
Abstract
In the current work, a new set of carbohydrazide linked benzofuran-isatin conjugates (5a–e and 7a–i) was designed and synthesised. The anticancer activity for compounds (5b–d, 7a, 7b, 7d and 7g) was measured against NCI-55 human cancer cell lines. Compound 5d was the most efficient, and thus subjected to the five-dose screen where it showed excellent broad activity against almost all tested cancer subpanels. Furthermore, all conjugates (5a–e and 7a–i) showed a good anti-proliferative activity towards colorectal cancer SW-620 and HT-29 cell lines, with an excellent inhibitory effect for compounds 5a and 5d (IC50 = 8.7 and 9.4 µM (5a), and 6.5 and 9.8 µM for (5d), respectively). Both compounds displayed selective cytotoxicity with good safety profile. In addition, both compounds provoked apoptosis in a dose dependent manner in SW-620 cells. Also, they significantly inhibited the anti-apoptotic Bcl2 protein expression and increased the cleaved PARP level that resulted in SW-620 cells apoptosis.
Graphical Abstract
1. Introduction
Cancer, a large family of diseases, is characterised by fast and uncontrolled cell division and differentiation mechanisms and has the potential to spread to or invade other body partsCitation1. For several decades, cancer is considered one of the major world public health problems, and it remains a serious reason of the death of human beings all over the worldCitation2. Despite the presence of a variety of cancer treatment strategies, the majority of which induces non-selective cell death by targeting the DNA synthesisCitation3–6 and/or the replication machineryCitation7–10. These early strategies are accompanied by severe side effects due to the unspecific cytotoxicity towards the cancer cells in addition to the resistance developed against themCitation4. Therefore, the development of safe and effective novel anticancer agents with increased selective treatment strategies towards cancer cells has received more attention and still ongoing active searchCitation11,Citation12.
Recent strategies for anticancer development are to target specific biomarkers required for cancer cells division and/or induction of cell apoptosis such as deregulated, mutated, or over expressed proteinsCitation13 and thus, affect cancer cells selectively with minimum influences on normal cellsCitation14. Among these targets are the anti-apoptotic protein Bcl2 and Poly ADP-ribose polymerase (PARP). In this regard, several reports stated that numerous of cancer cells are characterised by anti-apoptotic proteins (Bcl2) over-expression, which could lead to prevention of cell apoptosis as well as development of drug resistanceCitation15,Citation16. On the other hand, PARP is a family of proteins involved in numerous cellular functions such as DNA repair and genomic stabilityCitation17–19 and also, PARP was reported to activate programmed cell death, through cleavage into PAR (Poly ADP-ribose), which motivates mitochondria to produce apoptosis inducing factorsCitation20. Thus, the development of compounds that inhibit the antiapoptotic Bcl2 proteins and/or potentiate the cleavage of PARP could be a promising approach to identify new anticancer therapies.
Heterocyclic compounds in particular oxygen containing heterocycles represent an important class of compounds possessing interesting pharmacological and biological activitiesCitation21–23. Benzofuran nucleus, as a key functional scaffold, represents a basic structure in a diversity of biologically active synthetic and natural productsCitation24–26, with broad range of desirable activities including; anti-Alzheimer’sCitation27, antibacterialCitation28, anti-tubercularCitation29, antioxidantCitation30, anti-inflammatoryCitation31, as well as antitumor activitiesCitation32. Benzofuran derivatives exert their antiproliferative activity with diversified mechanisms such as inhibition of tubulin polymerisationCitation33,Citation34, HIF-1Citation35, Aurora B kinaseCitation36 and VEGFR-2 activityCitation37. Furthermore, some benzofurans mediate their antiproliferative activity via apoptosis induction in various human cancer cell linesCitation38–40. In addition, benzofuran-based conjugates were largely studied and were found to exert significant anticancer activity, such as conjugation of benzofuran with pyrazoleCitation41, indoleCitation42 and othersCitation43,Citation44.
On the other hand, isatin is identified as a privileged nucleus that included in many pharmacologically active small molecules, such as antiviralCitation45, antimicrobialCitation46, anticonvulsantCitation47, CNS-actingCitation48, as well as anticancerCitation49,Citation50 agents. Over the last few years, hybridisation of isatin nucleus with different heterocycles has been reported as a successful approach to develop efficient antitumor agents towards different cancer types through diverse enzymatic and cellular mechanismsCitation49,Citation50. To name just a few, isatin-phthalazine (compound I)Citation51, isatin-thiazolo[3,2-a]benzimidazole (compound II)Citation52, isatin-thiazolidinone (compound III)Citation53, isatin-indole (compound IV)Citation54 and isatin-quinazoline (compound V)Citation55 conjugates were reported to possess promising anticancer activities ().
Figure 1. Structures of some reported isatin-bearing conjugates (I–V), as well as structures for target benzofuran-isatin conjugates (5a–e and 7a–i).
