
Abstract
Identifying isoform-specific inhibitors for closely related kinase family members remains a substantial challenge. The necessity for achieving this specificity is exemplified by the RSK family, downstream effectors of ERK1/2, which have divergent physiological effects. The natural product, SL0101, a flavonoid glycoside, binds specifically to RSK1/2 through a binding pocket generated by an extensive conformational rearrangement within the RSK N-terminal kinase domain (NTKD). In modelling experiments a single amino acid that is divergent in RSK3/4 most likely prevents the required conformational rearrangement necessary for SL0101 binding. Kinetic analysis of RSK2 association with SL0101 and its derivatives identified that regions outside of the NTKD contribute to stable inhibitor binding. An analogue with an n-propyl-carbamate at the 4” position on the rhamnose moiety was identified that forms a highly stable inhibitor complex with RSK2 but not with RSK1. These results identify a SL0101 modification that will aid the identification of RSK2 specific inhibitors.
1. Introduction
Identifying inhibitors that are specific for individual members of highly related proteins remains a challenge. For example, the Serine/Threonine protein kinase RSK family have ∼90% identity in their N-terminal kinase domains (NTKD), which is the domain responsible for phosphorylation of target substrates. RSK is a downstream effector of the Ras-Raf-MEK1/2-ERK1/2 (MAP kinase), a signalling cascade that is an important oncogenic driver in various cancers and this pathway has been the subject of intensive drug development effortsCitation1. Treatments for cancers with an activated MAP kinase pathway include a B-Raf and MEK1/2 inhibitor combination but unfortunately, resistance inevitably occurs via reactivation of the pathwayCitation1. Interestingly, through an undefined mechanism, cell lines resistant to BRAF and MEK1/2 inhibitors remain dependent on RSKCitation2,Citation3. RSK is also associated with resistance against a diverse group of chemotherapeutic agents that include platinum compounds as well as sonic hedgehog, phosphoinositide 3-kinase, and heat shock protein 90 inhibitorsCitation4–7. Thus, targeting RSK activity independently of its upstream activators may provide novel therapeutic approaches for treatment-induced resistance.
RSK has two non-identical kinase domains, an NTKD and a C-terminal kinase domain (CTKD) (). The NTKD belongs to the protein A, G and C kinase familyCitation8 and the CTKD belongs to the Ca2+/calmodulin-dependent protein kinase II family and is necessary for autophosphorylationCitation8. SL0101, a flavonoid glycoside (), was the first specific RSK inhibitor described and demonstrates specificity for inhibiting the RSK1/2 NTKD Citation9–11. The covalent inhibitors, FmkCitation12,Citation13 and CN-NHiPRCitation14, are CTKD inhibitors and are fairly specific. Dimethyl fumarate also acts as a covalent inhibitor of the RSK2 CTKD but inhibits the related MSK familyCitation15. The CTKD is only required for RSK activation and CTKD inhibitors do not inhibit activated RSK. Additionally, RSK activation by the CTKD can be bypassedCitation12,Citation16. Therefore, inhibition of RSK via the CTKD is context dependent, suggesting that the clinical utility of CTKD inhibitors may be limited. The pan RSK inhibitors, BI-D1870Citation10,Citation17–19 and BIX02565Citation20,Citation21, are not RSK specific and those based on the 3,4-bi-aryl pyridylCitation22 or 7-azaindole scaffoldCitation23 have poor pharmacokinetics. Importantly, the functions of individual RSK family members are still not clearly understood, although RSK1/2 are implicated in tumorigenesis and RSK3/4 are thought to act as tumour suppressorsCitation5,Citation24.
Figure 1. SL0101 binding to RSK2 is time dependent. (A) Simplified schematic of RSK2 kinase domains and their activation sites and functions, adapted fromCitation39. (B) Chemical composition of SL0101 and analogues. (C) Recombinant, purified RSK1, RSK2, and RSK2 NTKD visualised with Coomassie Blue. (D) Schematic of FRET assay (upper). Kinase, Eu-labelled anti-His antibody, SL0101, and tracer are mixed, incubated for 5 min and fluorescence measured, or a kinase/antibody/SL0101 mixture incubated for 2 h before tracer addition and fluorescence determined. Amount of tracer bound to RSK2 relative to vehicle control (lower) (N = 5 in quadruplicate for 5 min incubation and n = 9 in quadruplicate for 2 h pre-incubation). (E) Relative kinase activity in ELISA-based assay of RSK2 pre-incubated with SL0101 for 5 min or 2 h (N = 3 each in triplicate). (F) Relative tracer bound to RSK2 incubated with inhibitor 1b for 5 min or 2 h (N = 2 in quadruplicate or 4 in quadruplicate, respectively). Data are plotted as mean and error bars represent SD.
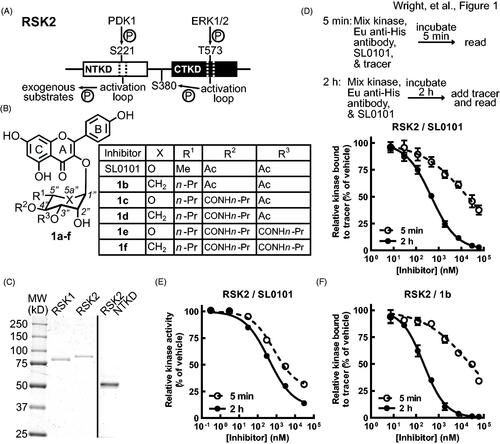
SL0101 (1a) was identified during a screen of botanical extracts for RSK2 inhibitionCitation9. The crystal structure for the RSK2NTKD in complex with SL0101 (RSK2NTKD-SL0101) identifies that the SL0101-binding pocket requires extensive re-arrangement of the N-lobe of the NTKDCitation25. There is partial overlap between residues that comprise ATP and SL0101 binding pockets, but the sites are distinct from each other. As expected, based on the interaction mechanism a detailed analysis of the specificity of the SL0101 derivative, C5”-n-propyl cyclitol SL0101 (1b), demonstrated that 1b primarily targeted RSK1/2 in a screen of 247 kinases, containing representatives from all kinase familiesCitation11. Furthermore, (1b) was ineffective at inhibiting proliferation in a cell-based assay when its targets, RSK1/2, were silencedCitation26. These data predict that SL0101-based derivatives will have limited off target effects and therefore, we have continued to evaluate and develop SL0101-based analogues.
It is now recognised that kinetic analysis of the mechanism of enzyme inhibition provides useful insights into the pharmacokinetic and pharmacodynamic properties of compounds, which can be used as a guide for further drug developmentCitation27. Therefore, in this study we analysed the binding kinetics of RSK1 and RSK2 with SL0101 and selected derivatives. RSK2 shows time dependent SL0101 binding and the SL0101-binding pocket is likely formed by random conformational sampling by the NTKD. In contrast RSK1 does not exhibit time dependent binding to SL0101. SL0101 analogues were identified that inhibited RSK2 but not RSK1 kinase activity in vitro for > 12 h. Because of the extremely slow off rate of these SL0101 analogues with RSK2 we speculate that in vivo RSK2 will be the primary target of inhibition in vivo and that the biological half-life of the derivatives will be less critical for effective in vivo inhibition. Despite the high degree of homology between RSK1 and RSK2 our data provide a solution to the identification of RSK2 specific inhibitors. RSK2-specific inhibitors would allow the functions of RSK2 kinase activity to be identified without contributions of the kinase activity of the other RSK isoforms or the ability of RSK2 to act as a scaffold to complicate the analysis.
