
Abstract
The ageing population is becoming a significant socio-economic issue. To address the expanding health gap, it is important to deepen our understanding of the mechanisms underlying ageing in various organisms at the single-cell level. The discovery of the antifungal, immunosuppressive, and anticancer drug rapamycin, which possesses the ability to extend the lifespan of several species, has prompted extensive research in the areas of cell metabolic regulation, development, and senescence. At the centre of this research is the mTOR pathway, with key roles in cell growth, proteosynthesis, ribosomal biogenesis, transcriptional regulation, glucose and lipid metabolism, and autophagy. Recently, it has become obvious that mTOR dysregulation is involved in several age-related diseases, such as cancer, neurodegenerative diseases, and type 2 diabetes mellitus. Additionally, mTOR hyperactivation affects the process of ageing per se. In this review, we provide an overview of recent insights into the mTOR signalling pathway, including its regulation and its influence on various hallmarks of ageing at the cellular level.
1. Introduction
Collecting soil samples on Easter Island (Rapa Nui) in the 1970s, no one could predict that bacterial cultures of Streptomyces hygroscopicus isolated from these samples would change the way we look at the cell cycle and help us better understand several signalling pathways and cellular processes. The isolated strain demonstrated inhibitory effects on Candida albicans, Microsporum gypseum, Trichophyton granulosum, as well as other gram-positive bacteria, while all gram-negative species were resistant. It was soon shown that S. hygroscopicus produces an antibiotic with antifungal properties, named rapamycin after its place of discovery (). This substance, also known by its generic name sirolimus, is a lipophilic macrolide with a white crystalline structure that is insoluble in water but readily soluble in organic solventsCitation1–3.
Figure 1. Chemical structure of rapamycin.
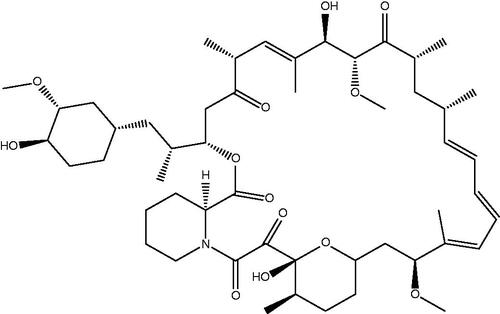
Rapamycin also shows antiproliferative effects in a wide range of taxa, from yeast to mammals. Moreover, Dr. Sehgal of the Ayerst Research Laboratories in Montreal (where the strain was first isolated) observed that the compound has anti-tumour activity, opening a new window for anticancer drug developmentCitation4.
The immunosuppressive activity of rapamycin was first evidenced by its ability to decrease humoral IgE production in rats and its preventive effects in two animal models of human autoimmune diseasesCitation5. Only the discovery of the immunosuppressive drug FK506 (tacrolimus) by the Fujisawa Pharmaceutical Laboratories drew more attention than rapamycin as an immunosuppressant. Both FK506 and rapamycin contain macrolactam rings with hemiketal-masked diketopipecolic acid amidic components that bind to the identical family of intracellular receptors. Despite these similarities, the mechanism of action of rapamycin is distinct from that of FK506. While FK506 binds to the immunophilin (peptidyl-prolyl cis/trans isomerase FKBP12, E.C. 5.2.1.8) receptor resulting in an immunophilin–drug complex with inhibitory effects against the activity of calcineurin (T cell antigen receptor activating enzyme), the rapamycin–immunophilin complex interferes with T cell growth factors. Nevertheless, both rapamycin and tacrolimus have significant clinical implications, since both are approved for use in prophylaxis for renal rejectionCitation6–9.
The exceptional antifungal, anticancer, and immunosuppressant properties of rapamycin have also been linked to its capacity to delay cell cycle progression at the G1 phaseCitation10. These effects were later related to the inhibition of large multiprotein complexes with serine/threonine kinase activity, called target of rapamycin (TOR)Citation11. In 2003, Vellai and colleagues revealed for the first time the role of TOR signalling in ageing. The downregulation of TOR signalling extends lifespan in Saccharomyces cerevisiae, Caenorhabditis elegans, and Drosophila melanogasterCitation12–14. In 2009, a study showed that the treatment of genetically heterogenous male and female mice with rapamycin extends mean and maximum lifespans, despite the initiation of treatment at an advanced age (corresponding to a human age of 60 years)Citation15. Several additional studies have shown a positive impact of rapamycin on lifespan in different genetically modified mouse strainsCitation16–20. Accordingly, rapamycin is a promising candidate for a mammalian longevity drug.
In this review, we summarise our current understanding of the roles of the mTOR pathway in ageing. In particular, we describe the structures, regulation, and functions of mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), with a focus on their effects on relevant cellular processes, as well as their associations with various age-related diseases.
2. Overview of mTOR complex 1 and mTOR complex 2
Cell proliferation and cell growth are two diverse yet connected processes involved in tissue, organ, and organismal development. Hartwell’s study of yeast mutants in the late 1960s, a clear distinction between these two processes was establishedCitation21. Owing to Hartwell’s intense focus on cell division, our understanding of the central regulator of cell proliferation—cyclin-dependent kinases (CDK)—is quite extensive. However, cell growth and its regulation were largely neglected until the late 1990s. Cell growth is not a mere accumulation of cell mass controlled by nutrients. Rather, it is a complex process of balanced macromolecular synthesis with a crucial role in cell physiology controlled by signalling pathways, among which TOR kinase plays a central roleCitation22.
Originally, TOR was identified from yeast mutants. However, it is highly conserved from yeast to mammals. In yeast, it is encoded by two genes, TOR1 and TOR2Citation23, while the human genome contains a single mTOR gene mapped to chromosomal band 1p36.2Citation24.
The mechanistic (formerly mammalian) target of rapamycin (mTOR), also known as RAFT, FRAP, or RAPT, was subsequently discovered by three independent groups in 1994 and 1995Citation11,Citation25–27. This 289-kDA serine/threonine kinase is a part of the phosphatidylinositol 3-kinase-related kinase (PIKK) family. This family includes several kinases involved in cell growth and cell cycle control, the maintenance of telomere length, DNA damage checkpoints, and recombinationCitation28. The mTOR protein consists of several domains, including at least 20 HEAT repeats (Huntingtin-Elongation factor 3-regulatory subunit of protein phospahase-2A, and TOR1) in the N-terminal region, the FAT domain (FKBP-associated protein/ataxia-telangiectasia mutated/transactivation-transformation domain-associated protein), which binds to Deptor (regulatory protein), the FRB domain (FKBP-rapamycin binding domain), which is the docking site for immunophilin-rapamycin inhibitory complex, kinase domain, and the FATC domain (FRAP/ATM/TRRAP/Carboxy terminal), which is involved in substrate recognitionCitation29.
The crystal structure of rapamycin-FKBP12 attached to the FRB domain of TOR revealed extensive interactions between the FRB domain and rapamycin, while the interaction between the FRB domain and immunophilin FKBP12 is limited, indicating that rapamycin is presented to TOR in a favourable conformation by FKBP12Citation30. The inhibitory effect of rapamycin on mTOR may be a consequence of the allosteric reduction of kinase domain-specific activity or a deficiency in the structural stability of the protein complex.
In eukaryotic cells, two large and functionally diverse multiprotein complexes have been identified: mTORC1 and mTORC2. Not only do the two complexes phosphorylate different substrates, contributing to various physiological functions, but their sensitivity to rapamycin differs. While mTORC1 is rapamycin-sensitive, mTORC2 is insensitive to acute rapamycin therapyCitation31. However, chronic exposure to the compound has a negative effect on mTORC2 activity. A recent proteomic analysis identified a novel mTOR complex containing GIT1 (GPCR kinase-interacting protein 1)Citation32. Focussing on the relatively well-established complexes, we provide detailed descriptions of the structures, upstream regulators, and downstream effectors of mTORC1 and mTORC2.
2.1. mTORC1—structure and function
mTORC1 multiprotein complex () consists of six protein components: mTOR, mammalian lethal SEC13 protein 8 (mLST8)Citation33, regulatory-associated protein of TOR (Raptor)Citation34, Tti1/Tel2 complexCitation35, Proline-rich Akt substrate 1 40 kDa (PRAS40)Citation36, and DEP domain-containing mTOR-interacting protein (Deptor)Citation37. mLST8 has a vital role in the phosphorylation of clearly established mTORC1 effectors: ribosomal S6 kinase 1 (S6K1) and the eukaryotic initiation factor 4E binding protein 1 (4EBP1)Citation33. Raptor and the Tti1/Tel2 complex are both involved in substrate recognition, regulation, and stabilisation of the mTORC1 assembly. Finally, PRAS40 and Deptor are regulatory proteins that inhibit the kinase activity of mTORC1Citation37. This multiprotein complex is an important regulator of cell growth and its effects are mediated by several signalling pathways, described in and the following text.
Figure 2. Structure and functions of mTOR complex 1. The best-known targets of mTORC1 phosphorylation S6K1 and 4EBP1 have a vital function in protein synthesis. Phosphorylated S6K1 consequently phosphorylates ribosomal protein S6 and commences cap-independent translation. Activated 4EBP1 relieves its inhibitory function towards eIF4E and initiates cap-dependent translation. Ribosomal biogenesis is enhanced by stimulated translation of 5′TOP mRNA via 4EBP1 and LARP1. Pol I and Pol III are activated by phosphorylation of TIF1A and inhibition of Maf1, respectively. Autophagy-related ULK1/ATG13/FIP200 protein complex is inhibited by mTORC1 phosphorylation, as well as WIPI2 (positive regulator of autophagy). On the other hand, autophagy suppressor DAP1 is activated. Protein expression of lipid and cholesterol homeostasis-involved genes is managed by transcription factors SREBP and PPAR-γ. mTORC1-mediated phosphorylation of lipin-1 alleviates SREBP inhibition. Several glycolytic enzyme genes are indirectly modulated by mTORC1 via transcription factors HIF1α and Myc. Expression of HIF1α is regulated either at the level of translation or via inhibited GSK3/Foxk1 pathway.