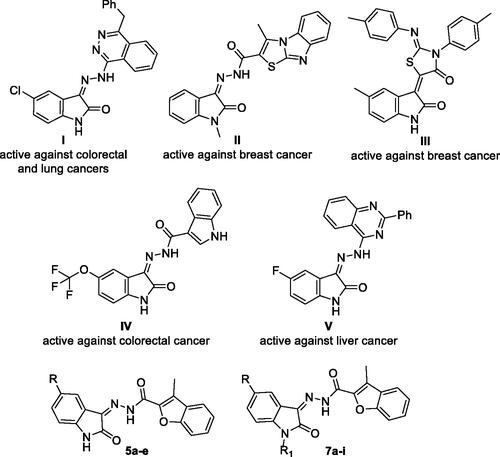
Encouraged by the aforementioned findings and considering the need to develop safe and effective novel anticancer agents, a new attempt to study the significance of utilisation of heterocycles hybridisation approach to furnish efficient anti-proliferative activity was reported herein. A novel series of benzofuran-isatin conjugates (5a–e and 7a–i, ) linked by a carbohydrazide group, was designed and synthesised. The new compounds were screened for their potential anticancer activity following NCI, USA protocol against fifty-five different cell lines under nine different cancer panels. In addition, the cytotoxic effect of these conjugates against SW-620 and HT-29 colorectal cancer cell lines was investigated and their ability to induce cell apoptosis was examined. Furthermore, the level of the mitochondrial antiapoptotic protein Bcl2 and the level of cleaved PARP in both SW-620 and HT-29 colorectal cancer cell lines were also determined.
2. Results and discussion
2.1. Chemistry
The adopted synthetic strategy to develop the target N-unsubstituted 3-methyl-N′-(oxoindolin-3-ylidene)benzofuran-2-carbohydrazide derivatives 5a–e was outlined in Scheme 1.
Scheme 1. Synthesis of target conjugates 5a–e; (i) Anhydrous CH3CN/potassium carbonate/reflux 8 h, (ii) Hydrazine hydrate/methanol/reflux 4 h, (iii) Ethanol/drops glacial acetic acid (Cat.)/reflux 3–6 h.
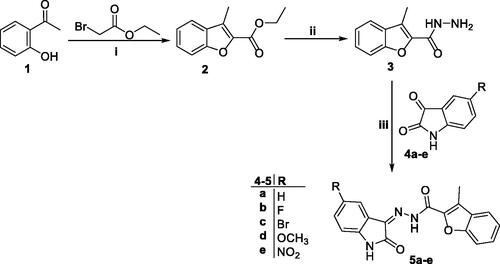
Key starting ester 3-methylbenzofuran-2-carboxylate 2, was prepared in 85% yield through cyclisation of 1–(2-hydroxyphenyl)ethan-1-one 1 and ethyl bromoacetate in anhydrous acetonitrile with the presence of potassium carbonate. Thereafter, heating of ester derivative 2 with hydrazine hydrate in methanol afforded the corresponding key intermediate 3-methylbenzofuran-2-carbohydrazide 3. Finally, carbohydrazide 3 was condensed with different indoline-2,3-dione derivatives 4a–e, through heating under reflux temperature in absolute ethyl alcohol and few drops of acetic acid, to give the desired benzofuran-based compounds 5a–e, respectively, in 72–89%yield.
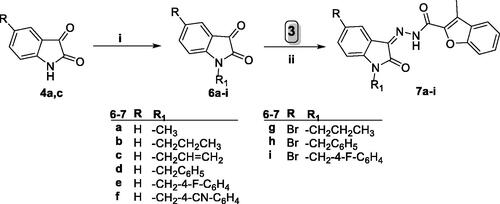
On the other hand, Scheme 2 illustrated the synthetic pathway utilised to synthesise N-substituted 3-methyl-N′-(oxoindolin-3-ylidene)benzofuran-2-carbohydrazide derivatives 7a–i. In this scheme, alkylation of indoline-2,3-diones 4a and 4c was accomplished via heating with different alkyl bromide or benzyl bromide derivatives in anhydrous acetonitrile to produce N-substituted indoline-2,3-dione derivatives 6a–i. Then indoline-2,3-diones 6a–i were condensed with the key intermediate carbohydrazide 3 producing target benzofurans 7a–i, respectively, in 75–87% yield.
Scheme 2. Synthesis of target benzofurans 7a–i; (i) (R-Br or Ar-Br)/Acetonitrile/KI (Cat.)/potassium carbonate/reflux 3 h, (ii) Ethanol absolute/drops glacial acetic acid (Cat.)/reflux 3–6 h.
Structures of the newly prepared benzofuran-based derivatives 5a–e and 7a–i were verified based on spectral and elemental analyses. 1H NMR spectra of 5a–e and 7a–i revealed the presence of two singlet peaks for the protons of C-3 CH3 of benzofuran ring and NH of the hydrazide linker at range δ (2.52–2.72) and (11.37–14.10) ppm, respectively. Moreover, structure of compounds 5a–e was confirmed via presence of another singlet D2O exchangeable signal attributable to the proton of NH for isatin moieties at δ 10.91–11.98 ppm. In addition, 1H NMR spectra of N-benzyl bearing derivatives 7d–f, 7h and 7i displayed the characteristic singlet signal of the benzylic protons at δ 4.98–5.07 ppm, while spectra for hybrids 7a, 7b and 7g revealed the presence of the aliphatic protons corresponding to the N-substituents in these derivatives at δ (3.28 ppm), (0.93, 1.66 and 3.76 ppm) and (0.97, 1.69 and 3.80 ppm), respectively.
On the other hand, 13 C NMR spectra for the novel compounds 5a–e and 7a–i showed one signal belonging to the carbon of CH3 of benzofuran ring at δ 8.12–9.49 ppm, also, they showed two signals belonging to the carbon of C = O functionalities for both the hydrazide linker and isatin moiety at range δ (161.15–163.62) and (164.08–167.02) ppm, respectively. In addition, the existence of benzylic carbon in N-benzyl bearing derivatives 7d–f, 7h and 7i was confirmed by a signal at δ 42.14–46.03 ppm, whereas, the carbons of propyl moiety in compounds 7b and 7g appeared as signals at range δ (11.68–13.00), (20.92–22.98) and (41.30–48.74) ppm.
