.29, 130.24, 128.37, 44.04, 34.42, 15.43, 15.20; Anal. Calcd. for C11H13ClN2O2 (240.68): C, 54.89; H, 5.44; N, 11.64. Found: C, 54.72; H, 5.20; N, 11.46%.
4.1.1.2. N-Butyl-4–(2-chloroacetamido)benzamide (18b)
White crystal (yield, 85%); m.p. = 170 − 172 °C; FT-IR (νmax, cm−1): 3369, 3263 (NH), 3185 (NH), 3099 (C–H aromatic), 2960 (C–H aliphatic), 1706, 1633 (C=O), 1606 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1H), 8.33 (s, 1H), 7.80 (dd, J = 8.8, 1.8 Hz, 2H), 7.66 − 7.61 (m, 2H), 4.27 (s, 2H), 3.25 − 3.18 (m, 2H), 1.51 − 1.43 (m, 2H), 1.33 − 1.26 (m, 2H), 0.89 − 0.84 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.93, 165.41, 141.30, 130.27, 128.67, 128.31, 119.43, 118.44, 44.44, 44.01, 31.74, 20.12, 14.18; MS (m/z): 269 (M+ + 1, 100%). Anal. Calcd. for C13H17ClN2O2 (268.74): C, 58.10; H, 6.38; N, 10.42. Found: C, 58.38; H, 6.61; N, 10.32%.
4.1.1.3. N-(Sec-butyl)-4-(2-chloroacetamido)benzamide (18c)
White crystals (yield 78%); m.p. = 180–182 °C; IR (KBr) ν cm−1: 3307, 3113 (NH), 2969, 2933 (CH aliphatic), 1684, 1625 (C=O); 1H NMR (400 MHz, DMSO-d6) δ 10.54 (s, 1H), 8.06 (d, J = 6.1 Hz, 1H), 7.82 (d, J = 6.3 Hz, 2H), 7.65 (d, J = 7.9 Hz, 2H), 4.28 (s, 2H), 3.89 (dq, J = 11.8, 6.8 Hz, 1H), 1.56 − 1.41 (m, 2H), 1.11 (d, J = 5.1 Hz, 3H), 0.84 (d, J = 4.7 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.46, 165.38, 141.25, 130.45, 128.64, 119.33, 118.74, 118.44, 46.76, 44.05, 29.31, 20.69, 11.29; Anal. Calcd. for C13H17ClN2O2 (268.74): C, 58.10; H, 6.38; N, 10.42. Found: C, 58.06; H, 6.65; N, 10.29%.
4.1.1.4. N-(Tert-butyl)-4-(2-chloroacetamido)benzamide (18d)
Yellow crystals (yield 68%); m.p. = 190–192 °C; IR (KBr) ν cm−1: 3314 (NH), 2975 (CH aliphatic), 1666, 1628 (C=O), 1606 (C=N); Anal. Calcd. for C13H17ClN2O2 (268.74): C, 58.10; H, 6.38; N, 10.42. Found: C, 58.31; H, 6.55; N, 10.59%.
4.1.1.5. 4-(2-Chloroacetamido)-N-cyclopentylbenzamide (18e)
White crystals (yield 82%); m.p. = 230–232 °C; IR (KBr) ν cm−1: 3290 (NH), 2961 (CH aliphatic), 1680, 1624 (C=O); 1H NMR (400 MHz, DMSO-d6) δ 10.77 (s, 1H), 8.20 (d, J = 7.3 Hz, 1H), 7.81 (d, J = 8.7 Hz, 2H), 7.66 (d, J = 8.7 Hz, 2H), 4.31 (s, 2H), 3.03 (qd, J = 7.2, 4.2 Hz, 1H), 1.87 − 1.82 (m, 2H), 1.66 (tt, J = 4.5, 2.5 Hz, 2H), 1.50 (qd, J = 7.7, 6.9, 3.9 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ 165.75, 165.42, 141.35, 130.26, 128.76, 119.28, 118.81, 118.36, 51.46, 44.00, 32.54, 31.05, 24.05, 8.93; MS (m/z): 281 (M+ + 1, 100%); Anal. Calcd. for C14H17ClN2O2 (280.75): C, 59.89; H, 6.10; N, 9.98. Found: C, 59.50; H, 6.04; N, 10.25%.
4.1.1.6. 4-(2-Chloroacetamido)-N-cyclohexylbenzamide (18f)
White crystals (yield 78%); m.p. = 220–222 °C; FT-IR ((νmax, cm−1): 3286 (NH), 3042 (C–H aromatic), 2938, 2854 (C–H aliphatic), 1681, 1624 (C=O), 1610 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H), 8.08 (d, J = 7.9 Hz, 1H), 7.83 − 7.77 (m, 2H), 7.65 − 7.59 (m, 2H), 4.26 (s, 2H), 3.72 (s, 1H), 1.78 (d, J = 7.1 Hz, 2H), 1.71 (d, J = 8.4 Hz, 2H), 1.58 (d, J = 12.8 Hz, 1H), 1.27 (dd, J = 11.4, 8.6 Hz, 4H), 1.12 (q, J = 10.8, 9.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 165.38, 165.15, 141.24, 130.41, 129.07, 128.80, 128.47, 119.35, 118.86, 118.41, 48.79, 48.66, 44.42, 44.02, 25.63; Anal. Calcd. for C15H19ClN2O2 (294.77): C, 61.12; H, 6.50; N, 9.50. Found: C, 60.88; H, 6.39; N, 9.25%.
4.1.1.7. N-(4-Acetylphenyl)-4-(2-chloroacetamido)benzamide (18g)
White crystals (yield 80%); m.p. = 250–252 °C; FT-IR (νmax, cm−1): 3326 (NH), 3062 (C–H aromatic), 1674, 1655 (C=O), 1596 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 1H), 10.46 (s, 1H), 7.98 − 7.90 (m, 6H), 7.72 (s, 2H), 4.29 (s, 2H), 2.53 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 197.08, 165.69(2 C), 165.57(2 C), 144.14, 142.15, 132.33, 129.90, 129.85, 129.60, 129.26, 119.97, 119.66, 118.65, 44.05, 27.16; Anal. Calcd. for C17H15ClN2O3 (330.76): C, 61.73; H, 4.57; N, 8.47. Found: C, 61.44; H, 4.39; N, 8.29%.
4.1.1.8. 4-(2-Chloroacetamido)-N-(3-chlorophenyl)benzamide (18h)
Yellow crystal (yield, 75%); m.p. = 190 − 192 °C; FT-IR (v max, cm−1): 3291 (NH), 1676, 1644 (C=O), 1593 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1H), 10.30 (s, 1H), 7.95 − 7.94 (m, 2H), 7.93 (d, J = 1.9 Hz, 1H), 7.74 − 7.68 (m, 3H), 7.36 (t, J = 8.1 Hz, 1H), 7.14 − 7.12 (m, 1H), 4.29 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 165.56, 165.53, 142.07, 141.20, 141.14, 133.33, 129.86, 129.41, 129.12, 120.24, 119.93, 119.03, 118.66, 118.59, 44.05; Anal. Calcd. for C15H12Cl2N2O2 (323.17): C, 55.75; H, 3.74; N, 8.67. Found: C, 55.64; H, 3.63; N, 8.45%.
