
Abstract
A series of 1,2,3-trisubstituted indolizines (2a–2f, 3a–3d, and 4a–4c) were screened for in vitro whole-cell anti-tubercular activity against the susceptible H37Rv and multidrug-resistant (MDR) Mycobacterium tuberculosis (MTB) strains. Compounds 2b–2d, 3a–3d, and 4a–4c were active against the H37Rv-MTB strain with minimum inhibitory concentration (MIC) ranging from 4 to 32 µg/mL, whereas the indolizines 4a–4c, with ethyl ester group at the 4-position of the benzoyl ring also exhibited anti-MDR-MTB activity (MIC = 16–64 µg/mL). In silico docking study revealed the enoyl-acyl carrier protein reductase (InhA) and anthranilate phosphoribosyltransferase as potential molecular targets for the indolizines. The X-ray diffraction analysis of the compound 4b was also carried out. Further, a safety study (in silico and in vitro) demonstrated no toxicity for these compounds. Thus, the indolizines warrant further development and may represent a novel promising class of InhA inhibitors and multi-targeting agents to combat drug-sensitive and drug-resistant MTB strains.
1. Introduction
Tuberculosis (TB) is a communicable infectious disease and a major cause of illness, particularly in low-income countries. It is caused by the opportunistic bacillus Mycobacterium tuberculosis (MTB) which primarily attacks the lungs (pulmonary) but may later affect other parts (extra-pulmonary) of the bodyCitation1. According to the World Health Organisation (WHO), TB is considered as one of the top 10 causes of death worldwide, and the leading cause of death from a single infectious agentCitation1. In 2019, TB resulted in nearly 1.4 million deaths, including 208,000 deaths among human immunodeficiency virus (HIV) positive patientsCitation2. HIV-infected patients are 19 times more likely to develop TB than HIV-negative subjectsCitation3,Citation4. Several factors have contributed to the continuous health threat of TB globally. This includes the development of drug resistance such as multidrug-resistant tuberculosis (MDR-TB), extensively drug-resistant tuberculosis (XDR-TB)Citation5, and totally drug-resistant tuberculosis (TDR-TB)Citation6; the co-morbidities with acquired immunodeficiency syndrome (AIDS)Citation7,Citation8 and the risks involved in developing diabetes mellitus among TB patientsCitation9,Citation10.
Several therapeutics have been approved by the US Food and Drug Administration (US-FDA) to enhance the treatment of TB. Bedaquiline (approved in late 2012 under the FDA’s accelerated review program) () was found to exhibit favourable anti-TB effects and promising anti-TB action against MDR and XDR-TB when combined with first-line or second-line anti-TB drugs, resulting in a shorter duration of treatment. However, bedaquiline suffered from various adverse effects such as nausea, elongation of QT interval, and drug interaction with Cytochrome P3A4 inducers and inhibitorsCitation11. Delamanid () was the second anti-TB agent approved by European Medicine Agency in late 2013. Nevertheless, resistant MTB strains against both bedaquiline and delamanid have recently been reported, which acquired resistance as a result of prolonged duration of therapyCitation12,Citation13. Pretomanid was the last drug candidate from the TB drug pipeline to be approved by the FDA in 2019 for the treatment of MDR and XDR TB ()Citation14,Citation15.
Figure 1. Chemical structures of approved anti-TB drugs (Bedaquiline, Delamanid and Pretomanid) and anti-TB compounds undergoing clinical trial (Q203, TBA-7371 and SQ109).
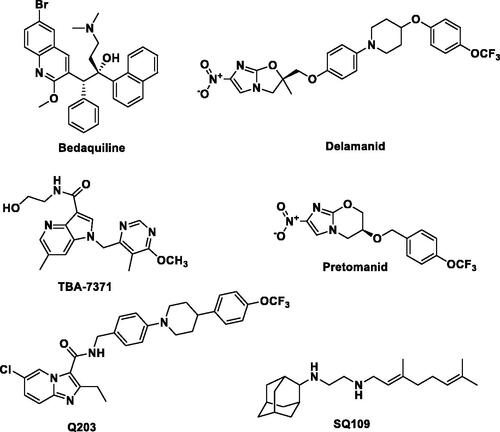
For more than 40 years, there has been no significant active research in the arena of anti-TB drug discovery despite the fact that currently available drugs are considered cytotoxic and have low efficacy due to the emergence of resistant bacterial strainsCitation16. A recent call for the development of novel potential therapeutic agents to overcome MDR, XDR and TDR-TB have led to the discovery of a limited number of anti-TB drug candidatesCitation17. Among them, TBA-7371 (an azaindole) and Q203 or telacebac (an imidazopyridine amide) are very promising drug candidates for the potential treatment of MDR and XDR TB (). However, considering the high attrition rate of drug candidates during clinical development, there is an urgent need to intensify and broaden the scope of the search for novel anti-TB lead structures by utilising validated new mycobacterial drug targets besides those targeted by current anti-TB therapies. A promising emerging approach to overcome MDR-TB is the development of multi-targeting compounds in which a single molecule has the ability to bind to different biological targetsCitation18. This concept is known as polypharmacology and has been proven promising in terms of efficacy, synergistic effect, and adverse events, and in preventing both drug-drug interaction and resistance insurgenceCitation18. For instance, SQ109, a drug candidate in the TB drug development pipeline, exhibits its anti-TB activity by acting on multiple targets (). SQ109 is a well-known MmpL3 inhibitor that also shows inhibitory activity against MenA and MenG MTB enzymes as wellCitation19. It is worth mentioning that recently several natural products have also been reported as lead molecules against clinical MDR strains of MTBCitation20. Therefore, the discovery of novel chemical entities having multiple modes of action is of paramount importance in the treatment of MDR, XDR, and TDR-TB infections.
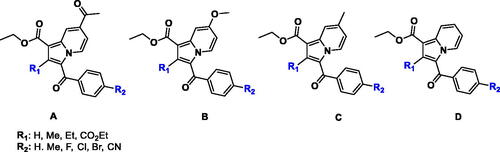
This study stems from our interest in designing new potential anti-TB therapeutics by focussing on structural modifications of the indolizine scaffold which has demonstrated potential anti-mycobacterial activity as previously reported by our groupCitation21–23 and other researchersCitation24–27. Previously, we identified a series of poly-substituted indolizine ACitation21, BCitation22, and CCitation23 as promising anti-TB agents against MDR-TB (). Based on our previously reported structure-activity relationship (SAR), the nature and the position of substituents on the indolizine scaffold were found to be very sensitive to retain the anti-mycobacterial activity against susceptible MTB strain as well as rifampicin and isoniazid-resistant clinical isolates of MTB. Approximately, two-thirds of the synthesised indolizines A, B, and C displayed moderate to good potency against susceptible H37Rv MTB strain, whilst half of the active derivatives demonstrated good inhibitory activity against MDR-MTB strain. Indolizine represents a privileged scaffold for the development of bioactive compounds. Several synthetic indolizines have also been reported to possess a broad spectrum of pharmacological activities such as analgesic, anti-inflammatory, anticancer, antidiabetic, antihistaminic, COX-2 inhibition, antileishmanic, antimicrobial, antimutagenic, antioxidant, antiviral, larvicidal, herbicidal and α7 nAChR inhibitors, anti-Alzheimer, antischizophrenic, anticonvulsant and inhibitors of various enzymesCitation28–30.
Figure 2. Indolizine structures with anti-TB activity developed by our group.
Despite the structural resemblance in the main bicyclic core of our designed compounds with Q203 and TBA-7371, to date, no indolizine-based anti-TB drug candidates have entered preclinical trials. With regards to the above considerations and as part of our continuous interest in the discovery of new potential anti-TB drugsCitation31–41 we report herein the anti-mycobacterial activity of a series of 1,2,3-trisubstituted indolizines of type D with the aim of expanding the current knowledge on our previously reported SAR by exploring the influence of substituents at 1, 2 and 3-position of the indolizine scaffold that might play a critical role in the anti-TB activity against H37Rv strain ATCC 25177 and MDR strains of MTB. Furthermore, the potential mechanism of action of the title compounds is suggested based on their anti-tubercular profile in parallel with the molecular modelling analyses. In addition, the ADMET and toxicity characteristics of the most promising indolizines are also presented.
