
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
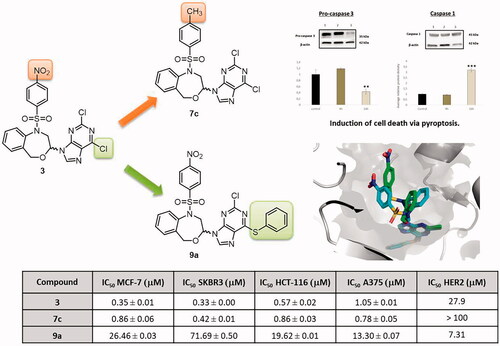
A series of 11 new substituted 1,5-dihydro-4,1-benzoxazepine derivatives was synthesised to study the influence of the methyl group in the 1-(benzenesulphonyl) moiety, the replacement of the purine by the benzotriazole bioisosteric analogue, and the introduction of a bulky substituent at position 6 of the purine, on the biological effects. Their inhibition against isolated HER2 was studied and the structure–activity relationships have been confirmed by molecular modelling studies. The most potent compound against isolated HER2 is 9a with an IC50 of 7.31 µM. We have investigated the effects of the target compounds on cell proliferation. The most active compound (7c) against all the tumour cell lines studied (IC50 0.42–0.86 µM) does not produce any modification in the expression of pro-caspase 3, but increases the caspase 1 expression, and promotes pyroptosis.
Graphical Abstract
Introduction
Cancer continues to be one of the leading causes of morbidity and mortality in the world, with approximately 18.1 million new cases in 2018. Population estimates indicate that the number of new cases will increase in the next two decades to 29.5 million a year in 2040. Breast cancer is the most commonly occurring cancer in women and the second most common cancer overall (11.6% of the total cases in 2018) followed by colorectal cancer (10.2%)Citation1. Although its incidence is much lower, melanoma also deserves special attention since it represents one of the fastest growing types of cancer, with advanced metastatic forms presenting high mortality ratesCitation2,Citation3.
The inherent heterogeneity of this disease, the dose-limiting adverse effects due to the unspecificity of classic chemotherapeutic agents and the appearance of chemoresistance are some of the drawbacks in current cancer treatments. To tackle these limitations, the development of new anticancer drugs is among the top priorities in medicinal chemistry.
Some of the most important signalling pathways involved in cancer are those linked to cell proliferation, differentiation and survival, all of which are promising targets for new antitumour drugs. As a result, the human epidermal growth factor receptor 2 (HER2) is often overexpressed in a variety of epithelial cancers in humansCitation4,Citation5. About 20% of breast cancer patients over-express HER2Citation6. HER2 is an essential protein for cell division and normal cell growth, its abnormal overexpression is related to cancerous processes associated with poor prognosis and an overall survival decrease in patientsCitation7.
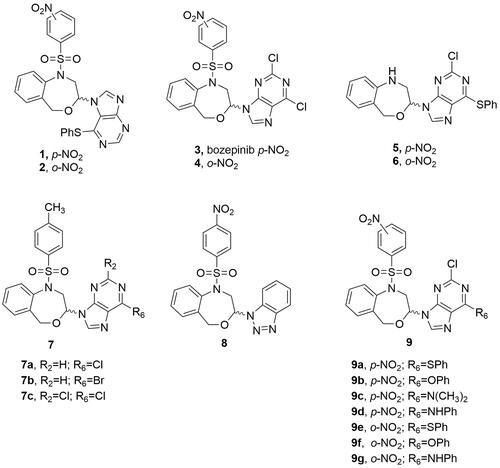
We previously described a series of 4,1-benzoxazepine-purines as potent antitumour compoundsCitation8–10. Compounds 1 and 2, with a phenylthio substituent in position 6 of the purine moiety () exhibited in vitro antiproliferative activities in the micromolar range (0.86 µM and 2.59 µM, respectively) against the MCF-7 human breast cancer cell lineCitation9. Later on, the N-9 regioisomers 3 and 4 () derived from the 2,6-dichloropurine also displayed relevant activities (0.355 µM and 0.383 µM, respectively), unlike analogues 5 and 6 which do not present the nitrobenzenesulphonyl group () notably decreased the cytotoxic effect of the compounds, with IC50 values of 9.71 µM (5) and 13.85 µM (6) against the MCF-7 cell line. (RS)-2,6-dichloro-9-[1-(p-nitrobenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-9H-purine (3, also named bozepinib, ) was the most potent and selective antitumour compound developed in our groupCitation11. The role of protein kinase-R (PKR) as a biological target for bozepinib in the apoptosis of breast and colon cancer cells was demonstrated. Furthermore, the inhibition of HER2, c-Jun N-terminal kinase (JNK) and extracellular signal-regulated protein kinases (ERKs), as well as antiangiogenic and antimigration activity, as well as the antitumour and antimetastatic activity in vivo, were reportedCitation11,Citation12. Very recently, we have demonstrated that bozepinib treatment was able to reduce glioblastoma cell viability by apoptosis and autophagy induction, without any change in interference on cell cycle progression or Akt activationCitation13. We also showed that one cycle of treatment with bozepinib selects resistant cells increasing the percentage of CD133+ and the activity of NF-ƙB cells and, however, two cycles of treatment eliminate the resistant cellsCitation13.
Figure 1. Chemical structures of the previously reported 4,1-benzoxazepine-purines (1-6) and molecular modifications employed for the design of the target molecules (7–9).
Taken together, these compounds may serve as prototypes for the development of even more potent structures endowed with a new mechanism of action.
Thus, we present here a new series of substituted 1,5-dihydro-4,1-benzoxazepines with the aim to develop antitumour agents. First, the methyl group was chosen to replace the nitro one of bozepinib in the present study. The different steps of the biotransformations that produce a primary amine from an aromatic nitro compound involve the nitro radical ion, the nitroso derivative, the nitroxyl radical, the hydroxylamine and then the primary amine. Each of these different intermediates could contribute to the toxicityCitation14. Then, the substitution of the nitro (3, ) by a methyl group (7, ) eliminates the above-mentioned toxic metabolites. In addition, this methyl group may be oxidised to an alcohol and further to a carboxylic acid, favouring its elimination from the body.
Purines are the most abundant and stable N-heterocyclic compounds found in nature. Many drug development programmes have focussed on structural modifications of natural purines, especially using bioisosteric ring systems. Thus, we also envisioned the benzotriazole analogue 8 for their close relationship with the purine fragment of compound 3. The benzotriazole can be considered as a privileged structure for its several pharmacological activitiesCitation15 being useful as scaffold for the design of new antitumour compoundsCitation16.
Finally, since the substituent size at the position 6 of the purine and the presence of the nitrobenzenesulphonyl group appear to be the essential characteristics for the antiproliferative effect, we decided to design molecules that combined both structural features (9, ) to complete the target compounds of the present study.
