
Abstract
A new series of sulphonamide derivatives bearing naphthalene moiety were synthesised and evaluated for their antiproliferative and tubulin polymerisation inhibitory activities. These new compounds were evaluated for their in vitro antiproliferative activity against MCF-7 and A549 by using CCK-8 method. Among all the tested compounds, compound 5c with naphthalen-1-yl moiety exhibited the most potent antiproliferative activity against MCF-7 and A549 cell line, with IC50 values of 0.51 ± 0.03 µM and 0.33 ± 0.01 µM, respectively. The results of tubulin polymerisation assay shown that 5c exhibited a significant ability to inhibit tubulin polymerisation with IC50 value of 2.8 μM. Consistent with its antitubulin activity, 5c can significantly arrest the cell cycle at G2/M phase and induce apoptosis in MCF-7 cancer cells. Molecular docking study indicated that compound 5c inhibited tubulin polymerisation through interacting at the colchicine-binding site of tubulin. Furthermore, 5c exhibited low cytotoxic activity on human normal cell line.
1. Introduction
Microtubules are crucial elements of the cytoskeleton in eukaryocyte, which are polymerised by α- and β-tubulin heterodimers in a head-to-tail manner to form hollow cylindrical filamentsCitation1. The microtubule system of eukaryotic cells plays important roles in numerous essential cellular functions, such as cell growth, division, motility, maintenance of cell shape, and intracellular vesicle transport Citation2,Citation3. There is an increasing evidence showing that the disruption of microtubule will result in the cycle arrest in G2/M phase and lead to the apoptosis of cell Citation4. Therefore, microtubule has become an attractive molecular target in anti-tumour drug discovery Citation5–7. Up to the present, a larger number of anti-tubulin agents have been developed and some of them (e.g., paclitaxel, docetaxel, vinblastine, and vincristine) have been approved by FDA for clinical treatment of cancer Citation8. However, these natural products and their derivatives are facing severe disadvantages, such as toxicity, drug resistance, and difficult synthesis Citation9. Therefore, the search of novel molecules with potent tubulin polymerisation inhibitory activity is still in progress.
In recent years, sulphonamide and its derivatives have attracted much attention for their versatile properties in medicinal chemistry and modern drug discovery, such as anticancer, antiviral, antifungal, anti-inflammatory, antimicrobial, antibacterial, anti-tuberculosis, antiparasitic, anticonvulsant, carbonic anhydrase inhibition, and antidepressant activities Citation10. Notably, several compounds containing sulphonamide moiety have been reported inhibit tubulin polymerisation, such as ABT-751, indisulam, HMN-214, I, III, and III ()Citation11–15. These previous literatures reveal that sulphonamide could be used as a useful pharmacophoric fragment for the design and development anti-tubulin agents.
Figure 1. Chemical structures of some tubulin polymerisation inhibitors containing sulphonamide or trimethoxyphenyl moiety.
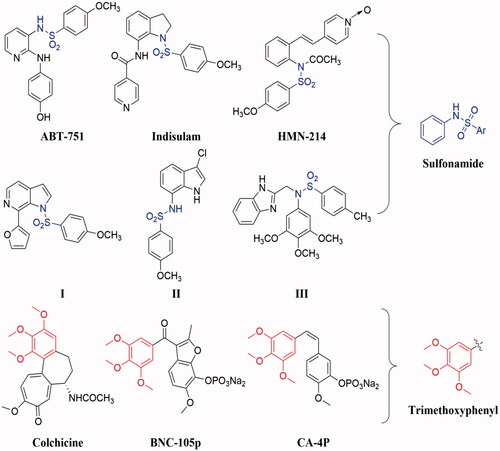
On the other hand, the trimethoxyphenyl has been proved to be an important pharmacophoric group of tubulin inhibitors by binding at the colchicine binding site of tubulin Citation16. Based on this, the trimethoxyphenyl moiety has been chosen as a core for the design and development of novel tubulin polymerisation inhibitors Citation16. In the last few decades, a large number of tubulin polymerisation inhibitors containing trimethoxyphenyl moiety have been reported in the literature, and some of them have entered clinical trials, such as BNC-105p, CA-4P, CKD-516, and AVE8062 () Citation17–20.