2. Materials and methods
2.1. Inhibitors
SL0101 was purchased from Cayman Chemical Company. Inhibitors 1b, 1d, and 1f and pyran 8 were synthesised and characterised as previously describedCitation26,Citation28,Citation29. Synthesis of inhibitors 1c and 1e are detailed below, starting from pyran 8 (Figure S1).
2.1.1. (2 s,3R,6S)-6-((5,7-bis(benzyloxy)-2–(4-(benzyloxy)phenyl)-4-oxo-4H-chromen-3-yl)oxy)-2-propyl-3,6-dihydro-2H-pyran-3-yl n-propylcarbamate (9)
To a solution of alcohol compound 8 (45 mg, 0.0646 mmol) in 0.13 ml acetonitrile, 1,8-diazabicyclo(5.4.0)undec-7-ene (0.8 μL, 6 μmol), triethylamine(46 μL, 0.33 mmol) and n-propyl isocyanate (20 μL, 0.2 mmol) were added successively. The resulting mixture was stirred at room temperature for 10 h. The reaction mixture was then washed with saturated sodium bicarbonate and brine, dried over magnesium sulphate, then concentrated on rotary evaporator. The resulting crude was purified by chromatography and flashed with solvent (hexanes: ethyl acetate = 4:1) to afford product as pale yellow solid, compound 9 (31 mg, 62%); Rf = 0.57 (hexanes: ethyl acetate = 1:1); m.p. 120 − 122 °C; [α]D20: −136.2 (DCM, c = 2.45); 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 8.7 Hz, 2H), 7.54 (d, J = 7.4 Hz, 2H), 7.49 − 7.31 (m, 12H), 7.29 (d, J = 7.3 Hz, 1H), 7.07 (d, J = 8.9 Hz, 2H), 6.55 (d, J = 1.9 Hz, 1H), 6.42 (d, J = 1.9 Hz, 1H), 6.23 (d, J = 10.2 Hz, 1H), 5.95 (d, J = 10.1 Hz, 1H), 5.84 (s, 1H), 5.26 (s, 2H), 5.13 (s, 2H), 5.06 (s, 2H), 4.95 (d, J = 9.2 Hz, 1H), 4.69 (t, J = 5.6 Hz, 1H), 3.46 − 3.36 (m, 1H), 3.14 (dt, J = 12.8, 6.5 Hz, 2H), 1.52 (m, 2H), 1.21 − 1.08 (m, 1H), 1.08 − 0.97 (m, 1H), 0.92 (t, J = 7.4 Hz, 3H), 0.62 (t, J = 8 Hz, 3H), 0.53 − 0.57 (m, 1H); 13 C NMR (100 MHz, CDCl3) δ 173.78, 162.93, 160.45, 159.97, 159.03, 155.94, 153.85, 138.49, 136.67, 136.64, 135.89, 131.17, 130.87, 128.97, 128.91, 128.83, 128.64, 128.39, 127.88, 127.84, 127.63, 127.22, 126.85, 124.17, 114.73, 110.22, 98.36, 95.28, 94.07, 77.59, 77.47, 77.27, 76.95, 70.91, 70.66, 70.30, 70.25, 69.56, 43.03, 34.46, 23.40, 17.76, 14.63, 11.44; HRMS (MALDI-TOF/CCA): Calcd. [C48H48NO9 + Na]+: 782.3324, Found: 782.3353; IR (thin film, cm−1) 2965, 1724, 1606, 1508, 1365, 1251, 1226, 1200, 963, 880, 787, 739, 493, 445, 396.
2.1.2. (2 s,3R,4S,5R,6S)-6-((5,7-bis(benzyloxy)-2–(4-(benzyloxy)phenyl)-4-oxo-4H-chromen-3-yl)oxy)-4,5-dihydroxy-2-propyltetrahydro-2H-pyran-3-yl n-propylcarbamate (10)
Acetate alkene 9 (65 mg, 0.083 mmol) was dissolved in 0.17 ml of a mixture of acetone and t-butyl alcohol (1:1), then osmium tetroxide (0.27 mg, 1.3 mol%) and 50% N-methylmorpholine N-oxide water solution (1.7 μL, 0.083 mmol) were added dropwise successively at 0 °C. The resulting mixture was stirred at room temperature overnight. After completion, the reaction mixture was washed with saturated sodium bicarbonate and brine, dried over magnesium sulphate, then concentrated on rotary evaporator. The resulting crude was purified by chromatography and flashed with solvent (hexanes: ethyl acetate = 1.5:1) to afford product as light yellow solid, compound 10 (60 mg, 72%); Rf = 0.21 (hexanes: ethyl acetate = 1:1); m.p. 79–81 °C; [α]D20: −131.4 (DCM, c = 1.92); 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.7 Hz, 2H), 7.52 (d, J = 7.4 Hz, 2H), 7.47 − 7.30 (m, 12H), 7.28 (d, J = 7.3 Hz, 1H), 7.07 (d, J = 8.8 Hz, 2H), 6.53 (d, J = 1.7 Hz, 1H), 6.42 (s, 1H), 5.69 (s, 1H), 5.25 (s, 2H), 5.13 (s, 2H), 5.05 (s, 2H), 4.81 (t, J = 5.8 Hz, 1H), 4.62 (t, J = 9.2 Hz, 1H), 4.42 (s, 1H), 4.01 –-3.90 (m, 1H), 3.82 (d, J = 5.1 Hz, 1H), 3.55 (s, 1H), 3.27 (t, J = 10.3 Hz, 1H), 3.09 (dd, J = 13.3, 6.4 Hz, 2H), 1.94 (s, 1H), 1.54 − 1.35 (m, 3H), 1.28 − 1.16 (m, 2H), 1.16 − 1.02 (m, 1H), 0.87 (t, J = 7.0 Hz, 3H), 0.83 − 0.68 (m, 1H), 0.64 (t, J = 7.2 Hz, 3H); 13 C NMR (100 MHz, CDCl3) δ 173.60, 163.07, 160.63, 160.00, 159.00, 157.46, 153.92, 137.62, 136.52, 135.81, 130.75, 128.97, 128.92, 128.83, 128.66, 128.43, 127.92, 127.81, 127.56, 126.91, 126.85, 123.60, 114.97, 110.08, 100.40, 98.44, 94.05, 74.69, 71.55, 70.95, 70.69, 70.63, 70.56, 70.35, 43.13, 33.84, 23.27, 18.26, 18.20, 14.42, 14.34, 11.40; HRMS (MALDI-TOF/CCA): Calcd. [C48H49NO11 + Na]+: 838.3198, Found: 838.3199; IR (thin film, cm−1) 3352, 2961, 2873, 1699, 1603, 1570, 1438, 1404, 1352, 1251, 1176, 1100, 1048, 1001, 950, 822, 735, 696, 622, 543, 465.
