
Abstract
Vascular endothelial growth factor receptor-2 (VEGFR-2) plays a critical role in cancer angiogenesis. Inhibition of VEGFR-2 activity proved effective suppression of tumour propagation. Accordingly, two series of new 3-methylquinoxaline derivatives have been designed and synthesised as VEGFR-2 inhibitors. The synthesised derivatives were evaluated in vitro for their cytotoxic activities against MCF-7and HepG2 cell lines. In addition, the VEGFR-2 inhibitory activities of the target compounds were estimated to indicate the potential mechanism of their cytotoxicity. To a great extent, the results of VEGFR-2 inhibition were highly correlated with that of cytotoxicity. Compound 27a was the most potent VEGFR-2 inhibitor with IC50 of 3.2 nM very close to positive control sorafenib (IC50 = 3.12 nM). Such compound exhibited a strong cytotoxic effect against MCF-7 and HepG2, respectively with IC50 of 7.7 and 4.5 µM in comparison to sorafenib (IC50 = 3.51 and 2.17 µM). In addition, compounds 28, 30f, 30i, and 31b exhibited excellent VEGFR-2 inhibition activities (IC50 range from 4.2 to 6.1 nM) with promising cytotoxic activity. Cell cycle progression and apoptosis induction were investigated for the most active member 27a. Also, the effect of 27a on the level of caspase-3, caspase-9, and BAX/Bcl-2 ratio was determined. Molecular docking studies were implemented to interpret the binding mode of the target compounds with the VEGFR-2 pocket. Furthermore, toxicity and ADMET calculations were performed for the synthesised compounds to study their pharmacokinetic profiles
1. Introduction
According to WHO reports, cancer is considered the second major cause of deathCitation1. By 2030, the incidence of cancer deaths will reach thirteen million worldwideCitation2. Although the high advances in the diagnosis and treatment of cancer diseases, the survival of patients remains poor due to the widespread adverse effects of anticancer agentsCitation3. So that, the discovery of new, effective, selective, and less toxic anticancer agents remains one of the most urgent needsCitation4.
Angiogenesis process plays an important role in the growth and regeneration of tissues. Such a role is crucial to prevent ischaemic necrosis and facilitate the survival of the damaged tissuesCitation5. During the normal state, angiogenesis is controlled by some protein kinases (PKs), which comprise VEGFRs, FGFRs, and EGFRsCitation6. PKs can be deregulated under pathological conditions, producing a disturbance in angiogenesis process. This leads to an increasing in the rate of cell division, creating tumour diseaseCitation7.
VEGFRs and their specific agonist (VEGF) are overexpressed in many human tumours, especially solid tumours as gliomas and carcinomasCitation8. Therefore, VEGFRs are considered as one the most important regulators of angiogenesis and consequently tumour growthCitation9. VEGFRs family comprises three subtypes including VEGFR-1, VEGFR-2, and VEGFR-3Citation10. VEGFR-1 controls embryonic vasculogenesisCitation11. VEGFR-2 regulates both embryonic vasculogenesis and tumour angiogenesisCitation12. On the other hand, VEGFR-3 is responsible for lymphangiogenesisCitation13. So that, VEGFR-2 is now the foremost target for antiangiogenic therapy, and its blocking is a relevant approach for the discovery of new drugs against angiogenesis–dependent malignanciesCitation14. VEGFR-2 inhibitors demonstrated effective suppression of tumour progression. ATP binding site is the main target of most VEGFR-2 inhibitorsCitation15.
The crystal structures of VEGFR-2 revealed the presence of many pockets at the ATP binding site. (i) Hinge region which locates on the front side and comprises two key amino acid residues (Cys919 and Glu917). These residues participate in H-bond interactions with the adenine ring of ATP. (ii) Gatekeeper region which separates between the hinge region and DFG-motif region. (iii) DFG-motif region which locates on the backside and contains two key amino acids (Glu885 and Asp1046). For maximum activity, VEGFR-2 inhibitors have to bind with these two residues via H-bonds. (iv) Allosteric hydrophobic region which consists of much hydrophobic amino acid residuesCitation16–19.
VEGFR-2 inhibitors can be classified into three types. Type I inhibitors (ATP competitive inhibitors), e.g. sunitinib 1 can bind to the adenine binding region of the ATP binding siteCitation15. Type II inhibitors, e.g. sorafenib 2 induce inactive activation of DFG-out confirmation of activation loop. Such type can bind additionally the allosteric hydrophobic pocket of ATPCitation20. Type III inhibitors, e.g. vatalanib 3 can form covalent interaction with cysteine amino acid residue at ATP binding siteCitation21,Citation22.
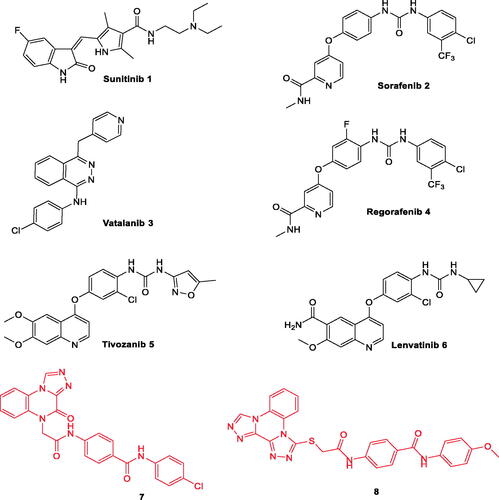
Many drugs targeting VEGFR-2 have been approved for clinical use in the treatment of different types of cancers ). Sorafenib 2 is a potent VEGFR-2 inhibitorCitation23. It is mainly used in the treatment of advanced renal cell carcinoma (RCC) and hepatocellular carcinoma (HCC)Citation24. Regorafenib 4, a fluoro derivative of sorafenib, has been developed to inhibits VEGFR1-3Citation25. Tivozanib 5, a VEGFR-2 inhibitor, has been approved by the FDA in March 2021 for the treatment of RCCCitation26–28. Sunitinib 1 is a VEGFR-2 kinase inhibitor that was approved for the treatment of RCC and of gastrointestinal stromal tumours (GIST)Citation29.
Figure 1. Some clinically used VEGFR-2 inhibitors as well as quinoxaline derivatives having VEGFR-2 inhibitory actions.
Binding mode sorafenib, a representative example of VEGFR-2 inhibitors, against VEGFR-2 active site (PDB: 4ASD) was reported in many publications. The heterocyclic (N-methylpicolinamide) moiety is buried in the hinge region forming two H-bonds with Cys919 through the N atom of the pyridine ring and NH group of acetamide moiety. In addition, the urea moiety binds to the receptor at the DFG motif through various H-bonding interactions with Glu885 and Asp1046. The terminal phenyl ring occupies the allosteric site forming hydrophobic interactions with the hydrophobic pocket created by the DFG flipping outCitation30–32.
Studying the SAR of VEGFR-2 inhibitors and analysing the binding mode of sorafenib revealed that the majority of potent and selective VEGFR-2 inhibitors have four common pharmacophoric features that facilitate their fitting with the active binding pocketCitation33–35. The first feature is a flat heteroaromatic ring system (ahead) incorporating at least one H-bond acceptor to interact with the crucial amino acid residue Cys919 in the hinge regionCitation30,Citation31. A second feature is a spacer group that gives the inhibitor enough lengthCitation36,Citation37, making the third feature (pharmacophore) near at DFG motif to bind with two crucial residues (Glu883 and Asp1044)Citation37,Citation38. A pharmacophore is a functional group that comprises at least one H-bond acceptor (HBA) and one H-bond donor (HBD) group (typically amide or urea)Citation31,Citation38. The fourth feature is a terminal hydrophobic moiety that can occupy the created allosteric hydrophobic binding siteCitation16,Citation39.
Apoptosis as a form of cellular suicide is one of the important mechanisms by which anticancer agents can affect cancer cellsCitation40. Apoptosis is regulated by several mediatorsCitation41. Among these are caspases, specifically caspase-3 and caspase-9Citation42,Citation43. Caspase-3 protease is a major mediator of apoptosis catalysing the cleavage of many vital cellular proteins leading to cell deathCitation44. Caspase-9 activates other executioner caspases as caspase-3, -6, and -7 initiating apoptosis as they cleave several other cellular targetsCitation45. In addition, the Bcl-2 family proteins are key regulators of apoptosis. This family comprised pro-apoptotic proteins as BAX that promote cell death and anti-apoptotic proteins as Bcl-2 which suppress cell deathCitation46,Citation47. The balance between pro-apoptotic and anti-apoptotic proteins (BAX/Bcl-2 ratio) regulates cell fateCitation48. Current evidence suggested that inhibition of angiogenesis, anti-angiogenic therapies have been shown to increase apoptosis in tumour cellsCitation49. VEGFR-2 inhibitors were found to induce and accelerate apoptosis in cancer cells which synergistically potentiates their antitumor effectCitation30,Citation49–51.
Moreover, the literature survey revealed that different scaffolds have been reported as excellent inhibitors of VEGFR-2. These scaffolds comprise quinoline (e.g. lenvatinib 6Citation52), quinazolineCitation53, and indazoleCitation54. Furthermore, quinoxaline, a bioisostere for the aforementioned scaffolds, is considered an important nucleus for anticancer drugsCitation3,Citation55–58. Many quinoxaline derivatives were reported to possess significant VEGFR-2 inhibitory activitiesCitation39,Citation59–62.
In our previous work, we developed [1,2,4]triazolo[4,3-a]quinoxaline containing derivatives as VEGFR-2 inhibitors. Compounds 8 and 9 were the most potent candidates exhibiting an excellent VEGFR-2 inhibitory activity with a promising cytotoxic efficacy against breast and hepatocellular carcinomaCitation63,Citation64. In continuation of our workCitation63–65 aimed at synthesising new anticancer agents targeting VEGFR-2 inhibition, new quinoxaline derivatives were designed and synthesised. The synthesised compounds were evaluated for their anti-proliferative activity. In addition, VEGFR-2 inhibitory activities were estimated for all compounds to hint at the potential mechanism of their cytotoxicity. Furthermore, deep investigations were performed on the most active member to assess its effect on apoptotic (caspase-3, caspase-9, and BAX) and anti-apoptotic (Bcl-2) mediators.
1.1. Rational of design
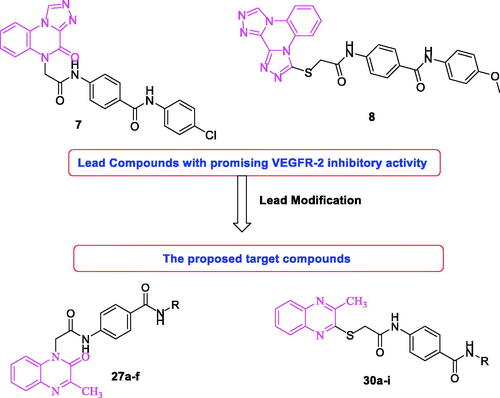
Considering compounds 7 and 8 as leading compoundsCitation63,Citation64. The design included the replacement of [1,2,4]triazolo[4,3-a]quinoxaline in compound 7 and/or bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline of compound 8 by 3-methylquinoxaline scaffold. 3-Methylquinoxaline is considered as bioisostere for N-methylpicolinamide moiety of sorafenibCitation61. Two quinoxaline moieties (3-methylquinoxalin-2(1H)-one and 3-methylquinoxaline-2-thiol) were used as a biological isostere. As in the lead compounds, N-phenylacetamide moiety was utilised as a linker. The pharmacophore (HBD/HBA) was designed to be an amide, diamide, and/or hydrazide groups. Amide pharmacophore served as hydrogen bond donor and acceptor in many reported VEGFR-2 inhibitorsCitation61,Citation63,Citation64,Citation66. Finally, different, aliphatic, and un/substituted aromatic derivatives were selected to be the terminal hydrophobic moieties to occupy the allosteric hydrophobic pocket (.
Figure 2. Rational design of new VEGFR-2 inhibitors.
