
Abstract
A series of [1]benzothieno[2,3-c]pyridines was synthesised. Most compounds were chosen by NCI-USA to evaluate their anticancer activity. Compounds 5a–c showed prominent growth inhibition against most cell lines. 5c was selected at five dose concentration levels. It exhibited potent broad-spectrum anticancer activity with a GI50 of 4 nM–37 µM. Cytotoxicity of 5a–c was further evaluated against prostate, renal, and breast cancer cell lines. 5c showed double and quadruple the activity of staurosporine and abiraterone, respectively, against the PC-3 cell line with IC50 2.08 µM. The possible mechanism of anti-prostate cancer was explored via measuring the CYP17 enzyme activity in mice prostate cancer models compared to abiraterone. The results revealed that 5c suppressed the CYP17 enzyme to 15.80 nM. Moreover, it was found to be equipotent to abiraterone in testosterone production. Cell cycle analysis and apoptosis were performed. Additionally, the ADME profile of compound 5c demonstrated both good oral bioavailability and metabolic stability.
Keywords:
1. Introduction
Prostate cancer remains a significant health problem that affects men worldwide, that is characterised by excessive uncontrolled growth of prostate gland cellsCitation1. It is considered the second leading cause of cancer deaths after lung cancerCitation2,Citation3. In 2019, the American Cancer Society estimated a 6% increase in prostate cancer cases and about a 7% increase in deaths from prostate cancer compared to the previous yearsCitation4.
Serum Prostate-Specific Antigen (PSA) with Digital Rectal Exam (DRE) is the most widely used first-line test in urology for the detection of the risk of prostate cancerCitation5. They can help to catch the disease at an early stage when treatment is thought to be more effective and potentially has fewer side effectsCitation6. However, no set cut-off point that can ensure whether a man has or hasn’t had prostate cancer, as high levels of PSA may be only observed after certain medical procedures, in the presence of infection or cases of non-cancerous overgrowth of the prostate, known as benign prostatic hyperplasia (BPH). Therefore, PSA tests may be useful as a signal for the need for a biopsy to examine the prostate cells and determine whether they are cancerousCitation7.
There is no obvious reason for prostate cancer, however, family history, race, age, hormonal disturbances, and diet are known to be the most common risk factorsCitation8. Therapeutic management of prostate cancer includes radiation and prostatectomy for early stages (I, II)Citation9,Citation10. Moreover, Androgen Deprivation Therapy (ADT) reduces the level of androgen hormones in the prostate, thereby preventing the growth of cancer cells. However, this approach should be accompanied by radiation therapy to ensure the eradication of cancer inside the prostateCitation11,Citation12. In stage III, the level of androgen hormones could be reduced by surgery (orchiectomy or surgical castration)Citation13, medication (chemical castration that reduces LH hormone production in the pituitary gland)Citation14,Citation15 and hormonal therapy or antiandrogens (that prevent androgen receptors (AR) in the prostate to bind to testosterone hormone)Citation16. Advanced prostate cancer cases that persist even after reduction of the testosterone level are known as castrate-resistant prostate cancer (CRPC)Citation17. This type of prostate cancer needs chemotherapeutic agents that work by total blockage of androgen biosynthesis via inhibition of CYP17.
Several categories of steroidal and non-steroidal CYP17 inhibitors were developed and characterised as an effective treatment of advanced prostate cancer casesCitation18. Such include abiraterone acetate I, a steroidal antiandrogen prodrug, which was described for the treatment of metastatic castration-resistant prostate cancer (mCRPC)Citation19, however, it showed undesirable side effects as a result of its non-selectivity. The solution to this problem came in the concomitant administration of prednisone. Galeterone II is an investigational steroidal antiandrogen drug used for the treatment of mCRPC cases that acts by a dual mechanismCitation20. The first involves AR antagonism, thereby preventing the testosterone from binding to its receptor, while the second mechanism is inhibition of CYP17 that reduces the synthesis of androgens, being more specific to 17α-lyase than 17α-hydrolyaseCitation21. CFG920 III is a new CYP17 inhibitor in phase I clinical trial used in CRPC patients who are abiraterone resistantCitation22. A very recent drug approved by FDA in 2018 is apalutamide IV, to be used for advanced prostate cancer cases by preventing testosterone hormone from binding to androgen receptorCitation23. Compounds Va–e bearing various bicyclic fused ring systems were reported to have potent CYP17 inhibitory activity with IC50 values ranging from 16–95 nM, except Vc displayed moderate inhibition. The most promising scaffold was Ve with a benzothiophene core in both vitro and vivo evaluationCitation24. The selective CYP17 inhibitors include YM116 VI, which has a tricyclic fused ring system, which showed a loss in rat prostatic weight by reducing androgen production in the testes and adrenal glandsCitation25. Literature survey on non-steroidal prostate cancer inhibitors demonstrated that the tetrahydrobenzo[4,5]thieno[2,3-c]pyridine derivatives, VIIa–d, exhibited a comparable potency to abiraterone in inhibiting rat CYP17 enzyme and were able to decrease plasma testosterone level in a dose-dependent mannerCitation26 ().
Figure 1. Certain active CYP17 inhibitors.
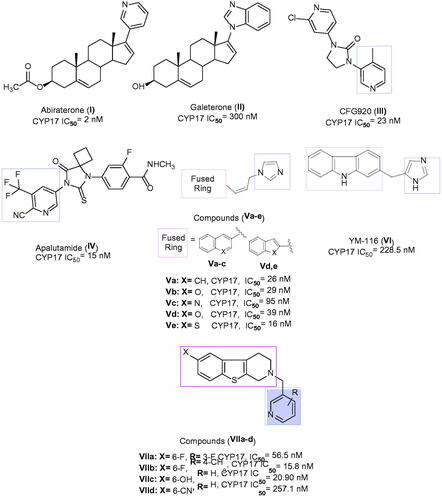
Due to the previously described side effects observed in steroidal drugsCitation27,Citation28, we were inspired to synthesise more specific and safer inhibitors against CYP17 lyase enzyme that could be able to block adrenal androgens and testosterone synthesis. SAR studies of some previously reported non-steroidal CYP17 inhibitors observe that most compounds belonging to this category consist of two parts essential for enzyme inhibition; one is the metal-binding atom or group, which should contain a lone pair of electrons, which is important for binding to haem iron in CYP17, while the second part involves the steroid mimetic scaffold, such as stilbene and biphenyl, or fused ring systems, such as naphthalene, benzothiophene, and 9H-carbazole occupying the pocket of CYP17 enzyme. Previously reported literature studies in this field, discovered the excellent CYP17 inhibitory effect of benzothienopyridine derivatives VIIa–d, making them promising leads for our design strategy ().