Experimental
Chemistry
General
Melting points were taken in open capillaries on a Stuart Scientific SMP3 electrothermal melting point apparatus and are uncorrected. Analytical thin layer chromatography (TLC) was performed using Merck Kieselgel 60 F254 aluminium plates and visualised by UV light or iodine. All evaporations were carried out in vacuo in a Büchi rotary evaporator and the pressure controlled by a Vacuubrand CVCII apparatus. Merck silicagel 60 (Merck, Kenilworth, NJ) with a particle size of 0.040–0.063 mm (230–400 mesh ASTM) was used for flash chromatography. Small-scale microwave-assisted synthesis was carried out in an Initiator 2.0 single-mode microwave instrument producing controlled irradiation at 2.450 GHz (Biotage AB, Upsala, Sweden). Reaction time refers to hold time at 110 °C or 160 °C, not to total irradiation time. These parameters were established following the basic principles of microwave assisted organic synthesis. The temperature was measured with an IR sensor on the outside of the reaction vessel. Nuclear magnetic resonance (NMR) spectra were recorded on a 500 MHz 1H and 125 MHz 13 C NMR Varian Direct Drive spectrometer at ambient temperature. Chemical shifts (δ) are quoted in parts per million (ppm) and are referenced to the residual solvent peak. Spin multiplicities are given as s (singlet), bs (broad singlet), d (doublet), dd (double doublet), ddd (double double doublet), pt (pseudo triplet) and m (multiplet). J values are given in Hz. Electrospray ionisation (ESI-TOF) mass spectra were carried out on a Waters LCT Premier Mass Spectrometer, respectively. Anhydrous CH2Cl2 and CH3CN were purchased from VWR International Eurolab (Barcelona, Spain). Anhydrous conditions were performed under argon. All reagents were purchased from Aldrich. Compounds 3Citation10, 4Citation10, 10Citation17 and 12Citation17 were synthesised as previously reported.
3-Methoxy-1-(p-methylbenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine (11)
Method a
To a solution of 10Citation17 (250 mg, 1.38 mmol) and p-methylbenzenesulphonyl chloride (532 mg, 2.79 mmol) in anhydrous CH2Cl2 (4.2 ml), was added triethylamine (0.582 ml, 4.18 mmol) at 0 °C under argon. The reaction mixture was microwave-irradiated at 110 °C for 30 min, then poured into water and extracted with CH2Cl2. The organic phase was washed with brine, dried over Na2SO4 and evaporated. The residue was purified by flash chromatography using EtOAc/hexane 1/7 as eluent.
Method b
To a solution of 10Citation17 (500 mg, 2.79 mmol) and pyridine (0.449 ml, 5.58 mmol) in anhydrous CH2Cl2 (25 ml), p-methylbenzenesulphonyl chloride (797 mg, 4.18 mmol) was added at 0 °C under argon. The reaction mixture was stirred for 3 days at room temperature, then poured into water and extracted with CH2Cl2. The organic phase were washed with 1 N HCl aq., water and brine, dried over Na2SO4 and evaporated. The residue was purified by flash chromatography using EtOAc/hexane 1/7 as eluent.
White solid, method a: 185 mg (40%), and method b: 620 mg (67%), mp: 106–107 °C. 1H NMR (CDCl3): δ (ppm) 7.56 (d, J = 8.3 Hz, 2H), 7.50 (d, J = 7.7 Hz, 1H), 7.30–7.14 (m, 5H), 4.66 (pt, J = 3.9, J = 3.8 Hz, 1H), 4.43 (d, J = 13.6 Hz, 1H), 4.10 (d, J = 13.6 Hz, 1H), 3.94–3.63 (bs, 2H), 3.38 (s, 3H), 2.40 (s, 3H). 13 C NMR (CDCl3): 144.35 (C), 140.53 (C), 138.64 (C), 138.20 (C), 130.15 (CH) (× 2), 130.04 (CH), 129.72 (CH), 129.66 (CH), 128.69 (CH), 128.51 (CH) (× 2), 101.68 (CH), 65.11 (CH2), 56.18 (CH3), 54.37 (CH2), 22.54 (CH3). HRMS (ESI-TOF) (m/z) calcd. for C17H19NO4SNa (M + Na)+ 356.0933, found 356.0932.
General procedure for the synthesis of 7a, 7b, 7c and 8
A suspension of 11 or 12Citation17 (1 equiv) and the corresponding purine derivative or benzotriazole (2.5 equiv) in anhydrous acetonitrile (5 ml/mmol) was prepared under argon and cooled to 0 °C and TMSCl (4 equiv), HMDS (4 equiv) and a 1 M solution of SnCl4 (4 equiv) in CH2Cl2 were subsequently added. The temperature was allowed to rise to 10 °C before microwave irradiating at 160 °C for 5 min. The reaction was quenched by cooling (ice/water bath) and by the addition of water. The pH was fixed to 7–8 with a saturated NaHCO3 solution and the aqueous phase extracted with EtOAc. The combined organic layers were dried (Na2SO4), filtered and evaporated. 7a–7c were purified by flash chromatography using EtOAc/hexane 1/5 as eluent. Compound 8 was obtained by washing the residue with CH2Cl2 and diethyl ether and the solid obtained was filtered.
6-Chloro-9-[1-(p-methylbenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-9H-purine (7a)
White solid, 25 mg (12%), mp: 178–180 °C. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.81 (s, 1H, H2pur), 8.19 (s, 1H), 7.76 (d, J = 8.0 Hz, 2H), 7.38–7.34 (m, 6H), 6.14 (d, J = 9.7 Hz, 1H), 4.78–4.68 (m, 2H, H), 4.56 (d, J = 13.6 Hz, 1H), 3.68 (dd, J = 14.8, 10.0 Hz, 1H), 2.46 (s, 3H). 13 C NMR (CDCl3, 75 MHz): 152.64 (CH), 151.67 (C), 151.11 (C), 144.70 (C), 143.06 (CH), 139.56 (C), 138.04 (C), 137.12 (C), 130.39 (CH) (×2), 130.14 (CH), 130.04 (CH), 129.22 (CH), 128.88 (CH), 127.62 (CH) (×2), 84.63 (CH), 71.81 (CH2), 54.52 (CH2), 21.88 (CH3). HRMS (ESI-TOF) (m/z) calcd. for C21H19N5O3SCl (M + H)+ 456.0897, found 456.0908.
6-Bromo-9-[1-(p-methylbenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-9H-purine (7b)
White solid, 15 mg (10%), mp: 169–171 °C. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.80 (s, 1H), 8.23 (s, 1H), 7.80 (d, J = 8.2 Hz, 2H, Hab), 7.41–7.37 (m, 6H), 6.16 (dd, J = 9.9, 1.4 Hz, 1H, H3), 4.82–4.72 (m, 2H), 4.57 (d, J = 13.6 Hz, 1H), 3.70 (dd, J = 14.8, 10.0 Hz, 1H), 2.50 (s, 3H). 13 C NMR (CDCl3, 75 MHz): 153.73 (CH), 151.01 (C), 145.86 (C), 144.08 (CH), 140.72 (C), 139.19 (C), 138.28 (C), 131.55 (CH) (×2), 131.30 (CH), 131.20 (CH), 130.38 (CH), 130.04 (CH), 128.78 (CH) (×2), 85.80 (CH), 72.98 (CH2), 55.68 (CH2), 23.04 (CH3). HRMS (ESI-TOF) (m/z) calcd. for C21H19N5O3SBr (M + H)+ 500.0392, found 500.0367.
2,6-Dichloro-9-[1-(p-methylbenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-9H-purine (7c)
White solid, 49.5 mg (28%), mp: 193–195 °C. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.17 (s, 1H), 7.82 (d, J = 8.3 Hz, 2H), 7.45–7.32 (m, 6H), 6.01 (dd, J = 10.0, 1,8 Hz, 1H), 4.73–4.69 (m, 2H), 4.56 (d, J = 13.6 Hz, 1H), 3.60 (dd, J = 14.9, 10.0 Hz, 1H), 2.48 (s, 3H). 13C NMR (CDCl3, 75 MHz): 153.29 (C), 152.12 (C), 151.99 (C), 144.61 (C), 143.28 (CH), 139.27 (C), 137.57 (C), 136.47 (C), 130.69 (C), 130.30 (CH) (×2), 129.88 (CH), 129.84 (CH), 129.30 (CH), 128.61 (CH), 127.49 (CH) (×2), 84.03 (CH), 71.72 (CH2), 54.08 (CH2), 21.68 (CH3). HRMS (ESI-TOF) (m/z) calcd. for C21H18N5O3SCl2 (M + H)+ 490.0507, found 490.0491.