2. Results and discussion
2.1. Chemistry
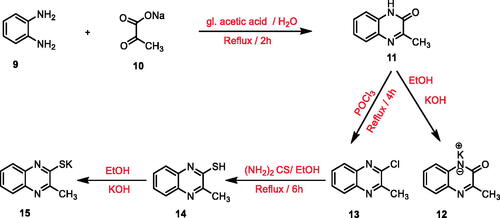
To reach the designed compounds, four Schemes 1–4 were adopted. The synthesis was initiated by the reaction of o- phenylenediamine 9 with sodium pyruvate 10 in glacial acetic acid according to the reported procedure to furnish 3-methylquinoxalin-2(1H)-one 11Citation67. This starting material 11 was subjected to subsequent treatment with potassium hydroxide to produce the corresponding potassium salt 12Citation67. Chlorination of compound 11 was achieved using phosphorous oxychloride to afford 2-chloro-3-methylquinoxaline 13Citation68. Refluxing the latter with thiourea in absolute ethanol resulted in 3-methylquinoxaline-2-thiol 14Citation69,Citation70 which was underwent heating with alcoholic potassium hydroxide to provide the corresponding potassium salt 15 (Scheme 1).
Scheme 1. General procedure for preparation of the key potassium salts 12 and 15.
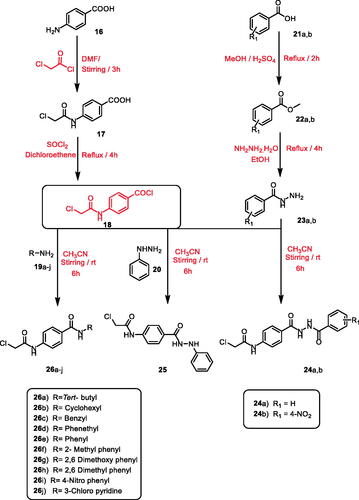
According to the literatureCitation71–73, stirring of commercially available p-amino benzoic acid 16 with chloroacetyl chloride in dry DMF in cold conditions yielded 4–(2-chloroacetamido)benzoic acid 17. Treatment of 17 with thionyl chloride in 1,2 dichloroethane and a catalytic amount of DMF gave the key intermediate 4–(2-chloroacetamido)benzoyl chloride 18Citation63,Citation74.
On the other hand, methyl esters of appropriate acid derivatives, namely benzoic acid 21a and 4-nitrobenzoic acid 21b were prepared by refluxing carboxylic acids in methanol with the presence of sulphuric acidCitation75. The ester derivatives 22a,b were treated with hydrazine hydrate to get the corresponding acid hydrazides 23a,bCitation76,Citation77. The produced acid hydrazides 23a,b were then allowed to stir with 4-(2-chloroacetamido)benzoyl chloride 18 in acetonitrile and TEA to furnish the intermediates 24a,b, respectively. Likewise, stirring of phenyl hydrazine 20 with 4-(2-chloroacetamido)benzoyl chloride 18 yielded the corresponding intermediate 2-chloro-N-(4–(2-phenylhydrazine-1-carbonyl)phenyl) acetamide 25. Compound 18 was stirred at room temperature in acetonitrile in the presence of a catalytic amount of TEA with appropriate amines 19a–j namely, tertiary butyl amine, cyclohexyl amine, benzyl amine, phenethyl amine, aniline, 2-methyl aniline, 2,6 dimethoxy aniline, 2,6 dimethyl aniline, 4-nitro-aniline, and 3-chloropyridine to give the corresponding chloroacetamide intermediates 26a–j, respectively (Scheme 2).
Scheme 2. General procedure for preparation of the intermediates 24a,b, 25, and 26a–j.
Synthesis of the final target compounds was illustrated in Schemes 3, 4. The 3-methylquinoxalin-2(1H)-one derivative (compounds 27a–f, 28, and 29) were obtained by heating of potassium salt 12 in dry DMF and a catalytic amount of KI with the previously prepared intermediates 26a,d,g–j, 24 b, and 25, respectively (Scheme 3).
Scheme 3. General procedure for preparation of the target compounds 27a–f, 28, and 29.
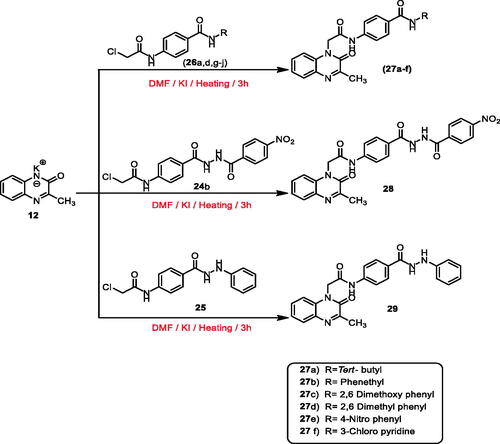
Furthermore, heating of potassium salt 15 with the intermediates 26a–g,i,j, 24a,b, and 25 in dry DMF and a catalytic amount of KI afforded the final target compounds 30a–i, 31a,b, and 32 (methylquinoxaline-2-thiol derivatives) (Scheme 4).
Scheme 4. General procedure for preparation of the target compounds 30a–i, 31a,b, and 32.
IR spectra of compounds 27a–f and 30a–i exhibited the presence of characteristic stretching bands at 3428–3215 cm−1 corresponding to NH groups. Furthermore, the NMR spectra of compounds 27a–f and 30a–i supported their assigned structures. 1H NMR charts of these compounds revealed the appearance of two singlet signals around δ 10.5 ppm attributed to the introduced two amidic NH protons. It also demonstrated increased integration of the aromatic protons corresponding to the additional phenyl ring. A characteristic up-field singlet signal equivalent to CH2 of acetamide moiety was detected at about δ 4.30 ppm. Additionally, a singlet signal of CH3 group of 3-methylquinoxalin-2(1H)-one moiety appeared around δ 2.49 ppm. 13C NMR spectra displayed the presence of two peaks at the aliphatic region attributed to the CH2 and CH3 groups around δ 40.0 and 22.5 ppm, respectively.
1H NMR spectra of compounds 28, 29, 31a,b, and 32 demonstrated the presence of the amidic protons of hydrazide moiety at a range of δ 10.02–12.71 ppm. Additionally, 13C NMR spectrum of compounds 31b as an example of these compounds revealed the appearance of two peaks at δ 35.45 and 22.18 ppm corresponding to CH2 and CH3 groups, respectively. Mass spectroscopic analysis displayed a base peak at 517.0 (m/z) corresponding to the exact mass of the compound 31b.
2.2. Biological testing
2.2.1. In vitro anti-proliferative activity
In vitro cytotoxic activities of the synthesised compounds were evaluated against two different cancer cell lines; breast cancer (MCF-7) and hepatocellular carcinoma (HepG2) using standard MTT colorimetric assay as described by MosmannCitation78,Citation79. Sorafenib was used as a reference cytotoxic drug. The results of cytotoxicity were expressed as growth inhibitory concentration (IC50) values and summarised in .
Table 1. In vitro cytotoxicity of the synthesised compounds against MCF-7 and HepG2 cell lines, their VEGFR-2 inhibitory activities on cancer HepG2 cell line, and cytotoxicity for compounds 27a and 30f against normal HepG2cell line.
General investigation of the cytotoxicity results clarified that the examined compounds had greater anti-proliferative activities against HepG2 than MCF-7 cells. In particular, compound 27a was found to be the most potent derivative. Such compound showed strong anti-proliferative activities against MCF-7 and HepG2 cancer cell lines with IC50 values of 7.7 and 4.5 µM, respectively. These values were close to that of the sorafenib (IC50 = 3.51 and 2.17 µM, respectively).
Additionally, compounds 28 (IC50 = 17.2 and 11.7 µM), 30f (IC50 = 18.1 and 10.7 µM), 30i (IC50 = 17.2 and 12.7 µM), and 31b (IC50 = 19.2 and 13.7 µM) demonstrated promising cytotoxicity against MCF-7 and HepG2, respectively. Compounds 27f, 29, 30h, and 31a showed moderate anti-proliferative activities against the two tested cell lines with IC50 values ranging from 17.5 to 23.5 µM. While compounds 27d, 27e, and 30e showed moderate activities against only HepG2 cells with IC50 values ranging from 24.1 to 27.5 µM.
On the other hand, compounds 30b, 30c, and 30d displayed weak cytotoxic activities against the two cell lines with IC50 values ranging from 32.4 to 43.8 µM. While compounds 27d, 27e, and 30e showed weak activities against only MCF-7 with IC50 values ranging from 30.7 to 42.3 µM. Compounds 27b, 27c, 30a, 30g, and 32 showed weak activities against only HepG2 with IC50 values ranging from 40.8 to 43.7 µM. In the contrast, these compounds were inactive against the MCF-7 cell line.
Comparing to the lead compounds 7 (IC50 = 7.2 and 4.1 µM) and 8 (IC50 = 4.4 and 3.3 µM), it was found that the most active compounds 27a (IC50 = 7.2 and 4.1 µM) was slightly less active than these compounds against MCF-7 and HepG2, respectively.
2.2.2. In vitro kinase inhibition assay
The newly prepared compounds have been further assayed for their inhibitory activity towards sorafenib's crucial target (VEGFR-2). The results were reported as 50% inhibition concentration values (IC50, expressed as nM) in comparison to sorafenib as a reference drug ().
To a great extent, the reported results were in good agreement with that of cytotoxicity. This may clarify the possible mechanism of cytotoxic action for the designed compounds. Most of the tested compounds exhibited excellent, moderate, to weak VEGFR-2 inhibitory activities with IC50 values ranging from 3.2 to 38.9 nM, compared to positive control sorafenib (IC50 = 3.12 nM).
Among them, compound 27a was the most potent VEGFR-2 inhibitor with an IC50 value of 3.2 nM. Besides, compounds 28, 29, 30b, 30f, 30i, and 31b showed strong VEGFR-2 inhibitory activities with IC50 values of 4.2, 9.8, 8.7, 4.9, 6.1, and 5.1 nM respectively. Furthermore, compounds 27c, 27f, 30c, 30d, 30h, and 31a displayed moderate activities with IC50 values ranging from 10.7 to 15.7 nM. Finally, compounds 27b, 27d, 27e, 30a, 30e, 30g, and 32 presented weak activities with an IC50 value range of 21.7–38.9 nM.
Comparing to the lead compounds 7 (IC50 = 3.4 nM) and 8 (IC50 = 3.2 nM), it was found that the most active compounds 27a (IC50 = 3.2 nM) had comparable VEGFR-2 inhibitory activity with these lead compounds.
2.2.3. Cytotoxicity against primary rat hepatocytes (normal hepatic cells)
It is of high importance for anticancer agents to have minimum side effects on normal cells. To assess the selectivity of the synthesised compounds against cancer cells over normal ones, the cytotoxicity of the most active anti-proliferative compounds 27a and 30f was evaluated in vitro against primary rat hepatocytes using sorafenib as referenceCitation80. The two compounds 27a and 30f showed cytotoxic activity against cancer HepG2 cell line (4.5-fold and 1.5-fold, respectively) more than cytotoxic activity against normal hepatic cells in comparison to sorafenib (8-fold) ().
2.2.4. Structure-activity relationship
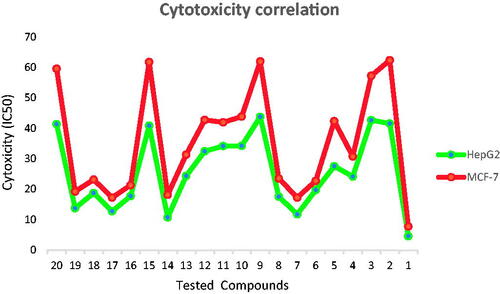
It was noticed that the synthesised compounds were more effective against HepG2 than the MCF-7 cell line (. The obtained data from VEGFR-2 inhibition was highly matched with that of cytotoxicity. So that, the SAR can be built on the results of cytotoxicity and/or VEGFR-2 inhibition. SAR of the newly synthesised compounds was studied along with the pharmacophoric features outlined in the rationale of molecular design.
Figure 3. Correlation of cytotoxicity of the synthesised compounds on the two tested cell lines; MCF-7 and HepG2 showing higher sensitivity against HepG2 than MCF-7 cell line.
Firstly, the effect of the flat heteroaromatic ring on biological activity was examined. Comparing the cytotoxicity and VEGFR-2 inhibitory activities of the synthesised compounds incorporating 3-methylquinoxalin-2(1H)one moiety (compounds 27a–f, 28, and 29) with that incorporating 3-methylquinoxaline-2-thiol moiety (compounds 30a–i, 31a,b, and 32), it was found that 3-methylquinoxalin-2(1H)one moiety was more valuable than methylquinoxaline-2-thiol nucleus.