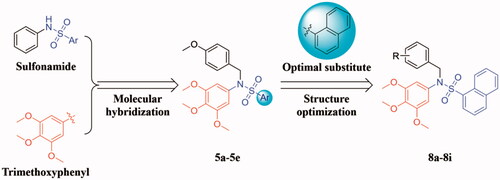
Hence, in continuation of our interest on the design and development of novel tubulin polymerisation inhibitors Citation21–24, a new series of sulphonamide derivatives (5a-5e and 8a-8i) were designed based on the molecular hybridisation approach () Citation25,Citation26. All the newly synthesised target compounds were evaluated for their antiproliferative activity to explore the preliminary structure-activity relationships (SAR). Tubulin polymerisation inhibition assay, cell cycle analysis, and cell apoptosis assay were performed to illuminate the pharmacologic mechanism. Additionally, molecular modelling was carried out to elucidate its possible binding mode in tubulin.
Figure 2. The design strategy of the target compounds in this study.
2. Results and discussion
2.1. Chemistry
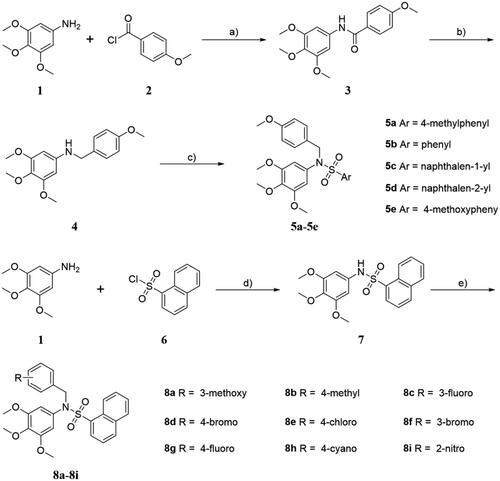
The sulphonamide derivatives (5a-5e and 8a-8i) were synthesised according to the synthetic route illustrated in Scheme 1. Firstly, 3,4,5-trimethoxyaniline 1 was condensed with 4-methoxybenzoyl chloride 2 in the presence of Et3N as base at room temperature to afford intermediate 3 in high yields, followed by the carbonyl reduction reaction with LiAlH4 to give the key intermediate 4 [Citation27]. Then, condensation of compound 4 with appropriate commercially available aryl sulphonyl chloride in the presence of Et3N and DMAP in THF to generate the title compounds (5a-5e) in high yields. On the other hand, treatment of 3,4,5-trimethoxyaniline 1 with naphthalene-1-sulphonyl chloride 6 in the presence of Et3N in CH2Cl2 to afforded key intermediate 7, which reacted with commercially available benzyl halide in the presence of KI and K2CO3 to provide the title compounds (8a-8i). All target derivatives were fully characterised by 1H NMR, 13 C NMR, HRMS, and elemental analysis (see Supporting Information).
Scheme 1. (a) Et3N, THF, room temperature, 4 h; (b) LiAlH4, THF, 0 °C 30 min to reflux, 2 h; (c) various aryl sulphonyl chloride, DMAP, Et3N, THF, room temperature, 12 h; (d) Et3N, CH2Cl2, room temperature, 6 h; (e) various benzyl halide, KI, K2CO3, acetone, reflux, 5 h.