2.1.3. (2 s,3R,4S,5S,6S)-2-((5,7-bis(benzyloxy)-2–(4-(benzyloxy)phenyl)-4-oxo-4H-chromen-3-yl)oxy)-3-hydroxy-6-propyl-5-((propylcarbamoyl)oxy)tetrahydro-2H-pyran-4-yl acetate (11)
To a solution of compound diol 10 (18.7 mg, 0.023 mmol) in 0.23 ml acetonitrile, 2-aminoethyl diphenylborinate (0.5 mg, 10 mol%), propyl isocyanate (2.5 μL, 0.035 mmol) and DIPEA (8 μL, 0.046 mmol) were added successively. The resulting mixture was stirred at room temperature for 12 h. Reaction mixture was then washed with saturated sodium bicarbonate and brine, dried over magnesium sulphate, then concentrated on rotary evaporator. The resulting crude was purified by HPLC and flashed with solvent (hexanes: ethyl acetate = 1.5:1) to afford product as yellow oil, compound 11 (12 mg, 57%): Rf = 0.32 (hexanes: ethyl acetate = 1:2); m.p. 115 − 117 °C; [α]D20: −130.0, (c = 1.1, DCM); 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.6 Hz, 2H), 7.54 (d, J = 7.6 Hz, 2H), 7.48 − 7.33 (m, 11H), 7.30 (d, J = 7.3 Hz, 1H), 7.16 − 7.08 (m, 2H), 6.56 (d, J = 2.2 Hz, 1H), 6.45 (d, J = 2.2 Hz, 1H), 5.71 (d, J = 2.3 Hz, 1H), 5.38 − 5.28 (m, 1H), 5.25 (s, 2H), 5.15 (s, 2H), 5.11 (d, J = 7.5 Hz, 1H), 5.07 (s, 2H), 4.89 (t, J = 9.4 Hz, 1H), 4.62 (t, J = 6.0 Hz, 1H), 4.52 (s, 1H), 3.23 (m, J = 9.2, 7.0 Hz, 1H), 3.11 (dt, J = 13.6, 6.9 Hz, 1H), 3.00 (dt, J = 13.3, 6.5 Hz, 1H), 2.80 (s, 1H), 2.09 (s, 3H), 1.44 (dt, J = 14.5, 7.3 Hz, 2H), 1.37 − 1.15 (m, 3H), 1.09 (dt, J = 13.6, 7.2 Hz, 1H), 0.87 (t, J = 7.4 Hz, 3H), 0.73 (m, J = 7.0 Hz, 1H), 0.64 (t, J = 7.1 Hz, 3H). 13 C NMR (100 MHz, CDCl3) δ 11.3, 14.3, 14.4, 18.1, 21.3, 23.4, 33.8, 42.9, 69.7, 70.4, 70.7, 71.0, 71.7, 72.2, 94.1, 98.5, 100.5, 110.1, 115.1, 123.6, 126.9, 127.6, 127.9, 128.0, 128.4, 128.7, 128.9, 128.9, 129.0, 130.8, 135.8, 136.5, 136.6, 137.5, 154.1, 155.7, 159.1, 160.0, 160.7, 163.1, 170.4, 173.5.; HRMS (MALDI-TOF/CCA): Calcd. [C50H51NO12 + Na]+: 880.3303, Found: 880.3315; IR (thin film, cm−1) 3434, 2962, 2930, 2871, 1726, 1573, 1486, 1454, 1437, 1353, 129, 1249, 1176, 1103, 1047, 1002, 954, 806, 736, 697.
2.1.4. (2 s,3R,4S,5S,6S)-2-((5,7-dihydroxy-2–(4-hydroxyphenyl)-4-oxo-4H-chromen-3-yl)oxy)-3-hydroxy-6-propyl-5-((propylcarbamoyl)oxy)tetrahydro-2H-pyran-4-yl acetate (1c)
To a solution of compound 11 (4.6 mg, 0.005 mmol) in 0.1 ml methanol, Pd/C (0.1 mg, 10 mol%) was added and resulting mixture was stirred under a H2 atmosphere for 24 h. The reaction mixture was loaded onto silica gel and eluted with hexane-EtOAc (1:2) to give inhibitor 1c (2.3 mg, 72%); Rf = 0.78 (EtOAc); m.p. 110 − 112 °C; [α]D19: −73.0 (MeOH, c = 0.3); 1H NMR (400 MHz, CD3OD) δ 7.85 (d, = 8.2 Hz, 2H), 6.95 (d, J = 8.5 Hz, 2H), 6.38 (d, J = 2.3 Hz, 1H), 6.20 (d, J = 2.3 Hz, 1H), 5.74 (s, 1H), 5.26 − 5.12 (m, 1H), 4.95 − 4.90 (m, 2H), 4.34 (d, J = 3.1 Hz, 1H), 3.06 − 2.96 (m, 2H), 2.95 − 2.79 (m, 1H), 2.06 (s, 3H), 1.44 (td, J = 14.4, 7.2 Hz, 3H), 1.32 − 1.03 (m, 5H), 0.87 (m, J = 6.3, 5.3 Hz, 4H), 0.64 (t, J = 7.2 Hz, 3H); 13 C NMR (100 MHz, CD3OD) δ 178.2, 171.0, 164.9, 162.0, 160.5, 157.3, 157.1, 157.0, 134.0, 130.8, 121.3, 115.6, 104.6, 100.5, 98.8, 93.6, 72.5, 72.1, 69.5, 68.4, 42.3, 33.4, 22.8, 19.7, 18.0, 13.1, 10.3; HRMS (MALDI-TOF/CCA): Calcd. [C29H33NO12 + Na]+:610.1895, Found: 610.1896; IR (thin film, cm−1) 3320, 3261, 2963, 2064, 1979, 1725, 1657, 1610, 1508, 1365, 1277, 1209, 1175, 1121, 975, 819, 657, 457.
2.1.5. (2 s,3S,4S,5R,6S)-6-((5,7-bis(benzyloxy)-2–(4-(benzyloxy)phenyl)-4-oxo-4H-chromen-3-yl)oxy)-5-hydroxy-2-propyltetrahydro-2H-pyran-3,4-diyl bis(n-propylcarbamate) (12)
To a solution of compound diol 10 (35 mg, 0.046 mmol) in 0.4 ml acetonitrile, 2-aminoethyl diphenylborinate (1 mg, 10 mol%), propyl isocyanate (8.7 μL, 0.092 mmol) and triethylamine (22 μL, 0.132 mmol) were added successively. The resulting mixture was stirred at room temperature for 12 h. Then reaction mixture was washed with saturated sodium bicarbonate and brine, dried over magnesium sulphate, then concentrated on rotary evaporator. The resulting crude was purified by HPLC and flashed with solvent (hexanes: ethyl acetate = 1.5:1) to afford product as yellow oil, compound 12 (16 mg, 40%); Rf = 0.34 (hexanes: ethyl acetate = 1:1.5); [α]D20: −144.9 (DCM, c = 0.73) ; 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 7.4 Hz, 2H), 7.48 − 7.26 (m, 11H), 7.13 (d, J = 8.6 Hz, 2H), 6.55 (d, J = 1.8 Hz, 1H), 6.44 (d, J = 1.6 Hz, 1H), 5.76 (s, 1H), 5.25 (s, 2H), 5.18 (s, 1H), 5.15 (s, 1H), 5.11 (s, 1H), 5.06 (s, 2H), 4.87 (t, J = 9.6 Hz, 1H), 4.67 (s, 1H), 4.53 (s, 1H), 3.17 (s, 1H), 3.10 (t, J = 11.0 Hz, 2H), 3.06 − 2.99 (m, 1H), 1.55 − 1.32 (m, 5H), 1.29 − 1.22 (m, 1H), 1.18 (d, J = 5.5 Hz, 2H), 1.08 (dd, J = 12.3, 6.4 Hz, 1H), 0.90 (t, J = 7.4 Hz, 3H), 0.84 (t, J = 7.4 Hz, 3H), 0.75 − 0.66 (m, 1H), 0.62 (t, J = 7.0 Hz, 3H); 13 C NMR (100 MHz, CDCl3) δ 173.52, 170.35, 163.10, 160.66, 160.03, 159.05, 155.70, 154.09, 137.51, 136.57, 136.51, 135.83, 130.83, 128.99, 128.92, 128.85, 128.68, 128.43, 127.94, 127.84, 127.58, 126.91, 123.56, 115.11, 110.10, 100.52, 98.46, 94.10, 72.17, 71.68, 70.98, 70.76, 70.71, 70.37, 69.67, 42.94, 33.83, 23.37, 21.28, 18.14, 18.10, 14.43, 14.32, 11.30; HRMS (MALDI-TOF/CCA): Calcd. [C52H56N2O12 + Na]+: 923.3725, Found: 923.3718; IR (thin film, cm−1) 3587, 2963, 2872, 1718, 1605, 1509, 1377, 1251, 1176, 997, 955, 889, 802, 736, 646.