In addition, the impact of pharmacophore (HBD/HBA) moiety was explored. Regarding 3-methylquinoxalin-2(1H)one derivatives, it was found that compounds comprising the amide group (compounds 27a–f) were more active than that containing diamide group (compound 28), which in turn was more active than that with hydrazide moiety (compound 29). Concerning for 3-methylquinoxaline-2-thiol series, despite maintaining the same order of activity, it was found that compounds bearing amide (compounds 30a–i), diamide (compounds 31a,b), and hydrazide (compound 32) groups showed decreased activity than that incorporating 3-methylquinoxalin-2(1H)one nucleus.
Furthermore, the structure of the terminal hydrophobic tail gave wide varieties of biological activity. In cornering methylquinoxalin-2(1H)one derivatives, it was noticed that hydrophobic aliphatic moiety was critical for activity as found in the most potent member 27a containing tertiary butyl tail.
For the hydrophobic aromatic tail, the heterocyclic ring (compound 27f) displayed high activity than non-heteroaromatic ones (compounds 27a–d). Then, the effect of the substitution on the aromatic rings was examined. it was observed that the cytotoxicity and VEGFR-2 inhibitory activities were highly fluctuated by substitution with electron-withdrawing (EWG) and electron-donating (EDG)groups. The unsubstituted phenyl ring (compound 27b) markedly decreased the biological activity. Substitution with EDG as a 2,6-dimethoxy group (compound 27c) increased the cytotoxicity. Changing the substitutions to be 2,6-dimethyl groups (compound 27d) markedly decreased the activity. Additionally, it was found that the cytotoxicity decreased upon substitution with the electron-withdrawing group as in compound 27e.
For 3-methylquinoxaline-2-thiol derivatives, it was found that aromatic tails (compound 30c-i) were more effective than aliphatic ones (compounds 30a and 30b). For aliphatic derivatives, the bulky tertiary butyl tail (compound 30a) showed cytotoxic activity higher than the alicyclic one (compound 30b). About aromatic tail, comparing the IC50 of compounds 30f (2-tolyl derivative), 30h (4-nitrophenyl derivative), and 30e (phenyl derivative), indicated that substitution with EDG group was more advantageous than substitution with EWG, which was more favourable than the unsubstituted one. Shifting the 2-tolyl into 5-chloro-2-pyridinyl (hetero-aromatic) moiety (compound 30i) produced a mild decrease in activity. While changing the 2-tolyl into 2,6-dimethoxy phenyl moiety (compound 30g) produced a dramatic decrease in activity.
Investigating the activity of compounds 30c, 30d, and 30e demonstrated that, insertion of carbon bridge between the phenyl moiety (hydrophobic tail) and the pharmacophore moiety produced an increase in activity with higher priority for a one-carbon bridge (compound 30c) over than two-carbon one (compound 30d).
2.2.5. Cell cycle analysis
Tissue homeostasis is tightly controlled by the balance between cell proliferation and cell deathCitation81. This creates a great link between the cell cycle and apoptosisCitation82. Therefore manipulation of the cell cycle efficiently affected apoptotic responseCitation83.
Flow cytometry analysis as described by Wand et al.Citation84,Citation85 was used to investigate the effect of the most active compound 27a on the cell cycle progression and apoptosis induction utilising HepG2 cell line. HepG2 cells were treated with 4.5 µM (IC50 of compound 27a) for 24 h, then analysed for its effect on cell cycle distribution. The cell cycle parameters of the incubated cells were compared with untreated control cells ( and ).
Figure 4. Flow cytometric analysis of cell cycle phases after treatment with compound 27a. (A) histograms showing the cell cycle distribution of control and treated cells. (B) Column graphs showing the percentage of cells in each phase of the cell cycle.
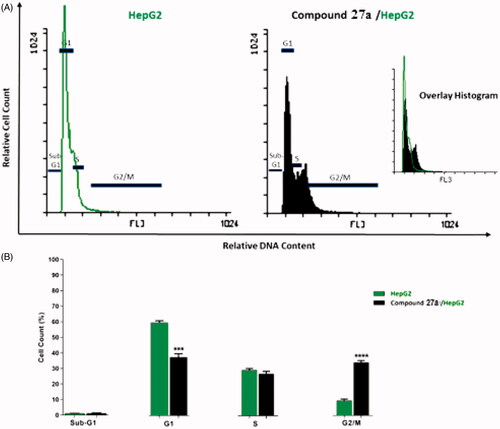
Table 2. Effect of compound 27a on cell cycle progression in HepG2 cells.
The data of flow cytometric analysis revealed the presence of massive accumulation of the treated HepG2 cells at the G2/M (34.14%, 3.5-fold) compared to control cells (9.62%). In addition, a slight difference was observed at the S phase in the percentages of the treated cells (26.96%) and control ones (29.32%). On other hand, the percentage of HepG2 cells increased in the sub G1 phase from 1.13% (control cell) to 1.24% (treated cell). Also, it decreased at the G1 phase from 59.93% (control cell) to 37.66% (treated cell). Such outcomes indicated that compound 27a had a high ability to hinder cell cycle progression of HepG2 cells at the G2/M phase.
2.2.6. Detection of apoptosis
As compound 27a produced a high accumulation of HepG2 cells at the G2/M phase, such compound was further investigated for its apoptotic effect using Annexin V and PI double staining assayCitation86. In such a procedure, HepG2 cells were treated with compound 27a at a concentration of 4.5 µM and incubated for 24 h.
The results revealed that compound 27a induced an increase of HepG2 cells at early (40.47%) and late (0.35%) stages of apoptosis by about five times more than the untreated cells (8.52 and 0.14%, respectively) ( and ).
Figure 5. Flow cytometric analysis of apoptosis in HepG2 cells exposed to compound 27a.
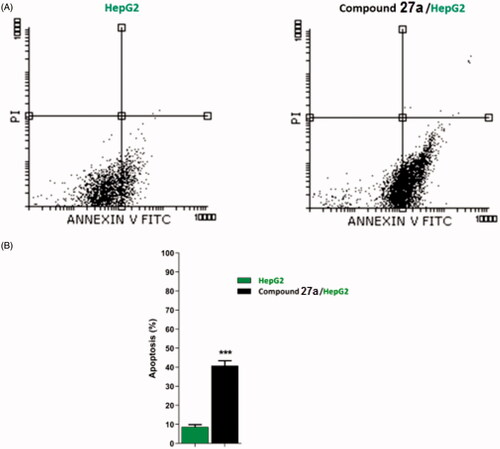
Table 3. Stages of the cell death process in HepG2 cells after treatment with compound 27a.
2.2.7. Effects on the levels of active caspase-3 and caspase-9
To analyse the effect of the most active compound 27a on protein expression levels of caspase-3 and caspase-9, Western blot analysis was utilisedCitation87. In this test, HepG2 cells were treated with compound 27a at its cytotoxic concentration (4.5 µM) for 24 h. The results displayed a marked increase in the level of caspase-3 (2.5-fold) and caspase-9 (3.43-fold) compared to the control cells ( and ). Such findings are consistent with previous reports declared that VEGFR-2 inhibitors can up-regulate both caspase-3 and caspase-9 to induce apoptosisCitation88,Citation89.
Figure 6. The immunoblotting of caspase-3, caspase-9, BAX, and Bcl-2 (normalised to β-actin). (A) Representative Western blot images show the effect of compound 27a (at its IC50 concentration) on the expression levels of BAX, Bcl-2, active caspases-9, and active caspases-3 proteins in HepG2 cells.
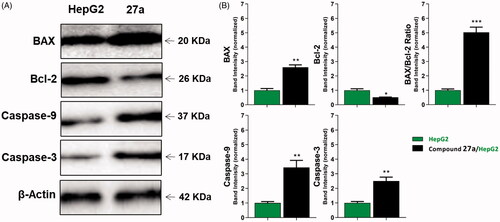
Table 4. Effect of compound 27a on the levels of active caspases-3, active caspases-9, BAX, and Bcl-2 proteins in HepG2 cells treated for 24 h.
2.2.8. Effects on the levels of Bcl-2 family and BAX/Bcl-2 ratio
In this study, BAX and Bcl-2 expression levels in the HepG2 cell line after the treatment with compound 27a were determined by quantitative Western blotting. The results revealed that compound 27a significantly affected the apoptosis pathway in HepG2 cells as it increased the BAX level by 2.6-fold compared to the control cell. Also, it decreased the Bcl-2 level by 2-fold less than the control cells. In addition, the BAX/Bcl-2 ratio was increased 5-fold in the treated cells compared to the control cell ( and ). Meanwhile, compound 27a increased BAX/Bcl-2 ratio, it could trigger apoptosis in the experienced cellsCitation90,Citation91.
2.3. In silico studies
2.3.1. Molecular docking studies
Computational docking studies were carried out against two VEGFR-2 crystal structures (PDB ID; 2OH4 and 4ASD)Citation92. Docking studies were performed to explore the binding mode of the synthesised compounds against the ATP binding pocket of VEGFR-2. The docking experiments were carried out using MOE.14 software. Sorafenib was used as a reference molecule. The binding free energies (ΔG) of the tested ligands and sorafenib against each protein were presented in .
Table 5. The calculated ΔG (binding free energies) of the synthesised compounds and reference drug against VEGFR-2 (PDB ID; 2OH4 and 4ASD) (ΔG in Kcal/mole).
At first, the co-crystallised ligands of each protein were re-docked into the active pockets of VEGFR-2. The resulted RMSD values between the original co-crystallised ligands and the re-docked ones were 1.04 and 1.15 Å. Such values approved the validation of docking processes ().
Figure 7. Alignment of the co-crystallised pose and the re-docked pose of the same ligand (PDB ID; 2OH4).
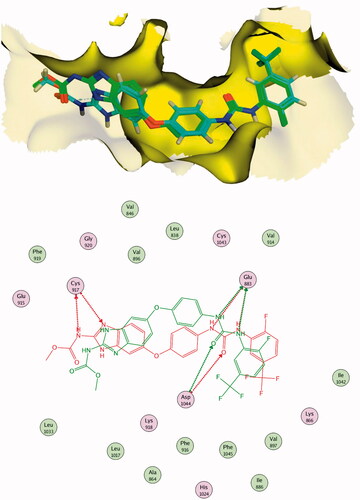
Figure 8. Alignment of the co-crystallised pose and the re-docked pose of the same ligand (PDB ID; 4ASD).
Docking of sorafenib as a reference compound was performed to compare its binding mode with those of the target compounds. The proposed binding mode of the docked sorafenib was the same in the two pockets. The urea moiety bound the receptor through three hydrogen bonding interactions with the crucial amino acids; Glu883 (Glu885) and Asp1044 (Asp1046). Moreover, the N-methylpicolinamide moiety occupied the hinge region, where the pyridine moiety formed one hydrogen bond with Cys917 (Cys919) ().
Figure 9. 2D binding mode of sorafenib into VEGFR-2 (PDB ID; 2OH4).
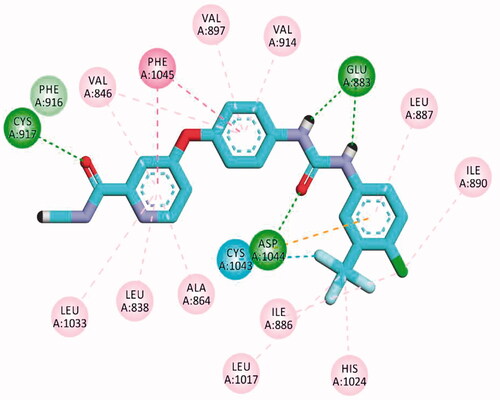
Figure 10. 2D binding mode sorafenib into VEGFR-2 (PDB ID; 4ASD).