Figure 2. Rational of molecular design of new CYP17 inhibitors.
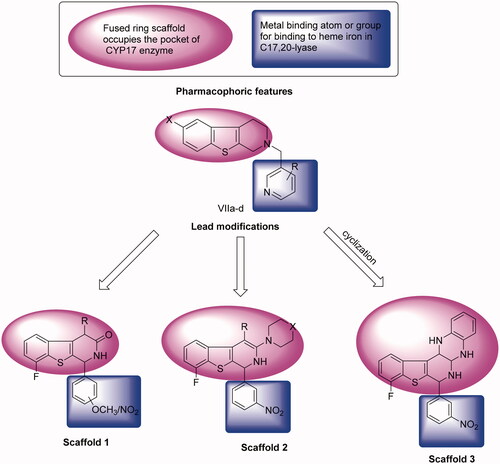
This work comprises the synthesis of new series of 1,2,3,4-tetrahydro[1]benzothieno[2,3-c]pyridines as a promising candidate for CYP17 inhibition. Our strategy involves the replacement of a steroid nucleus by 8-fluoro-1,2-dihydro[1]benzothieno[2,3-c]pyridine core bearing phenyl ring at position 1, as a structural analog for compounds VIIa–d. In addition, groups or substituents bearing lone pair of electrons were present on the phenyl ring at C-1, representing a fundamental requirement for the enzyme inhibition, as the free electron, lone pairs are essential to interact with haem iron in CYP17, besides the lone pair of electrons, which is present on the nitrogen atom of benzothienopyridine scaffold ().
Twenty-seven of the newly synthesised compounds were subjected to in-vitro anticancer screening by National Cancer Institute (USA) against 60 cell lines at one dose concentration followed by a five-dose screening for the most active candidate 5c. The most active compounds were selected for measuring their IC50 against prostate cancer cell line (PC-3), renal cancer cell line (UO-31), and breast cancer cell line (MCF-7). Moreover, compound 5c was evaluated in-vivo for inhibition of the CYP17 enzyme and gauging plasma testosterone level. Furthermore, the mechanism of action of compound 5c was studied on the cell cycle of PC-3 cells and induction of apoptosis. Finally, the ADME profile of compound 5c has been examined to investigate its potential as a promising drug candidate.
2. Experimental section
2.1. Chemistry
2.1.1. General
All chemicals and reagents were obtained from Aldrich (Sigma-Aldrich) and used without further purification. Reactions were monitored by TLC, performed on silica gel glass plates containing 60 GF-254, and visualised on TLC using UV light or iodine indicator. IR spectra were determined on Shimadzu IR 435 spectrophotometer (KBr, cm−1). 1H-NMR and 13C-NMR spectra were carried out using Bruker 400 and 100 MHz spectrophotometers, respectively, using TMS as internal standards. Chemical shifts were recorded in ppm on δ scale, Microanalytical Centre, Faculty of Pharmacy, Cairo University, Egypt. Mass spectra and elemental analyses were recorded on Shimadzu Qp-2010 plus spectrometer at the Regional Centre for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt. The results correspond to the calculated values within experimental error. Melting points were determined with the Stuart apparatus and are uncorrected.
2.1.2. General procedure for the synthesis of 2-arylidene-7-fluoro[1]benzothio-phen-3(2H)-ones (2a–d)
7-Fluoro[1]benzothiophen-3-(2H)one (1) (1.68 g, 0.01 mol) was added to a solution of an appropriate aromatic aldehyde (0.01 mmol) in glacial acetic acid (20 ml) containing anhydrous sodium acetate (0.82 g, 0.01 mol), and the reaction mixture was heated under reflux for 2 h. The solvent was concentrated under reduced pressure, and the resulting solid product was dried then crystallised from acetonitrile.
2.1.2.1. 2-(2,4-Dimethoxybenzylidene)-7-fluoro[1]benzothiophen-3(2H)-one (2a)
Yield 77%, mp 195–197 °C, IR (KBr, cm−1): 3062 (C-H aromatic), 2951, 2813 (C-H aliphatic), 1670 (C=O), 1604 (C=C). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.92 (s, 3H, CH3O), 3.96 (s, 3H, CH3O), 6.66 (d, 1H, J = 8.30 Hz, 2,4-(CH3O)2-C6H3-C5-H), 6.72 (s, 1H, 2,4-(CH3O)2-C6H3-C3-H), 6.80 (d, 1H, J = 8.30 Hz, 2,4-(CH3O)2-C6H3-C6-H), 7.43–7.52 (m, 1H, [1]benzothiophene-C5-H), 7.64–7.73 (m, 1H, [1]benzothiophene-C4-H), 7.76 (d, 1H, J = 7.56 Hz, [1]benzothiophene-C6-H), 8.27 (s, 1H, benzylidene-H).13C-NMR (DMSO-d6, 100 MHz, δ ppm): 56.28, 56.44 (OCH3)2, 98.63, 99.15, 107.24, 107.33 (d, J = 18 Hz, C-5) 115.22, 118.59, 123.26, 125.86, 129.66, 130.28, 131.51, 132.62, 161.32, 165.25 (d, J = 260 Hz, C-F) (ArCs), 187.68 (C=O). Anal. Calcd. (%) for C17H13FO3S (316): C, 64.54, H, 4.14. Found: C, 64.78, H 4.37.