2. Materials and methods
2.1. Chemistry
The chemicals reported here were obtained from Sigma-Aldrich Co. (St. Louis, MO, USA), while the solvents were obtained from Millipore Sigma (Burlington, MA, USA). Thin-layer chromatography (TLC) using silica gel (Sigma-Aldrich Co.) on aluminium foil was employed to observe the chemical reactions; n-hexane and ethyl acetate (4:6) were used as solvents. The reactions were visualised under an ultraviolet (UV)-light/iodine chamber. B-545 was used to measure the melting points (Büchi, Labortechnik, Flawil, Switzerland). Fourier-transform infrared (FT-IR) spectra were recorded on a Shimadzu FT-IR spectrophotometer. Furthermore, 1H- and 13C-NMR spectra were recorded on Bruker AVANCE III 400 MHz instruments using DMSO-d6 as a solvent. Chemical shifts (δ) were recorded in parts per million (ppm) downfield from tetramethylsilane, while the coupling constants (J) were recorded in Hz. The splitting pattern was documented as follows: s, singlet; d, doublet; q, quartette; m, multiplet. Liquid chromatography–mass spectrometry (LC–MS; Agilent 1100 series) was used to measure the mass spectra in conjunction with the MSD, as well as 0.1% aqueous trifluoroacetic acid in an acetonitrile system on the C18-BDS column. Then, elemental analysis was carried out using the analyser FLASH EA 1112 CHN (Thermo Finnigan LLC, New York, NY, USA). A single-crystal X-ray diffraction study was performed using a Bruker KAPPA APEX II DUO diffractometer equipped with a CCD detector; monochromated Mo Kα radiation (λ = 0.71073 Å) was used. Data collection was carried out at 173(2) K using an Oxford Cryostream cooling system featuring the Bruker Apex II software.
2.1.1. General synthetic procedure for the preparation of 1-(2-(substitutedphenyl)-2-oxoethyl)pyridin-1-ium bromides (1a-f) and ethyl 3-(substitutedbenzoyl)-2-methylindolizine-1-carboxylates (2a–2f and 3a–3d)
Compounds 1a–1f, 2a–2f, and 3a–3d were synthesised, purified by column chromatography, and well characterised by FT-IR, NMR, LC-MS, and elemental analysis as reported earlier from our research groupCitation42. The purity of the compounds was ≥99% as measured by HPLC.
2.1.2. General procedure for the preparation of diethyl-3-(4-substitutedbenzoyl)indolizine-1,2-dicarboxylates (4a–4c)
To a stirred solution of 1-(2-(4-fluoro/chloro/nitro-phenyl)-2-oxoethyl)pyridin-1-ium bromide (1a/1b/1d) (0.0016 mol), in water (10 mL), was added substituted diethyl 2-butynedioate (0.0016 mol), stirred at 80 °C for 3 h. Completion of reaction was monitored on TLC. The reaction mixture was diluted with ethyl acetate. The organic layer was separated, washed with brine and dried under sodium sulphate. The crude compound was purified by recrystallization method using hexane and ethyl acetate to afford 69–83% yield of diethyl-3-(4-substitutedbenzoyl)indolizine-1,2-dicarboxylate (4a–4c). The characterisation details of title compounds (4a–4c) are reported below, and their spectral data are shown in Figures S1–S9.
2.1.2.1. Diethyl-3-(4-fluorobenzoyl)indolizine-1,2-dicarboxylate (4a)
Appearance; Brown solid. Yield 72%, MP 138–139, FT-IR (KBr neat cm−1): 2985, 1733, 1695, 1635, 1608, 1228. 1H NMR (400 MHz, CDCl3) δ = 9.51–9.48 (d, J = 7.2 Hz, 1H), 7.78–7.70 (m, 3H), 7.16–7.11 (m, 2H), 6.82–6.77 (m, 1H), 4.36–4.30 (q, J = 7.2 Hz, 2H), 3.76–3.68 (q, J = 7.2 Hz, 2H), 1.37–1.33 (t, J = 7.2 Hz, 3H), 1.13–1.11 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz CDCl3) δ = 183.77, 165.17, 164.12, 163.62, 163.23, 159.92, 141.51, 136.09, 136.02, 132.21, 131.59, 131.23, 129.89, 119.59, 115.62, 114.87, 110.18, 102.38, 97.63, 61.66, 60.28, 55.87, 14.32, 13.52. LC-MS (ESI, Positive): m/z: (M + H)+: 384.38: Anal. calculated for: C21H18FNO5; C, 65.79; H, 4.73; N, 3.65; Found; C, 65.41; H, 4.50; N, 3.55.
2.1.2.2. Diethyl-3-(4-chlorobenzoyl)indolizine-1,2-dicarboxylate (4b)
Appearance; Brown solid. Yield 74%, MP 137–138, FT-IR (KBr neat cm−1): 2989, 1736, 1696, 1637, 1605, 1227. 1H NMR (400 MHz, CDCl3) δ = 9.53–9.49 (d, J = 7.2 Hz, 1H), 7.76–7.71 (m, 3H), 7.15–7.10 (m, 2H), 6.82–6.77 (m, 1H), 4.37–4.30 (q, J = 7.2 Hz, 2H), 3.72–3.68 (q, J = 7.2 Hz, 2H), 1.37–1.33 (t, J = 7.2 Hz, 3H), 1.13–1.10 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz CDCl3) δ = 183.51, 165.34, 164.15, 163.59, 163.30, 160.21, 141.49, 136.07, 135.89, 132.23, 131.28, 131.16, 129.91, 119.48, 115.49, 114.99, 110.24, 102.74, 97.69, 61.78, 60.36, 55.71, 14.42, 13.83. LC-MS (ESI, Positive): m/z: (M + H)+: 400.2: Anal. calculated for: C21H18ClNO2; C, 63.09; H, 4.54; N, 3.50; Found; C, 62.91; H, 4.48; N, 3.45.
2.1.2.3. Diethyl-3-(4-nitrobenzoyl)indolizine-1,2-dicarboxylate (4c)
Appearance; Light green colour. Yield 70%, MP 111–112, FT-IR (KBr neat cm−1): 2983, 1739, 1700, 1649, 1603, 1225. 1H-NMR (400 MHz CDCl3) δ = 9.49–9.47 (d, J = 7.2 Hz, 1H), 7.77–7.72 (m, 3H), 7.15–7.11 (m, 2H), 6.81–6.78 (m, 1H), 4.38–4.32 (q, J = 7.2 Hz, 2H), 3.75–3.69 (q, J = 7.2 Hz, 2H), 1.36–1.33 (t, J = 7.2 Hz, 3H), 1.13–1.11 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz CDCl3) δ = 184.77, 166.12, 165.08, 163.59, 163.29, 159.91, 142.47, 137.04, 136.03, 132.21, 131.25, 131.18, 129.95, 119.52, 116.59, 114.93, 109.15, 103.44, 98.59, 61.58, 60.29, 55.87, 14.31, 13.72. LC-MS (ESI, Positive): m/z: (M + H)+: 411.2: Anal. calculated for: C21H18N2O7; C, 61.46; H, 4.42; N, 6.83; Found; C, 61.31; H, 4.30; N, 6.55.
2.2. Crystallography
Single-crystal X-ray diffraction data of the compound 4b was collected on a Bruker KAPPA APEX II DUO diffractometer using graphite-monochromated Mo-Kα radiation (χ = 0.71073 Å). Data collectionCitation43 was carried out at 173(2) K. Temperature was controlled by an Oxford Cryostream cooling system (Oxford Cryostat). Cell refinement and data reduction were performed using the SAINT programCitation44. The data were scaled and absorption correction was performed using SADABSCitation44.
The structure was solved by direct methods using SHELXS-97Citation45 and refined by the full-matrix least-squares method based on F2 using SHELXL-2014Citation46. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms, were placed in idealised positions and refined in riding models with Uiso assigned 1.2 or 1.5 times the Ueq of their parent atoms, and the C–H bond distances were constrained to 0.95 Å for aromatic hydrogens and 1.00 Å for methylene and methyl hydrogens. All geometrical calculations were done using PLATONCitation47. The program WinGxCitation48 was used to prepare molecular graphic images.
2.3. Hirshfeld surface analysis and energy framework calculation
The Hirshfeld surfaces analysis is a technique for visualisation and investigation to illustrate the nature of intermolecular interactions. The Hirshfeld surfaces and two-dimensional fingerprint plot for compound 4b have been generated using the software Crystal Explorer 17.5. This software has been also used to assess the interaction energies of the compound 4b.