4.1.1.9. 4-(2-Chloroacetamido)-N-(4-chlorophenyl)benzamide (18i)
Yellow crystal (yield, 83%); m.p. = 228 − 230 °C; FT-IR (νmax, cm−1): 3324 (NH), 1666 (C=O amide), 1595 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.59 (s, 1H), 10.27 (s, 1H), 7.93 (d, J = 8.6 Hz, 2H), 7.79 (d, J = 8.9 Hz, 2H), 7.72 (d, J = 8.7 Hz, 2H), 7.38 (d, J = 8.8 Hz, 2H), 4.29 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 165.53 (2C), 165.36 (2C), 141.98, 138.67, 130.01, 129.36 (2C), 129.08, 128.58, 128.30, 127.56, 118.63, 44.05; Anal. Calcd. for C15H12Cl2N2O2 (322.17): C, 55.75; H, 3.74; N, 8.67. Found: C, 55.59; H, 3.57; N, 8.40%.
4.1.1.10. 4-(2-Chloroacetamido)-N-(2,5-dichlorophenyl)benzamide (18j)
Yellow crystal (yield, 77%); m.p. = 195 − 197 °C; FT-IR (νmax, cm−1): 3277, 3104 (NH), 3033 (C–H aromatic), 1770, 1679 (C=O), 1606 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.63 (s, 1H), 10.03 (s, 1H), 7.96 (d, J = 8.7 Hz, 2H), 7.73 (dd, J = 5.6, 3.0 Hz, 3H), 7.57 (d, J = 8.6 Hz, 1H), 7.34 (dd, J = 8.6, 2.5 Hz, 1H), 4.29 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 165.60 (2 C), 165.26, 142.26, 136.86, 131.87, 131.81, 130.80, 129.48 (2C), 129.19, 128.97 (2C), 118.75, 44.02; Anal. Calcd. for C15H11Cl3N2O2 (357.61): C, 50.38; H, 3.10; N, 7.83. Found: C, 50.42; H, 3.18; N, 7.64%.
4.1.1.11. 4-(2-Chloroacetamido)-N-(4-fluorophenyl)benzamide (18k)
White crystal (yield, 70%); m.p. = 238–240 °C; FT-IR (νmax, cm−1); 3325 (NH), 1666, 1643 (C=O), 1611 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 10.23 (s, 1H), 7.95 (d, J = 7.0 Hz, 2H), 7.82 − 7.70 (m, 4H), 7.17 (s, 2H), 4.31 (t, J = 2.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 165.54, 165.21, 141.91, 136.06, 136.02, 130.12, 129.28, 129.04, 122.15, 119.45, 119.02, 118.63, 116.11, 115.88, 44.06; Anal. Calcd. for C15H12ClFN2O2 (306.72): C, 58.74; H, 3.94; N, 9.13. Found: C, 58.55; H, 3.89; N, 9.05%.
4.1.1.12. 4-(2-Chloroacetamido)-N-(2-hydroxyphenyl)benzamide (18l)
Brown crystal (yield, 65%); m.p. = 192 − 194 °C; FT-IR (νmax, cm−1): 3262, 3190 (NH), 3064 (C–H aromatic), 2952 (C–H aliphatic), 1735, 1683 (C=O), 1600 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 10.00 (s, 1H), 9.49 (s, 1H), 8.11–8.02 (m, 2H), 8.02–7.91 (m, 2H), 7.12 − 6.73 (m, 4H), 4.32 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 165.77, 165.24, 165.07, 163.82 (2C), 149.77, 144.89, 143.80, 141.98, 131.61, 129.69 (2C), 126.39, 124.39, 44.07; Anal. Calcd. for C15H13ClN2O3 (304.72): C, 59.12; H, 4.30; N, 9.19. Found: C, 59.01; H, 4.14; N, 8.92%.
4.1.1.13. 4-(2-Chloroacetamido)-N-(4-hydroxyphenyl)benzamide (18m)
Brown crystal (yield, 85%); m.p. >300 °C; FT-IR (νmax, cm−1): 3309, 3104 (NH), 3033 (C–H aromatic), 1770, 1671 (C=O), 1607 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 10.29 (s, 1H), 9.95 (s, 1H), 8.11 (d, J = 9.2 Hz, 2H), 7.85 − 7.80 (m, 2H), 7.50 (d, J = 8.4 Hz, 2H), 6.72 (d, J = 8.3 Hz, 2H), 4.30 (d, J = 4.1 Hz, 2H); Anal. Calcd. for C15H13ClN2O3 (304.73): C, 59.12; H, 4.30; N, 9.19. Found: C, 59.32; H, 4.15; N, 8.95%.
4.1.1.14. 4-(2-Chloroacetamido)-N-(2-hydroxy-4-nitrophenyl)benzamide (18n)
Greenish yellow crystal (yield, 60%); m.p. = 198–200 °C; FT-IR (νmax, cm−1): 3596, 3409 (NH), 3074 (C–H aromatic), 1700, 1652 (C=O), 1602 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1H), 10.66 (s, 1H), 9.51 (s, 1H), 8.22 (d, J = 8.3 Hz, 1H), 7.96 (d, J = 8.3 Hz, 2H), 7.85 − 7.67 (m, 4H), 4.30 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 165.61, 165.05 (2C), 148.61 (2C), 143.65, 142.42, 133.57, 129.12 (2 C), 122.04, 118.83, 115.16, 109.46, 44.07; Anal. Calcd. for C15H12ClN3O5 (349.72): C, 51.52; H, 3.46; N, 12.02. Found: C, 51.33; H, 3.57; N, 11.87%.
4.1.2. General procedure for the synthesis of compounds 22a–c
A mixture of 17 (2 mmol) and the acid hydrazides 21a–c namely, 2-chlorobenzohydrazide, 3-chlorobenzohydrazide, and 2-hydroxybenzohyazide (2 mmol) was allowed to stir in acetonitrile (50 ml) in the presence of trimethylamine (1 ml) for 8 h. The precipitated products were filtered, dried, and crystalised from ethanol to give the diamide intermediated, 22a–c, respectively.
4.1.2.1. 2-Chloro-N-(4-(2-(2-chlorobenzoyl)hydrazine-1-carbonyl)phenyl)acetamide (22a)
White crystal (yield, 85%); m.p. = 230 − 232 °C; FT-IR (νmax, cm−1): 3409, 3249 (NH), 3074 (C–H aromatic), 1673, 1650 (C=O), 1603 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.65 (s, 1H), 10.55 (s, 1H), 10.38 (s, 1H), 7.92 (d, J = 7.5 Hz, 2H), 7.72 (d, J = 7.6 Hz, 2H), 7.56 − 7.46 (m, 4H), 4.33 − 4.28 (m, 2H); Anal. Calcd. for C16H13Cl2N3O3 (366.19): C, 52.48; H, 3.58; N, 11.47. Found: C, 52.72; H, 3.48; N, 11.34%.
4.1.2.2. 2-Chloro-N-(4-(2-(3-chlorobenzoyl)hydrazine-1-carbonyl)phenyl)acetamide (22b)
Yellow crystal (yield, 80%); m.p. = 235 − 237 °C; FT-IR (νmax, cm−1): 3409, 3283 (NH), 3074 (C–H aromatic), 1676, 1645 (C=O), 1604 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 10.63 (s, 2H), 10.51 (s, 1H), 7.91 (dd, J = 19.0, 9.7 Hz, 4H), 7.74 − 7.66 (m, 3H), 7.58 (d, J = 6.4 Hz, 1H), 4.30 (t, J = 2.4 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 165.67, 165.58 (2C), 165.00, 142.13, 134.92, 133.83, 129.11, 128.88, 127.83 (2C), 127.59, 119.58, 119.13, 118.75, 44.06; Anal. Calcd. for C16H13Cl2N3O3 (366.19): C, 52.48; H, 3.58; N, 11.47. Found: C, 52.71; H, 3.62; N, 11.60%.