2.1.6. (2 s,3S,4S,5R,6S)-6-((5,7-dihydroxy-2–(4-hydroxyphenyl)-4-oxo-4H-chromen-3-yl)oxy)-5-hydroxy-2-propyltetrahydro-2H-pyran-3,4-diyl bis(n-propylcarbamate) (1e)
To a solution of bis-carbamate compound 12 (7.5 mg, 0.008 mmol) in 0.05 ml methanol, Pd/C (0.4 mg, 10 mol%) was added and resulting mixture was stirred under a H2 atmosphere for 24 h. The reaction mixture was loaded onto silica gel and elution with hexane-EtOAc (1:1.5) to give inhibitor 1e (3.5 mg, 67%); Rf = 0.12 (hexanes-EtOAc = 1:2); [α]D20: −110.1 (MeOH, c = 0.15), m.p. 158 − 160 °C; 1H NMR (400 MHz, CD3OD-d4) 7.86 (d, J = 8.7 Hz, 2H), 6.97 (d, J = 8.5 Hz, 2H), 6.38 (d, J = 1.5 Hz, 1H), 6.20 (d, J = 1.6 Hz, 1H), 5.72 (s, 1H), 5.08 (dd, J = 10.2, 2.7 Hz, 1H), 4.35 (s, 1H), 3.08 (t, J = 6.8 Hz, 2H), 3.00 (t, J = 6.9 Hz, 2H), 1.48 (m, J = 29.4, 14.5, 7.2 Hz, 5H), 1.17 (m, J = 18.2, 16.9, 10.1 Hz, 4H), 0.93 (t, J = 7.4 Hz, 3H), 0.87 (t, J = 7.4 Hz, 4H), 0.64 (t, J = 7.1 Hz, 3H); 13 C NMR (100 MHz, Acetone-d6) δ 178.80, 164.69, 162.84, 160.62, 157.57, 156.34, 156.29, 131.38, 122.08, 116.18, 105.32, 101.65, 99.24, 94.22, 94.16, 73.18, 72.04, 69.59, 43.01, 42.94, 33.98, 23.71, 23.48, 18.36, 14.00, 11.24, 11.13; HRMS (MALDI-TOF/CCA): Calcd. [C31H38N2O12 + Na]+: 653.2317, Found: 653.2314; IR (thin film, cm−1) 3377, 2963, 2874, 1698, 1652, 1607, 1508, 1457, 1358, 1262, 1207, 1171, 967, 949, 841, 813, 768, 580, 521.
2.2. Fret kinase assay
The relative amount of inhibitor bound to RSK2 was determined using the LanthaScreen Eu Kinase Binding Assay for RPS6KA3 (Invitrogen). Kinase, Eu anti-His antibody, inhibitor or vehicle, and Kinase Tracer 236 (tracer) in kinase buffer were incubated as described in , with final assay concentrations of 5 nM kinase, 2 nM antibody, inhibitor or vehicle (0.95% DMSO), and 15 nM tracer in Proxiplate white 384-well plates (Perkin Elmer) with 15 μL total assay volume per well. For RSK1 and RSK2 NTKD, 2 nM Eu Streptavidin and 2 nM Biotin anti-His antibody (Invitrogen) and for RSK3/4, 2 nM Eu anti-GST antibody (Invitrogen) were used in place of the Eu anti-His antibody. Time resolved fluorescence was measured with a Synergy Neo 2 Multi Mode Plate Reader and Gen5 software (Biotek) with excitation fluorescence at 330 ± 80 nm and emission measured at 620 ± 10 nm (background fluorescence from Eu antibody) and at 665 ± 8 nm (emission from fluorescence resonance energy transfer, FRET, from tracer) with 100 μs delay and 200 μs collection time. For each time point, emission ratio (EMR) was calculated as emission at 665 nm divided by emission at 620 nm. The corresponding average EMR of wells without inhibitor or kinase was subtracted, and background-subtracted EMR was normalised to average vehicle control. Normalised EMR was calculated in MATLAB (Mathworks).
2.3. Protein expression and purification
The cDNA plasmids for human RSK1 and RSK2 were obtained from Addgene (RSK1: #70574; RSK2: #70578) as Gateway Entry Clones. Recombinant baculoviruses were generated by the Gateway cloning using the BaculoDirect Baculovirus Expression System (Invitrogen) according to the manufacturer's manual.The cDNA for RSK2 NTKD was amplified by conventional PCR, then cloned into pENTR-D-TOPO (pENTR Directional TOPO Cloning Kits, Invitrogen). The resulting Gateway Entry vectors were used for making the baculoviruses in the same way as RSK1 and RSK2 WT viruses. These constructs have 6xHis sequence in their C-termini and recombinant proteins were purified by Ni-NTA Agarose (Qiagen). The molecular weight and concentration of purified proteins were confirmed by standard SDS-PAGE and CBB R-250 staining.
2.4. Kinase activity assay
The activity of RSK2 was measured as previously reported Citation9. Briefly, 1 nM kinase, inhibitor or vehicle (1% DMSO), and 10 μM ATP in kinase buffer were dispensed into the wells of a 96-well plate with adsorbed glutathione S-transferase fusion protein containing the sequence RRRLASTNDKG. The reaction was stopped after 30 min, during the linear phase, with the addition of equal volume 500 mM EDTA, then phosphorylated peptide was measured by ELISA. Luminescence from wells with EDTA added before ATP was subtracted, then luminescence was normalised to the average vehicle luminescence. For the dilution reaction, 100 nM RSK2 was incubated with 5 μM SL0101 or 1.5 μM 1c in kinase buffer. After 2 h incubation, the kinase/inhibitor mix was diluted 100-fold in kinase buffer. As the control reaction, 1 nM RSK2 was incubated with 0.05 μM SL0101 or 0.015 μM 1c for 2 h. After the dilution, kinase/inhibitor solution and 3.3 μM ATP in kinase buffer were mixed to initiate the reaction. The reaction was stopped at the time indicated in the figures with the addition of EDTA, and amount of phosphorylated peptide was measured by ELISA.
