
Abstract
There is an urgent need to design new anticancer agents that can prevent cancer cell proliferation even with minimal side effects. Accordingly, two new series of 3-methylquinoxalin-2(1H)-one and 3-methylquinoxaline-2-thiol derivatives were designed to act as VEGFR-2 inhibitors. The designed derivatives were synthesised and evaluated in vitro as cytotoxic agents against two human cancer cell lines namely, HepG-2 and MCF-7. Also, the synthesised derivatives were assessed for their VEGFR-2inhibitory effect. The most promising member 11e were further investigated to reach a valuable insight about its apoptotic effect through cell cycle and apoptosis analyses. Moreover, deep investigations were carried out for compound 11e using western-plot analyses to detect its effect against some apoptotic and apoptotic parameters including caspase-9, caspase-3, BAX, and Bcl-2. Many in silico investigations including docking, ADMET, toxicity studies were performed to predict binding affinity, pharmacokinetic, drug likeness, and toxicity of the synthesised compounds. The results revealed that compounds 11e, 11g, 12e, 12g, and 12k exhibited promising cytotoxic activities (IC50 range is 2.1 − 9.8 µM), comparing to sorafenib (IC50 = 3.4 and 2.2 µM against MCF-7 and HepG2, respectively). Moreover, 11b, 11f, 11g, 12e, 12f, 12g, and 12k showed the highest VEGFR-2 inhibitory activities (IC50 range is 2.9 − 5.4 µM), comparing to sorafenib (IC50 = 3.07 nM). Additionally, compound 11e had good potential to arrest the HepG2 cell growth at G2/M phase and to induce apoptosis by 49.14% compared to the control cells (9.71%). As well, such compound showed a significant increase in the level of caspase-3 (2.34-fold), caspase-9 (2.34-fold), and BAX (3.14-fold), and a significant decrease in Bcl-2 level (3.13-fold). For in silico studies, the synthesised compounds showed binding mode similar to that of the reference compound (sorafenib).
1. Introduction
Cancer has been the most difficult and life-threatening illness to be treatedCitation1. After cardiovascular disease (CVD) cancer has been reported to be a significant cause of death worldwideCitation2. At the end of 2018, cancer caused 9.6 million deathsCitation3. The current anticancer therapy has many side effects arising from non-selectivity of the development of drug resistanceCitation4. Nonetheless, there is an urgent need to design new anticancer drugs that can prevent cancer cell proliferation even with minimal to no side effects on healthy cells.
At the level of molecular biology, protein tyrosine kinases have an important role in cell proliferation, migration, survival, and progressionCitation5. Tyrosine kinases phosphorylate the protein's tyrosine residues resulting in altered protein function. In some cases, tyrosine kinases become continuously active which ultimately leads to cancerCitation6. Tyrosine kinase receptors (RTKs) are a panel of cell surface receptors that transfer the signal to polypeptides, hormones, and growth factorsCitation7. There have been numerous known RTKs such as Vascular endothelial growth factor receptors (VEGFRs) and endothelial growth factor receptors (EGFRs)Citation8.
VEGFRs have been recognised as an outstanding medicinal target to discover new anticancer agentsCitation9,Citation10. The class of VEGFRs comprises three subtypes; VEGFR-1, VEGFR-2, and VEGFR-3Citation11. Among them, VEGFR-2 which has a crucial role in tumour angiogenesis. VEGFR-2 can be activated through binding with VEGF which starts the process of phosphorylation which boosts proliferation and migration of the endothelial cellsCitation12. VEGFR-2 is mainly overexpressed throughout endothelial cells of the tumour vasculature, with less expression in normal endothelial cellsCitation13. Hepatocellular carcinoma and breast cancer are well-known examples of tumours with overexpressed VEGFR-2Citation14–16.
VEGFR-2 inhibitors are small molecules that bind at the ATP binding site of VEGFR-2 to inhibit angiogenesis and lymphangiogenesisCitation17. Beside many VEGFR-2 inhibitors approved by FDA or under clinical trials, there are a lot of effort to discover new ones for the management of cancer. Sorafenib I is a bi-aryl urea, has inhibitory effect against tyrosine kinases involved in tumour development, including VEGFR-2Citation18. Sunitinib II is anti-tumour drug with dual activity against VEGFR-2 and PDGFR-βCitation19. Telatinib III is an orally active anti-VEGFR-2 small-moleculeCitation20. Nintedanib IV is a potent small-molecule tyrosine kinase inhibitor with oral activity. In addition, it has a triple angiokinase inhibitory effect as it inhibits the three major signalling pathways involved in angiogenesisCitation21. Acrizanib V is a VEGFR-2 inhibitor with limited systemic exposure after topical ocular administrationCitation22. Vorolanib VI is a novel indolinone-based kinase inhibitor that targets the VEGFR-2Citation23. It has fewer adverse effects and a wide therapeutic windowCitation24.
Essential Pharmacophoric features of VEGFR-2 inhibitors have been reported in many publicationsCitation25–30. The reported pharmacophore includes: i) a flat hetero aromatic moiety which binds Cys917 via a hydrogen bonding interactionCitation26, (ii) a spacer moiety which occupies the area between the ATP-binding domain and the DFG domainCitation31, (iii) a pharmacophore moiety which consists of H-bond acceptor (HBA) and one H-bond donor (HBD) groups (e.g. amide or urea). Both HBA and HBD have a key binding role, as they form hydrogen bonding interactions with two crucial residues (Glu883 and Asp1044) Citation32, and (iv) a terminal hydrophobic moiety can make many hydrophobic interactions in the allosteric hydrophobic pocket of VEGFR-2Citation33 ( and ).
Figure 1. Some reported VEGFR-2 inhibitors and their basic pharmacophoric features.
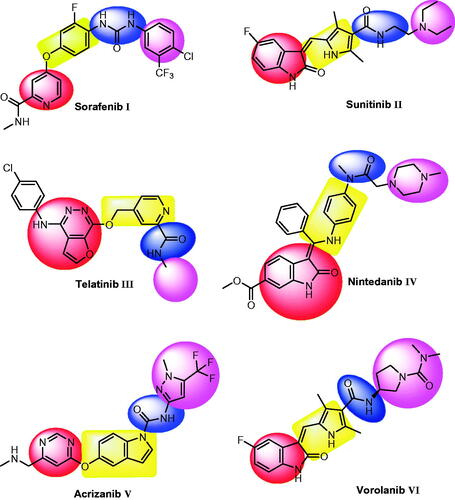
Figure 2. A) Different bio-isosteres that can occupy the ATP binding site of VEGFR-2. B) Representative examples of the new synthesised compounds having the same essential pharmacophoric features of VEGFR-2 inhibitor.
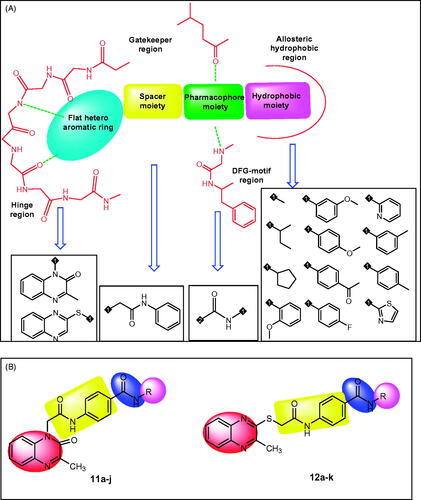
In the current work, ligand-based drug design approachCitation34–37 was used to synthesise two series of 3-methylquinoxalin-2(1H)-one and 3-methylquinoxaline-2-thiol derivatives. This work is an extension of the earlier activities of our team to synthesise effective anticancer agents targeting VEGFR-2Citation9,Citation38,Citation39. The synthesised derivatives were evaluated in vitro and in silico to assess their VEGFR-2 inhibitory activities.
1.1. Rationale of molecular design
VEGFR-2 inhibitors competitively block the ATP binding site which consists of four main regions. i) Hinge region which is occupied by the flat hetero aromatic ring of VEGFR-2 inhibitors. ii) Gatekeeper region which is occupied by the spacer moiety of VEGFR-2 inhibitors. iii) DFG-motif region which is occupied by the pharmacophore moiety of VEGFR-2 inhibitors. iv) Allosteric hydrophobic region which is occupied by the terminal hydrophobic moiety of VEGFR-2 inhibitorsCitation33,Citation40–42 ().
The main objective of our design was the synthesis of new compounds having the main pharmacophoric features of VEGFR-2 inhibitors. Such compounds comprise different bio-isosteres, each of them occupy a specific region at ATP binding site.
For the hinge region, two quinoxaline moieties were used; i) 3-methylquinoxalin-2(1H)-one (compounds 11a-j) and ii) 3-methylquinoxaline-2-thiol (compounds 12a-k). The bicyclic structure of quinoxaline moiety is suitable to the large size space of the ATP binding regionCitation43. In addition, the nitrogen atoms act as hydrogen-bond acceptors to facilitate the hydrogen bonding interaction with the hinge region. The Gatekeeper region was targeted to be occupied by N-phenylacetamide moiety as spacer group. Regarding the DFG-motif region, an amide group (pharmacophore moiety) was selected to be buried in it. Finally, the allosteric hydrophobic region can be occupied by different aliphatic and aromatic derivatives to study the structure-activity relationships ().
2. Results and discussion
2.1. Chemistry
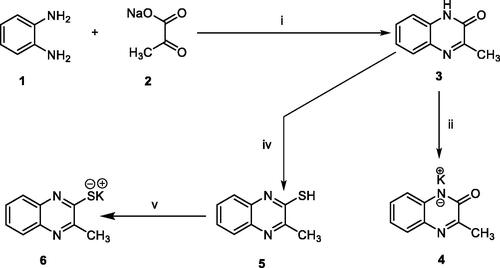
In order to synthesise the designed compounds, Schemes 1–4 were adopted. Initially, o-phenylenediamine 1 was refluxed with sodium pyruvate 2 to afford 3-methylquinoxalin-2(1H)-one 3 according to the reported procedureCitation44. Subsequent heating of compound 3 with alcoholic potassium hydroxide gave the corresponding potassium salt 4Citation44. To prepare 3-methylquinoxaline-2-thiol 5, the previously prepared 3-methylquinoxalin-2(1H)-one 3 was refluxed with P2S5 in pyridine as a solvent, then the reaction was acidified using HClCitation45,Citation46. Heating of compound 5 with alcoholic potassium hydroxide gave the corresponding potassium salt 6 (Scheme 1).
Scheme 1. synthesis of compound key potassium salts 4 and 5. Reagents and conditions: i) g. acetic acid/H2O/reflux/2 h, ii) Alc. KOH/reflux/30 min., iii) i) P2S5/pyridine/reflux/2 h then HCl, v) Alc. KOH/reflux/30 min.