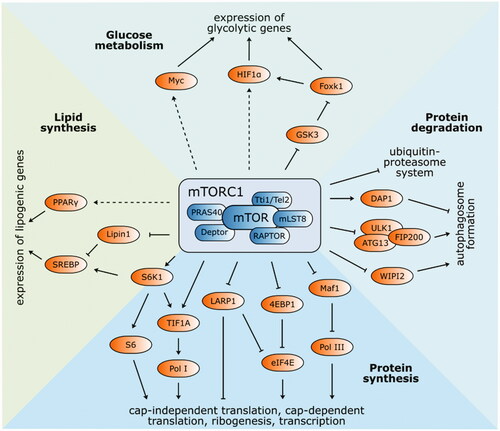
2.1.1. Protein synthesis
Containing a short sequence important for phosphorylation, called a TOR signalling (TOS) motif, both S6K1 and 4EBP1 bind to RaptorCitation38,Citation39. S6K1 is activated by mTORC1 via direct Thr389 phosphorylation, which leads to the phosphorylation of ribosomal S6, concluding in cap-independent translation.
The translation of mRNA containing a cap structure (m7GpppN moiety) at the 5′end is initiated by the mTORC1 phosphorylation of 4EBP1 on Thr37/46/70 and Ser65. The phosphorylation of 4EBP1 relieves its inhibitory activity towards eukaryotic initiation factor 4E (eIF4E), which might subsequently recognise the cap structure on mRNAs and commence cap-dependent translationCitation40.
2.1.2. Ribosome biogenesis and transcription
An additional mechanism by which the proteosynthesis capacity of cells is enhanced is the promotion of ribosomal biogenesis. Ribosomal proteins and other components of the translational apparatus are encoded by mRNAs carrying the 5′ terminal oligopyrimidine (5′TOP) motif with translational regulatory functionCitation41. The translation of these 5′TOP-capped mRNAs is enhanced by mTORC1 via two mechanisms, the phosphorylation of the previously mentioned effector 4EBP1 and La-related protein 1 (LARP1). Curiously, LARP1 may act as both an enhancer and repressor of 5′TOP mRNA translation. As a repressor, LARP1 prevents eIF4F complex formation by disrupting mRNA bindingCitation42. On the other hand, when phosphorylated by S6K1, LARP1 binds to the 3′UTR of 5′TOP mRNAs and increases translationCitation43.
In addition, the synthesis of rRNAs and tRNAs is positively controlled by mTORC1. Mammalian RNA Polymerase I (Pol I) transcribes precursor rRNA that sequentially forms three out of four mature rRNA species (5.8S, 18S, and 28S), while RNA Polymerase III (Pol III) transcribes the fourth RNA species, 5S rRNA, as well as tRNA. Pol I can be activated by the mTORC1 effector S6K1 or by the mTORC1-mediated phosphorylation of transcription initiation factor IA (TIF1A), which is then translocated to the nucleolus, where it can activate Pol ICitation44,Citation45. mTORC1 also phosphorylates (and consequently inhibits) the repressor of RNA polymerase III transcription MAF1 homolog (MAF1)Citation46. Moreover, the direct binding of mTORC1 to the promoter region of Pol I and Pol III increases gene expressionCitation47.
2.1.3. Autophagy
Autophagy, together with the ubiquitin-proteasome system (UPS), maintains the cytoplasmic protein quality in the cell. This catabolic process involving the sequestration and degradation of intracellular components is controlled by mTORC1 via the regulation of the ULK1/ATG13/FIP200 protein complex (unc-51-like kinase 1/autophagy-related gene13/focal adhesion kinase family-interacting protein of 200 kDa), which interacts with Raptor via ULK1. The phosphorylation of ULK1 and ATG13 inhibits their activity and suppresses autophagosome formation. Autophagy regulation is limited to mTORC1 since ULK1 does not bind to the mTORC2-specific component Rictor (rapamycin-insensitive companion of mTOR)Citation48,Citation49. mTORC1 also regulates autophagy by the activation of the autophagy suppressor death-associated protein 1 (DAP1)Citation50 or the phosphorylation of WD-repeat protein interacting with phosphoinositide 2 (WIPI2), a positive regulator of autophagosome formation prone to proteasomal degradation upon mTORC1 phosphorylationCitation51. Furthermore, mTORC1 is a repressor of the UPS. Upon rapamycin inhibition, the ubiquitination and subsequent proteasome degradation of long-lived proteins increasesCitation52. However, the biochemical mechanism by which mTORC1 suppresses UPS has not been identified and requires further studies.
2.1.4. Lipid and nucleotide synthesis, mitochondrial metabolism, and biogenesis
Lipid and cholesterol synthesis is promoted by mTORC1 via sterol regulatory element-binding protein (SREBP)Citation53,Citation54 and peroxisome proliferator-activated receptor-γ (PPAR-γ)Citation55. These transcription factors regulate the protein expression of lipid and cholesterol homeostasis-related genes. SREBP activation is promoted by low sterol levels in the cell or by the mTORC1-S6K1 pathwayCitation56. In addition, mTORC1 phosphorylates the SREBP suppressor lipin-1, which prevents its nuclear entry and attenuates SREBP inhibitionCitation57.
The mitochondrial oxidative function is balanced by mTORC1, as its inhibition by rapamycin decreases the activity of the peroxisome-proliferator-activated receptor coactivator (PGC)-1α and yin-yang1 (YY1) transcriptional complexCitation58. Furthermore, Schieke et al. observed decreases in mitochondrial membrane potential, reductions in oxygen consumption and cellular ATP levels, as well as modified mitochondrial phosphoproteomes as a result of mTORC1 inhibitionCitation59.
Lastly, mTORC1 intervenes in de novo pyrimidine synthesis by the S6K1-dependent activation of carbamoyl-phosphate synthetase (CAD)Citation60 and in purine synthesis by increasing the expression of MTHFD2 (mitochondrial tetrahydrofolate cycle component)Citation61.
2.1.5. Glucose metabolism
mTOR influences the glycolytic pathway at several points by the regulation of transcription factors, such as hypoxia-inducible factor 1-alpha (HIF1α) or Myc proto-oncogene protein (Myc)Citation62–64. The effects of mTORC1 are mediated by its ability to promote the transcription of HIF1α while inhibiting glycogen synthase kinase-3 (GSK3). Inhibited GSK3 represses the phosphorylation of forkhead box protein k1 (Foxk1) and subsequently results in the accumulation of hypophosphorylated Foxk1, which induces HIF1α transcriptionCitation65. HIF1α expression is also regulated at the level of translation, involving the mTORC1 target 4EBP1. Therefore, levels of genes encoding several glycolytic enzymes are indirectly modulated by mTORC1 via HIF1α. Another transcription factor influenced by mTORC1 is Myc, which stimulates genes involved in metabolism; however, its regulation by mTORC1 remains uncharacterisedCitation64,Citation66.
2.2. mTORC1—upstream regulators
Growth factors, such as insulin or insulin-like growth factor-1 (IGF-1), nutrients and amino acids, ATP, and stress regulate mTORC1 by different mechanisms, described in detail in . Growth factors increase the activity of mTORC1 via PI3K-AktCitation67–69 and RasCitation70 signalling pathways.mTORC1 amino acid-sensitive activation is independent of tuberous sclerosis complex (TSC1/2). The activation pathway relies on the interaction between four Rag proteins (Ras-related GTP-binding proteins; GTPases) and Raptor in the presence of amino acids (especially leucine and arginine) and the subsequent localisation of mTORC1 to the lysosomal surface containing its known activator Rheb (Ras homolog enriched in brain)Citation71. mTORC1 senses amino acids by two mechanisms (). Intra-lysosomal amino acids require the transporter SLC38A9 to interact with the Rag-Regulator-v-ATPase complex for the arginine-dependent activation of mTORC1Citation72,Citation73, while cytosolic amino acids activate GATOR1 and GATOR2 complexes in mTORC1 activation. GATOR1, tethered to the lysosomal surface by the KICSTOR complexCitation74, inhibits mTORC1 signalling, while GATOR2 acts as a positive regulator that interacts with GATOR1 at the lysosomal surface. In the presence of amino acids, the direct leucine sensor Sestrin2 and cytosolic arginine sensor CASTOR1 dissociate from GATOR2 and subsequently activate mTORC1Citation75–78.