2.1.2.2. 7-Fluoro-2-(4-methoxybenzylidene)[1]benzothiophen-3(2H)-one (2b)
Was previously reportedCitation29.
2.1.2.3. 7-Fluoro-2-(2-nitrobenzylidene)[1]benzothiophen-3(2H)-one (2c)
Yield 70%, mp 170–172 °C, IR (KBr, cm−1): 3059, 3022 (C-H aromatic), 1689 (C=O), 1593 (C=C), 1519, 1346 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 7.49–7.54 (m, 1H, [1]benzothiophene-C4-H), 7.72 (t, 1H, J = 8.76 Hz, [1]benzothiophene-C5-H), 7.76–7.78 (m, 1H, [1]benzothiophene-C6-H), 7.81 (d, 1H, J = 7 Hz, 2-NO2-C6H4-C6-H), 7.93–7.96 (m, 2H, 2-NO2-C6H4-C4,5-H), 8.15 (d, 1H, J = 7 Hz, 2-NO2-C6H4-C3-H), 8.26 (s, 1H, benzylidene C-H). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 122.95, 123.58, 124.78, 125.99, 128.92, 128.95 (d, J = 6 Hz, C-5), 129.61, 130.60, 131.87 (d, J = 255 Hz, C-F), 133.15, 134.65, 134.74, 134.99, 148.81 (ArCs), 190.41 (C=O). Anal. Calcd. (%) for C15H8FNO3S (301): C, 59.80, H, 2.68, S 10.64, N 4.65. Found: C, 59.63, H, 3.02, S, 10.75, N, 4.89.
2.1.2.4. 7-Fluoro-2-(3-nitrobenzylidene)[1]benzothiophen-3(2H)-one (2d)
Yield 83%, mp 200–202 °C, IR (KBr, cm−1): 3070, 3001 (C-H aromatic), 1681 (C=O), 1589 (C=C), 1527, 1354 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 7.52–8.00 (m, 3H, [1]benzothiophene-C4,5,6-H), 8.16–8.22 (m, 1H, 3-NO2-C6H4-C5-H), 8.23–8.42 (m, 2H, 3-NO2-C6H4-C4,6-H), 8.55 (s, 1H, 3-NO2-C6H4-C2-H), 8.70 (s, 1H, benzylidene C-H). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 123.56, 124.55, 125.86, 129.04, 131.44, 131.54 (d, J = 20 Hz, C-5), 132.41, 133.89 (d, J = 260 Hz, C-F), 135.38, 137.66, 139.38, 140.25, 148.76, 157.58 (ArCs), 192.32 (C=O). Anal. Calcd. (%) for C15H8FNO3S (301): C, 59.80, H, 2.68, N, 4.65. Found: C, 60.13, H, 2.89, N, 4.76.
2.1.3. General procedure for synthesis of 1-aryl-8-fluoro-1,2-dihydro[1]benzo-thieno[2,3-c]pyridin-3(4H)-ones (3a–d)
A mixture of α,β-unsaturated ketones 2a–d (1 mmol), acetamide (0.118 g, 0.002 mol), and potassium hydroxide (0.28 g, 0.005 mol) in 95% ethanol (25 ml) was heated under reflux for 8 h. The reaction mixture was allowed to cool and poured onto ice-cooled water. The precipitated solid was filtered and crystallised from ethanol to give the target compounds 3a–d.
2.1.3.1. 1-(2,4-Dimethoxyphenyl)-8-fluoro-[1]benzothieno[2,3-c]pyridin-3(4H)-one (3a)
Yield 55%, mp 70–72 °C, IR (KBr, cm−1): 3417 (OH, taut), 3313 (NH), 3078 (C-H aromatic), 2943–2889 (C-H aliphatic), 1681 (C=O), 1604 (C=N), 1570 (C=C). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.84 (s, 2H, CH2), 3.87 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.95 (s, 1H, C1-H), 6.65 (d, 1H, J = 8.20 Hz, 2,4-(OCH3)2-C6H3-C5-H), 6.69 (s, 1H, 2,4-(OCH3)2-C6H3-C3-H), 6.78 (d, 1H, J = 8.20 Hz, 2,4-(OCH3)2-C6H3-C6-H), 7.30 (d, 1H, J = 8.40 Hz, C5-H), 7.40–7.52 (m, 1H, C6-H), 7.67 (d, 1H, J = 8.40 Hz, C7-H), 8.26 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 31.72 (CH2), 55.92 (C-1), 56.28, 56.44 (OCH3)2, 98.63, 99.15, 99.23 (d, J = 17 Hz, C-6), 107.24, 115.21, 118.59, 122.97, 125.67, 128.46, 129.67, 130.28, 131.50, 165.25 (d, J = 260 Hz, C-F), 166.55 (ArCs), 187.86 (C=O). Anal. Calcd. (%) for C19H16FNO3S (357): C, 63.85, H, 4.51, N, 3.92. Found: C, 63.50, H, 4.74, N, 4.25.
2.1.3.2. 8-Fluoro-1-(4-methoxyphenyl)[1]benzothieno[2,3-c]pyridin-3(4H)-one (3b)
Yield 65%, mp 150–152 °C, IR (KBr, cm−1): 3336 (NH), 3070 (C-H aromatic), 2843 (C-H aliphatic), 1674 (C=O), 1604 (C=N), 1581 (C=C). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.79 (s, 2H, CH2), 3.84 (s, 1H, C1-H), 3.87 (s, 3H, OCH3), 7.16 (d, 2H, J = 8.56 Hz, 4-OCH3-C6H4-C3,5-H), 7.49 (d, 2H, J = 8.56 Hz, 4-OCH3-C6H4-C2,6-H), 7.68 (t, 1H, J = 8 Hz, C6-H), 7.77 (d, 1H, J = 8 Hz, C5-H), 7.82 (d, 1H, J = 8 Hz, C7-H), 8.01 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 36.75 (CH2), 55.77 (OCH3), 56.03 (C-1), 114.76, 115.52, 120.14, 121.85, 122.03, 122.99, 126.07, 126.18 (d, J = 23 Hz, C-6), 128.56, 132.36 (d, J = 266 Hz, C-F), 133.69, 135.38, (ArCs), 186.70 (C=O). Anal. Calcd. (%) for C18H14FNO2S (327): C, 66.04, H, 4.31, N, 4.28. Found: C, 65.87, H, 4.53, N, 4.67.