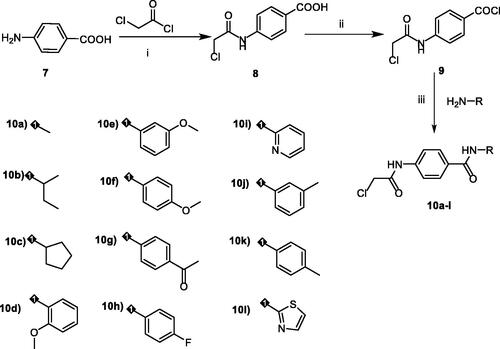
The key intermediates were synthesised as described in Scheme 2. The commercially available p-amino benzoic acid 7 was treated with chloroacetyl chloride in dry DMF in the presence of NaHCO3 to afford 4–(2-chloroacetamido)benzoic acid 8. Chlorination of 8 was achieved by its reflux with SOCl2 in dichloroethane in the presence of catalytic amount of dry DMF to give the key compound 4–(2-chloroacetamido)benzoyl chloride 9. At the end, in acetonitrile and TEA mixture, compound 9 was stirred at room temperature with appropriate amines namely, methylamine, sec-butylamine, cyclopentylamine, 2-methoxyaniline, 3-methoxyaniline, 4-methoxyaniline, 4-aminoacetophenone, 4-fluoroaniline, 2-aminopyridine, m-toluidine, p-toluidine, and 2-aminothiazole to give the corresponding key intermediates 10a-l, respectively. The IR spectra of the key intermediates 10a-l exhibited the appearance of absorption bands at the ranges of 3254 − 3326 cm−1 and 1702 − 1625 cm−1 due to the NH and 2C=O groups, respectively. In addition, 1H NMR analyses exhibited the appearance of characteristic singlet signals for amidic NHs around δ 10.00 ppm. Also, it showed singlet signals for CH2 protons of acetamide moiety around δ 4.30 ppm. Besides, such CH2 group was detected around δ 44.02 ppm in 13 C NMR spectra.
Scheme 2. synthesis of the key intermediates 10a-l. Reagents and conditions: i) DMF/NaHCO3/stirring, r.t./1h, ii) dichloroethane/SOCl2/DMF/reflux/1h, iii) CH3CN/stirring/r.t./3 h.
The first series of the target compounds was prepared as described in Scheme 3. The potassium salt of 3-methylquinoxalin-2(1H)-one 4 was heated in dry DMF with the keys intermediates in the presence of catalytic amount of KI to give the titled compounds 11a-j.
Scheme 3. synthesis of the target compounds 11a-j. Reagents and conditions: i) KI/DMF/heating/W.B./8h.
The second series of the target compounds was synthesised depending on the synthetic pathway described in Scheme 4. The potassium salt of methylquinoxaline-2-thiol 6 was heated in dry DMF with the keys intermediates 10a-k in the presence of catalytic amount of KI to give the titled compounds 12a-k.
Scheme 4. synthesis of the target compounds 12a-k. Reagents and conditions: i) KI/DMF/heating/W.B./8h.
1H NMR spectra exhibited the presence of singlet signals of CH3 group of 3-methylquinoxalin-2(1H)-one moiety around δ 2.49 ppm. In addition, 1H NMR spectra showed characteristic signals for additional aromatic and aliphatic protons at the expected chemical shift. Taken compound 11c and 12c as representative examples, it showed many characteristic signals at aliphatic region corresponding to cyclopentyl moiety. Moreover, 13 C NMR spectra of such compounds confirmed the previous results as the aliphatic protons of cyclopentyl moiety appeared at the aliphatic region.
2.2. Biological testing
2.2.1. In vitro cytotoxic activities
Cytotoxic activities of the synthesised compounds were evaluated against MCF-7 (human breast cancer cell line) and HepG2 (human liver carcinoma cell line) using MTT assayCitation47, using sorafenib as a reference standard (). Among the target compounds, 11e, 11g, 12e, 12g, and 12k exhibited promising cytotoxicity against the two cell lines with IC50 values ranging from 2.1 to 9.8 µM, comparing to sorafenib (IC50 = 3.4 and 2.2 µM against MCF-7 and HepG2, respectively). Compound 11e exhibited a superior activity against MCF-7 and HepG2 with IC50 values of 2.7 and 2.1, respectively. In addition, compounds 11f and 12f showed promising activity against HepG2 cells with IC50 values of 9.6 and 7.5, respectively. Compounds 11a, 11b, 11c, 12c, and 12d showed moderate activity against HepG2 cells with IC50 values of 16.5, 12.8, 18.7, 11.4, 18.7, and 17.6 µM, respectively. In addition, compounds 11f and 12f showed moderate activity against MCF-7 cells with IC50 values of 12.4 and 10.8 µM, respectively. On the other hand, compounds 11d, 11h, 11i, 11j, 12a, and 12b showed weak cytotoxic activity against the two cell lines.
Table 1. In vitro cytotoxic and VEGFR-2inhibitory activities.
2.2.2. Vegfr-2 inhibitory assay
VEGFR-2 inhibitory activity of the synthesised compounds was investigated using sorafenib as a reference drug. . Summarised the IC50 values of VEGFR-2 growth inhibitory concentration for all the synthesised members.
Compound 11e and 12k exhibited VEGFR-2 inhibitory activity (IC50 = 2.6 and 2.9 nM, respectively) higher than that of sorafenib (IC50 = 3.07 nM). Additionally, compounds 11b, 11f, 11g, 12e, 12f, 12g, and 12k showed promising activities with IC50 values ranging from 2.9 to 5.4 nM. On the other hand, compounds 11a, 11c, 11d, 11h, 11i, 11j, 12a, 12b, 12c, 12d, 12h, 12i, and 12j showed moderate to weak activity. Its IC50 values are ranging from 11.2 to 52.7 nM.
2.2.3. Statistical correlation between VEGFR-2 inhibition and cytotoxicity
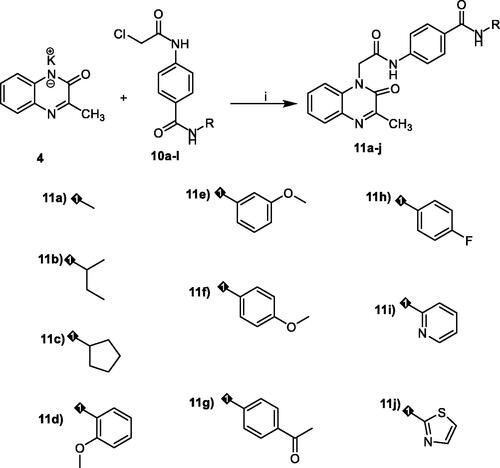
To study the relation between cytotoxicity and VEGFR-2 inhibition, we plotted the values of VEGFR-2 inhibition against the corresponding cytotoxicity results using simple linear regression analysis. Co-efficient of determination (R2) were calculated in this analysis. It was found that R2 of VEGFR-2 inhibition and MCF-7 cytotoxicity is 0.887 with p values >0.0001. In addition, R2 of VEGFR-2 inhibition and HepG2 cytotoxicity is 0.887 with p values >0.0001. The results indicated that there are high correlations between VEGFR-2 inhibition and cytotoxic activity on both cell lines, which reveals that the cytotoxicity may be a result of VEGFR-2 inhibition ().
Figure 3. Simple linear regression for the correlation between VEGFR-2 inhibition and cytotoxicity.
2.2.4. Structure-Activity relationship (SAR)
The results of different biological analyses (cytotoxic activity and VEGFR-2 inhibitory assay) gave a valuable SAR. Initially, the effect of the flat hetero aromatic ring on the activity was explored. Comparing the cytotoxic activity of compounds 11a-j (incorporating 3-methylquinoxalin-2(1H)-one) with compounds 12a-k (incorporating 3-methylquinoxaline-2-thiol) indicated that 3-methylquinoxalin-2(1H)-one moiety is more advantageous than 3-methylquinoxaline-2-thiol moiety for cytotoxic and VEGFR-2 inhibitory effects.
Then, we investigated the effect of the terminal hydrophobic moiety. Comparing the activity of compounds 11a-c and 12a-c having aliphatic hydrophobic moieties with compounds 11e-g and 12e-g having aromatic hydrophobic moieties indicated that aromatic moieties are beneficial for activity. For aliphatic moieties, there is no great variation in the activity among small size (compound 11a and 12a), bulk (compound 11c and 12c), and branched (compound 11b and 12b) aliphatic moieties.
In addition, the effect of the substitution on the aromatic hydrophobic moieties has been investigated. Comparing the activity of compound 12k (incorporating 4-methylphenyl moiety) and compounds 11f and 12f (incorporating 4-methoxyphenyl moiety) with compounds 11h and 12h (incorporating 4-fluorophenyl) and compounds 11g and 12g (incorporating acetophenone moiety), revealed that grafting an electron donating group is more preferred biologically than electron withdrawing one. For electron withdrawing groups, it was found that acetyl incorporating members (11g and 12g) are more active than fluoro incorporating one (11h and 12h).
Next, the effect of the substitution on the aromatic hydrophobic moieties with electron donating group has been examined. For methylquinoxalin-2(1H)-one derivatives, the activities reduced in the order of 3-methoxy (11e) > 4-methoxy (11f) > 2-methoxy (11d). With regard to 3-methylquinoxaline-2-thiol derivatives, the activities decreased in the order of 4-methyl (12k) > 3-methoxy (12e) > 4-methoxy (12f) > 2-methoxy (12d) > 3-methoxy (12j).
Finally, the decreased IC50 values of compound 11j (with thiazole moiety) against the tested cell lines and VEGFR-2 than the IC50 values of compound 11i (with pyridine moiety), indicated that five-membered hetero aromatic hydrophobic moiety is more efficient than six- membered one.
2.2.5. In vitro cytotoxicity against normal cell
The cytotoxicities of the most active compounds (11e and 12e) against primary rat hepatocytes (normal hepatic cells) were evaluated in vitro (). The results revealed that the tested compounds have low toxicity against the tested cells with IC50 values of 15.0 and 16.7 μM, respectively. Sorafenib as a reference drug showed IC50 value of 13.4 µM against the tested cells. These results revealed that the synthesised compounds have low toxicity against the normal cells as their toxicities are comparable to that of FDA approved drug (sorafenib).
Table 2. In vitro cytotoxicity of the most active compounds (11e and 12e) and sorafenib against normal cells (primary rat hepatocytes).
2.2.6. Cell cycle analysis
The cell cycle is a well-maintained process by which cells of eukaryotes replicate themselves. The homeostatic balance between cell loss and cell gain must be achieved to produce and conserve the complex architecture of tissues. One way in which this connection may be achieved is through the coupling of the cell cycle and apoptosisCitation48.