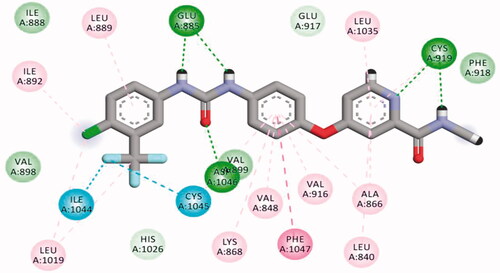
The results of docking studies against VEGFR-2 (PDB ID; 2OH4) showed that the tested ligands have binding modes similar to that of sorafenib against the VEGFR-2 active pocket. From each series, the most cytotoxic compound was selected to analyse its biding mode against the active pocket. Compound 27a as a representative example of 3-methylquinoxalin-2(1H)one series revealed an affinity value of −25.71 kcal/mol. The pharmacophore (amide) moiety bound to the key amino acids Asp1046 and Glu388, where the NH group formed a hydrogen bond with Glu388 while the C=O group formed another hydrogen bond with Asp1046. Four hydrophobic interactions took place between the phenyl ring (linker) and the amino acid residues Val897, Cys1043, Val914, and Lys866 in the linker region. Furthermore, the 3-methylquinoxalin-2(1H)one moiety occupied the hinge region and was involved in five hydrophobic interactions with Leu838, Leu1033, Phe1045, and Phe916. The tert-butyl moiety occupied the allosteric pocket ().
Figure 11. 2D binding mode compound 27a into VEGFR-2 (PDB ID; 2OH4).
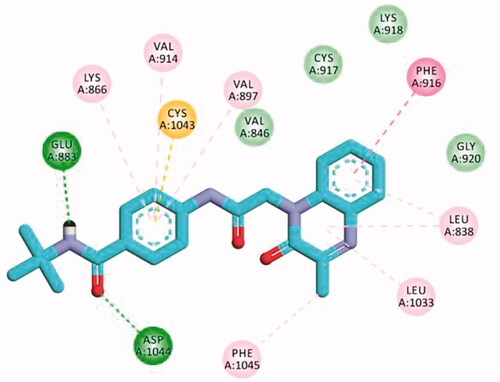
The proposed binding mode of 30f as an example of 3-methylquinoxaline-2-thiol series against VEGFR-2 (PDB ID; 2OH4) was like that of sorafenib. The docking score of such a compound was −23.22 kcal/mol. Compound 30f interacted with the key amino acid Asp1046 via its hydrogen bond acceptor (C=O) group of amide moiety, while the hydrogen bond donor (NH) group interacted with the carboxylate moiety of Glu388. Three hydrophobic interactions were observed between the spacer phenyl ring and amino acid residues Lys866, Val897, and Val914. In addition, 3-methylquinoxaline-2-thiol moiety formed seven hydrophobic interactions with Phe1045, Val846, Leu838, Phe916, and Leu1033. Finally, the terminal 2-tolyl moiety occupied the allosteric biding site forming two hydrophobic interactions with Ile868 ().
Figure 12. 2D binding mode compound 30f into VEGFR-2 (PDB ID; 2OH4).
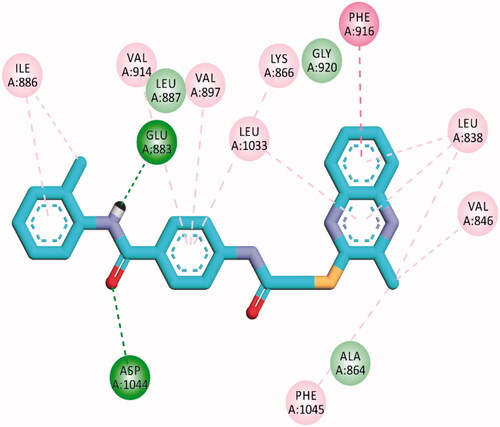
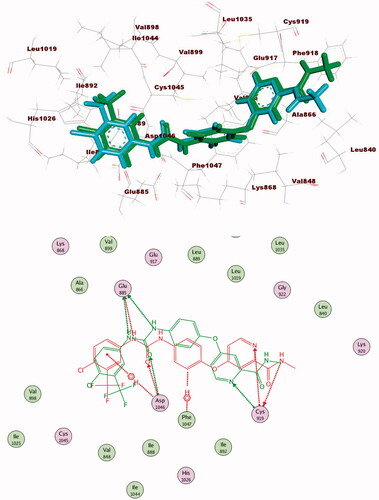
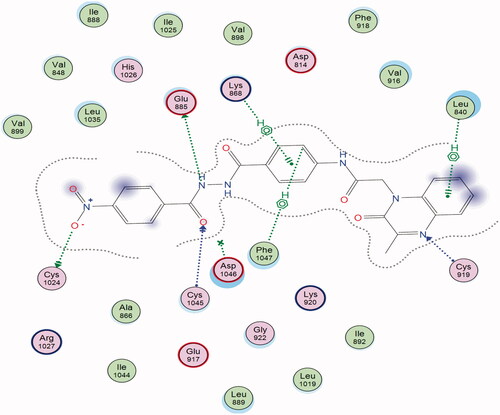
The proposed binding mode of 28 against VEGFR-2 (PDB ID; 4ASD) was like that of sorafenib. The docking score of such a compound was −29.46 kcal/mol. Compound 28 interacted with Cys1045 via its hydrogen bond acceptor (C=O) group of amide moiety, while the hydrogen bond donor (NH) group interacted with the carboxylate moiety of Glu885. Two hydrophobic interactions were observed between the spacer phenyl ring and amino acid residues Lys868, Phe1047. In addition, 3-methylquinoxaline moiety formed one hydrogen bond with the key amino acid Cys919 and one hydrophobic interaction with Leu840. Finally, the terminal p-nitrophenyl moiety occupied the allosteric biding site forming an extra hydrogen bond with Cys1024 ().
Figure 13. 2D binding mode of compound 28 into VEGFR-2. PDB ID; 4ASD.
2.3.2. In silico ADMET study
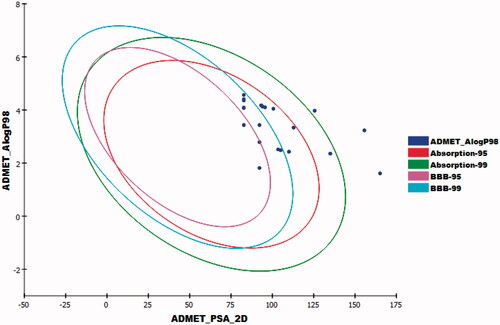
To predict the pharmacokinetics properties of the newly synthesised compounds, computer-aided ADME studies were performed using Discovery Studio 4.0 software. Sorafenib was used as a reference drug. These studies include the assessment of certain parameters as blood-brain barrier (BBB) penetration, absorption level, aqueous solubility, CYP2D6 binding, and plasma protein binding. Predictions of ADME properties for the studied compounds were listed in Supplementary Data).
The results showed that compounds 30a–f have medium BBB diffusion levels while the rest of the compounds showed low to very low levels. Consequently, the CNS side effects were anticipated to be minimal for the majority of the synthesised compounds. Regarding, aqueous solubility, compounds 27a–c, 27e, 28, and 29 exhibited good levels, while compounds 27d, 27f, 30a–i, 31a,b, and 32 showed poor levels. With respect to absorption parameter, all compounds demonstrated good absorption levels except compounds 27e showed moderate absorption level and compounds 28, 30h, and 31b which showed poor to very poor intestinal absorption levels. Moreover, the effect on cytochrome P450 2D6 was investigated. The results showed that all the tested compounds were non-inhibitors of CYP2D6. Consequently, hepatotoxicity is not expected upon their administration. The plasma protein binding model displayed that compounds 27a, 28, 30a–c, 30g,h, 31a,b, and 32 were anticipated to bind plasma protein <90%. On the other hand, compounds 27b–f, 29, 30d–f, and 30i were expected to bind plasma protein more than 90% ().
Figure 14. The expected ADMET study for the synthesised compounds.
2.3.3. In silico toxicity study
The toxicity profile of the synthesised compounds was determined based on the validated and constructed models in Discovery studio 4.0 softwareCitation93,Citation94. This includes the prediction of certain parameters as FDA rodent carcinogenicity, carcinogenic potency TD50, rat maximum tolerated dose, developmental toxicity potential, rat oral LD50, rat chronic lowest observed adverse effect level (LOAEL), ocular irritancy, and skin irritancy.
Regarding the FDA rodent carcinogenicity model compounds 27a–f, 28, 30b, 30g, and 30i were forecasted to be non-carcinogenic. For carcinogenic potency, TD50 mouse model compounds 27a,b, 29, 30a, 30c–f, 30h, i, 31a,b, and 32 showed TD50 values ranging from 15,600 to 225,175 mg/kg body weight, which is higher than sorafenib (14,244 mg/kg body weight), while compounds 27c–f, 28, 30b, and 30g showed carcinogenic potency TD50 lower than that of sorafenib. With respect to the rat maximum tolerated dose model (MTD), compounds 29, 30e,f, 30i, 31a, and 32 demonstrated maximum tolerated dose with a range of 0.096 to 0.194 g/kg body weight, which is higher than that of sorafenib (0.089 g/kg body weight). In Addition, all compounds were predicted to be non-toxic against the developmental toxicity potential model except compounds 27f, 30g, and 30i. For the rat oral LD50 model, all compounds revealed oral LD50 values in a range of 1.523 to 19.408 g/kg body weight which is higher than that of sorafenib (0.823 g/kg body weight). For the rat chronic LOAEL model, the examined members displayed LOAEL values ranging from 0.036 to 0.376 g/kg body weight. These values are higher than sorafenib (0.005 g/kg body weight). Additionally, all the tested compounds were predicted to be mild irritants against the ocular irritancy model and non-irritant against the skin irritancy model. The values of toxicity parameters were depicted in the Supplementary Data.
Citation3. Conclusion
New twenty quinoxaline derivatives were designed and synthesised as anticancer agents with VEGFR-2 inhibitory activity. The synthesised compounds were evaluated in vitro for their anti-proliferative activities against breast cancer (MCF-7) and hepatocellular carcinoma (HepG2). Compound 27a was the most potent derivative revealing strong anti-proliferative activity against MCF-7 and HepG2 cell lines with IC50 values of 7.7 and 4.5 µM, respectively, comparing to sorafenib (IC50 = 3.51 and 2.17 µM, respectively). In addition, compounds 28 (IC50 = 17.2 and 11.7 µM), 30f (IC50 = 18.1 and 10.7 µM), 30i (IC50 = 17.2 and 12.7 µM), and 31b (IC50 = 19.2 and 13.7 µM) demonstrated promising cytotoxicity against MCF-7 and HepG2, respectively. The results of the VEGFR-2 enzyme assay were highly correlated with that of cytotoxicity, where the most potent antiproliferative derivatives exhibited good VEGFR-2 inhibitory effects. Compounds 27a was the most potent VEGFR-2 inhibitor with an IC50 value of 3.2 nM in comparison to sorafenib (IC50 = 3.12 nM). SAR revealed that 3-methylquinoxalin-2(1H)one moiety was more efficient than 3-methylquinoxaline-2-thiol moiety as a heterocyclic ring. The amide moiety was more advantageous as a pharmacophore than the corresponding diamide and hydrazide moieties. In addition, the tert-butyl moiety was the most effective hydrophobic tail. Cell cycle analysis of compounds 27a revealed that such compound can arrest HepG2 cell growth at G2/M phase by 3.5-fold greater than control cell. Moreover, compound 27a induced apoptosis in HepG2 cells by five times more than the control cells. Furthermore, it increased the level of caspase-3 and caspase-9 by 2.5-folds and 3.43-fold, respectively. Also, compound 27a showed an increase in the BAX level (2.6-fold), decrease in the Bcl-2 level (2-fold), and elevation of BAX/Bcl-2 ratio (5-fold) for the control cell. The results of docking studies revealed that the proposed binding modes of the designed compounds were similar to that of sorafenib. Finally, this work presents compound 27a as a lead candidate that can be further optimised for the synthesis of a promising VEGFR 2 inhibitor.
4. Experimental
4.1. Chemistry and material
All the reagents, chemicals, and apparatus were presented in Supplementary Data. Compounds 11Citation67, 12Citation67, 13Citation68, 14Citation69,Citation70, 15Citation69,Citation70, and the intermediates (24a,b, 25, 26a–j)Citation63,Citation64 were prepared according to the reported procedures.
4.1.1. General procedure for the synthesis of target compounds 27a–f, 28, and 29
A mixture of potassium salt 12 (0.5 g, 2 mmol) and appropriate intermediates 26a,d,g–j, 24b, and 25 (1 mmol) in dry DMF (15 ml) with the presence of potassium iodide (0.2 g, 1.2 mmol), was heated over a water bath for 3 h. The reaction mixture was then cooled, poured into ice-cooled water (150 ml) with continuous stirring. The solid separated was filtered, washed with water several times, then dried and crystallised from the proper solvent to afford the final target compounds 27a-f, 28, and 29, respectively.