2.1.3.3. 8-Fluoro-1-(2-nitrophenyl)[1]benzothieno[2,3-c]pyridin-3(4H)-one (3c)
Yield 70% mp 160–162 °C, IR (KBr, cm−1): 3332 (NH), 3071 (C-H aromatic), 2951 (C-H aliphatic), 1681 (C=O), 1589 (C=N), 1570 (C=C), 1527, 1342 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.00 (s, 2H, CH2), 3.92 (s, 1H, C1-H), 6.67–7.28 (m, 3H, C5,6,7-H), 7.30–7.65 (m, 3H, 2-NO2-C6H4-C4,5,6-H), 7.67 (s, 1H, NH, D2O exchangeable), 7.97 (d, 1H, J = 8.28 Hz, 2-NO2-C6H4-C3-H). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 37.10 (CH2), 56.40 (C-1), 113.19, 115.46, 116.78, 121.10, 122.91, 124.12, 124.55, 127.07, 128.21, 129.76, 131.77, 132.79, 135.56 (d, J = 276 Hz, C-F), 136.94 (ArCs), 189.62 (C=O). Anal. Calcd. (%) for C17H11FN2O3S (342): C, 59.64, H, 3.24, N, 8.18, S, 9.37. Found: C, 59.87, H, 3.51, N, 7.95, S, 9.12.
2.1.3.4. 8-Fluoro-1-(3-nitrophenyl)[1]benzothieno[2,3-c]pyridin-3(4H)-one (3d)
Yield 80%, mp 110–112 °C, IR (KBr, cm−1): 3468 (OH, taut), 3360 (NH), 3075 (C-H aromatic), 2927 (C-H aliphatic), 1685 (C=O), 1616 (C=N), 1595 (C=C), 1527, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.93 (s, 2H, CH2), 4.19 (s, 1H, C1-H), 7.15–7.92 (m, 4H, C5,6,7-H and 3-NO2-C6H4-C5-H), 8.05 (d, 1H, J = 8 Hz, 3-NO2-C6H4-C6-H), 8.53 (d, 1H, J = 8 Hz, 3-NO2-C6H4-C4-H), 8.60 (s, 1H, 3-NO2-C6H4-C2-H), 8.63 (s, 0.5H, NH, D2O exchangeable, taut), 8.70 (s, 0.5H, OH, D2O exchangeable, taut). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 32.17 (C-1), 62.20 (C-4), 122.02, 124.44, 124.84, 125.93, 127.92, 128.80, 128.83 (d, J = 6 Hz, C-6), 130.06, 131.41, 132.40, 134.66 (d, J = 266 Hz, C-F), 135.99, 145.42, 148.23 (ArCs), 192.27 (C=O). MS: m/z (% relative abundance): 342 (M+., 20.52%) and 300 (100%). Anal. Calcd. (%) for C17H11FN2O3S (342): C, 59.64, H, 3.24, N, 8.18. Found: C, 59.79, H, 3.50, N, 8.40.
2.1.4. General procedure for synthesis of 1-aryl-4-chloro-8-fluoro-1,2-dihydro [1]benzothieno[2,3-c]pyridine-3(4H)-ones (4a–d)
To a solution of α,β-unsaturated ketones 2a–d (1 mmol) and potassium hydroxide (0.28 g, 0.005 mol) in 95% ethanol (25 ml), 2-chloroacetamide (0.187 g, 0.002 mol) was added, and the reaction mixture was heated under reflux for 8 h. After cooling, the mixture was poured on water, and the obtained solid was filtered off and crystallised from ethanol.
2.1.4.1. 4-Chloro-1-(2,4-Dimethoxyphenyl)-8-fluoro-1,2-dihydro[1]benzothieno [2,3-c]pyridine-3(4H)-one (4a)
Yield 60%, mp 120–122 °C, IR (KBr, cm−1): 3441 (OH, taut), 3332 (NH), 3078 (C-H aromatic), 2943 (C-H aliphatic), 1681 (C=O), 1604 (C=N), 1566 (C=C). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.82 (s, 1H, C1-H), 3.88 (s, 3H, CH3O), 3.92 (s, 3H, CH3O), 3.95 (s, 1H, C4-H), 6.65 (d, 1H, J = 8.20 Hz, 2,4-(CH3O)2-C6H3-C5-H), 6.69 (s, 1H, 2,4-(CH3O)2-C6H3-C3-H), 6.78 (d, 1H, J = 8.20 Hz, 2,4-(CH3O)2-C6H3-C6-H), 7.40–7.52 (m, 1H, C6-H), 7.67 (d, 1H, J = 8.50 Hz, C5-H), 7.74–7.76 (m, 1H, C7-H), 8.26 (s, 0.5H, NH, D2O exchangeable, taut), 10.18 (s, 0.5H, OH, D2O exchangeable, taut). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 56.20 (C-1), 56.27, 56.43 (OCH3)2, 56.55 (C-4), 98.84 (d, J = 47 Hz, C-6), 99.08, 107.34, 115.20, 118.58, 121.62, 122.90, 125.61, 128.33, 129.59, 130.27, 131.46, 162.76 (d, J = 290 Hz, C-F), 164.24, 166.53 (ArCs), 186.67 (C=O). Anal. Calcd. (%) for C19H15ClFNO3S (391): C, 58.24, H, 3.86, N, 3.57. Found: C, 58.53, H, 4.10, N, 3.80.