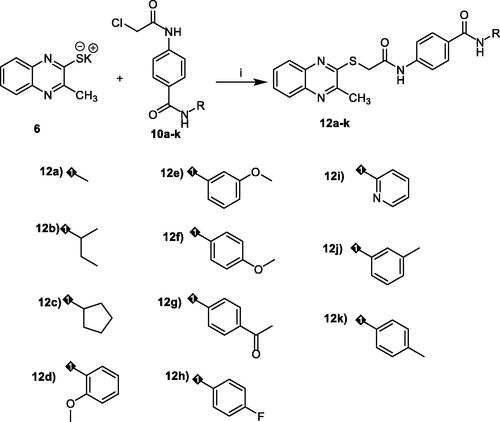
Since compound 11e effectively inhibited the growth of HepG2 cells, it was expected that this inhibitory effect was due to its capability to hinder the cell cycle progression. Therefore, cell cycle process was analysed after exposure of HepG2 cells to 11e with a concentration of 2.1 µM (IC50 value of compound 11e) after 24 h. HepG2 cells were used as a control without treatment by compound 11e. Flow cytometry data revealed that the percentage of cells arrested at the G2/M phase increased from 18.24% (in control cells) to 46.62 (in 11e treated cells). In addition, the percentage of HepG2 cells mild increased at the S phase from 25.48 to 31.80%. Oppositely, the percentage of HepG2 cells decreased at G1 phase from 55.03% to 20.34%. Such findings revealed that compound 11e arrested the HepG2 cell growth at G2/M phase ( and Supplementary Data).
Figure 4. Flow cytometric analysis of cell cycle phases post the compound 11e treatment.
2.2.7. Apoptosis analysis
To quantify the apoptosis triggered by 11e, Annexin-V/propidium iodide (PI) staining assay was conducted. In such procedure, compound 11e at a concentration of 2.1 µM was applied on HepG2 cells. Then, the media were incubated for 24 h. As shown in , the apoptotic effect of 11e in HepG2 cells was five times more than observed in control cells. In details, compound 11e induced apoptosis by 49.14% (early apoptosis = 48.87% & late apoptosis = 0.27%), compared to 9.71% in the control cells (early apoptosis = 9.58% & late apoptosis = 0.13%).
Table 3. Effect of compound 11e on stages of the cell death process in HepG2 cells after 24 h treatment.
2.2.8. Western blot analysis
Apoptosis is a programmed cell death characterised by some biological processes including condensation of nuclear chromatin, loss of plasma membrane phospholipid asymmetry, DNA cleavage into small fragments, and formation of membrane-bound apoptotic bodiesCitation49.
During intrinsic apoptosis, caspase-9 is activated to produce a subsequent activation of other effector caspases. It was reported that caspase-9 is a highly specific protease that only cleaves a few proteins, whereas caspase-3 contributes to the majority of cleavage that takes place during apoptosisCitation50,Citation51. Additionally, the mitochondrial apoptosis is largely mediated through Bcl-2 family proteins, which include. i) Pro-apoptotic members such as BAX that promote mitochondrial permeability and cell death. ii) Anti-apoptotic members such as Bcl-2 that inhibit the mitochondrial release of cyt c and suppress cell deathCitation52. According to these reports, a cell with a high BAX/Bcl-2 ratio will be more sensitive to some given apoptotic stimuli when compared to a similar cell type with a low BAX/Bcl-2ratioCitation53.
2.2.8.1. Caspase-3 and caspase-9 determination
To investigate the effect of the synthesised compounds on caspase-3 and caspase-9 levels, the most promising member 11e was applied on the most sensitive cells (HepG2) at a concentration of 2.1 µM for 24 h. Western blot analyses revealed that compound 11e produced a significant increase in the level of caspase-3 (2.34-fold) compared to the control cells. Moreover, compound 11e showed a significant increase in the level of caspase-9 (2.34-fold) compared to the control cells ( and Supplementary Data).
Figure 5. The immunoblotting of BAX, Bcl-2, Caspase-9, and Caspase-3 (Normalized to β-actin).
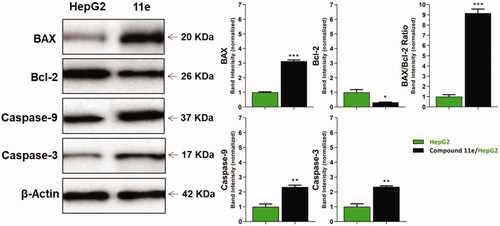
2.2.8.2. BAX and Bcl-2 determination
Compound 11e as a promising member was investigated to evaluate its effect on BAX and Bcl-2 after 24 h of its application on HepG2 cells using Western blot technique. The results showed that compound 11e produced an up-regulation of BAX and down-regulation of Bcl-2. In details, BAX level increased by 3.14-fold, while Bcl-2 level decreased by 3.13-fold. In addition, BAX/Bcl-2 ratio was 9.17, which indicated that compound 11e had a significant effect on apoptosis pathway ( and Supplementary Data).
2.3. In silico studies
2.3.1. Docking studies
In this work, the synthesised compounds were docked against VEGFR-2 using sorafenib as a reference drug. This investigational work was performed to get further insight into the binding modes of the synthesised compounds against VEGFR-2 binding site (PDB ID: 2OH4). The binding free energies (ΔG) for all the synthesised compounds against the target receptor were calculated and reported in . The reported key binding site of VEGFR-2 consists of Glu883 and Asp1044Citation33,Citation54. Validation of the docking procedure and the binding mode of the reference drug (sorafenib)Citation33,Citation54 were showed in Supplementary data.
Table 4. Binding free energies (ΔG in Kcal/mol) of the synthesised compounds and sorafenib against VEGFR-2
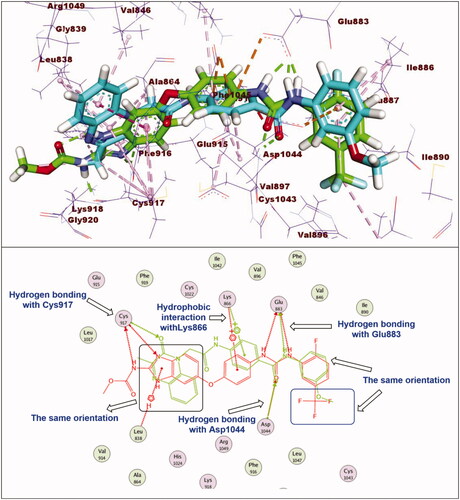
The synthesised compounds exhibited binding mode inside the binding sites of VEGFR-2 similar to that of sorafenib. Compound 11e was completely buried inside VEGFR-2 active site with similar binding mode to sorafenib. The docking score of such compound was −28.81 kcal/mol. The pharmacophore moiety (amide group) was incorporated in hydrogen bonding interaction forming a hydrogen bond with Glu883 and another one with Asp1044. The phenyl group (spacer) formed three hydrophobic interactions with Val914 and Cys1043. The 3-methylquinoxalin-2(1H)-one nucleus was inserted in hinge region of the binding pocket forming five hydrophobic interactions with Leu1033, Phe916, Ala864, and Leu838. In addition, the terminal methoxyphenyl group (hydrophobic tail) formed one hydrophobic bond with Leu887. Additionally, it formed two electrostatic interactions with Asp1044 ().
Figure 6. Superimposition of compound 11e and sorafenib inside the active sites of VEGFR-2. Compound 11e was completely buried inside VEGFR-2 active site with similar binding mode to sorafenib.
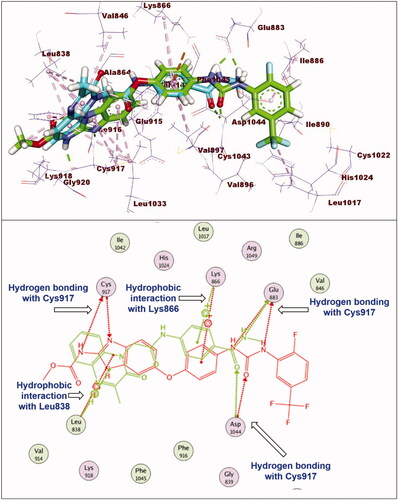
Regarding compound 11a (incorporating methyl group as hydrophobic tail) showed binding energy of −22.37 kcal/mol. It showed binding mode similar to that of sorafenib with some deviations. Firstly, due to lack of bulk aromatic moiety (as appeared in compound 11e), this led to disappearance of hydrophobic interactions at the allosteric binding pocket of VEGFR-2. In addition, the orientation of 3-methylquinoxalin-2(1H)-one nucleus at the hinge region prevent the hydrogen bonding interaction with Cys917 ().
Figure 7. Superimposition of compound 11a and sorafenib inside the active sites of VEGFR-2. Compound 11a showed binding mode similar to that of sorafenib with lack of hydrophobic interaction inside the allosteric binding pocket and absence of hydrogen bonding interaction with Cys917 at hinge region.
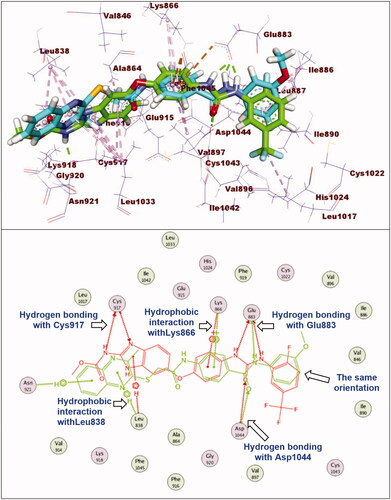
With respect to compound 12a, it exhibited a binding mode like that of sorafenib with binding energy of −27.98 kcal/mol. The different features of compounds 12a occupied the same regions which occupied by sorafenib. However, elongation of the linker moiety exerted mild change in the orientation of 3-methylquinoxaline-2-thiol nucleus at the hinge region preventing the hydrogen bonding interaction with Cys917 ().
Figure 8. Superimposition of compound 12e and sorafenib inside the active sites of VEGFR-2. Compound 12e showed binding mode similar to that of sorafenib with lack of hydrogen bonding interaction with Cys917 at hinge region.
2.3.2. In silico ADMET studies
The in silico ADMET parameters were assessed via Discovery studio 4.0 using Sorafenib as a reference molecule.
The results revealed that the tested compounds have low or very low BBB penetration levels except for compounds 12h, 12j, and 12k which were predicted to have medium levels. Accordingly, most compounds were anticipated to be safe against CNS. Furthermore, compounds 11a-g, 11i, and 11j were predicted to have good levels of aqueous solubility, while compounds 12a-k were predicted to have low levels. Moreover, intestinal absorption levels of all the tested compounds were predicted to be good. The cytochrome P4502D6 inhibition was predicted using CYP2D6 modelCitation55. All the tested compounds were predicted as non-inhibitors of CYP2D6. So that, these compounds are expected to be safe for the liver. The plasma protein binding (PPB) model predicts the degree of molecule binding to PP. If it is > = 90%, it means that a molecule can bind the PP at high concentrationCitation56. Compounds 11c-e, 11g-i, 12h, 12i, and 12k were expected to bind plasma protein over 90%, while compounds 11a, 11b, 11f, 11j, 12a-g, and 12i were predicted to bind plasma protein less than 90% (, ).