Figure 3. Upstream regulators of mTORC1 and mTORC2. Growth factors (insulin/IGF-1) bind to their receptors that phosphorylate IRS and activate PI3K. PI3K-IRS complex converts phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3), which recruits phosphoinositide-dependent kinase 1 (PDK1) to activate Akt. PIP2-PIP3 conversion is counteracted by PTEN. Akt inhibits the TSC complex that acts as GTPase-activating (GAP) protein for Rheb. mTORC1 is activated by GTP-bound Rheb protein. Thus, when Akt activity is stimulated by PIP3, it phosphorylates TSC1/2 and switches off its inhibiting activity towards Rheb. Akt activates mTORC1 directly via phosphorylation of PRAS40. Amino acids induce activation of Rag proteins, which mediates translocation of mTORC1 to lysosomal surface. The intra lysosomal amino acids activate mTORC1 in an arginine-dependent manner via interaction of transporter SLC38A9 with the Rag-Ragulator-v-ATPase complex. Cytosolic amino acids engage the negative regulator GATOR1 (tethered to the lysosome by KICSTOR) and GATOR2. A direct leucine sensor Sestrin2 and arginine sensor CASTOR1 dissociate from GATOR2 in the presence of amino acids and release its inhibitory effect on GATOR1, thus positively regulate the mTORC1 pathway. AMPK negatively regulates mTORC1 either directly by Raptor phosphorylation, or via TSC1/2-Rheb axis. mTORC2 activation is PI3K-sensitive. Regulators of the mTOR pathway are depicted in green (positive) and red (negative). Dashed lines indicate indirect regulation.
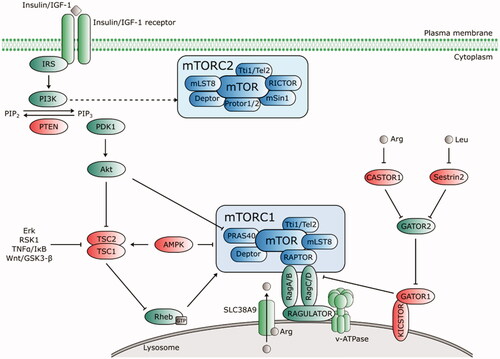
The processes controlled by mTORC1, such as the increase in cell mass, are energy-intensive. Under low cellular energy, AMP-activated protein kinase (AMPK) inhibits the mTORC1-dependent phosphorylation of S6K1 and 4EBP1 via the TSC1/2-Rheb axis, thus maintaining the energy balance of the cell. Additionally, AMPK reduces mTORC1 activity directly by Raptor phosphorylationCitation79,Citation80.
Induced expression of TSC2 and phosphatase and tensin homolog (PTEN) by DNA damage as well as AMPK activation by the induction of Sestrin 1/2 lead to decreased mTORC1 activityCitation81–83. In addition to DNA damage, hypoxia inhibits mTORC1 signalling and upregulates the inhibitory protein REDD1/2 (DNA damage-inducible transcript 4 protein), which activates TSC1/2Citation84. The mTORC1 pathway is coordinated by an S6K1-dependent negative feedback loop, which inhibits the PI3K-Akt axis upstream of PI3K; this pathway has profound implications for the treatment of tumorigenesis and metabolic diseases and the side effects of mTOR-based therapies (reviewed in Harrington et al. and ManningCitation85,Citation86).
2.3. mTORC2—structure, function, and regulation
Our understanding of mTORC1 activity is mainly based on experiments involving the rapamycin-induced inhibition of its signalling pathway. However, mTORC2 shows acute resistance to rapamycin and our understanding of its function is therefore rather limited. mTOR complex 2 contains the mTOR protein interacting with RictorCitation87, Protor 1 or 2, mammalian stress-activated protein kinase interacting protein 1 (mSin1), mLST8 The Role of mTOR in Age-related Diseases, Tti1/Tel2, and DeptorCitation88 (). The two latter components exert inhibitory effects, identical to the mTORC1 complex. Rictor stabilises mTORC2 assembly and activityCitation89, while mSin1 is involved in substrate recruitment and selectionCitation90. mLST8 is essential for mTORC2 function since its deletion completely suppresses complex activity. In contrast, mLST8 deletion has no clear effect on mTORC1Citation91. The last complex component Protor 1 is required for serum/glucocorticoid-regulated kinase 1 (SGK1) activation via mTORC2Citation92.mTORC2 mainly functions in the phosphorylation of several members of the AGC kinase family (e.g. PKCα, PKCβ, PKCγ, PKCε, PKCη/Λ, PKCδ, PKCθ, and SGK1)Citation93,Citation94. It regulates cell morphology by modulating actin polymerisation via protein kinase CαCitation95. mTORC2 alters the cytoskeletal structure via the Rho family of small GTPases, including Rac, Rho, and Cdc42, and activates Akt, which indirectly stimulates mTORC1 activity and other effectors in the PI3K-Akt axisCitation89.
Figure 4. Structure and functions of mTOR complex 2. mTORC2 mainly phosphorylate several kinases from AGC family (PKCα, PKCβ, PKCγ, PKCε, PKCη/Λ, PKCδ, PKCθ, and SGK1). It is involved in the cytoskeletal organisation by modulating actin polymerisation and cytoskeletal structure regulation via PKCα and Rho family of small GTPases (Rac, Rho, and CDC42), respectively. mTORC2 also activates Akt, an important enhancer and suppressor of several metabolic pathways engaging mTORC2 in metabolism regulation and cell survival.
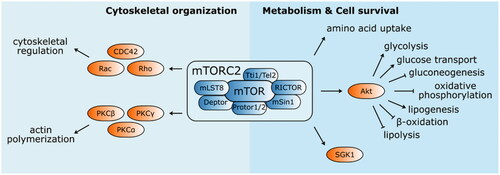
Recent studies have indicated that the localisation of mTORC2 near the plasma membrane is important for its activation. The activity of plasma membrane-associated mTORC2 is continuous and autonomous of PI3K signalling, different from mTORC1. However, the activity of endosomal mTORC2 is PI3K-sensitive, suggesting the existence of several mTORC2 subpopulationsCitation96. Upstream, mTORC2 activity is controlled by the ubiquitination of mLST8 and DeptorCitation97,Citation98 and the phosphorylation or acylation of Rictor on several residuesCitation99,Citation100. Additionally, ribosomesCitation101, small GTPasesCitation102, TSC complexCitation103,Citation104, and amino acidsCitation105 contribute to mTORC2 regulation. The mechanisms underlying these regulatory interactions as well as the sensitivity of mTORC2 to other mTORC1 upstream regulators demand further studies.mTORC2 dysregulation is related to several human diseases, such as cancer, type 2 diabetes mellitus (T2DM), and ageing. As mentioned above, mTORC2 activates Akt, an important enhancer of aerobic glycolysis (via the regulation of glycolytic enzymes, such as hexokinase or 6-phosphofructo-2-kinase) and inducer of GLUT1 expressionCitation106–108. Moreover, Akt inhibits the activity of pyruvate dehydrogenase (PDH), a mitochondrial respiration enzymeCitation109. mTORC2 upregulates glycolysis by inhibiting the phosphorylation of Class IIa histone deacetylases, leading to increased acetylation of forkhead box protein O1 (FoxO1) and FoxO3 and the subsequent upregulation of Myc, which activates various genes, such as lactate dehydrogenase (LDH) and the pyruvate kinase PKM2Citation110–112. The regulation of glucose metabolism by increasing glucose uptake and glycolysis and inhibiting gluconeogenesis and oxidative phosphorylation might explain the abnormal glucose metabolism in T2DM and might contribute to the Warburg effect in cancer cellsCitation113.
In addition, there is compelling evidence that mTORC2 has a role in lipid metabolism; in particular, it increases lipogenesis and suppresses lipolysis and fatty acid β-oxidation via Akt and SREBP pathwaysCitation114. mTORC2 dysregulation is linked to insulin resistance and lipogenesis-induced hepatocellular carcinoma via altered lipid metabolismCitation115–120.mTORC2 also regulates amino acid uptake by increasing the expression of surface amino acid transportersCitation121 and altering their activity by phosphorylationCitation122. Direct mTORC2 phosphorylation of glutamate/cystine antiporter (xCT) decreases cystine uptake, thereby reducing the synthesis of glutathione and subsequently decreasing the cellular reactive oxygen species (ROS) buffering capacityCitation122. The inhibition of mTORC2 by Torin 1 and Rictor silencing by siRNAs decrease nucleotide synthesisCitation123. The mTORC2-mediated activation of Akt regulates nucleotide synthesis by modulating phosphoribosylpyrophosphate (PRPP) production and by altering the activity of the bifunctional purine biosynthesis protein ATICCitation124.
The exact role of mTORC2 in ageing remains unknown. Longevity studies using murine models have yielded conflicting results. In mouse models with depleted or deleted Rictor, lifespan is reduced in males but not in females, and the deleterious effect was independent of glucose intoleranceCitation125. On the other hand, in heterozygous Akt1 mice, the lifespan was increasedCitation126, suggesting that the role of mTORC2 in ageing and age-related diseases is complex and influenced by several factors.
3. Age-related diseases and mTOR
Several disorders have age as a risk factor, including neurodegenerative diseases (e.g. Parkinson’s disease [PD] and Alzheimer’s disease [AD]), cancer, and T2DM. The hyperfunction of mTOR has been observed in some of these disorders. Furthermore, the inhibition with rapamycin results attenuates the pathological processes in these diseases. However, there are still unresolved issues, including the extent to which mTOR regulates ageing per se, the extent to which mTOR influences pathologies, and the effectiveness of mTOR inhibition for the treatment of these diseases and even for slowing ageing. Although there is no simple answer, the amelioration of age-related diseases by mTOR inhibition is still a satisfying result of rapamycin therapy, because this approach may extend the lifespan, delay these pathologies, and promote healthy ageing.
In the text below, we provide a short description of the effects of mTOR and its inhibition on age-related diseases.