2-[1-(p-Nitrobenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-2H-benzotriazole (8)
White pale solid, 38 mg (31%), mp: 209–211 °C. 1H NMR (DMSO-d6, 400 MHz,): δ (ppm) 8.43 (dd, J = 7.9, 2.0 Hz, 2H), 8.24 (dd, J = 7.9, 2.0 Hz, 2H), 8.13 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.64 (ddd, J = 8.1, 7.0, 0.8 Hz, 1H), 7.52–7.46 (m, 2H), 7.44–7.38 (m, 2H), 7.22–7.17 (m, 1H), 6.58 (dd, J = 9.9, 2.1 Hz, 1H), 4.98–4.83 (m, 3H), 4.26 (dd, J = 15.1, 9.9 Hz, 1H). 13 C NMR (DMSO-d6, 100 MHz) δ 150.65 (C), 146.18 (C), 145.54 (C), 139.36 (C), 137.53 (C), 132.89 (C), 129.94 (CH), 129.20 (CH) (×2), 128.91 (CH), 128.68 (CH) (×2), 127.43 (CH), 125.53 (CH) (×2), 125.25 (CH), 119.84 (CH), 111.67 (CH), 86.40 (CH), 69.70 (CH2), 53.50 (CH2). HRMS (ESI-TOF) (m/z) calcd. for C21H18N5O5S (M + H)+ 452.1029, found 452.1027.
General procedure for the synthesis of 9a and 9e (A)
To a solution of 1 or 2 (1.0 equiv) in DMF (5 ml/mmol), K2CO3 (1.6 equiv) and thiophenol (0.8 equiv) were added at room temperature and stirred for 1 h. The reaction mixture was concentrated and the residue was dissolved in CH2Cl2, washed with water and brine, dried (Na2SO4), filtered and evaporated. The crude products were purified by flash chromatography using EtOAc/hexane 3/1 as eluent.
2-Chloro-6-phenylthio-9-[1-(p-nitrobenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-9H-purine (9a)
White solid, 37 mg (72%), mp: 183–185 °C. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.44 (d, J = 8.5 Hz, 2H), 8.15 (d, J = 8.5 Hz, 2H), 8.03 (s, 1H), 7.67–7.61 (m, 2H), 7.50–7.46 (m, 3H), 7.46–7.32 (m, 4H), 5.94 (d, J = 9.3 Hz, 1H), 4.74 (t, J = 13.5 Hz, 2H), 4.55 (d, J = 13.5 Hz, 1H), 3.64 (dd, J = 14.2, 7.1 Hz, 1H). 13 C NMR (CDCl3, 100 MHz): 163.54 (C), 154.25 (C), 150.63 (C), 149.37(C), 146.18 (C), 141.02 (CH), 138.69 (C), 136.49 (C), 135.44 (CH) (×2), 130.27 (CH), 130.08 (CH), 129.91 (CH), 129.38 (CH) (×2), 129.20 (CH), 129.07 (CH), 129.03 (CH) (×2), 126.29 (C), 124.99 (CH) (×2), 84.31 (CH), 71.82 (CH2), 54.55 (CH2). HRMS (ESI-TOF) (m/z) calcd. for C26H20ClN6O5S2 (M + H)+ 595.0625, found 595.0652.
2-Chloro-6-phenylthio-9-[1-(o-nitrobenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-9H-purine (9e)
White solid, 20 mg (35%), mp: 212–214 °C. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.14 (dd, J = 7.1, 1.7 Hz, 1H), 8.03 (s, 1H), 7.85–7.74 (m, 3H), 7.68–7.62 (m, 2H), 7.51–7.43 (m, 3H), 7.42–7.38 (m, 2H), 7.36–7.30 (m, 1H), 7.16 (d, J = 7.7 Hz, 1H), 6.00 (dd, J = 10.0, 1.8 Hz, 1H), 5.01 (d, J = 13.7 Hz, 1H), 4.82 (d, J = 13.7 Hz, 1H), 4.72 (dd, J = 15.0, 1.8 Hz, 1H), 3.81 (dd, J = 10.0, 15.0 Hz, 1H). 13C NMR (CDCl3, 100 MHz): 163.23 (C), 154.06 (C), 149.48 (C), 147.76 (C), 141.03 (CH), 138.76 (C), 137.35 (C), 135.30 (CH) (×2), 134.53 (CH), 133.54 (C), 132.35 (CH), 132.31 (CH), 130.17 (CH), 129.69 (CH), 129.62 (CH), 129.22 (CH) (× 2), 129.10 (CH), 128.71 (CH), 126.42 (C), 124.63 (CH), 84.51 (CH), 71.81 (CH2), 54.65 (CH2). HRMS (ESI-TOF) (m/z) calcd. for C26H20ClN6O5S2 (M + H)+ 595.0625, found 595.0652.
General procedure for the synthesis of 9b, 9c, 9d, 9f and 9g (B)
A solution of 3 or 4 (1.0 equiv), phenol or aniline (2.0 equiv) and Et3N (2.0 equiv) in DMF (5 ml/mmol) was stirred at 80 °C (3 h for 9d and 9g, 6h for 9f, 20h for 9b and 9c). The reaction mixture was concentrated and the residue was dissolved in CH2Cl2, washed with water and brine, dried (Na2SO4), filtered and evaporated. The residue was purified by flash chromatography using EtOAc/hexane 1/1 for 9b and 9c, 2/1 for 9d and 9g and 4/1 for 9f as eluent.
2-Chloro-6-phenoxy-9-[1-(p-nitrobenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-9H-purine (9b)
Orange solid, 24 mg (48%), mp: 210–212 °C. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.45 (d, J = 8.7 Hz, 2H), 8.16 (d, J = 8.7 Hz, 2H), 8.07 (s, 1H), 7.48–7.35 (m, 6H), 7.33–7.25 (m, 3H), 5.98 (d, J = 9.4 Hz, 1H), 4.77 (d, J = 13.8 Hz, 2H); 4.56 (d, J = 13.8 Hz, 1H), 3.68 (dd, J = 14.7, 10.0 Hz, 1H). 13 C-NMR (CDCl3, 100 MHz): 160.41 (C), 153.66 (C), 152.11 (C), 150.65 (C), 146.25 (C), 140.83 (C), 138.71 (C), 136.52 (C), 130.28 (CH), 130.12 (CH), 129.71 (CH) (×2), 129.24 (CH), 129.10 (CH), 129.01 (CH) (×2), 126.17 (CH), 125.00 (CH) (×2), 121.53 (CH) (×2), 84.38 (CH), 71.83 (CH2), 54.53 (CH2). HRMS (ESI-TOF) (m/z) calcd. for C26H20ClN6O6S (M + H)+ 579.0854, found 579.0817.
