
Abstract
The non-nucleoside reverse transcriptase inhibitor VM1500A is approved for the treatment of HIV/AIDS in its N-acyl sulphonamide prodrug form elsulfavirine (Elpida®). Biochemical profiling against twelve human carbonic anhydrase (CA, EC 4.2.1.1) isoforms showed that while elsulfavirine was a weak inhibitor of all isoforms, VM1500A potently and selectively inhibited human (h) hCA VII isoform, a proven target for the therapy of neuropathic pain. The latter is a common neurologic complication of HIV infection and we hypothesise that by using Elpida® in patients may help alleviate this debilitating symptom.
Graphical Abstract
1. Introduction
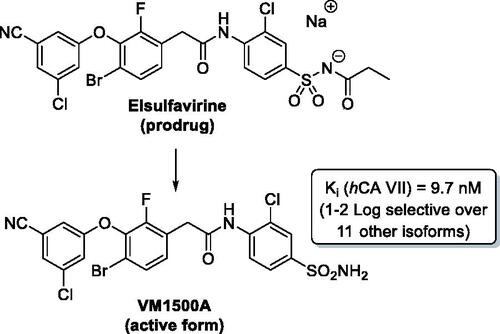
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) inhibit the human immunodeficiency virus (HIV) reverse transcriptase (RT), which is responsible for the production of double-stranded viral DNA from a single-stranded viral RNA genomeCitation1,Citation2. Elsufavirine (Elpida) is a propanoyl sulphonamide prodrug form of its active form, the new-generation NNRTI, VM1500A ().
Figure 1. Elsufavirine (Elpida) and VM1500A.
As a drug candidate, elsufavirine was discovered by Roche. In 2009, the San Diego based Viriom biotechnology company entered into a licencing agreement with Roche for the development and commercialisation of this drug for the treatment of HIV infectionCitation3. Elsulfavirine demonstrated excellent antiviral efficacy in treatment-naive patients during the clinical trials. The virologic response to elsulfavirine (viral load and HIV RNA reduction) was comparable to that of the patients treated with efavirenz; however, compared to the latter, elsufavirine showed superior tolerability and fewer side effectsCitation4. Elsulfavirine received its first global approval on 30 June 2017, in Russia, for the treatment of HIV-1 infections in combination with other antiretroviral drugsCitation2.
Elsulfavirine was shown to be rapidly converted (t1/2 ∼ 2 h) to VM1500A which is eliminated much more slowly (t1/2 > 5 days)Citation3. This pharmacokinetic profile is determined by the primary sulphonamide moiety being taken up into the red blood cells (RBC). In erythrocytes, VM1500A is accumulated through reversible binding to RBC carbonic anhydrase (CA). The CA-bound drug is thereafter slowly released to plasma and ultimately reaches the target cells. This phenomenon determines the need for infrequent dosing, improves patients’ compliance and will likely improve the long-term treatment and prevention of HIV/AIDSCitation5.
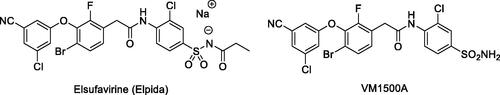
The primary benzene sulphonamide group ensures the affinity to various CA isoforms via binding, in its deprotonated form, to the zinc ion from the enzyme active siteCitation6. However, it is the molecular periphery (or the “tail”Citation7) of the primary sulphonamide warhead that determines the differential affinity of carbonic anhydrase inhibitors (CAIs) to CA isoforms, i.e. their isoform selectivityCitation8. Both polar and lipophilic periphery groups can enhance affinity to (and, hence, inhibitory potency of CAIs towards) a specific CA isoform as CAs, in general, have a very characteristic active site topology where polar and hydrophobic sides are clearly delineated ()Citation9.
Figure 2. Schematic representation of a sulphonamide-based CAI bound in the active site of CA.
The periphery of elsulfavirine and VM1500A is rather lipophilic if the tetrasubstituted diphenyl ether moiety is considered. At the same time, the benzene sulphonamide tail contains several polar moieties capable of participating in hydrogen bonding. Therefore, we were curious to establish the inhibitory profile of both agents against a comprehensive panel of recombinant CA isoforms. This, we reasoned, could help assess prospects of using elsulfavirine (VM1500A) for indications other than HIV infection/AIDS. Furthermore, considering the plethora of concomitant symptoms accompanying the advanced stages of the disease and the multifaceted therapeutic potential of isoform-selective CAIsCitation10, the inhibition data obtained in multiple CA isoform inhibition assays could strengthen the position of elsulfavirine as a first-line treatment of AIDS patients. Herein, we report on the inhibitory profile of elsulfavirine and VM1500A assessed with respect to all catalytically active human (hCA) isoforms I, II, III, IV, VA, VB, VI, VII, IX, XII, XIII and XIVCitation8.