2.2. Biological evaluation
2.2.1. Nci screening of anticancer activity
In the present investigation, the chemical structures for the novel benzofuran-isatin conjugates were presented to the Developmental Therapeutics Program at the National Cancer Institute (NCI), USA. Seven conjugates (5b–d,7a, 7b, 7d and 7g) were selected, according to NCI’s-DTP selection guidelinesCitation56, for evaluating their potential in vitro anticancer activity against a panel of fifty-five human cancer cell lines representing nine tumour panels according to the NCI, Bethesda, Drug Evaluation Branch protocolCitation57,Citation58.
2.2.1.1. Preliminary single high dose screening at 10 μM concentration
Firstly, the seven selected conjugates (5b–d,7a, 7b, 7d and 7g) were screened at a dose of 10 μM for their antiproliferative activity against a panel of fifty-five cancer cell lines. The mean percent growth inhibition values (GI%) for conjugates 5b–d,7a, 7b, 7d and 7g against NCI-55 cancer cell lines were displayed in (, ). The primary assay data analysis revealed that the new benzofuran-isatin hybrids showed weak to moderate inhibitory activity some of the subpanel cancer cell lines except for compound 5d that possessed excellent activity against nearly all the cancer cell lines. Although compound 5b, 7a, 7b, 7d and 7g proved inactive against most of the subpanels cancer cell lines with mean GI% = 0.75%, 0.09%, 5.18%, 6.01%, and −0.8%, respectively, they showed selective moderate anticancer activity against certain cell lines such as Ovarian-IGROV1, Non-small cell lung-EKVX, Renal-UO-31 and Breast/MCF7 cancer cell lines with GI% range 17–53% ().
Figure 2. Mean % growth inhibition of compounds 5b–d,7a, 7b, 7d and 7g against NCI-55 cancer cell line panel.
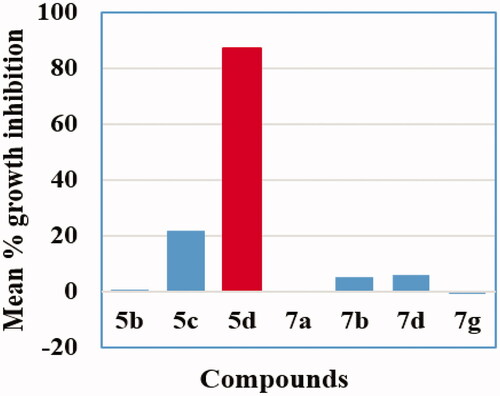
Table 1. In vitro Anticancer screening results of compounds 5b–d,7a, 7b, 7d and 7g against fifty-five human tumour cell lines with single dose assay (10−5 M concentration). Data was provided as cell growth inhibition percentage.
In particular, compound 5d was the most efficient anti-proliferative agent and exhibited excellent activity against almost all subpanel cancer cell lines with mean growth inhibitory activity of 87.33%. Remarkably, compound 5d exerted excellent growth inhibition properties against Non-small cell lung cancer (NCI-H23), CNS cancer (SF-295, U251), Melanoma (LOX IMVI, SK-MEL-28), Ovarian cancer (IGROV1), Prostate cancer (DU-145) and Breast cancer (MDA-MB-468) cell lines with GI% of 89.35, 94.95, 97.95, 83.71, 84.16, 93.67, 82.38 and 80.10%, respectively (). In addition, conjugate 5d showed good potency with GI% equals or greater than 60% towards Non-small cell lung cancer (EKVX), Colon cancer (COLO 205, HT-29 and SW-620), Melanoma (M14, MDA-MB-435 and UACC-62), Ovarian cancer (OVCAR-8), CNS cancer (SF-539), Renal cancer (SN12C), Breast cancer (MCF7, MDA-MB-231/ATCC and HS 578 T) and Prostate cancer (PC-3) cell lines with GI% of 62.73, 77.24, 72.67, 64.85, 71.32, 61.04, 65.68, 62.23, 62.58, 65.13, 77.14, 76.19, 74.50and 78.23% respectivly ().
It is worthy to mention that 5d exhibited a lethal cytotoxic impact with GI% >100 against Non-small cell lung cancer (HOP-62, A549/ATCC, HOP-92 and NCI-H460), Colon cancer (HCT-116), Melanoma (MALME-3M), Ovarian cancer (OVCAR-4 and OVCAR-3), Renal cancer (CAKI-1, 786–0, RXF 393, ACHN, TK-10 and UO-31) and Breast cancer (T-47D) cells with GI% values equals 124.09, 123.59, 143.99, 122.83, 128.55, 129.46, 152.85, 165.51, 151.66, 166.85, 136.15, 150.93, 193.95, 155.33 and 139.16%, respectively ().
On the other hand, compound 5c showed moderate to good activity against some cell lines with mean GI% = 21.99%. The best results of compound 5c was against cancer cell lines Non-small cell lung-HOP-62 (GI% = 41.41%), Non-small cell lung-NCI-H460 (GI% = 42.60%), Renal-UO-31 (GI% = 43.42%), Ovarian-IGROV1 (GI% = 44.59%), Breast-HS-578T (GI% = 46.27%), CNS-SF-539 (GI% = 57.88%) and Melanoma-MALME-3M (GI% = 64.93%) ().