2.1.4.2. 4-Chloro-8-fluoro-1-(4-methoxyphenyl)-1,2-dihydro[1]benzothieno[2,3-c] pyridine-3(4H)-one (4b)
Yield 50%, mp 154–156 °C, IR (KBr, cm−1): 3441 (OH, taut), 3380 (NH), 3039 (C-H aromatic), 2880 (C-H aliphatic), 1674 (C=O), 1604 (C=N), 1585 (C=C). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.78 (s, 1H, C1-H), 3.85 (s, 1H, C4-H), 3.87 (s, 3H, CH3O), 7.16 (d, 2H, J = 8.30 Hz, 4-CH3O-C6H4-C3,5-H), 7.49 (d, 2H, J = 8.30 Hz, 4-OCH3-C6H4-C2,6-H), 7.68 (t, 1H, J = 8 Hz, C6-H), 7.76 (d, 1H, J = 8 Hz, C5-H), 7.82 (d, 1H, J = 8 Hz, C7-H), 8.01 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 55.82 (C-1), 56.07 (OCH3), 56.16 (C-4), 114.98, 115.59, 121.93, 122.11, 123.07, 126.11, 126.22 (d, J = 22 Hz, C-6), 128.57, 132.27, 132.44 (d, J = 260 Hz, C-F), 135.46, 162.09 (ArCs), 191.78 (C=O). Anal. Calcd. (%) for C18H13ClFNO2S (361): C, 59.75, H, 3.62, N, 3.87. Found: C, 59.91, H, 3.84, N, 4.05.
2.1.4.3. 4-Chloro-8-fluoro-1-(2-nitrophenyl)-1,2-dihydro[1]benzothieno[2,3-c] pyridine-3(4H)-one (4c)
Yield 62%, mp 110–112 °C, IR (KBr, cm−1): 3332 (NH), 3070 (C-H aromatic), 2904, 2835(C-H aliphatic), 1681 (C=O), 1550 (C=C), 1527, 1342 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.95 (s, 1H, C1-H), 4.16 (s, 1H, C4-H), 7.21 (d, 1H, J = 8.40 Hz, C5-H), 7.18–7.25 (m, 1H, C6-H), 7.30 (d, 1H, J = 8.40 Hz, C7-H), 7.40–7.52 (m, 1H, 2-NO2-C6H4-C5-H), 7.56–7.60 (m, 1H, 2-NO2-C6H4-C4-H), 7.67 (d, 1H, J = 7.70 Hz, 2-NO2-C6H4-C6-H), 7.83 (d, 1H, J = 7.70 Hz, 2-NO2-C6H4-C3-H), 8.60 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, ppm): 54.61 (C-1), 56.17 (C-4), 124.00, 126.68, 127.92, 128.88, 129.14, 129.90, 130.04, 130.21 (d, J = 30 Hz, C-6), 130.90, 131.00, 131.05 (d, J = 223 Hz, C-F), 131.90, 131.99, 132.13 (ArCs), 181.68 (C=O). Anal. Calcd. (%) for C17H10ClFN2O3S (376): C, 54.11, H, 2.68, N, 7.43, S, 8.51. Found: C, 54.36, H, 2.85, N, 7.60, S, 8.64.
2.1.4.4. 4-Chloro-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro[1]benzothieno[2,3-c] pyridine-3(4H)-one (4d)
Yield 85%, mp 160–162 °C, IR (KBr, cm−1): 3390 (NH), 3078 (C-H aromatic), 2927(C-H aliphatic), 1685 (C=O), 1600 (C=N), 1560 (C=C), 1527, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.81 (s, 1H, C1-H), 3.93 (s, 1H, C4-H), 7.20–8.15 (m, 4H, C5,6,7-H and 3-NO2-C6H4-C5-H), 8.34 (d, 1H, J = 7.50 Hz, 3-NO2-C6H4-C6-H), 8.53 (d, 1H, J = 7.50 Hz, 3-NO2-C6H4-C4-H), 8.54 (s, 1H, 3-NO2-C6H4-C2-H), 8.70 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 51.51 (C-1), 58.68 (C-4), 118.11, 120.21, 122.00, 124.39, 124.59, 128.89 (d, J = 10 Hz, C-6), 130.03, 130.15 (d, J = 243 Hz, C-F), 131.37, 135.96, 136.05, 137.58, 145.71, 148.67 (ArCs), 192.19 (C=O). MS: m/z (% relative abundance): 378 (M+.+2, 6.38%), 376 (M+., 14.45%) and 329 (100%). Anal. Calcd. (%) for C17H10ClFN2O3S (376): C, 54.11, H, 2.68, N, 7.43. Found: C, 54.35, H, 2.43, N, 7.72.
2.1.5. General procedure for synthesis of 1-aryl-4-cyano-8-fluoro-1,2-dihydro [1]benzothieno[2,3-c]pyridin-3(4H)-ones (5a–d)
A mixture of α,β-unsaturated ketones 2a–d (1 mmol), 2-cyanoacetamide (0.168 g, 0.002 mol), and potassium hydroxide (0.28 g, 0.005 mol) in 95% ethanol (25 ml) was heated under reflux for 8 h. The reaction mixture was poured on water after cooling, and the produced solid was filtered and crystallised from ethanol to afford the target compounds 5a–d.
