
Abstract
A library of variously decorated N-phenyl secondary sulphonamides featuring the bicyclic tetrahydroquinazole scaffold was synthesised and biologically evaluated for their inhibitory activity against human carbonic anhydrase (hCA) I, II, IV, and IX. Of note, several compounds were identified showing submicromolar potency and excellent selectivity for the tumour-related hCA IX isoform. Structure–activity relationship data attained for various substitutions were rationalised by molecular modelling studies in terms of both inhibitory activity and selectivity.
Introduction
Carbonic anhydrases (CAs) are ubiquitous zinc enzymes that catalyse the reversible hydration of carbon dioxide to bicarbonate by a metal hydroxide nucleophilic mechanismCitation1. CAs play key roles in several physiological processes, such as pH and CO2 homeostasis, respiration, secretion of electrolytes in a variety of tissues/organs, biosynthetic reactions, including lipogenesis, gluconeogenesis, ureagenesis, calcification, bone resorption, and tumorigenicityCitation2–5. To date, 15 different isoforms of human CAs (hCAs) have been identified with different catalytic activities, cellular and subcellular localisation, and tissue distributionsCitation6: CA I, CA II, CA III, CA VII, and CA XIII isoforms are cytosolic, CA IV, CA IX, CA XII, CA XIV, and CA XV are transmembrane bound, CA VA and CA VB are mitochondrial and CA VI is secreted in saliva and milkCitation7,Citation8. Isoforms VIII, X, and XI are cytosolic, acatalytic, and preponderantly expressed within the brain, but their functions remain elusive.
Many hCA isoenzymes are considered promising therapeutic targets in the development of potentially valuable new small molecules for the treatment or prevention of many diseases such as glaucoma, epilepsy, obesity, and cancerCitation9,Citation10. Indeed, CA inhibitors (CAIs) have been in clinical use for many decades as diuretics and systemically acting antiglaucoma drugs; many CAIs are primary sulphonamides (acetazolamide (AAZ, ), ethoxzolamide, dichlorphenamide) coordinating the zinc ion present in the catalytic site through their deprotonated sulphonamide nitrogen atomCitation11. However, the high sequence homology between hCA isoforms, combined with their widespread and ubiquitous tissue localisation, makes the discovery of CAIs with functional selectivity for a specific target isoform a major challenge in the development of potential drugs, free from off-target side effectsCitation12.
In the last decades, several tricyclic/bicyclic scaffolds, decorated with a primary sulphonamide moiety, have been exploited by our research group, leading to the identification of several derivatives with high CA inhibitory activity and promising selectivity profilesCitation13–15. In particular, tricyclic primary benzenesulfonamides featuring the benzothiopyrano[4,3-c]pyrazole and pyridothiopyrano[4,3-c]pyrazole systems (see general formula I, ) were initially developed as geometrically constrained analogues of celecoxib (CLX, ) and valdecoxib (VLX, ), cyclooxygenase 2 (COX-2) specific inhibitors that act also as potent CAIsCitation6,Citation13. Based on the encouraging results obtained and the SAR outlined, our project on CAIs was pursued, always within primary benzenesulfonamide-based small molecules, following two different approaches: (i) investigation of three different, but structurally related, tricyclic scaffolds, that is benzo/pyrido-thiopyranopyrimidine II and pyrazolodihydroquinazoline III ()Citation14,Citation15; (ii) structural simplification of the core scaffolds of I and II to yield bicyclic tetrahydroindazole IV and tetrahydroquinazole V derivatives, respectively, in which the phenyl- or pyrido-fused moiety was removed ()Citation15.
More recently, the tetrahydroindazole scaffold of class IV was further exploited by the development of a library of N-phenyl-secondary sulphonamides, variously decorated at para- or meta-position with groups characterised by different electron-donor/acceptor capabilities (VI, )Citation16. Indeed, although secondary and tertiary sulphonamides have been recently reported as efficient and selective inhibitors of the cancer-related hCA IX and XII isoforms,Citation17–25 the chemical space of such CAIs remains so far rarely explored and their binding mode is still a subject of investigationCitation26.
For what concerns class VI, although, in general, the insertion of a secondary sulphonamide function on the tetrahydroindazole scaffold produced inhibitors with a moderate micromolar potency, lower than the primary sulphonamide parent compounds of series IV, some interesting structure–activity relationship (SAR) data were acquired and rationalised through theoretical studiesCitation16. Specifically, the inhibitory profile of the novel indazolyl-N-arylbenzenesulfonamides VI appears strictly dependent on the substitution pattern at 6-position, with the 6-phenyl group able to produce hCA II inhibitors with good potency and selectivityCitation16.
Pursuing our interest in the development of secondary sulphonamide CAIs, in the present study, we turned out our attention to the bicyclic tetrahydroquinazole scaffold of class V. Thus, derivatives 1–12 () were synthesised and biologically evaluated for their enzyme inhibitory activity against four physiologically relevant CA isoforms, the hCA I, II, IV, and IX. In the novel compounds, the secondary benzenesulfonamide moiety was decorated at para position with groups of various nature (R3=H, OCH3, NO2, CH3) to modulate the acidity of the sulphonamide itself, keeping in mind the groups conferring the best results, in terms of potency and selectivity, in series VI; in addition, the same substituents of the corresponding primary sulphonamides V described by us15 were maintained at 7-position (R1=R2=H; R1=H, R2=C6H5; R1=R2=CH3).