4.1.2.3. 2-Chloro-N-(4–(2-(2-hydroxybenzoyl)hydrazine-1-carbonyl)phenyl)acetamide (22c)
Yellow crystal (yield, 80%); m.p. = 233 − 235 °C; FT-IR (νmax, cm−1): 3272 (NH), 1687, 1661 (C=O), 1600 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H), 10.69 (d, J = 9.7 Hz, 2H), 10.63 (s, 1H), 7.93 (d, J = 8.0 Hz, 3H), 7.75 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.6 Hz, 1H), 6.96 (q, J = 8.4, 7.4 Hz, 2H), 4.32 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 168.25, 165.62, 165.47, 159.76, 159.72, 142.21, 129.14, 128.87, 128.65, 127.68, 119.58, 119.16, 118.79, 118.12, 114.98, 44.07; Anal. Calcd. for C16H14ClN3O4 (347.76): C, 55.26; H, 4.06; N, 12.08. Found: C, 55.45; H, 3.83; N, 11.90%.
4.1.3. General procedure for the synthesis of compounds 23a–n
A mixture of potassium salt of bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]quinoxaline-3-thiol 14 (0.5 g, 0.001 mol) and 4-(2-chloroacetamido)-N-(substituted)benzamide 18a–n (0.001 mol) in DMF (50 ml) was heated on a water bath for 6 h. After cooling to room temperature, the reaction mixture was poured on crushed ice. The obtained precipitates were collected by filtration, dried, and crystalised from ethanol to give the target compounds 23a–n.
4.1.3.1. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)acetamido)-N-ethylbenzamide (23a)
White crystal (yield, 70%); m.p. = 260–262 °C. FT-IR (νmax, cm−1): 3267 (NH), 1700, 1655 (C=O), 1596 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.69 (s, 1H), 10.12 (s, 1H), 8.35 (dq, J = 11.5, 3.7, 3.1 Hz, 2H), 7.86 (dd, J = 8.1, 3.1 Hz, 1H), 7.85 − 7.80 (m, 2H), 7.75 − 7.69 (m, 2H), 7.69 − 7.65 (m, 1H), 7.63 (dt, J = 10.1, 5.1 Hz, 1H), 4.41 (d, J = 3.4 Hz, 2H), 3.32 − 3.25 (m, 2H), 1.16 − 1.09 (m, 3H); 13C NMR (176 MHz, DMSO-d6) δ 166.67, 165.83, 151.65, 141.85 (2C), 138.29, 135.59, 129.91, 128.73, 128.52 (2C), 128.34, 128.32 (2C), 124.07, 118.76 (2C), 117.13, 34.45 (2C), 15.33; Anal. Calcd. for C21H18N8O2S (446.49): C, 56.49; H, 4.06; N, 25.10. Found: C, 56.21; H, 4.15; N, 24.95%.
4.1.3.2. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)acetamido)-N-butylbenzamide (23b)
Yellowish white crystal (yield, 70%); m.p. = 252–254 °C. FT-IR (νmax, cm−1): 3299 (NH), 2928 (CH aliphatic), 1676, 1631 (C=O); 1H NMR (700 MHz, DMSO-d6) δ 10.68 (s, 1H), 10.02 (s, 1H), 8.65 − 8.58 (m, 1H), 8.50 − 8.45 (m, 1H), 8.33 (q, J = 4.9 Hz, 1H), 7.86 − 7.79 (m, 2H), 7.75 (p, J = 7.6, 7.2 Hz, 2H), 7.65 (d, J = 8.4 Hz, 2H), 4.57 (s, 2H), 3.25 (p, J = 6.6, 5.4 Hz, 2H), 1.50 (q, J = 7.5 Hz, 2H), 1.33 (q, J = 8.0 Hz, 2H), 0.94 − 0.88 (m, 3H); 13C NMR (176 MHz, DMSO-d6) δ 165.93, 165.90, 147.72, 142.11, 141.56, 139.35, 138.78, 130.08, 128.54, 128.37, 128.35, 124.09, 123.20, 118.76, 118.58, 118.07, 39.29, 38.88, 31.78, 20.15, 14.22; MS (m/z): 475 (M+ + 1, 50%); Anal. Calcd. for C23H22N8O2S (474.54): C, 58.21; H, 4.67; N, 23.61. Found: C, 58.05; H, 4.60; N, 23.55%.
4.1.3.3. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)acetamido)-N-(sec-butyl)benzamide (23c)
White crystal (yield, 72%); m.p. = 245–247 °C. FT-IR (νmax, cm−1): 3266, 3127 (NH), 2965, 2910 (CH aliphatic), 1701, 1636 (C=O), 1605 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.68 (s, 1H), 10.01 (s, 1H), 8.58 (d, J = 8.1 Hz, 1H), 8.45 (d, J = 7.9 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 8.3 Hz, 2H), 7.73 (p, J = 7.6 Hz, 2H), 7.65 (d, J = 8.3 Hz, 2H), 4.57 (s, 2H), 3.90 (dd, J = 14.5, 7.5 Hz, 1H), 1.51 (ddq, J = 29.9, 15.4, 8.3, 7.7 Hz, 2H), 1.13 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 7.5 Hz, 3H); 13C NMR (176 MHz, DMSO-d6) δ 165.88, 165.48, 147.74, 142.07, 141.52, 139.32, 138.77, 130.27, 128.63 (2C), 128.34, 128.32, 124.04, 123.15, 118.69 (2C), 118.56, 118.03, 46.82, 38.89, 29.34, 20.77, 11.25; MS (m/z): 475 (M+ + 1, 60%); Anal. Calcd. for C23H22ClN8O2S (474.54): C, 58.21; H, 4.67; N, 23.61. Found: C, 57.98; H, 4.62; N, 23.57%.
4.1.3.4. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)acetamido)-N-(tert-butyl)benzamide (23d)
Faint yellow crystal (yield, 75%); m.p. = 248–250 °C. FT-IR (νmax, cm−1): 3439, 3104 (NH), 3051 (CH aromatic), 2968, 2929 (CH aliphatic), 1651 (C=O), 1600 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.67 (s, 1H), 10.03 (s, 1H), 8.61 (d, J = 8.0 Hz, 1H), 8.50 − 8.47 (m, 1H), 7.79 (t, J = 6.9 Hz, 3H), 7.76 (d, J = 8.1 Hz, 2H), 7.63 (d, J = 8.1 Hz, 2H), 4.57 (s, 2H), 1.38 (s, 9H); 13C NMR (176 MHz, DMSO-d6) δ 166.06, 165.85, 147.74, 142.13, 141.39, 139.37, 138.78, 131.16, 128.74 (2C), 128.38, 128.37, 124.12, 123.23, 118.60, 118.57 (2C), 118.08, 51.16, 38.87, 29.11 (3C); MS (m/z): 475 (M+ + 1, 100%); Anal. Calcd. for C23H22N8O2S (474.54): C, 58.21; H, 4.67; N, 23.61. Found: C, 58.05; H, 4.55; N, 23.44%.