2-Chloro-6-dimethylamino-9-[1-(p-nitrobenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-9H-purine (9c)
Yellow solid, 12 mg (26%), mp: 203–205 °C. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.46 (d, J = 8.2 Hz), 8.19 (d, J = 8.2 Hz), 7.91 (s, 1H), 7.49–7.41 (m, 2H), 7.39 (t, J = 7.2 Hz, 1H); 7.34 (d, J = 7.2 Hz, 1H), 5.88 (d, J = 6.7 Hz, 1H), 4.78–4.69 (m, 2H), 4.44 (d, J = 13.7 Hz, 1H), 4.12 (bs, 3H, N-CH3), 3.78 (bs, 3H, N-CH3), 3.55 (dd, J = 15.0, 10.1 Hz, 1H). 13C-NMR (CDCl3, 100 MHz): 154.72 (C), 153.59(C), 150.61 (C), 150.24(C), 146.17(C), 138.69(C), 136.32 (C), 135.95 (CH), 130.20 (CH), 130.00 (CH), 129.48 (CH), 129.19 (CH) (×2), 129.08 (CH), 125.02 (CH) (×2), 116.55 (C), 83.90 (CH), 71.87 (CH2), 54.52 (CH2), 44.11 (CH3), 43.53 (CH3). HRMS (ESI-TOF) (m/z) calcd. for C22H21ClN7O5S (M + H)+ 530.1013, found 530.1012.
2-Chloro-6-anilino-9-[1-(p-nitrobenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-9H-purine (9d)
Yellow solid, 44 mg (85%), mp: 182–183 °C. 1H NMR (CDCl3, 400 MHz): δ (ppm) 9.87 (bs, 1H), 8.54 (bs, 1H); 8.46 (d, J = 8.0 Hz, 2H), 8.20 (d, J = 8.0 Hz, 2H), 7.84 (d, J = 8.0 Hz, 2H), 7.46–734 (m, 6H), 7.19 (t, J = 7.3 Hz, 1H), 6.05 (s, 1H), 4.90 (d, J = 11.6 Hz, 1H), 4.83 (d, J = 13.3 Hz, 1H), 4.67 (d, J = 13.3 Hz, 1H), 3.58 (bs, 1H). 13C-NMR (CDCl3, 100 MHz): 157.03 (C), 150.71 (C), 150.34 (C), 148.24 (C), 145.86 (C), 138.78 (C), 137.41 (C), 136.22 (C), 135.95 (C), 130.33 (CH), 130.30 (CH), 129.28 (CH), 129.23 (CH) (×2), 129.16 (CH) (×2), 129.02 (CH), 125.15 (CH), 125.06 (CH) (×2), 120.72 (CH) (×2), 85.12 (CH), 72.14 (CH2), 54.22 (CH2). HRMS (ESI-TOF) (m/z) calcd. for C26H21ClN7O5S (M + H)+ 578.1013, found 578.0991.
2-Chloro-6-phenoxy-9-[1-(o-nitrobenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-9H-purine (9f)
Yellowish solid, 38 mg (47%), mp: 142–144 °C. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.11 (dd, J = 1.6, 7.3 Hz, 1H), 8.05 (s, 1H) 7.82–7.72 (m, 3H), 7.46–7.34 (m, 4H), 7.34–7.20 (m, 4H), 7.12 (d, J = 7.7 Hz, 1H), 6.03 (dd, J = 10.0, 1.8 Hz, 1H), 5.00 (d, J = 13.6 Hz, 1H), 4.80 (d, J = 13.6 Hz, 1H), 4.72 (dd, J = 14.9, 1.8 Hz, 1H), 3.81 (dd, J = 14.9, 10.0 Hz, 1H). 13C-NMR (CDCl3, 100 MHz): 160.41 (C), 153.59 (C), 153.38 (C), 152.21 (C), 147.92 (C), 141.20 (CH), 138.91 (C), 137.61 (C), 134.74 (CH), 133.73 (C), 132.55 (CH), 132.49 (CH), 130.38 (CH), 129.82 (CH), 129.74 (CH) (×2), 129.33 (CH), 128.85 (CH), 126.14 (CH), 124.84 (CH), 121.61 (CH) (×2), 120.72 (C), 84.83 (CH), 71.99 (CH2), 54.86 (CH2). HRMS (ESI-TOF) (m/z) calcd. for C26H20ClN6O6S (M + H)+ 579.0854, found 579.0817.
2-Chloro-6-anilino-9-[1-(o-nitrobenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-9H-purine (9g)
Yellowish solid, 43 mg (82%), mp: 152–157 °C. 1H NMR (CDCl3, 400 MHz): δ (ppm) 10.05 (bs, 1H), 8.54 (bs, 1H), 8.23–8.19 (m, 1H), 7.87 (d, J = 7.9 Hz, 2H), 7.85–7.80 (m, 2H), 7.79–7.75 (m, 1H), 7.43-7.31 (m, 5H), 7.21–7.15 (m, 2H), 6.08 (d, J = 9.3 Hz), 5.06 (d, J = 13.5 Hz, 1H), 4.89 (dd, J = 14.6, 5.1 Hz, 2H), 3.72 (dd, J = 14.6, 9.3 Hz, 1H). 13C-NMR (CDCl3, 100 MHz): 157.11 (C), 150,31 (C), 148.26 (C), 147.83 (C), 138.86 (C), 137.50 (C), 136.69 (C), 136.03 (C), 134.79 (CH), 133.38 (C), 132.61 (CH), 132.57 (CH), 130.29 (CH), 129.97 (CH), 129.28 (CH), 129.17 (CH) (×2), 128.89 (CH), 125.06 (CH), 124.83 (CH), 121.98 (CH), 120.81 (CH) (×2), 85.32 (CH), 72.29 (CH2), 54.69 (CH2). HRMS (ESI-TOF) (m/z) calcd. for C26H21ClN7O5S (M + H)+ 578.1013, found 578.0991.
Biology
HER2 inhibition assay
The IC50 profile of the target compounds was determined using isolated HER2. IC50 values were measured by testing 10 concentrations (1 × 10 −4 M to 3 × 10 −9 M) of each compound in singlicate. A radiometric protein kinase assay (33PanQinase® Activity Assay) was used for measuring the kinase activity of HER2. The assay was performed in 96-well FlashPlatesTM from PerkinElmer (Boston, MA) in a 50 µl reaction volume. The assay contained 70 mM HEPES-NaOH pH 7.5, 3 mM MgCl2, 3 mM MnCl2, 3 µM Na-orthovanadate, 1.2 mM DTT, 50 µg/ml PEG20000, ATP (corresponding to the apparent ATP-Km of the kinase), [γ-33P]-ATP (approx. 7.2 × 1005 cpm per well), HER2, and the target compound solutions in DMSO (the final DMSO concentration in the reaction cocktails was 1% in all cases). The reaction cocktails were incubated at 30 °C for 60 min. The reaction was halted with 50 µl of 2% (v/v) H3PO4, plates were aspirated and washed two times with 200 µl 0.9% (w/v) NaCl. Incorporation of 33Pi was determined with a microplate scintillation counter (Microbeta, Wallac, Waltham, MA).
Cell culture
MCF-7, HCT-116, A-375 and SKBR cells were grown at 37 °C in an atmosphere containing 5% CO2, with Dulbecco’s modified Eagle Medium (DMEM) (Gibco, Grand Island, NY) supplemented with 10% heat-inactivated foetal bovine serum (FBS) (Gibco), 2% l-glutamine, 2.7% sodium bicarbonate, 1% Hepes buffer, 40 mg/L gentamicin and 500 mg/L ampicillin.
Drug treatment
Compounds were dissolved in DMSO and stored at −20 °C. For each experiment, the stock solutions were further diluted in medium to obtain the desired concentrations. The final solvent concentration in cell culture was ≤ 0.1% v/v of DMSO, a concentration without any effect on cell replication. Parallel cultures of MCF-7, HCT-116, A375 and SKBR-3 cells in medium with DMSO were used as controls.