2. Materials and methods
2.1. Compounds for biological evaluation
Active pharmaceutical ingredients elsulfavirine (Elpida®) and VM1500A were provided by Viriom, Inc. (San Diego, CA, USA).
2.2. Stopped flow CO2 hydrase assay
An Applied Photophysics stopped-flow instrument has been used for assaying the CA-catalysed CO2 hydration activityCitation11. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as a buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionised water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were pre-incubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation12,Citation13 and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlier and their concentrations ranged between 4.1 and 9.7 nMCitation14–18.
2.3. In silico studies
The crystal structure of hCA VII (pdb code 3ML5) was retrieved from the Protein Data BankCitation19. Сrystal structure was prepared according to the Protein Preparation module in Maestro–Schrodinger suite, assigning bond orders, adding hydrogens, deleting water molecules, and optimising H-bonding networksCitation20,Citation21. Finally, energy minimisation with a root mean square deviation (RMSD) value of 0.30 was applied using an Optimised Potentials for Liquid Simulation (OPLS-3) force fieldCitation22. Input 3 D ligand structure was prepared by Maestro and its ionisation states were evaluated with EpikCitation23. OPLS-3 force field was used for energy minimisation. Glide with default parameters was used to generate the docking grid setting the centre of the co-crystallized ligand (AAZ) as grid centreCitation24. Docking was performed using the standard precision mode (SP) implemented in Glide. For each obtained docking pose, the Prime refinement was performed on all the residues located within 5 Å from the ligandCitation25. Key ligand-protein interactions were analysed for the minimum-energy docking pose of VM1500A.
3. Results and discussion
The inhibitory profile of VM1500A, elsulfavirine and the known CA inhibitor acetazolamide (AAZ) was determined using stopped-flow CO2 hydrase assay as described in Section 2.2. The inhibitory data (Ki) are summarised in .
Table 1. hCA isoform inhibitory profile of acetazolamide (AAZ), VM1500-A and elsulfavirine.
Acetazolamide (5-acetamido-1,3,4-thiadiazole-2-sulphonamide) is known for its pan-isoform CA inhibitory profile which is considered a source of the plethora of side effects associated with this classical CA inhibitory drugCitation26. Although potent and isoform-selective N-acyl sulphonamide CA inhibitors have been reportedCitation27, elsulfavirine itself demonstrated rather weak inhibition across the entire hCA panel, in the range of single- to double-digit micromolar concentrations with virtually no inhibition of hCA III and IV isoforms. To our delight, however, the primary sulphonamide VM1500A showed a much more effective inhibitory profile. The inhibition of the cytosolic hCA I and hCA II isoforms by VM1500A–abundantly present in erythrocytes and to which the drug presumably binds thus forming a slow-release depotCitation5–is at the level of 513.8 and 35.5 nM, respectively. However, a notable selectivity towards cytosolic hCA VII isoform (generally 1–2 orders of magnitude over the majority of other hCA isoforms) is rather promising. hCA VII is a validated target for the development of inhibitors to treat neuropathic painCitation28. Considering the fact that neuropathy related to HIV is affecting as many as 50% of all individuals infected with HIVCitation29–treatment of patients with elsulfavirine (Elpida®) may be beneficial for alleviating this most common neurologic complications of the disease.