2.2.1.2. In vitro 5 dose full NCI-55 cell panel screening.
The preliminary screening results showed that conjugate 5d (NSC: D-819833/1) was the most potent compound in the present study, and displayed effectiveness towards various cell lines represent numerous tumour subpanels (). Accordingly, 5d was promoted to the five-dose (0.01–100 µM) screening assay. Accordingly, three main response parameters (GI50, TGI and LC50) towards each of the examined cancer cell line were calculated for hybrid 5d and displayed in . Where, GI50 values represents molar concentration which produces 50% inhibitory effect in the net cell growth; TGI (cytostatic activity) is the molar concentration with total growth inhibition and LC50 is the cytotoxicity parameter that reflects the molar concentration that results in 50% net cell death. In addition, the mean graph midpoints (MG-MID), representing the GI50 average for the individual subpanels as well as the full panel cell lines were calculated giving an average potency parameter for the examined compound 5d, (). Furthermore, by dividing the full panel MID by their individual subpanel MID, the selectivity index of compound 5d was calculated and was used to measure the selectivity of 5d towards different cancer cell subpanels.
Table 2. NCI in vitro screening results (GI50, TGI, and LC50 (μM) of 5d (NSC: D-819833/1) in the five-dose test.
Table 3. Median growth inhibitory concentrationsa (GI50, µM) of in-vitro cancer cell lines subpanel for compound 5d.
Results displayed in , revealed that conjugate 5d exhibited powerful anti-proliferative activity at a single-digit micromolar level towards all the examined human cancer cell subpanels with GI50 values range: 1.25 − 6.07 µM, except for Melanoma HL60(TB) cell line (more than100 µM). Moreover, regarding the cytostatic activity, hybrid 5d exhibited excellent cytostatic activity with TGI values range 2.75–7.72 µM against numerous cell lines including NSCLC (HOP-62, NCI-H23 and HOP-92), CNS Cancer (SF-295, SF-539, SNB-75 and U251), Melanoma (MALME-3Mand SK-MEL-2), Ovarian Cancer (OVCAR-8), Renal Cancer (786–0, A498, CAKI-1, RXF 393 andTK-10) and Breast Cancer (MDA-MB-231/ATCC, HS 578 T and MDA-MB-468). On the other hand, while, compound 5d showed weak to moderate cytostatic activity towards CNS Cancer (SF-268 and SNB-19), and Breast Cancer (BT-549) with TGI = 56.8, 16.2 and 33.7 µM, respectively, it proved to have no cytostatic impact (TGI >100 µM) against entire Leukaemia, Colon cancer and Prostate Cancer and the remaining examined cancer cell lines (). Furthermore, compound 5d as revealed by the results could be considered as a non-lethal agent that exhibited LC50 values more than 100 µM for the all of cancer cell lines herein examined, except for three cancer cell lines; Non-Small Cell Lung Cancer (HOP-62), CNS Cancer (SNB-75) and Melanoma (MASK-MEL-2) which possessed a lethal effect of IC50 = 8.44, 6.04 and 54.7, respectively ().
On the other hand, as shown in , all tested subpanels were sensitive to compound 5d with MG-MID spinning between 1.92 and 3.18 µM and the most susceptible subpanels were Colon Cancer and Renal Cancer that exhibited MG-MID = 1.92 and 1.98 µM, respectively. Furthermore, it is well known that compounds with selectivity index between 3 and 6 are considered to be of a moderate selectivity, ratios more than six indicated high selectivity towards the corresponding cell line, while compounds not meeting either of these values are considered as non-selectiveCitation59. Therefore, as displayed in the , the calculated selectivity index for compound 5d ranged from 0.78 to 1.30 indicated that conjugate 5d has non-selective, broad spectrum antiproliferative activity against all tested subpanels cancer cells.
2.2.2. In vitro anti-cancer activity against SW-620 and HT-29colorectal cancer cell lines
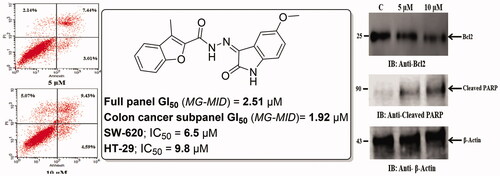
In the present investigation a new set of benzofuran–isatin hybrids (5a–e and 7a–i) was synthesised to be evaluated for their potential anticancer activity towards two human colorectal cancer cell lines, SW-620 and HT-29. The anticancer activity of the new conjugates was assessed using MTT assayCitation60, and the results were shown in . The most active compound in the NCI assay (5d), in addition to another one from untested compounds by NCI (5a), were selected to explore their activity. Both, SW-620 and HT-29 cells were treated with 10 µM of each compound for 24 h and the percent cell viability was calculated using MTT assay. Regarding impact of the target conjugates towards SW-620 cancer cells viability, compound 5d exhibited about 52% inhibition, whereas, compound 5a showed 46% inhibition. On the other hand, the results showed that seven compounds (5a, 5d, 7b, 7c, 7e, 7h and 7i) showed >50% inhibition of HT-29 cancer cells viability ().
Figure 3. Effect of benzofuran–isatin conjugates (5a–e and 7a–i) on the cell viability. (A) SW-620 with hybrids 5a–e and 7a–i, (B) HT-29 with hybrids 5a–e and 7a–i.
