
Abstract
Piperazine moiety is a cyclic molecule containing two nitrogen atoms in positions 1 and 4, as well as four carbon atoms. Piperazine is one of the most sought heterocyclics for the development of new drug candidates with a wide range of applications. Over 100 molecules with a broad range of bioactivities, including antitumor, antibacterial, anti-inflammatory, antioxidant, and other activities, were reviewed. This article reviewed investigations regarding piperazine groups for the modification of natural product derivatives in the last decade, highlighting parameters that affect their biological activity.
1. Introduction
The piperazine ring, an important class of N-heterocyclic bioactive natural products, is extensively present in biologically active compoundsCitation1. The piperazine scaffold has been recognised as a privileged structure in drug discovery and is widely distributed in biologically active compounds employed in several different therapeutic fields, including antitumorCitation2, antibacterialCitation3, anti-inflammatoryCitation4, antipsychoticCitation5,Citation6, anti-AlzheimerCitation7,Citation8, antifungalCitation9, and anti-diabeticsCitation10. The N-4 nitrogen of piperazine can be used as a basic amine, while the N-1 nitrogen can easily introduce hydrogen bond acceptors and hydrophobic groups through other heterocyclics without necessitating the addition of a stereocenterCitation11,Citation12.
Piperazine core has two primary nitrogen atoms which exert the improvement in pharmacokinetic features of drug candidates because of their appropriate pKaCitation13. These nitrogen sites lead to the essential increase in water solubility of the drug-like molecules thereby playing a crucial role in the bioavailability. Maintaining a balance between pharmacodynamic and pharmacokinetic profiles of drug-like molecules is the important factor in designing and developing of new drugs, thus, one of the goals in the drug discovery process is to design molecules with a high affinity for their targets and appropriate physicochemical properties. For this purpose, the characteristics of the piperazine template make this molecular subunit a useful and well positioned system in the rationale design of drugsCitation14.
Natural products are closely related to human development. Through the process of metabolic evolution, bacteria, fungi, and plants have co-evolved along with animals, greatly exploring the chemical space, and producing a large number of natural products that can alter the senses and behaviour of animalsCitation15. Designing hybrid molecules based on natural products has become an effective strategy, including combining two or more biologically active compounds from natural sources, and is expected to deliver these compounds to target organs simultaneously. Reportedly, this combination strategy has lower toxicity when compared with traditional single-target molecule co-administration methods. Additionally, hybrid molecules improved dose compliance, reduced drug interactions, and the cost of preclinical evaluationCitation16,Citation17. Natural products have always been an alternative source of chemical substances for exploring biological systems, as well as biological pre-verification prototypes for chemical probes and drug discovery, which are considered sources of new drug research and development owing to their unique chemical diversityCitation18,Citation19. A significant number of compounds have been developed from plants, microbial metabolites, and marine organisms. Between 1981 and 2019, approximately 23.5% of the new drugs approved globally, 33.6% of the approved small molecule drugs, and 75.6% of the drugs in the anti-infective field were derived from natural products and their derivativesCitation20. Given the importance and significant biological activity of the piperazine skeleton in natural products, in recent years, an increasing number of scientific articles, books, and patents have been based on piperazine ring modifications of natural products. A growing wealth of literature reflects the research interests and importance of this field, as well as the potential for developing drugs based on piperazine groupsCitation21–23. Therefore, this article combined the latest literature reports, especially from the perspective of biological activity, to introduce the research progress in this field. We anticipate that this review will provide novel concepts and research directions for the modification of natural products.
2. Biological effects of natural products containing the piperazinyl moiety
2.1. Antitumour activity
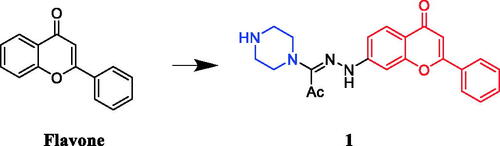
A series of N1-(flavon-7-yl)amidrazones incorporating N-piperazines were synthesised by Abu-AishehCitation24. Structure-activity relationships (SARs) shows that the presence of the phenyl group at C-2 does make a difference for the anti-K562 activity whereas none of the compounds having a methyl substituent at C-2 showed any activity against the K562 cells lines, more compounds of the flavone series (where there is a phenyl group linked to C-2) and only a few of the 2-methyl series displayed activity, and leaving the piperazine ring unsubstituted is better for the anti-K562 activity while for breast cancer, the anti-T47D activity resembles that of the K562 activity. Among them, compound 1 () was reportedly a potential antineoplastic agent against T47D cells (breast cancer cell line), with an IC50 value of 1.42 µM. Unfortunately, its activity is not as effective as the positive control drug doxorubicin (IC50 = 0.33 µM).
Figure 1. Chemical structures of flavone and its derivative.
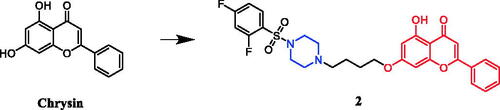
Anticancer properties of chrysin-based sulfonylpiperazines were reported by Pecere et al.Citation25 SARs show that the presence of halogen atom(s) was found essential to get better potency against SK-OV-3, HeLa, and A‐549 cell lines. Derivative 2 () with dual fluorine atom substitution on the sulfonylpiperazine ring exhibited the best activity against SK-OV-3 with least 12.67 ± 0.74 μg/ml of IC50 and was observed as potent as gefitinib (IC50 = 12.31 ± 0.33 μg/ml). Furthermore, in case of inhibition of HeLa cell line, again derivative with 2,4-difluoro substituent demonstrated remarkable 4.67 ± 1.42 μg/mL of IC50, which was better than gefitinib (IC50 = 17.92 ± 1.50 μg/ml). Patel et al. documented the synthesis of chrysin-piperazine conjugates and evaluated the in vitro anticancer activity against HeLa, CaSki, and SKOV-3 cancer cell lines. The findings revealed that the nature and position of the functional groups on the piperazine core may contribute to the expected antioxidant and anticancer effects from the perspective of structure-activityCitation26.
Figure 2. Chemical structures of chrysin and its derivative.
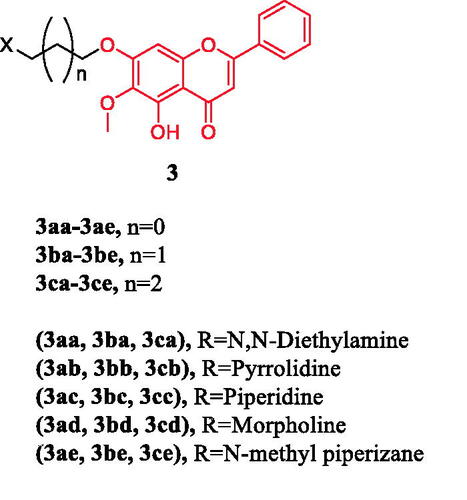
Oroxylin A (5,7-dihydroxy-6-methoxyflavone) is a naturally occurring monoflavonoid isolated from the root of Scutellaria baicalensis Georgi and exhibits potent anticancer activities in vitro and in vivo. Fu et al. synthesised three series of oroxylin derivatives by connecting a nitrogen-containing hydrophilic, heterocyclic ring to the C7-OH via a varying length of the carbon chain. Compounds (3ca–3cc) () containing a 4-carbon spacer were particularly potent with respect to cellular inhibition of all the three cell lines, with IC50 value ranging from 1.42 to 9.52 µM, approximately 5- to 20-fold more potent than oroxylin A, except for 3cd and 3ce. Compound 3cb and 3cc were found to be the most potent, with IC50 of 1.42 µM (HCT116) and 2.98 µM (HepG2), respectively. The results indicate that the length of the chain between the terminal heterocyclic substitutes and oroxylin A is an important factor for their potency. However, with a morpholinyl or N-methyl piperazinyl group at the terminal of 7-O-alkyl oroxylin A, compound 3cd and 3ce (with IC50 at 12.5–50.6 µM) displayed moderate levels of inhibition, which was similar to that of oroxylin A but over 4–25-fold (HepG2), 2–30-fold (HCT116), 3–6-fold (BCG823) less potent than 3ca–3cc. The morpholinyl or N-methyl piperazinyl substitution in the compounds 3ad–3ae (with IC50 at 15.4–67.6 µM) and 3bd–3be (with IC50 at 30.4–74.8 µM) also exhibited lower activities than substitutions with N, N-diethylamino (3aa and 3ba), pyrrolyl (3ab and 3bb) or a piperidyl (3ac and 3bc) group, respectively, regardless to the length of the carbon chain. It may be attributed to the volume of the side chain. Annexin V/PI staining experiments proved that compound 3cc has the highest in vitro efficacy on HepG2 cells among the derivatives. The difference of 3cc between the inhibition of cell proliferation and the apoptotic induction indicated that 3cc was more likely to be a necrosis-inducing agent or both apoptosis/necrosis inducer. Thus, the novel 7-O-alkylamino derivative of oroxylin A, compound 3cc, could be a promising antitumor candidate, and further in vitro and in vivo biological character evaluations are warrantedCitation27.
Figure 3. Chemical structures of oroxylin A and its derivatives.
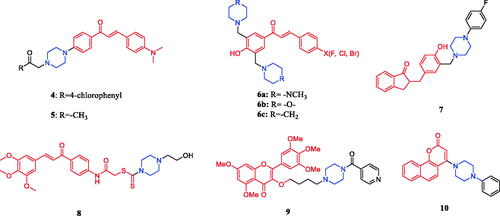
Novel hybrid compound 4 () between chalcone and piperazine demonstrated potent antitumor activity against A549 (a human lung cancer cell line), Hela and SGC7901 cells (IC50 = 5.24, 0.19, and 0.41 µM, respectively), which was better than positive control drug cisplatin (IC50 = 11.54 20.52, and 12.44 µM, respectively)Citation28. In addition, chalcone-piperazine derivative 5 () exhibited inhibitory activity against the A549 cell line, with an IC50 value of 0.19 µM, which was better than positive control drug cisplatin (IC50 = 11.54 µM)Citation29. Mannich bases 6a, 6b, 6c () were derivatives of chalcone. Though compounds showed a low inhibition potency towards hCA I (25–43%) and hCA II (6–25%) isoforms at an inhibitor concentration of 10 µM, they were more selective (1.5–5.2 times) towards hCA I isoenzyme. Compared with piperidine and N-methylpiperazine, Mannich bases 6b containing morpholine (substituted by fluorine, chlorine or bromine on the benzene ring) had the highest average SI value. The experimental results showed that the chalcone-piperazine hybrid compounds containing acetophenone had good anti-tumour activity. Among them, the substitution of halogen atoms on the benzene ring had a great impact on the anti-tumour activity, and the compounds substituted by fluorine atoms on the benzene ring showed the best activityCitation30. A series of chalcone-piperazine derivatives as mono Mannich bases were designed and synthesised by Tugrak and co-workers. The amine part was altered to N-phenylpiperazine, N-benzylpiperazine, 1–(2-fluorophenyl)piperazine, 1–(4-fluorophenyl) piperazine, and 1–(2-methoxyphenyl)piperazine. All synthesised compounds synthesised demonstrated a good inhibition profile towards hCA I and II isoenzymes, with Ki values between 29.6 and 58.4 nM and 38.1–69.7 nM, respectively. Additionally, compound 7 () revealed the highest tumour selectivity value (TS: 59.4), possibly by inducing necrotic cell death. The research showed cytotoxicity of some compounds containing piperazine in chalcone-Mannich base derivatives is stronger than other compounds containing different secondary amines (such as morpholine, piperidine, pyrrolidine, dimethylamine, diethylamine, dipropylamine, dibenzylamine)Citation31,Citation32. Novel chalcone-dithiocarbamate hybrids were designed, synthesised, and evaluated for their antiproliferative activity against select cancer cell lines (MGC803, MCF7, and PC3). Investigations evaluating modifications and structure-activity relationship (SAR) revealed that substituents on the piperazine unit are important for their inhibitory activity. Among these analogs, 8 showed the best inhibitory activity against PC3 cells (IC50 = 1.05 µM), which was better than positive control drug 5-FU (IC50 = 29.31 µM). Cellular mechanism studies elucidated that 8 () could inhibit colony formation, arrest the cell cycle at the G2/M phase, and induce DNA damage against PC3 cells. On replacing the piperazine ring with the morpholine or pyrrolidine group, the activity showed a particularly noticeable decreaseCitation33. The discovery of new chromen-4-one derivatives as telomerase inhibitors has been reported. Compound 9 () showed potent inhibitory activity against telomerase. Compound 9 exhibited high activity against Hela, SMMC-7721, SGC-7901, U87 and HepG2 cell lines with IC50s of 1.02, 1.33, 1.35, 2.50 and 4.12 µM, respectively, compared to the positive doxorubicin (IC50 = 0.21, 0.81, 0.98, 2.24, and 2.87 µM, respectively). The preliminary SARs showed that most compounds had good activity against SGC-7901 cells, but, almost all compounds possessed poor activity against HepG2 cells. And the substituent R showed a large effect for the activity, heterocycle is better than that benzene ring (compound 9). Further, the substituent number of benzene rings showed great influence on anticancer. Preliminary investigations revealed that compound 9 inhibited telomerase activity by decreasing the expression of dyskerin. Furthermore, the docking results showed that the target compound could strongly interact with residues LYS189 and Asp254 after introducing the piperazinphenyl methanone structure. In vivo experiments, compound 9 could evidently abate pathological changes of hepatic lobules in DEN-induced rats. The hepatic cells showed markedly reduced atypia, full cytoplasm and high differentiationCitation34. Reportedly, 4-amino- 2H-benzo[h] chromen-2-one analogs were synthesised and evaluated for their anticancer activity. Compound 10 () displayed potent androgen receptor binding affinity, as well as strong cytotoxic activity against LNCaP-hr cells (IC50 = 0.52 ± 0.11 µM), which was better than positive control drug finasteride (IC50 = 14.5 µM). The SAR results showed that the 4-position substituent of the piperazine ring and the ortho-substituted phenyl analogs had relatively good inhibitory activityCitation35.
Figure 4. Chemical structures of chalcone and its derivatives.
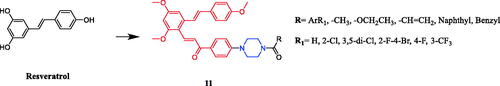
Novel resveratrol-chalcone-piperazine amide derivative 11 (R=–CH = CH2) () showed superior cytotoxic activity against A549 and HeLa cells (IC50 = 0.26 and 7.35 µmol/L, respectively), which was better than positive control drug cisplatin (IC50 = 11.54 and 20.52 µmol/L, respectively). And fluorescence-activated cell sorter (FACS) analysis showed that the compound effectively induced apoptosis in A549 cells. The structure of the compound had a significant effect on anticancer activity. The preliminary structure-activity relationship shows that the anticancer activity was related to the number and category of substituents on the benzene ring, but had nothing to do with the position of substituents, and the lack of electronic benzene ring led to positive activitiesCitation36.
Figure 5. Chemical structures of resveratrol and its derivatives.
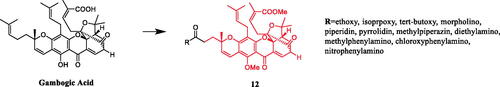
A gambogic acid derivative 12 () containing a methylpiperazine substituent was found to possess a most potent inhibitory activity against A549 and BGC-823 cells among all compounds tested. In A549 cell line, compound 12 exhibited the highest potency with an IC50 value of 0.12 µM, which was 10-fold more potent than gambogic acid. In BGC-823 cell line, compound 12 displayed the best potency with an IC50 value of 0.57 µM, which was almost 5-fold more potent than gambogic acid. The SARs at the C-37 site of gambogic acid shows that the removal of hydrophobic prenyl tail and introduction of hydrophilic carboxyl group slightly reduced the anti-tumour activity. Further introduction of hydrophobic motifs such as methyl, isoprenyl and tert-butyl groups had little effect on improving the potency. Inspiringly, substituted by aliphatic amines showed obviously increased activities. Meanwhile, the aliphatic amines also could improve the solubility and drug-like properties of the derivatives. Notably, compound 12 containing methylpiperazine substituents was the most excellent derivative with the IC50 values ranging from 0.12 to 3.10 µM against the five cell lines. Phenylamine groups were not a good moiety for improving the activity at the C-37 site. In addition, although compounds with the flexible amino acid side chains ought to have greatly improved solubility, they were found to be less active or even mostly inactive against all the five cell lines. The introduction of glycine group to C-37 site could increase the selectivity against HCT-116 and HepG2 cells. This study found that compounds with aliphatic amines had higher anti-tumour activity than aniline, improving the solubility and drug-like properties of derivativesCitation37.
Figure 6. Chemical structures of gambogic acid and its derivative.
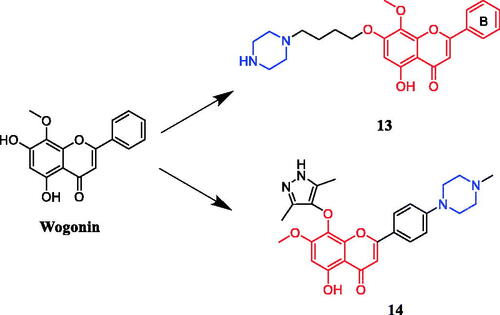
Li and co-workers revealed that wogonin-piperazine derivative 13 () possessed the highest potency against HepG2, A549, and BCG-823, with IC50 values of 1.07 µM, 1.74 µM, and 0.98 µM, respectively, which was better than positive control drug 5-FU (IC50 = 17.2, 16.1 and 10.1 µM, respectively) and parent compound wogonin (IC50 = 19.0, 15.8 and 16.5 µM, respectively). The results showed that the anti-tumour activity was enhanced when the 7-position of wogonin was occupied by aliphatic amines, in contrast, wogonin derivatives substituted at 8-position or B-ring were generally less potent than the control 5-fluorouracilCitation38. In another study, wogonin derivative 14 () showed excellent activity against MV4-11 (leukaemia cell line) (IC50 = 20 nM) and CDK9 (IC50 = 19.9 nM) cell growth. In addition, compound 14 showed much improved physicochemical properties, such as water solubility, compared with the parent compound wogonin. The follow-up studies showed that the compound 14 is selective towards CDK9-overexpressing cancer cells over normal cells. Preliminary mechanism studies on the anticancer effect indicated that 14 inhibited the proliferation of MV4-11 cells via caspase-dependent apoptosis. In addition, highlighted compound 14 showed significant antitumor activity in mouse acute myeloid leukaemia (AML) models without producing apparent toxic effects in vivo. Additionally, among the synthesised compounds, the piperazine and methylpiperazine substituted derivatives exhibited higher antiproliferative potency against the HepG2 cell line, while morpholine and pyrrolidine substituted analogs showed dramatically lower antiproliferative activityCitation39.
Figure 7. Chemical structures of wogonin and its derivative.