2.4. Anti-tubercular activity
The antitubercular activity of the designed compounds 2a–2f, 3a–3d, and 4a–4c were evaluated against two types of MTB strains, namely H37Rv and well characterised MDR strains using the colorimetric Resazurin Microplate Assay (REMA) methodCitation49. A 100 µL of Middelbrook 7H9 broth was aseptically prepared and dispensed in each of the wells of a 96 well flat-bottomed microtiter plate with lids (Lasec, South Africa). Each of the test compounds was accurately weighed, dissolved in the appropriate solvent, and filter sterilised using a 0.2 micron polycarbonate filter. Stock solutions of the test samples were aliquoted into cryovials and stored at −20 °C. A 100 µL of the test samples were added to each of the wells containing Middlebrook 7H9 broth supplemented with 0.1% Casitone, 0.5% glycerol, and 10% OADC (oleic acid, albumin, dextrose, and catalase). The test samples were then serially diluted two folds directly in the broth of the microtiter plate to the desired concentration ranging from 40–0.625 µg/mL.
Inoculums from clinical isolates were prepared fresh from Middlebrook 7H11 agar plates by scraping and re-suspending loopful of colonies into Middlebrook 7H9 broth containing glass beads. The inoculum turbidity was adjusted to a McFarland number 1 standard and further diluted to 1:10 in M7H9 broth prior to the addition of 100 µL to each of the test samples and drug-free wells. Growth control and a sterile control were also included for each isolate. Sterile M7H9 broth was added to all perimeter walls to avoid evaporation during the incubation. The plate was covered, sealed in a plastic bag, and incubated at 37 °C. After 8 days of incubation, 30 µL of 0.02% working solution of resazurin salt was inoculated into each microtiter well. The plates were then incubated overnight and read the following day. A positive reaction resulted in a colour change from blue to pink owing to the reduction of resazurin to rezarufin which confirmed MTB cell viability/growth and, hence, drug resistance. The MICs were defined as the minimum drug concentration to inhibit the growth of the organism with no colour changes present in the well.
2.5. Safety studies (in vitro)
The safety of the tested indolizines was evaluated by MTT assay. The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) cytotoxicity assay was used to evaluate the cytotoxic effect of the most promising compounds against peripheral blood mononuclear cells (PBMCs) according to the described protocolCitation50. Cells were pipetted (90 µL of cell culture, 1 × 105 cells/mL) into each well of 96-well microtiter plates, and the outer wells were filled with PBS (phosphate buffer saline) in order to prevent the medium from evaporation during incubation. Thereafter, plates were incubated at 37 °C for 24 h. Each well of the plate was then treated with 10 µL of the compounds (1000–5 µg/mL). In the control wells, the negative control DMSO (dimethyl sulfoxide) and media were added. Thereafter, the plates were incubated for 2 days at 37 °C in a humidified incubator that contained a 5% CO2 atmosphere. After the incubation time, 20 µL of MTT reagent (5 mg/mL) was further added to the individual well. The plate was then incubated for a further 4 h at 37 °C (5% CO2 incubator). The media was then removed, and an aliquot of 100 µL DMSO was added to each well in order to dissolve the formazan crystals that were formed in metabolically active cells. After that, plates were incubated for an extra hour. The absorbance of the formazan was evaluated at 590 nm using an ELISA plate reader (Thermo Scientific Multiskan GO).
2.6. Molecular modelling
Molecular docking of the designed compounds into the active site of proposed potential targets was performed using Accelrys Discovery Studio 4.0 (from BIOVIA Software Inc.). The X-ray crystal structures of mycobacterial enyol-ACP-reductase (InhA) and anthranilate phosphoribosyltransferase (trpD) enzymes in complex with their inhibitors (PDB accession codes 5G0S and 3R6C, respectively) were retrieved from the RSCB Protein Data Bank.
Crystal complexes were prepared using the clean protein tool and prepare protein protocol in DS to standardise atoms names, correct connectivity and bond order, protonate the ionisable residues at a pH of 7.4, add missing residues and correct incomplete ones, and remove water molecules. To validate the docking protocol, the co-crystallised ligand was extracted and re-docked into the active site of the enzyme to ensure proper definition of the binding site and to assess the accuracy of the docking algorithm in reproducing the pose of the co-crystallised ligand. C-Docker protocol was used to perform the docking studies of the tested indolizines into their potential targets. The ligands are docked into a rigid receptor while a set of ligand conformation is generated. Additional scoring functions PLP1, PLP2, Jain, and PMF were utilised to ensure the optimal ligand orientation into the active binding site of the target. The highest negative score of PLP1, PLP2, Jain, and PMFCitation51,Citation52, indicating the strongest receptor-ligand binding affinities, is considered for refining binding poses. The binding energy calculation was performed using C-Docker protocol and in situ ligand minimisation protocol.
2.7. ADMET calculation (in silico)
The ADMET Descriptors protocol and the Toxicity Prediction (TOPKAT) protocol in DS were used to calculate a range of absorption, distribution, metabolism, excretion, and toxicity (ADMET) related properties including, aqueous solubility (AS), blood–brain barrier (BBB) penetration, CYP2D6 inhibition, hepatotoxicity, human intestinal absorption (HIA), plasma protein binding (PPB), AlogP, and polar surface area (PSA); in addition, different toxicity parameters including rodent carcinogenicity (for both male and female rats and mice, CMR, CFR, CLM and CFM respectively), Ames mutagenicity (AM), skin irritation (SI), ocular irritancy (OI), aerobic biodegradability and developmental toxicity potential (AB) were also calculated.
3. Results and discussion
3.1. Chemistry
The synthesis of the title compounds (2a–2f, 3a–3d, 4a–4c) is portrayed in Scheme 1. The intermediates (1a–1f) were obtained by stirring a mixture of pyridine, and para-substituted phenacyl bromides in a dry acetone medium at 5 h. Compounds 1a–1f on further reaction with ethyl prop-2-ynoate/ethyl but-2-ynoate/diethyl 2-butynedioate in the presence of water with continuous stirring at 80 °C for 3 h resulted in the formation of title compounds 2a–2f, 3a–3d, 4a–4c. The resulting title compounds were purified using ethyl acetate and hexane as an eluent by column chromatography, and the compound purity was found to be more than 99% with a satisfactory yield (69% to 83%). Synthesis, purification, and characterisation details of compounds 1a–1f, 2a–2f, and 3a–3d were reported earlier from our research groupCitation42. The chemical structures of the newly synthesised compounds (4a-4c) have been ascertained with the help of spectroscopic techniques such as FT-IR, NMR (1H and 13C), LC-MS, and elemental analysis. In LC-MS, the molecular ion peaks of these novel compounds (4a–4c) were in good agreement with their proposed molecular masses. Elemental analysis results of the title compounds (4a–4c) were within ±0.4% of the calculated values.
Scheme 1. Synthesis of 1,2,3-trisubstiuted indolizine derivatives (2a–2f, 3a–3d, 4a–4c): Reagents and conditions (i) pyridine, dry acetone, stir at room temperature, 5 h; (ii) ethyl propialate/ethy1 2-butynoate/diethy1 2- butynedioate water, stir 80 °C, 3 h
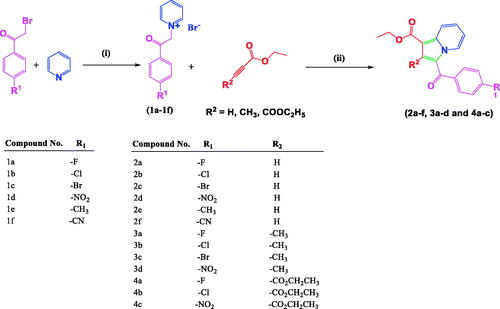
The primary goal of the current study was to investigate the impact of the substitution pattern, in terms of nature and position of substituents, at positions 1, 2, and 3 of the indolizine ring system on the anti-tubercular activity of the resultant analogues. The synthetic strategy for the development of the target compounds involved a 1.3-dipolar [3 + 2] cycloaddition as a key step, allowing the introduction of the substituent in the diverse position of the indolizine ring. The 1.3-dipolar [3 + 2] cycloaddition of pyridinium ylides with electron-deficient alkynes offered a convenient approach for the construction of an indolizine scaffold.