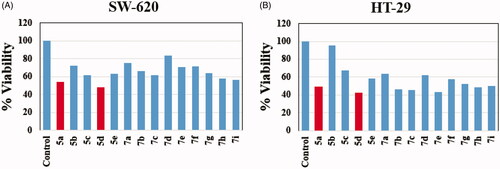
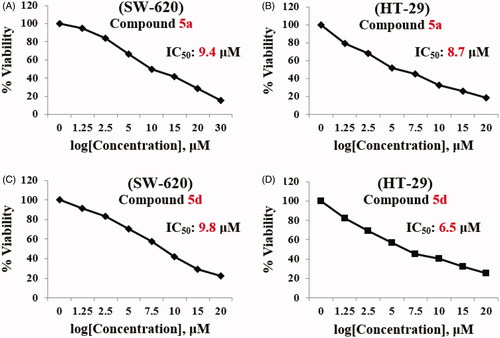
The results revealed that compounds 5a and 5d exhibited promising cytotoxic activity for both cell lines. For this reason, compounds 5a and 5d were pursued for further studies. Starting with determination of IC50s and cytotoxic selectivity studies. Serial concentrations of compounds 5a and 5d were used to examine their impact on cell viability using MTT protocol. Results of concentration vs percent viability were charted, and the IC50 was calculated for SW-620 and HT-29 cell lines using Graph Pad prism 8 (). Compound 5a was found to have IC50 = 9.4 µM and 8.7 µM against SW-620 and HT-29 cell lines, respectively. In addition, the IC50 for compound 5d equals 9.8 µM and 6.5 µM against SW-620 and HT-29 cell lines, respectively, compared to IC50 of Irinotecan, a reference drug, which was found to be 1.0 µM against SW-620 cell line and 6.18 µM against HT-29 cell line ().
Figure 4. IC50 of Compound 5a and 5d. (A) SW-620 with compound 5a, (B) HT-29 with 5a, (C) SW-620 with compound 5d, and (D) HT-29 with 5d.
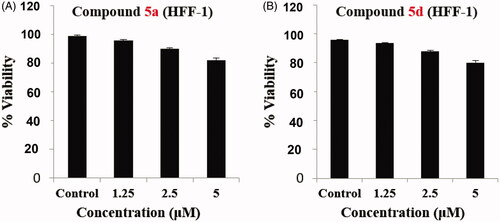
Furthermore, selective cytotoxicity of compounds 5a and 5d was studied on human skin fibroblast (HFF-1) normal cells. Both conjugates were found to possess a little effect on fibroblast normal cell viability (). These results revealed that compounds 5a and 5d possessed a selective cytotoxicity against SW-620 and HT-29 cancer cell lines with non-significant effect on normal fibroblast cells.
Figure 5. Impact of 5a and 5d on normal HFF-1 fibroblast cells, upon incubation for 24 h. (A) compound 5a and (B) compound 5d.
2.2.2. Annexin V-FITC/propidium iodide apoptosis assay
Further investigation for compounds 5a and 5d concerning their potential role of apoptosis induction, using Annexin V-FITC/PI double staining assayCitation61, was performed to evaluate their impact on both early and late apoptosis percentages in SW-620 cancer cell lines (). The assay findings showed that compounds 5a and 5d resulted in a dose dependent induction of apoptosis for SW-620 cancer cells. As shown, compound 5a induced approximately 1.7-folds and 3.8-folds total increase in apoptosis at concentration of 5 µM and 10 µM, respectively, in comparison to the control untreated SW-620 cell line ().
Figure 6. (A) AnnexinV/PI apoptosis assay for compound 5a. Tow concentrations (5 and 10 µM) of compound 5a, in addition untreated plate as a control were used to test the apoptotic effect by using Annexin V/PI in SW-620 cell line. Cells were treated with the compound 5a for 24 h. (B) AnnexinV/PI apoptosis assay for compound 5d. Tow concentrations (5 and 10 µM) of compound 5d, in addition untreated plate as a control were used to test the apoptotic effect by using Annexin V/PI in SW-620 cell line. Cells were treated with the compound 5d for 24 h.
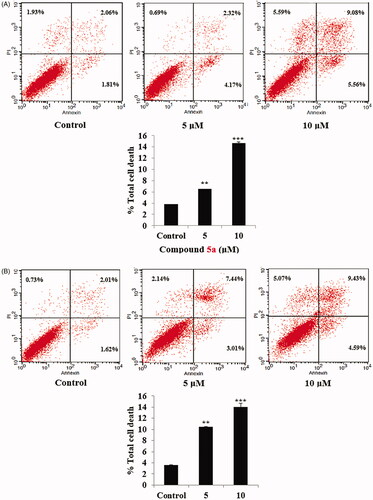
Similarly, compound 5d, at concentration of 5 µM and 10 µM approximately induced 2.9-folds and 3.8-folds total increase in apoptosis, respectively, when incubated with SW-620 cell line, compared to the untreated cells (). Encouraged by these results compounds 5a and 5d were further investigated for their effect on the anti-apoptotic mitochondrial protein Bcl2 and their effect on the level of cleaved PARP in SW-620 colorectal cancer cell line.
2.2.3. Effect of compounds 5a and 5d on the anti-apoptotic markers Bcl2 and the level of cleaved PARP
To further examine the possible mechanism of apoptosis, the effect of compounds 5a and 5d on certain apoptosis-related proteins was studied. Bcl2 protein as a critical component of the mitochondrial apoptotic pathway is reported to be overexpressed in numerous tumours causing survival of cancer cellCitation62. In addition, it was reported that caspase activation during apoptosis leads to proteolytic cleavage of several cellular substrates participating in DNA reparation including [poly (ADP-ribose) polymerase]Citation63. Therefore, the impact of compounds 5a and 5d on the anti-apoptotic protein Bcl2 and the level of cleaved PARP was examined (). The results showed that, Western blot analysis of the extracts prepared from SW-620 cells incubated with compound 5a (5 μM and 10 μM) for 24 h, resulted in a dose dependent inhibition of Bcl2 protein expression and significant increase in the level of cleaved PARP ().
Figure 7. (A) Effect of hybrid 5a on anti-apoptotic Bcl2 protein and the level of cleaved PARP. Statistical analysis was performed where the significance of data was assessed at a p values < 0.05. *** p < 0.001; ** p < 0.01 control vs treated. (B) Effect of hybrid 5d on anti-apoptotic Bcl2 protein and the level of cleaved PARP. Statistical analysis was performed where the significance of data was assessed at a p values < 0.05. *** p < 0.001; ** p < 0.01 control vs treated.