4.1.3.5. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)acetamido)-N-cyclopentylbenzamide (23e)
Yellow crystal (yield, 77%); m.p. = 244–246 °C. FT-IR (νmax, cm−1): 3262, 3124 (NH), 2950, 2905 (CH aliphatic), 1699, 1636 (C=O), 1607 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.68 (s, 1H), 10.02 (s, 1H), 8.61 (dd, J = 8.0, 1.6 Hz, 1H), 8.47 (dd, J = 7.8, 1.8 Hz, 1H), 8.17 (d, J = 7.3 Hz, 1H), 7.83 (d, J = 8.6 Hz, 2H), 7.76 (ddd, J = 8.9, 7.6, 1.6 Hz, 2H), 7.65 (d, J = 8.6 Hz, 2H), 4.57 (s, 2H), 4.21 (q, J = 7.1 Hz, 1H), 1.88 (dtd, J = 12.0, 8.8, 7.9, 4.4 Hz, 2H), 1.70 (dt, J = 9.6, 5.3 Hz, 2H), 1.56 − 1.51 (m, 4H); 13C NMR (176 MHz, DMSO-d6) δ 165.88, 165.73, 147.73, 142.12, 141.53, 139.36, 138.78, 130.15, 128.70 (2C), 128.37, 128.35, 124.10, 123.21, 118.66 (2C), 118.59, 118.06, 51.34, 38.87, 32.61 (2C), 24.09 (2C); MS (m/z): 487 (M+ + 1, 65%); Anal. Calcd. for C24H22N8O2S (486.55): C, 59.25; H, 4.56; N, 23.03. Found: C, 59.08; H, 4.47; N, 22.89%.
4.1.3.6. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)acetamido)-N-cyclohexylbenzamide (23f)
White crystal (yield, 70%); m.p. = 270–272 °C. FT-IR (νmax, cm−1): 3269, 3131 (NH), 2930 (CH aliphatic), 1700, 1633 (C=O), 1607 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.68 (s, 1H), 10.02 (s, 1H), 8.61 (dd, J = 7.9, 1.6 Hz, 1H), 8.47 (dd, J = 7.7, 1.8 Hz, 1H), 8.09 (d, J = 7.9 Hz, 1H), 7.82 (d, J = 8.6 Hz, 2H), 7.76 (dtd, J = 16.4, 7.5, 1.5 Hz, 2H), 7.65 (d, J = 8.7 Hz, 2H), 4.57 (s, 2H), 3.75 (ddt, J = 15.1, 11.0, 5.3 Hz, 1H), 1.81 (dt, J = 8.6, 4.1 Hz, 2H), 1.74 (dt, J = 11.4, 4.1 Hz, 2H), 1.64 − 1.59 (m, 1H), 1.30 (dd, J = 11.3, 8.6 Hz, 4H), 1.13 (qt, J = 8.4, 4.3 Hz, 1H); 13C NMR (176 MHz, DMSO-d6) δ 165.88, 165.14, 147.73, 142.12, 141.54, 139.36, 138.78, 130.21, 128.69 (2C), 128.37, 128.35, 124.10, 123.22, 118.67 (2C), 118.59, 118.07, 48.73, 38.87, 32.95 (2C), 25.76, 25.45 (2C); MS (m/z): 501 (M+ + 1, 100%); Anal. Calcd. for C25H24N8O2S (500.58): C, 59.99; H, 4.83; N, 22.39. Found: C, 59.85; H, 4.76; N, 22.11%.
4.1.3.7. N-(4-Acetylphenyl)-4-(2-(bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)acetamido)benzamide (23g)
Pale white crystal (yield, 80%); m.p. = 275–277 °C. FT-IR (νmax, cm−1): 3300 (NH), 1655 (C=O), 1593 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.77 (s, 1H), 10.14 (s, 1H), 7.97 (d, J = 8.6 Hz, 2H), 7.81 (dd, J = 8.0, 1.5 Hz, 1H), 7.78 − 7.76 (m, 2H), 7.72 (d, J = 8.7 Hz, 2H), 7.59 − 7.57 (m, 1H), 7.54 (dd, J = 8.5, 1.3 Hz, 1H), 7.40 − 7.37 (m, 1H), 7.36 − 7.34 (m, 2H), 7.10 (t, J = 7.4 Hz, 1H), 5.19 (s, 2H), 2.49 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ 165.80, 165.23, 157.97, 154.85, 142.00, 139.71, 133.48, 132.47 (2C), 130.20 (2C), 130.04, 129.29, 129.23 (2C), 129.05 (2C), 123.99, 123.94 (2C), 120.80 (2C), 118.83 (2C), 115.21, 45.80, 21.59; MS (m/z): 537 (M+ + 1, 40%), 354 (40%); Anal. Calcd. for C27H20N8O3S (536.57): C, 60.44; H, 3.76; N, 20.88. Found: C, 60.10; H, 3.66; N, 20.59%.
4.1.3.8. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)acetamido)-N-(3-chlorophenyl)benzamide (23h)
Yellowish white crystal (yield, 63%); m.p. = 280–282 °C. FT-IR (νmax, cm−1): 3267, 3109 (NH), 1701, 1647 (C=O), 1593 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.79 (s, 1H), 10.30 (s, 1H), 10.02 (s, 1H), 8.61 (dd, J = 8.1, 1.5 Hz, 1H), 8.47 (dd, J = 7.9, 1.7 Hz, 1H), 7.98 − 7.95 (m, 3H), 7.78 − 7.73 (m, 4H), 7.70 (ddd, J = 8.3, 2.1, 1.0 Hz, 1H), 7.38 (t, J = 8.1 Hz, 1H), 7.15 (ddd, J = 8.0, 2.1, 0.9 Hz, 1H), 4.59 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 166.08, 165.52, 147.71, 142.38, 142.12, 141.23, 139.34, 138.78, 133.38, 130.76, 129.63, 129.30 (2C), 128.37, 124.08, 123.65, 123.20, 120.10 (2C), 119.03, 118.86 (2C), 118.59, 118.06, 38.89; Anal. Calcd. for C25H17ClN8O2S (528.98): C, 56.77; H, 3.24; N, 21.18. Found: C, 56.37; H, 3.11; N, 20.99%.
4.1.3.9. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)acetamido)-N-(4-chlorophenyl)benzamide (23i)
Yellow crystal (yield, 65%); m.p. = 277–279 °C. FT-IR (νmax, cm−1): 3382, 3111 (NH), 1674 (C=O), 1595 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.78 (s, 1H), 10.28 (s, 1H), 10.03 (s, 1H), 8.62 (d, J = 8.0 Hz, 1H), 8.53 − 8.44 (m, 1H), 7.96 (d, J = 8.5 Hz, 3H), 7.82 (d, J = 8.5 Hz, 2H), 7.78 (d, J = 9.1 Hz, 1H), 7.74 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.6 Hz, 2H), 4.59 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 166.07, 165.39, 147.71, 142.29, 142.15, 139.37, 138.79, 138.72, 129.80, 129.26 (2C), 128.98 (2C), 128.38 (2C), 127.57, 124.12, 123.24, 122.27 (2C), 118.86 (2C), 118.61, 118.08, 38.88; Anal. Calcd. for C25H17ClN8O2S (528.98): C, 56.77; H, 3.24; N, 21.18. Found: C, 56.44; H, 3.19; N, 20.85%.