2.1.5.1. 1-(2,4-Dimethoxyphenyl)-4-cyano-8-fluoro-1,2-dihydro[1]benzothieno [2,3-c]pyridin-3(4H)-one (5a)
Yield 75%, mp 100–102 °C, IR (KBr, cm−1): 3379 (OH, taut), 3313 (NH), 3078 (C-H aromatic), 2943 (C-H aliphatic), 2210 (CN), 1681 (C=O), 1604 (C=N), 1570 (C=C). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.75 (s, 3H, CH3O), 3.78 (s, 3H, CH3O), 3.89 (s, 1H, C4-H), 3.95 (s, 1H, C1-H), 6.40 (s, 1H, 2,4-(CH3O)2-C6H3-C3-H), 6.45–6.53 (m, 1H, 2,4-(CH3O)2-C6H3-C5-H), 6.78–6.83 (m, 1H, 2,4-(CH3O)2-C6H3-C6-H), 7.19–7.39 (m, 1H, C6-H), 7.42–7.59 (m, 1H, C5-H), 7.71 (d, 1H, J = 8.50 Hz, C7-H), 8.27 (s, 0.5H, NH, D2O exchangeable, taut), 10.17 (s, 0.5H, OH, D2O exchangeable, taut). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 33.46 (C-4), 55.49 (C-1), 56.22, 56.57 (OCH3)2, 99.11, 107.39, 108.69, 115.63, 118.23, 119.44, 122.91, 125.68, 128.46, 129.64, 131.48, 132.44, 145.92 (d, J = 268 Hz, C-F), 149.26, 155.15 (ArCs and CN), 180.29 (C=O). Anal. Calcd. (%) for C20H15FN2O3S (382): C, 62.82, H, 3.95, N, 7.33. Found: C, 62.71, H, 4.23, N, 7.62.
2.1.5.2. 4-Cyano-8-fluoro-1-(4-methoxyphenyl)-1,2-dihydro[1]benzothieno[2,3-c] pyridin-3(4H)-one (5b)
Yield 64%, mp 103–105 °C, IR (KBr, cm−1): 3336 (NH), 3097 (C-H aromatic), 2904 (C-H aliphatic), 2206 (CN), 1674 (C=O), 1604 (C=N), 1581 (C=C). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.82 (s, 3H, CH3O), 3.84 (s, 1H, C4-H), 3.86 (s, 1H, C1-H), 6.91 (d, 2H, J = 8.30 Hz, 4-CH3O-C6H4-C3,5-H), 7.12 (d, 2H, J = 8.30 Hz, 4-CH3O-C6H4-C2,6-H), 7.35 (d, 1H, J = 8.50 Hz, C5-H), 7.66 (t, 1H, J = 8.50 Hz, C6-H), 7.80 (d, 1H, J = 8 Hz, C7-H), 7.99 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 33.11 (C-4), 55.83 (C-1), 56.07 (OCH3), 115.59, 119.51, 122.12, 123.08, 126.33, 128.57, 128.76 (d, J = 38 Hz, C-6), 129.92, 132.44 (d, J = 259 Hz, C-F), 133.74, 135.47, 162.09, 163.95 (ArCs and CN), 177.52 (C=O). Anal. Calcd. (%) for C19H13FN2O2S (352): C, 64.76, H, 3.72, N, 7.95. Found: C, 64.98, H, 3.86, N, 8.19.
2.1.5.3. 4-Cyano-8-fluoro-1-(2-nitrophenyl)-1,2-dihydro[1]benzothieno[2,3-c] pyridin-3(4H)-one (5c)
Yield 70%, mp 140–142 °C, IR (KBr, cm−1): 3194 (NH), 3067 (C-H aromatic), 2904(C-H aliphatic), 2199 (CN), 1720 (C=O), 1593 (C=N), 1565 (C=C), 1554, 1384 (NO2).1H-NMR (DMSO-d6, 400 MHz, δ ppm): 4.09 (s, 1H, C4-H), 4.31 (s, 1H, C1-H), 7.14 (d, 1H, J = 7.00 Hz, C5-H), 7.31–7.34 (m, 1H, C6-H), 7.44 (d, 1H, J = 7.00 Hz, C7-H), 7.62–7.66 (m, 2H, 2-NO2-C6H4-C4,5-H), 8.01–8.03 (m, 1H, 2-NO2-C6H4-C6-H), 8.44 (d, 1H, J = 8.00 Hz, 2-NO2-C6H4-C3-H), 9.50 (s, 1H, NH, D2O exchangeable). Anal. Calcd. (%) for C18H10FN3O3S (367): C, 58.85, H, 2.74, N, 11.44, S, 8.73. Found: C, 59.03, H, 2.61, N, 11.73, S, 8.94.
2.1.5.4. 4-Cyano-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro[1]benzothieno[2,3-c] pyridin-3(4H)-one (5d)
Yield 84%, mp 180–182 °C, IR (KBr, cm−1): 3417 (OH, taut), 3226 (NH), 3082 (C-H aromatic), 2974(C-H aliphatic), 2210 (CN), 1670 (C=O), 1620 (C=N), 1600 (C=C), 1527, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 4.21 (s, 1H, C4-H), 4.75 (s, 1H, C1-H), 7.07 (d, 1H, J = 8.50 Hz, C5-H), 7.15–8.40 (m, 5H, C6,7-H and 3-NO2-C6H4-C4,5,6-H), 8.60 (s, 1H, 3-NO2-C6H4-C2-H), 9.10 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 18.93 (C-4), 56.53 (C-1), 116.13, 117.98, 119.65, 120.62, 121.98, 123.13, 124.64, 128.53, 128.58 (d, J = 11 Hz, C-6), 130.98, 135.14, 136.79, 146.98 (d, J = 259 Hz, C-F), 156.03, 158.45 (ArCs and CN), 189.71 (C=O). MS: m/z (% relative abundance): 367 (M+., 25.61%) and 326 (50.23%). Anal. Calcd. (%) for C18H10FN3O3S (367): C, 58.85, H, 2.74, H, 11.44. Found: C, 59.11, H, 2.96, N, 11.78.
2.1.6. General procedure for synthesis of 4-substitutedamino-8-fluoro-1-(3-nitro phenyl)-1,2-dihydro[1]benzothieno[2,3-c]pyridin-3(4H)-ones (6a–c)
An equimolar mixture of 4d (0.376 g, 0.001 mol), an appropriate secondary amine (0.001 mol), and triethylamine (two drops) in absolute ethanol (25 ml) was heated under reflux for 5 h. After cooling, the precipitated solid was filtered and crystallised from ethanol.