Materials and methods
Chemistry
The uncorrected melting points were determined using a Reichert Köfler hot-stage apparatus. NMR spectra were obtained on a Bruker AVANCE 400 (1H, 400 MHz, 13C, 100 MHz) in DMSO-d6. Chemical shifts are expressed in δ (ppm) and coupling constants (J) in Hertz. Magnesium sulphate was used as the drying agent. Evaporations were made in vacuo (rotating evaporator). Analytical TLC have been carried out on Merck 0.2 mm precoated silica gel aluminium sheets (60 F-254). Silica gel 60 (230–400 mesh) was used for column chromatography. Microwave (MW) assisted reactions were carried out in BIOTAGE Initiator 2.5 microwave apparatus. The purity of the target inhibitors 1–12 was determined, using a Shimadzu LC-20AD SP liquid chromatograph equipped with a DDA Detector (Kyoto, Japan) at 220 nm (column C18 (250 mm × 4.6 mm, 5 µm, Shim-pack)). The mobile phase, delivered at isocratic flow, consisted of methanol (80%) and water (20%) and a flow rate of 1.0 ml/min. All the compounds showed percent purity values of >95%. Reagents, starting materials, and solvents were purchased from commercial suppliers and used as received. The following intermediates were obtained according to previously described procedures: 2-[(dimethylamino)methylene]cyclohexan-1,3-dione 14aCitation27, 2-[(dimethylamino)methylene]-5-phenylcyclohexan-1,3-dione 14bCitation27, 2-[(dimethylamino)methylene]-5,5-dimethylcyclohexan-1,3-dione 14cCitation27, and N-(4-aminosulfonyl)phenylguanidine carbonate 15Citation14.
General procedure for the synthesis of 4-((5-oxo-5,6,7,8-tetrahydroquinazolin-2-yl)amino)benzenesulfonamide derivatives 16a–c
A mixture of the proper 2-(dimethylamino)methylene-1,3-dione derivative 14a–c (0.45 mmol), N-(4-aminosulfonyl)phenylguanidine carbonate 15 (0.25 g, 0.90 mmol) and NaOH (54 mg, 1.35 mmol) in 5 ml of n-BuOH was irradiated at a T = 160 °C (pre-stirring time = 3 min) for 40 min. After cooling, the obtained crude solid compounds 16a–c were purified by recrystallisation from ethanol.
4-((5-oxo-5,6,7,8-tetrahydroquinazolin-2-yl)amino)benzenesulfonamide (16a). Yield 66%; m.p. 294–296 °CCitation15.
4-((5-oxo-7-phenyl-5,6,7,8-tetrahydroquinazolin-2-yl)amino)benzenesulfonamide (16b). Yield 70%; m.p. 214–215 °CCitation15.
4-((5-oxo-7,7-dimethyl-5,6,7,8-tetrahydroquinazolin-2-yl)amino)benzenesulfonamide (16c). Yield 72%; m.p. 258–260 °CCitation15.
General procedure for the synthesis of N-aryl-4-((5-oxo-5,6,7,8-tetrahydroquinazolin-2-yl)amino)benzenesulfonamide derivatives 1–12
To a solution of the proper 4-(5-oxo-7-substituted-5,6,7,8-tetrahydroquinazolin-2-yl)amino)benzenesulfonamide 16a–c (0.94 mmol) in dry MeCN (10 ml) at room temperature, CuI (8 mg, 0.04 mmol), anhydrous K2CO3 (0.28 g, 2.07 mmol), the proper aryl iodide (1.13 mmol), and DMEDA (0.04 ml, 0.38 mmol) were added, and the resulting mixture was heated at 100 °C for 8–24 h (TLC analysis). After cooling, the reaction mixture was diluted with water (20 ml), acidified with 2.0 N HCl to pH = 4, and extracted with EtOAc (3 × 20 ml). The organic phase was dried (MgSO4), filtered and evaporated under reduced pressure. The crude material was purified by flash chromatography using PE/EtOAc as eluting system, to afford compounds 1–12.
4-((5-Oxo-5,6,7,8-tetrahydroquinazolin-2-yl)amino)-N-phenylbenzenesulfonamide (1)
Compound 1 was obtained as a solid (0.093 g, 25%) starting from compound 16a (0.30 g) and iodobenzene (0.13 ml); m.p. 281–283 °C; 1H NMR (400 MHz, DMSO-d6): δ 2.04–2.07 (m, 2H), 2.57 (t, J= 6.4 Hz, 2H), 2.93 (t, J= 6.0 Hz, 2H), 6.99 (t, J= 7.2 Hz, 1H), 7.09 (d, J= 7.6 Hz, 2H), 7.19–7.23 (m, 2H), 7.70 (d, J= 8.8 Hz, 2H), 7.94 (d, J= 8.8 Hz, 2H), 8.84 (s, 1H), 10.19 (s, 1H), 10.67 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 21.0, 31.8, 38.0, 119.3 (2C), 119.5 (2C), 120.2, 124.4, 128.2 (2C), 129.6 (2C), 132.7, 138.1, 143.8, 158.2, 160.4, 174.2, 195.8.
N-(4-methoxyphenyl)-4-((5-oxo-5,6,7,8-tetrahydroquinazolin-2-yl)amino)benzenesulfonamide (2)
Compound 2 was obtained as a solid (0.080 g, 20%) starting from compound 16a (0.30 g) and 1-iodo-4-methoxybenzene (0.26 g); m.p. 233–235 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.05–2.08 (m, 2H), 2.58 (t, J= 6.2 Hz, 2H), 2.94 (t, J= 6.2 Hz, 2H), 3.66 (s, 3H), 6.79 (d, J= 9.2 Hz, 2H), 6.98 (d, J= 9.2 Hz, 2H), 7.62 (d, J= 8.8 Hz, 2H), 7.93 (d, J= 9.2 Hz, 2H), 8.85 (s, 1H), 9.81 (s, 1H), 10.67 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 20.8, 31.6, 37.8, 55.3, 114.4 (2C), 115.1, 119.0, 119.1 (2C), 123.3 (2C), 126.1, 127.9 (2C), 130.5, 132.6, 143.4, 156.5, 157.8, 160.5, 195.3.
N-(4-nitrophenyl)-4-((5-oxo-5,6,7,8-tetrahydroquinazolin-2-yl)amino)benzenesulfonamide (3)
Compound 3 was obtained as a solid (0.10 g, 25%) starting from compound 16a (0.30 g) and 1-iodo-4-nitrobenzene (0.28 g); m.p. 264–266 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.05–2.08 (m, 2H), 2.58 (t, J= 6.4 Hz, 2H), 2.93 (t, J= 6.2 Hz, 2H), 7.30 (d, J= 9.2 Hz, 2H), 7.83 (d, J= 8.8 Hz, 2H), 7.99–8.02 (m, 2H), 8.13 (d, J= 9.2 Hz, 2H), 8.85 (s, 1H), 10.74 (s, 1H), 11.20 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 21.0, 31.8, 38.0, 118.2 (2C), 119.4, 119.7 (2C), 125.8 (2C), 128.4 (2C), 131.9, 142.8, 144.4, 144.7, 158.1, 160.4, 174.2, 180.3, 195.9.