The molecular formula of compound 5c was determined to be C27H27NO6S by high-resolution mass spectrum (HRMS) peak at m/z 516.1432 as [M + Na]+. The 1H NMR spectrum (Table S1 and ) of 5c displayed four singlets at δ 3.33 (6H, s, OCH3-C-3′′, C-5′′), 3.53 (3H, s, OCH3-C-4′′), and 3.63 (3H, s, OCH3-C-4′) ppm due to four methoxy groups on the phenyl rings. The methylene protons of –CH2– was appeared as singlet at δ 4.69 ppm (s, 2H, H-11). Two doublet peaks at δ 6.75 and δ 7.09 ppm with coupling constant of 8.4 Hz were attributed to the C-3′,5′-H and C-2′,6′-H of 4-methoxybenzyl moiety, respectively. According to the aromatic proton signal at at δ 6.11 ppm (s, 2H, H-2′′, 6′′), there was a 3,4,5-trimethoxyphenyl in the structure. Seven aromatic hydrogen atoms of naphthalen-1-yl were appeared at δ 7.46–8.26 ppm (7H, H-2, 3, 4, 5, 6, 7, and 8). In 13 C NMR spectrum of 5c the signal at δ 53.37 ppm (C-11) was assigned to the carbon in methylene. The signals at δ 55.50 (OCH3-C-4′), 56.20 (OCH3-C-3′′, 5′′), and 60.61 (OCH3-C-4′′) ppm were attributed to the carbon in four methoxyl groups. The carbon of –CH2– was appeared at δ 53.37 ppm (C-11). In addition, there are 17 carbon signals at δ 107.41 (C-2′′, 6′′), 114.18 (C-3′, 5′), 125.24 (C-3), 125.36 (C-8), 127.40 (C-6), 128.15 (C-7), 128.59 (C-1′), 128.83 (C-9), 129.35 (C-5), 130.24 (C-2′, 6′), 131.17 (C-2), 133.95 (C-1), 134.08 (C-1′′), 134.32 (C-10), 135.17 (C-4), 137.59 (C-4′′), 152.80 (C-3′′, 5′′), 159.10 (C-4′).
Figure 3. Chemical structure and key HMBC correlations of compound 5c.
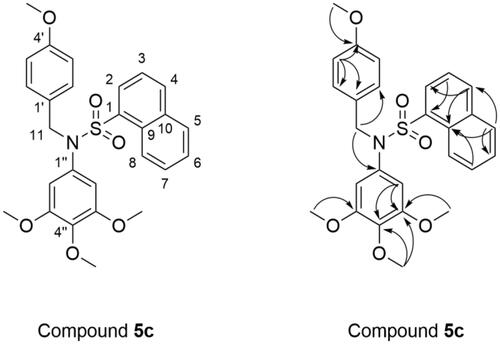
The HMBC correlations from the aromatic proton at δ 8.05 (H-5) to C-6, C-9 and C-4, from δ 7.65 (H-3) to C-1 and C-10, from δ 7.48 (H-7) to C-9 indicated the presence of a 1-naphthalene sulphonyl moiety. In addition, the correlations from the methoxy proton at δ 3.63 (H-OCH3-C-4′) to C-4′, from δ 3.33 (H-OCH3-C-3′′, 5′′) to C-3′′ and 5′′, from δ 3.53 (H-OCH3-C-4′′) to C-4′′ and C-3′′ showed four methoxy substituents connected to C-4′, C-3′′, 5′′, and C-4′′, respectively. Furthermore, key HMBC correlations between δ 4.69 (H-11) and C-2′, C-6′ and C-1′′ showed that 4-methoxybenzyl and 3,4,5-trimethoxyphenyl were linked through N. Therefore, the spectral data were in full agreement with the expected structure of the compound 5c ( and Table S2).
2.2. In vitro antiproliferative activities and SARs
The in vitro antiproliferative activities of these sulphonamide derivatives (5a-5e and 8a-8i) were evaluated against MCF-7 human breast cancer cells and A549 human non-small cell lung carcinoma cells by using CCK-8 method. Cisplatin, 5-fluorouracil (5-Fu), tamoxifen, and combretastatin A-4 (CA-4) were used as positive control. The results expressed as IC50 (µM) were summarised in . Among the series, compounds 5a, 5c, 5e, and 8 b showed potent anticancer activity against both MCF-7 and A549 cell lines with IC50 between 0.33 ± 0.01 and 5.34 ± 0.31 µM. All these compounds were more potent than cisplatin (IC50 = 11.15 ± 0.75 µM), 5-Fu (IC50 = 11.61 ± 0.60 µM), tamoxifen (IC50 = 14.28 ± 0.40 µM), and CA-4 (IC50 = 5.55 ± 0.11 µM) on MCF-7 cancer cell line. In particular, compound 5c with a methoxyl group at the para position of phenyl ring and naphthalen-1-yl at the sulphonamide exhibited the most potent anticancer activity against MCF-7 and A549 cell lines (IC50 = 0.51 ± 0.03 and 0.33 ± 0.01 µM, respectively).