Proliferation assays
The effect of the compounds on cell viability was assessed using the sulphorhodamine-B colorimetric assay. Cells suspension (1 × 103 cells/well) were seeded onto 24-well plates and incubated for 24 h. The cells were then treated with different concentrations of drugs in their respective culture medium and maintained with the treatment for 3 days. Three days later, wells were aspirated and fresh medium and drug were added. Then cell cultures were maintained for 3 additional days. Thereafter, a Titertek Multiscan (Flow, Irvine, CA) at 492 nm was used. The linearity of the SRB assay was evaluated with a cell number for each cell stock before each cell growth experiment. The IC50 values were calculated from semi-logarithmic dose–response curves by linear interpolation. All the experiments were plated in triplicate wells and were carried out twice.
Protein analysis
We performed a western blot to study differences in protein expression after treatment with 5 µM of 7c.
SKBR cells seeded on 6-well plates were treated during 4 h and 16, lysed using RIPA Lysis Buffer System (Santa Cruz Biotechnology, Santa Cruz, CA) and quantified by Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). Western blot was realised using 4× Laemmli sample buffer (Bio-Rad, Hercules, CA) and separated by SDS-polyacrylamide gel electrophoresis (SDS–PAGE) and then, transferred to PVDF membranes. Blocking was realised with milk in PBS 5%.
The following primary antibodies were used to detect protein expression,: caspase 3: ref-9662S (Cell Signalling, Beverly, MA), caspase 1(14F468): sc-56036 (Santa Cruz Biotechnology, Santa Cruz, CA), cytochrome-c (A-8): sc-13156 (Santa Cruz Biotechnology, Santa Cruz, CA), Bax(B-9): sc-7480 (Santa Cruz Biotechnology, Santa Cruz, CA) and β-actin: sc-47778 (Santa Cruz Biotechnology, Santa Cruz, CA). Secondary antibodies used included mouse anti-rabbit IgG-HRP:sc-2357(Santa Cruz Biotechnology, Santa Cruz, CA) and anti-mouse IgG (m-IgGκ BP-HRP):sc-516102 (Santa Cruz Biotechnology, Santa Cruz, CA). Protein–antibody complexes were made visible using enhanced chemiluminescence (ECL, Bonus, Amersham, Little Chalfont, UK) with the program IMAGE READER LAS-4000 in a LAS-4000 imaging system. Quantity One program were used to analyse the intensity of the signal. The values of each band were normalised with its β-actin, and were relative to the control sample to which the value 1 was assigned.
Statistical analysis
Data are shown as the mean ± standard deviation. Two-tailed Student’s t-test was used to analyse differences between groups. p < 0.05 was accepted as the statistical significance level.
Molecular modelling
Protein preparation and ligand docking
The 3D structure of the tyrosine kinase domain of the HER2 was retrieved from the protein databank (PDB code 3RCD)Citation18, and prepared for docking simulations with the protein preparation wizard tool from the Schrödinger suiteCitation19. This stage includes the modelling of missing loop regions (757–760, 867–883 and 992–999 and 1013–1015, mostly resolved in the alternative structure 3PP0) with Prime, addition of protons and evaluation of the protonation state of ionisable residues with Epik. A docking grid was defined on the basis of the co-crystallised pyrrolo[3,2-d]pyrimidine inhibitorCitation18 with GlideCitation20. After failed attempts to obtain reliable docking poses with the default flexible ligand docking procedure within Glide, an alternative strategy was conceived to enhance the conformational sampling, based on a 2-stepwise protocol: (i) the ligand bozepinib was built in a reliable 3D conformation within maestro, in independent R and S stereoisomeric configurations. (ii) each stereoisomer was subjected to an exhaustive conformational search using the Conformational Search (default options) algorithm within MacromodelCitation19, generating 76 conformers for each stereoisomer. iii) each conformer was subjected to rigid-docking with Glide-XPCitation20. Three poses for each stereoisomer were retained for further analysis, based on a double criteria combining the highest docking scores and the best shape and electrostatic superposition with the co-crystallised ligand pyrrolo[3,2-d]pyrimidine inhibitor and with the ATP analogue crystalised in the related EGFR kinase (ANP, PDB code 2EB3). Each of these binding modes for bozepinib in the HER2 structure was used as a starting point to manually dock all reported compounds within the series by means of the “Flexible Ligand Alignment" tool in Maestro.
Free energy perturbation calculations
The automated QligFEP protocol was used for free energy perturbation (FEP) calculationsCitation21. This protocol relies on molecular dynamics (MD) simulations obtained with the software QCitation22 under spherical boundary conditions, using A 25 Å sphere centred on the core of geometry of the ligand. Protein atoms in the boundary of the sphere had a positional restraint of 10 kcal/mol/Å2, while solvent atoms were subjected to polarisation and radial restrains using the surface constrained all-atom solvent (SCAAS)Citation21,Citation23 model to mimic the properties of bulk water at the sphere surface. Atoms lying outside the simulation sphere are tightly constrained (200 kcal/mol/Å2 force constant) and excluded from the calculation of non-bonded interactions. Long-range electrostatics interactions beyond a 10 Å cut off were treated with the local reaction field methodCitation24, except for the atoms undergoing the FEP transformation where no cut-off was applied. Solvent bond and angles were constrained using the SHAKE algorithmCitation25. All titratable residues outside the sphere were neutralised and histidine residues were assigned a hydrogen atom on the ε nitrogen. Residue parameters were translated from the OPLS-AA/M force fieldCitation26 and the parameters for the ligand and lipids were obtained with the FFLD software within Schrödinger suiteCitation19. The simulation sphere was warmed up from 0.1 to 298 K, during a first equilibration period of 0.61 nanoseconds, where an initial restraint of 25 kcal/mol/Å2 imposed on all heavy atoms was slowly released for all complexes. Thereafter the system was subjected to ten parallel replicates of unrestrained MD, where the FEP protocol is applied for each ligand transformation. Each of these MD replicates starts with a 0.25 nanosecond unbiased equilibration period, with different initial velocities. Thereafter the FEP protocol follows, which consists of 21 FEP λ-windows, distributed using a sigmoidal function and consisting of 10 ps each for every investigated ligand pair. To fulfil a thermodynamic cycle and calculate relative binding free energies, parallel FEP transformations are run in a sphere of water for each ligand pair. In these water simulations, the same parameters apply (i.e. sphere size, simulation time, etc.), and the relative binding free energy difference was estimated by solving the thermodynamic cycle utilising the Bennett acceptance ratio (BAR)Citation27.
Results and discussion
Chemistry
The target compounds (7a–7c, 8 and 9a–9g) were synthesised as described in Scheme 1. Compound 11 was obtained from the N-unsubstituted acetal 1017 by applying two different methods (i, Scheme 1). The best conditions were room temperature for 3 days using pyridine as a base. Although the use of microwave considerably reduces the reaction time, the yield is lower.
Scheme 1. Reagents and conditions: (i) p-CH3PhSO2Cl, 0 °C, anhydrous CH2Cl2, argon, Method a: TEA, 110 °C microwave irradiation, 30 min, Method b: pyridine, 3 days, rt; (ii) purine or benzotriazole, TMSCl, HMDS, SnCl4 (1 M solution in CH2Cl2), 160 °C microwave irradiation, 5 min; (iii) PhSH, DMF, K2CO3, 1 h, rt for 9a and 9e, PhOH or PhNH2, DMF, TEA, 80 °C, 3 h for 9d and 9g, 6 h for 9f, 20 h for 9b and 9c.