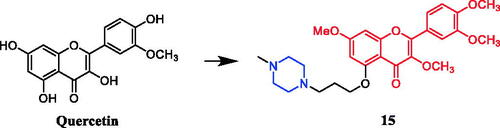
In an investigation targeting prostate cancer cells, 6 of the 48 quercetin derivatives synthesised contained piperazine moieties. Moreover, quercetin derivative 15 () demonstrated promising efficacy against the PC-3 cell line, with an IC50 value of 4.47 ± 0.92 µM, being over 2–13 times more potent than quercetin. The experimental results indicated that modification of the hydroxyl groups at positions 3 and 5 significantly improved their inhibitory activity against prostate cancer cells, besides, the 48 derivatives are generally more effective in inhibiting the proliferation of PC-3 and LNCaP cells than DU145 cellsCitation40.
Figure 8. Chemical structures of quercetin and its derivative.
A series of novel bile acid (including ursodeoxycholic acid, cholic acid, chenodeoxycholic acid) hybrid compounds comprising piperazine were designed for anticancer activity. Among them, compound 16 () as the most potent against KMS-11, a multiple myeloma cell line, with an LD50 value of 8.5 ± 0.5 µM. Concerning the structure–activity relationship, the best activity was obtained with compound 16 having cinnamylpiperazinyl group in the side chain and hydroxyl group at C-7 of the steroid skeleton. These results prompt us in a future study to incorporate other amino-substituted cinnamylpiperazinyl groups in the side chain of chenodeoxycholic acidCitation41,Citation42.
Figure 9. Chemical structures of chenodeoxycholic acid and its derivative.
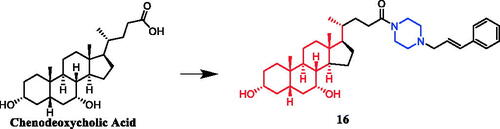
Currently, an ursolic acid (UA) derivative, obtained from the leaves and berries of many natural plants, is under development as a lead anticancer agent, which has also been modified by introducing piperazine rings to develop novel compounds. In 2015, Shao and co-workers revealed that substituted piperazine coupled to UA could induce cancer cell apoptosis and arrest cell cycle at the G0/G1 stage in HT-29, HepG2, and RL95-2 cellsCitation43. In the same year, UA derivative 17 () with piperazine was reported as a potential anticancer agent, inducing cell apoptosis by G1 cell cycle arrest. Compound 17 displayed the best proliferation among this series of compounds, with IC50 values of 9.82 µM, 18.97 µM, 13.64 µM, 5.40 µM and 11.06 µM against MGC-803, HCT-116, T24, HepG2 and A549 cancer cell lines, respectively. And compound 17 showed better potent antiproliferative activity than UA (as well as 5-FU) against these five cancer lines, with IC50 values of 27.08 µM, 38.78 µM, 29.29 µM, 30.21 µM and 35.79 µM. SARs indicated that the synchronous introduction of piperazine and thiourea at C-28 and an acyl group at C-3 may improve the antitumor activity of UA, and solely introduction of an acyl piperazine thiourea at C-28 may decrease the antitumor activity. Moreover, it could be concluded that para-substituents in the phenyl ring were important than meta-substituents to their antiproliferative activities. Western blot and qRT-PCR (quantitative real-time PCR) experiments demonstrated that compound 17 may induce apoptosis through both intrinsic and extrinsic apoptosis pathways. Moreover, 17 has shown a pronounced increase in Fas and caspase-8 activity. Therefore, these studies indicated that the target compound may induce apoptosis, arrest cell cycle progression at the S phase and increase the activity of caspase-3 to inhibit cell growth. It is worth noting that piperazine was used as a linker in this UA modificationCitation44. UA derivatives targeting hypoxia-inducible factor-1α (HIF-1α) by introducing a 1,3,4-oxadiazole moiety, triazolone, and piperazine ring were designed and synthesised. Compound 18 () demonstrated excellent activity inhibiting the expression of HIF-1α, with IC50 = 38.1 µM, possessing a lower appreciable cytotoxic activity (IC50 > 100 µmol/L) than that of UA (IC50 = 23.8 µmol/L). SAR indicating that the triazolone moiety was favourable for HIF-1α inhibition in the UA derivatives, and introducing a piperazine ring to the oxadiazole structure can also enhance the biological activity of the compoundsCitation45. In 2016, Tian et al. reported that the introduction of a piperazine moiety at the C-28 position of oleanolic acid (OA) and UA demonstrated a superior growth inhibition against MCF-7 cancer cells than the lead compounds or positive control gefitinib, including derivatives 19 (), 21 (), 23 () and 24 (), with IC50 values of 40.27 ± 4.88 µM, 56.85 ± 4.88 µM, 12.1 µM, and 14.2 µM, respectivelyCitation46,Citation47. In 2019, some UA derivatives containing a piperazine moiety presented significant antitumor activities against the cancer cell lines evaluated. The structure-activity relationship studies revealed that the introduction of thiazole on A ring and triazole or tetrazole moiety on C-28 has little help to improve antitumor activities. The introduction of piperazine or homopiperazine can significantly improve the antitumor activity. And 4-fluorobenzyl and piperazine moieties are the crucial anticancer functional groups of these compounds. Among them, compound 20 () was found most effective, demonstrating IC50 values of 2.6 µM and 2.1 µM against the HeLa and MKN45 cell lines, respectively. And compound 20 was better than positive control drug cisplatin (IC50 = 15.1 and 2.8 µM, respectively) and was better than parent compound UA (IC50 = 15.1 and 16.7 µM, respectively). Compound 20 decreased the apoptosis regulator (BCL2/BAX) ratio, which subsequently disrupted the mitochondrial potential and induced apoptosis and significantly suppressed the growth of Hela xenografts in nude miceCitation48. In 2012, UA derivative 22 () was described as a potent antitumor agent that induced cell apoptosis and inhibited growth against MGC-803 cells (gastric cancer cell), with an IC50 value of 2.50 ± 0.25 µM, which was better than positive control drug hydroxycamptothecin (IC50 > 20 µM) and parent compound UA (IC50 = 24.32 ± 0.57 µM). The SAR studies revealed that incorporation of an acyl piperazine moiety at C-28 while retaining the polar group at C-3 significantly improved the antitumor bioactivities of the compoundsCitation49. In 2015, Zhao et al. synthesised and investigated a series of piperazine-containing pentacyclic triterpene derivatives. OA derivative 25 () and asiatic acid derivative 26 () was reported as the most promising antitumor agents, with IC50 values in the range of 7.05–13.13 µM, when evaluated against MCF-7, HeLa, and A549 cell lines. The pronounced influence of the incorporation of piperazine on antiproliferative effects in the series of synthesised compounds could be explained on the capacity for the formation of hydrogen bonds, improvement of water-solubility, and adjustment of molecular physicochemical properties. Furthermore, 25 and 26 can induce cell cycle arrest in all of the three cellsCitation50.
Figure 10. Chemical structures of ursolic acid and its derivative.
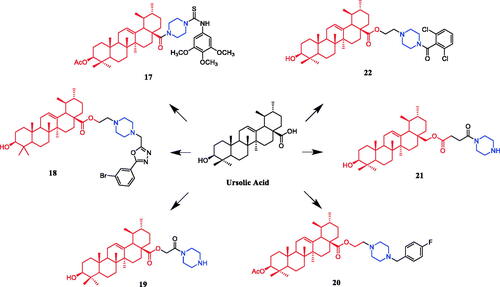
Figure 11. Chemical structures of oleanolic acid and its derivative.
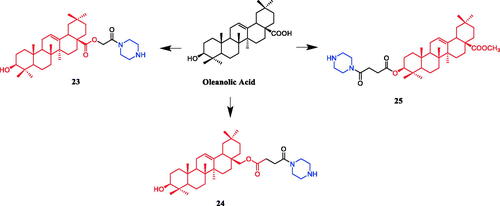
Figure 12. Chemical structures of asiatic acid and its derivatives.
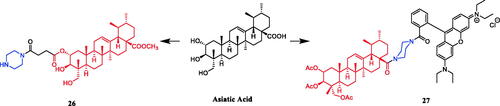
Asiatic acid piperazine derivative 27 () shows distinct cytotoxicity for several human tumour cell lines, e.g. EC50 (A2780) = 8 ± 2 nM, which was better than parent compound asiatic acid (IC50 = 28.2 µM). Interestingly, compound 27 showed non-linear, bimodal dose-response relationships against two human tumour cell lines (HT29 and 518A2)Citation51.
A series of C-28 amide derivatives of hederagenin were synthesised, with or without acetyl groups at the 3 and 23 positions of ring A, to develop effective cytotoxic agents. The amide derivatives were cytotoxic for a variety of human tumour cell lines. In general, the hydroxylated derivatives were less active than the acetylated derivatives. Among them, compound 28 () carrying a piperazinyl presented good cytotoxic activity against A2780 cells (EC50 = 1.9 µM), which was better than parent compound hederagenin (IC50 > 30 µM)Citation52.
Figure 13. Chemical structures of hederagenin and its derivatives.
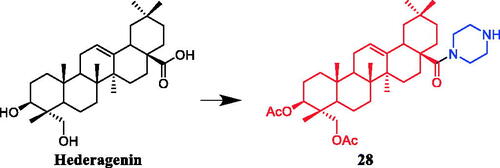
Li et al. synthesised a series of acetyl-11-keto-β-boswellic acid (AKBA) derivatives and evaluated their anticancer activity. Indicating that the cytotoxic activity of AKBA was improved effectively through modification of ring A and C-24. Introduction of 2-cyano-3-oxo-1-en system in ring A of boswellic acids increased cytotoxicity. The esterification of free carboxyl acid at C-24 resulted in a sharp decrease in the activity. AKBA derivative 29 () with 2-cyano-3-oxo-1-en in ring A and piperazine amide at C-24 was reported as the most active in inhibiting the growth of prostate cancer PC-3 (IC50 = 0.04 µM) and LNCaP (IC50 = 0.27 µM) cell lines, with 796-fold and 102-fold more potent than AKBA. The mechanism of action of 29 indicated that cell cycle arrest and apoptosis induction were related to its anti-proliferative effects. 29 could regulate the expression levels of cell cycle- and apoptosis-related proteins, such as p21, cyclin D1, Mcl-1 and NOXA, in a concentration-dependent manner. AKBA had a weak ability to inhibit Pin1 activity, which was improved by the modification on ring ACitation53.
Figure 14. The structure of pentacyclic triterpenoid AKBA.
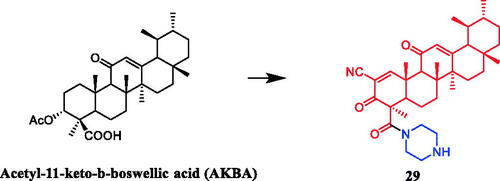
Zhao and co-workers designed and synthesised several series of dehydroabietic acid piperazine derivatives. The results indicated that compound 30 () with piperazine moiety showed selectivity for HepG-2 over MCF-7, with IC50 values of 23.56 and 62.55 µM, respectivelyCitation54. In another study, 1H-dibenzo[a, c]carbazole derivatives of dehydroabietic acid bearing different N-(piperazin-1-yl)alkyl side chains were designed, synthesised, and evaluated for their in vitro anticancer activities against three human hepatocarcinoma cell lines. SAR indicated that methyl, methoxyl, ethoxyl and chloro groups anchored on the indole moiety were more beneficial to the anticancer activity. In addition, the derivatives containing 12-Me and 12-OEt generally showed greater cytotoxic activities than their analogs with the same substituents at C-10, while compound with 10-OMe substituent was relatively more active than compound with 12-OMe. Especially, Compound 31 () with 12-OEt substituent showed strong cytotoxic activity against SMMC-7721, HepG2, and Hep3B cell lines, with IC50 values of 1.39 ± 0.13, 0.51 ± 0.09, and 0.73 ± 0.08 µM, respectively. Compared with lead compound and the positive control doxorubicin, it exhibited considerably more potent anticancer activities against three cancer cells and lower cytotoxicity to normal hepatocyte cell line QSG-7701 (IC50: 12.52 ± 0.58 µM). Compound 31 could significantly inhibit MEK1 kinase activity with IC50 of 0.11 ± 0.02 µM. Furthermore, compound 31 increased the level of intracellular reactive oxygen species (ROS), reduced the mitochondrial membrane potential, destroyed the integrity of the cell membrane, ultimately resulting in the proliferation and apoptosis of HepG2 cells. Therefore, it could be a potent MEK inhibitor and a potential anticancer drugCitation55.
Figure 15. Chemical structures of dehydroabietic acid and its derivatives.
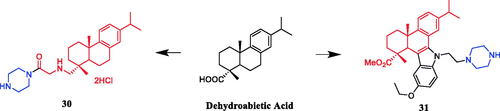
To explore the SAR improve drug-like properties of celastrol derivatives, 23 derivatives targeting the Hsp90-Cdc37 interaction were synthesised by Jiang et al. SAR indicated that the derivatives that incorporated polar groups on the C29 carboxyl showed promising Hsp90–Cdc37 disruption activities and compound 32 () containing the piperazine moiety exhibited the most significant Hsp90-Cdc37 activity disruption, with an IC50 value of 4.71 ± 0.14 µM, which was better than parent compound celastrol (IC50 = 6.49 ± 0.27 µM). Compound 32 improved solubility and permeability. Compound 32 also exhibited potent antitumor activities, with IC50 values of 1.01–1.12 µM against MCF-7, Panc-1 and A549, better than parent compound celastrol (IC50 = 2.02–2.48 µM). Compound 32 could induce the degradation of clients Akt and Cdk4 dose-dependently, suggesting that Compound 32 exerted their antitumor activities through Hsp90 inhibition in part. Moreover, Compound 32 induced cell cycle arrest at G0/G1 phase and physiological apoptosis in Panc-1 cells. Compound 32, with improved Hsp90–Cdc37 disruption activity, cytotoxicity and good druglikeCitation56. A series of 3-carbamate and 29-ester celastrol derivatives were synthesised by Shan et al. SAR indicated that the activity of derivatives with piperazine was better than that with aniline, Piperidine and morpholine. The substitution of piperazine and morpholine by piperidine and aniline weakened the activity, which might indicate that nitrogen atom and oxygen atom here are favourable as hydrogen bond acceptor but the alkyl substituents and hydrophobic groups not. The nitrogen atom is more suitable than the oxygen atom here and the groups on it make difference to the activity. The activity gets lower when the substituents get bigger. Meanwhile, we observed a significant loss of cytotoxicity when the size of the substituent group on C-3 hydroxyl become as big as 8–10 carbons length. However, when the size was smaller than that, it did not cause an obvious change in the activity. It indicated a threshold about the size of this position, which is located in front of C-3 hydroxyl. It might affect the activity and pharmacokinetic property of celastrol, and the stereo structure here may improve the selectivity. The carbamates were introduced nitrogen and oxygen atoms as new hydrogen bond acceptors and hydrophilic centre. The antitumor activity of compound 33 () against several cancer cell lines, with IC50 values of 5.18 and 1.02 µmol/L against A549 and Bel7402 cell lines. It is slightly less active than the lead compound celastrol (IC50 = 2.12 µmol/L and 0.44 µmol/L), but compound 33 revealed a dose-dependent inhibition and better inhibitory activity, with 70.96% inhibition of A549 cells at a dose of 12 mg/kg when compared with celastrol at 12 mg/kg (p < .05) in vivoCitation57. Piperazine-containing celastrol derivative 34 () was revealed a potential telomerase inhibitor against the MGC-803 cell line, presenting an IC50 value of 1.00 ± 0.17 µM, which was better than positive control drug 5-FU (IC50 = 3.58 ± 0.40 µM), AMD (IC50 = 1.19 ± 0.07 µM) and parent compound celastrol (IC50 = 1.55 ± 0.25 µM)Citation58. In 2017, a series of triterpenoic acid derivatives containing the piperazine moiety exhibited low EC50 values against several human tumour cell linesCitation59.
Figure 16. Chemical structures of celastrol and its derivatives.
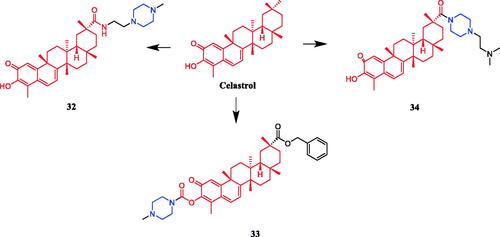
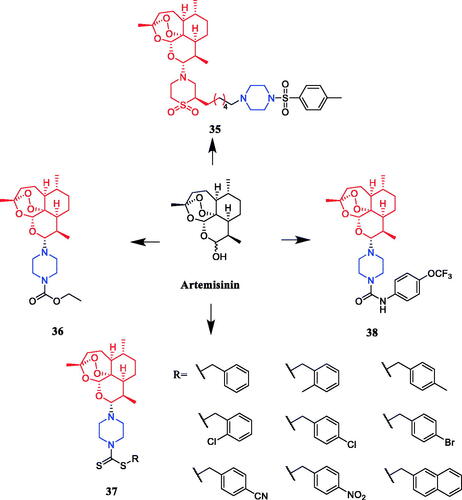
Reportedly, several artemisone-piperazine-sulphonamide derivatives have been synthesised. The antiproliferative activity of the new compounds was evaluated against human hepatoma cells. Compound 35 () showed activity against SMMC-7721 cell lines (IC50 = 0.09 µmol/mL), which was better than parent compound artemisone (IC50 = 0.44 µmol/mL)Citation60. In 2017, a series of novel artemisinin derivatives were designed and synthesised by Sun et al. and were evaluated using the MTT assay against ten cell lines. These derivatives were generally more effective in inhibiting cancer cell growth than artemisinin (IC50 > 40 µM). Among them, compound 36 () which bore an N-ethoxycarbonyl-piperazine group was the most active against HepG2 and PLC-PRF-5 cells (the IC50 values were 4.1 and 5.7 µM, respectively) and presented no cytotoxicity on L-02 cells, and 36 might be a valuable potential antitumor drug against HCC. SAR found that introducing the piperazine group into the C10 position of artemisinin could enhance the antiproliferative effect on certain cancer cells to some degree. The structure of compounds 36, which showed the highest activity against hepatocellular carcinoma cells, inspired to improve the oil solubility of the piperazine group into the C10 position. Furthermore, this substrate could induce cell cycle arrest in the G2/M phase and apoptosis in HepG2 cellsCitation61. Reportedly, Yu et al. synthesised twelve artemisinin-piperazine- dithiocarbamate derivatives, all of which had stronger in vitro antitumor activity than dihydroartemisinin. In particular, artemisinin derivative 37 (R = 4-cyanobenzyl) () showed promising inhibitory activity against the SMMC-7721 cell line, with an IC50 value of 0.0025 ± 0.04 µM, which was better than positive control drug vincristine (IC50 = 0.0103 ± 0.03 µM), cytosine arabinoside (IC50 = 0.0271 ± 0.04 µM) and dihydroartemisinin (IC50 ≫ 0.1000 µM) and lower toxicity against LO2 cell lines, presenting an IC50 of 0.18 ± 0.04 µMCitation62. In another study, artemisinin derivatives containing fluorine atoms synthesised by Li et al. were evaluated for their antiproliferative potencies against U87MG, SHSY5Y, MCF-7, MDA-MB-231, A549, and A375 cancer cell lines. Compound 38 (), the most active derivative containing the piperazine moiety, presented an IC50 value of 2.1 µM against the human breast cancer MCF-7 cells by inducing apoptosis and G1-phase arrest and showed lower cytotoxicity (IC50 = 6.7 µM) than doxorubicin (IC50 = 0.9 µM) against L02. SAR showed that the structural modification of artemisinin, i.e. introducing the group of piperazine with attached amide bond into C10 position of artemisinin, led to a dramatic increase of the inhibitory effect on certain cancer cells to some degreeCitation63.