4-((5-Oxo-5,6,7,8-tetrahydroquinazolin-2-yl)amino)-N-(4-tolyl)benzenesulfonamide (4)
Compound 4 was obtained as a solid (0.077 g, 20%) starting from compound 16a (0.30 g) and 1-iodo-4-methylbenzene (0.25 g); m.p. 272–274 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.05–2.08 (m, 2H), 2.17 (s, 3H), 2.58 (t, J= 6.8 Hz, 2H), 2.93 (t, J= 6.0 Hz, 2H), 6.96–7.03 (m, 4H), 7.66 (d, J= 9.2 Hz, 2H), 7.93 (d, J= 9.2 Hz, 2H), 8.85 (s, 1H), 10.01 (s, 1H), 10.67 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 20.6, 21.0, 31.8, 38.0, 119.2, 119.5 (2C), 120.8 (2C), 128.1 (2C), 129.9 (2C), 132.8, 133.7, 135.4, 143.6, 158.1, 160.4, 174.2, 195.9.
4-((5-Oxo-7-phenyl-5,6,7,8-tetrahydroquinazolin-2-yl)amino)-N-phenylbenzenesulfonamide (5)
Compound 5 was obtained as a solid (0.13 g, 30%) starting from compound 16b (0.37 g) and iodobenzene (0.23 ml); m.p. 111–113 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.69–2.73 (m, 2H), 2.97–3.10 (m, 2H), 3.52–3.59 (m, 1H), 6.99 (t, J= 7.2 Hz, 1H), 7.09 (d, J= 8.0 Hz, 2H), 7.19–7.28 (m, 3H), 7.34–7.40 (m, 4H), 7.69 (d, J= 8.8 Hz, 2H), 7.96 (d, J= 8.8 Hz, 2H), 8.90 (s, 1H), 10.19 (s, 1H), 10.75 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 38.3, 44.8, 118.6, 119.3, 119.9 (2C), 124.0, 127.0, 127.1 (2C), 127.9, 128.8 (2C), 129.3 (2C), 132.6, 138.0, 143.3, 143.5, 157.8, 160.5, 172.9, 194.7.
N-(4-methoxyphenyl)-4-((5-oxo-7-phenyl-5,6,7,8-tetrahydroquinazolin-2-yl)amino)benzenesulfonamide (6)
Compound 6 was obtained as a solid (0.094 g, 20%) starting from compound 16b (0.37 g) and 1-iodo-4-methoxybenzene (0.26 g); m.p. 103–105 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.67–2.73 (m, 2H), 2.97–3.01 (m, 2H), 3.53–3.57 (m, 1H), 3.65 (s, 3H), 6.79 (d, J= 9.2 Hz, 2H), 6.97 (d, J = 9.2 Hz, 2H), 7.26–7.28 (m, 1H), 7.34–7.40 (m, 4H), 7.61 (d, J= 8.8 Hz, 2H), 7.95 (d, J= 8.8 Hz, 2H), 8.91 (s, 1H), 9.82 (s, 1H), 10.75 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 38.4, 44.8, 55.2, 114.3 (2C), 116.8, 118.4, 119.1, 123.2 (2C), 127.0, 127.8 (2C), 128.7, 130.6 (2C), 132.6, 143.2, 156.4, 158.0, 160.5, 194.5.
N-(4-nitrophenyl)-4-((5-oxo-7-phenyl-5,6,7,8-tetrahydroquinazolin-2-yl)amino)benzenesulfonamide (7)
Compound 7 was obtained as a solid (0.14 g, 30%) starting from compound 16b (0.37 g) and 1-iodo-4-nitrobenzene (0.28 g); m.p. 118–120 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.67–2.73 (m, 2H), 2.97–3.04 (m, 2H), 3.54–3.56 (m, 1H), 7.26–7.30 (m, 2H), 7.34–7.40 (m, 3H), 7.82 (d, J= 8.8 Hz, 2H), 7.96–8.07 (m, 4H), 8.12 (d, J= 9.2 Hz, 2H), 8.91 (s, 1H), 10.82 (s, 1H), 11.21 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 38.3, 44.9, 104.5, 118.0 (2C), 118.7, 119.6 (2C), 125.2 (2C), 125.6 (2C), 127.0, 127.1 (2C), 128.2, 128.9 (2C), 139.0, 143.3, 144.0, 157.9, 160.6, 173.0, 194.8.
4-((5-Oxo-7-phenyl-5,6,7,8-tetrahydroquinazolin-2-yl)amino)-N-(4-tolyl)benzenesulfonamide (8)
Compound 8 was obtained as a solid (91 mg, 20%) starting from compound 16b (0.37 g) and 1-iodo-4-methylbenzene (0.25 g); m.p. 83–85 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.10 (s, 3H), 2.67–2.73 (m, 2H), 2.97–3.04 (m, 2H), 3.54–3.56 (m, 1H), 7.26–7.30 (m, 2H), 7.34–7.40 (m, 3H), 7.82 (d, J= 9.2 Hz, 2H), 7.96–8.07 (m, 4H), 8.12 (d, J= 9.2 Hz, 2H), 8.91 (s, 1H), 10.82 (s, 1H), 11.21 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 20.9, 38.5, 41.7, 45.0, 118.1, 119.4 (2C), 123.1 (2C), 126.4 (2C), 127.2, 127.3 (2C), 129.0 (2C), 130.7 (2C), 132.0, 138.2, 139.8, 142.7, 143.5, 158.1, 161.0, 173.1, 195.1.
4-((7,7-Dimethyl-5-oxo-5,6,7,8-tetrahydroquinazolin-2-yl)amino)-N-phenylbenzenesulfonamide (9)
Compound 9 was obtained as a solid (0.12 g, 30%) starting from compound 16c (0.32 g) and iodobenzene (0.23 ml); m.p. 215–217 °C. 1H NMR (400 MHz, DMSO-d6): δ 1.03 (s, 6H), 2.46 (s, 2H), 2.86 (s, 2H), 7.00 (t, J= 7.2 Hz, 1H), 7.09 (d, J= 8.8 Hz, 2H), 7.21–7.24 (m, 2H), 7.70 (d, J= 8.8 Hz, 2H), 7.95 (d, J= 8.8 Hz, 2H), 8.84 (s, 1H), 10.17 (s, 1H), 10.68 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 27.9, 32.4, 44.9, 51.2, 118.0, 119.2 (2C), 120.0, 124.0 (2C), 126.1, 127.9 (2C), 129.2 (2C), 129.8, 132.5, 143.5, 157.4, 160.7, 195.2.