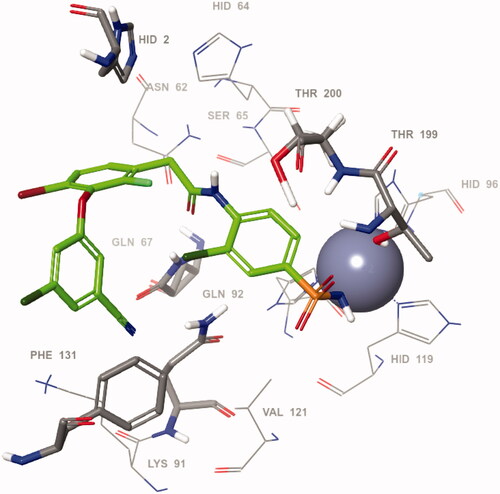
Taking into account the pronounced inhibitory activity of VM1500A towards hCA VII isoform, we proceeded to define its likely binding mode within the active site of the protein. To this end, VM1500A was docked into the hCA VII crystal structure (pdb code 3ML5)Citation30 and the key ligand-protein interactions were analysed for the minimum-energy docking pose shown in . Expectedly, the orientation of the benzenesulfonamide motif was found to be the same as for the majority of such ligands in the hCA active site, providing anchoring to the prosthetic zinc metal ion and hydrogen bonding to the Thr199 residue at the bottom of the catalytic cavityCitation31. In the meantime, the amide moiety attached to the benzenesulfonamide moiety forms contacts with amino acids Thr200, Gln67 and Gln92. Particularly, a hydrogen bond was observed between the NH group of the amide function and oxygen atom of the Thr200 side chain whereas the carbonyl group formed hydrogen bonds with NH moieties of the Gln67 and Gln92 residues. On the other hand, the ligand-protein contacts at the outer rim of the active side were mostly characterised by π–π stacking of the benzene rings with His2 and Phe131 sidechains and the bromine and chlorine substituents of these aromatic moieties were partially exposed to the bulk solvent.
Figure 3. Predicted binding pose of VM1500A (green) within the active site of hCA VII.
4. Conclusion
Biochemical profiling of the new anti-HIV prodrug elsulfavirine (Elpida®) and its active form primary sulphonamide VM1500A against a panel of twelve human carbonic anhydrase isoforms revealed that while elsulfavirine had a weak inhibitory profile across the panel of hCAs, VM1500A showed a potent (Ki = 9.6 nM) and selective inhibition of hCA VII, a neuropathic pain target. As neuropathic pain is common among HIV-infected individuals, concomitant inhibition of hCA VII may help alleviate this debilitating neurologic complication.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Zhan P, Pannecouque C, De Clercq E, Liu X. Anti-HIV drug discovery and development: current innovations and future trends. J Med Chem 2016;59:2849–78.
- Namasivayam V, Vanangamudi M, Kramer VG, et al. The journey of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) from lab to clinic. J Med Chem 2019;62:4851–83.
- Al-Salama ZT. Elsulfavirine: first global approval. Drugs 2017;77:1811–6.
- Wang Y, De Clercq E, Li G. Current and emerging non-nucleoside reverse transcriptase inhibitors (NNRTIs) for HIV-1 treatment. Expert Opin Drug Metab Toxicol 2019;15:813–29.
- Rai MA, Pannek S, Fichtenbaum CJ. Emerging reverse transcriptase inhibitors for HIV-1 infection. Expert Opin Emerg Drugs 2018;23:149–57.
- Nocentini A, Angeli A, Carta F, et al. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J Enzyme Inhib Med Chem 2021;36:561–80.
- (a) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72. (b) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Krasavin M, Korsakov M, Dorogov M, et al. Probing the ‘bipolar nature of the carbonic anhydrase active site: aromatic sulfonamides containing 1,3-oxazol-5-yl moiety as picomolar inhibitors of cytosolic CA I and CA II isoforms. Eur J Med Chem 2015;101:334–47.
- (a) Supuran CT. Therapeutic applications of the carbonic anhydrase inhibitors. Therapy 2007;4:355–78. (b) Supuran CT. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin Drug Discov 2020;15:671–86. (c) Supuran CT. Experimental carbonic anhydrase inhibitors for the treatment of hypoxic tumors. J Exp Pharmacol 2020;12:603–17.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase: I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Singer M, Lopez M, Bornaghi LF, et al. Inhibition of carbonic anhydrase isozymes with benzene sulfonamides incorporating thio, sulfinyl and sulfonyl glycoside moieties. Bioorg Med Chem Lett 2009;19:2273–6. (b) Angeli A, Carta F, Nocentini A, et al. Response to perspectives on the classical enzyme carbonic anhydrase and the search for inhibitors. Biophys J 2021;120:178–81.
- (a) Angeli A, Del Prete S, Osman SM, et al. Activation studies of the α- and β-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae with amines and amino acids. J Enzyme Inhib Med Chem 2018;33:227–33. (b) Bua S, Bozdag M, Del Prete S, et al. Mono- and di-thiocarbamate inhibition studies of the δ-carbonic anhydrase TweCAδ from the marine diatom Thalassiosira weissflogii. J Enzyme Inhib Med Chem 2018;33:707–13.