Figure 9. The expected ADMET study.
Table 5. Calculated ADMET descriptors
2.3.3. In silico toxicity studies
Discovery studio 4.0 was used to determine the expected toxicity potential of the synthesised compoundsCitation57,Citation58.
As shown in , most compounds showed in silico low toxicity profile against the tested models. Compounds 11a-j and 12a,b were predicted to have carcinogenic potency TD50 values ranging from 19.427 to 81.588 mg/kg body weight/day, which were higher than that of sorafenib (carcinogenic potency TD50 = 19.236 mg/kg body weight/day). While compounds 12c-k were estimated to have low carcinogenic potency TD50 values (from 7.026 to 17.638 mg/kg body weight/day). In addition, the rat maximum tolerated doses of compounds 11h, 12b, and 12h-k were estimated to be between 0.133 and 0.096 g/kg body weight, which were higher than that of sorafenib (rat maximum tolerated dose = 0.089 g/kg body weight). The other derivatives were predicted to have fewer rat maximum tolerated doses. Moreover, compounds 11a-c, 11g, 11i, 11j, 12a, 12c, 12g, 12i, and 12k were predicted to be non-toxic against developmental toxicity potential model. For rat oral LD50 model, the tested compounds showed oral LD50 values ranging from 2.102 to 18.807 g/kg body weight/day. Such values are far more than that of sorafenib (oral LD50 = 0.823 g/kg body weight/day). Moreover, all the tested compounds were predicted to be mild irritant against ocular irritancy model, and non-irritant against skin irritancy model.
Table 6. Toxicity properties of the synthesised compounds.
3. Conclusion
Twenty-two compounds were designed, synthesised, and evaluated as VEGFR-2 inhibitors. Such derivatives were assessed against MCF-7 and HepG-2 cell lines to estimate its antiproliferative activities. Compounds 11e, 11g, 12e, 12g, and 12k displayed promising cytotoxic activities against MCF-7 and HepG-2 with IC50 values ranging from 2.1 to 9.8 µM. Furthermore, compounds 11b, 11e, 11f, 11g, 12e, 12f, 12g, and 12k showed VEGFR-2 inhibitory activities with IC50 values of 5.3, 2.6, 4.8, 3.4, 3.8, 3.8, 5.4, and 2.9 nM, respectively. SAR revealed that 3-methylquinoxalin-2(1H)-one moiety is more beneficial than 3-methylquinoxaline-2-thiol moiety for cytotoxicity and VEGFR-2 inhibitory activities. Also, the terminal aromatic moieties were found to be more valuable than the terminal aliphatic ones. Compound 11e, the most potent member, arrested the HepG2 cell growth at G2/M phase and induced apoptosis by 49.14% compared to the control cells (9.71%). Additionally, such derivative showed a significant elevation in the level of caspase-3 (2.34-fold) and caspase-9 (2.34-fold). Moreover, it showed a marked increase in BAX (3.14-fold) and a significant reduction in Bcl-2 level (3.13-fold). The in silico studies revealed that the synthesised compounds showed binding interactions like that of sorafenib with good drug likeness profile.
4. Experimental
4.1. Chemistry
4.1.1. General
Reagents, solvent, and apparatus used in chemical synthesis were showed in Supplementary data. Compounds 3, 4, 5, 6, 8, 9, 10a-l were prepared according to reported proceduresCitation44–46,Citation59–64. Physical, elemental, and spectral data of the intermediate compounds 10a-l were depicted in Supplementary data.
General procedure for synthesis of compounds 11a-j
To a solution of the potassium salt of 3-methylquinoxalin-2(1H)-one 4 (320 mg, 0.002 mol) in DMF (20 ml) the appropriate 4–(2-chloroacetamido)-N-substituted-benzamides 10a-i and 10l (0.002 mol) was added. The mixture was heated on a water bath for 10 h. After cooling to room temperature, the reaction mixture was poured onto crushed ice. The precipitated solids were filtered, dried and crystalised from ethanol to give the target compounds 11a-j.
4.1.1.1. N-Methyl-4–(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)benzamide 11a
White powder (yield 75%); mp: 294–297 °C; FT-IR (v max, cm−1): 3288 (NH), 1674, 1655 (C=O), 1601 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.68 (s, 1H), 8.34 (q, J = 4.6 Hz, 1H), 7.83 − 7.79 (m, 3H), 7.64 (d, J = 8.8 Hz, 2H), 7.57 (t, J = 8.5 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.38 (t, J = 7.5 Hz, 1H), 5.16 (s, 2H), 2.76 (s, 3H), 2.49 (s, 3H); 13 C NMR (176 MHz, DMSO) δ 166.46, 165.68, 157.97, 154.84, 141.46, 133.46, 132.46, 130.18, 129.87, 129.27, 128.47 (2 C), 123.92, 118.81(2 C), 115.19, 45.75, 26.66, 21.59; MS (m/z): 351 (M+ + 1, 15% %), 201 (60%); Anal. Calcd. for C19H18N4O3 (350.38): C, 65.13; H, 5.18; N, 15.99; Found: C, 65.53; H, 5.06; N, 15.64%.
4.1.1.2. N-(sec-Butyl)-4–(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)benzamide 11b
White powder (yield 70%); mp: 319–321 °C; FT-IR (v max, cm−1): 3275 (NH), 2965 (CH aliphatic), 1660, 1634 (C=O), 1600 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.68 (s, 1H), 8.04 (d, J = 8.2 Hz, 1H), 7.85 − 7.82 (m, 2H), 7.81 (dd, J = 8.0, 1.5 Hz, 1H), 7.66 − 7.62 (m, 2H), 7.57 (ddd, J = 8.6, 7.1, 1.5 Hz, 1H), 7.53 (dd, J = 8.5, 1.3 Hz, 1H), 7.38 (ddd, J = 8.2, 7.1, 1.3 Hz, 1H), 5.16 (s, 2H), 3.91 (ddd, J = 14.1, 7.6, 6.2 Hz, 1H), 2.49 (s, 3H), 1.58 − 1.44 (m, 2H), 1.13 (d, J = 6.6 Hz, 3H), 0.86 (t, J = 7.4 Hz, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 165.66, 165.44, 157.96, 154.85, 141.39, 133.47, 132.46, 130.22, 130.18, 129.27, 128.65 (2 C), 123.92, 118.69 (2 C), 115.20, 46.81, 45.75, 29.32, 21.59, 20.77, 11.25; MS (m/z): 393 (M+ + 1, 80%); Anal. Calcd. for C22H24N4O3 (392.46): C, 67.33; H, 6.16; N, 14.28; Found: C, 66.94; H, 6.34; N, 13.95%.
4.1.1.3. N-Cyclopentyl-4–(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)benzamide 11c
Brown powder (yield 72%); mp: 298 − 300 °C; FT-IR (v max, cm−1): 3281 (NH), 2953 (CH aliphatic), 1631 (C=O), 1603 (C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.69 (s, 1H), 8.18 (d, J = 7.6 Hz, 1H), 7.84 (d, J = 8.2 Hz, 2H), 7.80 (d, J = 8.0 Hz, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.57 (t, J = 7.7 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.37 (t, J = 7.5 Hz, 1H), 5.16 (s, 2H), 4.22 (h, J = 6.6 Hz, 1H), 2.49 (s, 3H), 1.91 − 1.84 (m, 2H), 1.69 (m, 2H), 1.53 (m, J = 7.4 Hz, 4H); 13 C NMR (176 MHz, DMSO-d6) δ 165.70, 165.66, 157.95, 154.84, 141.41, 133.46, 132.46, 130.17, 130.09, 129.27 (2 C), 128.72, 123.90, 118.66 (2 C), 115.18, 51.35, 45.75, 32.61 (2 C), 24.09 (2 C), 21.58; MS (m/z): 405 (M+ + 1, 50% %), 330 (100%); Anal. Calcd. for C23H24N4O3 (404.47): C, 68.30; H, 5.98; N, 13.85; Found: C, 68.65; H, 6.13; N, 13.52%.
4.1.1.4. N-(2-Methoxyphenyl)-4–(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)benzamide 11d
Grey powder (yield 74%); mp: 265 − 267 °C; FT-IR (v max, cm−1): 3289, 3040 (NH), 2922 (CH aliphatic), 1659 (C=O), 1602 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.77 (s, 1H), 9.33 (s, 1H), 7.98 − 7.93 (m, 2H), 7.80 (ddd, J = 19.3, 7.9, 1.6 Hz, 2H), 7.75 − 7.70 (m, 2H), 7.58 (ddd, J = 8.5, 7.0, 1.5 Hz, 1H), 7.54 (dd, J = 8.5, 1.3 Hz, 1H), 7.39 (ddd, J = 8.1, 7.0, 1.3 Hz, 1H), 7.18 (td, J = 7.8, 1.7 Hz, 1H), 7.10 (dd, J = 8.3, 1.4 Hz, 1H), 6.97 (td, J = 7.7, 1.4 Hz, 1H), 5.18 (s, 2H), 3.84 (s, 3H), 2.49 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 165.81, 165.25, 159.87, 157.97, 154.85, 142.03, 140.91, 133.47, 132.47, 130.20, 130.01, 129.82, 129.29, 129.22 (2 C), 123.94, 118.82 (2 C), 115.21, 112.98, 109.49, 106.45, 55.46, 45.80, 21.59; MS (m/z): 443 (M+ + 40%); Anal. Calcd. for C25H22N4O4 (442.48): C, 67.86; H, 5.01; N, 12.66; Found: C, 68.03; H, 4.77; N, 12.36%.
4.1.1.5. N-(3-Methoxyphenyl)-4–(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)benzamide 11e
Grey powder (yield 70%); mp: 237 − 239 °C; FT-IR (v max, cm−1): 3273 (NH), 2922 (CH aliphatic), 1655 (C=O), 1604 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.77 (s, 1H), 10.11 (s, 1H), 7.98 − 7.94 (m, 2H), 7.81 (dd, J = 8.1, 1.5 Hz, 1H), 7.72 (d, J = 8.7 Hz, 2H), 7.58 (td, J = 7.7, 6.9, 1.5 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.47 (t, J = 2.2 Hz, 1H), 7.41 − 7.35 (m, 2H), 7.25 (t, J = 8.1 Hz, 1H), 6.68 (dd, J = 8.3, 2.5 Hz, 1H), 5.18 (s, 2H), 3.76 (s, 3H), 2.49 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 165.81, 165.25, 159.87, 157.97, 154.85, 142.03, 140.91, 133.47, 132.47, 130.20, 130.01, 129.82, 129.29, 129.22(2 C), 123.94, 118.82 (2 C), 115.21, 112.98, 109.49, 106.45, 55.46, 45.80, 21.59; MS (m/z): 443 (M+ + 1, 40% %); Anal. Calcd. for C25H22N4O4 (442.48): C, 67.86; H, 5.01; N, 12.66; Found: C, 67.50; H, 4.95; N, 12.25%.