4-((7,7-Dimethyl-5-oxo-5,6,7,8-tetrahydroquinazolin-2-yl)amino)-N-(4-methoxyphenyl)benzenesulfonamide (10)
Compound 10 was obtained as a solid (0.15 g, 35%) starting from compound 16c (0.32 g) and 1-iodo-4-methoxybenzene (0.26 g); m.p. 210–212 °C. 1H NMR (400 MHz, DMSO-d6): δ 1.03 (s, 6H), 2.86 (s, 2H), 3.65 (s, 3H), 6.79 (d, J= 8.8 Hz, 2H), 6.98 (d, J= 8.8 Hz, 2H), 7.62 (d, J= 8.8 Hz, 2H), 7.93 (d, J= 8.8 Hz, 2H), 8.84 (s, 1H), 9.80 (s, 1H), 10.67 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 27.8, 32.4, 51.2, 55.2, 114.3 (2C), 119.1 (2C), 123.2 (2C), 127.8 (2C), 129.2, 130.5, 138.1, 143.2, 153.0, 153.8, 160.6, 166.4, 195.1.
4-((7,7-Dimethyl-5-oxo-5,6,7,8-tetrahydroquinazolin-2-yl)amino)-N-(4-nitrophenyl)benzenesulfonamide (11)
Compound 11 was obtained as a solid (0.13 g, 30%) starting from compound 16c (0.32 g) and 1-iodo-4-nitrobenzene (0.28 g); m.p. 264–266 °C. 1H NMR (400 MHz, DMSO-d6): δ 1.04 (s, 6H), 2.87 (s, 2H), 7.32 (d, J= 8.8 Hz, 2H), 7.84 (d, J= 8.8 Hz, 2H), 8.02 (d, J= 9.2 Hz, 2H), 8.14 (d, J= 9.2 Hz, 2H), 8.85 (s, 1H), 10.74 (s, 1H), 11.19 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 27.7, 32.3, 44.8, 51.0, 117.7 (2C), 117.9, 119.3 (2C), 125.3 (2C), 127.9 (2C), 131.6, 142.3, 144.0, 144.4, 157.2, 160.5, 172.3, 195.0.
4-((7,7-Dimethyl-5-oxo-5,6,7,8-tetrahydroquinazolin-2-yl)amino)-N-(p-tolyl)benzenesulfonamide (12)
Compound 12 was obtained as a solid (0.10 g, 25%) starting from compound 16c (0.32 g) and 1-iodo-4-methylbenzene (0.25 g); m.p. 246–248 °C. 1H NMR (400 MHz, DMSO-d6): δ 1.03 (s, 6H), 2.17 (s, 3H), 2.86 (s, 2H), 6.96–7.03 (m, 4H), 7.66 (d, J= 8.8 Hz, 2H), 7.93 (d, J= 8.8 Hz, 2H), 8.84 (s, 1H), 10.01 (s, 1H), 10.67 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 20.4, 27.9, 32.4, 45.9, 51.2, 119.0, 119.2 (2C), 120.4 (2C), 126.1, 127.8 (2C), 129.6 (2C), 130.3, 143.4, 143.8, 144.4, 157.2, 157.4, 195.1.
CA inhibition assays
An Applied Photophysics stopped-flow instrument was used for assaying the CA catalysed CO2 hydration activityCitation28. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.5) as buffer, 0.1 M Na2SO4 (for maintaining constant ionic strength), following the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitors (10 mM) were prepared in distilled-deionised water with 10% of DMSO and dilutions up to 0.01 µM were done thereafter with the assay buffer. The inhibitor and enzyme solutions were preincubated together for 15 min (standard assay at room temperature) prior to assay, to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation and represent the mean from at least three different determinations. Enzyme concentrations in the assay system were in the range of 5–12 nM.
Molecular modelling methods
Docking calculations were executed using the latest AutoDockGPU docking softwareCitation29 along with its GUI AutoDockTools (ADT)Citation30. The high-resolution X-ray crystal structure of hCA IX having (PDB 5FL4), hCA I (PDB 6EVR), hCA II (PDB 3K34), and hCA IV (PDB 5JN9)Citation31–33 was downloaded and superimposed on the structure of hCA IX. Before docking, the co-crystal ligands of 5FL4, 6EVR, 3K34, and 5JN9 were removed. Later, the protein structures were prepared using the Protein Preparation Wizard of the Maestro suiteCitation34,Citation35. The Protein Preparation Wizard deletes water molecules, adds both bond orders and hydrogen atoms, and also produces the appropriate protonation states. Compound 3, (S)-5, and (R)-5 were constructed using the 2D sketcher tool of Maestro. The ligands were considered in all the possible flip conformations and appropriate protonation states were obtained. The ligand and protein files were then translated into the AD4 specific format (PDBQT) utilising the scripts prepare_ligand4.py and prepare_receptor4.py, respectively, with all the standard settings. Using AutoDock4Zn forcefield protocol, the script zinc_pseudo.py was applied to add the tetrahedral pseudo atoms to the receptor PDBQT fileCitation36. The grid boxes were centred on the active site of the protein. While creating the grid parameter files (GPF), the zinc-specific non-bonded pairwise potentials were also included. The dimensions of the grid boxes were defined with a set of grids of 60 Å×40 Å×50 Å along with the spacing of 0.375 Å. A total of 100 independent docking simulations were achieved for every possible conformation of compounds 3, (S)-5, and (R)-5. Each docking calculation comprised 20 million energy evaluations utilising the Lamarckian genetic algorithm local search (GALS) method. All the dockings were performed with a population size of 250 and 300 runs of Solis and Wets local search with a probability of 0.6. A rate of mutation of 0.02 and a crossover rate of 0.8 were used to produce new docking attempts for following generations, and the best individual from each generation was propagated over the following generation. The docking modes of every 100 independent docking calculations of the ligand binding to the metalloenzyme were clustered based on a 2 Å cut-off value based on Cartesian coordinates of the atom and were also ranked based on free energy binding (ΔGAD4). Compounds 3, (S)-5, and (R)-5 best conformations were selected based upon the predicted ΔGAD4 as well the cluster population.