3.1. Type 2 diabetes mellitus
T2DM is an epidemic endocrine disorder with multifactorial causes. The prediabetic stage is characterised by insulin resistance and hyperinsulinemia, involving overworked β-islets. Progression to the second stage is associated with hypoinsulinemia and hyperglycaemia due to the failure of β-cell compensation for insulin resistance. Moreover, hyperamylinemia is found in insulin-resistant patients. The oligomerisation of amylin has a toxic effect on pancreatic β-cells, in which misfolded proteins alter endoplasmic reticulum (ER) homeostasis and cause prolonged unfolded protein response (UPR), resulting in β-cell deathCitation127.
In contrast to its general role in organisms, autophagy increases pancreatic β-cell survival. Ebato et al. and Jung et al. used mouse models with the deletion of Atg-7 (autophagy-related 7) in pancreatic β-cellsCitation128,Citation129 and found that autophagy is necessary to maintain the structural and functional capacity of pancreatic β-cells in T2DM. Furthermore, autophagy protects against ER stress in insulin secretion-deficient β-cells and avoids the toxicity of human amylin (hIAPP)Citation127,Citation130.
The upstream regulation of autophagy by mTORC1 may be important in the progression of T2DM. In fact, an increase in pancreatic β-cell death and deterioration of autophagy has been observed in a mouse model of chronic mTORC1 overactivation. βTSC2−/− mice show an early increase in β-cell mass and elevated insulin levels; however, the opposite effects are observed in older mice, showing a drop in β-cell mass and lower insulin levels. Older mice also accumulate ER stress markers, sequestosome-1 (p62/SQSTM1), and apoptosis markersCitation131. In addition, an increase in mitochondrial mass and impaired mitophagy (mitochondrial autophagy) further support the link between autophagy impairment as a result of mTORC1 hyperactivation and mitochondrial dysfunction contributing to pancreatic β-cell failureCitation132,Citation133.
3.2. Alzheimer’s disease
AD is an age-related metabolic neurodegenerative disorder whose exact cause and pathogenesis remain unclear. It is defined by a progressive cognitive decline and formation of senile plaques and neurofibrillary tangles. Several theories for the aetiology of the disease include the β-amyloid (Aβ) cascade hypothesisCitation134–136 and tau hyperphosphorylation hypothesisCitation137–139. However, effective therapies based on these two theories are still lacking. Indeed, growing evidence indicates that dysfunctional cerebral glucose metabolism is a pathophysiological feature in ADCitation140–142. Glucose transportation (depending on the function of astrocytes and glucose transporters)Citation143 and intracellular oxidative catabolism (including glycolysis, the pentose phosphate pathway in the cytoplasm, Krebs cycle, and oxidative phosphorylation) are two main processes involved in cerebral glucose metabolism. In patients with AD, glucose transportation abnormalities and intracellular metabolic alterations occur due to insulin resistance and mitochondrial dysfunction, respectivelyCitation144,Citation145.
mTOR is highly expressed in the brain. mTORC1 and mTORC2 are fundamental for normal neuronal development (although the mechanism is still not fully understood) as well as for the maintenance of synaptic plasticityCitation146. In the aged brain, mTOR is involved in translation and autophagy regulation, preventing the accumulation of toxic protein aggregates and neuronal degeneration (reviewed in Takei et al.Citation147).
Analyses of both AD brains and mouse models have shown that mTOR signalling is upregulated during neurodegenerative development, with particularly high levels of 4EBP1 and S6K1 in the hippocampus and other areasCitation148. Caccamo et al. detected mTOR hyperactivity in cell lines transfected with mutant amyloid precursor protein (APP) and in an animal model of AD (3 × Tg-AD mice), in which Aβ-induced mTOR hyperactivity is mediated by PRAS40Citation149. Additionally, the continuous activation of the neuronal PI3K/Akt/mTOR axis in the AD brain causes insulin receptor substrate 1 (IRS1) inhibition, disabling normal insulin activation of this axis, providing a link between the pathology of AD and insulin resistanceCitation150,Citation151.
Autophagy is altered in the AD brain and animal models of AD. The activity of this well-characterised effector pathway downstream of mTOR is reduced in AD due to the hyperactivation of the PI3K/Akt/mTOR axis, resulting in the reduced clearance and accumulation of protein aggregates. Aβ monomers and oligomers increase the activity of the PI3K/Akt axis, leading to autophagy inhibition as well as insulin receptor (IR) internalisation and inactivation, resulting in a vicious cycleCitation152. Moreover, studies of mouse models and post-mortem analyses of AD brains suggest that there is an association between mTOR signalling and tau neuropathology, as evidenced by the observation that a dysfunctional autophagy-lysosome system promotes the formation of tau aggregatesCitation153,Citation154.
In fact, the inhibition of mTOR by rapamycin eradicates cognitive deficits and reduces levels of Aβ in a transgenic mouse model of AD, in which autophagy is strongly activated in the hippocampusCitation155. Several other studies have suggested that mTOR inhibition has positive effects on Aβ and tau levels in AD models. Cassano et al. found that everolimus-induced mTOR inhibition improves cognitive function and reduces APP/Aβ and tau levels in depressive-like phenotype in 3xTg-AD miceCitation156. Tramutola et al. suggested that rapamycin has a neuroprotective effect on the hippocampus in Ts65Dn (an established mouse model of Down syndrome); in particular, mTOR inhibition led to autophagy and insulin signalling recovery, reductions of APP and tau hyperphosphorylation levels, and reductions in the levels of oxidative stress markersCitation157.
3.3. Other neurodegenerative diseases
PD, the second most prevalent neurodegenerative disease, is characterised by the presence of Lewy bodies—aggregates composed of α-synuclein and poly-ubiquitinated proteins. The loss of dopamine neurons in the substantia nigra results in motor symptoms (muscular rigidity and tremor) as well as in non-motor somatic and psychological manifestationsCitation158. In addition to genetic mutations, ageing and dopaminergic neuron-specific toxins (e.g. 6-hydroxydopamine, rotenone, and 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine) are the main causes of disease progressionCitation159. mTOR signalling participates in several stages of PD in both active and inactive states. α-Synuclein accumulation has been observed in the cerebral cortex of patients with PD, who shows elevated levels of mTOR protein expressionCitation160. α-Synuclein aggregates eventually worsen disease progression by autophagy inhibition in response to increased mTOR activity. The rapamycin-induced inhibition of mTOR can restore the autophagy impairment, leading to the clearance of neurotoxic aggregatesCitation161,Citation162.
Nonetheless, the attenuation of mTOR activation, either by Akt phosphorylation or AMPK activation, has been described in different cellular models of PD. Since the mTOR pathway is important for cell proliferation and survival, Akt or AMPK-dependent mTOR downregulation by PD toxins leads to the impairment of protein synthesis and neuronal cell death. The overexpression of functional mTOR can partially restore this conditionCitation163,Citation164.
Considering the role of mTOR in the pathogenesis of PD, a balance between mTOR activation and inhibition should be maintained, as increased autophagy would ameliorate α-synuclein accumulation, while mTOR-regulated cellular functions (e.g. synaptic plasticity and memory formation) would remain intact.
Many neurodegenerative disorders are associated with pathological intra-neuronal protein aggregates. The importance of autophagy in neuronal health and the impact of autophagy dysfunction in neurodegeneration has been confirmed, thus emphasising the key role of mTORCitation165. For instance, the mutant form of the huntingtin protein (mHTT), responsible for the formation of pathogenic aggregates in Huntington’s disease, may mediate autophagy by different mechanisms. Both wild-type and mutant HTT interact with autophagy-associated proteins and influence the autophagy pathway, and both forms are degraded by autophagyCitation166,Citation167. Rapamycin-induced mTOR inhibition attenuates HTT toxicity in an animal model of HD and has a neuroprotective effect via autophagy activationCitation168,Citation169.
Moreover, there is a tight connection between SOD1 (superoxide dismutase; in which toxic gain-of-function mutations lead to aggregation in amyotrophic lateral sclerosis [ALS]) and autophagy dysfunctionCitation170. On the one hand, mutant SOD1 increases mTOR-dependent autophagyCitation171,Citation172; on the other hand, autophagy fails to degrade the products of mutant SOD1Citation173,Citation174. The autophagic degradation of mutant SOD1 is believed to be beneficial in ALS. However, rapamycin itself has shown both beneficial and harmful effects; for example, it promotes motor neuron degeneration and shortens the lifespan of miceCitation175 but simultaneously increases the survival of ALS mice deficient in mature lymphocytesCitation176. Owing to these contradictory results, the exact roles of mTOR-dependent autophagy and rapamycin in ASL require further research.
3.4. Cancer
During tumorigenesis, a high rate of proliferation demands an increased supply of nutrients and energy. Accordingly, one of the hallmarks of cancer cells is metabolic reprogramming, known as the Warburg effectCitation177,Citation178. A metabolic shift towards glycolysis and lactic acid fermentation (instead of oxidative metabolism) influences the tumour microenvironment and eventually suppresses antitumor immunity as well as the expression of cell surface markersCitation179. mTOR alters the metabolism of glucose, lipids, amino acids, and nucleotides according to the immediate demands of a cell, and the hyperactivation of its pathway may confer an advantage to cancer cells.
Mutations in the PI3K/Akt/mTOR signalling pathway contribute to the Warburg effect by increasing the expression of glucose transporters and glycolytic enzymes in cancer cells via the HIF-1α and Myc pathwaysCitation110,Citation180–182. The PI3K/Akt/mTOR signalling axis also increases lipid metabolism in tumours by the production of lipogenic enzymes via SREBP activityCitation183 and decreases antitumor immunity by reducing PGC1α expression in tumour-infiltrating lymphocytes, resulting in their exhaustionCitation184. The phosphorylation of the mTORC1 effector S6K1 promotes pyrimidine and purine synthesis, which is required for massive DNA replication in cancer cellsCitation60,Citation61. In addition, loss-of-function mutations in the tumour suppressor PTEN and mutations in the Ras signalling pathway might drive oncogenic changes in the mTOR pathwayCitation185.