Figure 17. Chemical structures of artemisinin and its derivatives.
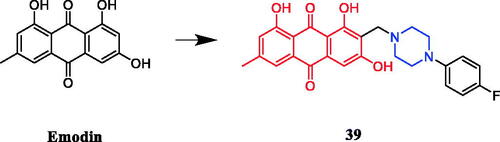
Narender et al. generated a variety of emodin derivatives and found that the piperazine-containing derivative 39 () strongly inhibited the proliferation of HepG2 and MDA-MB-231 cancer cell line with an IC50 of 10.44 µM and 5.027 µM, respectively, which is comparable to marketed drug epirubicin (IC50 of 4.6 µM and 7.7 µM, respectively). SAR indicated that O-alkylation and C-alkylation of emodin improve the activity in both cell lines, whereas esterification only improves the activity in the MDA-MB-231 cell line. The derivatives 39 was capable of arresting the cell cycle at the G1/S phase, demonstrating caspase-dependent apoptosis in the HepG2 cell line, as well as DNA intercalating activityCitation64.
Figure 18. Chemical structures of emodin and its derivatives.
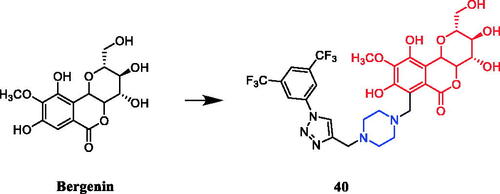
A series of bergenin-1,2,3-triazole hybrids were synthesised and evaluated for their potent activity against cancer cell lines. Among them, piperazine-containing compound 40 () exhibited potent activity against HeLa and A-549 cells, with IC50 values of 1.33 µM and 1.86 µM, respectively, and was reportedly equipotent to doxorubicin (IC50 values 1.98 µM and 1.34 µM respectively). Preliminary structure-activity relationship indicates that the presence of a substituent such as a chlorine atom or a cyano, floro and CF3 group on aromatic triazole partner enhanced activity with IC50 values ranging from 1.33 to 9.9 µM on A459 cell as well as HeLa cell lines within the studied series. In addition, flow cytometry analysis had demonstrated that compound 40 triggered cell cycle arrest in the G2/M phase and induced apoptosis in a dose and time-dependent manner. Taken together, compound 40 effectively inhibited tubulin polymerisation, disrupted intracellular tubulin-microtubule balance, resulting in prolonged G2/M cell cycle arrest. Docking studies also indicated a strong hydrophobic interaction with tubulin, thus leading to stable binding, consequently leading to apoptosis of cancer cellsCitation65.
Figure 19. Chemical structures of bergenin and its derivatives.
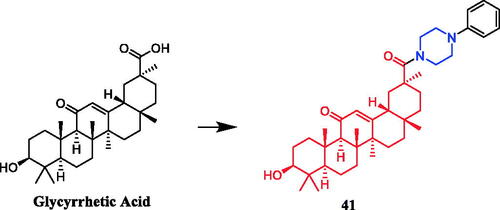
Yan et al. revealed that glycyrrhetic acid-piperazine derivative 41 () exhibited the best inhibitory activity against MCF-7 cells, with an IC50 value of 1.08 µM, which was better than the positive control vandetanib (IC50 = 1.40 µM). SAR can be concluded that a fragment of a phenylpiperazine skeleton plays a significant role in the antiproliferative effect in vitro. Among the thirteen glycyrrhetic acid derivatives containing phenylpiperazine skeleton, compound 41, which has smaller steric hindrance showed better inhibitory capability than the others, and compounds that contain alkylpiperazine had the same trend. Compound 41 also showed potent inhibitory activity against vascular endothelial growth factor receptor 2 (VEGFR2) tyrosine kinase, with an IC50 value of 0.35 µM. Docking simulations were performed to discover the binding mode, and the results indicated that compound 41 could bind well to VEGFR2 at the active siteCitation66.
Figure 20. Chemical structures of glycyrrhetic acid and its derivatives.
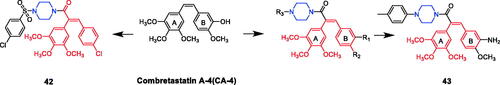
Combretastatin-A4 linked sulphonyl piperazine derivative 42 () exhibited remarkable cytotoxicity in all cancerous cell lines investigated. Compound 42 displayed an IC50 of 0.36 ± 0.02 µM against A549 cells, which was more active than the lead compound Combretastatin-A4 (IC50 = 0.43 ± 0.02 µM). Further investigations revealed that it could significantly inhibit tubulin assembly, effectively acting at the colchicine binding pocket of tubulin. The SARs of these derivatives indicated that the 4-chloro substitution on the benzene ring C4 of the sulphonamide showed effective activity, and the activity was significantly reduced when sulphonamide ring is substituted by other substituents such as hydrogen, methyl, methoxy and tert-butyl. In addition, the selectivity of compound 42 to A549 cells is almost 3 times that of normal HaCaT cellsCitation67. O’Boyle et al. reported the synthesis and anticancer activities of combretastatin-A4 piperazine conjugates. The SAR research pointed out that the impact on the viability of MCF-7 breast cancer cells was related to the substituents on the piperazine ring, and the general trend in potency was as follows: phenylpiperazine > acetylpiperazine ≈ cinnamylpiperazine > benzylpiperazine. Amino-substituted p-tolylpiperazine 43 () was the most potent of all piperazine derivatives, suggesting that an amino group is advantageous. Derivative 43 revealed an IC50 value of 83 nM against MCF-7 breast cancer cells, unfortunately, the activity of 43 is not as high as the lead compound combretastatin-A4 (IC50 = 3.9 nM). Furthermore, compound 43 was selectively toxic to MCF cells, inducing G2/M phase arrest and apoptosis of MCF-7 cells, but not in peripheral blood monocytes, and inducing cleavage of DNA repair enzyme poly adenosine diphosphate ribose polymerase (PARP) in MCF-7 cellsCitation68.
Figure 21. Chemical structures of combretastatin-A4 and its derivatives.
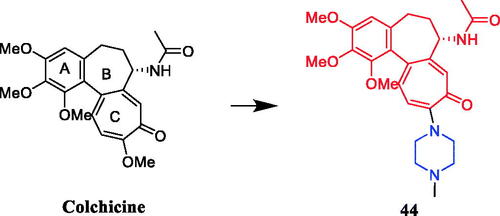
Singh et al. reported the synthesis and anticancer activities of colchicine derivatives. SAR indicated that A- and C-rings of colchicine scaffold are the minimum structural features necessary for high-affinity drug–tubulin binding, and C-ring modifications and B-ring substitutions (particularly, on acetamido group) are well tolerated for their anti-tubulin activity. Compound 44 () showed notably lower P-glycoprotein (Pgp) induction activity when compared with colchicine, and good antiproliferative activity against the Colo-205 cell line, with an IC50 value of 1.0 ± 0.006 µM. Unfortunately, the activity of 44 is not as high as the lead compound colchicine (IC50 = 0.032 µM)Citation69.
Figure 22. Chemical structures of colchicine and its derivatives.
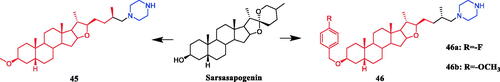
Sarsasapogenin-piperazine derivatives 45, 46a, and 46 b () displayed significant inhibitory effects against several tumour cell lines. Derivatives 45 revealing potent toxicity against the A549 cancer cell line, with an IC50 value of 1.70 µM, which was more active than positive control timosaponin A-III (IC50 = 11.92 µM) and 5-fluorouracil (IC50 = 42.15 µM). Compounds 46a and 46b exhibited significant cytotoxic activities against the six cell lines, being more potent than their parent compound sarsasapogenin. Furthermore, the p-fluorobenzyloxy series of compounds generally exhibited stronger cytotoxicities against all the tested cancer cells compared with the benzyloxy and p-methoxybenzyloxy series, and the substitution of pyrrolidinyl and piperazinyl groups at the C26 position was the preferred option for these compounds to display antitumor activities. Compound 46b exhibited excellent cytotoxic activity against MCF-7 cell line (IC50 = 2.95 µM), and was 16.7-fold more potent than sarsasapogenin. Further studies of the cellular mechanism of 46b showed that it arrested MCF-7 cells at the G2/M phase and induced apoptosis and necrosisCitation70,Citation71.
Figure 23. Chemical structures of sarsasapogenin and its derivatives.
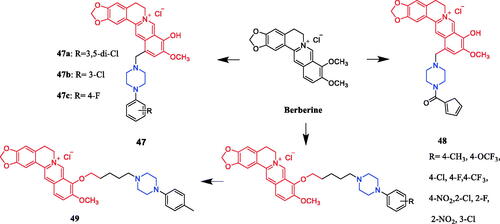
In recent years, several berberine derivatives were synthesised by Mistry et al., exerting anticancer activity against various human cancer cell lines. Conversely, a series of derivatives were synthesised by introducing various piperazine ring systems into 12-position of berberine. Mistry and co-workers reported that the berberine-piperazine derivative 47a () demonstrated potent anticancer effects against HeLa cells, with an IC50 of 7.327 ± 0.08 µM, and was more potent than the lead compound berberine (IC50 = 13.42 ± 0.23 µM). The analysis of the results suggested that the derivatives may enhance their ability to bind to the target of the drug mainly through hydrophobic interaction, conjugation and hydrogen bonding on the 9-hydroxyl group. On further preparation, analog 47b (), demonstrating a substitution at the 3-position of the phenyl ring of piperazine moiety, was found to be the most effective, with an IC50 value of 3.983 ± 0.07 µg/mL against HeLa cells, and was more potent than the lead compound berberine (IC50 = 4.499 ± 0.08 µg/mL). In another report, compound 47c () with a 3,4-dichlorophenyl piperazine entity was found to be the most active among derivatives tested, with an IC50 of 5.782 ± 0.55 µg/mL and CC50 of 320.7 ± 1.04 µg/mL, and the highest TI of 55.47 against HeLa cell lines. The potency of this compound was double that of the parent berberine which showed a TI of 28.16. The results clearly showed that the piperazine-based berberine analogues had stronger anti-tumour activity on the Caski cell line than Hela. It appeared that introduction of electron-withdrawing substituents (e.g. chloro- or fluoro-substituent) on the piperazine-linked benzene ring enhanced anti-tumour activityCitation72–74. In 2016, berberine derivative 48 () with a furoyl piperazine substituent exhibited the highest anticancer activity against the CaSki cell line, with an IC50 value of 6.11 µM, CC50 value of 344.5 µM, and selectivity index of 56.39. The potency of this compound was double that of the parent berberine which showed a TI of 29.15. Conversely, a series of derivatives were synthesised by introducing various piperazine ring systems into the 9-position of berberine. Concerning the activity regarding the functional group attached to the piperazine ring, the order falls in the way alkyl > chloro > fluoro > nitroCitation75. Analog 49 () with a 4-methylpiperazine substituent showed excellent anticancer property, with a therapeutic index (TI) of 58.53 (HeLa) and 48.76 (CaSki), better than that of a parent compound berberine with 27.41 and 25.84 of TI, and IC50 values of 5.595 ± 0.02 µg/mL and 6.716 ± 0.05 µg/mL, respectivelyCitation76.
Figure 24. Chemical structures of berberine and its derivatives.
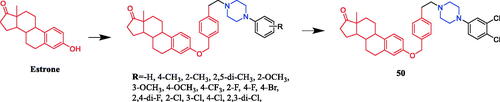
Chen reported the synthesis of oestrone derivatives containing the piperazine moiety and evaluated their antitumor activities against several classical prostate cancer cell lines, including PC3, LNCaP, and DU145. Derivative 50 (R = 3,4-di-Cl) () showed significant cytotoxic actions against the LNCaP cell line (IC50 = 0.78 ± 0.34 µM), better than that of positive control finasteride (IC50 = 14.53 µM). and exhibited better α1-adrenergic receptor (AR) subtype selectivity over α1B (α1B/α1A ratio = 14.7). The SAR results showed that the ortho-substituted phenyl derivatives and the para-substituted phenyl derivatives were more effective on the cytotoxic activity of LNCaP and/or DU145 cellsCitation77.
Figure 25. Chemical structures of oestrone and its derivatives.
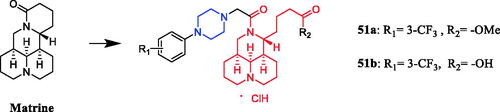
The in vivo antitumor assay revealed that matrine derivatives containing a piperazine moiety possessed improved therapeutic efficacy. Compound 51a and 51b () showed strong antitumor activity against human hepatoma Bel-7402 and colorectal carcinoma RKO cells (IC50 = 50.4 ± 1.4 µM and 48.3 ± 3.1 µM, 48.3 ± 2.5 µM and 51.3 ± 3.3 µM, respectively), proved to be of much better therapeutic efficacy than parent compound matrine (IC50 = 86.6 ± 2.6 µM and 84.3 ± 2.5 µM, respectively) and positive control cisplatin (IC50 = 92.5 ± 1.6 µM and 68.0 ± 1.3 µM, respectively). The SAR analysis indicated that the introduction of a piperazine moiety on matrine might significantly improve its antiproliferative activity, and the substituents and their position on phenyl of R1 and the variation of R2 could take a great role in antitumor activityCitation78.
Figure 26. Chemical structures of matrine and its derivatives.
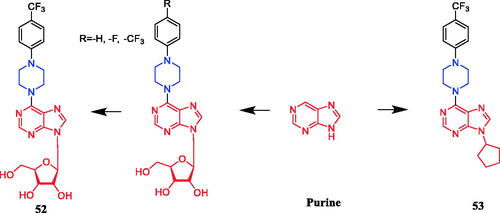
Tuncbilek et al. developed purine ribonucleoside analogs and evaluated their antitumor activity. Compound 52 (), containing 4-substituted piperazine in the substituent at N6, reportedly exhibited good antitumor activity against six different human tumour cell lines, with IC50 values between 5.2 and 9.2 µM. Further experiments also found that compound 52 may interfere with cellular ATP reserves by affecting cell kinase activityCitation79. A series of purine derivatives were synthesised and evaluated against liver Huh7, breast T47D, and colon HCT116 carcinoma cells. Compound 53 () containing a piperazine moiety had better cytotoxic activities (IC50 ≤ 1 µM) than the nucleobase 5-FU and nucleosides fludarabine, cladribine, and pentostatineCitation80.
Figure 27. Chemical structures of purine and its derivatives.
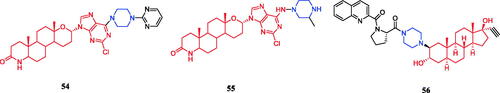
Huang et al. synthesised novel C6-piperazine substituted purine steroid-nucleosides analogs. Among them, compounds 54 and 55 () showed remarkable cytotoxicity against the PC-3 cell line (IC50 = 1.84 µM and 2.90 µM, respectively), better than that of parent compound purine nucleosides analogues with 11.19 µM of IC50. SAR suggesting that the substituents at C2- and C6-position of the purine ring and N4-position of piperazine moiety had a significant influence on the cytotoxicityCitation81,Citation82. Among the amino steroid derivatives synthesised by Ayan et al., compound 56 () with a piperazine-proline side-chain displayed potent antiproliferative activity among the five cancer cell lines evaluated (IC50 = 0.1, 0.1, 0.1, 2.0, and 1.1 µM for HL-60, MCF-7, T-47D, LNCaP, and WEHI-3, respectively), which was equivalent to the positive control doxorubicin (IC50 = 0.2, > 10, 0.7, 0.1, and 0.02 µM). Moreover, compound 56 showed extremely low cytotoxicity towards normal cells. In addition, compound 56 was injected subcutaneously in rats, plasmatic concentration was 151 ng/mL and 15.7 ng/mL, respectively at the 3 h and 12 hCitation83.
Figure 28. Chemical structures of steroid derivatives.
Piperazine carbamate was used to link camptothecin derivatives to prevent rapid hydrolysis. Compound 57 () demonstrated a potent affinity to integrin receptors and a potent ability to inhibit A2780 cells, with an IC50 value of 0.033 µM. Importantly, a significant increase in t1/2 for 57 vs irinotecan in rat plasma (13 h vs 45 min, respectively)Citation84. Yang et al. reported the synthesis and cytotoxic activity of camptothecin derivatives incorporating piperazinyl-sulfonylamidine moieties. All derivatives were evaluated for their activity against five tumour cell lines (A-549, MDA-MB-231, MCF-7, KB and KB-VIN). SAR indicated that an aromatic group in R1 substituents is much less favourable than a short aliphatic group. The conjugation of the aromatic (R1) substituents with the sulphonyl moiety might disturb the electronic properties of ring B to a greater extent than alkyl groups. Moreover, the potency of the compounds with the aromatic substituents (R1) depended significantly upon the nature of the R2 substituent on the aromatic ring. Compound 58 () displayed the highest cytotoxicity against the MDR KB-VIN cell line (IC50 = 0.38 µM), while irinotecan lost activity completely against KB-VIN (IC50 > 20 µM)Citation85. A series of 7-(N-[(substituted-sulphonyl) piperazinyl]-methyl)-camptothecin derivatives were designed and synthesised by Zhu and co-workers, assessed for their in vitro cytotoxicity against A-549, MDA-MB-231, KB, KB-VIN, and MCF-7 cell lines. Remarkably, compound 59 ((, IC50 = 1.2 nM) and 60 ((, IC50 = 20.2 nM) exhibited the highest level of cytotoxicity against the multidrug-resistant (MDR) KB-VIN cell line, which was equivalent to camptothecin (IC50 = 15.7 nM). SAR study suggested that both aromatic and aliphatic substituents on the sulphonyl-piperazinyl side chain at C-7 can promote potency to some extent, while selected variations of these substituents can greatly affect the activity. This study revealed that the integration of the sulfonylpiperazine modify into the 7-position of camptothecin is a potent strategy for discovering effective cytotoxic camptothecin derivativesCitation86.
Figure 29. Chemical structures of camptothecin and its derivatives.