2.1.6.1. 4-(4-Ethylpiperazin-1-yl)-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro[1]benzo thieno[2,3-c]pyridin-3(4H)-one (6a)
Yield 53%, mp 240–241 °C, IR (KBr, cm−1): 3417 (NH), 3070 (C-H aromatic), 2916 (C-H aliphatic), 1680 (C=O), 1620 (C=N), 1600 (C=C), 1527, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 0.99 (t, 3H, J = 7 Hz, CH3-CH2), 1.16–1.30 (m, 4H, piperazine-C3,5-H), 2.38 (q, 2H, J = 7 Hz, CH3-CH2), 2.90–3.09 (m, 4H, piperazine-C2,6-H), 3.89 (s, 1H, C1-H), 4.01 (s, 1H, C4-H), 7.10–8.40 (m, 6H, C5,6,7-H and 3-NO2-C6H4-C4,5,6-H), 8.43 (s, 1H, 3-NO2-C6H4-C2-H), 8.70 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 12.02 (CH3), 15.60 (CH2), 29.12 (piperazine-C-3,5), 49.41 (piperazine-C-2,6), 51.49 (C-1), 52.53 (C-4), 97.60, 113.90, 120.98, 122.74, 123.95, 125.84, 127.94, 130.43, 131.06, 135.03 (d, J = 275 Hz, C-F), 136.41, 136.78, 144.13, 149.34 (ArCs), 187.21 (C=O). Anal. Calcd. (%) for C23H23FN4O3S (454): C, 60.78, H, 5.10, N, 12.33. Found: C, 60.61, H, 5.34, N, 12.49.
2.1.6.2. 8-Fluoro-4-morpholino-1(3-nitrophenyl)-1,2-dihydro[1]benzothieno[2,3-c]pyridin-3(4H)-one (6b)
Yield 55%, mp 250–251 °C, IR (KBr, cm−1): 3417 (NH), 3074 (C-H aromatic), 2900 (C-H aliphatic), 1700 (C=O), 1590 (C=N), 1575 (C=C), 1531, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 2.93–3.19 (m, 4H, morpholine-C3,5-H), 3.68–3.80 (m, 4H, morpholine-C2,6-H), 4.06 (s, 1H, C1-H), 4.83 (s, 1H, C4-H), 7.24–7.39 (m, 2H, C5,6-H), 7.44 (d, 1H, J = 7.16 Hz, C7-H), 7.77–8.34 (m, 1H, 3-NO2-C6H4-C5-H), 8.10 (d, 1H, J = 7.00 Hz, 3-NO2-C6H4-C6-H), 8.34 (d, 1H, J = 7.00 Hz, 3-NO2-C6H4-C4-H), 8.44 (s, 1H, 3-NO2-C6H4-C2-H), 8.70 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 19.00 (morpholine C-3,5), 56.49 (morpholine C-2,6), 68.50 (C-1), 70.68 (C-4), 120.70, 122.86, 129.36, 130.69, 132.78 (d, J = 250 Hz, C-F), 134.03, 135.86, 139.36, 142.03, 145.36, 148.87, 156.20, 161.20, 169.20 (ArCs), 180.54 (C=O). MS: m/z (% relative abundance): 427 (M+., 33.23%) and 62 (100%). Anal. Calcd. (%) for C21H18FN3O4S (427): C, 59.01, H, 4.24, N, 9.83. Found: C, 58.79, H, 4.47, N, 10.11.
2.1.6.3. 8-Fluoro-1-(3-nitrophenyl)-4-(piperidin-1-yl)-1,2-dihydro[1]benzothieno [2,3-c]pyridin-3(4H)-one (6c)
Yield 61%, mp 230–232 °C, IR (KBr, cm−1): 3350 (NH), 3070 (C-H aromatic), 2943 (C-H aliphatic), 1670 (C=O), 1580 (C=N), 1556 (C=C), 1531, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 1.50–1.57 (m, 2H, piperdine-C4-H), 1.58–1.70 (m, 4H, piperdine-C3,5-H), 2.90–3.07 (m, 4H, piperdine-C2,6-H), 3.93 (s, 1H, C1-H), 4.06 (s, 1H, C4-H), 7.20–7.32 (m, 1H, C6-H), 7.34 (d, 1H, J = 6.35 Hz, C5-H), 7.44 (d, 1H, J = 6.35 Hz, C7-H), 7.90 (t, 1H, J = 7.90 Hz, 3-NO2-C6H4-C5-H), 8.34 (d, 1H, J = 7.90 Hz, 3-NO2-C6H4-C6-H), 8.53 (d, 1H, J = 7.90 Hz, 3-NO2-C6H4-C4-H), 8.54 (s, 1H, 3-NO2-C6H4-C2-H), 8.69 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, ppm): 22.45 (piperdine-C-4), 22.92 (piperdine-C-3,5), 38.12 (piperdine-C-2,6), 44.11 (C-1) 56.51 (C-4), 122.22, 123.95, 124.64, 125.51, 127.59, 128.88, 130.94 (d, J = 11 Hz, C-6), 131.00, 134.33 (d, J = 212 Hz, C-F), 135.39, 136.95, 138.68, 148.39, 156.37 (ArCs), 191.73 (C=O). Anal. Calcd. (%) for C22H20FN3O3S (425): C, 62.10, H, 4.74, N, 9.88. Found: C, 62.37, H, 4.90, N, 10.09.
2.1.7. General procedure for synthesis of 3,4-dichloro-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro[1]benzothieno[2,3-c]pyridine (7a) and 3-chloro-4-cyano-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro[1]benzothieno[2,3-c]pyridine (7b)
To a solution of phosphorus oxychloride (10 ml) and pyridine (2 ml), 4d or 5d (0.002 mol) was added, and the mixture was heated under reflux for 1 h. After cooling, the reaction mixture was poured cautiously to the crushed ice (40 g), and the solid product was filtered, washed with water, dried, and crystallised from acetonitrile.