Mutations that constitutively hyperactivate mTOR contribute to cancerCitation186. Mutations have been reported in the mTOR protein and mTOR complex components, e.g. Rictor, in breast cancer and lung cancerCitation187,Citation188. The dysregulation of the mTORC1 downstream effectors 4EBP1/eIF4E promotes the translation of pro-oncogenic proteins involved in angiogenesis, cell survival, metabolism, and metastasisCitation189.
The mTOR signalling pathway is the second most frequently altered pathway in human cancers (after the p53 pathway)Citation190; accordingly, extensive research has focussed on the role of mTOR in cancer. Furthermore, the development and application of appropriate mTOR inhibitors or compounds targeting the dysregulated PI3K/Akt/mTOR axis are key areas of research. Rapalogs, ATP-competitive inhibitors, PI3K/mTOR dual inhibitors, and RapaLink have all been evaluated in pre-clinical and clinical studies of various types of tumoursCitation191. A detailed description of this topic is beyond the scope of this review; for a more comprehensive overview, see the reviews by Magaway et al.Citation191 and Mossmann et al.Citation192
4. mTOR in ageing
Ageing can be defined as functional decline causing age-related diseases and eventually death. Several theories regarding the mechanism underlying ageing have been proposed, among which the Hayflick limit and the ROS theory are the most widely known. The Hayflick limit is based on a 1965 study showing that fibroblasts have a limited replicative lifespan due to telomere shortening during each replicationCitation193. However, this theory is not universally supported, since a clear correlation between telomere length and the maximal lifespan has not been observedCitation194–196. The ROS theory was proposed by Harman in 1956Citation197,Citation198, who stated that “there had to be some common, some basic cause which is killing everything. Free radicals cause random damage, and depending on the type of radical, they can cause all kinds of damage from day one.” However, clinical trials have disproven this theory, since antioxidants have no effect on age-related diseases or mortality and do not extend lifespan. Indeed, lifespan can be extended without reductions in ROSCitation199,Citation200.
Blagosklonny proposed an alternative theory in which cell senescence is considered a quasi-program of post-development. Development is a highly regulated program, and senescence is a continuance of this program and is constantly switched on, eventually becoming hyper-functional and damaging. Ageing and age-related diseases are, therefore, initially described as cellular hyperfunctions (apoptosis resistance, hypertrophy, and large cell morphology), involving increased growth and increased nutrient intake, and secondly as a loss of function. Age-related diseases, such as hypertension, cancer, or atherosclerosis, result from hyperfunction and hypertrophy (e.g. atherosclerosis involves smooth muscle cell, macrophage, and platelet hyperfunction)Citation201. Cell senescence might be induced by different stimuli that positively regulate the CDK inhibitors p21 and p16Citation202–204. Such stimuli include oxidative stress, microtubule-stabilising agents, retinoids, DNA-damaging agents, oncogenes, and mitogenic signalling via mitogen-activated protein kinase (MAPK) cascades that block the cell cycle but do not block cell growth, resulting in a hypertrophic phenotype typical of senescent cellsCitation205–211.
López-Otín et al. proposed nine tentative hallmarks of ageing, including telomere attrition, epigenetic alterations, genomic instability, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication; these appear to be common features of ageing across taxaCitation212. Although there are obvious links between hallmarks of ageing and previously described theories, telomere attrition or genomic instability alone are not a cause of ageing but rather a component of a complex mechanism.
Some of the mentioned hallmarks, namely a loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, and stem cell exhaustion, are affected by mTOR hyperactivation or dysregulation. Hence, as a key factor in the process of ageing, the roles of mTOR in these hallmarks are briefly described below.
4.1. Loss of proteostasis
The term proteostasis describes several cellular processes that maintain the appropriate proteome within the cell. Proteostasis includes the initial synthesis of nascent proteins, correct folding, transport and secretion of mature proteins, and degradation of damaged proteins. A decrease in proteostasis is related to ageing and some age-associated pathologies (e.g. AD)Citation213. By the regulation of cap-dependent and cap-independent mRNA translation, mTORC1 can potentiate the synthesis of translation/ribosomal-related proteins. mTORC1 inhibition, leading to a general decrease in proteosynthesis, might slow down ageing by reducing oxidative and proteotoxic stress (however, direct evidence for this effect has not been obtained)Citation214. In fact, protein quality control and translation are interconnected via mTOR, whose activity is also regulated by chaperone availabilityCitation215. Moreover, lifespan might be prolonged by the deletion of the translational regulator S6K1Citation216. In addition to being an important regulator of protein synthesis, the function of mTOR as an autophagy regulator has a crucial role in the maintenance of proteostasis (along with the UPS). Thus, the inhibition of mTORC1 inhibits the age-related decline in autophagy and loss of proteostasisCitation217.
4.2. Deregulated nutrient sensing
The insulin/PI3K/Akt signalling pathway upstream of mTOR responds to growth factors and nutrients and is deregulated in ageing organisms. Increased mTOR activity is linked to insulin resistance, commonly observed in ageing organisms (negative feedback with insulin/PI3K/Akt signalling) and age-related obesity (evidenced by the increased activity of mTOR in hypothalamic neurons in ageing mice)Citation218,Citation219. Therefore, it is unsurprising that a caloric restriction decreases mTORC1 activity and promotes lifespan and health in all investigated eukaryotes to dateCitation220–222.
4.3. Cellular senescence
mTOR, as a central regulator of nutrient conversion into biomass, plays an important role in cell senescence and the hypertrophic phenotype. Its activity is associated with ageing as well as senescence-associated diseases (metabolic syndrome, hereditary tumours, osteoporosis, AD, and macular degeneration). The loss of proteostasis, accumulation of mitochondrial and lysosomal mass, shifts in metabolism, and failure of autophagy are all associated with senescent cells and constitutive mTORC1 signallingCitation223. Moreover, the senescence-associated secretory phenotype (SASP) contributes to ageing by the secretion of proinflammatory mediators and is promoted by mTORC1Citation224,Citation225. Senescent cells steadily accumulate with age as a result of proinflammatory and pro-oxidant signal production, inducing inflammation and suppressing apoptosis. In fact, the inhibition of mTORC1 activity, as well as the activity of its effectors, results in SASP inhibition, lifespan extension, and the amelioration of several age-related diseasesCitation226.
4.4. Mitochondrial dysfunction
As mentioned above, mTOR is involved in the control of cellular energy metabolism by regulating mitochondrial functions and mitochondrial biogenesis. An increase in mitochondria is associated with ageing and age-related diseasesCitation227. In fact, mutations activating mTOR signalling increase the mitochondrial DNA number copy as well as oxidative metabolism-related gene expression, suggesting that the ageing-associated hyperactivation of mTOR also increases the number of mitochondria and alters mitochondrial function (via PGC-1 and YY1)Citation58,Citation228. Additionally, mTORC1 inhibition not only attenuates these increases in mitochondria but also stimulates autophagy (in particular, mitophagy), thereby clearing old and dysfunctional mitochondria. Thus, preserved number and function of mitochondria via mTOR inhibition might slow down ageing and alleviate age-related diseasesCitation227,Citation229.
4.5. Stem cell exhaustion
Age-related dysfunctions in tissues might be caused by declines in the amount and function of stem cells, thus lowering the tissue regenerative potential. The upstream hyperactivation of mTOR due to the deletion of PTEN or TSC1 or the constitutive activation of Akt lessens the number and function of haematopoietic stem cells (HSCs)Citation230–232. Rapamycin treatment restores the self-renewal capacity of mouse HSCsCitation233, enhances intestinal stem cell function in Paneth cellsCitation234, and generally promotes stem cell function by reprogramming somatic cells to generate induced pluripotent stem cellsCitation235. Taken together, mTOR inhibition might reverse the ageing phenotype by inducing stem cell rejuvenation, and this might expand lifespan and restore pathophysiological changes in some age-related diseases.
5. Expert opinion
The discovery of a pharmacological approach to slow ageing is considered the Holy Grail of medicine. The finding that mTOR inhibition prolongs lifespan and postpones the onset of age-associated diseases in mammals prompted substantial interest in the development of mTOR inhibitors as drugs to augment human longevity. However, interventions targeting the mTOR signalling pathway, a central switch in cellular metabolism, might be both a valuable and dangerous strategy. Since the pharmacological inhibition of mTOR by rapamycin and rapalogs is an FDA-approved clinical principle, there is substantial information about side effects. The most common side effects are immunosuppression, hyperglycaemiaCitation236, dyslipidemiaCitation237, as well as interstitial pneumonitisCitation238. Most of these side effects are dosage-dependent and may regress with lower dosages. Moreover, rapalogs are limited by the lack of tissue specificity and unwanted disruption of mTORC2. Thus, further efforts should focus on the development of mTOR-targeting therapeutics outside of these two modalities, such as truly mTORC1-specific inhibitors for use in diabetes, neurodegeneration, and life-span extension or tissue-specific mTORC1 agonists for use in muscle wasting diseases and immunotherapy. It is also worth noting that rapamycin and rapalogs have mostly been tested in combination with other drugs, such as steroids or proliferation inhibitors, in patients with severe disease; preventive mTOR inhibitor therapy in the general healthy population might be more tolerable. Considering that the mTOR signalling pathway is involved in multiple biological processes, it is an attractive candidate for research. We should learn how to take advantage of its beneficial effects alone while minimising side effects. Additionally, the discovery of such specific mTORC1 therapeutics might shed light on key biological questions regarding the link between metabolism, immune responses, and ageing.