2.5. Molecular modeling
Sequences of the RSK family NTKD were aligned in Uniprot (Uniprot Consortium). Single acid mutations of the RSK2NTKD in complex with SL0101 (Protein Databank Accession 3UBD) were simulated in DynaMut (University of Melbourne)Citation30. Briefly, Dynamut uses normal mode analysis to predict the impact of mutations on protein conformation and stability. The Pymol (Schrodinger) session generated by Dynamut was used to visualise changes in bonding and steric interference. Dynamut removes SL0101 during modelling, resulting in some rotamer positions that would interfere with SL0101. Those rotamer positions were edited to the next lowest strain conformation that would not interfere with SL0101. The mutagenesis wizard was used to visualise steric clashes between side chains, which were then marked ( yellow dashes). Hydrogen bonds ( red dashes) were generated by Dynamut. To determine favorability for π-bond stacking, the distance and angle between the A ring of SL0101 and RSK2NTKD-SL0101 Phe79 was measured using the measurement wizard. Pseudoatoms were placed in each ring centre, then distance was measured between the pseudoatoms. Angle was measured between the plane of SL0101 A ring and the γ carbon of Phe79.
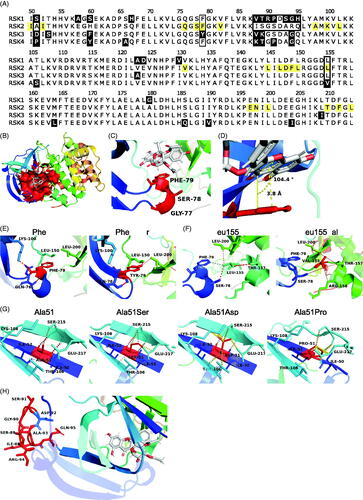
Figure 2. Specificity of SL0101 for RSK1/2 cannot be explained by SL0101-interacting residues. (A) Alignment of the RSK family NTKD catalytic core. Numbering and highlighting are relative to RSK2. Black = non-identical amino acids. Yellow = amino acids composing the SL0101-binding pocket. Black outline = residues discussed. (B) Crystal structure of RSK2NTKD-SL0101 (PDB Accession 3UBD) showing SL0101 (white) in its binding pocket (red surface). (C,D) Position of the P-loop relative to SL0101 in RSK2NTKD-SL0101. Distance measured between centre of Phe79 side chain phenol and A ring of SL0101, and angle measured between plane of A ring of SL0101 and Phe79. (E) Phe79 showing hydrogen bonding (red) to nearby residue side chains (left) and computational mutation of Phe79 to Tyr with additional hydrogen bonds to Leu150 and Leu200 (right). (F) Leu155 showing hydrogen bonding (red) and hydrophobic interactions (grey) to nearby residue side chains (left); computational mutation of Leu155 to Val with additional hydrogen bonds to Arg158 and Thr157 and loss of hydrophobic interaction with Phe79 (right). (G) Ala51 positioned in the middle β strand of the de novo β sheet formed in RSK2NTKD-SL0101 (left). Computational mutations to Ser (second to left), Asp (second to right), and to Pro (right) showing hydrogen bonding (red) or clashes between van der Waals radii (yellow). (H) Ile88 - Ser91 and Ala93 - His95 in RSK2NTKD-SL0101 in an unstructured region (red) clustered away from the SL0101 (white) binding pocket.
2.6. Quantification and statistical analysis
IC50 was determined by four parameter logistic curve, with top constrained to equal 100% and minimum greater than or equal to 0% but otherwise unconstrained. Hill slope and IC50 were unconstrained. Changes in IC50 over time were fit by simple linear regression during the linear portion, less than 15 min, or log-log line over longer periods. Curve fitting and statistics were calculated in Prism 9.0.2 (Graphpad). Statistical test methods are reported in the figure legends. Statistical significance is considered using α = 0.01.
3. Results
3.1. Time dependence of RSK2 binding to SL0101
To further understand the mechanism of RSK1/2 binding to SL0101 (1a) we expressed and purified His-tagged recombinant proteins, wild type (WT) RSK2, WT RSK1 and the isolated RSK2NTKD domain, containing residues 1–389 (). Initially, a fluorescence resonance energy transfer (FRET) assay was used to measure SL0101 binding. This assay uses an ATP-mimetic tracer labelled with an acceptor fluorophore and a recombinant His-tagged kinase, which is indirectly labelled with a europium donor chelated to an anti-His antibody. Interaction of the tracer with the kinase generates a FRET signal. A tracer concentration of 15 nM was used in all FRET assays, which generated a robust signal-to-noise for all constructs (). For IC50 determination the maximum signal was set to 100% as defined by the vehicle controls. The IC50 for the recombinant, purified RSK2 with SL0101 was ∼ 0.4 µM, which is based on a 2 h pre-incubation time of the inhibitor before tracer addition ( and ). GST-tagged RSK3 and RSK4 were purchased and tested with a labelled anti-GST antibody in the FRET assay. SL0101 did not inhibit RSK3 and RSK4 (), which is in agreement with the results from the ZLYTE screen we previously reported Citation11. RSK3 and RSK4 were inhibited by the pan-RSK inhibitor, BI-D1870, demonstrating that the assay was functional (). Unexpectedly, decreasing the pre-incubation time with SL0101 from 2 h to 5 min increased the IC50 by ∼ 40-fold for RSK2 ( and ). In contrast to SL0101, the efficacy of BI-D1870 to inhibit RSK2 showed no time dependence (). These data are consistent with observations showing that BI-D1870 is an ATP competitive inhibitorCitation31. To determine whether the IC50 time dependence was unique to the FRET assay we also measured the IC50 in an ELISA-based assay that was used in the original screen of botanical extractsCitation9. In the activity assay the kinase is pre-incubated with or without the inhibitor in solution with a substrate that is adsorbed onto a hydrophilic, protein-binding surface. The substrate contains the RSK-phosphorylation motif contained in oestrogen receptor alpha (ERα) in which Ser-167 ERα is targeted for phosphorylationCitation32. ATP is used to initiate the reaction and phosphorylation is measured by an anti-phospho-Ser-167 antibody. The reaction is stopped during the linear phase. In agreement with previous reports using the ELISA assay the IC50 for SL0101 (1a) inhibition of RSK2 was ∼ 0.4 µM, which is based on a 2 h pre-incubation time of the inhibitor before initiating the kinase reaction. Pre-incubation of RSK2 with SL0101 for 5 min resulted in a more modest increase in the IC50 (∼ two-fold) than that obtained in the FRET assay, most likely because the kinase continues to be incubated with SL0101 over the 30 min kinase assay resulting in further inhibitor binding () and ). The time dependence of RSK2 for SL0101 binding is consistent with the requirement of the extensive conformational change required to form the SL0101-binding pocket and provides support for the ability of RSK2 to form this pocket in solutionCitation25. Support for this hypothesis is provided by the observations that RSK2 has a higher affinity for 1b after a 2 h incubation compared to SL0101 in both the FRET and ELISA-based assays ( and ). Inhibitor 1b () was designed to take advantage of a hydrophobic pocket present in the SL0101-binding pocket and therefore, its higher binding affinity than SL0101 is consistent with our assumption.
Table 1. Binding characteristics of SL0101 and selected analogues.