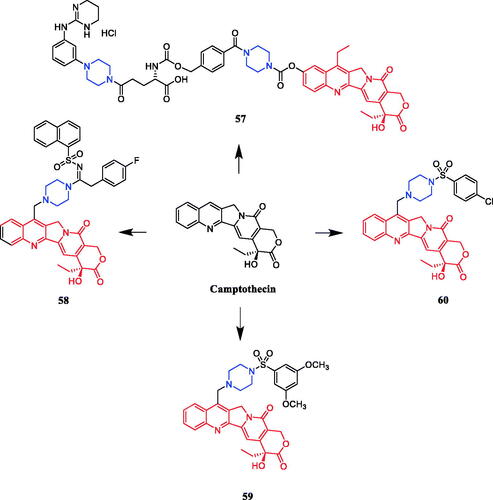
New analogs of benzo-α-pyrone containing piperazine derivatives have been synthesised. Compound 61 () displayed the greatest activity against the WM266-4 (BRAFV600E) cell line, with an IC50 value of 1.35 µM. Unfortunately, the activity of 61 was not as good as that of the positive control vemurafenib (IC50 = 0.07 µM). Following the biological evaluation assays indicated that 61 could result in cellular apoptosis, marked DNA fragmentation, and G0/G1 phase arrest in melanoma cells. Moreover, docking simulation showed that compound 61 could bind well with the crystal structure of BRAFV600E at the active site. Compared with the compound in which piperazine is connected to the alkyl substituent, the aromatic-substituted compound inhibited BRAFV600 more strongly. Studies pointed out that the change in the position of the substituents on the piperazine-linked benzene ring may have little effect on the inhibitory activity, besides, the compounds substituted by the electron-donating groups on the benzene ring showed better inhibitory activity than the electron-withdrawing groupsCitation87.
Figure 30. Chemical structures of benzo-α-pyrone and its derivatives.
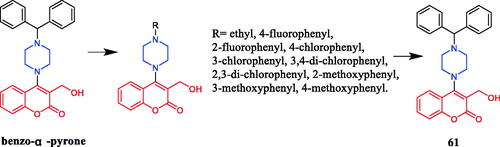
Myricetin piperazine derivative 62 () exerted a moderate inhibitory effect against MDA-MB-231 cells (human breast adenoma cell) (IC50 = 3.87 ± 0.21 µM), was slightly lower than the positive control doxorubicin (IC50 = 2.01 ± 0.17 µM). The SAR indicated that all examined myricetin derivatives showed good activity against human breast cancer cell lines (MDA-MB-231 and Bcap-37) but poor activity against human gastric cancer cells (SGC-7901 and MGC-803 cell lines)Citation88. Ruan et al. reported that myricetin derivative 63 () containing a piperazine moiety showed potent inhibitory activity against a human breast cancer cell line (MDA-MB-231) (Inhibition rate= 86.7% in 10 µmol/L), which was close to epirubicin (94.6%), In addition, through the analysis of the morphological effect on the target tumour, it can be seen that although the cytotoxicity of myricetin is small, its inhibitory effect on the tumour cells is not obvious; and the myricetin derivative after the introduction of piperazine amide has low cytotoxicity and can effectively inhibit the proliferation of tumour cells, which has good anti-tumour research valueCitation89.
Figure 31. Chemical structures of myricetin and its derivatives.
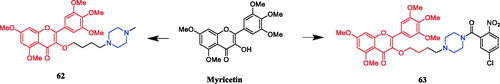
Reportedly, derivative 64 () (1-p-nitrophenylpiperazinyl substituted derivative of deoxypodophyllotoxin) demonstrated the most active cytotoxicity against A-549, HeLa, and SiHa cells (IC50 = 0.102, 0.180, and 0.0195 µM, respectively), better than that of parent compound deoxypodophyllotoxin (IC50 = 0.470, 36.0, and 10.2 µM, respectively). Moreover, flow cytometric analysis indicated that the compound could arrest the cell cycle at the G1 phase in A-549 cellsCitation90. In 2013, demethylepipodophyllotoxins carbamate derivative 65 () was reported as the most promising compound synthesised in their study, resulting in cell cycle arrest in the G2/M phase and apoptosis in HeLa cells, presenting an IC50 value of 0.074 µM, better than that of positive control etoposide (IC50 = 2.91 µM). SAR indicated that the substituent of the amines markedly affected the activity profiles of this compound class, and the compounds with piperazines incorporated appear to be more potent than those with alkyl amines. Second, the compounds with amino acids incorporated appear to be less potent than those with p-nitrobenzylpiperzine directly and the compounds with L-amino acids incorporated appear to be more potent than those with D-amino acid. Lastly, compounds that contain only one substituent at the C-4 position showed superior activities in comparison with compounds simultaneously substituted at the C-4 and C-4′ position of 4′-demethylepipodophyllotoxinCitation91. Five bis-epipodophyllotoxin compounds were designed using a structure-based design approach, showing strong evidence that these compounds targeted topoisomerase II in a variety of assays. Compound 66 () containing a linker with two piperazine rings exhibited the highest activity (IC50 = 0.016 µM), with an IC50 value 10-fold lower than etoposide (IC50 = 0.163 µM) against K562 cells. The IC50 values systematically decreased with an increase in methylene linker length and reached a minimum with compound 66 which contained eight methylenes. The IC50 values then coordinately increased when the linker length was increased to ten and twelve methylenes. These results suggested that an optimum chain length was in the order of eight methylene groups, and thus may be the optimum chain length needed for the bis-epipodophyllotoxins to achieve bisintercalation in the DNA within the topoisomerase IIbDNA ternary structureCitation92. In 2017, Sun reported that podophyllotoxin piperazine derivative 67 () manifested prominent cytotoxicity against the MCF-7 cell line (IC50 = 2.78 ± 0.15 µM). Furthermore, compound 67 inhibited tubulin assembly without causing damage to non-cancerous cells, inducing apoptosis and cell cycle arrest at the G0/M phase in MCF-7 cells. It can be seen from the experimental data that almost all synthesised compounds showed anti-cancer activity, the substituents on the aromatic ring had a significant effect on the activity. According to the SAR, it was clearly found that compounds with substitution on the phenyl ring of the phenylpiperazine group in general exhibited inhibitory activities, electron-withdrawing groups (F, Cl, NO2, CF3) > electron-donating groups (OMe, Me), however, the modification of the 2 or 4 positions of the benzene ring resulted in a decrease in potencyCitation93.
Figure 32. Chemical structures of podophyllotoxin and its derivatives.
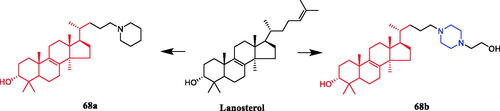
The synthesis of a range of novel 24-amino-25,26,27-trinorlanost-8-ene derivatives including 24-piperadino-trinorlanost-8-enes, 24-piperazino-trinorlanost-8-enes, 24-morpholino-trinorlanost-8- enes, and 24-diethylamino-trinorlanost-8-enes is reported and their cytotoxic and apoptotic potential evaluated in U937 cell lines. SAR indicated that morpholine, aniline, piperidine or diethylamine functionality was introduced at C-24 with acetate or hydroxyl functionality at C-3. The order of potency of these derivatives was piperidine > diethylamine > morpholine > aniline, with hydroxyl functionality at C-3 proving more potent than the acetylated counterpart. The piperidine derivative 68a () was revealed as a potential anticancer agent against human leukemic monocyte lymphoma U937 cells, presenting an IC50 value of 1.9 µM, which was equivalent to the positive control etoposide (IC50 = 1.5 µM). Besides, 68b () with piperazine moiety had an IC50 value of 2.7 µMCitation94.
Figure 33. Chemical structures of lanosterol and its derivatives.
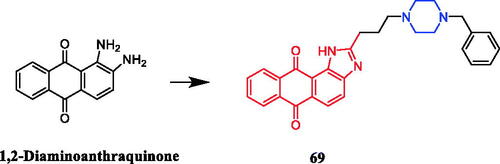
Chen and co-workers synthesised a series of anthraquinone derivatives to determine their potential telomerase inhibitory activities. It was found that compound 69 () with benzylpiperazine was found to show the best selectivity towards repressing hTERT expression in H1299 cells among these tested compounds. And compound 69 showed moderate potency against PC-3 with IC50 = 16.0 µM. Furthermore, SAR analysis indicated that the piperazine linker might be a significant moiety for telomerase inhibition or hTERT repressionCitation95.
Figure 34. Chemical structures of anthraquinone and its derivatives.
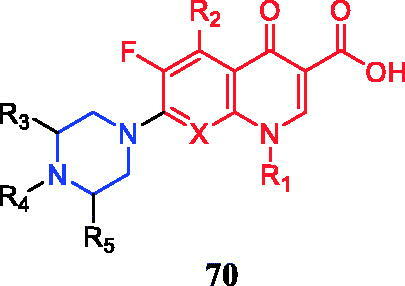
Piperazinylquinolone 70 () was considered a starting point for the design and development of new anticancer agents. Chemical modification of N-4 in the piperazine ring and the carboxylic acid at C-3 could present sufficient flexibilityCitation96.
Figure 35. Chemical structures of piperazinylquinolone skeleton.
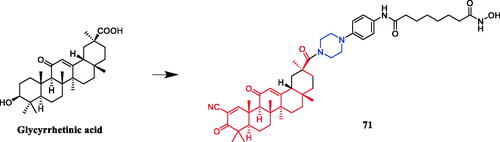
Pterins are a class of fused N-heterocyclic compounds containing pyrimidine and pyrazine rings, which widely existed in natural products and bioactive moleculesCitation97. In a study presented by Huang et al., a piperazine linker was identified as the best hybrid approach for glycyrrhetinic acid and hydroxamic acid. Conjugate 71 () displayed effective cytotoxicity against cancer cells inducing apoptosis, with IC50 values of 0.47 ± 0.09 µM and 0.37 ± 0.07 µM, respectively, against PC-3 and HL-60 cell lines. Notably, compound 71 showed better potency than SAHA (IC50 = 1.81 ± 0.15 µM and 0.42 ± 004 µM), while exhibiting superior inhibitory efficacy comparing to the parental compound CDODA-Me (IC50 = 1.68 ± 0.28 µM and 1.35 ± 0.16 µM) as well. The preliminary pharmacokinetic (PK) property of compound 71 was evaluated in male Sprague-Dawley (SD) rats. Compound 71 at 5 mg/kg was intravenously (iv) administered to rats (n = 3). Compound 71 demonstrated reasonable PK profiles with a high half-life (T1/2) of 16.75 h and maximum concentration (Cmax) of 10753.33 ng/mL. The results suggested that the piperazine substituted 5,8-dihydropteridine-6,7-dione frameworks may be regarded as new chemotypes for designing effective antitumor agents against gastric cancer cellsCitation98.
Figure 36. Chemical structures of glycyrrhetinic acid and its derivatives.
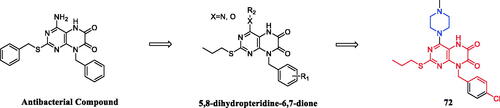
Geng et al. reported the synthesis and preliminary biological evaluation of 5,8- dihydropteridine-6,7-diones. The SARs studies highlighted the importance of the piperazine substituted 5,8-dihydropteridine-6,7-dione frameworks for the activity and revealed essential structural elements. Among these derivatives, compound 72 () displayed the most potent and broad-spectrum proliferative inhibition against evaluated cell lines (HeLa, CaSki, MGC-803, and SKOV-3 cancer cell lines) and was sensitive to the MGC-803 cell line, with an IC50 value of 8.78 µM, slightly more potent than 5-fluorouracil (IC50 = 14.15 µM). Preliminary mechanistic studies showed that compound 72 could inhibit the colony formation and migration of MGC-803 cells. Besides, flow cytometry analysis showed that compound 72 concentration-dependently induced apoptosis of MGC-803 cellsCitation99.
Figure 37. Chemical structures of 5,8-dihydropteridine-6,7-dione and its derivatives.
A series of disubstituted C20’-urea derivatives of vinblastine were prepared by Barker et al. and were examined across a panel of 15 human tumour cell lines. N-methylpiperazine was introduced to increase the polarity of designed compoundsCitation100.
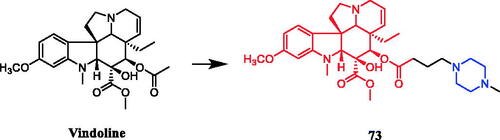
Vindoline conjugated with N-methylpiperazine (compound 73, ) demonstrated a significant antiproliferative effect, with IC50 values of 9.36 µM, 14.10 µM and 25.49 µM, respectively, against HeLa, MCF-7 and MDA-MB-231 cell lines, although the monomer vindoline was inactive (IC50 > 30 µM). In addition, the experimental results suggested that the compound of ventolin and amino acid, tryptophan methyl piperazine and N-methyl piperazine have significant anti-proliferative effects. Concerning structure–activity relationships, it seems that the 1,2,3‐triazole and morpholine moieties could not improve the efficacy of vindoline at allCitation101.
Figure 38. Chemical structures of vindoline and its derivative.
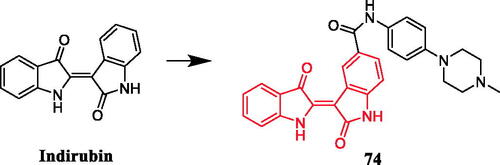
A series of indirubin derivatives were designed to improve water solubility and antitumor activity. Among them, compounds containing a piperazine moiety exhibited significant inhibitory activity against human large cell lung cancer cells (LXFL529L cells). Compound 74 () carrying a methylpiperazino-phenylamino substituent attached to 5-carboxyindirubin, with a water solubility of 520 mg/L, displayed the most potent antitumor activities, with an IC50 value of 0.54 µM, better than that of Positive control 5-carboxyindirubin (IC50 = 7 µM). When the N-methylpiperazinyl substituent on the benzene ring was changed to dimethylamino, although the water solubility increased, the activity of inhibiting cell proliferation decreased. In addition, the phenyl moiety of 74 replaced by an aliphatic chain also produced a similar effectCitation102.
Figure 39. Chemical structures of indirubin and its derivative.
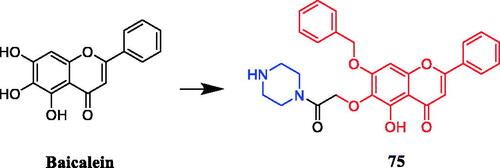
Several baicalein-piperazine derivatives showed moderate anti-angiogenic activity, with weak toxicity on normal tissues. Notably, compound 75 (), with the piperazine acetamide group at 6-position, was the most active compound on human umbilical vein endothelial cell (HUVEC) proliferation, migration, and tube formation in vitro. Moreover, consistent with the anti-angiogenic activity, compound 75 significantly inhibited the growth of A549 cells (IC50= 4.73 µM). The data indicated the introduction of an alkyl alcohol substituent at the 6 positions could increase the anti-angiogenic activity, and shortening the chain length reduced toxicity. In addition, regarding the 6-O-acetamide substituted scaffold, the substitution of a benzyl group for the phenolic hydroxyl group did not significantly improve the effect on inhibiting angiogenesis, while the compound 75 with a piperazine group showed the strongest anti-angiogenic activityCitation103.
Figure 40. Chemical structures of baicalein and its derivative.
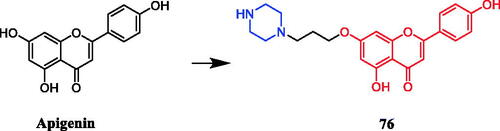
Liu et al. reported that apigenin-piperazine derivative 76 () possessed active antiproliferative activity with the lowest IC50 values of 16 µg/mL, 11 µg/mL, 25 µg/mL and 32 µg/mL against A549, HeLa, HepG2, and MCF-7 cancer cells, more potent than parent compound apigenin (IC50 = 1740 µg/mL, 450 µg/mL, 460 µg/mL and > 2000 µg/mL). SAR analysis indicated that compared with the compounds with 2-carbon spacer, 3-carbon-spacer-containing compounds, showed a slight increase in antiproliferative activity against the four cancer cell lines. This result may have been caused by the elongation of the alkyl side chain from two to three C-atoms that retained the modulating activity, with C3-bridge derivatives being the most active. The compounds containing N-heterocyclic-ring amino side chains displayed better activities against the four cancer cell lines than those containing alkyl amino side chains. This result may be attributed to the increasing lipophilicity of these compounds, which could influence their ability to reach the target via transmembrane diffusionCitation104.
Figure 41. Chemical structures of apigenin and its derivative.
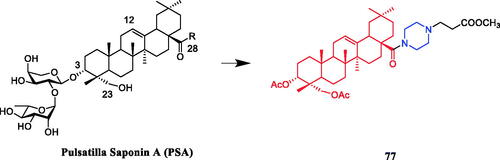
Twenty Pulsatilla saponin A (PSA) derivatives with C ring, C-28, or C-3 modifications were synthesised and evaluated for cytotoxic activities against A549, MCF-7, MDA-MB-231, KB, SMMC-7721, BGC-823, and KB-VIN cell lines. Compound 77 () with the piperazine ring at the 28-COOH position exhibited better cytotoxic activity (IC50= 4.68 µM) and lower haemolytic toxicity (HD50 > 500 µM) against the A549 cell line when compared with PSA (IC50= 22.87 µM, HD50 =3.86 µM). The data indicated that the carbon ring (especially the C-12/C-13 double bond) might play an important role in the cytotoxicity and haemolytic toxicity of prostate-specific antigen. The 28-COOH may cause haemolysis of red blood cells. In addition, the fully acetylated sugar chain at position 1 showed weaker cytotoxicity than PSA, indicating that the sugar hydroxyl group is important for the cytotoxicity of PSA. Further molecular biology studies showed that compound 77 might cause G1 cell cycle arrestCitation105.
Figure 42. Chemical structures of pulsatilla saponin A and its derivatives.
2.2. Antibacterial activity
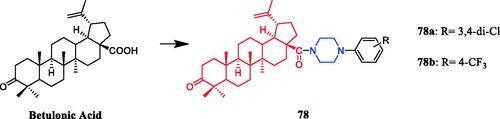
Piperazine derivatives developed by Chue et al. were assessed for their antibacterial potential, demonstrating the synthesis of betulonic acid amides with N-derivatives of piperazine and their antibacterial activities. Compounds 78a and 78b () inhibited bacterial growth in all tested cultures. The diameter of the growth inhibition zone was in the range of 13 − 16 mm for five of the studied test cultures, which is better than the positive control gentamicin (16 − 21 mm)Citation106.
Figure 43. Chemical structures of betulonic acid and its derivatives.
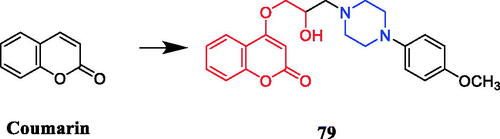
Wang synthesised coumarin derivatives containing a piperazine skeleton as potential antibacterial agents. Compound 79 () demonstrated the most potent antibacterial activity [minimum inhibitory concentration (MIC) = 0.236 µg/mL for Bacillus subtilis; MIC = 0.355 µg/mL for Staphylococcus aureus] compared with the positive control Penicillin G (MIC = 0.752 µg/mL and 1.685 µg/mL). What’s more, it showed the most potent activity against SaFabI with IC50 of 0.57 µM. The SAR revealed the activity of compounds with substituents on the benzene ring is better than that of unsubstituted compounds, moreover, the para-substituted compounds showed stronger antibacterial activity than the meta- and ortho-substituted compoundsCitation107.
Figure 44. Chemical structures of coumarin and its derivatives.