4.1.1.1. N-(tert-butyl)-4-(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)benzamide (27a)
The product was crystallised from EtOH/DCM mixture (50:50) as dark green crystal (0.3 g, 80%); m. p. = 230–232 °C; FT-IR (ν max, cm−1): 3291 (NH), 3077 (CH aromatic), 2967 (CH aliphatic), 1648 (C=O), 1601 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm):10.66 (s, 1H), 7.81–7.79 (m, 3H), 7.64–7.61 (m, 3H), 7.58–7.56 (m, 1H), 7.53 (dd, J = 8.5, 1.3 Hz, 1H), 7.38 (s, 1H), 5.16 (s, 2H), 2.49 (s, 3H), 1.38 (s, 9H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 166.00, 165.63, 157.96, 154.85, 141.25, 133.48, 132.46, 131.07, 130.18(2 C), 129.27(2 C), 128.76, 123.92, 118.55(2 C), 115.20, 51.16, 45.76, 29.11(3 C), 21.59; MS (m/z): 393.19 (M+ +1, base beak, 100%); Anal. Calcd. for C22H24N4O3 (392.46): C, 67.33; H, 6.16; N, 14.28; Found C, 67.74; H, 6.32; N, 14.39%.
4.1.1.2. 4-(2-(3-Methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)-N-phenethylbenzamide (27b)
The product was crystallised from EtOH/DCM mixture (50:50) as white crystal (0.34 g, 85%); m. p. = 283–285 °C; FT-IR (ν max, cm-1): 3274 (NH), 3038 (CH aromatic), 2921 (CH aliphatic), 1676, 1656, 1630 (C=O), 1604 (C=N); 1H NMR (700 MHz, DMSO d6) δ (ppm):10.69 (s, 1H), 10.39 (s, 1H), 8.48 (t, J = 5.6 Hz, 1H), 7.82–7.81 (m, 1H), 7.80 (q, J = 1.7 Hz, 1H), 7.67–7.62 (m, 2H), 7.57 (ddd, J = 8.5, 7.1, 1.5 Hz, 1H), 7.52 (dd, J = 8.5, 1.3 Hz, 1H), 7.38 (ddd, J = 8.1, 7.1, 1.2 Hz, 1H), 7.30 (tt, J = 7.7, 1.7 Hz, 2H), 7.26–7.23 (m, 2H), 7.22–7.19 (m, 1H), 5.16 (s, 2H), 3.47 (ddd, J = 8.6, 7.5, 5.9 Hz, 2H), 2.86–2.82 (m, 2H), 2.49 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 165.97, 165.69, 157.97, 154.84, 141.52, 140.03, 133.46, 132.46, 130.18(2 C), 129.28(2 C), 129.11(2 C), 128.81(2 C), 128.56, 126.55, 123.92, 118.79(2 C), 115.19, 45.76, 41.32, 35.64, 21.59; MS (m/z): 441.2 (M+ +1, 28.68%); Anal. Calcd. for C26H24N4O3 (440.50): C, 70.89; H, 5.49; N, 12.72; Found C, 70.99; H, 5.13; N, 12.64%.
4.1.1.3. N-(2,6-Dimethoxyphenyl)-4-(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)-benzamide (27c)
The product was crystallised from ethanol/gl.acetic acid mixture (80:20) as yellow crystal (0.31 g, 80%); m. p. < 300 °C; FT-IR (ν max, cm−1): 3422, 3293 (NH), 3058 (CH aromatic), 2929 (CH aliphatic), 1661 (C=O), 1602 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm):10.76 (s, 1H), 10.52 (s,1H), 7.97 (d, J = 6.5 Hz, 1H), 7.94–7.93 (m, 2H), 7.83 (d, J = 6.7 Hz, 1H), 7.77 (d, J = 8.7 Hz, 2H), 7.73–7.71 (m, 1H), 7.68 (t, J = 1.4 Hz, 1H), 7.57 (m, 2H), 7.48 (d, J = 1.3 Hz, 1H), 4.33 (s, 2H), 3.32 (s, 6H), 2.68 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm):167.11, 166.26, 165.46, 155.45, 151.97, 142.71, 140.82, 139.36, 135.19, 131.97, 130.93, 130.38, 130.05, 129.89, 129.05(2 C), 128.91, 128.68, 127.64, 127.43, 127.38, 118.85, 35.46(3 C), 22.19; Anal. Calcd. for C26H24N4O5 (472.50): C, 66.09; H, 5.12; N, 11.86; Found C, 66.01; H, 5.15; N, 11.64%.
4.1.1.4. N-(2,6-Dimethylphenyl)-4-(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)-benzamide (27d)
The product was crystallised from EtOH/DCM mixture (50:50) as yellowish white crystal (0.35 g, 85%); m. p. = 240–242 °C; FT-IR (ν max, cm−1): 3429 (NH), 3067 (CH aromatic), 2934 (CH aliphatic), 1656, (C=O), 1603 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.76 (s, 1H), 9.67 (s, 1H), 7.99 (d, J = 8.5 Hz, 2H), 7.81 (dd, J = 8.0, 1.5 Hz, 1H), 7.72 (d, J = 8.6 Hz, 2H), 7.58 (ddd, J = 8.5, 7.0, 1.5 Hz, 1H), 7.54 (dd, J = 8.6, 1.3 Hz, 1H), 7.42–7.35 (m, 1H), 7.12 (s, 3H), 5.19 (s, 2H), 2.50 (s, 3H), 2.18 (s, 6H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 165.77, 164.83, 157.98, 154.86, 141.88, 136.12, 135.89, 133.48, 132.48, 130.19(2 C), 129.64(2 C), 129.29(2 C), 129.02(2 C), 128.16, 127.07, 123.94, 118.95, 115.21, 45.79, 21.60, 18.56(2 C); MS (m/z): 440.18 (M+, 27.13%); Anal. Calcd. for C26H24N4O3 (440.50): C, 70.89; H, 5.49; N, 12.72; Found C, 70.54; H, 5.39; N, 12.52%.
4.1.1.5. 4-(2-(3-Methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)-N-(4-nitrophenyl)benzamide (27e)
The product was crystallised from ethanol/gl.acetic acid mixture (80:20) as brown crystal (0.29 g, 78%); m. p. > 300 °C; FT-IR (ν max, cm−1): 3428 (NH), 3060 (CH aromatic), 2955 (CH aliphatic), 1653, (C=O), 1599 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.83 (s, 1H), 10.72 (s, 1H), 8.29–8.26 (m, 2H), 8.08–8.06 (m, 2H), 8.02–8.00 (m, 2H), 7.81 (dt, J = 8.0, 1.8 Hz, 1H), 7.77–7.74 (m, 2H), 7.59–7.53 (m, 2H), 7.39 (ddd, J = 8.2, 7.0, 1.4 Hz, 1H), 5.19 (s, 2H), 2.49 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 165.92, 157.96, 154.85, 146.10, 142.82, 142.58, 133.46, 130.20(2 C), 129.62(2 C), 129.29(2 C), 129.18(2 C), 125.27(2 C), 123.95, 120.24(2 C), 118.90, 115.21, 45.82, 21.59. Anal. Calcd. for C24H19N5O5 (457.45): C, 63.02; H, 4.19; N, 15.31; Found C, 63.17; H, 4.13; N, 15.64%.
4.1.1.6. N-(5-Chloropyridin-2-yl)-4-(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetamido)-benzamide (27f)
The product was crystallised from ethanol/gl.acetic acid mixture (80:20) as dark brown crystal (0.36 g, 90%); m. p. < 300 °C; FT-IR (ν max, cm−1): 3414 (NH), 3030 (CH aromatic), 2962 (CH aliphatic), 1660 (C=O), 1600 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.78 (s, 1H), 10.21 (s, 1H), 7.97 (d, J = 8.4 Hz, 2H), 7.81–7.79 (m, 2H), 7.73 (d, J = 8.5 Hz, 2H), 7.58 (td, J = 7.6, 6.9, 1.5 Hz, 1H), 7.56–7.53 (m, 1H), 7.40–7.36 (s, 1H), 7.19 (t, J = 8.9 Hz, 2H), 5.19 (s, 2H), 2.49 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 165.82, 165.15, 158.00, 157.96, 154.85, 142.05, 136.06, 133.46, 132.48, 130.18, 129.86(2 C), 129.21(2 C), 123.93, 122.62, 122.58, 118.86, 115.68, 115.55, 115.19, 45.80, 21.59; MS (m/z): 448.1 (M+ +1, base beak, 100%); Anal. Calcd. for C23H18ClN5O3 (447.88): C, 61.68; H, 4.05; N, 15.64; Found C, 61.17; H, 4.13; N, 15.36%.
4.1.1.7. 2-(3-Methyl-2-oxoquinoxalin-1(2H)-yl)-N-(4-(2-(4-nitrobenzoyl)hydrazine-1-carbonyl) phenyl)acetamide (28)
The product was crystallised from EtOH/DCM mixture (50:50) as white crystal (0.28 g, 74%); m. p. = 257–259 °C; FT-IR (ν max, cm−1): 3271 (NH), 3055 (CH aromatic), 2944 (CH aliphatic), 1656 (C=O), 1601(C=N); 1H NMR (700 MHz, DMSO-d6) δ 10.82 (s, 1H), 10.56 (s, 2H), 8.01 (m, 2H), 7.94 (m, 2H), 7.81 (d, J = 7.9 Hz, 2H), 7.73 (d, J = 8.7 Hz, 2H), 7.56 (dd, J = 22.6, 8.2 Hz, 3H), 7.39 (d, J = 7.6 Hz, 1H), 5.19 (s, 2H), 2.49 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ 165.86, 164.81 (2C), 157.97, 154.85, 149.86, 142.31, 138.75, 133.45, 132.47(2 C), 130.20 (2C), 129.46(2C), 129.06(3 C), 124.25(2C), 119.01(2C), 115.20, 45.81, 21.59. Anal. Calcd. for C25H20N6O6 (500.47): C, 60.00; H, 4.03; N, 16.79; Found C, 60.78; H, 4.54; N, 16.56%.
4.1.1.8. 2-(3-Methyl-2-oxoquinoxalin-1(2H)-yl)-N-(4-(2-phenylhydrazine-1-carbonyl) phenyl) acetamide (29)
The product was crystallised from ethanol/gl.acetic acid mixture (80:20) as greenish yellow crystal (0.3 g, 75%); m. p. < 300 °C; FT-IR (ν max, cm−1): 3291 (NH), 3041 (CH aromatic), 2967 (CH aliphatic), 1648 (C=O), 1601 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.70 (s, 1H), 10.31 (s, 1H), 10.02 (s, 1H), 8.33 (dd, J = 8.1, 1.4 Hz, 1H), 8.26–7.75 (m, 4H), 7.75–7.72 (m, 2H), 7.72–7.69 (m, 1H), 7.63 (d, J = 8.5 Hz, 1H), 7.59–7.56 (m, 1H), 7.49–7.46 (m, 1H), 7.38 (t, J = 8.1 Hz, 1H), 7.17–7.14 (m, 1H), 5.22 (s, 2H), 2.74 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 157.97, 155.40, 154.86, 133.47, 132.47(2C), 130.20(3 C), 129.78, 129.29(2C), 129.05(3 C), 128.34(2C), 123.95, 123.50(2C), 115.69, 115.20, 45.79, 21.59; Anal. Calcd. for C24H21N5O3 (427.46): C, 67.44; H, 4.95; N, 16.38; Found C, 67.73; H, 4.76; N, 16.54%.
4.1.2. General procedure for the synthesis of target compounds 30a–i, 31a,b, and 32
Equimolar amounts of potassium salt 15 (0.3 g, 1 mmol) and appropriate intermediates 26a–g,i,j, 24a,b, and 25 (1 mmol) in dry DMF (15 ml) with the presence of potassium iodide (0.2 g, 1.2 mmol) was heated on a water-bath for 3 h. After cooling, the reaction mixture was poured into an ice-water (150 ml) with continuous stirring. The resulted precipitate was filtered and crystallised from the proper solvent to give the final target compounds 30a–i, 31a,b, and 32, respectively.