3.2. Modelling of RSK2 interactions with SL0101
To gain further insight into the specificity of RSK1/2 for SL0101 we performed a pairwise alignment of RSK1, RSK3, and RSK4 NTKD to that of RSK2, and found that they have ∼ 90% identity (). The crystal structure of the RSK2NTKD in complex with SL0101 (RSK2NTKD-SL0101) shows the hydrophobic SL0101 binding pocket comprises 22 amino acid residues, and 11 of these residues interact with SL0101 by Van der Waals force or hydrogen bonding ()Citation25. To explain the specificity of RSK1/2 for SL0101, single amino acid mutations were introduced computationally into the crystal structure of the RSK2NTKD-SL0101 complex representing amino acids from RSK1/3/4. In the RSK family, the SL0101-interacting amino acids are identical in RSK1 and differ by one amino acid each in RSK3 and RSK4 compared to RSK2 at Phe79 and Leu155, respectively. Phe79 lies at the tip of the P-loop comprising Gly77, Ser78, and Phe79 at the end of two β strands in RSK2NTKD-SL0101 (). In RSK2NTKD-SL0101, the tip of the P-loop is positioned such that the aglycone A ring of SL0101 and the aromatic ring of Phe79 lie 3.8 Å apart and 14.9° above parallel, a conformation favourable for π-bond stacking (). In this position, the aromatic ring of a Tyr residue, as in RSK3, could participate in π-bond stacking, and the ortho-hydroxyl could form additional hydrogen bonds with the side chains of Leu150 and Leu200 on the other side of the SL0101-binding pocket, and therefore, the Tyr residue seems unlikely to interfere with binding of SL0101 (). Leu155 lies at the end of an α helix and forms a hydrogen bond with the B ring of SL0101 (). Substitution of Leu155 with a Val residue, as in RSK4, results in additional weak hydrogen bonds with Thr147 and Arg158 and loses hydrophobic interaction with Phe79. However, these changes appear unlikely to result in destabilisation of the α helix or loss of interaction with SL0101. Taken together, it is unlikely that differences between RSK2 and RSK3/4 at Phe79 and Leu155 result in the observed differences in specificity for SL0101.
In addition to differences within residues that directly interact with SL0101 in RSK2 we investigated contributions of residues outside of the SL0101-binding pocket. These residues are mostly located in unstructured or undefined regions in the RSKNTKD-SL0101 crystal structure or would appear to not result in changes in charge or hydrophilicity. However, Ala51 is of interest as it is located the middle β strand (β1’) of a β sheet that uniquely forms in RSK2NTKD-SL0101, and its identity differs between all four RSK isoforms (). Substitution of Ala51 with a Ser residue, as in RSK1, adds a hydrogen bond with the nearby Glu217 side chain, but also sterically interferes with the Ser215 side chain on the adjacent β strand but neither of these changes appear likely to destabilise the β sheet. In contrast, an Asp residue in place of Ala51, as in RSK3, is sterically strained with the side chains of Thr106, Lys108, Ser215, and Glu217. Substitution of Ala51 with a Pro residue, as in RSK4, destabilises the entire de novo β-sheet by loss of main chain interactions with Ser215 on the adjacent β strand and steric interference with the side chains of Ser215 and Glu217. Previously, we found that mutation of either Ile50 or Ile52, the SL0101-interacting residues adjacent to Ala51, resulted in loss of interaction with either the nucleotide analogue AMP-PNP or SL0101, suggesting the formation of the de novo β sheet is necessary for the formation and stability of the SL0101-binding pocketCitation25. Based on these observations we propose that Ala51 is important in the stability of the de novo β sheet formed in the RSK2NTKD-SL0101 complex and thus the replacement of this residue as occurs in RSK3/4 accounts for their inability to interact with SL0101.
3.3. SL0101 binding is consistent with conformational sampling
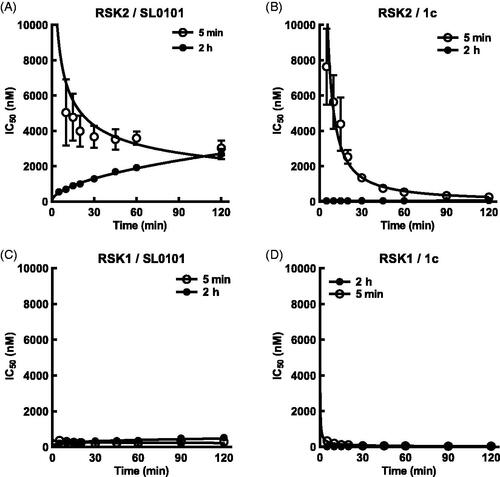
To investigate whether the time dependent binding of SL0101 was unique to RSK2 the interaction of SL0101 with RSK1 was investigated. In the FRET assay the affinity of RSK1 for SL0101 is ∼ two-fold higher than RSK2 ( and ). However, in contrast to RSK2 the length of pre-incubation of RSK1 with SL0101 did not substantially alter the IC50. Incubation of RSK1 with SL0101 for 2 h compared to 5 min had only a three-fold higher affinity compared to the > 40-fold difference observed with RSK2. Furthermore, a 5 min incubation with SL0101 is sufficient for 80% inhibition of RSK1 in contrast to RSK2 where only ∼ 50% inhibition occurs (compare , ). These data suggest that RSK1 is predominately in the conformation that facilitates the interaction with SL0101 compared to RSK2. In support of this suggestion RSK1 differs from RSK2 in an unstructured region comprising 7 non-identical amino acids () and although this region does not interact with SL0101, it undergoes significant conformational rearrangement upon SL0101 binding the RSK2NTKDCitation25. Therefore, changes in flexibility could profoundly affect the formation and stability of the SL0101-binding pocket; although, the current state of protein modelling is unable to assess such a claim.
Figure 3. SL0101 binds differentially to RSK1 and isolated RSKNTKD compared to RSK2. (A,B) Amount of tracer bound to RSK1 or RSK2NTKD after 5 min or 2 h incubation with SL0101 relative to vehicle control (N = 4 in quadruplicate each). (C) Amount of tracer bound to isolated RSK2 after 5 min or 2 h incubation with inhibitor 1c relative to vehicle control (N = 5 and 3, respectively, each in quadruplicate). Data are plotted as mean and error bars represent SD.
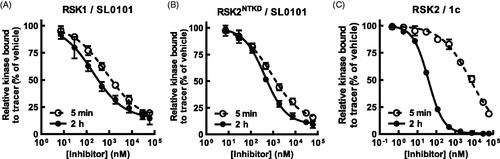
We also analysed the binding kinetics of the isolated NTKD (RSK2NTKD) and observed in the FRET-based assay that the IC50 of RSK2NTKD for SL0101 with a 2 h pre-incubation was similar to that obtained for WT RSK2 ( and ). But like RSK1, the IC50 of RSK2NTKD for SL0101 increased by only ∼ two-fold and ∼ 80% inhibition achieved with a 5 min incubation, and we conclude from these data that domains outside of the NTKD inhibit the formation of the SL0101-binding pocket in RSK2.
The formation of the SL0101-binding pocket in RSK2 could occur by an induced fit mechanism or alternatively the pocket could be created during spontaneous movements that occur in the kinase independent of the presence of SL0101. In the induced fit model, we predict that there would be an initial interaction of SL0101 with the NTKD most likely through the ATP-binding pocket to generate a low affinity interaction (Figure S3A). This low affinity binding could induce a conformational change to generate the high affinity site. In the spontaneous model SL0101 would bind after conformational sampling by the NTKD generated the SL0101-binding pocket (Figure S3B). Support for the pocket occurring by conformational sampling is provided by observations with 1c (). Inhibitor 1c achieves the lowest IC50 at 5 min pre-incubation among all the analogues evaluated ( and ). However, an 8-fold higher concentration than the IC50, which should result in occupancy of the low affinity sites, results in only 75% SL0101 inhibition. It would be expected in the induced fit model that high concentrations of SL0101 should completely occupy the low affinity sites resulting in 100% inhibition. The inability to obtain complete inhibition with saturating concentrations of inhibitor is consistent with a model in which the formation of the SL0101-binding pocket occurs by conformational sampling.