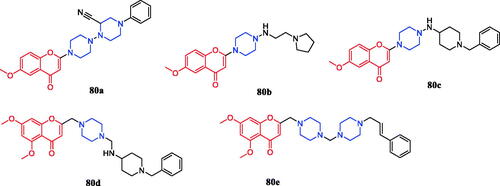
Flavone derivatives 80a–80e () with piperazine rings were found to demonstrate more potent antibacterial activity, showing even 2 to 2.5-fold more potency than that of standard ciprofloxacin and miconazole at the same MIC value of 10 µg/mL. It was observed that the presence of amino alkyl, cyano or alkenyl alkyl group on piperazine ring seemed to be favourable for the high antimicrobial activity, however, the mere presence of electron donor group on the piperazine ring had very little or nothing to do with antimicrobial activityCitation108.
Figure 45. Chemical structures of flavone derivatives.
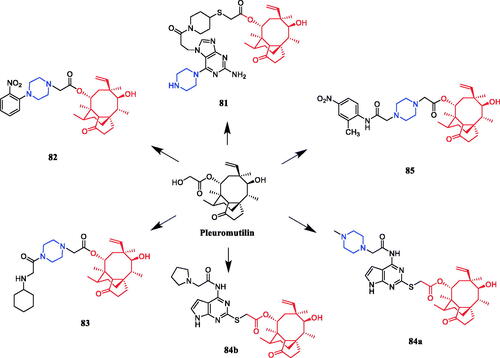
In recent years, several pleuromutilin derivatives containing piperazine moieties were designed and synthesised. Pleuromutilin analog 81 () was designed and synthesised with a purine ring as a polar and water solubilising group. In further investigation, analog 81 with piperazine displayed superior in vitro and in vivo antibacterial activity against some Gram-positive strains, including drug-resistant pathogens. Analog 81 with good solubility in water showed remarkable antibacterial activity against all Gram-positive pathogens, including methicillin-resistant S. aureus (MRSA), penicillin-resistant Streptococcus pneumoniae (PRSP), and vancomycin-resistant enterococci (VRE), except for Enterococcus faecalis. Importantly, analog 81 exhibited the most potent antibacterial activity against susceptible and resistant S. pneumoniae, with a MIC of 0.063 µg/mL, appeared as a possible alternative to the weak marketed antibacterial agents macrolide erythromycin (MIC > 128 µg/mL), tetracycline minocycline (MIC = 8 µg/mL), and quinolone levofloxacin (MIC = 8 µg/mL). Interestingly, 81 and glycopeptide vancomycin (VCM) exhibited superior ED50 values (about 1–2 mg/kg) when given intravenously regardless of the frequency of dosing (once or twice), indicating the potential of 81 as an antibacterial agent with efficacy equal to that of VCMCitation109. A series of novel pleuromutilin derivatives having a piperazine ring were synthesised under mild conditions. In the ortho-, meta- and para-position of the benzene ring, electron withdrawing and donating groups were introduced to explore SAR. The ortho-NO2 substituted derivative 82 () was found to be the most active compound against S. aureus (MIC = 0.0625 µg/mL) among a series of pleuromutilin derivatives, better than the positive control tiamulin (MIC = 0. 25 µg/mL). The results revealed ortho-substituted benzene derivatives have the greatest impact on activity compared with para and meta substitutions, for example, replacement of the nitro group in compound 82 with chlorine group at the same position, which resulted in dramatic decrease in antibacterial activity. The meta-substituted benzene derivatives with CH3O–, CH3–, and NO2– substituents showed enhanced antibacterial activities, all the para-substituted benzene derivatives bearing methoxyl, methyl, hydroxyl, nitro, and chlorine substituents exhibited moderate antibacterial activities. The compound-bearing nitro group in the para-position showed higher activity compared to other para-position substituted derivatives. The para-position appeared to have less influence on the antibacterial activity of these pleuromutilin derivatives bearing a phenyl piperazine110. In 2015, derivative 83 () was revealed remarkable activity against S. aureus (ATCC25923; MIC of 0.125 µg/mL), which is equal to the control compound tiamulin. The antibacterial activities of 83 to Streptococcus suis (MIC of 2 µg/mL), Streptococcus agalactiae (MIC of 0.5 µg/mL), and Streptococcus dysgalactiae (MIC of 0.5 µg/mL) were also excellent compared with the control drug erythromycin (MIC of > 128 µg/mL). Further, the results preliminary confirmed that the activity of secondary amine is better than that of a tertiary amine. A molecular docking study indicated that 83 could bind to the active pocket of the ribosome with seven hydrogen bondsCitation111. In 2018, pleuromutilin-piperazine derivative 84a () exhibited moderate antibacterial activity against methicillin-sensitive S. aureus (MSSA) with MIC of 0.125 µg/mL, that even more active than the comparator valnemulin (MIC = 0.25 µg/mL). However, the activity against P. multocida was also examined, and the most efficient compound 84b proffered identical MIC (16 µg/mL) as valnemulin. In addition, the results provided by the dose-response study demonstrated 84b could supply infected mice with a 70% survival rate at a dose of 40 mg/kg via intragastric (i.g.) administrationCitation112. Furthermore, in 2020, pleuromutilin derivative 85 () was revealed as the most potent derivative against S. aureus and Staphylococcus epidermidis, with a MIC value of 0.0625 µg/mL, showed an almost two-fold increase in activity in comparison with that of reference drugs, and an eight-fold increase in activity against Enterococcus faecalis compared to that of tiamulin. Furthermore, the docking study revealed that derivative 85 could interact with the ribosome pocket with more bonds when compared with tiamulinCitation113.
Figure 46. Chemical structures of pleuromutilin and its derivatives.
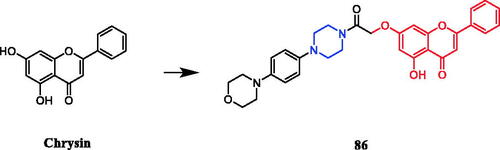
Reportedly, a new series of piperazinyl-substituted chrysin derivatives have been designed and synthesised. Among the synthesised compounds, analogs accommodating morpholine, piperidine, and electron-donating groups at the benzene ring showed better antibacterial activities than those that possess electron-withdrawing substituents. The chrysin containing morpholine compound 86 () displayed the most potent inhibitory activity against Escherichia coli FabH (IC50 = 5.78 ± 0.24 µM), comparing to the control positive drugs penicillin G (7.56 ± 0.30 µM). In the antibacterial activity assay, 86 was found to be the most active against S. aureus and E. coli (MIC = 1.25 ± 0.01 and 1.15 ± 0.12 µg/mL, respectively), compared with the control positive drugs (MIC = 1.39 ± 0.02 and 1.82 ± 0.47 µg/mL, respectively)Citation114.
Figure 47. Chemical structures of chrysin and its derivatives.
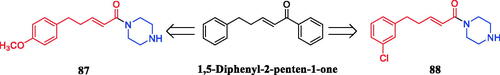
A series of novel 1,5-diphenyl-2-penten-1-one analogs with a piperazine moiety were synthesised and evaluated for their antifungal and larvicidal activities. Among them, the most potent compound 87 () demonstrated 100% mortality against mosquito larvae at a concentration of 1 mg/L. Compound 88 (), with a 3-position chlorine substituted phenyl group, exhibited the highest activity with an EC50 value of 0.075 mM against Pythium aphanidermatum, superior to lead compound 1,5-Diphenyl-2-penten-1-one (0.39 mM). Initial SAR analysis showed that a methyl group had significant effects on the biological activities of these compounds, the compounds with N’-unsubstituted piperazine exhibited much better antifungal activities and larvicidal activity against mosquitoes than the compounds with N’-methylated piperazine. In addition, the larvicidal activity against mosquitoes had a sharp decline when the substituent on the benzene ring was changed from 4-position to 2 or 3-positionCitation115.
Figure 48. Chemical structures of 1,5-diphenyl-2-penten-1-one and its analogs.
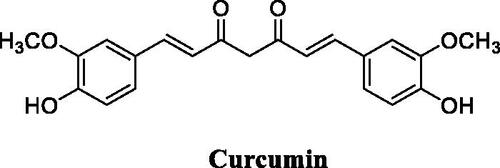
Eight novel curcumin derivatives were synthesised using the Mannich reaction and evaluated for their antibacterial activities; two derivatives containing a piperazine ring showed remarkable mildew resistance ()Citation116.
Figure 49. Chemical structures of curcumin.
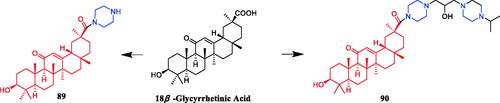
In 2011, glycyrrhetinic acid derivative 89 () containing a piperazine ring displayed excellent anti-mycobacterial properties against the drug-susceptible and drug-resistant Mycobacterium bovis. More importantly, it exhibited synergistic effects with the first‐line drugs isoniazid (INH), rifampicin (RIF) and streptomycin (SM) against clinical M. bovis isolates, including drug‐resistant strains. In the presence of 89, MICs for the first‐line agents resulted in a 4–16‐fold decrease for INH ((fractional inhibitory concentration index (FICI) 0.094–0.266), RIF (FICI 0.114–0.313) and SM (FICI 0.094–0.281). Additionally, the MICs of PGA alone showed significant decreases ranging from 8‐ to 16‐, 8‐ to 64‐ and 8‐ to 128‐fold in the presence of INH, RIF and SM, respectively. These findings indicate that 89 might serve as potential therapeutic compounds for future antimycobacterial drug developmentCitation117. Xiang et al. synthesised a series of novel 18β-glycyrrhetinic piperazine amides to evaluate in vitro and in vivo antibacterial activities against phytopathogens Xanthomonas oryzae pv. oryzae (Xoo) and X. axonopodis pv. citri (Xac), as well as induced apoptotic behaviours. Screening results revealed that all designed compounds were bioactive against Xoo and Xac. Among them, compound 90 () bearing an isopropyl tail presented the most potential antibacterial efficiency, with EC50 values of 2.28 and 1.42 µg/mL against Xoo and Xac, respectively, which is far better than lead compound 18β-glycyrrhetinic acid (EC50 > 400 µg/mL), positive control bismerthiazol (EC50 = 92.6 µg/mL and NT) and thiodiazole copper (EC50 = 121.8 µg/mL and 77.0 µg/mL). Pharmacophore studies suggested that the synergistic effect of glycyrrhetinic acid skeleton and N-containing scaffolds was essential for the antibacterial activity. Further studies revealed that compound 90 could induce the generation of excessive ROS in tested pathogensCitation118.
Figure 50. Chemical structures of glycyrrhetinic acid and its derivatives.
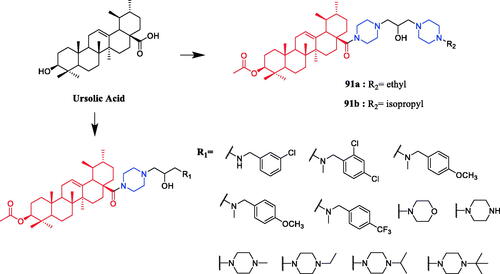
In another study, piperazine-tailored ursolic acid hybrids 91a () exhibited remarkable antimicrobial activity against Xoo and Xac (EC50 values were 0.37 and 1.08 µg/mL, respectively), which is far better than lead compound ursolic acid (EC50 > 400 µg/mL), positive control bismerthiazol (EC50 = 92.6 µg/mL and NT) and thiodiazole copper (EC50 = 121.8 µg/mL and 77.0 µg/mL). Studies have shown that the hydroxyl group in the derivative structure had a positive role in promoting antibacterial action, on the contrary, the elimination of the N-ethyl piperazinyl or N-isopropyl piperazinyl group dramatically reduced antibacterial activity. Importantly, compounds 91a and 91b () could manage bacterial blight in vivo in pot experiments. A preliminary finding revealed that the synthesised compounds could disrupt the equilibrium of the redox system in tested pathogens, leading to excessive ROS accumulation, which induced the apoptosis-like effect in tested pathogensCitation119.
Figure 51. Chemical structures of ursolic acid and its derivatives.
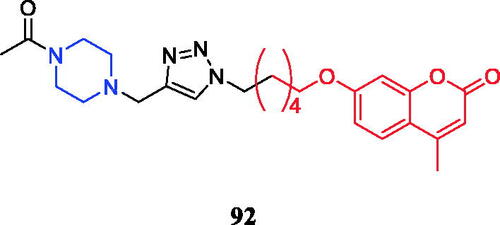
Amongst a series of novel 2H-chromen-2-one derivatives bearing 1,2,3-triazole moiety, compound 92 () containing N-acetylpiperazine moiety was found to be most effective against Pseudomonas aeruginosa (zone of inhibition in 17.1 mm), which is equivalent to the standard (zone of inhibition in 18 mm). Based on, in silico pharmacokinetic studies, compounds 92 was identified as lead compounds for future investigation due to their lower toxicity, high drug score values and good oral bioavailability as per OECD guidelinesCitation120.
Figure 52. Chemical structures of chromen derivatives.
2.3. Anti-inflammatory activity
Inflammation is a non-specific immune response to infection, tissue stress, or some other form of injuryCitation121. Almost all modern human diseases are related to chronic inflammationCitation122. Moreover, glucocorticoids and non-steroidal anti-inflammatory drugs currently in clinical use have serious gastrointestinal side effectsCitation123,Citation124. Therefore, researchers have developed a series of anti-inflammatory piperazine derivatives based on natural products.
A series of flavone piperazine derivatives were synthesised and evaluated for their in vitro anti-inflammatory activity. Among all the compounds screened, compounds 93a–93b () showed promising TNF-α and IL-6 inhibitory activity. Furthermore, Compound 93ba () exhibited excellent inhibition against tumour necrosis factor-α (TNF-α) and interleukin (IL)-6 (up to 65–87% TNF-a and 70–93% IL-6 inhibitory activity) at a concentration of 10 µM with reference to standard dexamethasone (71% TNF-a and 84% IL-6 inhibitory activities at 1 µM. Notably, the presence of highly electron-rich groups such as methoxy, pyrimidyl, morpholine on piperazine as well as homologation of chromone and piperazine moiety had strong relevance to the anti-inflammatory activityCitation108.
Figure 53. Chemical structures of flavone derivatives.
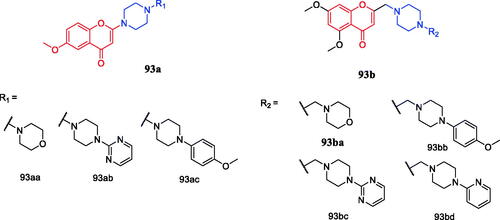
Compound 94 () demonstrated significant activity against β-glucuronidase (inhibition rate = 75.43% at 1 mM concentration), which was better than that of natural chalcone (NC, (E)-1–(2-hydroxy-4,6-dimethoxyphenyl)-3-phenylprop-2-eb-1-one, inhibition rate = 28.42% at 1 mM concentration). Further results indicating that chalcones with N-methyl piperazine methyl and piperidine methyl substitutions appear to be crucial for β-glucuronidase inhibition, and N-methyl piperazine group substitution at 3 positions of terminal benzene ring was found to be more suitable for the reduction of DPPH. Compound 94 was revealed the highest antioxidant activity in the DPPH free radical scavenging assay at 1 mM concentration (DPPH reduction rate = 37%) in the synthesised compound, far exceeding NC (DPPH reduction rate = 16.67%) but still weaker than quercetin (DPPH reduction rate = 86.30%)Citation125.
Figure 54. Chemical structures of chalcones derivative.
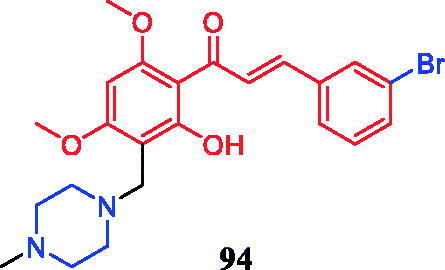
Novel chalcone-biscoumarin hybrids were synthesised by exploiting the anti-inflammatory potential of natural chalcone derivatives. All hybrid molecules were evaluated for in vivo anti- inflammatory activity using the in vivo para-xylene-induced mice ear-swelling model. Particularly, hybrid 95 () showed the most active anti-inflammatory activity (inhibition rate, 75.46%), which exceeded that of aspirin (inhibition rate, 64.91%) and was equal to that of celecoxib (inhibition rate, 77.03%). Preliminary SAR analysis indicated that substitution of aryl-piperazine or aryl-sulphonyl-piperazine with an electron-withdrawing group demonstrated an advantage over the electron-donating group, improving the anti-inflammatory activityCitation126.
Figure 55. Chemical structures of chalcone-biscoumarin derivative.
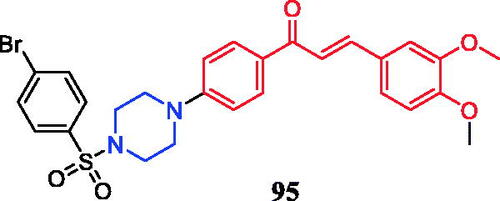
In a study performed by Li et al., compound 96 () with a piperazine unit exhibited the most potent nitric oxide (NO) and IL-6 inhibitory activity (IC50 = 13.3 µM) in the tested unsaturated glycyrrhetic acid derivatives. Moreover, compound 96 significantly reduced lipopolysaccharide (LPS)-induced inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2 expression, as well as IL-6 production, through the MAPK and NF-κB signalling pathwaysCitation127.
Figure 56. Chemical structures of glycyrrhetic acid and its derivative.
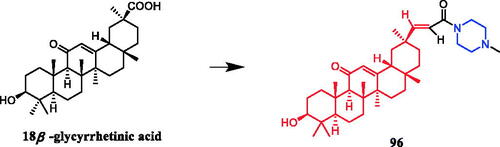
2.4. Antioxidant activity
In 2019, smilagenin derivative 97 () was evaluated for its potential neuroprotective effects, and revealed improved cell viability against H2O2-induced damage, inhibited NO production, and demonstrated strong antioxidant activity. The protective effect of compound 100 at a concentration of 10 µM (Cell viability = 40.5 ± 2.3%) against the oxidative stress triggered by H2O2 in SH-SY5Y cells was more effective than that of positive control N-acetyl-L-cysteine at 1 mM (Cell viability = 37.1 ± 2.5%). The activity results revealed benzyl-substituted piperazine formate derivatives showed the potent neuroprotective activityCitation128.
Figure 57. Chemical structures of smilagenin and its derivatives.
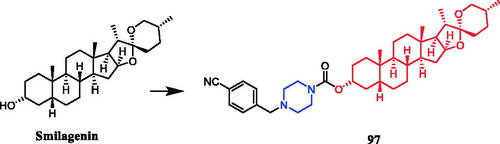
β-elemene piperazine derivatives were synthesised and evaluated for their inhibitory abilities. The data results indicated that these derivatives induced apoptosis through ROS production and reduced c-FLIP levels, activating both death receptor-mediated and mitochondrial-mediated apoptotic pathwaysCitation129.