4.1.2.1. N-(tert-Butyl)-4-(2-((3-methylquinoxalin-2-yl)thio)acetamido)benzamide (30a)
The product was crystallised from ethanol as brown crystal (0.15 g, 70%); m. p. = 231–233 °C; FT-IR (ν max, cm−1): 3315 (NH), 3048 (CH aromatic), 2969 (CH aliphatic), 1673, 1631 (C=O), 1599 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.64 (s, 1H), 7.97 (d, J = 8.2 Hz, 1H), 7.82 (d, J = 7.9 Hz, 1H), 7.78 (d, J = 8.3 Hz, 2H), 7.73–7.71 (m, 1H), 7.70–7.68 (m, 1H), 7.67 (d, J = 8.3 Hz, 2H), 7.62 (s, 1H), 4.30 (s, 2H), 2.67 (s, 3H), 1.37 (s, 9H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 166.88, 166.11, 155.47, 151.98, 141.71, 140.81, 139.35, 130.97, 130.03(2C), 128.91(2C), 128.72, 128.68(2C), 127.36, 118.51, 51.14, 35.39, 29.11(3 C), 22.19; MS (m/z): 409.1 (M+ +1, base beak, 100%); Anal. Calcd. for C22H24N4O2S (408.52): C, 64.68; H, 5.92; N, 13.71; Found C, 64.56; H, 5.89; N, 13.32%.
4.1.2.2. N-Cyclohexyl-4-(2-((3-methylquinoxalin-2-yl)thio)acetamido)benzamide (30b)
The product was crystallised from ethanol as dark brown crystal (0.18 g, 85%); m. p. = 238–240 °C; FT-IR (ν max, cm−1): 3289 (NH), 3055 (CH aromatic), 2928, 2851 (CH aliphatic), 1671, 1628 (C=O), 1607 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.66 (s, 1H), 8.07 (d, J = 7.9 Hz, 1H), 7.96 (s, 1H), 7.84–7.81 (m, 3H), 7.71 (ddd, J = 8.3, 6.9, 1.6 Hz, 1H), 7.70–7.67 (m, 3H), 4.30 (s, 2H), 3.77–3.72 (m, 1H), 2.67 (s, 3H), 1.83–1.72 (m, 4H), 1.63–1.59 (m, 1H), 1.33–1.27 (m, 4H), 1.12 (s, 1H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 166.92, 165.21, 155.47, 151.98, 141.88, 140.82, 139.36, 130.04(2C), 128.91(2C), 128.68(2C), 127.37, 118.62(2C), 48.71, 35.38, 32.95(2C), 25.75, 25.44(2C), 22.18; MS (m/z): 435.1 (M+ +1, base beak, 100%); Anal. Calcd. for C24H26N4O2S (434.56): C, 66.33; H, 6.03; N, 12.89; Found C, 66.39; H, 6.09; N, 12.98%.
4.1.2.3. N-benzyl-4-(2-((3-methylquinoxalin-2-yl)thio)acetamido)benzamide (30c)
The product was crystallised from ethanol as yellow crystal (0.16 g, 75%); m. p. = 235–237 °C; FT-IR (ν max, cm−1): 3287 (NH), 3071 (CH aromatic), 2933 (CH aliphatic), 1674, 1633, (C=O), 1607 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.69 (s, 1H), 8.94 (s, 1H), 7.96 (dd, J = 8.0, 1.5 Hz, 1H), 7.88 (d, J = 8.5 Hz, 2H), 7.82 (dd, J = 8.3, 1.6 Hz, 1H), 7.73–7.70 (m, 3H), 7.68 (td, J = 7.6, 1.5 Hz, 1H), 7.32 (d, J = 6.3 Hz, 4H), 7.24 (tt, J = 5.9, 2.4 Hz, 1H), 4.47 (d, J = 5.9 Hz, 2H), 4.31 (s, 2H), 2.67 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 166.97, 166.10, 155.45, 151.97, 142.13, 140.81, 140.27, 139.35, 130.04(2C), 129.49(2C), 128.90(2C), 128.73(2C), 128.69, 128.67, 127.67, 127.38, 127.16, 118.77, 43.02, 35.41, 22.18; MS (m/z): 443.1 (M+ +1, 79.98%); Anal. Calcd. for C25H22N4O2S (442.54): C, 67.85; H, 5.01; N, 12.66; Found C, 67.73; H, 5.09; N, 12.78%.
4.1.2.4. 4-(2-((3-Methylquinoxalin-2-yl)thio)acetamido)-N-phenethylbenzamide (30d)
The product was crystallised from ethanol as brown crystal (0.18 g, 85%); m. p. = 220–222 °C; FT-IR (ν max, cm−1): 3299 (NH), 3026 (CH aromatic), 2925 (CH aliphatic), 1664, 1630 (C=O), 1607 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.67 (s, 1H), 8.47 (s, 1H), 7.98–7.94 (m, 1H), 7.85–7.79 (m, 3H), 7.72 (ddd, J = 8.4, 7.0, 1.7 Hz, 1H), 7.71–7.66 (m, 3H), 7.32–7.27 (m, 2H), 7.26–7.23 (m, 2H), 7.23–7.18 (m, 1H), 4.31 (s, 2H), 3.47 (ddd, J = 8.7, 7.5, 5.9 Hz, 2H), 2.84 (t, J = 7.5 Hz, 2H), 2.67 (s, 3H); 13C NMR (176 MHz, DMSO d6) δ (ppm): 166.95, 166.05, 155.45, 151.97, 141.98, 140.81, 140.04, 139.35, 130.03(2C), 129.76 (2C), 129.11(2C), 128.90, 128.81, 128.68(2C), 128.53, 127.38, 126.54, 118.75, 41.32, 35.66, 35.40, 22.18; MS (m/z): 457.2 (M+ +1, base beak, 100%); Anal. Calcd. for C26H24N4O2S (456.56): C, 68.40; H, 5.30; N, 12.27; Found C, 68.39; H, 5.09; N, 12.32%.
4.1.2.5. 4-(2-((3-Methylquinoxalin-2-yl)thio)acetamido)-N-phenylbenzamide (30e)
The product was crystallised from ethanol as brown crystal (0.18 g, 85%); m. p. = 218–220 °C; FT-IR (ν max, cm−1): 3268 (NH), 3039 (CH aromatic), 2970, 2908 (CH aliphatic), 1666, 1640 (C=O), 1597 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.75 (s, 1H), 10.14 (s, 1H), 7.98–7.95 (m, 3H), 7.83 (dd, J = 8.0, 1.6 Hz, 1H), 7.80–7.76 (m, 4H), 7.72 (ddd, J = 8.3, 6.9, 1.6 Hz, 1H), 7.68 (ddd, J = 8.4, 7.0, 1.6 Hz, 1H), 7.37–7.33 (m, 2H), 7.09 (tt, J = 7.4, 1.2 Hz, 1H), 4.33 (s, 2H), 2.67 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 167.07, 165.34, 155.44, 151.97, 142.46, 140.82, 139.74, 139.36, 130.02(2C), 129.93, 129.20(2C), 129.04(2C), 128.89, 128.68(2C), 127.37, 123.97, 120.79, 118.79, 35.44, 22.18; MS (m/z): 429.1 (M+ +1, base beak, 100%); Anal. Calcd. for C24H20N4O2S (428.51): C, 67.27; H, 4.70; N, 13.08; Found C, 67.04; H, 4.89; N, 13.42%.
4.1.2.6. 4-(2-((3-Methylquinoxalin-2-yl)thio)acetamido)-N-(o-tolyl)benzamide (30f)
The product was crystallised from EtOH/DCM mixture (50:50) as white crystal (0.2 g, 90%); m. p. = 247–249 °C; FT-IR (ν max, cm−1): 3262 (NH), 3056 (CH aromatic), 2911 (CH aliphatic), 1659, 1641 (C=O), 1607 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.75 (s, 1H), 9.77 (s, 1H), 7.97 (dd, J = 8.4, 4.1 Hz, 3H), 7.84 (d, J = 8.1 Hz, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.73 (t, J = 7.5 Hz, 1H), 7.69 (t, J = 7.5 Hz, 1H), 7.34 (d, J = 7.8 Hz, 1H), 7.27 (d, J = 7.5 Hz, 1H), 7.22 (t, J = 7.5 Hz, 1H), 7.17 (t, J = 7.4 Hz, 1H), 4.33 (s, 2H), 2.68 (s, 3H), 2.24 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 167.05, 165.12, 155.46, 151.98, 142.42, 140.82, 139.36, 136.99, 134.13, 130.75, 130.05, 129.57(2C), 129.14(2C), 128.91, 128.69, 127.38, 127.03, 126.44, 126.33, 118.82, 35.44, 22.19, 18.40; MS (m/z): 443.1 (M+ +1, base beak, 100%); Anal. Calcd. for C25H22N4O2S (442.54): C, 67.85; H, 5.01; N, 12.66; Found C, 67.73; H, 5.09; N, 12.78%.
4.1.2.7. N-(2,6-Dimethoxyphenyl)-4-(2-((3-methylquinoxalin-2-yl)thio)acetamido) benzamide (30g)
The product was crystallised from EtOH/DCM mixture (50:50) as light brown crystal (0.17 g, 80%); m. p. = 258–260 °C; FT-IR (ν max, cm−1): 3298 (NH), 3033 (CH aromatic), 2931 (CH aliphatic), 1665 (C=O), 1607 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.77 (s, 1H), 9.37 (s, 1H), 7.98 (t, J = 8.3 Hz, 4H), 7.93 (d, J = 8.1 Hz, 1H), 7.84 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 8.3 Hz, 2H), 7.75–7.72 (m, 1H), 7.71–7.67 (m, 1H), 7.35–7.33 (m, 1H), 4.34 (s, 2H), 3.99–3.97 (s, 3H), 2.69 (s, 6H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 167.09, 164.78, 155.46, 151.99, 151.84, 142.57, 140.83, 139.37, 137.68, 130.06, 129.45(2C), 129.05, 129.00, 128.92, 128.69, 127.39, 126.88, 124.35, 119.00, 118.90, 110.11, 56.46(2C), 35.46, 22.20; Anal. Calcd. for C26H24N4O4S (488.56): C, 63.92; H, 4.95; N, 11.47; Found C, 63.56; H, 4.45; N, 11.56%.
4.1.2.8. 4-(2-((3-Methylquinoxalin-2-yl)thio)acetamido)-N-(4-nitrophenyl)benzamide (30h)
The product was crystallised from ethanol as pale yellow crystal (0.12 g, 60%); m. p. = 200–202 °C; FT-IR (ν max, cm−1): 3316 (NH), 3028 (CH aromatic), 2921 (CH aliphatic), 1682, 1650 (C=O), 1596 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.76 (s, 1H), 10.26 (s, 1H), 7.96 (dd, J = 9.2, 2.3 Hz, 3H), 7.82 (td, J = 7.6, 7.0, 1.8 Hz, 3H), 7.82–7.76 (m, 2H), 7.71 (ddd, J = 8.3, 6.9, 1.6 Hz, 1H), 7.68 (ddd, J = 8.3, 6.9, 1.6 Hz, 1H), 7.43–7.38 (m, 2H), 4.33 (s, 2H), 2.67 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 167.10, 165.42, 155.43, 151.96, 142.61, 140.81, 139.36, 138.73, 130.02, 129.61(3 C), 129.25(2C), 128.96(2C), 128.89, 128.68, 127.55, 127.36, 122.24, 118.80, 35.43, 22.18; Anal. Calcd. for C24H19N5O4S (473.51): C, 60.88; H, 4.04; N, 14.79; Found C, 60.39; H, 4.58; N, 14.66%.
4.1.2.9. N-(5-Chloropyridin-2-yl)-4-(2-((3-methylquinoxalin-2-yl)thio)acetamido)benzamide (30i)
The product was crystallised from ethanol as brown crystal (0.19 g, 90%); m. p. = 190–192 °C; FT-IR (ν max, cm−1): 3274 (NH), 3041 (CH aromatic), 2945 (CH aliphatic), 1679 (C=O), 1599 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.86 (s, 1H), 10.78 (s, 1H), 8.44 (t, J = 2.6 Hz, 1H), 8.24–8.23 (m, 1H), 8.04–8.02 (m, 2H), 7.96 (d, J = 3.3 Hz, 2H), 7.91 (d, J = 4.8 Hz, 2H), 7.82 (d, J = 6.6 Hz, 1H), 7.76 (s, 1H), 7.72–7.71 (m, 1H), 4.32 (s, 2H), 2.67 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 167.35, 167.16, 155.42, 151.96, 151.48, 146.72, 140.80, 139.35, 138.27, 130.95, 130.04(2C), 129.69(2C), 128.91, 128.67(2C), 127.38, 118.86, 118.71, 116.18, 35.45, 22.18; MS (m/z): 464.1 (M+ +1, 68.32%),; Anal. Calcd. for C23H18ClN5O2S (463.94): C, 59.54; H, 3.91; N, 15.10; Found C, 59.56; H, 3.59; N, 15.35%.