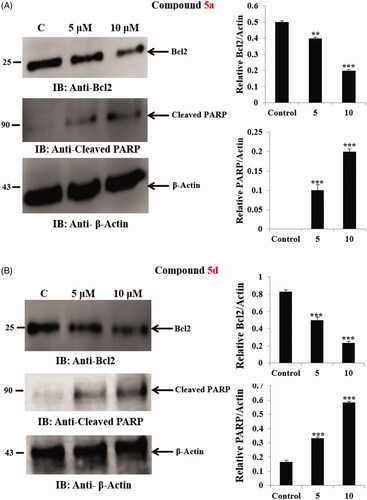
Similarly, compound 5d was found to follow the same pattern with significant inhibition of the anti-apoptotic Bcl2 protein expression and significant increase in the level of cleaved PARP in SW-620 cancer cells (). These findings indicated that both compounds 5a and 5d inhibited SW-620 cells viability by deregulating apoptosis-related proteins (anti-apoptotic Bcl2 and cleaved PARP) resulting in the induction of apoptosis.
3. Conclusions
In summary, a novel series of benzofuran-isatin conjugates linked by a carbohydrazide group, (5a–e and 7a–i) was designed and synthesised. Seven compounds (5b–d and 7a,b,d,g) were selected according to NCI’s DTP selection guidelines for the assessment of their antitumor activity against NCI-55 human cancer cell lines. All compounds proved effective against diverse cell lines among which compound 5d was promoted to the five-dose screen and showed good to excellent growth inhibitory activity against almost all subpanel cancer cell lines. In addition, the novel conjugates (5a–e and 7a–i) showed good anti-proliferative activity against two human colorectal cancer cell lines, SW-620 and HT-29, with excellent inhibitory activity for compounds 5a and 5d that showed IC50 = 8.7 µM and 9.4 µM for 5a and IC50 = 6.5 µM and 9.8 µM for 5d against SW-620 and HT-29 cell lines, respectively, and proved to have selective cytotoxicity with increased safety profile to fibroblast (HFF-1) normal cells. Further mechanistic studies revealed that both compounds 5a and 5d were able to induce apoptosis in a dose dependent manner with an approximately 1.7–3.8 folds and 2.9–3.8 folds total increase in apoptosis for compounds 5a and 5d, respectively, compared to the control untreated SW-620 cell line. Furthermore, both conjugates significantly inhibited the expression of the anti-apoptotic Bcl2 protein and increased the level of the cleaved PARP and resulted in SW-620 cells apoptosis. Collectively, the significant potency and high selective cytotoxicity of this series specially compounds 5a and 5d suggested that these conjugates might serve as starting point for additional optimisation to develop potential anticancer agents and apoptotic inducers.
4. Experimental
4.1. Chemistry
4.1.1. General
Solvents of HPLC grade have been used and purchased from Thermo Fisher. Follow up of reactions has been performed utilising precoated TLC F254 Merck plates. Schimadzu FT-IR spectrometer has been used for functional groups analysis for the synthesised derivatives. NMR spectrometric analyses have been conducted using Bruker-Avance 400 NMR spectrometer (100 MHz for 13CNMR and 400 MHz for 1H NMR). Chemical shifts have been recorded in ppm. Multiplicities have been reported with their 1st order J coupling constants (Hz) for doublets (d); Stuart apparatus has been used to determine the melting points. FLASH 2000 CHNS/O analyser has been adopted to perform the elemental analysis. Compounds 3Citation64, and 6a–iCitation65,Citation66 have been reported previously.
4.1.2. Synthesis of target derivatives 5a–e and 7a–i
To stirred hot solution of 3-methylbenzofuran-2-carbohydrazide 3 (0.25 g, 1.3 mmol) in 13 ml of absolute EtOH with catalytic drops of ethanoic acid, equivalent amount of appropriate indoline-2,3-dione compounds 4a–e or 6a–i has been added. The reaction mixture has been then refluxed for (3–6) h. The produced precipitate, after cooling, was collected by filtration, washed with water then recrystallized from glacial acetic acid to produce target derivatives 5a–e and 7a–i, respectively in a good yield (70–87%).
Full characterisation (NMR, IR, and elemental analysis) data for target compounds (5a–e and 7a–i) have been presented in the Supporting Materials.
4.2. Biological evaluation
All in vitro biological assays in this study; NCI anticancer screeningCitation67,Citation68, MTT cell viability assayCitation54, Annexin V-FITC/PI assayCitation54 and Western blot analysisCitation54 were performed as reported earlier. All experimental procedures were provided in the Supporting materials.
Supplemental Material
Download PDF (1.3 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J. Clin 2020;70:7–30.
- Cheung-Ong K, Giaever G, Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol 2013;20:648–59.
- DeVita VT Jr., Chu E. A history of cancer chemotherapy. Cancer Res 2008;68:8643–53.
- Fischhaber PL, Gall AS, Duncan JA, Hopkins PB. Direct demonstration in synthetic oligonucleotides that N,N′-bis(2-chloroethyl)-nitrosourea cross links N1 of deoxyguanosine to N3 of deoxycytidine on opposite strands of duplex DNA. Cancer Res 1999;59:4363–8.
- Goodman LS, Wintrobe MM. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J Am Med Assoc 1946;132:126–32.
- Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med 1948;238:787–93.