Table 1. In vitro cell growth inhibitory effects of compounds (5a-5e and 8a-8i).
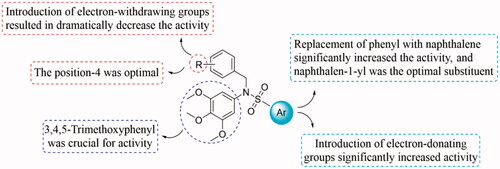
To study the structure-activity relationships (SAR) of this class of compounds, the aryl substituents on sulphonamides were discussed firstly. Based on the antiproliferative activity of compounds 5a-5e, it can be seen that the aryl substituents on sulphonamides affected on antiproliferative activity of this class of compounds. Among these molecules, 5 b with a phenyl ring displayed low antiproliferative activity with IC50 value of > 30.0 µM. Introduction of electron-donating (5a and 5e) groups into the phenyl ring, results in significantly increased the antiproliferative activity. The replacement of phenyl ring with naphthalen-1-yl or naphthalen-2-yl led to compounds 5c and 5d, resulting in significantly increased the antiproliferative activity. In particular, compound 5c with naphthalen-1-yl moiety was found to be the most active compound in this series. These inhibitory results indicate that naphthalen-1-yl group seems to be the optimal substituent on the position.
To explore the effect of the substituents in phenyl ring on the inhibitory activity, various substituents including 3-methoxy, 4-methyl, 3-fluoro, 4-bromo, 4-chloro, 3-bromo, 4-fluoro, 4-cyano, and 2-nitro were introduced into the targeted compounds (8a-8i). Shifting methoxy group to the 3- position (8a) decreased the inhibitory activity. The replacement of the 4-methoxy (5c) with 4-methyl group (8 b) resulted in a slightly decrease of the inhibitory activity. Introduction of electron-withdrawing groups such as 3-fluoro (8c), 4-bromo (8d), 4-chloro (8e), 3-bromo (8f), 4-fluoro (8 g), 4-cyano (8 h), and 2-nitro (8i) into the phenyl ring, resulted in dramatically decrease the inhibitory activity. Compared the inhibitory activity of 5c (4-methoxy) and 8d (4-bromo) with 8a (3-methoxy) and 8f (3-bromo), the results were shown that the substituent group on the 4-position of the phenyl ring is more beneficial to the antiproliferative activity. The detailed SARs were summarised in . The information of SARs provided us a guideline to improve the inhibitory activity in the future structural modification.
Figure 4. Summarised SARs of this class of compounds.
In order to verify the safety profile of this class of compounds, the most potent compound 5c was selected to test its cytotoxicity against human normal liver cell line (LO2). The result was shown that compound 5c exhibited moderate cytotoxic activity against human normal liver cell (LO2) with IC50 value of 12.73 ± 3.26 µM. While, 5c displayed potent anticancer activity against MCF-7 and A549 cell lines with IC50 value of 0.51 ± 0.03 and 0.33 ± 0.01 µM, respectively. Hence, we could conclude that these compounds have good safety for potential application in the treatment of tumour cells.