4.1.2.10. N-(4-(2-benzoylhydrazine-1-carbonyl)phenyl)-2-((3-methylquinoxalin-2-yl)thio) acetamide (31a)
The product was crystallised from ethanol as brown crystal (0.13 g, 65%); m. p. = 195–197 °C; FT-IR (ν max, cm−1): 3262 (NH), 3038 (CH aromatic), 2921 (CH aliphatic), 1657 (C=O), 1601 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.76 (s, 1H), 10.48 (s, 1H), 10.42–10.41 (s, 1H), 7.94 (d, J = 6.8 Hz, 4H), 7.83 (d, J = 6.8 Hz, 1H), 7.77 (d, J = 4.6 Hz, 1H), 7.72 (d, J = 1.6 Hz, 1H), 7.69–7.67 (m, 1H), 7.60 (d, J = 6.1 Hz, 2H), 7.53 (m, 3H), 4.33 (s, 2H), 2.68 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 167.11, 166.36, 165.77(2C), 155.45, 151.97, 142.68, 140.82, 139.36, 133.09, 132.32, 130.05(2C), 129.00(3 C), 128.98(2C), 128.68(2C), 127.93, 127.38, 118.90, 35.45, 22.19; MS (m/z): 472.1 (M+ +1, 53.18%); Anal. Calcd. for C25H21N5O3S (471.54): C, 63.68; H, 4.49; N, 14.85; Found C, 63.88; H, 4.68; N, 14.99%.
4.1.2.11. 2-((3-Methylquinoxalin-2-yl)thio)-N-(4-(2-(4-nitrobenzoyl)hydrazine-1-carbonyl) phenyl) acetamide (31b)
The product was crystallised from ethanol as brownish red crystal (0.15 g, 70%); m. p. = 196–198 °C; FT-IR (ν max, cm−1): 3261 (NH), 3044 (CH aromatic), 2911 (CH aliphatic), 1664 (C=O), 1601 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 10.81 (s, 2H), 10.55 (s, 1H), 8.39 (t, J = 8.7 Hz, 4H), 8.17 (s, 2H), 7.96 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 3.8 Hz, 1H), 7.78 (d, J = 8.3 Hz, 1H), 7.72 (t, J = 7.6 Hz, 1H), 7.68 (t, J = 7.5 Hz, 1H), 4.33 (s, 2H), 2.67 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm): 165.70, 164.87, 164.85, 164.75, 155.44, 149.98, 149.89, 139.36, 138.67, 130.08(2C), 129.52(2C), 129.47(3 C), 127.38(3 C), 124.32(2C), 119.10, 118.93, 35.45, 22.18; MS (m/z): 517.0 (M+ +1, base beak, 100%); Anal. Calcd. for C25H20N6O5S (516.53): C, 58.13; H, 3.90; N, 16.27; Found C, 58.21; H, 3.98; N, 16.48%.
4.1.2.12. 2-((3-Methylquinoxalin-2-yl)thio)-N-(4-(2-phenylhydrazine-1-carbonyl)phenyl) acetamide (32)
The product was crystallised from ethanol as white crystal (0.18 g, 85%); m. p. = 185–187 °C; FT-IR (ν max, cm−1): 3272 (NH), 3083 (CH aromatic), 2931 (CH aliphatic), 1676 (C=O), 1598 (C=N); 1H NMR (700 MHz, DMSO-d6) δ (ppm): 12.71 (s, 1H), 10.76 (s, 1H), 10.20 (s, 1H), 7.92–7.88 (m, 2H), 7.84–7.79 (m, 2H), 7.76–7.73 (m, 2H), 7.69–7.65 (m, 2H), 7.38–7.31 (m, 2H), 7.15 (t, J = 7.7 Hz, 1H), 6.78 (d, J = 12.6, 8.0, 7.3 Hz, 2H), 4.32 (s, 2H), 2.69 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ (ppm):155.42, 151.96, 143.54, 140.81, 139.35, 131.08, 130.96, 130.03(2C), 129.79(3 C), 129.19(3 C), 128.9(2C)0, 119.77, 118.91, 118.87, 112.78, 112.01, 35.45, 22.18; Anal. Calcd. for C24H21N5O2S (443.53): C, 64.99; H, 4.77; N, 15.79; Found C, 64.54; H, 4.79; N, 15.88%.
4.2. Biological testing
4.2.1. In vitro anti-proliferative activity
The in vitro antiproliferative activities were assessed using the MTT assay protocolCitation78,Citation79,Citation95. As shown in Supplementary Data.
Citation4.Citation2.Citation2. In vitro VEGFR-2 enzyme inhibition assay
The synthesised compounds were estimated for their in vitro inhibition on human VEGFR-2 in HepG2 cell line; using ELISA kitCitation96 as described in Supplementary Data.
Citation4.Citation2.Citation3. Flow cytometry analysis for cell cycle
Cell cycle analysis for the most potent candidate 27a has been carried out through Flow cytometric analysis as described in Supplementary DataCitation84,Citation85.
4.2.4. Flow cytometry analysis for apoptosis
Apoptosis analysis for compound 27a in HepG2 cells was carried out using Annexin V-FITC as shown in Supplementary DataCitation30,Citation86.
4.2.5. Determination of active caspase-3 and caspase-9 levels
Quantitative assay of caspase-3 and caspase-9 activation was performed using Caspase- Invitrogen Caspase-3 ELISA Kit (KHO1091) and Invitrogen Caspase 9 Human ELISA Kit (BMS2025)Citation87,Citation97–99 as shown in Supplementary Data.
Citation4.Citation3. In silico studies
4.3.1. Docking studies
The docking studies were performed utilising MOE.14 software as described in Supplementary DataCitation57,Citation100,Citation101. The final figures were visualised using Discovery studio 4.0Citation102.
4.3.2. ADMET study
ADMET descriptors were determined using Discovery studio 4.0. as described in Supplementary DataCitation101,Citation103.
4.3.3. Toxicity study
The toxicity parameters were calculated using Discovery studio 4.0. as described in Supplementary DataCitation102.
Acknowledgement
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group no (RG-1441–333).
Disclosure statement
The authors declare that no conflict of interest exists. The authors give valuable appreciation to Dr. Mohamed R. Elnagar, Department of Pharmacology and Toxicology, Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt for his valuable technical assistance.
References
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424.
- Barbosa IR, Souza D. LB d, Bernal MM, Costa Í. d CC. Cancer mortality in Brazil: temporal trends and predictions for the year 2030. Medicine 2015;94:e746.
- El Newahie AM, Ismail NS, Abou El Ella DA, Abouzid KA. Quinoxaline-based scaffolds targeting tyrosine kinases and their potential anticancer activity. Arch Pharm 2016;349:309–26.
- Nam NH, Parang K. Current targets for anticancer drug discovery. Curr Drug Targets 2003;4:159–79.
- Chu H, Wang Y. Therapeutic angiogenesis: controlled delivery of angiogenic factors. Ther Deliv 2012;3:693–714.
- Ghith A, Ismail NS, Youssef K, Abouzid KA. Medicinal attributes of thienopyrimidine based scaffold targeting tyrosine kinases and their potential anticancer activities. Arch Pharm 2017;350:1700242.
- Sajib S, Zahra FT, Lionakis MS, et al. Mechanisms of angiogenesis in microbe-regulated inflammatory and neoplastic conditions. Angiogenesis 2018;21:1–14.
- Hamerlik P, Lathia JD, Rasmussen R, et al. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med 2012;209:507–20.
- Guo S, Colbert LS, Fuller M, et al. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim Biophys Acta Rev Cancer 2010;1806:108–21.
- Stuttfeld E, Ballmer-Hofer K. Structure and function of VEGF receptors. IUBMB Life 2009;61:915–22.
- Shibuya M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis. Angiogenesis 2006;9:225–30.
- Takahashi S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biol Pharm Bull 2011;34:1785–8.
- Flister MJ, Wilber A, Hall KL, et al. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1. Blood 2010;115:418–29.
- Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal 2007;19:2003–12.
- Modi SJ, Kulkarni VM. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med Drug Discov 2019;2:100009.
- Dietrich J, Hulme C, Hurley LH. The design, synthesis, and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: a structural analysis of the binding interactions of Gleevec, Nexavar, and BIRB-796. Bioorg Med Chem 2010;18:5738–48.
- Zhang L, Shan Y, Ji X, et al. Discovery and evaluation of triple inhibitors of VEGFR-2, TIE-2 and EphB4 as anti-angiogenic and anti-cancer agents. Oncotarget 2017;8:104745–60.
- Abdel-Mohsen HT, Abdullaziz MA, Kerdawy AME, et al. Targeting receptor tyrosine kinase VEGFR-2 in hepatocellular cancer: rational design, synthesis and biological evaluation of 1, 2-disubstituted benzimidazoles. Molecules 2020;25:770.
- Rampogu S, Baek A, Park C, et al. Discovery of small molecules that target vascular endothelial growth factor receptor-2 signalling pathway employing molecular modelling studies. Cells 2019;8:269.
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2006;2:358–64.
- Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA 2005;102:7665–70.
- Ghorab MM, Alsaid MS, Soliman AM, Ragab FA. VEGFR-2 inhibitors and apoptosis inducers: synthesis and molecular design of new benzo[g]quinazolin bearing benzenesulfonamide moiety. J Enzyme Inhib Med Chem 2017;32:893–907.
- Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 2006;5:835–44.
- Woo HY, Heo J. Sorafenib in liver cancer. Expert Opin Pharmacother 2012;13:1059–67.
- Wilhelm S, Dumas J, Ladouceur G, et al. Diaryl ureas with kinase inhibiting activity, Google Patents; 2007.
- Albiges L, Barthélémy P, Gross-Goupil M, et al. TiNivo: safety and efficacy of tivozanib-nivolumab combination therapy in patients with metastatic renal cell carcinoma. Ann Oncol 2021;32:97–102.
- Bedke J, Albiges L, Capitanio U, et al. Updated European Association of Urology guidelines on renal cell carcinoma: nivolumab plus cabozantinib joins immune checkpoint inhibition combination therapies for treatment-naïve metastatic clear-cell renal cell carcinoma. Eur Urol Suppl 2021;79:339–42.
- Szarek M, Needle MN, Rini BI, et al. Q-TWiST analysis of tivozanib versus sorafenib in patients with advanced renal cell carcinoma in the TIVO-3 study. Clin Genitourin Cancer 2021.
- Roskoski R Jr., Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun 2007;356:323–8.
- Zeidan MA, Mostafa AS, Gomaa RM, et al. Design, synthesis and docking study of novel picolinamide derivatives as anticancer agents and VEGFR-2 inhibitors. Eur J Med Chem 2019;168:315–29.
- Hassan A, Badr M, Hassan HA, et al. Novel 4-(piperazin-1-yl)quinolin-2(1H)-one bearing thiazoles with antiproliferative activity through VEGFR-2-TK inhibition. Bioorg Med Chem 2021;40:116168.
- McTigue M, Murray BW, Chen JH, et al. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc Natl Acad Sci USA 2012;109:18281–9.
- Xie Q-Q, Xie H-Z, Ren J-X, et al. Pharmacophore modeling studies of type I and type II kinase inhibitors of Tie2. J Mol Graph Model 2009;27:751–8.
- Abdullaziz MA, Abdel-Mohsen HT, El Kerdawy AM, et al. Design, synthesis, molecular docking and cytotoxic evaluation of novel 2-furybenzimidazoles as VEGFR-2 inhibitors. Eur J Med Chem 2017;136:315–29.
- Lee K, Jeong K-W, Lee Y, et al. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur J Med Chem 2010;45:5420–7.
- Machado VA, Peixoto D, Costa R, et al. Synthesis, antiangiogenesis evaluation and molecular docking studies of 1-aryl-3-[(thieno[3,2-b]pyridin-7-ylthio)phenyl]ureas: discovery of a new substitution pattern for type II VEGFR-2 Tyr kinase inhibitors. Bioorg Med Chem 2015;23:6497–509.