3.2. Crystallography
Compound 4b was crystallised in the triclinic centrosymmetric space group P-1 with one molecule in the asymmetric unit. The parameters for crystal data collection and structure refinement, and the list of intramolecular and intermolecular interactions in the crystal structure of diethyl 3-(4-chlorobenzoyl)indolizine-1,2-dicarboxylate (4b) are given in Table S1 and , respectively. The side chain ethoxy groups do not exhibit any orientational disorder. The molecular conformation is locked via C-H···O H-bonds (involving H1/O5 and H4/O2) forming pseudo-six membered rings (). The crystal packing is stabilised via C–H···O H-bonds (involving H21 and O5), forming dimers, and these are connected with centro symmetrically related dimers formed via C-H···O H-bonds (involving H17/H18 with O3) via additional C–H···O H-bonds (involving H3 and O3), thus leading to the overall formation of a “hexamer” in the solid-state (). Thus, O3 functions as a “trifurcated acceptor” which is a unique feature in the packing of this molecule in the crystal. In addition, the molecules are connected via a π···π stacking motif, involving the 6-membered ring (Cg1: centre of gravity of ring N1/C1-C5), the stacking distance being 3.654 Å. Finally, a short contact between O1 = C7···C5–N1 of distance 3.359 Å, involving electrophilic C7 and relatively less electrophilic C5 is present in the crystalline lattice ().
Figure 3. The crystal structure of diethyl 3-(4-chlorobenzoyl)indolizine-1,2-dicarboxylate (4b). ORTEP drawn with 50% ellipsoidal probability. Intra-molecular C–H···O·H bonds are shown as dotted lines.
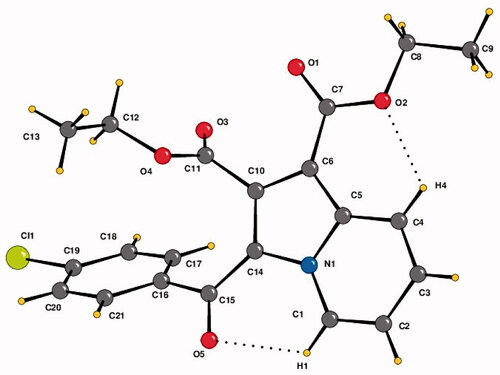
Figure 4. Crystal packing depicting C–H···O·H-bonds, π–π stacking interaction and O1 = C7···C5–N1 short contact in the compound 4b. Cg1 is the centre of gravity of N1/C1–C5 ring. The dotted lines represent intermolecular interactions. Non-interacting hydrogens have been omitted for clarity.
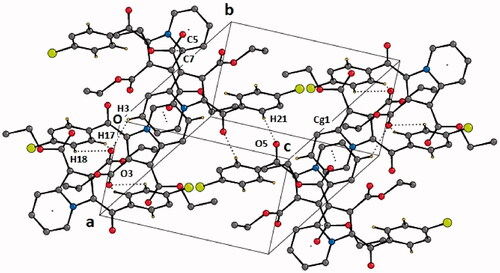
Table 1. List of intramolecular and intermolecular interactions in the crystal structure of diethyl 3–(4-chlorobenzoyl)indolizine-1,2-dicarboxylate (4b).
3.3. Hirshfeld surface analysis and energy framework calculation
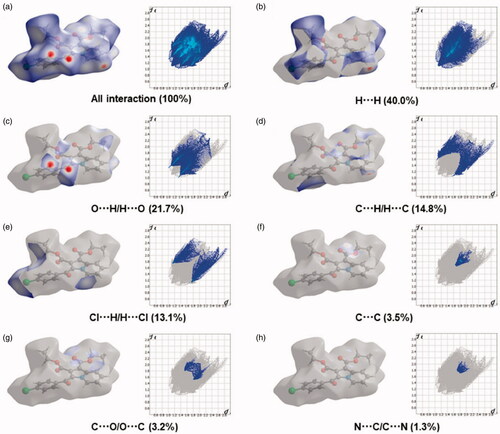
The Hirshfeld surfaces of the title compound 4b were mapped over de, dnorm, shape index, curvedness, and fragment patches as shown in . The contribution of individual intermolecular interactions on the Hirshfeld surface can be identified through the colour codes. If di is greater than de, it is shown by the dark surface area that exists because of the acceptor of the hydrogen bond. The red colour spot indicates shorter molecular contacts on the dnorm surface, and the blue colour on the surface of the dnorm reflects longer molecular contacts. The white colour on the dnorm surface indicates the interaction around the van der Waals radii. The red colour spot indicates the hydrogen bonding H···O contacts in the dnorm surfaces, while the blue surface region represents the H···H contacts (). The characteristics of the de surface appear as a relatively flat green region where the distances of contact are similar (). The strong hydrogen bonding interactions present in the molecule () are also demonstrated by the adjacent highlighted red and yellow regions on the shape index surface. Whereas, the H···H interactions () are shown by the blue curved and yellow regions on the curvedness surfaces. The colour patches are mapped differently on the Hirshfeld surface depending on the closeness to the neighbouring molecules (). The 2D fingerprint plots indicate the sharp spike peak that reflects the strong hydrogen bonding present in the molecule (). The fingerprint plot shows that the H···H contacts have a comparatively larger contribution of 40.0% relative to other interactions and implies that van der Waals radii and hydrogen bonding play a key role in the crystal packing. The H···O contacts contribute 21.7% of the total interactions. In addition, the percentage contribution in this crystal structure of other intermolecular interactions are as follows: C···H/H···C (14.8%), H···Cl/Cl···H (13.1%), C···C (3.5%), C···O/O···C (3.2%), N···C/C···N (1.3%), O···Cl/Cl···O (1.1%), N···H/H···N (0.7%), O···N/N···O (0.4%), Cl···C/C···Cl (0.1%).
Figure 5. Hirshfeld surfaces mapped with (a) dnorm (b) de (c) shape-index, (d) curvedness and (e) fragment patches for the compound 4b.
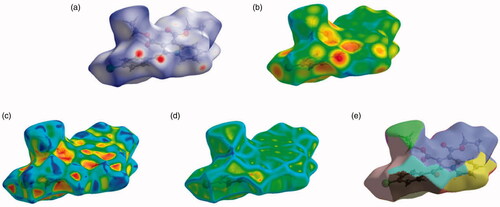
Figure 6. Two-dimensional fingerprint plot for the compound 4b showing the contributions of individual types of interactions: (a) all intermolecular contacts, (b) H···H contacts, (c) O···H/H···O contacts, (d) C···H/H···C contacts, (e) Cl···H/H···Cl contacts, (f) C···C contacts, (g) C···O/O···C contacts, (h) N···C/C···N. The outline of the full fingerprint is indicating in grey. The related surface patches associated with the specific contacts with the dnorm mapped are illustrated by surfaces to the left.
Systematic and theoretical energy was measured in terms of electrostatic, dispersion, and total energy using the program Crystal Explorer software, which further facilitated to depict the 3D topological images for the compound 4b (detailed information is provided in the supplementary section Figures S10-S13 and Table S2).
3.4. Anti-tubercular activity
The anti-tubercular activity of the target compounds (2a–2f, 3a–3d, and 4a–4c) was evaluated (in vitro) against two different MTB strains, namely susceptible H37Rv MTB strain, and rifampicin and isoniazid-resistant MTB strain (). Rifampicin and isoniazid were also used as positive controls (). The anti-mycobacterial activity showed that compound 3a, substituted with a fluorine atom at the 4-position of the benzoyl group and a methyl group at the 2-position of the indolizine ring, is the most potent molecule demonstrating the highest inhibitory action against the susceptible H37Rv MTB strain with MIC value of 4 µg/mL. Five Compounds (2b, 2d, 3d, 4a, and 4b) were found to be equipotent, with a MIC value of 8 µg/mL. Compounds 2c, 3b, and 4c showed moderate anti-TB activity with a MIC value of 32 µg/mL against the same strain. Meanwhile, no activity was observed for the remaining indolizines against the H37Rv MTB strain.
Table 2. In vitro anti-mycobacterial activity of 1,2,3-trisubstituted indolizine derivatives (2a–2f, 3a–3d, and 4a–4c) against H37Rv and MDR strains of Mycobacterium tuberculosis. 