2.3. Inhibition of tubulin polymerisation
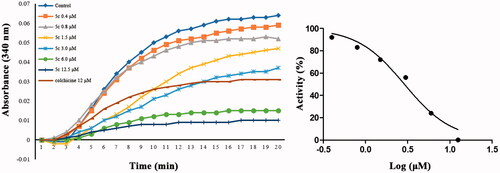
To examine whether tubulin is the target of this class of compounds, the in vitro tubulin polymerisation inhibitory activity of 5c was evaluated using tubulin polymerisation assay Citation28. Meanwhile, tubulin polymerisation inhibitor colchicine was used as positive control. As shown in , compared with the control, the absorbance values at 340 nm of tubulin gradually decreased after incubation with different concentrations of 5c or colchicine. The results shown that 5c exhibited a significant ability to inhibit tubulin polymerisation in a concentration-dependent manner with IC50 values of 2.8 µM, as compared to colchicine (IC50 = 9.3 µM). Besides, 5c and colchicine have similar effects on inhibit tubulin polymerisation, indicating that 5c was a microtubulin-destabilizing agent.
Figure 5. Tubulin polymerisation inhibitory activities of compound 5c and colchicine.
2.4. Cell cycle arrest
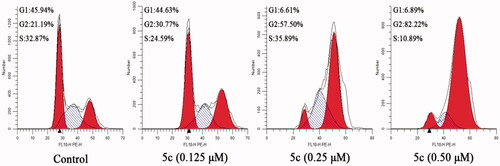
Due to microtubules play an important role in eukaryotic cell division, tubulin polymerisation inhibitors can disrupt regulated cell cycle distribution and block the cell cycle in G2/M phase Citation21,Citation29. Therefore, the effect of compound 5c on the cell cycle of MCF-7 cancer cells was evaluated by using flow cytometry. As shown in , after treated with different concentrations of compound 5c (0.125 µM, 0.25 µM or 0.5 µM), the G2/M population in MCF-7 cancer cells increased from 21.19% (control) to 30.77% (0.125 µM), 57.50% (0.25 µM), and 82.22% (0.5 µM), respectively. The results indicate that compound 5c can arrest cell cycle at G2/M phase in a dose-dependent manner.
Figure 6. Effect of compound 5c on cell cycle arrest in MCF-7 cells.
2.5. Cell apoptosis
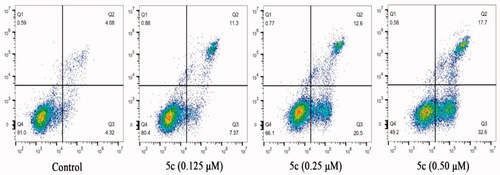
Since many literatures have reported that tubulin polymerisation inhibitors are able to induce cellular apoptosis Citation22,Citation30, the Annexin V-FITC/PI assay was carried out to examine the influence of compound 5c on cell apoptosis in MCF-7 cancer cells. As shown in , when the cells were treatment with compound 5c in the concentration of 0.125 µM, 0.25 µM, or 0.5 µM, the total numbers of early (the lower right quadrant) and late apoptotic cells (the upper right quadrant) were 18.67%, 33.1% and 50.3%, respectively, whereas that of control was only 8.4%. These results indicated that compound 5c effectively induced cell apoptosis in MCF-7 cells in a dose-dependent manner.
Figure 7. Effect of compound 5c on cell apoptosis in MCF-7 cells.
2.6. Molecular docking
To elucidate the binding mode of this class of compounds, molecular docking simulations of compound 5c with tubulin were performed. Colchicine was first docked into the colchicine binding site of tubulin. The result was shown that co-crystallized conformation of colchicine was reproduced approximately (RMSD: 1.10 Å), indicating that this protocol of molecular docking is credible. Then, the theoretical binding mode between 5c and tubulin was investigated, and the estimated binding energy was −9.6 kcal·mol−1. As shown in , Compound 5c adopted an “Y-shaped” conformation in the colchicine pocket of tubulin. Compound 5c located at the hydrophobic pocket, surrounded by the residues A/Ala-180, A/Val-181, B/Leu-248, B/Ala-250, B/Leu-255, B/Ala-316, B/Val-318, and B/Ala-354, forming a strong hydrophobic binding. Detailed analysis showed that the phenyl and naphthyl groups of 5c formed cation-π interactions with the residues Lys-254 and Lys-352, respectively. All these interactions helped 5c to anchor in the colchicine binding site of tubulin.