Coumarin derivative 98 () exhibited significant activity (IC50 = 2.1 µM) and good selectivity for AChE with respect to butyrylcholinesterase. Structure-activity relationship studies have shown that the anti-acetylcholinesterase activity of the compound was affected by the type of cyclic amine on the 2-oxo- or 4-oxoalkoxycoumarinbackboneCitation130. Coumarin piperazine derivative 99 () was found to be a potent AChE inhibitor, with an IC50 value of 4.9 µmol/L against human AChE enzyme (hAChE), while the IC50 values of reference drug ensaculin and doneprazil were 0.36 ± 0.01 µmol/L and 0.11 ± 0.01 µmol/L, respectively. Among the newly synthesised analogs, the most effective compound was 102, having a methoxy group at the meta position. If the methyl group is attached in the ortho and para positions, the activity was further reduced. In addition, when the ortho-substituted methyl group of the compound was shifted to the para and meta positions, the inhibitory activity declined. Compounds showed better inhibitory activity when nitro was substituted at the meta position, or remarkably decreased if nitro shifted to the ortho and para positions. Subsequently, a SAR study using the molecular field method revealed that the inhibitory mechanism may be attributed to the hydrophobic field and positive charge centre conferred by the coumarin and piperazine moietiesCitation131.
Figure 58. Chemical structures of coumarin and its derivatives.
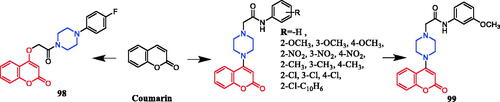
In the previous section, it was stated that a series of berberine derivatives were successively reported by Mistry et al, simultaneously presenting the anticancer and antioxidant activities. Compound 100 () showed remarkable antioxidant ability in the ABTS assay, with an IC50 of 4.644 µg/mL. In another report, analog 101 () exhibited the highest scavenger activity in the DPPH free radical scavenging assay, with an IC50 of 17.25 µg/mL. Furthermore, compound 102 (), demonstrating two electron-withdrawing chlorine atoms on the piperazine entity attached to the berberine ring, revealed remarkable radical scavenging potential in the DPPH assay, with an IC50 of 19.80 ± 0.94 µM, comparable to that of control drug ascorbic acid at 61.03 ± 0.66 µM and far better than berberine at 89.18 ± 1.60 µM. Compound 103 (), with a naphthyl piperazine substituent, demonstrated the notable potency in the ABTS•+ analysis, with an IC50 value of 8.94 µM while the parent scaffold berberine with IC50 = 232.1 ± 2.11 µM and ascorbic acid with IC50 = 27.02 ± 0.05 µM. Compound 50 () showed significant antioxidant activity with an IC50 value of 11.08 and 4.76 µg/mL in DPPH and ABTS bioassays, respectively, while the parent scaffold berberine with IC50 = 34.29 µg/mL and 82.17 µg/mL and ascorbic acid with IC50 = 10.75 µg/mL and 5.528 µg/mL in the DPPH and ABTS bioassays, respectivelyCitation72,Citation74–76.
Figure 59. Chemical structures of berberine and its derivatives.
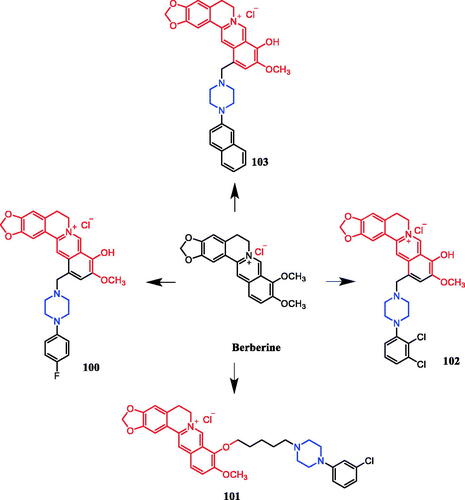
A series of 2-(2-hydroxyphenyl) pyrimidine/benzothiazole piperazinyl-substituted flavones were synthesised and evaluated for their in vivo biological properties. The results indicated that all tested compounds containing flavonoid-piperazine moieties were found to possess free radical scavenging potential. The SAR study showed that the antioxidant activity was decreased with methoxy group substitution on the flavone unit. Furthermore, the analogue 104 () benzothiazole substituted piperazine ring showed better antioxidant activity. Hydroxyl free radical scavenging capacity of analogue 104 at 1.25 mM (scavenging ratio = 48.0%) was comparable to positive control thiourea (scavenging ratio = 46.0%), and higher than α-tocopherol (scavenging ratio = 18.7%)Citation132.
Figure 60. Chemical structures of piperazinyl flavone derivatives.
2.5. Nervous system activity
In 2011, by introducing N-methylpiperazine on aromatic compounds, it was expected that the hydrogen bond substitution and additional electrostatic interaction would significantly enhance the binding affinity with the Aβ peptide. Curcumin analog 105 () containing a piperazine ring reportedly demonstrated the most significant activity, with an IC50 value of 2.5 ± 1.2 µM and 90.2 ± 3.2% inhibition of Aβ aggregation at a concentration of 50 µM, a much higher activity than curcumin (IC50 value of 12.1 ± 1.2 µM and 71.3 ± 1. 3% inhibition of Aβ aggregation at a concentration of 50 µM)Citation133.
Figure 61. Chemical structures of curcumin and its derivative.
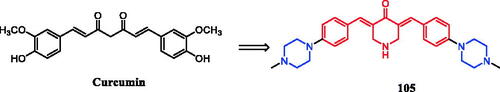
In 2013, tacrine-flavonoid hybrids were connected via piperazine side-arm alkyl groups. Among the series of compounds, compound 106 () displayed balanced inhibitory effects against acetylcholinesterase (AChE) and self-induced Aβ1−42 aggregation (IC50 = 133 nM for AChE, IC50 = 6.5 µM for aggregation), better than tacrine (IC50 = 260 ± 8 nM for AChE) and curcumin (IC50 = 20.3 ± 1.2 µM for aggregation). Furthermore, it exhibited moderate metal chelating activity and low cell toxicity. A molecular docking study indicated that the piperazine in the structure could bind to the AChE binding site in the middle of the canyon through a cation-π interaction (5.45 Å) between its protonated nitrogen atom and Tyr334 in the middle of the canyonCitation134.
Figure 62. Chemical structures of tacrine and its derivative.
In 2018, a series of multifunctional 3-piperazinecarboxylate sarsasapogenin derivatives were designed and synthesised against AD. The protective effect of compound 107 () at a concentration of 10 µM (Cell viability = 43.9 ± 2.8%) against the oxidative stress triggered by H2O2 in PC12 cells was stronger than that of positive control N-acetyl-L-cysteine at 1 mM (Cell viability = 35.3 ± 2.5%). Further SAR analysis indicated that benzyl, electron-donating groups, and intramolecular hydrogen bonding may help increase its neuroprotective activity. The report indicated that N-substituted piperazine carboxylate can be used as a promising structural unit for designing anti-AD drugs. Significantly, compound 107 protected PC12 cells from Aβ-induced damage and improved learning and memory impairments in Aβ-injected mice with low-dose administration at 6 mg/kgCitation135.
Figure 63. Chemical structures of sarsasapogenin and its derivatives.
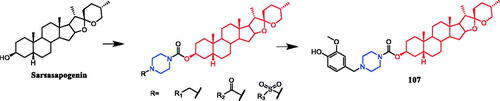
In 2019, genipin analog 108 () with a benzylpiperazine modification at C-8 position showed a higher inhibition rate (%Inhibition = 22.29) on protecting PC12 cells against injured by Aβ1–42 than the positive reference donepezil (%Inhibition = 17.65). For compounds containing methyl substituents, replacement at 3- position of the terminal benzene ring was more beneficial to the enzymatic activity than at 4-position substitution. In addition, the methoxy substitution at the terminal phenyl moiety was more advantageous than the methyl group. When an electron-withdrawing groups were introduced at the end of the phenyl ring, the increase in activity was consistent with the enhancement of the electron-withdrawing performance of the substituent. In the entire series, genipin derivatives with ligustrazine as the terminal aryl group were identified as the two most effective compounds. In terms of neuroprotective activity, compounds with benzene as the terminal aryl group revealed better neuroprotective effects than those with ligustrazine as the terminal aryl groupCitation136.
Figure 64. Chemical structures of genipin and its analog.
However, in 2020, Yang et al. found that the introduction of piperazine reduced the anti-AD effect of diosgenin carbamate derivativesCitation137.
Coumarin derivative 109 () demonstrated clear neuroprotection in the middle cerebral artery occlusion rat model by reducing the infarct size and brain-water content, improving neurological function, and suppressing neuronal loss and neuropathological changes in the cortex and hippocampus. Compound 109 at doses of 3.0 and 6.0 mg/kg significantly reduced the percentage of the infarct area within the ipsilateral hemispheres compared with the vehicle control group (20.36 ± 7.9% and 18.89 ± 5.83%, respectively). The same result was observed for Edaravone at 6.0 mg/kg, with which a significant reduction of brain-water content also occurred. Further pharmacokinetic studies have shown that compound 109 could penetrate the blood-brain barrier of ratsCitation138. Ostrowska and co-workers designed 7-hydroxy-4-methyl coumarin derivatives to increase the affinity to 5-hydroxytryptamine (5-HT) receptors. Compound 110 and 111 () showed higher affinities to 5-HT1A receptors (Ki value of 0.8 ± 0.009 and 0.50 ± 0.05 nM, respectively) than serotonin (1.3 ± 0.1 nM). Reportedly, substituents at ortho or meta positions in the phenyl group of piperazine play a decisive role in the affinity to 5-HT1A receptorsCitation139,Citation140.
Figure 65. Chemical structures of coumarin and its derivatives.
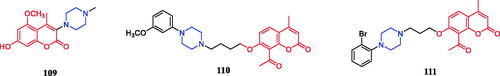
The synthesis and evaluation of novel, potential antipsychotic coumarin derivatives were undertaken by Yin Chen et al. Compound 112 () displayed a high affinity for 5-HT1A, 5-HT2A, and dopamine (D2) receptors (Ki = 0.2, 3.9 and 5.6 nM, respectively). Based on the results, compound 112 exhibited a higher affinity for the 5-HT1A receptor when compared with risperidone (Ki = 190.2 nM). Moreover, compound 112 demonstrated lower affinity for H1 receptors (Ki = 699.1 nM) when compared with risperidone (Ki = 22.9 nM) and clozapine (Ki = 6.8 nM)141. Chen et al. reported a series of coumarin piperazine (piperidine) derivatives as potential multireceptor atypical antipsychotics. The SAR has shown that several factors affected the binding affinity for the D2, 5-HT1A, and 5-HT2A receptors of these compounds. (1) Bearing a 6-fluorobenzo[d]isoxazol-3- yl)piperidine moiety exhibited higher affinities to three receptors than those with other amine moieties, (2) the substitution on the 4-position (R1) with methyl as the favoured substituent, (3) the substitution on the 3-position (R2) with unsubstituted or substitution (methyl) preferred, and (4) the substitution on the 8-position (R3) with electron-withdrawing (chloro) as the preferred substituent; (5) the substitution on the 5-position (R5) or 6-position (R4) did lower the affinity for the three receptors, (6) the importance of the double bond for affinity to D2, 5-HT1A and 5-HT2A receptors, and (7) straight four-carbon chain alkyl was superior to another alkyl linker. Notably, piperazine derivative 113 () was considered among the most promising derivatives investigated, displaying the strongest affinity for 5-HT1A receptors. According to the results, compound 113 exhibited a higher affinity for D2 and 5-HT1A receptors (D2, Ki =1.4 nM; 5-HT1A, Ki = 0.012 nM) when compared with risperidone (D2, Ki = 3.7 nM; 5-HT1A, Ki = 180 nM)Citation142.
Figure 66. Chemical structures of coumarin and its derivatives.
Generally, the introduction of the piperazine group improved the neuroprotective activity when compared with tropolone according to a study reported by Koufaki et al. Compound 114 () showed the most potent protective activity against glutamate-challenged HT22 cells, with an EC50 value of 0.08 ± 0.02 µM (the EC50 value of positive control drug tropolone >10 µM, β-thujaplicin = 4.80 ± 0.42 µM)Citation143.
Figure 67. Chemical structures of tropolone and its derivative.
A series of flavone derivatives were synthesised and evaluated for their H3R inhibitory activity. Among them, a compound containing piperazine ring 115a () exhibited good inhibitory activity against H3R. The H3R inhibition rate treated with compound 115a at doses of 25 µmol/ml was 81.32 ± 4.3% while positive control thiazole amide at 5 µmol/mL was 60.92 ± 3.5%. The results revealed that the hydroxyl at C6 also showed a significant effect by comparing compound 115a with 115 b (H3R inhibition rate = 12.31 ± 4.5% at doses of 25 µmol/mL) ()Citation144.
Figure 68. Chemical structures of baicalein and its derivative.
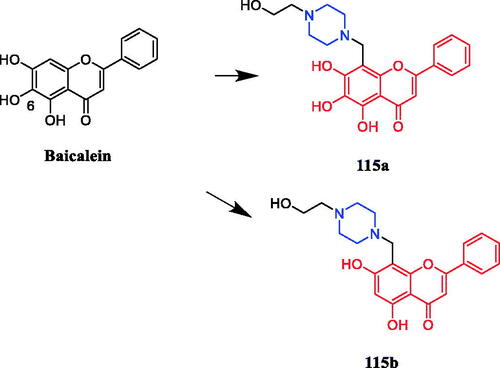
Waszkielewicz et al. reported that a xanthone derivative with piperazine moiety 116 () showed excellent anticonvulsant activity, with an ED50 value of 105 mg/kg in the maximal electroshock (MES) seizure assay. In the anticonvulsant test, the mice were administered 0.5 or 4 h before the test, and the doses of the drug were 30, 100 and 300 mg/kg, respectively. Finally, derivative 116 proved to have better performance because it showed no neurotoxicity at both test time points, but both 100 mg/kg and 300 mg/kg were active in MES at 4 h after administration. In another study, xanthone derivative 117a–117d () containing a piperazine moiety was evaluated for interactions with major classes of G-protein coupled receptors (GPCRs), uptake systems, and ion channels, revealing that it binds to 5-HT2A, 5-HT2B receptors, and sodium channels. And all derivatives synthesised were assessed using the SwissADME platform, the results revealed most of the compounds fulfilled Lipinski’s rule of five along with good BBB (blood-brain barrier) penetration. In the for-plate test compound 117a–117d significantly increased the number of punished crossings at dose 5 mg/kg by 66.6% and at the dose 10 mg/kg by 50.1%, respectively. Furthermore, derivative 117a–117d were evaluated by measuring mean plasma and brain concentrations after intragastric administration in mice at a dose of 5 mg/kg. Among the molecules, compound 117b had the longest half-life, ca. 3 h compared to compound 117a (ca. 1 h). The compound 117d showed the largest apparent volume of distribution (41.3 l/kg) that indicated the ability for penetration to the deep compartment and binding to tissues. After i.p. administration, all compounds quickly, within 15 min achieve maximum concentration (Cmax) in the blood. The molecules were characterised by a high permeability to the brain, with the highest brain to plasma ratio (31.6) for compound 117d. As for the SAR, the partial substitution of piperazine at the 4 positions of the heteroanthrone structure is more conducive to enhancing the antidepressant and anti-anxiety activities in vivo than the 2 positions. Besides, the presence of methoxy or chloro substituent at 6 or 7 positions had a favourable effect on the antidepressant activity and anti-anxiety activityCitation145,Citation146.
Figure 69. Chemical structures of xanthone and its derivatives.
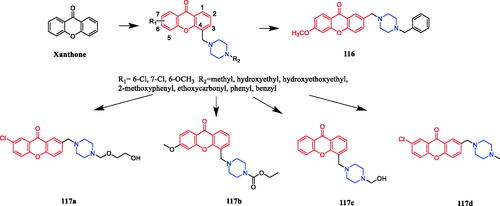
2.6. Cardiovascular activity
A series of xanthone derivatives with piperazine were synthesised and evaluated for their cardiovascular activity. Compound 118 () exhibited significant antiarrhythmic activity, with an ED50 value of 0.69 mg/kg in adrenaline-induced arrhythmia, while ED50 values of Propranolol, Urapidil, Carvedilol were 1.05, 1.26, 0.25 mg/kg, respectively. From the structure-activity relationship, it can be considered that the most active compound contained a 2-methoxyphenylpiperazine moiety. Compounds containing 4-methoxyphenylpiperazine moieties are less effective in in vivo tests than their 2-substituted analogs. In vivo experiments showed that the binding affinity of all test compounds to the α2-receptors and β1-adrenoceptors were within the µM-range, and revealed the structural relationship between them. The introduction of chlorine substituents on the xanthone ring also reduced the binding affinity of the α1-adrenergic receptors, indicating that the increase in lipophilicity is undesirableCitation147.
Figure 70. Chemical structures of xanthone and its derivatives.
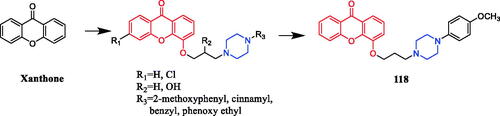
In a series of polyphenolic natural product analogs bearing an arylpiperazine moiety developed by Xie et al., compound 119 () acted as a potential antihypertensive agent in spontaneously hypertensive rats (SHRs). In biological investigations, compound 119 reduced the systolic and diastolic blood pressure in SHRs comparable to naftopidil. The studies have shown that the mean arterial pressure (MAP) of 119 on SHR was reduced by nearly 23.7% at 4 h, which is better than the 16.1% of naftopidil at 2 h, demonstrating compound 119 may possess no effects on the basal heart rate. The SAR analysis showed that the substitution position of the linker had an effect on vasodilation activity. In general, most analogs with C6 substitution are more effective than corresponding analogues with C7 and C8 substitutionCitation148.
Figure 71. Chemical structures of 7,8-dihydroxy-3-methyl-isochromanone-4 (XJP) and its derivatives.
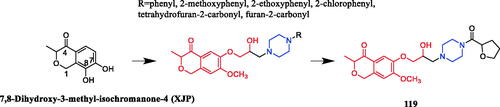
2.7. Other biological activity
16 semisynthetic C-10 pyrrole Mannich artemisinin derivatives were first evaluated in vitro against the chloroquine-sensitive 3D7 strain of P. falciparum, several of which are more than three times more effective than the natural product artemisinin. According to the analysis of the optimal Mannich side-chain substitution for in vitro and in vivo activity, the morpholine and N-methylpiperazine in the Mannich side-chain substitution 120 () displayed the best activity among these derivatives. According to the SAR analysis, morpholine, N-methylpiperazine, and sulfonylmorpholine heterocyclics could provide molecules with increased potency both in vitro and in vivo. Furthermore, compound 120 had significantly better antimalarial activity in vivo versus the P. Berghei N strain (ED50 = 1.99 mg/kg, ED90 = 5.34 mg/kg, respectively) than water-soluble sodium artesunate (ED50 = 3.23 mg/kg, ED90 > 10 mg/kg, respectively)Citation149.
Figure 72. Chemical structures of artemisinin and its derivatives.
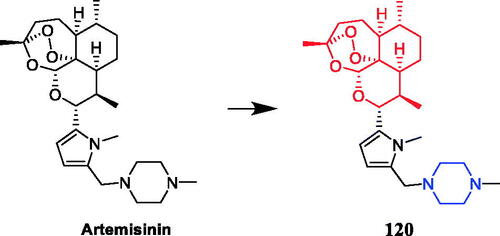
To improve solubility and maintain some rigidity, pentacyclic triterpene echinocystic acid dimer 121 () was designed and synthesised by replacing the phenyl group of the linker with piperazine, demonstrating significant anti-hepatitis C (HCV) entry activity, with an IC50 value of 2.87 nmol/L, two orders of magnitudes more potent than EA (1.4 µmol/L). In addition, the haemolysis of the synthesised compound is eliminated under the concentration gradient of 0, 6.25, 12.5, 25 and 50 mmol/L, while EA had obvious haemolytic side effects when the CC50 was 15 mmol/LCitation150.