- Sobhy MK, Mowafy S, Lasheen DS, et al. 3D-QSAR pharmacophore modelling, virtual screening and docking studies for lead discovery of a novel scaffold for VEGFR 2 inhibitors: design, synthesis and biological evaluation. Bioorg Chem 2019;89:102988.
- Wang Z, Wang N, Han S, et al. Dietary compound isoliquiritigenin inhibits breast cancer neoangiogenesis via VEGF/VEGFR-2 signaling pathway. PLOS One 2013;8:e68566.
- Shahin MI, Abou El Ella DA, Ismail NS, Abouzid KA. Design, synthesis and biological evaluation of type-II VEGFR-2 inhibitors based on quinoxaline scaffold. Bioorg Chem 2014;56:16–26.
- Reed JC. Apoptosis-targeted therapies for cancer. Cancer Cell 2003;3:17–22.
- Reed JC. Mechanisms of apoptosis. Am J Pathol 2000;157:1415–30.
- Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem 1999;68:383–424.
- Cohen GM. Caspases: the executioners of apoptosis. Biochem J 1997;326:1–16.
- Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ 1999;6:99–104.
- Kuida K. Caspase-9. Int J Biochem Cell Biol 2000;32:121–4.
- Cotter T, Daniel PT, Schulze-Osthoff K, et al. Guardians of cell death: the Bcl-2 family proteins. Essays BIOCHEM 2003;39:73–88.
- Chen H-C, Kanai M, Inoue-Yamauchi A, et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol 2015;17:1270–81.
- Hsu SY, Kaipia A, McGee E, et al. Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc Natl Acad Sci USA 1997;94:12401–6.
- Harmey JH, Bouchier-Hayes D. Vascular endothelial growth factor (VEGF), a survival factor for tumour cells: implications for anti-angiogenic therapy. Bioessays 2002;24:280–3.
- Ling Y, Lu N, Gao Y, et al. Endostar induces apoptotic effects in HUVECs through activation of caspase-3 and decrease of Bcl-2. Anticancer Res 2009;29:411–7.
- Mahdy HA, Ibrahim MK, Metwaly AM, et al. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4(3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorg Chem 2020;94:103422.
- Scott LJ. Lenvatinib: first global approval. Drugs 2015;75:553–60.
- Plé PA, Jung F, Ashton S, et al. Discovery of AZD2932, a new quinazoline ether inhibitor with high affinity for VEGFR-2 and PDGFR tyrosine kinases. Bioorg Med Chem Lett 2012;22:262–6.
- Cetin B, Yılmaz GE, Armagan B, et al. Pazopanib-induced hepatotoxicity in an experimental rat model. Chemotherapy 2018;63:39–45.
- Pinheiro AC, Mendonça Nogueira TC, VN de Souza M. Quinoxaline nucleus: a promising scaffold in anti-cancer drug discovery. Anti-Cancer Agent 2016;16:1339–52.
- Kaushal T, Srivastava G, Sharma A, Negi AS. An insight into medicinal chemistry of anticancer quinoxalines. Bioorg Med Chem 2019;27:16–35.
- El-Adl K, El-Helby A-GA, Sakr H, Elwan A. Design, synthesis, molecular docking and anti-proliferative evaluations of [1,2,4]triazolo[4,3-a]quinoxaline derivatives as DNA intercalators and topoisomerase II inhibitors. Bioorg Chem 2020;105:104399.
- El-Adl K, El-Helby A-GA, Sakr H, Elwan A. [1,2,4] Triazolo [4,3-a] quinoxaline and [1,2,4] triazolo [4,3-a] quinoxaline-1-thiol-derived DNA intercalators: design, synthesis, molecular docking, in silico ADMET profiles and anti-proliferative evaluations. New J Chem 2021;45:881–97.
- Whatmore JL, Swann E, Barraja P, et al. Comparative study of isoflavone, quinoxaline and oxindole families of anti-angiogenic agents. Angiogenesis 2002;5:45–51.
- Weng Q, Zhang J, Cao J, et al. Q39, a quinoxaline 1,4-Di-N-oxide derivative, inhibits hypoxia-inducible factor-1α expression and the Akt/mTOR/4E-BP1 signaling pathway in human hepatoma cells. Investigational New Drugs 2011;29:1177–87.
- Khandan M, Sadeghian-Rizi S, Khodarahmi G, Hassanzadeh F. Synthesis and cytotoxic evaluation of some novel quinoxalinedione diarylamide sorafenib analogues. Res Pharm Sci 2018;13:168–76.
- Sadeghian-Rizi S, Khodarahmi GA, Sakhteman A, et al. Biological evaluation, docking and molecular dynamic simulation of some novel diaryl urea derivatives bearing quinoxalindione moiety. Res Pharm Sci 2017;12:500–9.
- Alsaif NA, Dahab MA, Alanazi MM, et al. New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: design, molecular modeling, and synthesis. Bioorg Chem 2021;110:104807.
- Alanazi MM, Mahdy HA, Alsaif NA, et al. New bis ([1,2,4] triazolo)[4,3-a:3′,4′-c] quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: design, synthesis, in silico studies, and anticancer evaluation. Bioorg Chem 2021;112:104949.
- El-Metwally SA, Abou-El-Regal MM, Eissa IH, et al. Discovery of thieno [2,3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg Chem 2021;112:104947.
- Abdallah AE, Eissa SI, Al Ward MMS, et al. Design, synthesis and molecular modeling of new quinazolin-4(3H)-one based VEGFR-2 kinase inhibitors for potential anticancer evaluation. Bioorg Chem 2021;109:104695.
- Ibrahim M-K, Abd-Elrahman AA, Ayyad RR, et al. Design and synthesis of some novel 2-(3-methyl-2-oxoquinoxalin-1 (2H)-yl)-N-(4-(substituted) phenyl) acetamide derivatives for biological evaluation as anticonvulsant agents. Bull Fac Pharm Cairo Univ 2013;51:101–11.
- Singh DP, Deivedi SK, Hashim SR, Singhal RG. Synthesis and antimicrobial activity of some new quinoxaline derivatives. Pharmaceuticals 2010;3:2416–25.
- Morrison D, Furst A. Quinoxaline-2-thiols. J Org Chem 1956;21:470–1.
- Saoudi B, Teniou A, Debache A, et al. Cyclisation reaction between 3-methylquinoxaline-2-thione and benzaldehydes into 3-benzyl-2-aryl-thieno [2,3-b] quinoxaline promoted by Brønsted acids. C R Chim 2015;18:808–15.
- Jacobs WA, Heidelberger M. Method for the acylation of aromatic amino compounds and ureas, with special reference to chloroacetylation. J Am Chem Soc 1917;39:1439–47.
- Panga S, Podila NK, Asres K, Ciddi V. Synthesis and anticancer activity of new isatin-benzoic acid conjugates. Ethiop Pharm J 2016;31:75–92.
- Abdel-Latif E, Fahad MM, Ismail MA. Synthesis of N-aryl 2-chloroacetamides and their chemical reactivity towards various types of nucleophiles. Synth Commun 2020;50:289–314.
- Newahie AE, Nissan YM, Ismail NS, et al. Design and synthesis of new quinoxaline derivatives as anticancer agents and apoptotic inducers. Molecules 2019;24:1175.
- Liu Y, Lotero E, Goodwin JG. Jr, Effect of carbon chain length on esterification of carboxylic acids with methanol using acid catalysis. J Catal 2006;243:221–8.
- Yadagiri B, Holagunda UD, Bantu R, et al. Rational design, synthesis and anti-proliferative evaluation of novel benzosuberone tethered with hydrazide-hydrazones. Bioorg Med Chem Lett 2014;24:5041–4.
- Abd Alla MSM, Hegab MI, Abo Taleb NA, et al. Synthesis and anti-inflammatory evaluation of some condensed [4-(3,4-dimethylphenyl)-1(2H)-oxo-phthalazin-2-yl]acetic acid hydrazide. Eur J Med Chem 2010;45:1267–77.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Scudiero DA, Shoemaker RH, Paull KD, et al. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res 1988;48:4827–33.
- Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol 1976;13:29–83.
- Fulda S, Debatin K-M. Resveratrol modulation of signal transduction in apoptosis and cell survival: a mini-review. Cancer Detect Prev 2006;30:217–23.
- Alenzi FQ. Links between apoptosis, proliferation and the cell cycle. Br J Biomed Sci 2004;61:99–102.
- Pucci B, Kasten M, Giordano A. Cell cycle and apoptosis. Neoplasia 2000;2:291–9.
- Wang J, Lenardo MJ. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J. Cell Sci 2000;113:753–7.
- Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc 2006;1:1458–61.
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods 1995;184:39–51.
- Hirano S. Western blot analysis. In: Reineke J, ed. Nanotoxicity. New York: Springer; 2012:87–97.
- Abdelhameid MK, Zaki I, Mohammed MR, Mohamed KO. Design, synthesis, and cytotoxic screening of novel azole derivatives on hepatocellular carcinoma (HepG2 Cells). Bioorg Chem 2020;101:103995.
- Mohammed AE-SM. Synthesis of some novel thieno[2,3-d]pyrimidine derivatives of expected anticancer activity [CU theses]; 2019.
- Al-Qathama A, Gibbons S, Prieto JM. Differential modulation of Bax/Bcl-2 ratio and onset of caspase-3/7 activation induced by derivatives of Justicidin B in human melanoma cells A375. Oncotarget 2017;8:95999–6012.
- Sakinah SS, Handayani ST, Hawariah LA. Zerumbone induced apoptosis in liver cancer cells via modulation of Bax/Bcl-2 ratio. Cancer Cell Int 2007;7:1–11.
- Hasegawa M, Nishigaki N, Washio Y, et al. Discovery of novel benzimidazoles as potent inhibitors of TIE-2 and VEGFR-2 tyrosine kinase receptors. J Med Chem 2007;50:4453–70.
- Xia X, Maliski EG, Gallant P, Rogers D. Classification of kinase inhibitors using a Bayesian model. J Med Chem 2004;47:4463–70.
- Ho J, Sciuscio D, Kogel U, et al. Evaluation of toxicity of aerosols from flavored e-liquids in Sprague-Dawley rats in a 90-day OECD inhalation study, complemented by transcriptomics analysis. Arch Toxicol 2020;94:2179–206.
- Al-Rashood ST, Hamed AR, Hassan GS, et al. Antitumor properties of certain spirooxindoles towards hepatocellular carcinoma endowed with antioxidant activity. J Enzyme Inhib Med Chem 2020;35:831–9.
- Abou-Seri SM, Eldehna WM, Ali MM, Abou El Ella DA. 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: synthesis and in vitro biological evaluation. Eur J Med Chem 2016;107:165–79.
- Scientific TF. CellROX® reagents and kits. Available from: http://www.thermofisher.com/us/en/home/brands/molecular-probes/key-molecular-probes-products/cellrox-reagentskits-ros-detection.html#flow [last accessed 26 Jul 2020].
- Aborehab NM, Elnagar MR, Waly NE. Gallic acid potentiates the apoptotic effect of paclitaxel and carboplatin via overexpression of Bax and P53 on the MCF‐7 human breast cancer cell line. J Biochem Mol Toxicol 2020;35:e22638.
- Elnagar MR, Walls AB, Helal GK, et al. Functional characterization of α7 nicotinic acetylcholine and NMDA receptor signaling in SH-SY5Y neuroblastoma cells in an ERK phosphorylation assay. Eur J Pharmacol 2018;826:106–13.
- Nasser AA, Eissa IH, Oun MR, et al. Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFRWT and EGFRT790M. Org Biomol Chem 2020;18:7608–34.
- El-Zahabi MA, Elbendary ER, Bamanie FH, et al. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of phthalimide-sulfonylurea hybrids as PPARγ and SUR agonists. Bioorg Chem 2019;91:103115.
- El-Naggar AM, Eissa IH, Belal A, El-Sayed AA. El-Sayed, Design, eco-friendly synthesis, molecular modeling and anticancer evaluation of thiazol-5(4H)-ones as potential tubulin polymerization inhibitors targeting the colchicine binding site. RSC Adv 2020;10:2791–811.
- Ibrahim MK, Eissa IH, Alesawy MS, et al. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of quinazolin-4(3H)-one derivatives as potential PPARγ and SUR agonists. Bioorg Med Chem 2017;25:4723–44.