3.4. SL0101 analogues interact with regions outside the SL0101-binding pocket
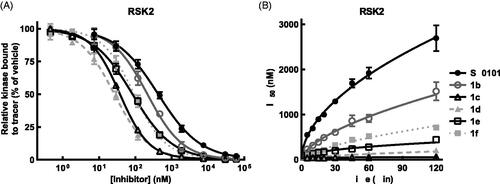
In previous SAR analysis we found that addition of the n-propyl at the 5” position in combination with an n-propyl-carbamate at the 4” position (1d) or with n-propyl-carbamate groups at both the 3” and 4” positions (1f) () improved the affinity of RSK2 for these analogues, which were confirmed in this study using the FRET assayCitation28 ( and ). These results were unexpected because in the crystal structure of the RSK2NTKD in complex with SL0101 (RSK2NTKD-SL0101) the acetyl groups of SL0101 interact with the solventCitation25; however, it is possible that these residues might be available to interact with other regions of the polypeptide in the full length kinase. This hypothesis is supported by our observations that acetylation of the hydroxy groups on the rhamnose increased the affinity of RSK2 for this analogue, which would not be expected to improve interactions with the solvent because of their hydrophobic natureCitation33. In compounds 1d and 1f the ring oxygen in the rhamnose was replaced with a methylene group to create a cyclitol as this change could improve in vivo stability by removing electron density in the O-glycosidic bondCitation34. For this study compounds 1c and 1e () were synthesised to investigate whether the ring oxygen influenced the in vitro affinity, but the IC50s were similar to the cyclitol versions ( and ).
Figure 4. SL0101 analogues have improved affinity for RSK2 and dissociate more slowly. (A) Amount of tracer bound to RSK2 after 2 h pre-incubation with SL0101 analogues (N = at least 3 each in quadruplicate); Data are plotted as mean and error bars represent SD. (B) Relative off-rate of SL0101 analogues (N = at least 3 each in quadruplicate). Data are plotted as IC50 and error bars represent SE of the IC50. Change in IC50 over time is plotted as log-log linear.
3.5. SL0101 analogues show differential interaction with RSK2 compared to RSK1
To further examine the interaction of SL0101 and its analogues with RSK2 and RSK1 we added tracer simultaneously with inhibitor or after a 2 h pre-incubation with inhibitor and monitored the IC50 over time. The results from the pre-incubation experiments provide an indirect measure of the inhibitor off rate whereas the simultaneous addition of inhibitor and tracer provide an indirect measure of the inhibitor on rate. Measuring the slope of the rate of IC50 change for both the pre-incubation and simultaneous experiments shows that the off rate of RSK2 for SL0101 is greater than the on rate, with a ratio of the absolute value of the off/on equal to ∼ 1.3 ( and ). This same analysis for inhibitor 1c shows the inhibitor on rate for RSK2 is greater than the inhibitor off rate, with an off/on ratio equal to ∼ 0.1 ( and ). The off rate for 1c was more than three-fold lower than that of 1d, 1e, and 1f, which is not reflective of their relative IC50s ( and ). The mechanism that accounts for the unique properties of RSK2 interacting with 1c are unknown. RSK1 has a higher off than on rate for SL0101, with an off/on ratio of ∼2.7, suggesting that the RSK1 complex with SL0101 is less stable than that of RSK2 ( and ). RSK1 also has a higher off/on ratio for 1c than does RSK2, suggesting that the RSK1 complex is less stable than the RSK2 complex with 1c ( and ).
Figure 5. SL0101 and analogues interact differentially with RSK2 compared to RSK1. (A) Relative off-rate (change in IC50 after 2 h pre-incubation, N = 6 in quadruplicate) and on-rate (change in IC50 after 5 min incubation, N = 3 in quadruplicate) between SL0101 and RSK2, (B) inhibitor 1c and RSK2 (N = 5 and 2, respectively, each in quadruplicate), (C) SL0101 and RSK1 (N = 4 in quadruplicate each), or (D) inhibitor 1c and RSK1 (N = 4 and 2, respectively, each in quadruplicate). Data are plotted as IC50 with error bars representing SE of the IC50. Change in IC50 over time is fit as log-log-linear.
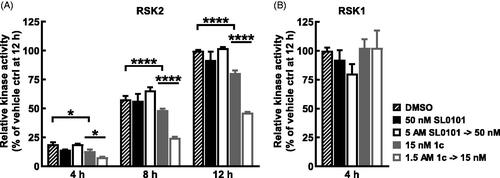
To further analyse the stability of RSK2 with 1c the ELISA assay was used as an orthogonal approach. For these experiments an inhibitor concentration ∼ 15-fold above the respective IC50 obtained in the ELISA assay was incubated with RSK2 for 2 h and then diluted 100-fold. Inhibition of kinase activity was measured at differing time points and compared to that obtained with a concentration of inhibitor equal to the concentration after dilution. SL0101 was unable to inhibit RSK2 at 4 h after the dilution whereas inhibitor 1c maintained ∼50% inhibition over a 12 h time course (). In agreement with the analysis of the stability of RSK1 with 1c RSK1 was not inhibited by 1c at 4 h after the dilution () in the ELISA assay. These results confirm the FRET results that RSK2 and inhibitor 1c form a stable complex.
Figure 6. Long term inhibition of RSK2 compared to RSK1 with inhibitor 1c. (A,B) RSK2 or RSK1 were pre-incubated 2 h with SL0101, inhibitor 1c, or vehicle at the assay concentration shown (control) or 100X assay concentration and then diluted with kinase buffer (dilution). After dilution, ATP was added and the reaction proceeded for the times indicated, then the reaction was stopped with EDTA and the amount of phosphorylated substrate was measured by ELISA. RSK2 activity is relative to vehicle control at 12 h and relative RSK1 activity is relative to vehicle at 4 h. (N = 2 in duplicate each, 2-way ANOVA with Fisher’s Uncorrected LSD for multiple comparisons between control and dilution for each timepoint, p-values: * < 0.01, **** < 10−5). Data are plotted as mean and error bars represent SD.
4. Discussion
Members of the RSK family share high homology, which has made it difficult to identify isoform specific RSK inhibitors. In support of this statement SL0101 is currently the only identified RSK inhibitor that specifically targets RSK1/2 but not RSK3/4. In this report using modelling studies we identified a key residue outside of the SL0101-binding pocket that appears to account for the specificity of RSK1/2 compared to RSK3/4. We also identified an analogue of SL0101 with 4” n-propyl-carbamate substitutions on the rhamnose that forms a highly stable complex with RSK2 but not RSK1, which raises the possibility of developing a novel series of compounds that would result in preferential inhibition of RSK2 in vivo. Unfortunately, these analogues were ineffective at inhibiting RSK in cell-based assays and we propose that it is likely due to the inability of the compounds to enter the cell.