4.1.3.10. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]uinoxaline-3-ylthio)acetamido)-N-(2,5-dichlorophenyl)benzamide (23j)
White crystal (yield, 78%); m.p. = 260–262 °C. FT-IR (νmax, cm−1): 3429 (NH), 1672 (C=O), 1511 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.80 (s, 1H), 10.03 (s, 2H), 8.62 (dd, J = 8.0, 1.5 Hz, 1H), 8.48 (dd, J = 7.9, 1.7 Hz, 1H), 7.99 − 7.97 (m, 2H), 7.77 (dt, J = 5.0, 2.2 Hz, 2H), 7.76 (m, 2H), 7.75 (s, 1H), 7.61 (d, J = 8.6 Hz, 1H), 7.38 (dd, J = 8.6, 2.6 Hz, 1H), 4.60 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 166.11, 165.23, 147.70, 142.59, 142.15, 139.37, 138.79, 136.95, 131.89, 131.35, 129.39 (2C), 128.76, 128.39, 128.37, 128.20, 127.90, 127.39, 124.11, 123.23, 118.94 (2C), 118.60, 118.08, 38.89; MS (m/z): 563 (M+, 30%), 420 (65%); Anal. Calcd. For C25H16Cl2N8O2S (563.42): C, 53.30; H, 2.86; N, 19.89. Found: C, 53.01; H, 2.77; N, 19.66%.
4.1.3.11. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]uinoxaline-3-ylthio)acetamido)-N-(4-fluorophenyl)benzamide (23k)
White powder (yield, 80%); m.p. = 250–252 °C. FT-IR (νmax, cm−1): 3289, 3108 (NH), 1645 (C=O), 1604 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.77 (s, 1H), 10.21 (s, 1H), 10.02 (s, 1H), 8.79 − 8.57 (m, 1H), 8.55 − 8.41 (m, 1H), 7.96 (t, J = 9.5 Hz, 2H), 7.78 (dt, J = 35.0, 12.1 Hz, 5H), 7.26 − 7.14 (m, 3H), 4.59 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 166.04, 165.20, 147.72, 142.11, 139.34, 138.78, 129.17 (2C), 128.36 (2C), 124.08, 123.19, 122.61 (2C), 122.57 (2C), 118.84 (2C), 118.58, 118.06, 115.68 (2C), 115.55 (2C), 38.90; MS (m/z): 513 (M+ + 1, 25%), 372 (100%); Anal. Calcd. For C25H17FN8O2S (512.52): C, 58.59; H, 3.34; N, 21.86. Found: C, 58.34; H, 3.22; N, 21.69%.
4.1.3.12. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]uinoxaline-3-ylthio)acetamido)-N-(2-hydroxyphenyl)benzamide (23l)
Off white crystal (yield, 71%); m.p. = 282–284 °C. FT-IR (νmax, cm−1): 3263 (NH), 1646 (C=O), 1600 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.78 (s, 1H), 10.02 (s, 1H), 9.76 (s, 1H), 9.46 (s, 1H), 8.60 (dd, J = 8.1, 1.4 Hz, 1H), 8.46 (dd, J = 8.0, 1.7 Hz, 1H), 7.97 − 7.96 (m, 2H), 7.75 − 7.73 (m, 3H), 7.69 (dt, J = 8.1, 2.1 Hz, 2H), 7.04 − 7.03 (m, 1H), 6.93 − 6.92 (m, 1H), 6.84 − 6.83 (m, 1H), 4.60 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 166.05, 165.10, 149.68, 147.72, 142.24, 142.10, 139.33, 138.77, 129.49, 129.08 (2C), 128.36, 128.34, 126.46, 126.03, 124.43, 124.06, 123.17, 119.53 (2C), 118.95, 118.57, 118.06, 116.50, 38.93; MS (m/z): 511 (M+ + 1, 70%), 293 (100%); Anal. Calcd. For C25H18N8O3S (510.53): C, 58.82; H, 3.55; N, 21.95. Found: C, 58.78; H, 3.50; N, 21.88%.
4.1.3.13. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]uinoxaline-3-ylthio)acetamido)-N-(4-hydroxyphenyl)benzamide (23m)
Pale yellow crystal (yield, 75%); m.p. = 285–287 °C. FT-IR (νmax, cm−1): 3414 (NH), 1601 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.75 (s, 1H), 10.01 (s, 1H), 9.93 (s, 1H), 9.26 (s, 1H), 8.63 − 8.54 (m, 1H), 8.51 − 8.39 (m, 1H), 7.93 (d, J = 8.7 Hz, 2H), 7.72 (dq, J = 14.7, 7.1, 6.6 Hz, 4H), 7.55 − 7.49 (m, 2H), 6.79 − 6.70 (m, 2H), 4.59 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 165.99, 164.72, 154.09 (2C), 147.74, 142.05, 139.30, 138.77, 131.21, 130.33, 128.98 (2C), 128.33 (2C), 124.01, 123.12, 122.74 (2C), 118.81(2C), 118.55, 118.02, 115.42 (2C), 38.92; MS (m/z): 511(M+ + 1, 30%); Anal. Calcd. For C25H18N8O3S (510.53): C, 58.82; H, 3.55; N, 21.95. Found: C, 58.63; H, 3.48; N, 21.73%.
4.1.3.14. 4-(2-(Bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]uinoxaline-3-ylthio)acetamido)-N-(2-hydroxy-4-nitrophenyl)benzamide (23n)
Red crystal (yield, 68%); m.p. = 265–267 °C. FT-IR (νmax, cm−1): 3294, 3101 (NH), 2927 (CH aliphatic), 1653 (C=O), 1603 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 11.14 (s, 1H), 10.82 (s, 1H), 10.02 (s, 1H), 9.49 (s, 1H), 8.61 (dd, J = 8.1, 1.5 Hz, 1H), 8.47 (dd, J = 7.8, 1.8 Hz, 1H), 8.25 (d, J = 8.9 Hz, 1H), 7.98 − 7.96 (m, 2H), 7.79 (dd, J = 8.9, 2.6 Hz, 1H), 7.78 − 7.75 (m, 3H), 7.74 (dd, J = 6.7, 2.1 Hz, 2H), 4.60 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 166.14, 165.06, 148.55, 147.70, 143.66, 142.72, 142.13, 139.35, 138.78, 133.63, 129.27 (2C), 128.90, 128.38, 128.36, 124.08, 123.20, 121.83, 119.05 (2C), 118.59, 118.07, 115.57, 109.89, 38.91; MS (m/z): 554 (M+ − 1, 70%); Anal. Calcd. For C25H17N9O5S (555.53): C, 54.05; H, 3.08; N, 22.69. Found: C, 53.85; H, 2.97; N, 22.43%.
4.1.4. General procedure for the synthesis of compounds 24a–c
A mixture of potassium salt of bis([1, 2, 4]triazolo)[4,3-a:3′,4′-c]quinoxaline-3-thiol 14 (0.5 g, 0.001 mol) and 2-chloro-N-(4–(2-(substituted)hydrazine-1-carbonyl)phenyl)acetamide 22a–c (0.001 mol) in DMF (50 ml) was heated on a water bath for 6 h. After cooling to room temperature, the reaction mixture was poured on crushed ice. The obtained precipitates were collected by filtration, dried, and crystalised from ethanol to give the target compounds 24a–c.