The structure–activity relationship (SAR) analysis revealed the presence of a hydrogen atom, a methyl or an ester substituent at the 2-position of the indolizine core was favourable to attain good anti-tubercular activity. The presence of the halogen and the nitro group at the para position of the benzoyl ring were found to be tolerated well to maintain the activity (except compounds 2a and 3c) against the H37Rv MTB strain, whilst the substitution with methyl and cyano group in compounds 2e and 2f, respectively, completely abolished the activity against both the MTB strains. In the case of nitro derivatives, compounds 2d and 3d with a hydrogen and methyl group, respectively, at the 2-position of the indolizine ring showed four-fold increased activity as compared with the compound 4c with an ester group at the same position.
The structural alteration at the 2-position of the indolizine scaffold resulted in a profound impact on the anti-mycobacterial activity against MDR-MTB. The indolizine derivatives that were active (2b–2d, 3a, 3b, and 3d) against the susceptible H37Rv MTB strain were found to be inactive against MDR-MTB, except for compounds 2d and 3a which exhibited 8-fold reduced MDR-MTB activity compared to H37Rv MTB strain (). However, compounds 4a-4c with an ethyl ester group at the 4-position of the benzoyl ring showed moderate activity against MDR-TB, with a two-fold decrease in potency as compared with the cellular activity against susceptible H37Rv MTB strain. The anti-tubercular activity profile of the tested compounds against MDR-MTB provided a clue for their mode of action. Indeed, the results demonstrated that the first and second series of indolizines (2a–2f and 3b–3d) did not show activity against MDR-MTB except for compounds 2d and 3a, while the third series of compounds 4a–4c clearly inhibited the MDR-MTB strain. These findings suggest that the three designed series are likely to be inhibiting different MTB molecular drug targets; such that the first and second series of indolizine derivatives are inhibiting a common target that is different from that inhibited by the third series. Alternatively, the compounds that retained activity against MDR-MTB strain (third series) could be interacting with the same molecular target as those of the first two series, yet, might be interacting differently with the target binding site.
The above findings and observations supported the view that the first and second series of indolizine were more likely to inhibit the mycobacterial cell growth by interacting with the InhA enzyme. With regards to the mode of action of the ester indolizine series 3, their differences in inhibitory activity between susceptible strains and clinical isolates of MDR-MTB strains suggested that the derivatives 4a–4c were interacting with another molecular target. Thus, further molecular modelling study was carried out to elucidate the potential mode of action of the tested compounds as well as to rationalise their SAR as inhibitors of InhA which represents one of the potential MTB therapeutic targets, and also to identify the second molecular target for the ester indolizine series 3.
3.5. Toxicity study
The promising activities of the designed compounds are very encouraging to undertake prospective optimisation cycles. However, it is highly recommended to optimise the ADMET properties for a lead compound early in the development stage to reduce later problems due to unfavourable ADMET characteristicsCitation53,Citation54. Based on the in silico calculated ADME and toxicity parameters (), all the compounds are showing acceptable ADMET profile which makes them worthy of further optimisation towards achieving the desired drug-like properties.
Table 3. Calculated ADMET descriptors and toxicity parameters of indolizine derivatives (2a–2f, 3a–3d, and 4a–4c).
Moreover, the toxicity profile for compounds 2d, 3a, 4a, 4b, and 4c that showed promising activity against MDR-MTB was further assessed against peripheral blood mononuclear cells (PBMCs) and no signs of toxicity were observed with concentrations up to 256 µg/mL.
3.6. Computational molecular modelling
A molecular docking study was carried out to gain insight into the mechanism of action of the indolizine derivatives (2a–2f, 3a–3d, and 4a–4c). In light of the MTB inhibitory action, InhA appeared to be a potential molecular target for the title compounds; therefore these compounds were docked against InhA enzyme in an attempt to corroborate their mode of action with their observed anti-mycobacterial activity.
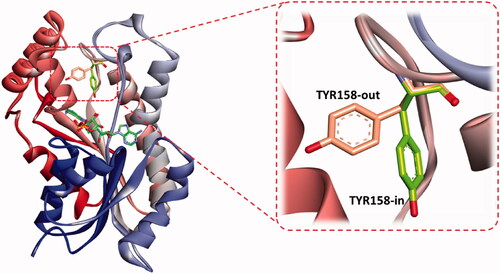
The binding feature common to all InhA inhibitors is the active participation of NAD, as a cofactor, contributing strong interaction with substrates through the H-bonding involvement from its ribose hydroxyl moiety. Additionally, the residue LYS 165 participates in the binding network by interacting with the hydroxyl oxygen of the NAD ribose through hydrogen bonding, and the residue ILE 91 is engaged in H-bonding with the amide functionality of the nicotinamide part of NAD. These residues greatly contributed to the stability of the cofactor. Hence, the latter tends to play a critical role in molecular interaction with the substrate and other residues in the InhA binding domain. Consequently, the presence of NAD during a docking study may have a profound effect not only on the ligand interaction but also on the occupancy size of the active site that may affect ligand orientation. Besides, a great majority of reported InhA inhibitors make a stable hydrogen bonding to the backbone hydroxyl group of the main chain TYR 158, responsible for blocking the enoyl-acyl carrier protein reductase functionCitation55. The role of TYR 158 in the catalytic mechanism of InhA has been the focus of many studies which revealed that its side chain can adopt different conformations when direct InhA inhibitors bind the catalytic site. Mainly two conformations were identified, an “in” and “out” conformations. The former is resembling the substrate-NAD-InhA ternary complex, and the latter resembles the NAD-InhA binary complex (); however, in the majority of available InhA-inhibitor complexes TYR158 is in the “in” conformation and the inhibitor is forming interactions with the cofactorCitation56–58. However, a novel binding mode of a benzimidazole-based InhA inhibitor has been recently disclosed, occupying an extended hydrophobic pocket formed by the amino acid residues Phe149, Met155, Pro156, Ala157, and Ile215 in the in-Tyr 128 conformationCitation57. Certain InhA inhibitors were also reported to co-crystallize with Tyr 158-out active site residueCitation59–65.
Figure 7. The two reported “in and out” conformations of Tyr158 in mycobacterial InhA enzyme. PDB ID for the in conformation is 5G0S, and that for the out conformation is 5G0U.
Accordingly, the binding mode of the indolizine 2d was investigated by docking it into the active site of the InhA enzyme utilising crystal structures of the in and out conformations, namely 5G0S and 5G0U respectively .
Figure 8. Comparative binding mode of indolizine 2d into InhA binding domain with (a) in-Tyr 158 (PDB ID: 5G0S) and (b) out-Tyr 158 conformations (PDB ID: 5G0U), respectively. The ligand, NAD and the receptor were represented as sticks in solmon, yellow and cyan colours respectively. The hydrogen bonds are shown as green dotted lines.
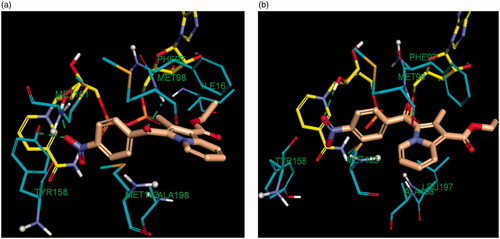
The docking results revealed that the estimated binding energy of 2d was −107.46 kcal/mol and −144.02 kcal/mol with the InhA binding site 5G0U (Tyr 158-out) and 5G0S (Tyr 158-in) respectively, indicating that the indolize 2d interacted more favourably with the in Tyr 158 conformation active residue. Indeed, the binding pose of 2d showed three hydrogen bond contacts with active site’s residues in the “in” conformation (pdb 5G0S), while only one with the “out” conformation (pdb 5G0U) (). The binding mode of 2d revealed that the benzoyl ring occupies the same location in both the crystal involving H-bond interaction with the co-factor NAD but differ on the orientation of the indolizine ring (). In light of the comparative docking analysis the InhA crystal structure of 5G0S, “in” conformation, appeared as more appropriate to investigate the binding patterns. Hence, docking of the remaining derivatives was performed with the Tyr 158-in conformation InhA active site (PDB 5G0S). The docking results, reported in , indicated that the active indolizines 2b–2d, 3a, 3b, and 4a–4c displayed greater binding energy as compared to that of inactive congeners 2a, 2e, 2f, and 3c. In general, the predicted docking energy of indolizines was consistent with the observed MTB inhibitory activity of the tested compounds, except for the compounds 2d, 3d, and 4c with a nitro substitution (). However, the order of the potency in the second indolizine series was 3a (F) > 3b (Cl) ≫ 3c (Br) which correlates with the docking scores. A similar correlation between the activity and docking scores was also observed in the first indolizine series in which the order of bioactivity was 2b (Cl) > 2c (Br) ≫ 2a (F), 2f (CN), 2e (CH3). Among the compounds 2a and 3a, the most active compound 3a (F) displayed a higher binding energy score than the inactive compound 2a (F). The compound 2b (Cl) being more active than compound 3b (Cl) demonstrated a higher energy score. A similar trend was also observed for the bromine derivatives 2c and 3c. The nitro compounds 2d and 3d were equipotent and showed comparable docking scores. As for the third series, the indolizines 4a–4c showed very high docking scores. The equipotent indolizines 4a (F) and 4b (Cl) showed comparable docking scores, whilst the nitro derivative 4c showing a higher docking score was four-fold less potent than the compounds 4a and 4b.