- Nitiss JL. DNA topoisomerases in cancer chemotherapy: using enzymes to generate selective DNA damage. Curr Opin Investig Drugs 2002;3:1512–6.
- Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 2009;9:338–50.
- Wadler S, Fuks JZ, Wiernik PH. Phase I and II agents in cancer therapy: I. Anthracyclines and related compounds. J Clin Pharmacol 1986;26:491–509.
- Espinosa E, Zamora P, Feliu J, González Barón M. Classification of anticancer drugs–a new system based on therapeutic targets. Cancer Treat. Rev 2003;29:515–23.
- Mansoori B, Mohammadi A, Davudian S, et al. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull 2017;7:339–48.
- Baudino TA. Targeted cancer therapy: the next generation of cancer treatment. Curr Drug Discov Technol 2015;12:3–20.
- Topcul M, Cetin I. Endpoint of cancer treatment: targeted therapies, Asian Pac. Asian Pac J Cancer Prev 2014;15:4395–403.
- Modugno M, Banfi P, Gasparri F, et al. Mcl-1 antagonism is a potential therapeutic strategy in a subset of solid cancers. Exp Cell Res 2015;332:267–77.
- Placzek WJ, Wei J, Kitada S, et al. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis 2010;1:e40.
- Bai P. Biology of Poly(ADP-Ribose) polymerases: the factotums of cell maintenance. Mol Cell 2015;58:947–58.
- Herceg Z, Wang ZQ. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat Res 2001;477:97–110.
- Langelier MF, Pascal JM. PARP-1 mechanism for coupling DNA damage detection to poly(ADP-ribose) synthesis. Curr Opin Struct Biol 2013;23:134–43.
- Yu SW, Andrabi SA, Wang H, et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA 2006;103:18314–9.,
- DeSimone RW, Currie KS, Mitchell SA, et al. Privileged structures: applications in drug discovery. Comb Chem High Throughput Screen 2004;7:473–94.
- Khanam H. Shamsuzzaman, Bioactive Benzofuran derivatives: a review. Eur. J. Med. Chem 2015;97:483–504.
- Nevagi RJ, Dighe SN, Dighe SN. Biological and medicinal significance of benzofuran. Eur J Med Chem 2015;97:561–81.
- Dawood KM. An update on benzofuran inhibitors: a patent review. Expert Opin Ther Pat 2019;29:841–70.
- Miao Y-h, Hu Y-h, Yang J, et al. Natural source, bioactivity and synthesis of benzofuran derivatives. RSC Adv 2019;9:27510–40.
- Radadiya A, Shah A. Bioactive benzofuran derivatives: an insight on lead developments, radioligands and advances of the last decade. Eur J Med Chem 2015;97:356–76.
- Goyal D, Kaur A, Goyal B. Benzofuran and Indole: promising scaffolds for drug development in Alzheimer’s Disease. ChemMedChem 2018;13:1275–99.
- Hiremathad A, Patil MR, K. R C, et al. Benzofuran: an emerging scaffold for antimicrobial agents. RSC Adv 2015;5:96809–28.
- Xu Z, Zhao S, Lv Z, et al. Benzofuran derivatives and their anti-tubercular, anti-bacterial activities. Eur J Med Chem 2019;162:266–76.
- Chand, Rajeshwari K, Hiremathad A, Singh M, et al. A review on antioxidant potential of bioactive heterocycle benzofuran: natural and synthetic derivatives. Pharmacol. Rep 2017;69:281–95.
- Alizadeh M, Jalal M, Hamed K, et al. Recent updates on anti-inflammatory and antimicrobial effects of furan natural derivatives. J Inflamm Res 2020;13:451–63.
- Kwiecień H, Goszczyńska A, Rokosz P. Benzofuran small molecules as potential inhibitors of human protein kinases: a review. Curr Pharm Des 2016;22:879–94.
- Flynn BL, Gill GS, Grobelny DW, et al. Discovery of 7-hydroxy-6-methoxy-2-methyl-3-(3,4,5-trimethoxybenzoyl)benzo[b]furan (BNC105), a tubulin polymerization inhibitor with potent antiproliferative and tumor vascular disrupting properties. J Med Chem 2011;54:6014–27.
- Romagnoli R, Baraldi PG, Carrion MD, et al. Design, synthesis and structure-activity relationship of 2-(3′,4′,5′-trimethoxybenzoyl)-benzo[b]furan derivatives as a novel class of inhibitors of tubulin polymerization. Bioorg Med Chem 2009;17:6862–71.
- Xia Y, Jin Y, Kaur N, et al. HIF-1α inhibitors: synthesis and biological evaluation of novel moracin O and P analogues. Eur J Med Chem 2011;46:2386–96.
- Xie F, Zhu H, Zhang H, et al. In vitro and in vivo characterization of a benzofuran derivative, a potential anticancer agent, as a novel Aurora B kinase inhibitor. Eur J Med Chem 2015;89:310–9.
- Abdelhafez OM, Amin KM, Ali HI, et al. Design, synthesis and anticancer activity of benzofuran derivatives targeting VEGFR-2tyrosine kinase. RSC Adv 2014;4:11569–79.
- Choi MJ, Jung KH, Kim D, et al. Anti-cancer effects of a novel compound HS-113 on cell growth, apoptosis, and angiogenesis in human hepatocellular carcinoma cells. Cancer Lett 2011;306:190–6.
- Gao C, Sun X, Wu Z, et al. A novel Benzofuran derivative Moracin N induces autophagy and apoptosis through ROS Generation In Lung Cancer. Front Pharmacol 2020;11:391.
- Manna SK, Bose JS, Gangan V, et al. Novel derivative of benzofuran induces cell death mostly by G2/M cell cycle arrest through p53-dependent pathway but partially by inhibition of NF-kappaB. J Biol Chem 2010;285:22318–27.