4.1.1.6. N-(4-Methoxyphenyl)-4–(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)benzamide 11f
Yellow crystals (yield 76%); mp: 324 − 227 °C; FT-IR (v max, cm−1): 3269 (NH), 2957, 2834 (CH aliphatic), 1655 (C=O), 1601 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.76 (s, 1H), 10.03 (s, 1H), 7.97 − 7.95 (m, 2H), 7.81 (dd, J = 8.0, 1.5 Hz, 1H), 7.73 − 7.70 (m, 2H), 7.68 − 7.66 (m, 2H), 7.58 (ddd, J = 8.5, 7.0, 1.5 Hz, 1H), 7.54 (dd, J = 8.5, 1.3 Hz, 1H), 7.38 (ddd, J = 8.1, 7.1, 1.3 Hz, 1H), 6.95 − 6.90 (m, 2H), 5.18 (s, 2H), 3.75 (s, 3H), 2.51 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 165.77, 164.81, 157.97, 155.92, 154.85, 141.84, 133.47, 132.76, 132.47, 130.19, 130.14, 129.28 (2 C), 129.08, 123.94, 122.41(2 C), 118.82(2 C), 115.20, 114.18(2 C), 55.63, 45.79, 21.59; MS (m/z): 443 (M+ + 1, 100%); Anal. Calcd. for C25H22N4O4 (442.48): C, 67.86; H, 5.01; N, 12.66; Found: C, 67.33; H, 4.97; N, 12.22%.
4.1.1.7. N-(4-Acetylphenyl)-4–(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)benzamide 11g
White crystals (yield 71%); mp: 313 − 315 °C; FT-IR (v max, cm−1): 3281 (NH), 2921 (CH aliphatic), 1659 (C=O), 1603 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.82 (s, 1H), 10.46 (s, 1H), 8.02 − 7.96 (m, 4H), 7.95 (d, J = 9.0 Hz, 2H), 7.81 (dd, J = 7.9, 1.5 Hz, 1H), 7.79 − 7.73 (m, 2H), 7.58 (ddd, J = 8.5, 6.9, 1.5 Hz, 1H), 7.54 (dd, J = 8.6, 1.5 Hz, 1H), 7.38 (ddd, J = 8.2, 7.0, 1.4 Hz, 1H), 5.19 (s, 2H), 2.55 (s, 3H), 2.49 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 197.05, 165.87, 165.64, 157.96, 154.85, 144.19, 142.34, 133.46, 132.47, 132.35, 130.19, 129.76 (2 C), 129.57, 129.46 (2 C), 129.29, 123.93, 119.84(2 C), 118.87 (2 C), 115.20, 45.81, 26.94, 21.59; Anal. Calcd. for C26H22N4O4 (454.49): C, 68.71; H, 4.88; N, 12.33; Found: C, 69.03; H, 4.71; N, 11.92%.
4.1.1.8. N-(4-Fluorophenyl)-4–(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)benzamide 11h
White powder (yield 72%); mp: 260 − 262 °C; FT-IR (v max, cm−1): 3274 (NH), 3038 (CH aromatic), 2945 (CH aliphatic), 1666, 1640 (C=O), 1605 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.88 (s, 1H), 10.63 (s, 1H), 8.44 (d, J = 8 Hz, 1H), 8.24 (d, J = 8 Hz, 1H), 8.03 (m, 2H), 7.83 − 7.74 (m, 2H), 7.73 (s, 1H), 7.63 (t, J = 10.3 Hz, 2H), 7.63 − 7.51 (m, 1H), 7.41 − 7.34 (m, 2H), 4.94 (s, 2H), 2.49 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 165.84, 165.43, 157.96, 154.85, 151.48, 147.63, 146.74, 139.02, 138.28, 133.46, 132.46, 131.23, 131.02, 130.20, 129.28, 125.86, 123.95, 119.16, 118.88, 118.78, 116.21, 115.18, 63.52, 21.58; MS (m/z): 431 (M+ + 1, 30% %), 201 (100%); Anal. Calcd. for C24H19FN4O3 (430.44): C, 66.97; H, 4.45; N, 13.02; Found: C, 66.57; H, 4.34; N, 12.74%.
4.1.1.9. 4–(2-(3-Methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)-N-(pyridin-2-yl)benzamide 11i
Buff powder (yield 70%); mp: 292 − 294 °C; FT-IR (v max, cm−1): 3438 (NH), 1660 (C=O), 1600 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.78 (s, 1H), 8.31 (s, 1H), 7.90 (ddd, J = 4.9, 1.9, 0.9 Hz, 1H), 7.89 (td, J = 7.7, 2.0 Hz, 1H), 7.80 (dd, J = 8.0, 1.5 Hz, 1H), 7.74 − 7.70 (m, 2H), 7.63 − 7.59 (m, 2H), 7.56 (ddd, J = 8.6, 7.1, 1.5 Hz, 1H), 7.52 − 7.48 (m, 2H), 7.37 (ddd, J = 8.1, 7.2, 1.2 Hz, 1H), 7.27 (ddd, J = 7.5, 4.8, 1.0 Hz, 1H), 5.14 (s, 2H), 2.48 (s, 3H), 13 C NMR (176 MHz, DMSO-d6) δ 172.54, 165.96, 157.95, 154.81, 154.18, 149.22, 142.85, 139.16, 133.41, 132.44, 130.93, 130.19 (2 C), 129.56, 129.27, 123.94, 122.89, 122.73, 118.99 (2 C), 115.17, 45.81, 21.57; Anal. Calcd. for C23H19N5O3 (413.44): C, 66.82; H, 4.63; N, 16.94; Found: C, 66.97; H, 4.46; N, 16.79%.
4.1.1.10. 4–(2-(3-Methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)-N-(thiazol-2-yl)benzamide 11j
Brown crystals (yield 71%); mp: 230 − 232 °C; FT-IR (v max, cm−1): 3413 (NH), 1660 (C=O), 1602 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 12.51 (s, 1H), 10.94 (s, 1H), 8.14 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 8.7 Hz, 1H), 7.82 − 7.75 (m, 2H), 7.73 (d, J = 8.5 Hz, 1H), 7.61 − 7.55 (m, 3H), 7.29 − 7.25 (m, 1H), 7.06 − 7.04 (m, 1H), 5.19 (d, J = 6.4 Hz, 2H), 2.49 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 172.05, 167.52, 166.18, 165.96, 157.97, 142.04, 133.45, 132.14, 130.34, 130.20, 129.85(2 C), 129.82, 129.04, 118.90, 118.60(2 C), 115.20, 114.21, 108.50, 45.84; MS (m/z): 420 (M+ + 1, 100%); Anal. Calcd. for C21H17N5O3S (419.46): C, 60.13; H, 4.09; N, 16.70; Found: C, 60.55; H, 3.68; N, 16.32%.
4.1.2. General procedure for synthesis of compounds 12a-k
To a solution of the potassium salt of 3-methylquinoxaline-2-thiol 4 (352 mg, 0.002 mol) in DMF (20 ml) the appropriate 4–(2-chloroacetamido)-N-substituted-benzamides 10a-k (0.002 mol) was added. The mixture was heated on a water bath for 6 h. After cooling to room temperature, the reaction mixture was poured onto crushed ice. The precipitated solids were filtered, dried, and crystalised from ethanol to give the target compounds 12a-k.
4.1.2.1. N-Methyl-4–(2-((3-methylquinoxalin-2-yl)thio)acetamido)benzamide 12a
Brown powder (yield 65%); mp: 219 − 221 °C; FT-IR (v max, cm−1): 3298 (NH), 2923 (CH aliphatic), 1649 (C=O), 1604 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.71 (s, 1H), 8.38 (q, J = 4.6 Hz, 1H), 7.85 − 7.80 (m, 3H), 7.68 (d, J = 8.4 Hz, 2H), 7.59 (t, J = 8.4 Hz, 1H), 7.55 (d, J = 8.4 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 5.19 (s, 2H), 2.79 (s, 3H), 2.30 (s, 3H); 13 C NMR (176 MHz, DMSO) δ 166.48, 165.70, 157.99, 154.86, 141.48, 133.48, 132.47, 130.19, 129.89, 129.28, 128.48 (2 C), 123.95, 118.85(2 C), 115.20, 45.76, 26.69, 21.61; Anal. Calcd. for C19H18N4O2S (366.44): C, 62.28; H, 4.95; N, 15.29; Found: C, 62.53; H, 5.42; N, 15.88%.
4.1.2.2. N-(sec-Butyl)-4–(2-((3-methylquinoxalin-2-yl)thio)acetamido)benzamide 12b
Brown powder (yield 65%); mp: 165–167 °C; FT-IR (v max, cm−1): 3286 (NH), 2965 (CH aliphatic), 1631 (C=O), 1608 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.66 (s, 1H), 8.05 (d, J = 8.2 Hz, 1H), 8.02 (d, J = 8.2 Hz, 1H), 7.96 (dd, J = 8.1, 1.5 Hz, 1H), 7.83 (d, J = 8.5 Hz, 2H), 7.82 (s, 1H), 7.72 (d, J = 6.9 Hz, 1H), 7.69 (d, J = 8.7 Hz, 2H), 4.30 (s, 2H), 3.91 (m, J = 7.1 Hz, 2H), 2.67 (s, 3H), 1.51 (dt, J = 22.9, 7.2 Hz, 2H), 1.13 (d, J = 6.6 Hz, 3H), 0.86 (t, J = 7.4 Hz, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 166.91, 165.53, 155.45, 151.96, 141.86, 140.81, 139.35, 130.09, 130.02, 128.89, 128.67, 128.62(2 C),127.37, 118.65(2 C), 46.79, 35.38, 29.34, 22.18, 20.78, 11.24; MS (m/z): 409 (M+ + 1, 100%, base beak); Anal. Calcd. for C22H24N4O2S (408.52): C, 64.68; H, 5.92; N, 13.71; Found: C, 64.99; H, 5.77; N, 13.50%.