Figure 73. Chemical structures of echinocystic acid and its derivatives.
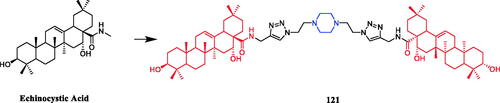
Li et al. synthesised a series of berberrubine derivatives by introducing various aminomethyl groups into 12-position of berberrubine and evaluated their anti-diabetic activity against type 2 diabetes mellitus. Compound 122 () with an N-methyl piperazine-4-methyl group at C-12 showed the most potent anti-diabetic activity and the sensitisation reached 1.26 fold of rosiglitazone. Even if the concentration dropped to 1 µmol/ml, its activity was still comparable to the positive control rosiglitazone. The research suggested compound 122 had a certain effect on promoting glucose uptake on insulin-resistant 3T3-L1 adipocytes and L6 myotubesCitation151.
Figure 74. Chemical structures of berberrubine and its derivative.
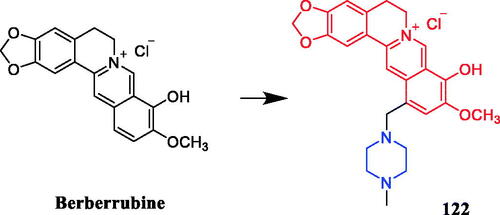
Based on the relationship between results of tyrosinase inhibition and molecular docking, kojic acid derivative 123 () was found to be a promising antityrosinase agent, and the hydroxymethyl group in the structure provided a metal complex of copper ions and chlorine atoms, allowing conventional hydrogen bonds to interact with tyrosinase, with an IC50 value of 86.2 µM. Compound 123 showed the antidiphenolase activity of mushroom tyrosinase to be almost 5-fold higher than that of control agent kojic acid (IC50 = 418.2 µM)Citation152.
Figure 75. Chemical structures of kojic acid and its derivative.
Nie et al. synthesised a series of α-glucosidase inhibitors based on OA. The piperazine fragment was introduced to link the cinnamic amide unit with OA at C-28. Compound 124 () with 3, 28-disubstituted OA exhibited potent inhibitory activity, with an IC50 value of 1.90 µM. Interestingly, the different substituents on the benzene ring of the cinnamic amide unit had a significant effect on the activity. For example, fluorine atoms can reduce the activity, and the expansion of the hydrophobic surface could also enhance the inhibitory activity of α-glucosidase. However, compared with oleanolic acid (IC50 = 98.5 mM), the introduction of cinnamamide at position C-28 did not result in a significant increase in α-glucosidase inhibitory activity. The activity of 3-OH derivatives had stronger effects than that of 3-OAc derivatives, possibly due to its good steric binding to enzymes. The 3,28-disubstituted oleanolic acid derivatives significantly improved the inhibitory activity of α-glucosidase than oleanolic acidCitation153.
Figure 76. Chemical structures of oleanolic acid and its derivatives.
Reportedly, adenosine analog with piperazine 125 () exhibited better inhibitory activity against Coxsackievirus B3 (CVB3), with a lower IC50 value of 5.1 ± 2.3 mg/L and a higher selectivity index (TI = 41.0) than ribavirin (IC50 = 36.8 ± 9.6 mg/L, TI = 29.1, TI, selectivity index represented by the TC50/IC50 ratio)). The studies have shown that in this series of compounds, the phenyl substitution of piperazine moiety may be more effective than pyridine substitution to enhance antiviral activity. This may be due to the lone pair of electrons in the aromatic ring. In addition, the calculated log p values of 125 were 0.8 and met the log P criteria, and the acute toxicity level was within 50 ≤ LD50 < 500 mg/kg, with no carcinogenic and mutagenic toxicity riskCitation154.
Figure 77. Chemical structures of adenosine and its analog.
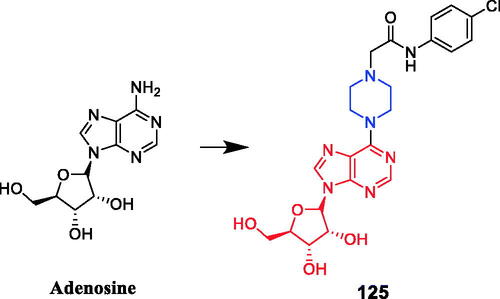
3. Conclusion
Piperazine is a part of numerous natural and synthetic molecules, with broad therapeutic potential. In addition to being a pharmacophore, piperazine is used as a linker/bridge between natural products and active structural molecules. This review summarises the research progress in the synthesis of natural product-piperazine derivatives in the past ten years, aiming to discover natural product piperazine hybrid compounds that may possess abundant biological activities. This will aid the scientific community in rationally design and develop novel, targeted, optimised, and diversified natural products-piperazine drugs for the treatment of multifactorial diseases. In conclusion, the piperazine group is widely used in for drug synthesis, attracting great attention from researchers worldwide, and its role cannot be ignored.
Acknowledgements
Thanks for the language editing service provided by the Cactus Communications (Shanghai) Co., Ltd. for the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Shaquiquzzaman M, Verma G, Marella A, et al. Piperazine scaffold: a remarkable tool in generation of diverse pharmacological agents. Eur J Med Chem 2015;102:487–529.
- Chen F-H, Zhang L-B, Qiang L, et al. Reactive oxygen species-mitochondria pathway involved in LYG-202-induced apoptosis in human hepatocellular carcinoma HepG(2) cells. Cancer Lett 2010;296:96–105.
- Foley TL, Rai G, Yasgar A, et al. 4-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4- methoxypyridin-2-yl)piperazine-1-carbothioamide (ML267), a potent inhibitor of bacterial phosphopantetheinyl transferase that attenuates secondary metabolism and thwarts bacterial growth. J Med Chem 2014;57:1063–78.
- Migliore M, Pontis S, Fuentes de Arriba AL, et al. Second-generation non-covalent NAAA inhibitors are protective in a model of multiple sclerosis. Angew Chem-Int Edit 2016;55:11193–7.
- Cao X, Zhang Y, Chen Y, et al. Synthesis and biological evaluation of fused tricyclic heterocycle piperazine (piperidine) derivatives as potential multireceptor atypical antipsychotics. J Med Chem 2018;61:10017–39.
- Moussa IA, Banister SD, Beinat C, et al. Design, synthesis, and structure-affinity relationships of regioisomeric N-benzyl alkyl ether piperazine derivatives as sigma-1 receptor ligands. J Med Chem 2010;53:6228–39.
- He Y, Xie F, Ye J, et al. 1-(4-[F]Fluorobenzyl)-4-[(tetrahydrofuran-2-yl)methyl]piperazine: a novel suitable radioligand with low lipophilicity for imaging σ receptors in the brain. J Med Chem 2017;60:4161–72.
- Sergeant N, Vingtdeux V, Eddarkaoui S, et al. New piperazine multi-effect drugs prevent neurofibrillary degeneration and amyloid deposition and preserve memory in animal models of Alzheimer’s disease. Neurobiol Dis 2019;129:217–33.
- Ji Q, Deng Q, Li B, et al. Design, synthesis and biological evaluation of novel 5-(piperazin-1-yl)quinolin-2(1H)-one derivatives as potential chitin synthase inhibitors and antifungal agents. Eur J Med Chem 2019;180:204–12.
- Tamayo NA, Norman MH, Bartberger MD, et al. Small molecule disruptors of the glucokinase-glucokinase regulatory protein interaction: 5. A novel aryl sulfone series, optimization through conformational analysis. J Med Chem 2015;58:4462–82.
- Clark RB, Lamppu D, Libertine L, et al. Discovery of novel 2-((pyridin-3-yloxy)methyl)piperazines as α7 nicotinic acetylcholine receptor modulators for the treatment of inflammatory disorders. J Med Chem 2014;57:3966–83.
- Brito AF, Moreira LKS, Menegatti R, et al. Piperazine derivatives with central pharmacological activity used as therapeutic tools. Fundam Clin Pharmacol 2019;33:13–24.
- Lacivita E, Leopoldo M, Giorgio PD, et al. Determination of 1-aryl-4-propylpiperazine pKa values: the substituent on aryl modulates basicity. Bioorg Med Chem 2009;17:1339–44.
- Maia R, Tesch R, Fraga C. Phenylpiperazine derivatives: a patent review (2006 – present). Expert Opin Ther Pat 2012;22:1169–78.
- Pluskal T, Weng J-K. Natural product modulators of human sensations and mood: molecular mechanisms and therapeutic potential. Chem Soc Rev 2018;47:1592–637.
- Choudhary S, Singh PK, Verma H, et al. Success stories of natural product-based hybrid molecules for multi-factorial diseases. Eur J Med Chem 2018;151:62–97.
- Borsari C, Trader DJ, Tait A, et al. Designing chimeric molecules for drug discovery by leveraging chemical biology. J Med Chem 2020;63:1908–28.
- Rodrigues T, Sieglitz F, Bernardes GJ. Natural product modulators of transient receptor potential (TRP) channels as potential anti-cancer agents. Chem Soc Rev 2016;45:6130–7.
- Tietze LF, Bell HP, Chandrasekhar S. Natural product hybrids as new leads for drug discovery. Angew Chem-Int Edit 2003;42:3996–4028.
- Newman DJ, Cragg GM. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat Prod 2020;83:770–803.
- Nishimura N, Norman MH, Liu L, et al. Small molecule disruptors of the glucokinase-glucokinase regulatory protein interaction: 3. Structure-activity relationships within the aryl carbinol region of the N-arylsulfonamido-N'-arylpiperazine series. J Med Chem 2014;57:3094–116.
- Mahmoudi Y, Badali H, Hashemi SM, et al. New potent antifungal triazole alcohols containing N-benzylpiperazine carbodithioate moiety: synthesis, in vitro evaluation and in silico study. Bioorg Chem 2019;90:103060.
- Amujuri D, Siva B, Poornima B, et al. Synthesis and biological evaluation of Schizandrin derivatives as potential anti-cancer agents. Eur J Med Chem 2018;149:182–92.
- Abu-Aisheh MN, Mustafa MS, El-Abadelah MM, et al. Synthesis and biological activity assays of some new N1-(flavon-7-yl)amidrazone derivatives and related congeners. Eur J Med Chem 2012;54:65–74.
- Patel RV, Mistry BM, Syed R, et al. Sulfonylpiperazines based on a flavone as antioxidant and cytotoxic agents. Arch Pharm 2019;352:1900051.
- Patel RV, Mistry B, Syed R, et al. Chrysin-piperazine conjugates as antioxidant and anticancer agents. Eur J Pharm Sci 2016;88:166–77.
- Fu W, Wang J, Yu L, et al. Synthesis and biological evaluation of 7-O-modified oroxylin A derivatives. Bioorg Med Chem Lett 2012;22:1118–21.
- Mao Z, Zheng X, Qi Y, et al. Synthesis and biological evaluation of novel hybrid compounds between chalcone and piperazine as potential antitumor agents. RSC Adv 2016;6:7723–7.
- Lin Y, Hu C, Zheng X, et al. Synthesis and anti-tumor activities of novel 4'-(N-substitued-1-piperazinyl) chalcone derivatives. Chin J Org Chem 2017;37:237–41.
- Yamali C, Gul HI, Sakagami H, et al. Synthesis and bioactivities of halogen bearing phenolic chalcones and their corresponding bis Mannich bases. J Enzym Inhib Med Chem 2016;31:125–31.
- Tugrak M, Gul HI, Bandow K, et al. Synthesis and biological evaluation of some new mono Mannich bases with piperazines as possible anticancer agents and carbonic anhydrase inhibitors. Bioorg Chem 2019;90:103095.
- Tugrak M, Yamali C, Sakagami H, et al. Synthesis of mono Mannich bases of 2-(4-hydroxybenzylidene)-2,3-dihydroinden-1-one and evaluation of their cytotoxicities. J Enzym Inhib Med Chem 2016;31:818–23.
- Fu DJ, Li JH, Yang JJ, et al. Discovery of novel chalcone-dithiocarbamates as ROS-mediated apoptosis inducers by inhibiting catalase. Bioorg Chem 2019;86:375–85.
- Quan Wang J, Di Yang M, Chen X, et al. Discovery of new chromen-4-one derivatives as telomerase inhibitors through regulating expression of dyskerin. J Enzym Inhib Med Chem 2018;33:1199–211.
- Chen H, Zhang J, Hu P, et al. Synthesis, biological evaluation and molecular docking of 4-amino-2H-benzo[h]chromen-2-one (ABO) analogs containing the piperazine moiety. Bioorg Med Chem 2019;27:115081.
- Gao H, Zheng X, Qi Y, et al. Synthesis and cytotoxic activity of novel resveratrol-chalcone amide derivatives. Chin J Org Chem 2018;38:648–55.
- Li X, Zhang X, Sun H, et al. Synthesis and anti-tumor evaluation of novel C-37 modified derivatives of gambogic acid. Chin J Chem 2012;30:1083–91.
- Bian J, Li T, Weng T, et al. Synthesis, evaluation and quantitative structure-activity relationship (QSAR) analysis of Wogonin derivatives as cytotoxic agents. Bioorg Med Chem Lett 2017;27:1012–6.
- Wang J, Li T, Zhao T, et al. Design of wogonin-inspired selective cyclin-dependent kinase 9 (CDK9) inhibitors with potent in vitro and in vivo antitumor activity. Eur J Med Chem 2019;178:782–801.
- Rajaram P, Jiang Z, Chen G, et al. Nitrogen-containing derivatives of O-tetramethylquercetin: synthesis and biological profiles in prostate cancer cell models. Bioorg Chem 2019;87:227–39.
- El Kihel L, Clement M, Bazin MA, et al. New lithocholic and chenodeoxycholic piperazinylcarboxamides with antiproliferative and pro-apoptotic effects on human cancer cell lines. Bioorg Med Chem 2008;16:8737–44.
- Brossard D, El Kihel L, Clément M, et al. Synthesis of bile acid derivatives and in vitro cytotoxic activity with pro-apoptotic process on multiple myeloma (KMS-11), glioblastoma multiforme (GBM), and colonic carcinoma (HCT-116) human cell lines. Eur J Med Chem 2010;45:2912–8.
- Yang X, Li Y, Jiang W, et al. Synthesis and biological evaluation of novel ursolic acid derivatives as potential anticancer prodrugs. Chem Biol Drug Des 2015;86:1397–404.
- Hua SX, Huang RZ, Ye MY, et al. Design, synthesis and in vitro evaluation of novel ursolic acid derivatives as potential anticancer agents. Eur J Med Chem 2015;95:435–52.
- Chi KQ, Wei ZY, Wang KS, et al. Design, synthesis, and evaluation of novel ursolic acid derivatives as HIF-1alpha inhibitors with anticancer potential. Bioorg Chem 2017;75:157–69.
- Tian T, Liu X, Lee ES, et al. Synthesis of novel oleanolic acid and ursolic acid in C-28 position derivatives as potential anticancer agents. Arch Pharm Res 2017;40:458–68.
- Chen Y, Hou X, Zhi D, et al. Synthesis, characterization and anticancer activity of oleanolic acid and ursolic acid derivatives. Chin J Org Chem 2016;36:795.
- Wang W, Lei L, Liu Z, et al. Design, synthesis, and biological evaluation of novel nitrogen heterocycle-containing ursolic acid analogs as antitumor agents. Molecules 2019;24:877.
- Liu MC, Yang SJ, Jin LH, et al. Synthesis and cytotoxicity of novel ursolic acid derivatives containing an acyl piperazine moiety. Eur J Med Chem 2012;58:128–35.
- Zhao CH, Zhang CL, Shi JJ, et al. Design, synthesis, and biofunctional evaluation of novel pentacyclic triterpenes bearing O-[4-(1-piperazinyl)-4-oxo-butyryl moiety as antiproliferative agents. Bioorg Med Chem Lett 2015;25:4500–4.
- Kahnt M, Wiemann J, Fischer L, et al. Transformation of asiatic acid into a mitocanic, bimodal-acting rhodamine B conjugate of nanomolar cytotoxicity. Eur J Med Chem 2018;159:143–8.
- Rodríguez-Hernández D, Barbosa LCA, Demuner AJ, et al. Hederagenin amide derivatives as potential antiproliferative agents. Eur J Med Chem 2019;168:436–46.
- Li K, Li L, Wang S, et al. Design and synthesis of novel 2-substituted 11-keto-boswellic acid heterocyclic derivatives as anti-prostate cancer agents with Pin1 inhibition ability. Eur J Med Chem 2017;126:910–9.
- Yang X, Qin X, Wang Q, et al. Synthesis and antitumor activities of piperazine- and cyclen-conjugated dehydroabietylamine derivatives. Heterocycl Commun 2015;21:233–7.
- Chen H, Qiao C, Miao T-T, et al. Synthesis and biological evaluation of novel-(piperazin-1-yl)alkyl-1-dibenzo[,]carbazole derivatives of dehydroabietic acid as potential MEK inhibitors. J Enzym Inhib Med Chem 2019;34:1544–61.
- Jiang F, Wang H-J, Bao Q-C, et al. Optimization and biological evaluation of celastrol derivatives as Hsp90–Cdc37 interaction disruptors with improved druglike properties. Bioorg Med Chem 2016;24:5431–9.
- Shan WG, Wang HG, Chen Y, et al. Synthesis of 3- and 29-substituted celastrol derivatives and structure-activity relationship studies of their cytotoxic activities. Bioorg Med Chem Lett 2017;27:3450–3.
- Tang WJ, Wang J, Tong X, et al. Design and synthesis of celastrol derivatives as anticancer agents. Eur J Med Chem 2015;95:166–73.
- Sommerwerk S, Heller L, Kerzig C, et al. Rhodamine B conjugates of triterpenoic acids are cytotoxic mitocans even at nanomolar concentrations. Eur J Med Chem 2017;127:1–9.
- Xu J, Wei M, Li G, et al. Synthesis and anti-tumor activities of novel artemisone- piperazine-sulfonamide derivatives. Chem J Chin Univ-Chin 2015;36:919–26.
- Sun Q, Wang J, Li Y, et al. Synthesis and evaluation of cytotoxic activities of artemisinin derivatives. Chem Biol Drug Des 2017;90:1019–28.
- Yu JY, Li XQ, Wei MX. Synthesis and biological activities of artemisinin-piperazine- dithiocarbamate derivatives. Eur J Med Chem 2019;169:21–8.
- Li S, Li G, Yang X, et al. Design, synthesis and biological evaluation of artemisinin derivatives containing fluorine atoms as anticancer agents. Bioorg Med Chem Lett 2018;28:2275–8.
- Narender T, Sukanya P, Sharma K, et al. Preparation of novel antiproliferative emodin derivatives and studies on their cell cycle arrest, caspase-dependent apoptosis and DNA binding interaction. Phytomedicine 2013;20:890–6.