4.1.4.1. 2-(Bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)-N-(4–(2-(2-chlorobenzoyl)hydrazine-1-carbonyl)phenyl)acetamide (24a)
White crystal (yield, 65%); m.p. = 220–222 °C. FT-IR (νmax, cm−1): 3185 (NH), 1698 (C=O), 1603 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.77 (s, 1H), 10.54 (s, 1H), 10.37 (s, 1H), 10.03 (s, 1H), 8.63 (dd, J = 8.1, 1.5 Hz, 1H), 8.48 (dd, J = 7.9, 1.7 Hz, 1H), 7.93 (d, J = 8.7 Hz, 2H), 7.77 (dtd, J = 17.1, 7.5, 1.5 Hz, 2H), 7.72 (d, J = 8.7 Hz, 2H), 7.57 (ddd, J = 8.0, 4.0, 1.5 Hz, 2H), 7.53 (td, J = 7.6, 1.7 Hz, 1H), 7.48 (td, J = 7.4, 1.3 Hz, 1H), 4.60 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 166.25, 166.08, 165.40, 147.70, 142.36, 142.15, 139.37, 138.79, 135.18, 131.98, 130.93, 130.38, 129.90, 129.06 (2C), 128.37 (2C), 127.64 (2C), 124.12, 123.23, 118.91(2C), 118.60, 118.10, 38.94. MS (m/z): 572 (M+, 100%); Anal. Calcd. for C26H18ClN9O3S (572): C, 54.60; H, 3.17; N, 22.04. Found: C, 54.39; H, 3.09; N, 21.88%.
4.1.4.2. 2-(Bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)-N-(4–(2-(3-chlorobenzoyl)hydrazine-1-carbonyl)phenyl)acetamide (24b)
White crystal (yield, 63%); m.p. = 232–234 °C. FT-IR (νmax, cm−1): 3260, 3108 (NH), 1639 (C=O), 1603 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.78 (s, 1H), 10.62 (s, 1H), 10.49 (s, 1H), 10.02 (s, 1H), 8.62 (dd, J = 8.0, 1.5 Hz, 1H), 8.47 (dd, J = 7.8, 1.7 Hz, 1H), 7.96 (t, J = 1.9 Hz, 1H), 7.92 (d, J = 8.5 Hz, 2H), 7.91 − 7.88 (m, 1H), 7.78 − 7.76 (m, 1H), 7.75 − 7.71 (m, 3H), 7.70 − 7.69 (m, 1H), 7.59 (t, J = 7.9 Hz, 1H), 4.60 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 166.09, 165.67, 164.99, 147.71, 142.40, 142.13, 139.35, 138.78, 135.00, 133.84, 132.23, 131.11, 129.03 (2C), 128.38, 128.36, 127.72 (2C), 127.66, 126.66, 124.09, 123.21, 118.96, 118.59, 118.07, 38.92; MS (m/z): 572 (M+, 100%); Anal. Calcd. for C26H18ClN9O3S (572): C, 54.60; H, 3.17; N, 22.04. Found: C, 54.45; H, 3.11; N, 21.95%.
4.1.4.3. 2-(Bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxalin-3-ylthio)-N-(4–(2-(2-hydroxybenzoyl)hydrazine-1-carbonyl)phenyl)acetamide (24c)
Yellow crystal (yield, 60%); m.p. = 270–272 °C. FT-IR (νmax, cm−1): 3282 (NH), 1648, 1619 (C=O), 1599 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 11.96 (s, 1H), 10.78 (s, 1H), 10.67 (s, 1H), 10.59 (s, 1H), 10.02 (s, 1H), 8.62 (dd, J = 8.0, 1.5 Hz, 1H), 8.47 (dd, J = 7.8, 1.7 Hz, 1H), 7.93 (dd, J = 10.4, 7.6 Hz, 3H), 7.77 (ddt, J = 10.8, 8.3, 3.9 Hz, 2H), 7.74 (dd, J = 7.3, 4.8 Hz, 2H), 7.47 (ddd, J = 8.7, 7.2, 1.7 Hz, 1H), 7.00 − 6.95 (m, 2H), 4.59 (s, 2H); 13C NMR (176 MHz, DMSO-d6) δ 168.26, 166.09, 165.44, 159.79, 147.71, 142.45, 142.13, 139.35, 138.78, 134.65, 129.07 (2 C), 128.73, 128.38, 128.36, 127.52, 124.09, 123.21, 119.52, 118.96 (2C), 118.59, 118.07, 117.88, 115.01, 38.91; MS (m/z): 554 (M+ + 1, 100%); Anal. Calcd. for C26H19N9O4S (553.56): C, 56.41; H, 3.46; N, 22.77. Found: C, 56.20; H, 3.39; N, 22.58%.
4.2. Biological testing
4.2.1. In vitro anti-proliferative activity
MTT assay protocolCitation55–57,Citation74 was applied as described in Supplementary data.
4.2.2. In vitro VEGFR-2 kinase assay
All the synthesised compounds were tested for their inhibitory activity against VEGFR-2 as described in Supplementary dataCitation75.
4.2.3. Flow cytometry analysis for cell cycle
Cell cycle analysis was performed using propidium iodide (PI) staining and flow cytometry analysis for compound 23j as described in Supplementary dataCitation76,Citation77.
4.2.4. Flow cytometry analysis for apoptosis
Apoptotic effect was assessed for compound 23j as described in Supplementary dataCitation78,Citation79.
4.2.5. Western blot analysis
Western blot technique was performed for compound 23j to determine its effect against caspase3, caspase9, BAX, and Bcl-2 as described in Supplementary dataCitation80–82.
4.3. In silico studies
4.3.1. Docking studies
Docking studies were carried out against VEEGFR-2 (PDB ID: 2OH4) using Discovery Studio 4.0 as described in Supplementary dataCitation83–85,Citation86,Citation87.
4.3.2. Admet studies
ADMET studies were carried out as described in Supplementary dataCitation85,Citation87.
4.3.3. Toxicity studies
Toxicity studies were performed as described in Supplementary data.
Supplemental Material
Download PDF (7.3 MB)Acknowledgements
The authors extend their appreciation to the Deanship ofScientific Research at King Saud University for funding this workthrough research group no [RG-1441–364].
Disclosure statement
No potential conflict of interest was reported by the author(s). The authors would like to acknowledge Dr. Mohamed R. Elnagar, Department of Pharmacology and Toxicology, Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt for his valuable suggestion and technical assistance.
Additional information
Funding
Related Research Data
References
- Madani I, De Neve W, Mareel M. Does ionizing radiation stimulate cancer invasion and metastasis? Bulletin du Cancer 2008;95:292–300.
- Konsoulova A. Principles of cancer immunobiology and immunotherapy of solid tumors, Immunopathology and immunomodulation. Rijeka (Croatia): InTech; 2015:77–99.
- Jadala C, Sathish M, Reddy TS, et al. Synthesis and in vitro cytotoxicity evaluation of β-carboline-combretastatin carboxamides as apoptosis inducing agents: DNA intercalation and topoisomerase-II inhibition. Bioorg Med Chem 2019;27:3285–98.
- Jafari F, Baghayi H, Lavaee P, et al. Design, synthesis and biological evaluation of novel benzo-and tetrahydrobenzo-[h] quinoline derivatives as potential DNA-intercalating antitumor agents. Eur J Med Chem 2019;164:292–303.
- El-Adl K, El-Helby A-GA, Sakr H, et al. Design, synthesis, molecular docking and anticancer evaluations of 5-benzylidenethiazolidine-2,4-dione derivatives targeting VEGFR-2 enzyme . Bioorg Chem 2020;102:104059.
- Shen F-Q, Shi L, Wang Z-F, et al. Design, synthesis, biological evaluation of benzoyl amide derivatives containing nitrogen heterocyclic ring as potential VEGFR-2 inhibitors. Bioorg Med Chem 2019;27:3813–24.
- Fan H, Wei D, Zheng K, et al. Discovery of dioxino[2,3-f]quinazoline derivative VEGFR-2 inhibitors exerting significant antipro-liferative activity in HUVECs and mice . Eur J Med Chem 2019;175:349–56.