4.1.2.3. N-Cyclopentyl-4–(2-((3-methylquinoxalin-2-yl)thio)acetamido)benzamide 12c
Grey powder (yield 72%); mp: 220 − 222 °C; FT-IR (v max, cm−1): 3285 (NH), 2954, 2866 (CH aliphatic), 1668, 1629 (C=O), 1607 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.66 (s, 1H), 8.15 (d, J = 7.2 Hz, 1H), 7.96 (dd, J = 8.1, 1.6 Hz, 1H), 7.85 − 7.80 (m, 3H), 7.71 (ddd, J = 8.3, 6.9, 1.6 Hz, 1H), 7.68 (dq, J = 9.8, 3.0, 2.2 Hz, 3H), 4.30 (s, 2H), 4.25 − 4.18 (m, 1H), 2.67 (s, 3H), 1.91 − 1.84 (m, 2H), 1.74 − 1.65 (m, 2H), 1.57 − 1.48 (m, 4H); 13 C NMR (176 MHz, DMSO-d6) δ 166.91, 165.79, 155.45, 151.97, 141.86, 140.81, 139.35, 130.02, 129.97, 128.90, 128.69, 128.67(2 C), 127.37, 118.61(2 C), 51.33, 35.38, 32.61(2 C), 24.09(2 C), 22.18; MS (m/z): 421 (M+ + 1, 100%); Anal. Calcd. for C23H24N4O2S (420.53): C, 65.69; H, 5.75; N, 13.32; Found: C, 65.23; H, 5.50; N, 13.02%.
4.1.2.4. N-(2-Methoxyphenyl)-4–(2-((3-methylquinoxalin-2-yl)thio)acetamido) benzamide 12d
Buff powder (yield 72%); mp: 185–187 °C; FT-IR (v max, cm−1): 3420 (NH), 1698, 1645 (C=O), 1602 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.75 (s, 1H), 9.31 (s, 1H), 7.99 − 7.93 (m, 3H), 7.85 − 7.82 (m, 1H), 7.80 (dd, J = 7.8, 1.6 Hz, 1H), 7.78 − 7.75 (m, 2H), 7.71 (dddd, J = 25.1, 8.4, 7.0, 1.5 Hz, 2H), 7.18 (ddd, J = 8.2, 7.4, 1.7 Hz, 1H), 7.10 (dd, J = 8.3, 1.3 Hz, 1H), 6.97 (td, J = 7.6, 1.4 Hz, 1H), 4.33 (s, 2H), 3.84 (s, 3H), 2.68 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 167.07, 164.77, 155.46, 151.98, 151.77, 142.51, 140.82, 139.36, 130.06, 129.49, 128.99, 128.92(2 C), 128.69, 127.41, 127.38, 125.97, 124.53, 120.67, 118.88(2 C), 111.80, 56.19, 35.44, 22.19; MS (m/z): 459 (M+ + 1, 70% %), 217 (100%); Anal. Calcd. for C25H22N4O3S (458.54): C, 65.49; H, 4.84; N, 12.22; Found: C, 65.11; H, 4.62; N, 12.56%.
4.1.2.5. N-(3-Methoxyphenyl)-4–(2-((3-methylquinoxalin-2-yl)thio)acetamido) benzamide 12e
Reddish crystals (yield 70%); mp: 232 − 234 °C; FT-IR (v max, cm−1): 3277 (NH), 3039 (CH aromatic), 2989, 2926, 2827 (CH aliphatic), 1678, 1650 (C=O); 1H NMR (700 MHz, DMSO-d6) δ 10.76 (s, 1H), 10.11 (s, 1H), 7.96 (d, J = 8.7 Hz, 3H), 7.83 (dd, J = 8.2, 1.5 Hz, 1H), 7.80 − 7.76 (m, 2H), 7.70 (dddd, J = 25.4, 8.4, 6.9, 1.5 Hz, 2H), 7.48 (t, J = 2.3 Hz, 1H), 7.40 − 7.35 (m, 1H), 7.25 (t, J = 8.1 Hz, 1H), 6.70 − 6.65 (m, 1H), 4.33 (s, 2H), 3.76 (s, 3H), 2.67 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 167.08, 165.36, 159.88, 155.43, 151.96, 142.49, 140.94, 140.82, 139.36, 130.02, 129.90, 129.81, 129.19 (2 C), 128.89, 128.68, 127.37, 118.78 (2 C), 112.97, 109.46, 106.44, 55.45, 35.44, 22.18; MS (m/z): 459 (M+ + 1, 100%); Anal. Calcd. for C25H22N4O3S (458.54): C, 65.49; H, 4.84; N, 12.22; Found: C, 65.83; H, 4.57; N, 11.94%.
4.1.2.6. N-(4-Methoxyphenyl)-4–(2-((3-methylquinoxalin-2-yl)thio)acetamido) benzamide 12f
White powder (yield 71%); mp: 250–252 °C; FT-IR (v max, cm−1): 3300 (NH), 3054 (CH aromatic), 2909, 2834 (CH aliphatic), 1677, 1644 (C=O), 1600 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.74 (s, 1H), 10.02 (s, 1H), 7.96 (t, J = 9.1 Hz, 3H), 7.83 (dd, J = 8.0, 2.8 Hz, 1H), 7.76 (dd, J = 8.6, 2.9 Hz, 2H), 7.72 (t, J = 7.5 Hz, 1H), 7.67 (dd, J = 9.1, 3.1 Hz, 3H), 6.93 (dd, J = 9.2, 3.0 Hz, 2H), 4.33 (s, 2H), 3.75 (s, 3H), 2.67 (d, J = 2.9 Hz, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 167.04, 164.91, 155.91, 155.45, 151.97, 142.30, 140.82, 139.36, 132.79, 130.03 (2 C), 129.05 (2 C), 128.89, 128.68, 127.37, 122.40 (2 C), 118.77 (2 C), 114.18 (2 C), 55.63, 35.43, 22.18; MS (m/z): 459 (M+ + 1, 100%); Anal. Calcd. for C25H22N4O3S (458.54): C, 65.49; H, 4.84; N, 12.22; Found: C, 65.02; H, 4.57; N, 11.99%.
4.1.2.7. N-(4-Acetylphenyl)-4–(2-((3-methylquinoxalin-2-yl)thio)acetamido)benzamide 12g
Brown powder (yield 77%); mp: 270 − 273 °C; FT-IR (v max, cm−1): 3300 (NH), 2918 (CH aliphatic), 1672, 1649 (C=O), 1590 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.78 (s, 1H), 10.45 (s, 1H), 7.99 (d, J = 2.3 Hz, 1H), 7.98 (d, J = 2.4 Hz, 2H), 7.97 (d, J = 1.9 Hz, 2H), 7.95 (s, 1H), 7.94 (d, J = 2.0 Hz, 1H), 7.83 (dd, J = 8.1, 1.6 Hz, 1H), 7.80 (d, J = 2.0 Hz, 1H), 7.79 (d, J = 1.9 Hz, 1H), 7.72 (ddd, J = 8.3, 6.9, 1.6 Hz, 1H), 7.68 (ddd, J = 8.3, 6.9, 1.6 Hz, 1H), 4.33 (s, 2H), 2.67 (s, 3H), 2.55 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 197.05, 167.14, 165.74, 155.43, 151.97, 144.22, 142.79, 140.81, 139.36, 132.33, 130.03, 129.76 (2 C), 129.43(2 C), 128.90, 128.69, 127.37, 119.84, 119.82(2 C), 118.81(2 C), 35.44, 26.93, 22.18; MS (m/z): 471 (M+ + 1, 100%); Anal. Calcd. for C26H22N4O3S (470.55): C, 66.37; H, 4.71; N, 11.91; Found: C, 65.94; H, 4.66; N, 11.58%.
4.1.2.8. N-(4-Fluorophenyl)-4–(2-((3-methylquinoxalin-2-yl)thio)acetamido)benzamide 12h
White powder (yield 72%); mp: 250 − 252 °C; FT-IR (v max, cm−1): 3261 (NH), 3042 CH aromatic), 2912 (CH aliphatic), 1659, 1640 (C=O), 1608 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.77 (s, 1H), 10.20 (s, 1H), 7.96 (d, J = 8.9 Hz, 3H), 7.83 (d, J = 8.1 Hz, 1H), 7.79 (t, J = 9.1 Hz, 4H), 7.74 − 7.65 (m, 2H), 7.19 (t, J = 8.7 Hz, 2H), 4.33 (s, 2H), 2.67 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 167.08, 165.26, 155.43, 151.96, 142.52, 140.82, 139.36, 130.01, 129.72, 129.17(2 C), 128.88, 128.68, 127.37, 122.60(2 C), 122.56, 118.80(2 C), 115.67(2 C), 115.54, 35.43, 22.18; MS (m/z): 447 (M+ + 1, 70% %); Anal. Calcd. for C24H19FN4O2S (446.50): C, 64.56; H, 4.29; F, 4.25; N, 12.55; Found: C, 64.12; H, 3.83; N, 12.14%.
4.1.2.9. 4–(2-((3-Methylquinoxalin-2-yl)thio)acetamido)-N-(pyridin-2-yl)benzamide 12i
Grey powder (yield 70%); mp: 215 − 217 °C; FT-IR (v max, cm−1): 3311 (NH), 1683 (C=O), 1593 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.77 (s, 1H), 10,20 (s, 1H), 8.30 (ddd, J = 4.8, 1.9, 0.8 Hz, 1H), 7.96 (dd, J = 8.0, 1.7 Hz, 1H), 7.88 (td, J = 7.8, 2.0 Hz, 1H), 7.81 − 7.77 (m, 1H), 7.73 − 7.71 (m, 2H), 7.69 (td, J = 7.8, 1.6 Hz, 2H), 7.67 − 7.65 (m, 2H), 7.49 (dt, J = 8.1, 1.0 Hz, 1H), 7.26 (ddd, J = 7.4, 4.8, 1.0 Hz, 1H), 4.29 (s, 2H), 2.66 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 172.58, 167.24, 155.37, 154.21, 151.94, 149.20, 143.34, 140.78, 139.35, 139.15, 130.95, 130.04, 129.28, 128.92 (2 C), 128.67, 127.37, 122.84, 122.68, 118.94 (2 C), 35.42, 22.17; MS (m/z): 430 (M+ + 1, 80%), 119 (100%); Anal. Calcd. for C23H19N5O2S (429.50): C, 64.32; H, 4.46; N, 16.31; Found: C, 64.61; H, 4.84; N, 15.92%.