- Abd El-Karim SS, Anwar MM, Mohamed NA, et al. Design, synthesis, biological evaluation and molecular docking studies of novel benzofuran-pyrazole derivatives as anticancer agents. Bioorg Chem 2015;63:1–12.
- Siddiqui SK, SahayaSheela VJ, Kolluru S, et al. Discovery of 3-(benzofuran-2-ylmethyl)-1H-indole derivatives as potential autophagy inducers in cervical cancer cells. Bioorg Med Chem Lett 2020;30:127431.
- Mao ZW, Zheng X, Lin YP, et al. Design, synthesis and anticancer activity of novel hybrid compounds between benzofuran and N-aryl piperazine. Bioorg Med Chem Lett 2016;26:3421–4.
- Xu K, Liu Y, Wang R, et al. Design, synthesis, and anticancer activities of Benzofuran–isatin hybrids tethered by pentylene and hexylene. J. Hetero. Chem 2019;56:2052–5.
- De Moraes G, Teixeira PA, Pena LJ, Leite ACL. Isatin derivatives and their antiviral properties against arboviruses: a review. Mini Rev Med Chem 2019;19:56–62.
- Guo H. Isatin derivatives and their anti-bacterial activities. Eur J Med Chem 2019;164:678–88.
- Mathur G, Nain S. Recent advancement in synthesis of isatin as anticonvulsant agents, a review. Med Chem 2014;4:417–27.
- Phogat P, Singh P. A mini review on central nervous system potential of isatin derivatives. Cent Nerv Syst Agents Med Chem 2015;15:28–31.
- Ding Z, Zhou M, Zeng C. Recent advances in isatin hybrids as potential anticancer agents. Arch Pharm 2020;353:e1900367
- Hou Y, Shang C, Wang H, Yun J. Isatin-azole hybrids and their anticancer activities. Arch Pharm 2020;353:e1900272.
- Abdel-Aziz HA, Eldehna WM, Keeton AB, et al. Isatin-benzoazine molecular hybrids as potential antiproliferative agents: synthesis and in vitro pharmacological profiling. Drug Des Devel Ther 2017;11:2333–46.
- Eldehna WM, El Hassab MA, Abo-Ashour MF, et al. Development of isatin-thiazolo[3,2-a]benzimidazole hybrids as novel CDK2 inhibitors with potent in vitro apoptotic anti-proliferative activity: synthesis, biological and molecular dynamics investigations. Bioorg Chem 2021;110:104748.
- El-Naggar M, Eldehna WM, Almahli H, et al. Novel Thiazolidinone/Thiazolo[3,2-a]Benzimidazolone-Isatin conjugates as apoptotic anti-proliferative agents towards breast cancer: one-pot synthesis and in vitro biological evaluation. Molecules 2018;23:1420.
- Eldehna WM, Abo-Ashour MF, Al-Warhi T, et al. Development of 2-oindolin-3-ylidene-indole-3-carbohydrazide derivatives as novel apoptotic and anti-proliferative agents towards colorectal cancer cells. J Enzy Inhib Med Chem 2021;36:319–28.
- Fares M, Eldehna WM, Abou-Seri SM, et al. Design, synthesis and in vitro antiproliferative activity of novel isatin-quinazoline hybrids. Arch Pharm 2015;348:144–54.
- Montreal Q. Operating Environment (MOE), 10. Montreal: Chemical Computing Group Inc; 2009.
- Boyd MR, Paull KD. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev. Res 1995;34:91–109.
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006;6:813–23.
- Acton EM, Narayanan VL, Risbood PA, et al. Anticancer specificity of some ellipticinium salts against human brain tumors in vitro. J Med Chem 1994;37:2185–9.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Rieger AM, Nelson KL, Konowalchuk JD, Barreda DR. Modified annexin V/propidium iodide apoptosis assay for accurate assessment of cell death. J. Vis. Exp 2011;(50):2597–603.
- Marone M, Ferrandina G, Macchia G, et al. Bcl-2, Bax, Bcl-x(L) and Bcl-x(S) expression in neoplastic and normal endometrium. Oncology 2000;58:161–8.
- Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ 2003;10:76–100.
- Eldehna WM, Nocentini A, Elsayed ZM, et al. Benzofuran-based carboxylic acids as carbonic anhydrase inhibitors and antiproliferative agents against breast cancer. ACS Med Chem Lett 2020;11:1022–7.
- Al-Warhi T, El Kerdawy AM, Aljaeed N, et al. Synthesis, biological evaluation and in silico studies of certain oxindole-indole conjugates as anticancer CDK inhibitors. Molecules 2020;25:2031–9.
- Elsayed ZM, Eldehna WM, Abdel-Aziz MM, et al. Development of novel isatin-nicotinohydrazide hybrids with potent activity against susceptible/resistant Mycobacterium tuberculosis and bronchitis causing-bacteria. J Enzyme Inhib Med Chem 2021;36:384–93.
- Al-Rashood ST, Hamed AR, Hassan GS, et al. Antitumor properties of certain spirooxindoles towards hepatocellular carcinoma endowed with antioxidant activity. J Enzyme Inhib Med Chem 2020;35:831–9.
- Al-Warhi T, Abo-Ashour MF, Almahli H, et al. Novel [(N-alkyl-3-indolylmethylene)hydrazono]oxindoles arrest cell cycle and induce cell apoptosis by inhibiting CDK2 and Bcl-2: synthesis, biological evaluation and in silico studies. J Enzyme Inhib Med Chem 2020;35:1300–9.