The conformational change required to generate the SL0101-binding pocket in RSK2 is constrained by regions outside the NTKD in comparison to RSK1. This hypothesis is based on the observations that RSK1 and the RSK2NTKD do not show a time dependence for binding to SL0101 and its analogues as observed with WT RSK2. The physiologic relevance for the greater flexibility of RSK1 to form the SL0101-binding pocket compared to RSK2 is not known. However, unlike RSK1, RSK2 is able to translocate to the nucleus and most likely requires other protein factors to aid in this translocationCitation35. It is possible that the conformational change that generates the SL0101-binding pocket regulates the interaction with other proteins and that control of the stability of this conformation is important in regulating the functions of RSK1 versus those of RSK2.
The residence time the inhibitor interacts with the target has been shown to be a better predictor of in vivo efficacy than affinityCitation27,Citation36,Citation37. Thus, identifying that the off rate for RSK2 and SL0101 analogues with 4” n-propyl-carbamate substitutions are extremely slow suggests that pharmacokinetic properties should not be a major driver for further drug development.
Supplemental Material
Download PDF (3.7 MB)Disclosure statement
The authors D.A.L. and G.A.O. have a patent related to this work.
Additional information
Funding
References
- Roskoski R. Jr. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol Res 2019;142:151–68.
- Theodosakis N, Micevic G, Langdon CG, et al. p90RSK blockade inhibits dual BRAF and MEK inhibitor-resistant melanoma by targeting protein synthesis. J Invest Dermatol 2017;137:2187–96.
- Kosnopfel C, Sinnberg T, Sauer B, et al. Human melanoma cells resistant to MAPK inhibitors can be effectively targeted by inhibition of the p90 ribosomal S6 kinase. Oncotarget 2017;8:35761–75.
- Pambid MR, Berns R, Adomat HH, et al. Overcoming resistance to Sonic Hedgehog inhibition by targeting p90 ribosomal S6 kinase in pediatric medulloblastoma. Pediatr Blood Cancer 2014;61:107–15.
- Serra V, Eichhorn PJ, Garcia-Garcia C, et al. RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer. J Clin Invest 2013;123(6):2551–2563.
- Chatterjee S, Huang EH, Christie I, et al. Acquired resistance to the Hsp90 inhibitor, Ganetespib, in KRAS-mutant NSCLC Is mediated via reactivation of the ERK-p90RSK-mTOR signaling network. Mol Cancer Ther 2017;16:793–804.
- Moyano-Galceran L, Pietila EA, Turunen SP, et al. Adaptive RSK-EphA2-GPRC5A signaling switch triggers chemotherapy resistance in ovarian cancer. EMBO Mol Med, e11177 2020;12(4):e11177.
- Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol 2008;9:747–58.
- Smith JA, Poteet-Smith CE, Xu Y, et al. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res 2005;65:1027–34.
- Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J 2007;408:297–315.
- Ludwik KA, Campbell JP, Li M, et al. Development of a RSK inhibitor as a novel therapy for triple-negative breast cancer. Mol Cancer Ther 2016;15:2598–608.
- Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nature Chem Biol 2007;3:156–60.
- Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005;308:1318–21.
- Serafimova IM, Pufall MA, Krishnan S, et al. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat Chem Biol 2012;8:471–6.
- Andersen JL, Gesser B, Funder ED, et al. Dimethyl fumarate is an allosteric covalent inhibitor of the p90 ribosomal S6 kinases. Nat Commun 2018;9:4344.
- Yang ZH, Wu XN, He P, et al. A Non-canonical PDK1-RSK Signal Diminishes Pro-caspase-8-Mediated Necroptosis Blockade. Mol Cell 2020;80:296–310 e296.
- Sapkota GP, Kieloch A, Lizcano JM, et al. Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essential for LKB1 to suppress cell vrowth. J Biol Chem 2001;276:19469–82.
- Edgar AJ, Trost M, Watts C, Zaru R. A combination of SILAC and nucleotide acyl phosphate labelling reveals unexpected targets of the Rsk inhibitor BI-D1870. Biosci Rep 2014;34:e00091.
- Neise D, Sohn D, Stefanski A, et al. The p90 ribosomal S6 kinase (RSK) inhibitor BI-D1870 prevents gamma irradiation-induced apoptosis and mediates senescence via RSK- and p53-independent accumulation of p21WAF1/CIP1. Cell Death Dis 2013;4:e859.
- Kirrane TM, Boyer SJ, Burke J, et al. Indole RSK inhibitors. Part 2: optimization of cell potency and kinase selectivity. Bioorg Med Chem Lett 2012;22:738–42.
- Fryer RM, Muthukumarana A, Chen RR, et al. Mitigation of off-target adrenergic binding and effects on cardiovascular function in the discovery of novel ribosomal S6 kinase 2 inhibitors. J Pharmacol Exp Ther 2012;340:492–500.
- Jain R, Mathur M, Lan J, et al. Discovery of potent and selective RSK inhibitors as biological probes. J Med Chem 2015;58(17):6766–6783.
- Jain R, Mathur M, Lan J, et al. Design and synthesis of potent RSK inhibitors. Bioorg Med Chem Lett 2018;28:3197–201.
- Ludwik KA, Lannigan DA. Ribosomal S6 kinase (RSK) modulators: a patent review. Expert Opin Ther Pat 2016;26:1061–78.
- Utepbergenov D, Derewenda U, Olekhnovich N, et al. Insights into the Inhibition of the p90 Ribosomal S6 Kinase (RSK) by the Flavonol Glycoside SL0101 from the 1.5 Å crystal structure of the N-terminal domain of RSK2 with bound inhibitor. Biochemistry 2012;51:6499–510.
- Li M, Li Y, Ludwik KA, et al. Stereoselective synthesis and evaluation of C6″-Substituted 5a-Carbasugar analogues of SL0101 as inhibitors of RSK1/2. Org Lett 2017;19:2410–3.
- Bernetti M, Cavalli A, Mollica L. Protein-ligand (un)binding kinetics as a new paradigm for drug discovery at the crossroad between experiments and modelling. Med Chem Comm 2017;8:534–50.
- Li Y, Sandusky ZM, Vemula R, et al. Regioselective Synthesis of a C-4'' Carbamate,C-6'' n-Pr Substituted Cyclitol Analogue of SL0101. Org Lett 2020;22:1448–52.
- Maloney DJ, Hecht SM. Synthesis of a potent and selective inhibitor of p90 Rsk. Org Lett 2005;7:1097–9.
- Rodrigues CH, Pires DE, Ascher DB. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res 2018;46:W350–W355.
- Sapkota GP, Cummings L, Newell FS, et al. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem J 2007;401:29–38.
- Joel PB, Smith J, Sturgill TW, et al. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol Cell Biol 1998;18:1978–84.
- Smith JA, Maloney DJ, Hecht SM, Lannigan DA. Structural basis for the activity of the RSK-specific inhibitor, SL0101. Bioorg Med Chem 2007;15:5018–34.
- Li M, Li Y, Mrozowski RM, et al. Synthesis and structure-activity relationship study of 5a-carbasugar analogues of SL0101. ACS Med Chem Lett 2015;6:95–9.
- Ludwik KA, McDonald OG, Brenin DR, Lannigan DA. ERalpha-mediated nuclear sequestration of RSK2 is required for ER + breast cancer tumorigenesis. Cancer Res 2018;78:2014–25.
- Copeland RA. Drug-target interaction kinetics: underutilized in drug optimization? Future Med Chem 2016;8:2173–5.
- Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nature Reviews. Drug Discovery 2006;5:730–9.
- Eisinger-Mathason TS, Andrade J, Lannigan DA. RSK in tumorigenesis: connections to steroid signaling. Steroids 2010;75:191–202.