Results and discussion
Chemistry
The key intermediates for the synthesis of all the new target compounds 1–12 were the bicyclic tetrahydroquinazoline derivatives 16a–c already described by us15 belonging to class V (). However, the synthetic procedure to obtain compounds 16a–c has been here improved by exploiting MW irradiation as a valuable alternative to conventional heating, allowing to reach higher yields and shorter reaction times (40 min vs. 16 h). In detail, the commercially available 5-substituted-1,3-cyclohexandiones 13a–c were reacted with an excess of N,N-dimethylformamide-dimethyl acetal (DMF-DMA) at 100 °C for 1 h, to furnish the intermediates 14a–c. Then, MW-assisted condensation reaction between the proper bielectrophyle 2-[(dimethylamino)methylene]cyclohexan-1,3-dione 14a–cCitation15 and the binucleophile guanidine 15Citation14 was performed to obtain crude compounds 16a–c which were purified by recrystallisation from ethanol (Scheme 1).
Scheme 1. Reagents and conditions: (i) DMF-DMA, 100 °C, 1 h; (ii) conc HCl/50% cyanamide water solution, 100 °C, 0.5 h; (iii) n-butanol, NaOH, M.W. (40 min, 160 °C); (iv) MeCN, K2CO3, DMEDA, CuI, 100 °C, 8–24 h.
The quinazolyl-N-arylbenzenesulfonamides 1–12 were then obtained by reacting compounds 16a–c with the proper p-substituted phenyliodide in acetonitrile (MeCN) in the presence of K2CO3, N,N′-dimethylethylendiamine (DMEDA), and CuI. The reaction mixture was heated at 100 °C and stirred for 8–24 h (TLC analysis); after cooling, the suspension obtained was filtered under reduced pressure and the solid was purified by flash chromatography, yielding the pure target derivatives 1–12.
CA inhibition assays and structure–activity relationships
All the newly synthesised compounds 1–12 were investigated for their enzyme inhibitory ability against four physiologically relevant CA isoforms, namely the human hCA I, II, IV, and IX (), by a stopped-flow CO2 hydrase assay using acetazolamide (AAZ, 5-acetamido-1,3,4-thiadiazole-2-sulphonamide, ) as the standard drugCitation28. also reports the data referring to the parent primary sulphonamides 16a–cCitation15 for comparison purposes.
Table 1. Inhibition of hCA isoforms I, II, IV, and IX with tetrahydroquinazoline derivatives 1–12, 16a–c, and AAZ as the reference standard by a stopped-flow CO2 hydrase assay.
In particular, the inhibitory profile of the tetrahydroquinazolin-2-yl-amino-benzenesulfonamides 1–12 appears strictly dependent on the substitution pattern at 7-position, with few exceptions. Unsubstituted compounds (1–4) exhibit high potency against hCA IX (Ki values ranging from 0.42 μM to 1.04 μM), combined with an excellent selectivity with respect to the other isoforms tested. This trend appears to be independent of the decoration of the pendant phenyl ring (R3).
The inhibitory potency of 7-phenyl derivatives (5–8) appears to be closely related to the decoration on the benzenesulfonamide moiety (R3). hCA IX inhibition potency was maintained high when position 1 is unsubstituted (hCA IX, 5 Ki 0.45 vs. 1 Ki 0.42), and slightly decreased when R3=OCH3 or NO2 (hCA IX, 6 Ki 1.73 and 7 Ki 1.33 vs. 2 Ki 0.46 and 3 Ki 0.42, respectively). The hCA IX-selectivity profile varied from excellent to good moving from R3=H to R3=NO2 and R3=OCH3. Specifically, the best performing compound of the subseries in terms of hCA IX inhibitory potency and selectivity is the unsubstituted (R3=H) compound 5; the p-nitrophenyl-substituted analogue 7 (R3=NO2) results in a quite potent (Ki 1.33 µM) and selective (Ki ratio CAI/CAIX >75.2, CAII/CAIX 50.7, CAIV/CAIX >75.2) hCA IX inhibitor; the p-methoxy-substituted 6 (R3=OCH3), retains the hCA IX inhibitory potency of the same order of magnitude of 7 (Ki 1.73 µM), but the selectivity towards this isoform is considerably reduced (Ki ratio CAI/CAIX >19.6, CAII/CAIX 6.6, CAIV/CAIX >57.8). The presence of a methyl group at R3 produced a marked reduction in hCA IX inhibitory potency (hCA IX: 8 Ki 15.7 vs. 4 Ki 1.04), and a loss of selectivity, being 8 equally potent on hCA II and hCA IX.
Insertion of a double methyl substitution at position 7 produced a dramatic drop in hCA IX inhibition potency and a general gain in activity towards the other hCAs tested, particularly for hCA II. The 7,7-dimethyl-quinazolinyl-N-arylbenzenesulfonamides 9–12 inhibited all the CAs tested with micromolar potency (with few exceptions), showing a moderate selectivity for the hCA II isoform (Ki values from 5.1 to 22.4 µM). Only compound 12 (R3=CH3) did not register a gain in selectivity for the hCA II isoform showing comparable Ki values for all the tested hCA isoforms.
All in all, these data highlighted the identification of several hCA IX inhibitors with high potency and selectivity, featuring a secondary benzenesulfonamide on the tetrahydroquinazoline scaffold. The best-performing compounds are represented by 3 (R1=R2=H, R3=NO2) and 5 (R1=H, R2=C6H5, R3=H), that are the most potent (3 Ki 0.42 µM; 5 Ki 0.45 µM) and selective (Ki ratio CAI/CAIX, CAII/CAIX, CAIV/CAIX >238.1 and >222.2 for 3 and 5, respectively) hCA IX inhibitors of this new series of derivatives.