Figure 8. Compound 5c was docked to the binding pocket of the tubulin (α: green; β: cyan). (A) Overall structure of tubulin with 5c. (B) Binding pose of 5c at colchicine binding site. (C) Superimposed pose of 5c (rose red) and colchicine (yellow-orange) in the binding site. (D) Binding pose of 5c in the surface of colchicine binding pocket.
2.7. Molecular dynamics (MD) simulations
To explore the potential binding mode between 5c and tubulin, molecular docking and molecular dynamics simulations were performed using the AutoDock vina 1.1.2 and Amber12 software package. The preferential binding mechanism of tubulin with 5c was determined by 30-ns molecular dynamics simulations based on the docking results. To explore the dynamic stability of the models and to ensure the rationality of the sampling strategy, the root-mean-square deviation (RMSD) value of the protein backbone based on the starting structure along the simulation time was calculated and plotted in . The result was shown that the protein structure of the system was stabilised during the simulation.
Figure 9. Molecular docking and molecular dynamics refinement of compound 5c with tubulin. (A) The root-mean-square deviation (RMSD) of all the atoms of tubulin-5c complex with respect to its initial structure as function of time. (B) Molecular dynamics results of tubulin-5c complex.
The theoretical binding mode between 5c and tubulin was shown in . Compound 5c adopted a compact conformation in the pocket of tubulin. The naphthyl group of 5c located at the hydrophobic pocket, surrounded by the residues A/Ala-180, A/Val-181, B/Val-238, B/Leu-248, B/Ala-250, B/Leu-255, B/Ala-316, B/Ala-317, B/Val-318 and B/Ala-354, forming a strong hydrophobic binding. Detailed analysis showed that the phenyl group of 5c formed a cation-π interaction with the residue Lys-254. It was shown that the residue Lys-254 (bond length: 2.7 Å) formed a hydrogen bond with 5c, which was the main interaction between 5c and tubulin. All in all, the above molecular dynamics simulation gave us rational explanation of the interaction between 5c and tubulin, which provided valuable information for further development of tubulin polymerisation inhibitors.
3. Conclusion
In summary, a new series of sulphonamide derivatives bearing naphthalene moiety have been synthesised and and characterised by 1H NMR, 13 C NMR, HRMS, and elemental analysis. All of the title compounds were screened for antiproliferative activity against human breast cancer cells (MCF-7) and human non-small cell lung carcinoma cells (A549) by using CCK-8 method. Among all synthesised compounds, compound 5c with naphthalen-1-yl moiety exhibited the most potent antiproliferative activity against MCF-7 and A549 cell line, with IC50 values of 0.51 ± 0.03 µM and 0.33 ± 0.01 µM, respectively. SAR studies suggested that the naphthalen-1-yl and 4-methoxybenzyl at the sulphonamide played an important role for the potent antiproliferative activity. Tubulin polymerisation assay revealed that compound was a microtubulin-destabilizing agent with IC50 value of 2.8 µM. Further mechanism evaluation demonstrated that 5c can significantly arrest the cell cycle at G2/M phase and induce apoptosis in MCF-7 cancer cells. Additionally, molecular modelling results showed that 5c binds well to the colchicine-binding site of tubulin. Hence, these results suggest that 5c could be used as a promising lead compound for further investigation in anticancer drug development.
4. Experimental
4.1. 4-Methoxy-N-(3,4,5-trimethoxyphenyl)benzamide (3)
To a solution of 3,4,5-trimethoxyaniline 1 (10 mmol) and Et3N (20 mmol) in THF (50 ml) were added 4-methoxybenzoyl chloride 2 (10 mmol) and the reacting mixture was stirred at room temperature for 4 h. Then, the solvent was removed under reduced pressure, and water was added to the reaction mixture and extracted 3 times with ethyl acetate. The combined organic layers were dried, filtered, and concentrated. The residue was purified by chromatography on silica gel with EtOAc/petroleum ether = 1:1 to give compound 3 as white solid (yield 88%).