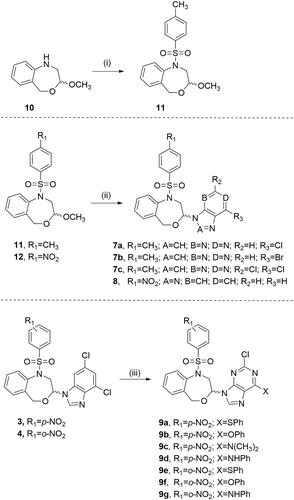
The preparation of 7a–7c and 8 was achieved by the microwave-assisted Vorbrüggen one-pot condensation of the acetals 11 and 12Citation16 and the commercially available 6-chloro-, 6-bromo- and 2,6-dichloro-purines or benzotriazole using chlorotrimethylsilane (TMSCl), 1,1,1,3,3,3-hexamethyldisilazane (HMDS) and tin(IV) chloride as the Lewis acid in anhydrous acetonitrile. The reaction mixture was microwave-irradiated at a temperature of 160 °C for 5 min (ii, Scheme 1).
The nucleophilic substitution of the chlorine in position 6 of 3Citation10 and 4Citation10 by phenylthio (9a and 9e), phenoxy (9b and 9f), or phenylamine (9d and 9g) was carried out in DMF using K2CO3 or TEA and applying different reaction conditions (see Experimental Part for details). Compound 9c was obtained along with 9b. The increase of the reaction time up to 20 h decomposes the DMF and the resulting dimethylamine competes as nucleophile as previously reportedCitation28.
HER2 inhibition studies
Human epidermal growth factor receptor 2 (HER2) is overexpressed in about 20% of all breast cancersCitation29. Bozepinib has proven to be a selective inhibitor of HER2 as we previously demonstrated by a kinase assay and then verified by immunoblot analysis in the HER2 positive SKBR-3 breast cancer cell lineCitation12. This prompted us to evaluated compounds 7–9 for their in vitro HER2 inhibitory effect. The IC50 profile of the target compounds was determined against isolated HER2 using a radiometric assay ().
Table 1. HER2 inhibition effect for compounds 3 and 7–9.
The nitro is crucial for the inhibition of the enzyme. Compounds that incorporate this group show IC50 ≤ 50 µM with the only exception of 9g.
The replacement of the 2,6-dichloropurine by the benzotriazole moiety does not modify the inhibitory activity of the structures (3, IC50 HER2 = 27.9 µM versus 8, IC50 HER2 = 22.2 µM).
The substitution of the chlorine at position 6 of the purine by the bulky groups phenylthio (9a and 9e, IC50 7.31 and 11.5 µM respectively) and phenyloxy (9b and 9f, IC50 13.3 and 24.6 µM respectively) favours the inhibition of the enzyme in contrast to the phenylamine substitution that leads to a decrease of the inhibition potency (9d and 9g, IC50 55.5 and > 100 µM, respectively). Compound 9c that presents the dimethylamine substituent displays higher inhibition against HER2 than their corresponding phenylamine counterparts 9d and 9g.
Finally, the p-nitro substitution in the structures leads to higher HER2 inhibition compared with the corresponding o-nitro derivatives (IC50 9a < 9e, 9b < 9f and 9d < 9g). In general, the p-nitro derivatives are twofold more active than the o-isomers.
The most potent compound against isolated HER2 is 9a with an IC50 of 7.31 µM. This structure bears a p-nitrobenzenesulphonyl moiety and the phenylthio substituent in the purine fragment.
Computational studies
To explore the SAR of the new series on HER2, we conducted a structure-based strategy as follows: (i) an exhaustive docking of the parent compound 3 on HER2 determined the most probable binding modes of this scaffold; (ii) the binding modes determined was used as a basis for parallel free energy perturbation (FEP) simulations, comparing the relative affinities of this compound with five selected ligands (7a–7c, 9a and 9c), representing different substitutions with varied effects on experimental binding affinity. The binding mode of 3 on HER2 was explored taking advantage of the crystallised tyrosine-kinase domain of the enzyme in complex with an ATP-competitive inhibitor (PDB code 3RCDCitation18). The selected structure presented a more open P-loop, as compared to the alternative structure 3PP0, allowing a better fitting of 3 (docking studies on that structure did not yield any reliable binding mode, data not shown). After independent docking conducted on the R and S stereoisomers of 3, three poses for each stereoisomer were retained for further analysis. We have previously demonstrated that both enantiomers are equally potentCitation10. Each of these occupied the ATP binding site, overlaying the ANP analogue co-crystalised in the related EGFR kinase, as well as the unrelated pyrrolo[3,2-d]pyrimidine co-crystallised with HER2 (see Methods). The six binding poses were the starting point of independent series of free energy perturbation (FEP) calculations aimed to explain the observed SAR within the series of bozepinib analogues ().
Table 2. Calculated and experimental free energy binding differences between selected derivatives and bozepinib (3) using the binding modes illustrated in .
In each case, we evaluated the pairwise binding free energy difference between bozepinib and five analogues, exploring different structural features: (i) the detrimental effect or removing the nitro group, with independence of the position of the halogen(s) (7a, 7b, 7c). (ii) The increase in affinity as a result of replacing one halogen by a phenylthio group (9a) or a dimethylamino group (9c). From the six docking alternatives filtered in the docking stage, only one pose per stereoisomer resulted in a qualitative correlation with the observed SAR of these compounds, the data shown in . Noteworthy, the orientation of the purine ring is equivalent between the two isomers, while the rest of the molecule is still bound in a similar area within the binding site, as shown in . Hence, the purine ring matches the position of the purine on ANP in EGFR, with the chlorine group in position 6 pointing towards the most buried region of the binding site (overlaying to the exocyclic amino group of ATP/ANP). This is precisely the position that can be replaced by either dimethylamino (9c) or a phenylthio group (9a), in the last case occupying the region that was accommodating the aminoaryl substituent of the co-crystallised pyrrolo[3,2-d]pyrimidine within residues Ala751, Ser783, Thr798 and Leu852 (see ).
Figure 2. Binding mode of compound 9a obtained by docking. Both R (left, blue) and S isomers (right, green) were docked independently, and used as a basis for parallel FEP simulations.
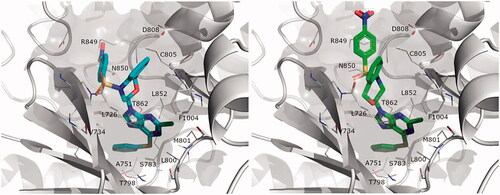
Our FEP calculations show indeed an enhancement of affinity by these two substitutions, in agreement with the experimental data (). Conversely, the replacement of the O or S atoms in 9a and 9c, respectively, by an NH functionality (9d) seems to be unfavourable for the binding affinity (). The corresponding FEP simulation of this compound is actually non-conclusive, since the results for either stereoisomer are contradictory (i.e. only the S-isomer is found to bind with less affinity than the reference compound), and the resulting small increase in binding affinity predicted for the racemic mixture is within the error bars of the calculation (−0.95 ± 1.03 kcal/mol, see ).