6. Conclusions
mTOR is a key factor in several hallmarks of ageing. Its signalling pathway directly or indirectly affects common determinants of ageing in various taxa. However, our understanding of ageing and the underlying mechanisms is still limited. Most experiments are based on the inhibition of mTOR or its effectors either genetically or by rapamycin. Future studies of ageing and age-related disease face several challenges. First, the mechanism underlying lifespan extension has not been characterised in a broad range of taxa. Theories for the roles of mTOR, though persuasive, require further investigation. Second, the life-prolonging effect of mTOR inhibition by rapamycin in mammals might not be directly applicable to humans. Rapamycin is an FDA-approved immunosuppressant; however, its chronic use results in several side effects (e.g. hyperglycaemia, dyslipidemia, and interstitial pneumonitis). Moreover, the mTOR signalling pathway is highly complex, and its inhibition might result in the undesirable activation or inhibition of other related pathways, leading to the development or worsening of pathologies (e.g. insulin resistance or tumorigenesis). Available mTOR inhibitors tested on animal models or in clinical trials do not show sufficient effects on lifespan or age-related disorders as a result of their physicochemical properties or side-effects.
The enigma of ageing is still unresolved, and successful lifespan-extending interventions represent a challenging task for scientists, with important implications in the context of the expanding social-economic problem that ageing is becoming. It is important to set reasonable goals aimed at (i) a better understanding of the mTOR signalling pathway; (ii) the discovery of effective inhibitors with appropriate properties; and (iii) the development of interventions that would not only add years to the lifespan but would also support healthy ageing and delay age-related disorders.
Author contributions
EN, ZC, and KK: conceptualisation. ZC and EN: writing. EN and KK: review and editing. EN and KK: supervision. All authors have read and agreed to the published version of the manuscript.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Vézina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot 1975;28:721–6.
- Sehgal SN, Baker H, Vézina C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot 1975;28:727–32.
- Singh K, Sun S, Vézina C. Rapamycin (AY-22,989), a new antifungal antibiotic. IV. Mechanism of action. J Antibiot 1979;32:630–45.
- Garber K. Rapamycin's resurrection: a new way to target the cancer cell cycle. J Natl Cancer Inst 2001;93:1517–9.
- Martel RR, Klicius J, Galet S. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can J Physiol Pharmacol 1977;55:48–51.
- Dumont FJ, Staruch MJ, Koprak SL, et al. The immunosuppressive and toxic effects of FK-506 are mechanistically related: pharmacology of a novel antagonist of FK-506 and rapamycin. J Exp Med 1992;176:751–60.
- Dumont FJ, Staruch MJ, Koprak SL, et al. Distinct mechanisms of suppression of murine T cell activation by the related macrolides FK-506 and rapamycin. J Immunol 1990;144:251–8.
- Bowman LJ, Brennan DC. The role of tacrolimus in renal transplantation. Expert Opin Pharmacother 2008;9:635–43.
- Morath C, Arns W, Schwenger V, et al. Sirolimus in renal transplantation. Nephrol Dial Transplant 2007;22:viii61–5.
- Eng CP, Sehgal SN, Vézina C. Activity of Rapamycin (AY-22,989) against transplanted tumors. J Antibiot 1984;37:1231–7.
- Brown EJ, Albers MW, Shin TB, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994;369:756–8.
- Vellai T, Takacs-Vellai K, Zhang Y, et al. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 2003;426:620.
- Kapahi P, Zid BM, Harper T, et al. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 2004;14:885–90.
- Kaeberlein M, Powers RW, Steffen KK, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005;310:1193–6.
- Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009;460:392–5.
- Miller RA, Harrison DE, Astle CM, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 2011;66:191–201.
- Anisimov VN, Zabezhinski MA, Popovich IG, et al. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol 2010;176:2092–7.
- Anisimov VN, Zabezhinski MA, Popovich IG, et al. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle 2011;10:4230–6.
- Wilkinson JE, Burmeister L, Brooks SV, et al. Rapamycin slows aging in mice. Aging Cell 2012;11:675–82.
- Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal 2009;2:ra75.
- Hartwell LH. Macromolecule synthesis in temperature-sensitive mutants of yeast. J Bacteriol 1967;93:1662–70.
- Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell 2000;103:253–62.
- Heitman J, Movva N, Hall M. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991;253:905–9.
- Lench NJ, Macadam R, Markham AF. The human gene encoding FKBP-rapamycin associated protein (FRAP) maps to chromosomal band 1p36.2. Hum Genet 1997;99:547–9.
- Sabers CJ, Martin MM, Brunn GJ, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem 1995;270:815–22.
- Sabatini DM, Erdjument-Bromage H, Lui M, et al. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994;78:35–43.
- Chiu MI, Katz H, Berlin V. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc Natl Acad Sci USA 1994;91:12574–8.
- Keith CT, Schreiber SL. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science 1995;270:50.
- Baretić D, Williams RL. The structural basis for mTOR function. Semin Cell Dev Biol 2014; 36:91–101.
- Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996;273:239–42.
- Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 2002;10:457–68.
- Smithson LJ, Gutmann DH. Proteomic analysis reveals GIT1 as a novel mTOR complex component critical for mediating astrocyte survival. Genes Dev 2016;30:1383–8.
- Kim DH, Sarbassov DD, Ali SM, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell 2003;11:895–904.
- Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002;110:177–89.
- Kaizuka T, Hara T, Oshiro N, et al. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem 2010;285:20109–16.
- Sancak Y, Thoreen CC, Peterson TR, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 2007;25:903–15.
- Peterson TR, Laplante M, Thoreen CC, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009;137:873–86.
- Nojima H, Tokunaga C, Eguchi S, et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem 2003;278:15461–4.
- Schalm SS, Fingar DC, Sabatini DM, Blenis J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr Biol 2003;13:654–8.
- Gingras AC, Raught B, Gygi SP, et al. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev 2001;15:2852–64.
- Levy S, Avni D, Hariharan N, et al. Oligopyrimidine tract at the 5′ end of mammalian ribosomal protein mRNAs is required for their translational control. Proc Natl Acad Sci USA 1991;88:3319–23.
- Lahr RM, Fonseca BD, Ciotti GE, et al. La-related protein 1 (LARP1) binds the mRNA cap, blocking eIF4F assembly on TOP mRNAs. Elife 2017;6:e24146.
- Hong S, Freeberg MA, Han T, et al. LARP1 functions as a molecular switch for mTORC1-mediated translation of an essential class of mRNAs. Elife 2017;6:e25237.
- Mayer C, Zhao J, Yuan X, Grummt I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev 2004;18:423–34.
- Hannan KM, Brandenburger Y, Jenkins A, et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol 2003;23:8862–77.
- Shor B, Wu J, Shakey Q, et al. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. J Biol Chem 2010;285:15380–92.
- Tsang CK, Liu H, Zheng XFS. mTOR binds to the promoters of RNA polymerase I- And III-transcribed genes. Cell Cycle 2010;9:953–7.
- Jung CH, Jun CB, Ro S-H, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009;20:1992–2003.
- Ganley IG, Lam DH, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009;284:12297–305.
- Koren I, Reem E, Kimchi A. DAP1, a novel substrate of mTOR, negatively regulates autophagy. Curr Biol 2010;20:1093–8.
- Wan W, Liu W. MTORC1 regulates autophagic membrane growth by targeting WIPI2. Autophagy 2019;15:742–43.
- Zhao J, Zhai B, Gygi SP, Goldberg AL. MTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci USA 2015;112:15790–7.
- Eid W, Dauner K, Courtney KC, et al. mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc Natl Acad Sci USA 2017;114:7999–8004.
- Porstmann T, Santos CR, Griffiths B, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 2008;8:224–36.
- Kim JE, Chen J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004;53:2748–56.
- Düvel K, Yecies JL, Menon S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 2010;39:171–83.
- Peterson TR, Sengupta SS, Harris TE, et al. MTOR complex 1 regulates lipin 1 localization to control the srebp pathway. Cell 2011;146:408–20.
- Cunningham JT, Rodgers JT, Arlow DH, et al. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007;450:736–40.
- Schieke SM, Phillips D, McCoy JP, et al. The mammalian target of Rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem 2006;281:27643–52.
- Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013;339:1323–8.
- Ben-Sahra I, Hoxhaj G, Ricoult SJH, et al. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016;351:728–33.
- Toschi A, Lee E, Gadi N, et al. Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J Biol Chem 2008;283:34495–9.
- Hudson CC, Liu M, Chiang GG, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 2002;22:7004–14.
- West MJ, Stoneley M, Willis AE. Translational induction of the c-myc oncogene via activation of the FRAP/TOR signalling pathway. Oncogene 1998;17:769–80.
- He L, Gomes AP, Wang X, et al. mTORC1 promotes metabolic reprogramming by the suppression of GSK3-dependent Foxk1 phosphorylation. Mol Cell 2018;70:949–60.e4.
- Dang CV. A time for MYC: metabolism and therapy. Cold Spring Harb Symp Quant Biol 2016;81:79–83.
- Inoki K, Li Y, Zhu T, et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002;4:648–57.
- Tee AR, Manning BD, Roux PP, et al. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 2003;13:1259–68.
- Long X, Lin Y, Ortiz-Vega S, et al. Rheb binds and regulates the mTOR kinase. Curr Biol 2005;15:702–13.