It should be outlined that, comparing the most interesting new compounds 1–5 with the parent primary tetrahydroquinazole sulphonamides 16a–c, despite the lower hCA IX inhibitory potency of the secondary 1–5, their hCA IX selectivity is greater than that observed for primary sulphonamides 16a–c. This greater selectivity is especially evident with respect to isoform II with Ki ratios CAII/CAIX ranging from 7.6 to 9.45 for 16a–c and from 85.8 to >238 for 1–5.
Furthermore, a comparison of the obtained results for 1–4 with those of the corresponding tetrahydroindazole-based secondary sulphonamides of series VI (VIa–d in ) recently described by us16, highlighted that, by keeping the same R3-substituents (R3=H, p-OCH3, p-NO2, p-CH3) and changing the scaffold from tetrahydroindazole (VIa–d) to tetrahydroquinazole (1–4), the good potency (Ki 1.4–5.7 µM) and selectivity towards the hCA I isoform shifted to high potency (Ki 0.42–1.04 µM) and excellent selectivity towards the hCA IX isoform. In particular, by comparing the p-nitro-substituted derivatives 3 and VIc (R1=R2=H, R3=NO2) which are the best performing compounds in the two subseries, a Ki value of 5.7 µM for VIc against hCA I with slight selectivity (Ki ratio CAII/CAI 8.8, CAIV/CAI 13.3, CAIX/CAI 14.7), and a Ki value of 0.42 µM against the hCA IX isoform with a high selectivity (Ki ratio CAI/CAIX, CAII/CAIX, CAIV/CAIX >238.1) for 3 are highlighted.
Table 2. Inhibition of hCA isoforms I, II, IV, and IX with tetrahydroquinazoline (1–4) tetrahydroindazole (VIa–d)Citation16 and secondary sulphonamides.
Molecular modelling studies
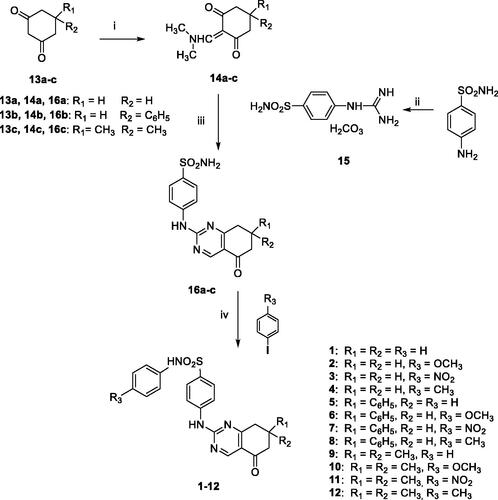
To rationalise the SAR data attained for various substitutions of newly designed secondary sulphonamides (), a series of molecular modelling studies were performed on compounds 3 and 5, as they are the most potent inhibitors of hCA IX as well as the most selective over the other tested hCAs (I, II, and IV) of the whole set. In this process, the AutoDock4Zn docking protocol, devised for docking experiments on zinc-chelating ligands, was employed as implemented in our previous reports on hCAIsCitation15,Citation37. The best docking solution, i.e. the one having the lowest predicted binding free energy, achieved for compound 3 in the hCA IX (PDB 5FL4)Citation31 binding site is shown in .
Chart 1. Structures of celecoxib (CLX), valdecoxib (VLX), acetazolamide (AAZ); structures of previously described (I-VI) and newly synthesized (1–12) CAIs.]
![Chart 1. Structures of celecoxib (CLX), valdecoxib (VLX), acetazolamide (AAZ); structures of previously described (I-VI) and newly synthesized (1–12) CAIs.]](/cms/asset/7188df13-76a4-46d8-8939-9ca36355eba0/ienz_a_1956913_f0005_b.jpg)
Figure 1. 3/hCA IX theoretical complex (PDB 5FL4) computed by docking calculations. The protein is shaded in salmon and all the critical residues are labelled. The ligand is shown in pink colour while the H-bonds are shown in black dashed lines.
Mainly, the sulphonamide chelates the catalytic zinc ion through its negatively charged nitrogen atom in a geometry that is consistent with the other CAIs. Additionally, one of the sulphonamide oxygen atoms is well-positioned to accept an H-bond from the backbone NH of Thr201. Apart from these interactions, which were consistent with our previous reports on hCAsCitation15,Citation37, it is also interesting to see that the phenyl ring, attached to the sulphonamide negative nitrogen, forms a π–π interaction with His98 and places its p-NO2 group in a hydrophilic protein region at H-bonding distance to multiple residues such as Asn66, His68, and Gln71. These latter interactions might explain why compound 4, featuring the lipophilic p-CH3 group, by establishing less favourable interactions with the enzyme counterpart, is less active than compounds 1–3. Interestingly, the same trend was recorded for the phenyl-substituted analogues 5–8. The benzene ring of the benzenesulfonamide forms van der Waals interactions with Val121 and Leu199 while the attached pyrimidine ring of the tetrahydroquinazoline scaffold forms an H-bond with Gln92 residue. This latter interaction might explain why the tetrahydroquinazolines described in the present work are generally more potent than the previously reported tetrahydroindazolesCitation16. Moreover, the oxygen atom enclosed in tetrahydroquinazoline is pointing towards the “selectivity hotspot” of the hCA IX binding site.
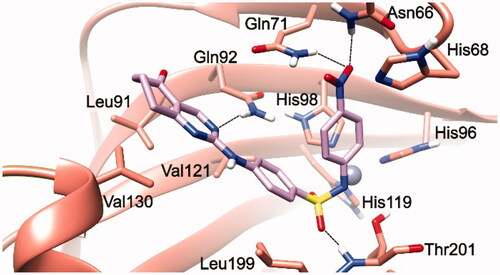
Dockings of both isomers of 5 allow postulating the absence of a stereoselective recognition by hCAIX as, in both cases, the predicted lowest energy conformation places the negatively charged sulphonamide nitrogen to chelate the Zn2+ () with an overall orientation resembling the one achieved for 3.
Figure 2. (S)-5/hCA IX (A) and (R)-5/hCA IX theoretical complexes (PDB 5FL4) computed by docking calculations. The protein is shaded in salmon and all the critical residues are labelled. The ligand is shown in yellow and cyan, respectively, while the H-bonds are shown in black dashed lines.