Along the compounds studied by molecular modelling, we consistently observed that the region occupied by the nitro group is somehow different between the R and S stereoisomers. In both cases, however, it is facilitated by the more open conformation of the P-loop observed as an induced-fit effect in the structure selected for docking and MD purposesCitation18. The β-hairpin encompassing this loop would accommodate the sulphonamide group, through mainchain or sidechain (Lys753) interactions. Though partially solvent exposed, the p-nitro group is placed between Asp808 and Arg849, offering a plausible justification for its key role in the affinity of this series. On the basis of these results, where both the R and S isomers show a similar orientation and predicted SAR in line with the experimental data, we postulate that the ATP binding site of the HER2 shows tolerance for the stereogenic centre, in other words that the binding of these compounds would not be stereoselective. shows an estimation of the relative free energy shift for each compound, as compared to bozepinib, for the racemic mixture estimated as an arithmetic average between the values independently obtained from the R and S isomers. We have previously shown no significant difference in activity of bozepinib enantiomers in MCF-7 and MDA-MB-231 tumour cellsCitation10.
Biological studies
We then investigated the effects of the target compounds on cell proliferation on the human breast cancer cell lines MCF-7 and SKBR-3, the human colon carcinoma cell line HCT-116 and the human malignant melanoma cell line A-375. The antiproliferative activity of 7–9 was evaluated using sulphorhodamine-B colorimetric assay after 72 h of treatment ().
Table 3. Antiproliferative activities for compounds 3 and 7–9 against the cancerous cell lines MCF-7, SKBR3, HCT-116 and A-375.
Compounds that bear halogen-substituted purines (7a and 7c) show IC50 values ≤ 1 μM against the four cell lines, with the exception of compound 7b with IC50 values ranging from 2.30 µM to 9.57 µM. The similarity of these compounds in terms of antiproliferative effects did not allow us to determine a clear structure–activity relationship.
All experiments were conducted in duplicate and gave similar results. The data are mean ± SD of three independent determinations. MCF-7: human breast cancer cell line; SKBR3: human breast cancer cell line; HCT-116: human colon cancer cell line and A-375: human melanoma cancer cell line.
The nitro and methyl groups, in general maintain the biological effect against the four cell lines (7a, 7b and 7c versus 3). The 6-chloro (7a) and 2,6-dichloropurine (7c) derivatives show IC50 values in the sub-micromolar range.
The substitution of the purine by benzotriazole considerable decreases the antiproliferative effect against MCF-7, SKBR3 and HCT-116 (8 versus 3).
Surprisingly, our results show that the substitution of the chlorine atom at position 6 of the purine by the bulky groups phenylthio (9a and 9e), phenoxy (9b and 9f), or phenylamine (9d and 9g) considerably decreases the antiproliferative effect. This effect is less pronounced in MCF-7 cells with IC50 values ranging from 13.80 µM (9d) to 26.46 µM (9a). In comparing the cytotoxicity of this series of compounds, 9a is the most potent structure against SKBR3, HCT-116 and A375.
Compound 7c displays the best antiproliferative effect against all the tumour cell lines studied (IC50 0.42–0.86 µM).
We selected 7c to decipher the mechanism by which it induces cell death. For this purpose, we studied HER2, cytochrome c, bax and caspase 3.
After normalisation and comparison to the relative value of the control, a clear decrease in the expression of the HER2 was observed reaching values of 0.48 and 0.07 after 4 h and 16 h of treatment respectively (). Since 7c has not shown inhibition of isolated HER2 in the kinase assay (), these results suggest that, unlike bozepinib, 7c does not act directly on the HER2 receptor but instead upstream. Trastuzumab, used currently as first-line clinical treatment for metastatic HER2 positive breast tumours, has improved overall survival in patients, although there are still a considerable percentage of patients with metastatic disease that ultimately developed resistance to this drugCitation30. Thus, 7c may be used in the future as a possible strategy to overcome treatment resistance.
Figure 3. Western blot analysis of (A) HER2, (B) Bax, (C) cytochrome c, (D) pro-caspase 3, (E) caspase 1. Band 1. Control, band 2. after 4 h treatment, band 3. after 16 h treatment. Relative quantification of the western blot proteins normalised with β-actin signal and relative to mock-treated cells (value 1) (a, b, c and d). Data expressed as a mean ± SD from three independent experiments performed in triplicated (*p < 0.05 versus control, **p < 0.01 versus control and ***p < 0.001 versus control).
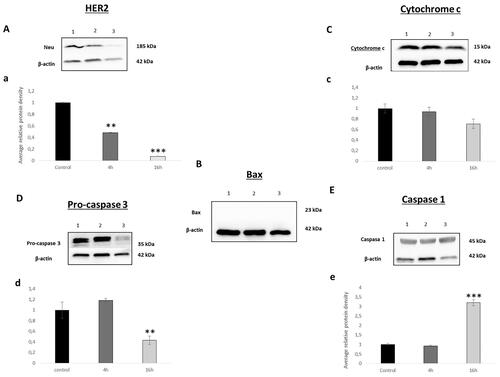
Bcl-2 family proteins play a key role in the mitochondrial cell death pathway by disrupting mitochondrial membrane potential and the release of apoptogenic factors, such as cytochrome c from mitochondria, in the cytosol. Bax is a member of the Bcl‐2 family and core regulators of the intrinsic pathway of apoptosis. Our results demonstrate none expression of Bax neither in the control nor in the treated cells (). These results were in accordance with what was obtained by analysing cytochrome c. As shown in , the normalised values were 0.93 and 0.70 after 4 h and 16 h of treatment. 7c has not modified the expression of cytochrome c, an electron acceptor protein located in the mitochondrial intermembrane and identified as a key molecule in intrinsic pathway of apoptosis indicating that there is no activation of the intrinsic apoptotic pathway mediated by mitochondrial effectsCitation31,Citation32
Compared with other members of the caspases family, caspase-3 is at the end of the caspase cascade and is activated by both the intrinsic and extrinsic death pathways in apoptosisCitation32,Citation33. In our analysis, and at our surprise, we have not observed any modification statistically significant in the expression of pro-caspase 3 at 4 h, while at 16 h this expression was approximately halved. Thus, the band corresponding to 4 h and 16 h after treatment had a value of 1.18 and 0.43, respectively (). This result indicates that cell death induced by 7c is not performed via apoptosis as we supposed a priori.
In recent years, researchers began to conduct in-depth investigations on cell death modes other than apoptosis, and the newly discovered pyroptosis gradually came into the field. Different studies have proved that pyroptosis can also shrink tumours and inhibit cells proliferationCitation34,Citation35. Traditionally pyroptosis is defined as caspase-1-mediated cell death, for that we studied the effect of 7c treatment on SKBR cell line. Our results demonstrated an increase in caspase 1 after 16 h of 7c exposition with normalised values reaching 3.1 (). Furthermore, recent study demonstrated that the apoptotic effector caspase, caspase-3 is capable of triggering pyroptosisCitation36,Citation37. In fact, Rogers et al. demonstrate that the decrease of pro-caspase-3 and increase of caspase 1 expression was related to the induction of secondary necrosis/pyroptosisCitation38. This can explain the decrease of pro-caspase 3 expression observed after 16 h of 7c exposition. For all the above, our finding suggests that 7c unlike 3, induces cell death via pyroptosis pathway.
Conclusions
We have designed and synthesised a series of 1,5-dihydro-4,1-benzoxazepines. The inhibitory activity against isolated HER-2 and the antiproliferative effects of the target compounds against the cancerous cell lines MCF-7, SKBR3, HCT-116 and A-375 have been determined. Our results have demonstrated that the presence of the nitro group is essential to inhibit HER2 but not cytotoxicity, since the substitution of the nitro for the methyl group maintains the toxicity against cancer cells. 7c shows the best activity against all the tumour cell lines studied (IC50 0.42–0.86 µM). This compound does not act directly on the HER2 receptor but acts upstream. To elucidate the mechanism by which this compound induces cell death we have studied the expression of cytochrome c, bax and caspase 3. Although 7c does not modified the expression of any of these key factors in the intrinsic pathway of apoptosis, an increase in caspase 1 levels was observed. These findings suggest that this compound induces cell death via pyroptosis. More studies should be carried out in order to delve deeper into this pathway and we plan to conduct in vivo studies in the near future.