- (a) Menchise V, De Simone G, Alterio V, et al. Carbonic anhydrase inhibitors: stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J Med Chem 2005;48:5721–7. (b) Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors – part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents in rabbits. Eur J Med Chem 1998;33:247–54. (c) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8. (d) Guglielmi P, Rotondi G, Secci D, et al. Novel insights on saccharin- and acesulfame-based carbonic anhydrase inhibitors: design, synthesis, modelling investigations and biological activity evaluation. J Enzyme Inhib Med Chem 2020;35:1891–905. (e) Supuran CT. Coumarin carbonic anhydrase inhibitors from natural sources. J Enzyme Inhib Med Chem 2020;35:1462–70.
- (a) Krall N, Pretto F, Decurtins W, et al. A small‐molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5. (b) Mishra CB, Tiwari M, Supuran CT. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev 2020;40:2485–565. (c) Bonardi A, Nocentini A, Bua S, et al. Sulfonamide inhibitors of human carbonic anhydrases designed through a three-tails approach: improving ligand/isoform matching and selectivity of action. J Med Chem 2020;63:7422–44. (d) Dubois L, Peeters S, Lieuwes NG, et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 2011;99:424–31. (e) Blandina P, Provensi G, Passani MB, et al. Carbonic anhydrase modulation of emotional memory. Implications for the treatment of cognitive disorders. J Enzyme Inhib Med Chem 2020;35:1206–14. (f) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal β-class (Cab) and γ-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–8.
- (a) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors: part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8. (b) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun 2010;46:8371–3. (c) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. (d) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11.
- (a) Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. J Enzyme Inhib Med Chem 2016;31:894–9. (b) Margheri F, Ceruso M, Carta F, et al. Overexpression of the transmembrane carbonic anhydrase isoforms IX and XII in the inflamed synovium. J Enzyme Inhib Med Chem 2016;31:60–3. (c) Bua S, Di Cesare Mannelli L, Vullo D, et al. Design and synthesis of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the treatment of rheumatoid arthritis. J Med Chem 2017;60:1159–70.
- (a) Alper Türkoğlu E, Şentürk M, Supuran CT, Ekinci D. Carbonic anhydrase inhibitory properties of some uracil derivatives. J Enzyme Inhib Med Chem 2017;32:74–7. (b) Soydan E, Olcay AC, Bilir G, et al. Investigation of pesticides on honey bee carbonic anhydrase inhibition. J Enzyme Inhib Med Chem 2020;35:1923–7.
- Berman HM, Battistuz T, Bhat TN, et al. The protein data bank. Acta Crystallogr D Biol Crystallogr 2002;58:899–907.
- Sastry GM, Adzhigirey M, Day T, et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comp Aided Mol Des 2013;27:221–34.
- Schrödinger Release 2021-1: Semiempirical NDDO protocol; Jaguar. New York (NY): MOPAC, Schrödinger, LLC; 2021.
- Harder E, Damm W, Maple J, et al. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J Chem Theory Comput 2016;12:281–96.
- Greenwood JR, Calkins D, Sullivan AP, Shelley JC. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J Comp Aided Mol Des 2010;24:591–604.
- Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J Med Chem 2006;49:6177–96.
- Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol 2002;320:597–608.
- Cuffaro D, Nuti E, Rossello A. An overview of carbohydrate-based carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2020;35:1906–22.
- Bianco G, Meleddu R, Distinto S, et al. N-Acylbenzenesulfonamide Dihydro-1,3,4-oxadiazole hybrids: seeking selectivity toward carbonic anhydrase isoforms. ACS Med Chem Lett 2017;8:792–6.
- Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother 2016;16:961–8.
- Schütz SG, Robinson-Papp J. HIV-related neuropathy: current perspectives. HIV AIDS 2013;5:243–51.
- Di Fiore A, Truppo E, Supuran CT, et al. Crystal structure of the C183S/C217S mutant of human CA VII in complex with acetazolamide. Bioorg Med Chem Lett 2010;20:5023–6.
- Kalinin S, Kopylov S, Tuccinardi T, et al. Lucky switcheroo: dramatic potency and selectivity improvement of imidazoline inhibitors of human carbonic anhydrase VII. ACS Med Chem Lett 2017;8:1105–9.