- Ma L, Chen Z, Erdjument-Bromage H, et al. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005;121:179–93.
- Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008;320:1496–501.
- Rebsamen M, Pochini L, Stasyk T, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015;519:477–81.
- Jung J, Genau HM, Behrends C. Amino acid-dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9. Mol Cell Biol 2015;35:2479–94.
- Wolfson RL, Chantranupong L, Wyant GA, et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017;543:438–42.
- Saxton RA, Knockenhauer KE, Wolfson RL, et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 2016;351:53–8.
- Wolfson RL, Chantranupong L, Saxton RA, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016;351:43–8.
- Saxton RA, Chantranupong L, Knockenhauer KE, et al. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 2016;536:229–33.
- Chantranupong L, Scaria SM, Saxton RA, et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 2016;165:153–64.
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003;115:577–90.
- Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008;30:214–26.
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 2005;102:8204–9.
- Stambolic V, MacPherson D, Sas D, et al. Regulation of PTEN Transcription by p53. Mol Cell 2001;8:317–25.
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008;134:451–60.
- Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 2004;18:2893–904.
- Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci 2005;30:35–42.
- Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol 2004;167:399–403.
- Sarbassov DD, Ali SM, Kim D-H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 2004;14:1296–302.
- Stuttfeld E, Aylett CH, Imseng S, et al. Architecture of the human mTORC2 core complex. Elife 2018;7:e33101.
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science 2005;307:1098–1101.
- Jacinto E, Facchinetti V, Liu D, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006;127:125–37.
- Guertin DA, Stevens DM, Thoreen CC, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 2006;11:859–71.
- Pearce LR, Sommer EM, Sakamoto K, et al. Protor-1 is required for efficient mTORC2-mediated activation of SGK1 in the kidney. Biochem J 2011;436:169–79.
- Ikenoue T, Inoki K, Yang Q, et al. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. Embo J 2008;27:1919–31.
- García-Martínez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J 2008;416:375–85.
- Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004;6:1122–8.
- Ebner M, Sinkovics B, Szczygieł M, et al. Localization of mTORC2 activity inside cells. J Cell Biol 2017;216:343–53.
- Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell 2011;44:304–16.
- Wang B, Jie Z, Joo D, et al. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature 2017;545:365–9.
- Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol 2009;29:5657–70.
- Glidden EJ, Gray LG, Vemuru S, et al. Multiple site acetylation of rictor stimulates mammalian target of rapamycin complex 2 (mTORC2)-dependent phosphorylation of Akt protein. J Biol Chem 2012;287:581–8.
- Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by Association with the Ribosome. Cell 2011;144:757–68.
- Hatano T, Morigasaki S, Tatebe H, et al. Fission yeast Ryh1 GTPase activates TOR complex 2 in response to glucose. Cell Cycle 2015;14:848–56.
- Cai W, Andres DA, Reiner DJ. MTORC2 Is required for Rit-mediated oxidative stress resistance. PLOS One 2014;9:e115602.
- Saci A, Cantley LC, Carpenter CL. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol Cell 2011;42:50–61.
- Byun J-K, Choi Y-K, Kim J-H, et al. A positive feedback loop between Sestrin2 and mTORC2 is required for the survival of glutamine-depleted lung cancer cells. Cell Rep 2017;20:586–99.
- Gottlob K, Majewski N, Kennedy S, et al. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev 2001;15:1406–18.
- Deprez J, Vertommen D, Alessi DR, et al. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem 1997;272:17269–75.
- Barthel A, Okino ST, Liao J, et al. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem 1999;274:20281–6.
- Cerniglia GJ, Dey S, Gallagher-Colombo SM, et al. The PI3K/Akt pathway regulates oxygen metabolism via pyruvate dehydrogenase (PDH)-E1α phosphorylation. Mol Cancer Ther 2015;14:1928–38.
- Masui K, Tanaka K, Akhavan D, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab 2013;18:726–39.
- Sun Q, Chen X, Ma J, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA 2011;108:4129–34.
- Zha X, Wang F, Wang Y, et al. Lactate dehydrogenase B is critical for hyperactive mTOR-mediated tumorigenesis. Cancer Res 2011;71:13–8.
- Masui K, Cavenee WK, Mischel PS. Mischel PS. mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol Metab 2014;25:364–73.
- Guri Y, Colombi M, Dazert E, et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 2017;32:807–23.e12.
- Tang Y, Wallace M, Sanchez-Gurmaches J, et al. Adipose tissue mTORC2 regulates ChREBP-driven de novo lipogenesis and hepatic glucose metabolism. Nat Commun 2016;7:11365.
- Kumar A, Harris TE, Keller SR, et al. Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances Basal glycogen synthase activity. Mol Cell Biol 2008;28:61–70.
- Hagiwara A, Cornu M, Cybulski N, et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 2012;15:725–38.
- Betz C, Stracka D, Prescianotto-Baschong C, et al. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci USA 2013;110:12526–34.
- Yuan M, Pino E, Wu L, et al. Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J Biol Chem 2012;287:29579–88.
- Puigserver P, Rhee J, Donovan J, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003;423:550–5.
- Rosario FJ, Kanai Y, Powell TL, Jansson T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J Physiol 2013;591:609–25.
- Gu Y, Albuquerque CP, Braas D, et al. mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Mol Cell 2017;67:128–38.e7.
- Saha A, Connelly S, Jiang J, et al. Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol Cell 2014;55:264–76.
- Wang W, Fridman A, Blackledge W, et al. The phosphatidylinositol 3-kinase/Akt cassette regulates purine nucleotide synthesis. J Biol Chem 2009;284:3521–8.
- Lamming DW, Mihaylova MM, Katajisto P, et al. Depletion of Rictor, an essential protein component of mTORC2, decreases male lifespan. Aging Cell 2014;13:911–7.
- Nojima A, Yamashita M, Yoshida Y, et al. Haploinsufficiency of Akt1 prolongs the lifespan of mice. PLOS One 2013;8:e69178.
- Shigihara N, Fukunaka A, Hara A, et al. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J Clin Invest 2014;124:3634–44.
- Ebato C, Uchida T, Arakawa M, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 2008;8:325–32.
- Jung HS, Chung KW, Won Kim J, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 2008;8:318–24.
- Bartolome A, Guillen C, Benito M. Autophagy plays a protective role in endoplasmic reticulum stress-mediated pancreatic β cell death. Autophagy 2012;8:1757–68.
- Shigeyama Y, Kobayashi T, Kido Y, et al. Biphasic Response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol 2008;28:2971–9.
- Hernández MG, Aguilar AG, Burillo J, et al. Pancreatic β cells overexpressing hIAPP impaired mitophagy and unbalanced mitochondrial dynamics. Cell Death Dis 2018;9:481.
- Bartolomé A, Kimura-Koyanagi M, Asahara SI, et al. Pancreatic β-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes 2014;63:2996–3008.
- Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron 1991;6:487–98.
- Hardy J, Higgins G. Alzheimer's disease: the amyloid cascade hypothesis. Science 1992;256:184–85.
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–6.
- Grundke-Iqbal I, Iqbal K, Tung YC, et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 1986;83:4913–7.
- Ihara Y, Nukina N, Miura R, Ogawara M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer's disease. J Biochem 1986;99:1807–10.
- Patrick GN, Zukerberg L, Nikolic M, et al. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999;402:615–22.
- Cunnane S, Nugent S, Roy M, et al. Brain fuel metabolism, aging, and Alzheimer's disease. Nutrition 2011;27:3–20.
- Reijmer YD, van den Berg E, Ruis C, et al. Cognitive dysfunction in patients with type 2 diabetes. Diabetes Metab Res Rev 2010;26:507–19.
- Small GW, Mazziotta JC, Collins MT, et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. J Am Med Assoc 1995;273:942.
- Duelli R, Kuschinsky W. Brain glucose transporters: relationship to local energy demand. News Physiol Sci 2001;16:71–6.
- Liu Y, Liu F, Grundke-Iqbal I, et al. Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J Pathol 2011;225:54–62.
- Norambuena A, Wallrabe H, Cao R, et al. A novel lysosome‐to‐mitochondria signaling pathway disrupted by amyloid‐β oligomers. Embo J 2018;37:e100241.
- Josselyn SA, Frankland PW. mTORC2: actin on your memory. Nat Neurosci 2013;16:379–380.
- Takei N, Nawa H. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci 2014;7:28.
- Griffin RJ, Moloney A, Kelliher M, et al. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem 2005;93:105–17.
- Caccamo A, Maldonado MA, Majumder S, et al. Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J Biol Chem 2011;286:8924–32.
- Gupta A, Dey CS. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Mol Biol Cell 2012;23:3882–98.
- O'Neill C, Kiely AP, Coakley MF, et al. Insulin and IGF-1 signalling: longevity, protein homoeostasis and Alzheimer's disease. Biochem Soc Trans 2012;40:721–7.
- O’ Neill C. PI3-kinase/Akt/mTOR signaling: impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp Gerontol 2013;48:647–53.
- An W-L, Cowburn RF, Li L, et al. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol 2003;163:591–607.
- Hamano T, Gendron TF, Causevic E, et al. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci 2008;27:1119–30.
- Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLOS One 2010;5:e9979.
- Cassano T, Magini A, Giovagnoli S, et al. Early intrathecal infusion of everolimus restores cognitive function and mood in a murine model of Alzheimer's disease. Exp Neurol 2019;311:88–105.