Supplemental Material
Download PDF (1.5 MB)Acknowledgments
A. C. -G. is thankful to Consejería de Economía, Conocimiento, Empresas y Universidad of the Junta de Andalucía (Excellence Research Project P18-RT-1679) and the Oficina de Transferencia de Resultados de Investigación of the University of Granada (PR/17/006 project) for financial support. J. A. M. and H. B. thanks Instituto de Salud Carlos III (RTI2018-101309-B-C22), Fundación Mutua Madrileña (project FMM-AP16683-2017), Consejería de Salud Junta de Andalucía (PI-0089-2017) for financial support.
Disclosure statement
The authors report that they have no conflict of interest.
References
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424.
- Rozeman EA, Dekker TJA, Haanen J, Blank CU. Advanced melanoma: current treatment options, biomarkers, and future perspectives. Am J Clin Dermatol 2018;19:303–17.
- Couto GK, Segatto NV, Oliveira TL, et al. The melding of drug screening platforms for melanoma. Front Oncol 2019;9:512.
- Hendriks BS, Opresko LK, Wiley HS, Lauffenburger D. Coregulation of epidermal growth factor receptor/human epidermal growth factor receptor 2 (HER2) levels and locations: quantitative analysis of HER2 overexpression effects. Cancer Res 2003;63:1130–7.
- Yarden Y. The EGFR family and its ligands in human cancer. Signalling mechanisms and therapeutic opportunities. Eur J Cancer 2001;37:3–8.
- Victorino VJ, Campos FC, Herrera ACSA, et al. Overexpression of HER-2/neu protein attenuates the oxidative systemic profile in women diagnosed with breast cancer. Tumour Biol 2014;35:3025–34.
- Puhalla S, Brufsky A. Treatment of HER2-positive breast cancer: looking backwards briefly. Lancet Oncol 2013;14:1250–1.
- Díaz-Gavilán M, Gómez-Vidal JA, Choquesillo-Lazarte D, et al. Synthesis and reactivity of (RS)-6-chloro-7- or 9-(1,2,3,5-tetrahydro-4,1-benzoxazepin-3-yl)-7H- or 9Hpurines bearing a nitrobenzenesulfonyl group on the nitrogen atom. Tetrahedron 2007;63:5274–86.
- Díaz-Gavilán M, Gómez-Vidal JA, Rodríguez-Serrano F, et al. Anticancer activity of (1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl)-pyrimidines and -purines against the MCF-7 cell line: preliminary cDNA microarray studies. Bioorg Med Chem Lett 2008;18:1457–60.
- López-Cara LC, Conejo-García A, Marchal JA, et al. New (RS)-benzoxazepin-purines with antitumour activity: the chiral switch from (RS)-2,6-dichloro-9-[1-(p-nitrobenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepin-3-yl]-9H-purine. Eur J Med Chem 2011;46:249–58.
- Marchal JA, Carrasco E, Ramírez A, et al. Bozepinib, a novel small antitumour agent, induces PKR-mediated apoptosis and synergizes with IFNα triggering apoptosis, autophagy and senescence. Drug Des Dev Ther 2013;7:1301–13.
- Ramírez A, Boulaiz H, Morata-Tarifa C, et al. HER2-signaling pathway, JNK and ERKs kinases, and cancer stem-like cells are targets of Bozepinib small compound. Oncotarget 2014;5:3590–606.
- Dias AdF, Scholl JN, Moritz CEJ, et al. New insights into cytotoxic mechanisms of bozepinib against glioblastoma. Eur J Pharm Sci 2021;162:105823.
- Macherey AC, Dansette PM. Chapter 33: Biotransformations leading to toxic metabolites: chemical aspects. In: Camille Georges Wermuth, ed. The practice of medicinal chemistry. 3rd ed. New York, NY: Elsevier Academic Press; 2008: 674–696.
- Briguglio I, Piras S, Corona P, et al. Benzotriazole: an overview on its versatile biological behavior. Eur J Med Chem 2015;97:612–48.
- Beauchard A, Jaunet AI, Murillo L, et al. Synthesis and antitumoral activity of novel thiazolobenzotriazole, thiazoloindolo[3,2-c]quinoline and quinolinoquinoline derivatives . Eur J Med Chem 2009;44:3858–65.
- Díaz-Gavilán M, Rodríguez-Serrano F, Gómez-Vidal JA, et al. Synthesis of tetrahydrobenzoxazepine acetals with electron-withdrawing groups on the nitrogen atom. Novel scaffolds endowed with anticancer activity against breast cancer cells. Tetrahedron 2004;60:11547–57.
- Ishikawa T, Seto M, Banno M, et al. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J Med Chem 2011;54:8030–50.
- Schrödinger. Schrödinger. New York, NY: LLC; 2009.
- Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 2004;47:1750–9.
- Jespers W, Esguerra M, Åqvist J, Gutiérrez-de-Terán H. QligFEP: an automated workflow for small molecule free energy calculations in Q. J Cheminform 2019;11:26.
- Marelius J, Kolmodin K, Feierberg I, Aqvist J. Q: a molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J Mol Graph Model 1998;16:213–25.
- King G, Warshel A. A surface constrained all-atom solvent model for effective simulations of polar solutions. J Chem Phys 1989;91:3647–61.
- Lee FS, Warshel A. A local reaction field method for fast evaluation of long-range electrostatic interactions in molecular simulations. J Chem Phys 1992;97:3100–7.
- Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 1977;23:327–41.
- Robertson MJ, Tirado-Rives J, Jorgensen WL. Improved peptide and protein torsional energetics with the OPLSAA Force Field . J Chem Theory Comput 2015;11:3499–509.
- Bennett CH. Efficient estimation of free energy differences from Monte Carlo data. J Comput Phys 1976;22:245–68.
- Núñez MC, Pavani MG, Díaz-Gavilán M, et al. Synthesis and anticancer activity studies of novel 1-(2,3-dihydro-5H-1,4-benzodioxepin-3-yl)uracil and (6’-substituted)-7 or 9-(2,3-dihydro-5H-1,4-benzodioxepin-3-yl)-7H-or 9H-purines. Tetrahedron 2006;62:11724–33.
- Kunte S, Abraham J, Montero AJ. Novel HER2-targeted therapies for HER2-positive metastatic breast cancer. Cancer 2020;126:4278–88.
- Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res 2006;8:215.
- Javid J, Mir R, Julka PK, et al. Extracellular cytochrome c as a biomarker for monitoring therapeutic efficacy and prognosis of non-small cell lung cancer patients. Tumour Biol 2015;36:4253–60.
- Cáceres B, Ramirez A, Carrillo E, et al. Deciphering the mechanism of action involved in enhanced suicide gene colon cancer cell killer effect mediated by Gef and Apoptin. Cancers 2019;11:264.
- Jiang M, Qi L, Li L, Li Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov 2020;6:112.
- Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 2009;10:241–7.
- Ramos-Junior ES, Morandini AC. Gasdermin: a new player to the inflammasome game. Biomed J 2017;40:313–6.
- Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017;547:99–103.
- Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ 2019;26:99–114.
- Rogers C, Fernandes-Alnemri T, Mayes L, et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 2017;8:14128.