Figure 9. Predicted binding interaction of indolizines (2a, 2b, 2d, 3a, 3b, 3d, 4a–4c) with InhA binding domain (PDB 5G0S). Ligand and receptor were represented as solmon and cyan respectively. Hydrogen bonding contact is shown with green dotted lines. π–π, π–sulphur and hydrophobic interactions are shown with magenta, gold and violet, respectively. For displaying key bingind interaction, NAD has been omitted.
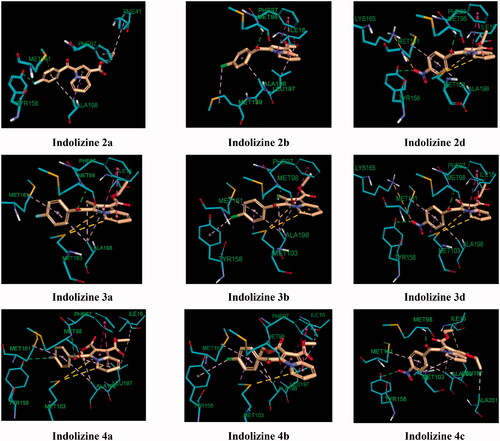
Table 4. Docking results of the indolizine derivatives 2a–2f, 3a–3d and 4a–4c into the InhA binding domain (PDB 5G0S).
The observed MTB inhibitory activity pattern of the indolizines can be accounted for their distinctive feature of binding mode into the InhA active site. The key binding interaction of the title compounds was highlighted in and . The binding poses of the title compounds showed that the benzoyl ring is located near to the co-factor binding site with a sandwich orientation between the nicotinamide ring of NAD and the main chain Tyr 158 which is consistent with the binding mode of known InhA inhibitors. The active indolizines 2b–2d, 3a, 3b, and 4a–4c were predicted to adopt a similar conformation in the binding domain by forming strong hydrogen bonding with the residue Met 98 and the carbonyl moiety of the benzoyl group, whereas the carbonyl moiety of the benzoyl group for the inactive indolizines 2a, 2e, 2f, and 3c was orientated away from the residue Met 98 preventing the H-bonding interaction (). The conformation alteration from the rotation of the benzoyl group could provide a rationale for the lack of potency observed for these derivatives. However, the interaction of the residue Met 98 with the native ligand and certain InhA inhibitors is reported as wellCitation61,Citation62. It was also demonstrated that interacting with the Met 98 residue through H-bonding was the key interaction to retain the MTB inhibitory activity when the main chain Tyr 158 was not involved in any hydrogen bonding with the ligand.Citation59 Derivatives 2d, 3a, 3d, 4a, and 4c were able to make additional hydrogen bonding contacts with residues in the active site. For instance, the para nitrobenzoyl indolizines 2d, 3d, and 4c showed two H-bonding interactions between the nitro group and the residue Tyr 158 and the hydrogen atom of the hydroxyl group from the NAD ribose (). Similarly, the derivative 4a having a fluorine atom in the para position of the benzoyl moiety showed a moderate hydrogen bonding contact with the hydroxyl group of the NAD ribose and a weak H-bond with the residue Tyr 158, while compound 3a demonstrated only one strong H-bond with the hydroxyl of the NAD ribose but exempting from any H-bond interaction with the residue Tyr 158 (). The strength of H-bonding involvement with NAD ribose may explain the greater potency observed for the derivative 3a (2.19 Å) as compared to the compound 4a (2.82 Å).
Figure 10. Predicted binding interaction of indolizines (4a, 4c) with InhA binding domain (PDB 5G0S). Ligand, NAD and receptor were represented as solmon, yellow and cyan respectively. Hydrogen bonding contact is shown with green dotted lines.
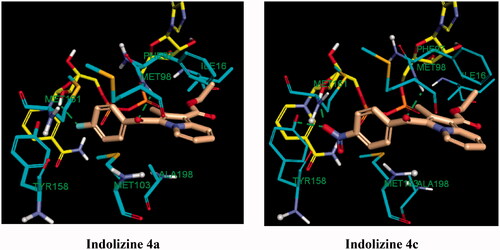
Another important contribution to the cellular activity of the bioactive compounds is the hydrophobic interaction implicated by the residues Phe 97 and Met 103 ( and ), Where Phe 97 formed T-shaped pi-stacking interaction with the indolizine ring, except for compound 4c, and the residue Met 103 was observed to interact with the indolizine ring through pi-sulfur interactions with the exception of compounds 2b and 4c. The lack of hydrophobic interaction with the residues Phe 97 and Met 103 for the compound 4c (NO2) provides an explanation for its reduced potency in comparison with its nitro congeners 2d and 3d ().
The molecular modelling analysis indicated that the observed MTB potency of indolizines was in good agreement with the predicted binding mode and binding energy predictions. These computational results strongly support our assumption that the molecular target of the designed indolizine is enoyl-acyl carrier protein reductase, and the key interactions with the residues Met 98 and Phe 91 were highlighted as being mainly responsible for the observed activity of the compounds. These findings could provide a valuable guide to designing more potent indolizines as potential InhA inhibitors through a structure-based design approach.
We next focussed on the prospective identification of the cellular target of the ester derivatives 4a–4c. According to the cellular activity profile against MDR MTB, the ester indolizine series 4a–4c, identified as potential InhA inhibitors, may have more than one molecular MTB target. In the quest to identify the second potential target for the indolizines 4a–4c, again, computational molecular docking studies were carried out. Based on the MTB activity of derivatives 4a–4c against both susceptible and resistant MTB strains, we hypothesised that the presence of the ester functional group at 2-position of the indolizine may strongly contribute to the bioactivity through the formation of hydrogen bonding contact. Furthermore, it has been observed that the indolizine 4a and 4b, having halogen substituent in the para position of the benzoyl moiety, had exhibited identical anti-TB activity and were more active than the nitro derivative 4c, indicative of probable involvement of halogen bonding interaction with the receptor. With regards to the above considerations, a large number of known TB molecular targets were screened. The molecular target anthranilate phosphoribosyltransferase appeared to be the appropriate enzyme target for indolizines 4a–4c. The docking scores, presented in , were also found to be consistent with the MTB activity of the compounds in which the equipotent indolizines 4a and 4b displayed similar binding energy, while the less active indolizine 4c showed a lower docking score. The binding interaction of the indolizines 4a–4c with the anthranilate phosphoribosyl enzyme was illustrated in . The most active compounds 4a and 4b were found to adopt similar orientation in the anthranilate phosphoribosyl binding domain showing two strong hydrogen bonding contacts between the carbonyl group of benzoyl and ester at 2-position of indolizine ring with the amino acid residues Arg 193 and Asn 138, respectively. Among the numerous hydrophobic interactions, the fluorine atom of compound 4a showed halogen interaction with the residues Gly 107 and Thr 108, similarly, the chlorine atom of compound 4b exhibited interaction with the residue Gly 107. Both indolizines 4a and 4b were also involved in pi-cation interaction between the benzoyl ring of the indolizine and the residue Arg 193. The binding mode of the indolizine 4c differed from its congeners demonstrating only one H-bond contact between the residue Asn 138 and the carbonyl of ester group at the 2-position of indolizine ring, explaining the lower observed MTB activity. However, although the above computational results reasonably explained the observed anti-tubercular activities of the tested compounds, yet, they need to be experimentally verified and validated via biomolecular in vitro testing. Thus, it could be the subject of prospective research due to its importance in confirming the in silico results that is imperative to guide prospective optimisation of these lead compounds towards designing more potent and selective drug candidates.