In particular, the (S)-5 docking suggests that the negative nitrogen connected to the secondary sulphonamide is chelating the zinc ion, while a sulphonamide oxygen is forming an H-bond with the backbone NH of Thr201. If compared to the binding pose achieved for 3, the presence of the phenyl ring at position 7 of the tetrahydroquinazoline ring seems to induce a partial relocation of the ligand inducing the loss of the H-bond interaction with Gln92, thereby explaining why compounds 5–8 are less active than their unsubstituted counterparts 1–4. In (S)-5, this loss seems to be counterbalanced by the presence of several van der Waals contacts between the phenyl ring and Leu91, Gln92, and Val130 enzyme residues in the “selectivity hotspot”. (R)-5 docking calculations indicate the presence of a similar interaction pattern ().
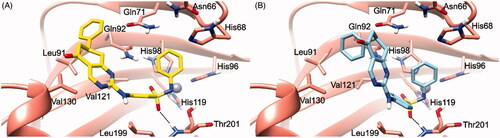
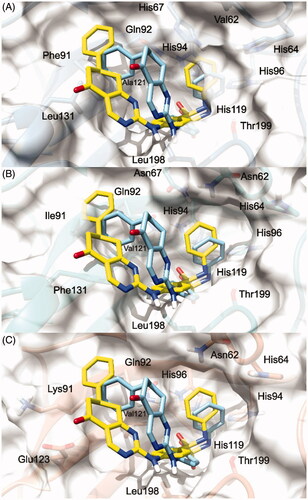
To explain the selectivity profiles of 3, (S)-5, and (R)-5, docking calculations of these compounds were also attempted employing the published structures of hCA I (PDB 6EVR), hCA II (PDB 3K34), and hCA IV (PDB 5JN9)Citation32,Citation33. Interestingly, analysis of the obtained results revealed a certain difficulty in predicting a binding pose featuring the chelation of the Zn2+ ion. To explain these theoretical results, the binding pose obtained for 3, (S)-5, and (R)-5 into the hCA IX structure and conducive of the Zn2+ chelation, was rigidly translated into the aligned binding sites of hCA I, hCA II, and hCA IV. From this analysis, it is clear that such a ligand orientation is unfeasible into the hCA I, hCA II as it would give rise to multiple steric clashes ( and ). Concerning hCA IV, no steric clashes were evidenced, while unfavourable electrostatic interactions between the hydrophilic “selectivity hotspot” of hCA IV and the ligands could be detected ( and ). This should explain while compounds 3 and 5 are unable to efficiently inhibit hCA I, hCA II, and hCA IV.
Figure 3. 3/hCA IX theoretical binding pose translated into hCA I (PDB 6EVR, A), hCA II (PDB 3K34, B), and hCA IV (PDB 5JN9, C) structures. The proteins are shown in blue, sea-green, and coral ribbons and sticks, respectively, with their molecular surface shaded in white. The ligand is depicted in pink.
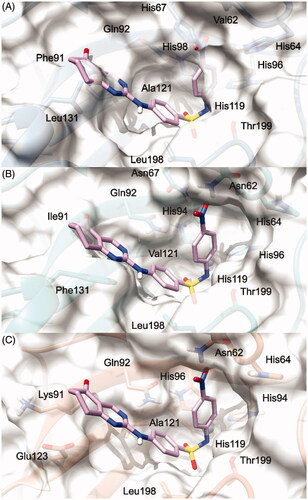
Figure 4. (S)-5 and (R)-5/hCA IX theoretical binding pose translated into hCA I (PDB 6EVR, A), hCA II (PDB 3K34, B), and hCA IV (PDB 5JN9, C) structures. The proteins are shown in blue, sea-green, and coral ribbons and sticks, respectively, with their molecular surface shaded in white. (S)-5 and (R)-5 are depicted in yellow and cyan sticks. All the pictures were rendered using UCSF chimera molecular visualisation softwareCitation40.
Conclusions
Within our project aimed at investigating the chemical space of secondary sulphonamide CAIs, we herein reported the design, synthesis, and biological evaluation of a focussed library of variously decorated N-phenyl secondary sulphonamides featuring the bicyclic tetrahydroquinazole scaffold. Exploiting SAR studies from our previous work, we identified several compounds showing submicromolar potency and excellent selectivity for hCA IX, an isoform that has attracted significant interest as a putative cancer therapeutic target. SAR studies highlighted a strict correlation between the activity/selectivity profile and the substitution patterns at the 7-position of the central scaffold and the para-position of the benzenesulfonamide moiety of the target derivatives. Molecular modelling studies rationalised the SARs in terms of both inhibitory activity and selectivity profile. Overall, this study provided further information on the structural requirements for efficient and selective inhibition of the cancer-related hCA IX isoform and it could be useful for the design of novel anticancer agents. The class of secondary sulphonamides, although less investigated for their CA inhibitory properties, may lead to relevant medicinal chemistry discoveries. Moreover, very recently Kennedy et al.Citation38 investigated inflammasome and pyroptotic pathways and identified a novel photoaffinity alkyne-tagged probe for such processes, compound belonging to the secondary sulphonamide class, which is also targeting efficiently CA II, confirming thus our earlier dataCitation16,Citation25,Citation39 that secondary sulphonamides effectively inhibit these enzymes, by a mechanism which starts to be elucidated through this and the above-mentioned works.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74.
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–80.
- Supuran CT. Emerging role of carbonic anhydrase inhibitors. Clin Sci 2021;135:1233–49.
- Angeli A, Abdel-Aziz AAM, Nocentini A, et al. Synthesis and carbonic anhydrase inhibition of polycyclic imides incorporating N-benzenesulfonamide moieties. Bioorg Med Chem 2017;25:5373–9.
- Kurt BZ, Sonmez F, Ozturk D, et al. Synthesis of coumarin-sulfonamide derivatives and determination of their cytotoxicity, carbonic anhydrase inhibitory and molecular docking studies. Eur J Med Chem 2019;183:111702.
- Kurt BZ, Dag A, Doğan B, et al. Synthesis, biological activity and multiscale molecular modeling studies of bis-coumarins as selective carbonic anhydrase IX and XII inhibitors with effective cytotoxicity against hepatocellular carcinoma. Bioorg Chem 2019;87:838–50.