- Yuan X, Yang Q, Liu T, et al. Design, synthesis and in vitro evaluation of 6-amide-2-aryl benzoxazole/benzimidazole derivatives against tumor cells by inhibiting VEGFR-2 kinase . Eur J Med Chem 2019;179:147–65.
- Ribatti D, Vacca A, Dammacco F. The role of the vascular phase in solid tumor growth: a historical review. Neoplasia 1999;1:293–302.
- Nagy J, Chang S, Dvorak A, Dvorak H. Why are tumour blood vessels abnormal and why is it important to know? Br J Cancer 2009;100:865–9.
- Zirlik K, Duyster J. Anti-angiogenics: current situation and future perspectives. Oncol Res Treat 2018;41:166–71.
- Sullivan LA, Brekken RA. The VEGF family in cancer and antibody-based strategies for their inhibition, MAbs. London, UK: Taylor & Francis; 2010: 165–175.
- Bagnasco L, Piras D, Parodi S, et al. Role of angiogenesis inhibitors in colorectal cancer: sensitive and insensitive tumors. Curr Cancer Drug Targets 2012;12:303–15.
- Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis 2010;13:1–14.
- Modi SJ, Kulkarni VM. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med Drug Discov 2019;2:100009.
- Yadav L, Puri N, Rastogi V, et al. Tumour angiogenesis and angiogenic inhibitors: a review. J Clin Diagn Res 2015;9:XE01–XE05.
- Sobhy MK, Mowafy S, Lasheen DS, et al. 3D-QSAR pharmacophore modelling, virtual screening and docking studies for lead discovery of a novel scaffold for VEGFR 2 inhibitors: design, synthesis and biological evaluation. Bioorg Chem 2019;89:102988.
- Niu G, Chen X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr Drug Targets 2010;11:1000–17.
- Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes & Cancer 2011;2:1097–105.
- Roskoski R Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol/Hematol 2007;62:179–213.
- Marzouk AA, Abdel-Aziz SA, Abdelrahman KS, et al. Design and synthesis of new 1,6-dihydropyrimidin-2-thio derivatives targeting VEGFR-2: molecular docking and antiproliferative evaluation . Bioorg Chem 2020;102:104090.
- Ge Y-L, Zhang X, Zhang J-Y, et al. The mechanisms on apoptosis by inhibiting VEGF expression in human breast cancer cells. Int Immunopharmacol 2009;9:389–95.
- Ghorab MM, Alsaid MS, Soliman AM, Ragab FA. VEGFR-2 inhibitors and apoptosis inducers: synthesis and molecular design of new benzo[g]quinazolin bearing benzenesulfonamide moiety. J Enzyme Inhib Med Chem 2017;32:893–907.
- El-Gazzar MG, El-Hazek RM, Zaher NH, El-Ghazaly MA. Design and synthesis of novel pyridazinoquinazoline derivatives as potent VEGFR-2 inhibitors: in vitro and in vivo study. Bioorg Chem 2019;92:103251.
- Fukumura D, Kloepper J, Amoozgar Z, et al. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol 2018;15:325–40.
- Zaman S, Wang R, Gandhi V. Targeting the apoptosis pathway in hematologic malignancies. Leuk Lymphoma 2014;55:1980–92.
- Ragab FA, Nissan YM, Seif EM, et al. Synthesis and in vitro investigation of novel cytotoxic pyrimidine and pyrazolopyrimidne derivatives showing apoptotic effect. Bioorg Chem 2020;96:103621.
- AbdelHaleem A, Mansour AO, AbdelKader M, Arafa RK. Selective VEGFR-2 inhibitors: synthesis of pyridine derivatives, cytotoxicity and apoptosis induction profiling. Bioorg Chem 2020;103:104222.
- Lee K, Jeong K-W, Lee Y, et al. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur J Med Chem 2010;45:5420–7.
- Xie Q-Q, Xie H-Z, Ren J-X, et al. Pharmacophore modeling studies of type I and type II kinase inhibitors of Tie2. J Mol Graph Model 2009;27:751–8.
- Eskander RN, Tewari KS. Incorporation of anti-angiogenesis therapy in the management of advanced ovarian carcinoma-mechanistics, review of phase III randomized clinical trials, and regulatory implications. Gynec Oncol 2014;132:496–505.
- Machado VA, Peixoto D, Costa R, et al. Synthesis, antiangiogenesis evaluation and molecular docking studies of 1-aryl-3-[(thieno [3, 2-b] pyridin-7-ylthio) phenyl] ureas: discovery of a new substitution pattern for type II VEGFR-2 Tyr kinase inhibitors. Bioorg Med Chem 2015;23:6497–509.
- Wang Z, Wang N, Han S, et al. Dietary compound isoliquiritigenin inhibits breast cancer neoangiogenesis via VEGF/VEGFR-2 signaling pathway. PLOS One 2013;8:e68566.
- Dietrich J, Hulme C, Hurley LH. The design, synthesis, and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: a structural analysis of the binding interactions of Gleevec®, Nexavar®, and BIRB-796. Bioorg Med Chem 2010;18:5738–48.
- Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 2006;5:835–44.
- Lintnerová L, García-Caballero M, Gregáň F, et al. A development of chimeric VEGFR2 TK inhibitor based on two ligand conformers from PDB: 1Y6A complex-medicinal chemistry consequences of a TKs analysis. Eur J Med Chem 2014;72:146–59.
- Matsui J, Funahashi Y, Uenaka T, et al. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res 2008;14:5459–65.
- Peng F-W, Liu D-K, Zhang Q-W, et al. VEGFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012-2016). Expert Opin Ther Pat 2017;27:987–1004.
- Zhang Y, Zou J-Y, Wang Z, Wang Y. Fruquintinib: a novel antivascular endothelial growth factor receptor tyrosine kinase inhibitor for the treatment of metastatic colorectal cancer. Cancer Manage Res 2019;11:7787–803.
- Mahdy HA, Ibrahim MK, Metwaly AM, et al. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4(3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers . Bioorg Chem 2020;94:103422.
- Eissa IH, El-Helby A-GA, Mahdy HA, et al. Discovery of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors: design, synthesis, and anti-proliferative evaluation . Bioorg Chem 2020;105:104380.
- El-Zahabi MA, Sakr H, El-Adl K, et al. Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents. Bioorg Chem 2020;104:104218.
- Elmetwally SA, Saied KF, Eissa IH, Elkaeed EB. Design, synthesis and anticancer evaluation of thieno[2,3-d]pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers . Bioorg Chem2019;88:102944.
- Ibrahim M, Taghour M, Metwaly A, et al. Design, synthesis, molecular modeling and anti-proliferative evaluation of novel quinoxaline derivatives as potential DNA intercalators and topoisomerase II inhibitors. Eur J Med Chem 2018;155:117–34.
- Eldehna WM, Abo-Ashour MF, Nocentini A, et al. Novel 4/3-((4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylidene)amino) benzenesulfonamides: Synthesis, carbonic anhydrase inhibitory activity, anticancer activity and molecular modelling studies . Eur J Med Chem 2017;139:250–62.
- Eissa IH, El-Naggar AM, El-Hashash MA. Design, synthesis, molecular modeling and biological evaluation of novel 1H-pyrazolo [3, 4-b] pyridine derivatives as potential anticancer agents. Bioorg Chem 2016;67:43–56.
- El-Naggar AM, Abou-El-Regal MM, El-Metwally SA, et al. Synthesis, characterization and molecular docking studies of thiouracil derivatives as potent thymidylate synthase inhibitors and potential anticancer agents. Mol Divers 2017;21:967–83.