4.1.2.10. 4–(2-((3-Methylquinoxalin-2-yl)thio)acetamido)-N-(m-tolyl)benzamide 12j
Buff powder (yield 73%); mp: 242–244 °C; FT-IR (v max, cm−1): 3287 (NH), 1674, 1643 (C=O), 1591 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.74 (s, 1H), 10.05 (s, 1H), 7.99 − 7.93 (m, 3H), 7.84 (dd, J = 8.1, 1.6 Hz, 1H), 7.79 − 7.75 (m, 2H), 7.71 (dddd, J = 24.1, 8.3, 7.0, 1.5 Hz, 2H), 7.61 (t, J = 2.0 Hz, 1H), 7.58 − 7.54 (m, 1H), 7.23 (t, J = 7.8 Hz, 1H), 6.94 − 6.90 (m, 1H), 4.33 (s, 2H), 2.68 (s, 3H), 2.31 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 167.06, 165.25, 155.45, 151.98, 142.42, 140.82, 139.65, 139.36, 138.16, 130.04, 129.96, 129.16 (2 C), 128.92, 128.88, 128.69, 127.38, 124.67, 121.33, 118.77 (2 C), 117.97, 35.43, 22.19, 21.70; MS (m/z): 443 (M+ + 1, 100%); Anal. Calcd. for C25H22N4O2S (442.54): C, 67.85; H, 5.01; N, 12.66; Found: C, 67.53; H, 4.94; N, 12.38%.
4.1.2.11. 4–(2-((3-Methylquinoxalin-2-yl)thio)acetamido)-N-(p-tolyl)benzamide 12k
White powder (yield 77%); mp: 259 − 261 °C; FT-IR (v max, cm−1): 3291, (NH), 3036 (CH aromatic), 2982, 2922 (CH aliphatic), 1661, 1641 (C=O), 1596 (C = N); 1H NMR (700 MHz, DMSO-d6) δ 10.74 (s, 1H), 10.05 (s, 1H), 7.96 (dd, J = 10.2, 8.4 Hz, 3H), 7.83 (dd, J = 8.1, 1.5 Hz, 1H), 7.77 (d, J = 8.5 Hz, 2H), 7.73 − 7.70 (m, 1H), 7.68 (ddd, J = 8.3, 7.0, 1.5 Hz, 1H), 7.65 (d, J = 8.2 Hz, 2H), 7.15 (d, J = 8.2 Hz, 2H), 4.33 (s, 2H), 2.67 (s, 3H), 2.28 (s, 3H); 13 C NMR (176 MHz, DMSO-d6) δ 167.05, 165.13, 155.45, 151.97, 142.37, 140.82, 139.36, 137.20, 132.90, 130.03, 130.01, 129.43(2 C), 129.12 (2 C), 128.90, 128.68, 127.37, 120.81 (2 C), 118.77 (2 C), 35.43, 22.19, 20.96; MS (m/z): 443 (M+ + 1, 100%); Anal. Calcd. for C25H22N4O2S (442.54): C, 67.85; H, 5.01; N, 12.66; Found: C, 67.45; H, 4.89; N, 12.21%.
4.2. Biological testing
4.2.1. In vitro cytotoxic activity
In vitro cytotoxicity was carried out using MTT assay protocolCitation47,Citation65–67 as described in Supplementary data.
4.2.2. In vitro VEGFR-2 kinase assay
In vitro VEGFR-2 inhibitory activity was assessed against. Human VEGFR-2 ELISA kit as described in Supplementary dataCitation68,Citation69.
4.2.3. In vitro cytotoxicity against normal cell
The toxicity of compounds 11e and 12e was assessed against normal cell lines (primary rat hepatocytes) according to method of two-steps in situ collagenase perfusion as described by SeglenCitation70 (Supplementary data).
4.2.4. Cell cycle analysis
The effect of compound 11e on cell cycle distribution was performed using propidium iodide (PI) staining technique as described in Supplementary dataCitation71–73.
4.2.5. Apoptosis analysis
The effect of compound 11e on cell apoptosis was investigated as described in Supplementary dataCitation74–76.
4.2.6. Western blot analysis
Western blot technique was applied to assess the potential effect of compound 11e on the expression of caspase‐9, caspase‐3, BAX, and Bcl‐2 as reported in Supplementary dataCitation77–79.
4.3. In silico studies
4.3.1. Docking studies
Crystal structure of VEGFR-2 [PDB ID: PDB ID: 2OH4, resolution: 2.05 Å] was obtained from Protein Data Bank. The docking investigation was accomplished using MOE2014 software. At first, the crystal structure of VEGFR-2 was prepared by removing water molecules. Only one chain was retained beside the co-crystallized ligand (sorafenib). Then, the selected chain was protonated and subjected to minimisation of energy process. Next, the active site of the target protein was defined.
Structures of the synthesised compounds and sorafenib were drawn using ChemBioDraw Ultra 14.0 and saved as MDL-SD format. Such file was opened using MOE to display the 3 D structures which were protonated and subjected to energy minimisation. Formerly, validation of the docking process was performed by docking the co-crystallized ligand against the isolated pocket of active site. The produced RMSD value indicated the validity of process. Finally, docking of the tested compounds was done through the dock option inserted in compute window. For each docked molecule, 30 docked poses were produced using ASE for scoring function and force field for refinement. The results of the docking process were then visualised using Discovery Studio 4.0 softwareCitation34,Citation80–83.
4.3.2. ADMET studies
ADMET descriptors were determined using Discovery studio 4.0 as according the reported methodCitation34,Citation80,Citation84 (Supplementary data).
4.3.3. Toxicity studies
Discovery studio 4.0 software was used to predict the toxicity potential of the synthesised compounds as reported in Supplementary dataCitation85.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group no (RG-1441–333).
Disclosure statement
There is no any conflict of interest. Authors thank Dr. Mohamad Elnagar, Pharmacology and Toxicology Department, Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt for his support in biological testing.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Baselga J, Bhardwaj N, Cantley LC, et al. AACR cancer progress report 2015. Clin Cancer Res. 2015;21:S1–S128.
- Daher IN, Daigle TR, Bhatia N, Durand J-B. The prevention of cardiovascular disease in cancer survivors. Tex Heart Inst J2012;39:190–8.
- WHO. Cancer. Available from: https://www.who.int/health-topics/cancer#tab=tab_1. [accessed May 2020].
- DE, Thurston Chemistry and pharmacology of anticancer drugs. 2nd ed. Boca Raton, FL: CRC Press; 2006.
- Roskoski R. Jr, The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res 2014;79:34–74.
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature 2001;411:355–65.
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000;103:211–25.
- Modi SJ, Kulkarni VM. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med Drug Discov 2019;2:100009.
- Mahdy HA, Ibrahim MK, Metwaly AM, et al. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4(3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorg Chem 2020;94:103422
- El-Adl K, El-Helby A-GA, Sakr H, et al. Design, synthesis, molecular docking and anticancer evaluations of 5-benzylidenethiazolidine-2, 4-dione derivatives targeting VEGFR-2 enzyme. Bioorg Chem 2020;102:104059.
- Olsson A-K, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol 2006;7:359–71.
- Melincovici CS, Boşca AB, Şuşman S, et al. Vascular endothelial growth factor (VEGF)-key factor in normal and pathological angiogenesis. Rom J Morphol Embryol 2018;59:455–67.
- Padro T, Bieker R, Ruiz S, et al. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia. Leukemia 2002;16:1302–10.
- Yang C, Qin S. Apatinib targets both tumor and endothelial cells in hepatocellular carcinoma. Cancer Med 2018;7:4570–83.
- Guo S, Colbert LS, Fuller M, et al. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim Biophys Acta 2010;1806:108–21.
- Martins SF, Garcia EA, LUZ MAM, et al. Clinicopathological correlation and prognostic significance of VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3 expression in colorectal cancer. Cancer Genomics Proteomics 2013;10:55–67.
- Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal 2007;19:2003–12.
- Morabito A, De Maio E, Maio MD, et al. Tyrosine kinase inhibitors of vascular endothelial growth factor receptors in clinical trials: current status and future directions. Oncologist 2006;11:753–64.
- Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol 2007;25:884–96.
- Steeghs N, Gelderblom H, Wessels J, et al. Pharmacogenetics of telatinib, a VEGFR-2 and VEGFR-3 tyrosine kinase inhibitor, used in patients with solid tumors. Invest New Drugs 2011;29:137–43.
- Roth GJ, Binder R, Colbatzky R, et al. Nintedanib: from discovery to the clinic. Washington, DC: ACS Publications; 2015.
- Adams CM, Anderson K, Artman Iii G, et al. The Discovery of N-(1-Methyl-5-(trifluoromethyl)-1 H-pyrazol-3-yl)-5-((6-((methylamino) methyl) pyrimidin-4-yl) oxy)-1 H-indole-1-carboxamide (Acrizanib), a VEGFR-2 inhibitor specifically designed for topical ocular delivery, as a therapy for neovascular age-related macular degeneration. J Med Chem 2018;61:1622–35.
- Sheng X, Yan X, Tang B, et al. A phase I clinical trial of CM082 (X-82) in combination with everolimus for treatment of metastatic renal cell carcinoma. Am Soc Clin Oncol 2017;35:4575–5.
- Moore KN, Jones SF, Kurkjian C, et al. Phase I, first-in-human trial of an oral VEGFR tyrosine kinase inhibitor (TKI) x-82 in patients (pts) with advanced solid tumors. Am Soc Clin Oncol 2012;30:3041.
- Xie Q-Q, Xie H-Z, Ren J-X, et al. Pharmacophore modeling studies of type I and type II kinase inhibitors of Tie2. J Mol Graph Model 2009;27:751–8.
- Lee K, Jeong K-W, Lee Y, et al. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur J Med Chem 2010;45:5420–7.
- Eskander RN, Tewari KS. Incorporation of anti-angiogenesis therapy in the management of advanced ovarian carcinoma-mechanistics, review of phase III randomized clinical trials, and regulatory implications. Gynecol Oncol 2014;132:496–505.
- Eissa IH, Ibrahim MK, Metwaly AM, et al. Design, molecular docking, in vitro, and in vivo studies of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors with potential activity against hepatocellular carcinoma. Bioorg Chem 2021;107:104532.
- El-Metwally SA, Abou-El-Regal MM, Eissa IH, et al. Discovery of thieno[2,3-d]pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg Chem 2021;112:104947.
- Alsaif NA, Dahab MA, Alanazi MM, et al. New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: design, molecular modeling, and synthesis. Bioorg Chem 2021;110:104807.
- Machado VA, Peixoto D, Costa R, et al. Synthesis, antiangiogenesis evaluation and molecular docking studies of 1-aryl-3-[(thieno[3,2-b]pyridin-7-ylthio)phenyl]ureas: discovery of a new substitution pattern for type II VEGFR-2 Tyr kinase inhibitors. Bioorg Med Chem 2015;23:6497–509.
- Wang Z, Wang N, Han S, et al. Dietary compound isoliquiritigenin inhibits breast cancer neoangiogenesis via VEGF/VEGFR-2 signaling pathway. PloS One 2013;8:e68566.