4.2. 3,4,5-Trimethoxy-N-(4-methoxybenzyl)aniline (4)
A mixture of LiAlH4 (2 mmol) in anhydrous THF (10 ml) was stirred in an ice bath for 10 min. Then, a solution of compound 3 (1 mmol) in 10 ml of THF was added dropwise to the mixture. After completion of dropwise addition, the mixture was stirred at 0 °C for 30 min then refluxed at 70–80 °C for 2 h. After completion of the reaction, the solvent was evaporated and the organic product was extracted with ethyl acetate. The combined organic layers were dried over sodium sulphate and evaporated to afford black oil, which was used directly for the next step without further purification.
4.3. General procedure for the synthesis of 5a-5e
To a solution of compound 4 (2 mmol), DMAP (0.2 mmol), and Et3N (2 mmol) in THF (50 ml) was added commercially available aryl sulphonyl chloride (2 mmol) and the reacting mixture was stirred at room temperature for 12 h. The solvent was evaporated and water was added to the reaction mixture. The organic material was extracted with ethyl acetate, and the organic layer was dried over Na2SO4 and concentrated under vacuum. The residue was purified by chromatography to give the title product 5a-5e.
4.4. N-(3,4,5-Trimethoxyphenyl)naphthalene-1-sulphonamide (7)
To a solution of 3,4,5-trimethoxyaniline 1 (10 mmol) and Et3N (10 mmol) in CH2Cl2 (100 ml) was added naphthalene-1-sulphonyl chloride 6 (10 mmol) and the mixture was stirred at room temperature for 6 h. Then, the solution was diluted with CH2Cl2 and washed with water. After the concentration of the solution, the residue was purified by silica gel column chromatography to give compound 7 as white solid (yield = 43%).
4.5. General procedure for the synthesis of 8a-8i
A mixture of 7 (1.0 mmol), K2CO3 (2.0 mmol), KI (1.0 mmol), and commercially available benzyl halide (1.0 mmol) in acetone (10 ml) was stirred at reflux for 5 h. After completion of the reaction, the mixture was concentrated under reduced pressure and the residue was purified by chromatography to give the title compounds 8a-8i.
Supplemental Material
Download PDF (2.1 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004;4:253–65.
- Akhmanova A, Steinmetz MO. Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol 2015;16:711–26.
- Janke C. The tubulin code: molecular components, readout mechanisms, and functions. J Cell Biol 2014;206:461–72.
- Pasquier E, André N, Braguer D. Targeting microtubules to inhibit angiogenesis and disrupt tumour vasculature: implications for cancer treatment. Curr Cancer Drug Targets 2007;7:566–81.
- Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 2010;9:790–803.
- Haider K, Rahaman S, Yar MS, Kamal A. Tubulin inhibitors as novel anticancer agents: an overview on patents (2013-2018). Expert Opin Ther Pat 2019;29:623–41.
- Stanton RA, Gernert KM, Nettles JH, Aneja R. Drugs that target dynamic microtubules: a new molecular perspective. Med Res Rev 2011;31:443–81.
- Mukhtar E, Adhami VM, Mukhtar H. Targeting microtubules by natural agents for cancer therapy. Mol Cancer Ther 2014;13:275–84.
- Attard G, Greystoke A, Kaye S, De Bono J. Update on tubulin-binding agents. Pathol Biol 2006;54:72–84.
- Khan FA, Mushtaq S, Naz S, et al. Sulfonamides as potential bioactive scaffolds. Curr Org Chem 2018;22:818–30.
- Yoshino H, Ueda N, Niijima J, et al. Novel sulfonamides as potential, systemically active antitumor agents. J Med Chem 1992;35:2496–7.
- Tanaka H, Ohshima N, Ikenoya M, et al. Hmn-176, an active metabolite of the synthetic antitumor agent hmn-214, restores chemosensitivity to multidrug-resistant cells by targeting the transcription factor nf-y. Cancer Res 2003;63:6942–7.
- Lee HY, Pan SL, Su MC, et al. Furanylazaindoles: potent anticancer agents in vitro and in vivo. J Med Chem 2013;56:8008–18.