Figure 11. Predicted binding interaction of indolizines (4a–4d) with anthranilate phosphoribosyl binding domain (PDB 3R6C). Ligand and receptor were represented as solmon and cyan respectively. Hydrogen bonding contact is shown with green dotted lines. Halogen bonding and hydrophobic intreactions are shown with cyan and violet respectively.
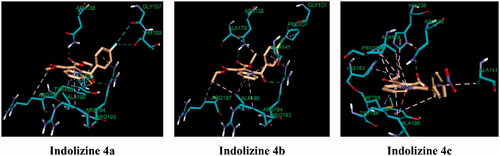
Table 5. Docking results of the indolizine derivatives 4a–4c into the anthranilate phosphoribosyl binding domain (PDB 3R6C).
4. Conclusion
In the current study, three novel series of 1,2,3-trisubstituted indolizine derivatives (2a–2f, 3a–3d, and 4a–4c) were evaluated for their anti-TB activity against susceptible and MDR-MTB strains. The majority of tested compounds showed good to excellent anti-tubercular activity with MIC ranging from 4 to 64 µg/mL. The inhibition profile of indolizines and molecular docking strongly supported that InhA is the principal drug target for the investigated compounds. Molecular modelling insight indicated that the conformation changes resulting from the benzoyl group rotation provided a rationale for the MTB cellular activity of the indolizines as InhA inhibitor. Furthermore, the cellular activity profile of the ester indolizine derivatives (4a-4c) against both the strains revealed their inhibitory action in multiple molecular targets in which anthranilate phosphoribosyltransferase might be a potential additional drug target. Our results highlighted the importance of indolizines as a novel promising class of multi-targeting agents for MTB with favourable toxicity profile. Such compounds might serve as attractive leads for further development of potential drug candidates for combating both the drug-sensitive and drug-resistant tuberculosis strains.
The compound 4b was crystallised in a triclinic centrosymmetric crystal system with space group P-1. In stabilising the molecular structure, the C–H···O and other short contact have played an important role. Hirschfeld surface analysis with 2D fingerprint plots was performed to provide insight into the stability of the crystal structure in order to understand and visualise the contribution of distinct intermolecular interactions. Systematic and theoretical energy was measured in terms of electrostatic, dispersion, and total energy using the program Crystal Explorer software, which further depicts 3D topological images.
Supplemental Material
Download PDF (978.8 KB)Acknowledgements
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number 1058, King Faisal University, Kingdom of Saudi Arabia for support and encouragement; the Department of Microbiology, National Health Laboratory Services, Inkosi Albert Luthuli Central Hospital, Durban South Africa for providing the facility to carry out in vitro anti-TB activity and Dr. Hong Su, Center for Supramolecular Chemistry Research, Department of Chemistry, University of Cape Town, South Africa, for single-crystal X-ray data collection. D.C. thanks IISER Bhopal for research facilities and infrastructure. R.D.N and S.K.N. thank the Department of Chemistry, VNIT, Nagpur for support and encouragement. The authors wish to thank Mr. Tameem M. Alyahian for providing technical support in this project.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- World Health Organization. Global tuberculosis report. Available from: http://www.who.int/tb/publications/global_report/en/ [last accessed 9 June 2020].
- World Health Organization. Global tuberculosis report 2019 (Executive summary). Available from: https://www.who.int/tb/publications/global_report/tb19_Exec_Sum_12Nov2019.pdf?ua=1 [last accessed 9 September 2020].
- World Health Organization. TB⁄HIV facts. Available from: http://wwwwhoint/tb/challenges/hiv/factsheet_hivtb_2011pdf [last accessed 8 March 2016].
- World Health Organization. Global tuberculosis report. Available from: http://www.who.int [Last accessed 1 May 2017].
- Campbell PJ, Morlock GP, Sikes RD, et al. Molecular detection of mutations associated with first- and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis. Antimicrob Agents Chemother 2011;55:2032–41.
- Velayati AA, Masjedi MR, Farnia P, et al. Emergence of new forms of totally drug-resistant tuberculosis bacilli: super extensively drug-resistant tuberculosis or totally drug-resistant strains in Iran. Chest 2009;136:420–5.
- Dheda K, Shean K, Zumla A, et al. Early treatment outcomes and HIV status of patients with extensively drug-resistant tuberculosis in South Africa: a retrospective cohort study. Lancet 2010;375:1798–807.
- Singh P, Raj R, Kumar V, et al. 1,2,3-Triazole tethered beta-lactam-chalcone bifunctional hybrids: synthesis and anticancer evaluation. Eur J Med Chem 2012;47:594–600.
- Baker MA, Lin HH, Chang HY, Murray MB. The risk of tuberculosis disease among persons with diabetes mellitus: a prospective cohort study. Clin Infect Dis 2012;54:818–25.
- Patpi SR, Pulipati L, Yogeeswari P, et al. Design, synthesis, and structure–activity correlations of novel dibenzo[b,d]furan, dibenzo[b,d]thiophene, and N-methylcarbazole clubbed 1,2,3-triazoles as potent inhibitors of Mycobacterium tuberculosis. J Med Chem 2012;55:3911–22.
- van Heeswijk RP, Dannemann B, Hoetelmans RM. Bedaquiline: a review of human pharmacokinetics and drug-drug interactions. J Antimicrob Chemother 2014;69:2310–8.
- Nguyen TVA, Anthony R, Bañuls A-L, et al. Bedaquiline resistance: its emergence, mechanism and prevention. Clin Infect Dis 2018;66:1625–30.
- Fujiwara M, Kawasaki M, Hariguchi N, et al. Mechanisms of resistance to delamanid, a drug for Mycobacterium tuberculosis. Tuberculosis 2018;108:186–94.
- Keam SJ. Pretomanid: first approval. Drugs 2019;79:1797–803.
- Tetali SR, Kunapaeddi E, Mailavaram RP, et al. Current advances in the clinical development of anti-tubercular agents. Tuberculosis 2020;125:101989.,
- Yee D, Valiquette C, Pelletier M, et al. Incidence of serious side effects from first-line antituberculosis drugs among patients treated for active tuberculosis. Am J Resp Crit Care Med 2003;167:1472–7.
- Grüber G. Introduction: novel insights into TB research and drug discovery. Prog Biophys Mol Biol 2020;152:2–5.
- Stelitano G, Sammartino JC, Chiarelli LR. Multitargeting compounds: a promising strategy to overcome multi-drug resistant tuberculosis. Molecules 2020;25:1239.
- Li W, Upadhyay A, Fontes FL, et al. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob Agents Chemother 2014;58:6413–23.
- Ganihigama DU, Sureram S, Sangher S, et al. Antimycobacterial activity of natural products and synthetic agents: pyrrolodiquinolines and vermelhotin as anti-tubercular leads against clinical multidrug resistant isolates of Mycobacterium tuberculosis. Eur J Med Chem 2015;89:1–12.
- Khedr MA, Pillay M, Chandrashekharappa S, et al. Molecular modeling studies and anti-TB activity of trisubstituted indolizine analogues; molecular docking and dynamic inputs. J Biomol Struct Dyn 2018;36:2163–78.
- Venugopala KN, Chandrashekharappa S, Pillay M, et al. Computational, crystallographic studies, cytotoxicity and anti-tubercular activity of substituted 7-methoxy-indolizine analogues. PLoS One 2019;14:e0217270.,
- Venugopala KN, Tratrat C, Pillay M, et al. Anti-tubercular activity of substituted 7-methyl and 7-formylindolizines and in silico study for prospective molecular target identification. Antibiotics 2019;8:247–16.
- Dannhardt G, Meindl W, Gussmann S, et al. Anti-mycobacterial 7-hydroxy-2,3-dihydro-1H-indolizin-5-ones. Eur J Med Chem 1987;22:505–10.
- Weide T, Arve L, Prinz H, et al. 3-Substituted indolizine-1-carbonitrile derivatives as phosphatase inhibitors. Bioorg Med Chem Lett 2006;16:59–63.