- Dietrich J, Hulme C, Hurley LH. The design, synthesis, and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: a structural analysis of the binding interactions of Gleevec, Nexavar, and BIRB-796. Bioorg Med Chem 2010;18:5738–48.
- Ibrahim MK, Eissa IH, Alesawy MS, et al. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of quinazolin-4(3H)-one derivatives as potential PPARγ and SUR agonists. Bioorg Med Chem 2017;25:4723–44.
- Eldehna WM, Abo-Ashour MF, Nocentini A, et al. Novel 4/3-((4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylidene)amino) benzenesulfonamides: synthesis, carbonic anhydrase inhibitory activity, anticancer activity and molecular modelling studies. Eur J Med Chem 2017;139:250–62.
- El-Zahabi MA, Sakr H, El-Adl K, et al. Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents. Bioorg Chem 2020;104:104218.
- Nasser AA, Eissa IH, Oun MR, et al. Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFRWT and EGFRT790M. Org Biomol Chem 2020;18:7608–34.
- El‐Helby AGA, Sakr H, Eissa IH, et al. Benzoxazole/benzothiazole‐derived VEGFR‐2 inhibitors: design, synthesis, molecular docking, and anticancer evaluations. Archiv Der Pharmazie 2019;352:1900178.
- El‐Helby AGA, Sakr H, Eissa IH, et al. Design, synthesis, molecular docking, and anticancer activity of benzoxazole derivatives as VEGFR‐2 inhibitors. Archiv Der Pharmazie 2019;352:1900113.
- Zhang L, Shan Y, Ji X, et al. Discovery and evaluation of triple inhibitors of VEGFR-2, TIE-2 and EphB4 as anti-angiogenic and anti-cancer agents. Oncotarget 2017;8:104745–60.
- Abdel-Mohsen HT, Abdullaziz MA, Kerdawy AME, et al. Targeting receptor tyrosine kinase VEGFR-2 in hepatocellular cancer: rational design, synthesis and biological evaluation of 1, 2-disubstituted benzimidazoles. Molecules 2020;25:770.
- Rampogu S, Baek A, Park C, et al. Discovery of small molecules that target vascular endothelial growth factor receptor-2 signalling pathway employing molecular modelling studies. Cells 2019;8:269.
- Hou T, Zhu L, Chen L, Xu X. Mapping the binding site of a large set of quinazoline type EGF-R inhibitors using molecular field analyses and molecular docking studies. J Chem Inf Comput Sci 2003;43:273–87.
- Ibrahim M-K, Abd-Elrahman AA, Ayyad RR, et al. Design and synthesis of some novel 2-(3-methyl-2-oxoquinoxalin-1 (2H)-yl)-N-(4-(substituted) phenyl) acetamide derivatives for biological evaluation as anticonvulsant agents. Bull Fac Pharm Cairo Univ 2013;51:101–11.
- Saoudi B, Teniou A, Debache A, et al. Cyclisation reaction between 3-methylquinoxaline-2-thione and benzaldehydes into 3-benzyl-2-aryl-thieno [2, 3-b] quinoxaline promoted by Brønsted acids. Comptes Rendus Chimie 2015;18:808–15.,
- Morrison D, Furst A. Quinoxaline-2-thiols. J Org Chem 1956;21:470–1.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Vermeulen K, Berneman ZN, Van Bockstaele DR. Cell cycle and apoptosis. Cell Prolif 2003;36:165–75.
- Naseri MH, Mahdavi M, Davoodi J, et al. Up regulation of Bax and down regulation of Bcl2 during 3-NC mediated apoptosis in human cancer cells. Cancer Cell Int 2015;15:55.
- Slee EA, Harte MT, Kluck RM, et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol 1999;144:281–92.
- Brentnall M, Rodriguez-Menocal L, De Guevara RL, et al. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol 2013;14:32.
- Antonsson B, Conti F, Ciavatta A, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science 1997;277:370–2.
- Perlman H, Zhang X, Chen MW, et al. An elevated bax/bcl-2 ratio corresponds with the onset of prostate epithelial cell apoptosis. Cell Death Differ 1999;6:48–54.
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2006;2:358–64.
- Roy PP, Roy K. QSAR studies of CYP2D6 inhibitor aryloxypropanolamines using 2D and 3D descriptors. Chem Biol Drug Design 2009;73:442–55.
- Ghafourian T, Amin Z. QSAR models for the prediction of plasma protein binding. BioImpacts 2013;3:21.
- Xia X, Maliski EG, Gallant P, Rogers D. Classification of kinase inhibitors using a Bayesian model. J Med Chem 2004;47:4463–70.
- BIOVIA, QSAR, ADMET and Predictive Toxicology. https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html [Accessed May 2020].
- Eissa IH, El-Helby A-GA, Mahdy HA, et al. Discovery of new quinazolin-4 (3H)-ones as VEGFR-2 inhibitors: design, synthesis, and anti-proliferative evaluation. Bioorg Chem 2020;105:104380.
- El-Helby A-GA, Sakr H, Ayyad RR, et al. Design, synthesis, molecular modeling, in vivo studies and anticancer activity evaluation of new phthalazine derivatives as potential DNA intercalators and topoisomerase II inhibitors. Bioorg Chem 2020;103:104233.
- Kanhed AA, Mehere AP, Pandey KR, Mahapatra DK. Mahapatra, 4-(2-chloroacetamido) Benzoic acid derivatives as local anesthetic agents: design, synthesis, and characterization. UK J Pharm Biosci 2016;4:35–44.
- Alanazi MM, Mahdy HA, Alsaif NA, et al. New bis ([1, 2, 4] triazolo)[4, 3-a: 3', 4'-c] quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: design, synthesis, in silico studies, and anticancer evaluation. Bioorg Chem 2021;112:104949.
- Alsaif MSTNA, Alanazi MM, Obaidullah AJ, et al. Discovery of new VEGFR-2 inhibitors based on bis([1, 2, 4]triazolo)[4,3-a:3',4'-c]quinoxaline derivatives as anticancer agents and apoptosis inducers. J Enzyme Inhib Med Chem 2021;36:1093–114.
- Abbass EM, Khalil AK, Mohamed MM, et al. Design, efficient synthesis, docking studies, and anticancer evaluation of new quinoxalines as potential intercalative Topo II inhibitors and apoptosis inducers. Bioorg Chem 2020;104:104255.
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986;89:271–7.
- Thabrew MI, Hughes RD, Mcfarlane IG. Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay. J Pharm Pharmacol 2011;49:1132–5.
- Al-Rashood ST, Hamed AR, Hassan GS, et al. Antitumor properties of certain spirooxindoles towards hepatocellular carcinoma endowed with antioxidant activity. J Enzyme Inhib Med Chem 2020;35:831–9.
- Abou-Seri SM, Eldehna WM, Ali MM, Abou El Ella DA. 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: synthesis and in vitro biological evaluation. Eur J Med Chem 2016;107:165–79.
- Sharma K, Suresh P, Mullangi R, Srinivas N. Quantitation of VEGFR2 (vascular endothelial growth factor receptor) inhibitors-review of assay methodologies and perspectives. Biomed Chromatogr 2015;29:803–34.
- Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol 1976;13:29–83.
- Wang J, Lenardo MJ. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci 2000;113:753–7.
- Eldehna WM, Hassan GS, Al-Rashood ST, et al. Synthesis and in vitro anticancer activity of certain novel 1-(2-methyl-6-arylpyridin-3-yl)-3-phenylureas as apoptosis-inducing agents. J Enzyme Inhib Med Chem 2019;34:322–32.
- Al-Warhi T, Abo-Ashour MF, Almahli H, et al. Novel [(N-alkyl-3-indolylmethylene)hydrazono]oxindoles arrest cell cycle and induce cell apoptosis by inhibiting CDK2 and Bcl-2: synthesis, biological evaluation and in silico studies. J Enzyme Inhib Med Chem 2020;35:1300–9.
- Lo KK-W, Lee TK-M, Lau JS-Y, et al. Luminescent biological probes derived from ruthenium(II) estradiol polypyridine complexes. Inorg Chem 2008;47:200–8.
- Sabt A, Abdelhafez OM, El-Haggar RS, et al. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: synthesis, in vitro biological evaluation, and QSAR studies. J Enzyme Inhib Med Chem 2018;33:1095–107.
- Al-Sanea MM, Al-Ansary GH, Elsayed ZM, et al. Development of 3-methyl/3-(morpholinomethyl)benzofuran derivatives as novel antitumor agents towards non-small cell lung cancer cells. J Enzyme Inhib Med Chem 2021;36:987–99.
- Balah A, Ezzat O, Akool E-S. Vitamin E inhibits cyclosporin A-induced CTGF and TIMP-1 expression by repressing ROS-mediated activation of TGF-β/Smad signaling pathway in rat liver. Int Immunopharmacol 2018;65:493–502.
- Aborehab NM, Elnagar MR, Waly NE. Gallic acid potentiates the apoptotic effect of paclitaxel and carboplatin via overexpression of Bax and P53 on the MCF‐7 human breast cancer cell line. J Biochem Mol Toxicol 2021;35:e22638.
- Elnagar MR, Walls AB, Helal GK, et al. Functional characterization of α7 nicotinic acetylcholine and NMDA receptor signaling in SH-SY5Y neuroblastoma cells in an ERK phosphorylation assay. Eur J Pharmacol 2018;826:106–13.
- El-Zahabi MA, Elbendary ER, Bamanie FH, et al. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of phthalimide-sulfonylurea hybrids as PPARγ and SUR agonists. Bioorg Chem 2019;91:103115.
- Ibrahim MK, Eissa IH, Abdallah AE, et al. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of novel quinoxaline derivatives as potential PPARγ and SUR agonists. Bioorg Med Chem 2017;25:1496–513.
- El-Gamal KM, El-Morsy AM, Saad AM, et al. Synthesis, docking, QSAR, ADMET and antimicrobial evaluation of new quinoline-3-carbonitrile derivatives as potential DNA-gyrase inhibitors. J Mol Struct 2018;1166:15–33.
- El-Naggar AM, Eissa IH, Belal A, El-Sayed AA. Design, eco-friendly synthesis, molecular modeling and anticancer evaluation of thiazol-5 (4 H)-ones as potential tubulin polymerization inhibitors targeting the colchicine binding site. RSC Adv 2020;10:2791–811.
- El-Demerdash A, Metwaly AM, Hassan A, et al. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against SARS-CoV-2 (COVID-19). Biomolecules 2021;11:460.
- Eissa IH, Dahab MA, Ibrahim MK, et al. Design and discovery of new antiproliferative 1, 2, 4-triazin-3 (2H)-ones as tubulin polymerization inhibitors targeting colchicine binding site. Bioorg Chem 2021;112:104965.