- Alsaif NA, Dahab MA, Alanazi MM, et al. New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: design, molecular modeling, and synthesis. Bioorg Chem 2021;110:104807.
- Romer DR. Synthesis of 2, 3‐dichloroquinoxalines via Vilsmeier reagent chlorination. J Heterocycl Chem 2009;46:317–9.
- Sarges R, Howard HR, Browne RG, et al. 4-Amino[1,2,4]triazolo[4,3-a]quinoxalines. A novel class of potent adenosine receptor antagonists and potential rapid-onset antidepressants . J Med Chem 1990;33:2240–54.
- Könnecke A, Lippmann E. Kondensierte Chinoxaline aus 2‐Chlor‐3‐hydrazinochinoxalin. Zeitschrift Für Chemie 2010;18:92–3.
- Kanhed AA, Mehere AP, Pandey KR, Mahapatra DK. 4-(2-chloroacetamido) Benzoic acid derivatives as local anesthetic agents: design, synthesis, and characterization. UK J Pharm Biosci 2016;4:35–44.
- El-Helby A-GA, Ayyad RR, El-Adl K, Elkady H. Phthalazine-1,4-dione derivatives as non-competitive AMPA receptor antagonists: design, synthesis, anticonvulsant evaluation, ADMET profile and molecular docking . Mol Divers 2019;23:283–98.
- El‐Helby AGA, Ayyad RR, Zayed MF, et al. Design, synthesis, in silico ADMET profile and GABA‐A docking of novel phthalazines as potent anticonvulsants. Archiv Der Pharmazie 2019;352:1800387.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986;89:271–7.
- Thabrew M, HUGHES RD, MCFARLANE IG. Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay. J Pharm Pharmacol 2011;49:1132–5.
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2006;2:358–64.
- Klopman G, Stefan LR, Saiakhov RD. ADME evaluation. 2. A computer model for the prediction of intestinal absorption in humans. Eur J Pharm Sci 2002;17:253–63.
- Mannhold R, Kubinyi H, Folkers G Pharmacokinetics and metabolism in drug design. Weinheim, Germany: John Wiley & Sons; 2012.
- Roy PP, Roy K. QSAR studies of CYP2D6 inhibitor aryloxypropanolamines using 2D and 3D descriptors. Chem Biol Drug Des 2009;73:442–55.
- Ghafourian T, Amin Z. QSAR models for the prediction of plasma protein binding. BioImpacts: BI 2013;3:21.
- Van De Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov 2003;2:192–204.
- Xia X, Maliski EG, Gallant P, Rogers D. Classification of kinase inhibitors using a Bayesian model. J Med Chem 2004;47:4463–70.
- BIOVIA. QSAR, ADMET and Predictive Toxicology. https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html [last accessed May 2020]
- Peto R, Pike M C, Bernstein L, et al. The TD50: a proposed general convention for the numerical description of the carcinogenic potency of chemicals in chronic-exposure animal experiments. Environ Health Perspect 1984; 58, 1–8.
- Goodman G, Wilson R. Comparison of the dependence of the TD50 on maximum tolerated dose for mutagens and nonmutagens. Risk Anal 1992;12:525–33.
- Council NR. Correlation between carcinogenic potency and the maximum tolerated dose: implications for risk assessment, issues in risk assessment. USA: National Academies Press; 1993.
- Louisse J, Bosgra S, Blaauboer BJ, et al. Prediction of in vivo developmental toxicity of all-trans-retinoic acid based on in vitro toxicity data and in silico physiologically based kinetic modeling. Arch Toxicol 2015;89:1135–48.
- EPA. Guidelines for Developmental Toxicity Risk Assessment. https://www.epa.gov/sites/production/files/2014-11/documents/dev_tox.pdf [last accessed May 2020].
- Diaza RG, Manganelli S, Esposito A, et al. Comparison of in silico tools for evaluating rat oral acute toxicity. SAR QSAR Environ Res 2015;26:1–27.
- Pizzo F, Benfenati E. In silico models for repeated-dose toxicity (RDT): prediction of the no observed adverse effect level (NOAEL) and lowest observed adverse effect level (LOAEL) for drugs, in silico methods for predicting drug toxicity. New York: Springer; 2016:163–176.
- Venkatapathy R, Moudgal CJ, Bruce RM. Assessment of the oral rat chronic lowest observed adverse effect level model in TOPKAT, a QSAR software package for toxicity prediction. J Chem Inform Comput Sci 2004;44:1623–9.
- Al-Rashood ST, Hamed AR, Hassan GS, et al. Antitumor properties of certain spirooxindoles towards hepatocellular carcinoma endowed with antioxidant activity. J Enzyme Inhib Med Chem 2020;35:831–9.
- Abou-Seri SM, Eldehna WM, Ali MM, Abou El Ella DA. 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: synthesis and in vitro biological evaluation. Eur J Med Chem 2016;107:165–79.
- Wang J, Lenardo MJ. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci 2000;113:753–7.
- Eldehna WM, Hassan GS, Al-Rashood ST, et al. Synthesis and in vitro anticancer activity of certain novel 1-(2-methyl-6-arylpyridin-3-yl)-3-phenylureas as apoptosis-inducing agents. J Enzyme Inhib Med Chem 2019;34:322–32.
- Lo KK-W, Lee TK-M, Lau JS-Y, et al. Luminescent biological probes derived from ruthenium(II) estradiol polypyridine complexes. Inorg Chem 2008;47:200–8.
- Sabt A, Abdelhafez OM, El-Haggar RS, et al. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: synthesis, in vitro biological evaluation, and QSAR studies. J Enzyme Inhib Med Chem 2018;33:1095–107.
- Balah A, Ezzat O, Akool E-S. Vitamin E inhibits cyclosporin A-induced CTGF and TIMP-1 expression by repressing ROS-mediated activation of TGF-β/Smad signaling pathway in rat liver. Int Immunopharmacol 2018;65:493–502.
- Aborehab NM, Elnagar MR, Waly NE. Gallic acid potentiates the apoptotic effect of paclitaxel and carboplatin via overexpression of Bax and P53 on the MCF‐7 human breast cancer cell line. J Biochem Mol Toxicol 2021;35:e22638.
- Elnagar MR, Walls AB, Helal GK, et al. Functional characterization of α7 nicotinic acetylcholine and NMDA receptor signaling in SH-SY5Y neuroblastoma cells in an ERK phosphorylation assay. Eur J Pharmacol 2018;826:106–13.
- Ibrahim MK, Eissa IH, Abdallah AE, et al. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of novel quinoxaline derivatives as potential PPARγ and SUR agonists. Bioorg Med Chem 2017;25:1496–513.
- El-Helby AGA, Ayyad RR, Sakr HM, et al. Design, synthesis, molecular modeling and biological evaluation of novel 2,3-dihydrophthalazine-1,4-dione derivatives as potential anticonvulsant agents. J Mol Struct 2017;1130:333–51.
- Ibrahim MK, Eissa IH, Alesawy MS, et al. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of quinazolin-4(3H)-one derivatives as potential PPARγ and SUR agonists . Bioorg Med Chem 2017;25:4723–44.
- El-Gamal KM, El-Morsy AM, Saad AM, et al. Synthesis, docking, QSAR, ADMET and antimicrobial evaluation of new quinoline-3-carbonitrile derivatives as potential DNA-gyrase inhibitors. J Mol Struct 2018;1166:15–33.
- El-Zahabi MA, Elbendary ER, Bamanie FH, et al. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of phthalimide-sulfonylurea hybrids as PPARγ and SUR agonists. Bioorg Chem 2019;91:103115.