- Chang JY, Hsieh HP, Chang CY, et al. 7-aroyl-aminoindoline-1-sulfonamides as a novel class of potent antitubulin agents. J Med Chem 2006;49:6656–9.
- Jian S, Gao QL, Wu BW, et al. Novel tertiary sulfonamide derivatives containing benzimidazole moiety as potent anti-gastric cancer agents: design, synthesis and sar studies. Eur J Med Chem 2019;183:111731.
- Li L, Jiang SB, Li XX, et al. Recent advances in trimethoxyphenyl (tmp) based tubulin inhibitors targeting the colchicine binding site. Eur J Med Chem 2018;151:482–94.
- Rischin D, Bibby DC, Chong G, et al. Clinical, pharmacodynamic, and pharmacokinetic evaluation of bnc105p: a phase i trial of a novel vascular disrupting agent and inhibitor of cancer cell proliferation. Clin Cancer Res 2011;17:5152–60.
- Kirwan IG, Loadman PM, Swaine DJ, et al. Comparative preclinical pharmacokinetic and metabolic studies of the combretastatin prodrugs combretastatin a4 phosphate and a1 phosphate. Clin Cancer Res 2004;10:1446–53.
- Lee J, Bae S, Lee SH, et al. Discovery of a potent tubulin polymerization inhibitor: synthesis and evaluation of water-soluble prodrugs of benzophenone analog. Bioorg Med Chem Lett 2010;20:6327–30.
- von Pawel J, DISRUPT Investigators, Gorbounova V, Reck M, et al. Disrupt: a randomised phase 2 trial of ombrabulin (ave8062) plus a taxane-platinum regimen as first-line therapy for metastatic non-small cell lung cancer. Lung Cancer 2014;85:224–9.
- Wang G, Liu W, Gong Z, et al. Synthesis, biological evaluation, and molecular modelling of new naphthalene-chalcone derivatives as potential anticancer agents on mcf-7 breast cancer cells by targeting tubulin colchicine binding site. J Enzyme Inhib Med Chem 2020;35:139–44.
- Wang G, Liu W, Gong Z, et al. Design, synthesis, biological evaluation and molecular docking studies of new chalcone derivatives containing diaryl ether moiety as potential anticancer agents and tubulin polymerization inhibitors. Bioorg Chem 2020;95:103565.
- Wang G, Liu W, Peng Z, et al. Design, synthesis, molecular modeling, and biological evaluation of pyrazole-naphthalene derivatives as potential anticancer agents on mcf-7 breast cancer cells by inhibiting tubulin polymerization. Bioorg Chem 2020;103:104141.
- Wang G, Liu W, Tang J, et al. Design, synthesis, and anticancer evaluation of benzophenone derivatives bearing naphthalene moiety as novel tubulin polymerization inhibitors. Bioorg Chem 2020;104:104265.
- Viegas-Junior C, Danuello A, VdS B, et al. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr Med Chem 2007;14:1829–52.
- Wang XD, Wei W, Wang PF, et al. Novel 3-arylfuran-2(5h)-one-fluoroquinolone hybrid: design, synthesis and evaluation as antibacterial agent. Bioorg Med Chem 2014;22:3620–8.
- Xiao ZP, Peng ZY, Dong JJ, et al. Synthesis, structure-activity relationship analysis and kinetics study of reductive derivatives of flavonoids as helicobacter pylori urease inhibitors. Eur J Med Chem 2013;63:685–95.
- Wang G, Qiu J, Xiao X, et al. Synthesis, biological evaluation and molecular docking studies of a new series of chalcones containing naphthalene moiety as anticancer agents. Bioorg Chem 2018;76:249–57.
- Ji YT, Liu YN, Liu ZP. Tubulin colchicine binding site inhibitors as vascular disrupting agents in clinical developments. Curr Med Chem 2015;22:1348–60.
- Mustafa M, Anwar S, Elgamal F, et al. Potent combretastatin a-4 analogs containing 1,2,4-triazole: synthesis, antiproliferative, anti-tubulin activity, and docking study. Eur J Med Chem 2019;183:111697.