- Tramutola A, Lanzillotta C, Barone E, et al. Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl Neurodegener 2018;7:1–22.
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron 2003;39:889–909.
- Liu J, Liu W, Lu Y, et al. Piperlongumine restores the balance of autophagy and apoptosis by increasing BCL2 phosphorylation in rotenone-induced Parkinson disease models. Autophagy 2018;14:845–61.
- Crews L, Spencer B, Desplats P, et al. Selective molecular alterations in the autophagy pathway in patients with lewy body disease and in models of alpha-synucleinopathy. PLOS One 2010;5:e9313.
- Gao S, Duan C, Gao G, et al. Alpha-synuclein overexpression negatively regulates insulin receptor substrate 1 by activating mTORC1/S6K1 signaling. Int J Biochem Cell Biol 2015;64:25–33.
- Jiang TF, Zhang YJ, Zhou HY, et al. Curcumin ameliorates the neurodegenerative pathology in A53T α-synuclein cell model of Parkinson's disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J Neuroimmune Pharmacol 2013;8:356–69.
- Selvaraj S, Sun Y, Watt JA, et al. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J Clin Invest 2012;122:1354–67.
- Xu Y, Liu C, Chen S, et al. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson's disease. Cell Signal 2014;26:1680–9.
- Meng T, Lin S, Zhuang H, et al. Recent progress in the role of autophagy in neurological diseases. Cell Stress 2019;3:141–61.
- Martin DDO, Ladha S, Ehrnhoefer DE, Hayden MR. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci 2015;38:26–35.
- Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 2002;11:1107–17.
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 2009;16:46–56.
- Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004;36:585–95.
- Sheng Y, Chattopadhyay M, Whitelegge J, Selverstone Valentine J. SOD1 Aggregation and ALS: role of metallation states and disulfide status. Curr Top Med Chem 2012;12:2560–72.
- Li A, Zhang X, Le W. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 2008;4:290–3.
- Morimoto N, Nagai M, Ohta Y, et al. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res 2007;1167:112–7.
- Rudnick ND, Griffey CJ, Guarnieri P, et al. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc Natl Acad Sci USA 2017;114:E8294–303.
- An T, Shi P, Duan W, et al. Oxidative stress and autophagic alteration in brainstem of SOD1-G93A mouse model of ALS. Mol Neurobiol 2014;49:1435–48.
- Zhang X, Li L, Chen S, et al. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 2011;7:412–25.
- Staats KA, Hernandez S, Schönefeldt S, et al. Rapamycin increases survival in ALS mice lacking mature lymphocytes. Mol Neurodegener 2013;8:31.
- Warburg O. On the Origin of Cancer Cells. Science 1956;123:309–14.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74.
- Ramapriyan R, Caetano MS, Barsoumian HB, et al. Altered cancer metabolism in mechanisms of immunotherapy resistance. Pharmacol Ther 2019;195:162–71.
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008;7:11–20.
- Buller CL, Loberg RD, Fan M-H, et al. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Physiol Cell Physiol 2008;295:C836–43.
- Tran Q, Lee H, Park J, et al. Targeting cancer metabolism – revisiting the Warburg effects. Toxicol Res 2016;32:177–93.
- Porstmann T, Santos CR, Lewis C, et al. A new player in the orchestra of cell growth: SREBP activity is regulated by mTORC1 and contributes to the regulation of cell and organ size. Biochem Soc Trans 2009;37:278–83.
- Scharping NE, Menk AV, Moreci RS, et al. Delgoffe correspondence GM. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 2016;45:374–88.
- Zhang Y, Kwok-Shing Ng P, Kucherlapati M, et al. A pan-cancer proteogenomic atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell 2017;31:820–32.e3.
- Grabiner BC, Nardi V, Birsoy K, et al. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov 2014;4:554–63.
- Cheng H, Zou Y, Ross JS, et al. RICTOR amplification defines a novel subset of patients with lung cancer who may benefit from treatment with mTORC1/2 inhibitors. Cancer Discov 2015;5:1262–70.
- Joly MM, Hicks DJ, Jones B, et al. Rictor/mTORC2 drives progression and therapeutic resistance of HER2-amplified breast cancers. Cancer Res 2016;76:4752–64.
- Laplante M, Sabatini DM. MTOR signaling in growth control and disease. Cell 2012;149:274–93.
- Klempner SJ, Myers AP, Cantley LC. What a tangled web we weave: emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov 2013;3:1345–54.
- Magaway C, Kim E, Jacinto E. Targeting mTOR and metabolism in cancer: lessons and innovations. Cells 2019;8:1584.
- Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer 2018;18:744–57.
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 1965;37:614–36.
- Raices M, Maruyama H, Dillin A, Karlseder J. Uncoupling of longevity and telomere length in C. elegans. PLOS Genet 2005;1:e30.
- Parrinello S, Samper E, Krtolica A, et al. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 2003;5:741–7.
- Tang DG, Tokumoto YM, Apperly JA, et al. Lack of replicative senescence in cultured rat oligodendrocyte precursor cells. Science 2001;291:868–71.
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956;11:298–300.
- Kitani K, Ivy GO. “I thought, thought, thought for four months in vain and suddenly the idea came” – an interview with Denham and Helen Harmanmitochondrial dysfunction and ageing. Biogerontology 2003;4:401–12.
- Andziak B, O’Connor TP, Buffenstein R. Antioxidants do not explain the disparate longevity between mice and the longest-living rodent, the naked mole-rat. Mech Ageing Dev 2005;126:1206–12.
- Corona M, Hughes KA, Weaver DB, Robinson GE. Gene expression patterns associated with queen honey bee longevity. Mech Ageing Dev 2005;126:1230–8.
- Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle 2006;5:2087–102.
- McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol 1998;8:351–4.
- Itahana K, Dimri G, Campisi J. Regulation of cellular senescence by p53. Eur J Biochem 2001;268:2784–91.
- Jacobs JJL, De Lange T. p16INK4a as a second effector of the telomere damage pathway. Cell Cycle 2005;4:1364–8.
- Serrano M, Lin AW, McCurrach ME, et al. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997;88:593–602.
- Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev 1998;12:2997–3007.
- Lin AW, Barradas M, Stone JC, et al. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 1998;12:3008–19.
- Deng Q, Liao R, Wu B-L, Sun P. High intensity Ras signaling induces premature senescence by activating p38 pathway in primary human fibroblasts. J Biol Chem 2004;279:1050–9.
- Klein LE, Freeze BS, Smith AB, III, Horwitz SB. The microtubule stabilizing agent discodermolide is a potent inducer of accelerated cell senescence. Cell Cycle 2005;4:501–7.
- Roninson IB, Dokmanovic M. Induction of senescence-associated growth inhibitors in the tumor-suppressive function of retinoids. J Cell Biochem 2003;88:83–94.
- Wang X, Wong SCH, Pan J, et al. Evidence of cisplatin-induced senescent-like growth arrest in nasopharyngeal carcinoma cells. Cancer Res 1998;58:5019–22.
- López-Otín C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell 2013;153:1194–217.
- Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res Rev 2011;10:205–15.
- Walters H, Cox L. mTORC inhibitors as broad-spectrum therapeutics for age-related diseases. Int J Mol Sci 2018;19:2325.
- Qian S-B, Zhang X, Sun J, et al. mTORC1 links protein quality and quantity control by sensing chaperone availability. J Biol Chem 2010;285:27385–95.
- Selman C, Tullet JMA, Wieser D, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009;326:140–4.
- Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 2008;14:959–65.
- Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes 2012;61:1315–22.
- Yang S-B, Tien A-C, Boddupalli G, et al. Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron 2012;75:425–36.
- Fontana L, Partridge L, Longo VD. Extending healthy life span-from yeast to humans. Science 2010;328:321–6.
- Mattison JA, Roth GS, Beasley TM, et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 2012;489:318–21.
- Colman RJ, Anderson RM, Johnson SC, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009;325:201–4.
- Carroll B, Nelson G, Rabanal-Ruiz Y, et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J Cell Biol 2017;216:1949–57.
- Laberge R-M, Sun Y, Orjalo AV, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 2015;17:1049–61.
- Herranz N, Gallage S, Mellone M, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 2015;17:1205–17.
- Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 2015;21:1424–35.
- Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine 2017;21:7–13.
- Morita M, Gravel S-P, Chénard V, et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab 2013;18:698–711.
- Lerner C, Bitto A, Pulliam D, et al. Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell 2013;12:966–77.
- Yilmaz ÖH, Valdez R, Theisen BK, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006;441:475–82.
- Chen C, Liu Y, Liu R, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med 2008;205:2397–408.
- Gan B, Sahin E, Jiang S, et al. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci USA 2008;105:19384–9.
- Jang Y-Y, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007;110:3056–63.
- Yilmaz ÖH, Katajisto P, Lamming DW, et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 2012;486:490–5.
- Chen T, Shen L, Yu J, et al. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell 2011;10:908–11.
- Gyurus E, Kaposztas Z, Kahan BD. Sirolimus therapy predisposes to new-onset diabetes mellitus after renal transplantation: a long-term analysis of various treatment regimens. Transplant Proc 2011;43:1583–92.
- Holdaas H, Potena L, Saliba F. mTOR inhibitors and dyslipidemia in transplant recipients: a cause for concern? Transplant Rev 2015;29:93–102.
- Nishino M, Boswell EN, Hatabu H, et al. Drug-related pneumonitis during mammalian target of rapamycin inhibitor therapy: radiographic pattern-based approach in Waldenström macroglobulinemia as a paradigm. Oncologist 2015;20:1077–83.