- Pavan Kumar P, Siva B, Venkateswara Rao B, et al. Synthesis and biological evaluation of bergenin-1,2,3-triazole hybrids as novel class of anti-mitotic agents. Bioorg Chem 2019;91:103161.
- Yan TL, Bai LF, Zhu HL, et al. Synthesis and biological evaluation of glycyrrhetic acid derivatives as potential VEGFR2 inhibitors. ChemMedChem 2017;12:1087–96.
- Jadala C, Sathish M, Anchi P, et al. Synthesis of Combretastatin-A4 Carboxamidest that Mimic Sulfonyl Piperazines by a Molecular Hybridization Approach: in vitro Cytotoxicity Evaluation and Inhibition of Tubulin Polymerization. ChemMedChem 2019;14:2052–60.
- O’Boyle NM, Ana G, Kelly PM, et al. Synthesis and evaluation of antiproliferative microtubule-destabilising combretastatin A-4 piperazine conjugates. Org Biomol Chem 2019;17:6184–200.
- Singh B, Kumar A, Joshi P, et al. Colchicine derivatives with potent anticancer activity and reduced P-glycoprotein induction liability. Org Biomol Chem 2015;13:5674–89.
- Yin Y, Zhao X-C, Wang S-J, et al. Synthesis and biological evaluation of novel sarsasapogenin derivatives as potential anti-tumor agents. Steroids 2015;93:25–31.
- Wang W, Wang D, Wang Z, et al. Synthesis of new sarsasapogenin derivatives with cytotoxicity and apoptosis-inducing activities in human breast cancer MCF-7 cells. Eur J Med Chem 2017;127:62–71.
- Mistry B, Keum Y-S, Noorzai R, et al. Synthesis of piperazine based N-Mannich bases of berberine and their antioxidant and anticancer evaluations. J Iran Chem Soc 2016;13:531–9.
- Mistry B, Patel RV, Keum YS, et al. Synthesis of N-Mannich bases of berberine linking piperazine moieties revealing anticancer and antioxidant effects. Saudi J Biol Sci 2017;24:36–44.
- Mistry B, Keum Y-S, Kim DH. Synthesis and biological evaluation of berberine derivatives bearing 4-aryl-1-piperazine moieties. J Chem Res 2015;39:470–4.
- Mistry B, Patel RV, Keum YS, et al. Synthesis of Mannich base derivatives of berberine and evaluation of their anticancer and antioxidant effects. J Chem Res 2016; 40:73–7.
- Mistry B, Keum YS, Pandurangan M, et al. Synthesis of berberine-piperazine conjugates as potential antioxidant and cytotoxic agents. Med Chem Res 2016;25:2461–70.
- Chen H, Liang X, Sun T, et al. Synthesis and biological evaluation of estrone 3-O-ether derivatives containing the piperazine moiety. Steroids 2018;134:101–9.
- Xu Y, Liang P, Rashid H, et al. Design, synthesis, and biological evaluation of matrine derivatives possessing piperazine moiety as antitumor agents. Med Chem Res 2019;28:1618–27.
- Tuncbilek M, Guven EB, Onder T, et al. Synthesis of novel 6-(4-substituted piperazine-1-yl)-9-(β-D-ribofuranosyl)purine derivatives, which lead to senescence-induced cell death in liver cancer cells. J Med Chem 2012;55:3058–65.
- Demir Z, Guven EB, Ozbey S, et al. Synthesis of novel substituted purine derivatives and identification of the cell death mechanism. Eur J Med Chem 2015;89:701–20.
- Huang LH, Xu HD, Yang ZY, et al. Synthesis and anticancer activity of novel C6-piperazine substituted purine steroid-nucleosides analogues. Steroids 2014;82:1–6.
- Huang L-H, Li Y, Xu H-D, et al. Synthesis and biological evaluation of novel C6-cyclo secondary amine substituted purine steroid-nucleosides analogues. Steroids 2014;85:13–7.
- Ayan D, Maltais R, Hospital A, et al. Chemical synthesis, cytotoxicity, selectivity and bioavailability of 5alpha-androstane-3alpha,17beta-diol derivatives. Bioorg Med Chem 2014;22:5847–59.
- Alloatti D, Giannini G, Vesci L, et al. Camptothecins in tumor homing via an RGD sequence mimetic. Bioorg Med Chem Lett 2012;22:6509–12.
- Yang CJ, Song ZL, Goto M, et al. Design, synthesis, and cytotoxic activity of novel 7-substituted camptothecin derivatives incorporating piperazinyl-sulfonylamidine moieties. Bioorg Med Chem Lett 2017;27:3959–62.
- Zhu G-X, Cheng P-L, Goto M, et al. Design, synthesis and potent cytotoxic activity of novel 7-(N-[(substituted-sulfonyl)piperazinyl]-methyl)-camptothecin derivatives. Bioorg Med Chem Lett 2017;27:1750–3.
- Chen L-W, Wang Z-F, Zhu B, et al. Design, synthesis and biological evaluation of novel benzo-α-pyrone containing piperazine derivatives as potential BRAF V600E inhibitors. Bioorg Med Chem Lett 2016;26:4983–91.
- Xue W, Song B-A, Zhao HJ, et al. Novel myricetin derivatives: design, synthesis and anticancer activity. Eur J Med Chem 2015;97:155–63.
- Ruan X, Zhao H, Zhang C, et al. Syntheses and bioactivities of myricetin derivatives containing piperazine acidamide moiety. Chem J Chin Univ-Chin 2018;39:1197–204.
- Jin Y, Liu J, Huang WT, et al. Synthesis and biological evaluation of derivatives of 4-deoxypodophyllotoxin as antitumor agents. Eur J Med Chem 2011;46:4056–61.
- Sang C-Y, Liu J-F, Qin W-W, et al. Synthesis and evaluation of the apoptosis inducing and CT DNA interaction properties of a series of 4β-carbamoyl 4′-O-demethylepipodophyllotoxins. Eur J Med Chem 2013;70:59–67.
- Yadav AA, Chee GL, Wu X, et al. Structure-based design, synthesis and biological testing of piperazine-linked bis-epipodophyllotoxin etoposide analogs. Bioorg Med Chem 2015;23:3542–51.
- Sun W-X, Ji Y-J, Wan Y, et al. Design and synthesis of piperazine acetate podophyllotoxin ester derivatives targeting tubulin depolymerization as new anticancer agents. Bioorg Med Chem Lett 2017;27:4066–74.
- O’Keeffe R, Kenny O, Brunton NP, et al. Synthesis of novel 24-amino-25,26,27-trinorlanost- 8-enes: cytotoxic and apoptotic potential in U937 cells. Bioorg Med Chem 2015;23:2270–80.
- Chen CL, Chang DM, Chen TC, et al. Structure-based design, synthesis and evaluation of novel anthra[1,2-d]imidazole-6,11-dione derivatives as telomerase inhibitors and potential for cancer polypharmacology. Eur J Med Chem 2013;60:29–41.
- Ahadi H, Emami S. Modification of 7-piperazinylquinolone antibacterials to promising anticancer lead compounds: synthesis and in vitro studies. Eur J Med Chem 2020;187:111970.
- Marín-Yaseli MR, Mompeán C, Ruiz-Bermejo M. A prebiotic synthesis of pterins. Chemistry 2015;21:13531–4.
- Huang M, Xie X, Gong P, et al. A 18beta-glycyrrhetinic acid conjugate with Vorinostat degrades HDAC3 and HDAC6 with improved antitumor effects. Eur J Med Chem 2020;188:111991.
- Geng P-F, Wang C-C, Li Z-H, et al. Design, synthesis and preliminary biological evaluation of 5,8-dihydropteridine-6,7-diones that induce apoptosis and suppress cell migration. Eur J Med Chem 2018;143:1959–67.
- Barker TJ, Duncan KK, Otrubova K, et al. Potent vinblastine C20' ureas displaying additionally improved activity against a vinblastine-resistant cancer cell line. ACS Med Chem Lett 2013;4:985–8.
- Keglevich A, Dányi L, Rieder A, et al. Synthesis and cytotoxic activity of new vindoline derivatives coupled to natural and synthetic pharmacophores. Molecules 2020;25:1010.
- Cheng X, Rasqué P, Vatter S, et al. Synthesis and cytotoxicity of novel indirubin-5-carboxamides. Bioorg Med Chem 2010;18:4509–15.
- Jiang X, Zhou J, Lin Q, et al. Anti-angiogenic and anticancer effects of baicalein derivatives based on transgenic zebrafish model. Bioorg Med Chem 2018;26:4481–92.
- Liu R, Zhang H, Yuan M, et al. Synthesis and biological evaluation of apigenin derivatives as antibacterial and antiproliferative agents. Molecules 2013;18:11496–511.
- Tong X, Han L, Duan H, et al. The derivatives of Pulsatilla saponin A, a bioactive compound from Pulsatilla chinensis: their synthesis, cytotoxicity, haemolytic toxicity and mechanism of action. Eur J Med Chem 2017;129:325–36.
- Synthesis and antibacterial activity of betulonic acid amides with piperazine derivatives. Chem Nat Compd 2011;47:759–63.
- Wang SF, Yin Y, Wu X, et al. Synthesis, molecular docking and biological evaluation of coumarin derivatives containing piperazine skeleton as potential antibacterial agents. Bioorg Med Chem 2014;22:5727–37.
- Hatnapure GD, Keche AP, Rodge AH, et al. Synthesis and biological evaluation of novel piperazine derivatives of flavone as potent anti-inflammatory and antimicrobial agent. Bioorg Med Chem Lett 2012;22:6385–90.
- Hirokawa Y, Kinoshita H, Tanaka T, et al. Water-soluble pleuromutilin derivative with excellent in vitro and in vivo antibacterial activity against Gram-positive pathogens. J Med Chem. 2008;51:1991–4.
- Luo J, Yang Q-E, Yang Y-Y, et al. Design, synthesis, and structure-activity relationship studies of novel pleuromutilin derivatives having a piperazine ring. Chem Biol Drug Des 2016;88:699–709.
- Liu H, Xiao S, Zhang D, et al. Synthesis and antibacterial activity of novel pleuromutilin derivatives. Biol Pharm Bull 2015;38:1041–8.
- Deng Y, Wang X-Z, Huang S-H, et al. Antibacterial activity evaluation of synthetic novel pleuromutilin derivatives in vitro and in experimental infection mice. Eur J Med Chem 2019;162:194–202.
- Liu HX, Ma DL, Cui G, et al. Design, synthesis and antibacterial activities of pleuromutilin derivatives. J Asian Nat Prod Res 2021;23:123–37.
- Li HX, Wang ZC, Qian YM, et al. Design, synthesis, and biological evaluation of chrysin derivatives as potential FabH inhibitors. Chem Biol Drug Des 2017;89:136–40.
- Xu G, Yang X, Jiang B, et al. Synthesis and bioactivities of novel piperazine-containing 1,5-Diphenyl-2-penten-1-one analogues from natural product lead. Bioorg Med Chem Lett 2016;26:1849–53.
- Liu Z, Wang Y, Zeng H, et al. Synthesis and antibacterial activities of Mannich bases of curcumin derivatives. Chin J Org Chem 2014;34:2345–9.
- Zhou X, Zhao L, Liu X, et al. Antimycobacterial and synergistic effects of 18beta-glycyrrhetinic acid or glycyrrhetinic acid-30-piperazine in combination with isoniazid, rifampicin or streptomycin against Mycobacterium bovis. Phytother Res 2012;26:253–8.
- Xiang M, Song Y-L, Ji J, et al. Synthesis of novel 18β-glycyrrhetinic piperazine amides displaying significant in vitro and in vivo antibacterial activities against intractable plant bacterial diseases. Pest Manag Sci 2020;76:2959–71.
- Wang P-Y, Xiang M, Luo M, et al. Novel piperazine-tailored ursolic acid hybrids as significant antibacterial agents targeting phytopathogens Xanthomonas oryzae pv. oryzae and X. axonopodis pv. citri probably directed by activation of apoptosis. Pest Manag Sci 2020;76:2746–54.
- Kushwaha K, Kaushik N, Lata, et al. Design and synthesis of novel 2H-chromen-2-one derivatives bearing 1,2,3-triazole moiety as lead antimicrobials. Bioorg Med Chem Lett 2014;24:1795–801.
- Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell 2010;140:771–6.
- Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell 2015;160:816–27.
- van der Velden VH. Glucocorticoids: mechanisms of action and anti-inflammatory potential in asthma. Mediat Inflamm 1998;7:229–37.
- Pereira-Leite C, Nunes C, Jamal SK, et al. Nonsteroidal anti-inflammatory therapy: a journey toward safety. Med Res Rev 2017;37:802–59.
- Bandgar BP, Patil SA, Gacche RN, et al. Synthesis and biological evaluation of nitrogen-containing chalcones as possible anti-inflammatory and antioxidant agents. Bioorg Med Chem Lett 2010;20:730–3.
- Li J, Li D, Xu Y, et al. Design, synthesis, biological evaluation, and molecular docking of chalcone derivatives as anti-inflammatory agents. Bioorg Med Chem Lett 2017;27:602–6.
- Li B, Cai S, Yang YA, et al. Novel unsaturated glycyrrhetic acids derivatives: design, synthesis and anti-inflammatory activity. Eur J Med Chem 2017;139:337–48.
- Zhang C, Wu Y, Li J, et al. Synthesis and biological evaluation of 3-carbamate smilagenin derivatives as potential neuroprotective agents. Bioorg Med Chem Lett 2019;29:126622.
- Yu Z, Wang R, Xu L, et al. beta-Elemene piperazine derivatives induce apoptosis in human leukemia cells through downregulation of c-FLIP and generation of ROS. PLOS One 2011;6:e15843.
- Razavi SF, Khoobi M, Nadri H, et al. Synthesis and evaluation of 4-substituted coumarins as novel acetylcholinesterase inhibitors. Eur J Med Chem 2013;64:252–9.
- Modh RP, Kumar SP, Jasrai YT, et al. Design, synthesis, biological evaluation, and molecular modeling of coumarin-piperazine derivatives as acetylcholinesterase inhibitors. Arch Pharm (Weinheim) 2013;346:793–804.
- Kladna A, Berczynski P, Dundar OB, et al. Synthesis and in vitro antioxidant activity of new pyrimidin/benzothiazol-substituted piperazinyl flavones. Future Med Chem 2018;10:2293–308.
- Chen SY, Chen Y, Li YP, et al. Design, synthesis, and biological evaluation of curcumin analogues as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg Med Chem 2011;19:5596–604.
- Li S-Y, Wang X-B, Xie S-S, et al. Multifunctional tacrine-flavonoid hybrids with cholinergic, β-amyloid-reducing, and metal chelating properties for the treatment of Alzheimer’s disease. Eur J Med Chem 2013;69:632–46.
- Yang G-X, Ge S-L, Wu Y, et al. Design, synthesis and biological evaluation of 3-piperazinecarboxylate sarsasapogenin derivatives as potential multifunctional anti-Alzheimer agents. Eur J Med Chem 2018;156:206–15.
- Huang W, Wang Y, Li J, et al. Design, synthesis, and evaluation of genipin derivatives for the treatment of Alzheimer’s disease. Chem Biol Drug Des 2019;93:110–22.
- Yang GX, Huang Y, Zheng LL, et al. Design, synthesis and evaluation of diosgenin carbamate derivatives as multitarget anti-Alzheimer’s disease agents. Eur J Med Chem 2020;187:111913.
- Sun M, Hu J, Song X, et al. Coumarin derivatives protect against ischemic brain injury in rats. Eur J Med Chem 2013;67:39–53.
- Ostrowska K, Grzeszczuk D, Gluch-Lutwin M, et al. 5-HT1A and 5-HT2A receptors affinity, docking studies and pharmacological evaluation of a series of 8-acetyl-7-hydroxy-4-methylcoumarin derivatives. Bioorg Med Chem 2018;26:527–35.
- Ostrowska K, Mlodzikowska K, Gluch-Lutwin M, et al. Synthesis of a new series of aryl/heteroarylpiperazinyl derivatives of 8-acetyl-7-hydroxy-4-methylcoumarin with low nanomolar 5-HT1A affinities. Eur J Med Chem 2017;137:108–16.
- Chen Y, Lan Y, Wang S, et al. Synthesis and evaluation of new coumarin derivatives as potential atypical antipsychotics. Eur J Med Chem 2014;74:427–39.
- Chen Y, Wang S, Xu X, et al. Synthesis and biological investigation of coumarin piperazine (piperidine) derivatives as potential multireceptor atypical antipsychotics. J Med Chem 2013;56:4671–90.
- Koufaki M, Theodorou E, Alexi X, et al. Synthesis of tropolone derivatives and evaluation of their in vitro neuroprotective activity. Eur J Med Chem 2010;45:1107–12.
- Wen G, Liu Q, Hu H, et al. Design, synthesis, biological evaluation, and molecular docking of novel flavones as H3R inhibitors. Chem Biol Drug Des 2017;90:580–9.
- Waszkielewicz AM, Gunia A, Szkaradek N, et al. Synthesis and evaluation of pharmacological properties of some new xanthone derivatives with piperazine moiety. Bioorg Med Chem Lett 2013;23:4419–23.
- Zelaszczyk D, Jakubczyk M, Pytka K, et al. Design, synthesis and evaluation of activity and pharmacokinetic profile of new derivatives of xanthone and piperazine in the central nervous system. Bioorg Med Chem Lett 2019;29:126679.
- Szkaradek N, Rapacz A, Pytka K, et al. Synthesis and preliminary evaluation of pharmacological properties of some piperazine derivatives of xanthone. Bioorg Med Chem 2013;21:514–22.
- Xie S, Li X, Yu H, et al. Design, synthesis and biological evaluation of isochroman-4-one hybrids bearing piperazine moiety as antihypertensive agent candidates. Bioorg Med Chem 2019;27:2764–70.
- Pacorel B, Leung SC, Stachulski AV, et al. Modular synthesis and in vitro and in vivo antimalarial assessment of C-10 pyrrole mannich base derivatives of artemisinin. J Med Chem 2010;53:633–40.
- Meng L, Wang Q, Tang T, et al. Design, synthesis and biological evaluation of pentacyclic triterpene dimers as HCV entry inhibitors. Chin J Chem 2017;35:1322–8.
- Li R, Wu J, He Y, et al. Synthesis and in vitro evaluation of 12-(substituted aminomethyl) berberrubine derivatives as anti-diabetics. Bioorg Med Chem Lett 2014;24:1762–5.
- Karakaya G, Türe A, Ercan A, et al. Synthesis, computational molecular docking analysis and effectiveness on tyrosinase inhibition of kojic acid derivatives. Bioorg Chem 2019;88:102950.
- Nie W, Luo J-G, Wang X-B, et al. Synthesis of new a-glucosidase inhibitors based on oleanolic acid incorporating cinnamic amides. Chem Pharm Bull 2011;59:1051–6.
- Hao Y, Zhou G, Wu W, et al. Synthesis and antiviral evaluation of novel N-6 substituted adenosine analogues. Tetrahedron Lett 2017;58:190–3.