- Awadallah FM, Bua S, Mahmoud WR, et al. Inhibition studies on a panel of human carbonic anhydrases with N1-substituted secondary sulfonamides incorporating thiazolinone or imidazolone-indole tails. J Enzyme Inhib Med Chem 2018;33:629–38.
- Awadallah FM, El-Waei TA, Hanna MM, et al. Synthesis, carbonic anhydrase inhibition and cytotoxic activity of novel chromone-based sulfonamide derivatives. Eur J Med Chem 2015;96:425–35.
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16.
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Marini AM, Maresca A, Aggarwal M, et al. Tricyclic sulfonamides incorporating benzothiopyrano[4,3-c]pyrazole and pyridothiopyrano[4,3-c]pyrazole effectively inhibit α- and β-carbonic anhydrase: X-ray crystallography and solution investigations on 15 isoforms. J Med Chem 2012;55:9619–29.
- Barresi E, Salerno S, Marini AM, et al. Sulfonamides incorporating heteropolycyclic scaffolds show potent inhibitory action against carbonic anhydrase isoforms I, II, IX and XII. Bioorg Med Chem 2016;24:921–7.
- Salerno S, Barresi E, Amendola G, et al. 4-Substituted benzenesulfonamides incorporating bi/tricyclic moieties act as potent and isoform-selective carbonic anhydrase II/IX inhibitors. J Med Chem 2018;61:5765–70.
- Salerno S, Amendola G, Angeli A, et al. Inhibition studies on carbonic anhydrase isoforms I, II, IV and IX with N-arylsubstituted secondary sulfonamides featuring a bicyclic tetrahydroindazole scaffold. Eur J Med Chem 2021;220:113490.
- Wilkinson BL, Bornaghi LF, Houston TA, et al. A novel class of carbonic anhydrase inhibitors: glycoconjugate benzene sulfonamides prepared by “click-tailing”. J Med Chem 2006;49:6539–48.
- Güzel-Akdemir Ö, Akdemir A, Isik S, et al. O-Benzenedisulfonimido-sulfonamides are potent inhibitors of the tumor-associated carbonic anhydrase isoforms CA IX and CA XII. Bioorg Med Chem 2013;21:1386–91.
- Liu F, Martin-Mingot A, Lecornué F, et al. Carbonic anhydrases inhibitory effects of new benzenesulfonamides synthesized by using superacid chemistry. J Enzyme Inhib Med Chem 2012;27:886–91.
- Métayer B, Mingot A, Vullo D, et al. New superacid synthesized (fluorinated) tertiary benzenesulfonamides acting as selective HCA IX inhibitors: toward a new mode of carbonic anhydrase inhibition by sulfonamides. Chem Commun 2013;49:6015–7.
- Coviello V, Marchi B, Sartini S, et al. 1,2-Benzisothiazole derivatives bearing 4-, 5-, or 6-alkyl/arylcarboxamide moieties inhibit carbonic anhydrase isoform IX (CAIX) and cell proliferation under hypoxic conditions. J Med Chem 2016;59:6547–52.
- Ammazzalorso A, Carradori S, Angeli A, et al. Fibrate-based N-acylsulphonamides targeting carbonic anhydrases: synthesis, biochemical evaluation, and docking studies. J Enzyme Inhib Med Chem 2019;34:1051–61.
- D'Ascenzio M, Guglielmi P, Carradori S, et al. Open saccharin-based secondary sulfonamides as potent and selective inhibitors of cancer-related carbonic anhydrase IX and XII isoforms. J Enzyme Inhib Med Chem 2017;32:51–9.
- Moeker J, Peat TS, Bornaghi LF, et al. Cyclic secondary sulfonamides: unusually good inhibitors of cancer-related carbonic anhydrase enzymes. J Med Chem 2014;57:3522–31.
- Alp C, Maresca A, Alp NA, et al. Secondary/tertiary benzenesulfonamides with inhibitory action against the cytosolic human carbonic anhydrase isoforms I and II. J Enzyme Inhib Med Chem 2013;28:294–8.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- Schenone P, Mosti L, Menozzi G. Reaction of 2-dimethylaminomethylene-1,3-diones with dinucleophiles. I. Synthesis of 1,5-disubstituted 4-acylpyrazoles. J Heterocycl Chem 1982;19:1355–61.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Santos-Martins D, Solis-Vasquez L, Tillack AF, et al. Accelerating AutoDock4 with GPUs and gradient-based local search. J Chem Theory Comput 2021;17:1060–73.
- Morris G, Ruth H, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91.
- Leitans J, Kazaks A, Balode A, et al. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J Med Chem 2015;58:9004–9.
- Chiaramonte N, Bua S, Ferraroni M, et al. 2-Benzylpiperazine: a new scaffold for potent human carbonic anhydrase inhibitors. Synthesis, enzyme inhibition, enantioselectivity, computational and crystallographic studies and in vivo activity for a new class of intraocular pressure lowering agents. Eur J Med Chem 2018;151:363–75.
- Behnke CA, Le Trong I, Godden JW, et al. Atomic resolution studies of carbonic anhydrase II. Acta Crystallogr D Biol Crystallogr 2010;66:616–27.
- Schrödinger Release 2020-2: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2020; Impact, Schrödinger, LLC, New York, NY; Prime, Schrödinger, LLC, New York, NY, 2020.
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J Comput Aid Mol Des 2013; 27:221–34.
- Santos-Martins D, Forli S, Ramos MJ, Olson AJ. AutoDock4(Zn): an improved AutoDock force field for small-molecule docking to zinc metalloproteins. J Chem Inf Model 2014;54:2371–9.
- Milite C, Amendola G, Nocentini A, et al. Novel 2-substituted-benzimidazole-6-sulfonamides as carbonic anhydrase inhibitors: synthesis, biological evaluation against isoforms I, II, IX and XII and molecular docking studies. J Enzyme Inhib Med Chem 2019;34:1697–710.
- Kennedy CR, Goya Grocin A, Kovačič T, et al. A probe for NLRP3 inflammasome inhibitor MCC950 identifies carbonic anhydrase 2 as a novel target. ACS Chem Biol 2021;16:982–990.
- Nocentini A, Angeli A, Carta F, et al. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J Enzyme Inhib Med Chem 2021;36:561–80.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12.