- Olaru A-M, Vasilache V, Danac R, Mangalagiu II. Antimycobacterial activity of nitrogen heterocycles derivatives: 7-(pyridine-4-yl)-indolizine derivatives. Part VII8–12. J Enzyme Inhib Med Chem 2017;32:1291–8.
- Gundersen L-L, Charnock C, Negussie AH, et al. Synthesis of indolizine derivatives with selective antibacterial activity against Mycobacterium tuberculosis. Eur J Pharma Sci 2007;30:26–35.
- Dawood KM, Abbas AA. Inhibitory activities of indolizine derivatives: a patent review. Expert Op Therap Patents 2020;30:695–714.
- Singh GS, Mmatli EE. Recent progress in synthesis and bioactivity studies of indolizines. Eur J Med Chem 2011;46:5237–57.
- Sandeep C, Venugopala KN, Mohammed AK, et al. Review on chemistry of natural and synthetic indolizines with their chemical and pharmacological properties. J Basic Clin Pharma 2016;8:49–61.
- Venugopala KN, Uppar V, Sandeep C, et al. Cytotoxicity and antimycobacterial properties of pyrrolo[1,2-a]quinoline derivatives: molecular target identification and molecular docking studies. Antibiotics 2020;9:233–14.
- Venugopala KN, Tratrat C, Pillay M, et al. In silico design and synthesis of tetrahydropyrimidinones and tetrahydropyrimidinethiones as potential thymidylate kinase inhibitors exerting anti-TB activity against Mycobacterium tuberculosis. Drug Design Dev Ther 2020;14:1027–39.
- Venugopala KN, Tratrat C, Chandrashekharappa S, et al. Anti-tubercular potency and computationally-assessed drug-likeness and toxicology of diversely substituted indolizines. Ind J Pharma Educ Res 2019;53:545–52.
- Venugopala KN, Khedr MA, Pillay M, et al. Benzothiazole analogs as potential anti-TB agents: computational input and molecular dynamics. J Biomol Struct Dyn 2019;37:1830–42.
- Venugopala KN, Chandrashekharappa S, Pillay M, et al. Synthesis and structural elucidation of novel benzothiazole derivatives as anti-tubercular agents: in-silico screening for possible target identification. Medicinal Chem 2019;15:311–26.
- Chandrashekharappa S, Venugopala KN, Venugopala R, Padmashali B. Qualitative anti-tubercular activity of synthetic ethyl 7-acetyl-2-substituted-3-(4-substituted benzoyl) indolizine-1-carboxylate analogues. J Appl Pharma Sci 2019;9:124–8.
- Alveera S, Venugopala KN, Khedr MA, et al. Antimycobacterial, docking and molecular dynamic studies of pentacyclic triterpenes from Buddleja saligna leaves. J Biomol Struct Dyn 2017;35:2654–64.
- Venugopala KN, Dharma Rao GB, Bhandary S, et al. Design, synthesis, and characterization of (1-(4-aryl)- 1H-1,2,3-triazol-4-yl)methyl, substituted phenyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates against Mycobacterium tuberculosis. Drug Design Dev Ther 2016;10:2681–90.
- Venugopala KN, Nayak SK, Pillay M, et al. Synthesis and antitubercular activity of 2-(substituted phenyl/benzyl-amino)-6-(4-chlorophenyl)-5-(methoxycarbonyl)-4-methyl-3,6-dihydropyrimidin-1-ium chlorides. Chem Biol Drug Des 2013;81:219–27.
- Venugopala KN, Albericio F, Coovadia YM, et al. Total synthesis of a depsidomycin analogue by convergent solid-phase peptide synthesis and macrolactonization strategy for antitubercular activity. J Peptide Sci 2011;17:683–9.
- Salgado-Moran G, Ramirez-Tagle R, Glossman-Mitnik D, et al. Docking studies of binding of ethambutol to the C-Terminal domain of the arabinosyltransferase from Mycobacterium tuberculosis. J Chem 2013;:601270.
- Sandeep C, Venugopala KN, Gleiser RM, et al. Greener synthesis of indolizine analogues using water as a base and solvent: study for larvicidal activity against Anopheles arabiensis. Chem Biol Drug Des 2016;88:899–904.
- Bruker. APEX2. Madison (WI): Bruker AXS Inc.; 2012.
- Bruker. SAINT and SADABS. Madison (WI): Bruker AXS Inc.; 2008.
- Sheldrick GM. A short history of SHELX. Acta Crystallographica Section A 2008;64:112–22.
- Sheldrick G. Crystal structure refinement with SHELXL. Acta Crystallographica Section C 2015;71:3–8.
- Spek A. checkCIF validation ALERTS: what they mean and how to respond. Acta Crystallographica Section E 2020;76:1–11.
- Farrugia L. WinGX and ORTEP for Windows: an update. J Appl Crystallogr 2012;45:849–54.
- Martin A, Morcillo N, Lemus D, et al. Multicenter study of MTT and resazurin assays for testing susceptibility to first-line anti-tuberculosis drugs. Int J Tuberc Lung Dis 2005;9:901–6.
- Mossman T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth 1983;65:55–63.
- Chandrashekharappa S, Venugopala KN, Tratrat C, et al. Efficient synthesis and characterization of novel indolizines: exploration of in vitro COX-2 inhibitory activity and molecular modelling studies. New J Chem 2018;42:4893–901.
- Christophe T. Novel thiazole-based thiazolidinones as potent anti-infective agents: in silico PASS and toxicity prediction. Synth Biol Eval Mol Model Combin Chem High Throughput Screen 2020;23:126–40.
- Deb PK, Al-Attraqchi O, Mohammed NA-Q, et al. Applications of computers in pharmaceutical product formulation. In: Tekade RK, ed. Dosage form design considerations: advances in pharmaceutical product development and research series. Vol. II. Waltham (MA): Academic Press; 2018:665–703.
- Deb PK, El-Rabie D, Ahmad J, et al. In silico binding mode analysis (molecular docking studies), and ADME prediction of some novel inhibitors of aurora kinase a in clinical trials. Asian J Chem 2014;26:6221–6.
- Chollet A, Maveyraud L, Lherbet C, Bernardes-Genisson V. An overview on crystal structures of InhA protein: Apo-form, in complex with its natural ligands and inhibitors. Eur J Med Chem 2018;146:318–43.
- Parikh S, Moynihan DP, Xiao G, Tonge PJ. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. BioChem 1999;38:13623–34.
- Kamsri P, Hanwarinroj C, Phusi N, et al. Discovery of new and potent InhA inhibitors as antituberculosis agents: structure-based virtual screening validated by biological assays and X-ray crystallography. J Chem Info Model 2020;60:226–34.
- Rozwarski DA, Vilchèze C, Sugantino M, et al. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD + and a C16 fatty acyl substrate*. J Biol Chem 1999;274:15582–9.
- Guardia A, Gulten G, Fernandez R, et al. N-Benzyl-4-((heteroaryl)methyl)benzamides: a new class of direct NADH-dependent 2-trans enoyl–acyl carrier protein reductase (InhA) inhibitors with antitubercular activity. ChemMedChem 2016;11:687–701.
- Martínez-Hoyos M, Perez-Herran E, Gulten G, et al. Antitubercular drugs for an old target: GSK693 as a promising InhA direct inhibitor. EBioMedicine 2016;8:291–301.
- Shirude PS, Madhavapeddi P, Naik M, et al. Methyl-thiazoles: a novel mode of inhibition with the potential to develop novel inhibitors targeting INHA in Mycobacterium tuberculosis. J Med Chem 2013;56:8533–42.
- Encinas L, O’Keefe H, Neu M, et al. Encoded library technology as a source of hits for the discovery and lead optimization of a potent and selective class of bactericidal direct inhibitors of Mycobacterium tuberculosis InhA. J Med Chem 2014;57:1276–88.
- Spagnuolo LA, Eltschkner S, Yu W, et al. Evaluating the contribution of transition-state destabilization to changes in the residence time of triazole-based InhA inhibitors. J Am Chem Soc 2017;139:3417–29.
- Mtb InhA complex with Pyradizinone compound 14. https://www.rcsb.org/structure/4D0S [last accessed 25 March 2021].
- Discovery of pyrimidine isoxazoles InhA in complex with compound 23. https://www.rcsb.org/structure/4UVI [last accessed 25 March 2021].