2.1.7.1. 3,4-Dichloro-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro[1]benzothieno[2,3-c] pyridine (7a)
Yield 87%, mp 120–122 °C, IR (KBr, cm−1): 3360 (NH), 3078 (C-H aromatic), 2947 (C-H aliphatic), 1608 (C=N), 1580 (C=C), 1531, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 4.06 (s, 1H, C1-H), 4.93 (s, 0.5 H, C4-H), 7.18–7.38 (m, 2H, C5,6-H), 7.46 (d, 1H, J = 7.80 Hz, C7-H), 7.70 (t, 1H, J = 8.00 Hz, 3-NO2-C6H4-C5-H), 8.05 (d, 1H, J = 8.00 Hz, 3-NO2-C6H4-C6-H), 8.21 (d, 1H, J = 8.00 Hz, 3-NO2-C6H4-C4-H), 8.34 (s, 1H, 3-NO2-C6H4-C2-H), 8.43 (s, 0.5H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 46.00 (C-1), 72.72 (C-4), 123.65, 123.85, 128.81, 128.83 (d, J = 6 Hz, C-6), 129.12, 130.19, 130.55 (d, J = 286 Hz, C-F), 130.88, 130.99, 131.98, 135.94, 140.36, 148.37, 156.03, 158.87 (ArCs). MS: m/z (% relative abundance): 399 (M+.+4, 1.82%), 397 (M+.+2, 3.41%), 395 (M+., 7.76%) and 352 (100%). Anal. Calcd (%) for C17H9Cl2FN2O2S (395): C, 51.66, H, 2.30, N, 7.09. Found: C, 51.92, H, 2.62, N, 7.36.
2.1.7.2. 3-Chloro-4-cyano-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro[1]benzothieno [2,3-c]pyridine (7b)
Yield 79%, mp 140–142 °C, IR (KBr, cm−1): 3367 (NH), 3078 (C-H aromatic), 2958 (C-H aliphatic), 2229 (CN), 1639 (C=N), 1593 (C=C), 1531, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 4.06 (s, 1H, C1-H), 4.66 (s, 0.5H, C4-H, taut), 7.08–7.39 (m, 3H, C5,6,7-H), 7.53 (t, 1H, J = 8.00 Hz, 3-NO2-C6H4-C5-H), 7.68 (d, 1H, J = 8.00 Hz, 3-NO2-C6H4-C4-H), 7.86 (d, 1H, J = 8.00 Hz, 3-NO2-C6H4-C6-H), 7.97 (s, 1H, 3-NO2-C6H4-C2-H), 8.31 (s, 0.5H, NH, D2O exchangeable, taut). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 18.97 (C-1), 56.51 (C-4), 114.54, 124.07, 124.89, 128.57, 128.71 (d, J = 29 Hz, C-6), 130.62, 130.87, 130.98, 131.56, 133.62 (d, J = 275 Hz, C-F), 135.50, 136.59, 147.72, 151.29, 158.28, 164.17 (CN and ArCs). MS: m/z (% relative abundance): 387 (M+.+2, 14.61%), 385 (M+., 38.03%) and 95 (100%). Anal. Calcd (%) for C18H9ClFN3O2S (385): C, 56.04, H, 2.35, N, 10.89. Found: C, 56.33, H, 2.59, N, 11.08.
2.1.8. General procedure for synthesis of 3-substitutedamino-4-chloro-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro[1]benzothieno[2,3-c]pyridines (8a–c) and 3-substitutedamino-4-cyano-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro[1]benzo-thieno[2,3-c]pyridines (8d–f)
A mixture of 7a or 7b (0.001 mol), an appropriate secondary amine (0.001 mol), and triethylamine (two drops) in absolute ethanol (25 ml) was heated under reflux for 5 h. The solvent was concentrated under reduced pressure, and the precipitated solid was filtered, washed with water, dried, and crystallised from acetonitrile.
2.1.8.1. 4-Chloro-3–(4-ethylpiperzin-1-yl)-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro [1]benzothieno[2,3-c]pyridine (8a)
Yield 55%, mp 221–223 °C, IR (KBr, cm−1): 3390 (NH), 3074 (C-H aromatic), 2935 (C-H aliphatic), 1608 (C=N), 1580 (C=C), 1531, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 0.99 (t, 3H, J = 7.16 Hz, CH3-CH2), 2.39 (q, 2H, J = 7.16 Hz, CH3-CH2), 2.54–2.60 (m, 4H, piperazine-C3,5-H), 3.02–3.09 (m, 4H, piperazine-C2,6-H), 4.06 (s, 1H, C1-H), 4.23 (s, 0.5H, C4-H, taut), 7.18–7.38 (m, 2H, C5,6-H), 7.42–7.52 (m, 1H, C7-H), 7.63–7.72 (m, 1H, 3-NO2-C6H4-C5-H), 7.86–7.90 (m, 1H, 3-NO2-C6H4-C6-H), 8.08 (d, 1H, J = 7.88 Hz, 3-NO2-C6H4-C4-H), 8.35 (s, 1H, 3-NO2-C6H4-C2-H), 9.02 (s, 0.5H, NH, D2O exchangeable, taut). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 12.31 (CH3), 22.02 (CH2), 27.74 (piperazine C-3,5), 49.58 (piperazine C-2,6), 51.77 (C-1), 76.80 (C-4), 118.23, 123.78, 124.47, 125.16, 126.23, 127.76, 128.88, 130.19, 130.90, 131.91, 135.04, 138.16 (d, J = 313 Hz, C-F), 139.73, 148.39, 161.22 (ArCs). Anal. Calcd (%) for C23H22ClFN4O2S (472): C, 58.41, H, 6.69, N, 11.85. Found: C, 58.59, H, 6.75, N, 11.99.
2.1.8.2. 4-Chloro-3-(4-morpholino)-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro [1]benzothieno[2,3-c]pyridine (8b)
Yield 74%, mp 175–177 °C, IR (KBr, cm−1): 3387 (NH), 3074 (C-H aromatic), 2927 (C-H aliphatic), 1608 (C=N), 1580 (C=C), 1531, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 3.02–3.10 (m, 4H, morpholine-C3,5-H), 3.70–3.78 (m, 4H, morpholine-C2,6-H), 4.07 (s, 1H, C1-H), 4.20 (s, 0.5H, C4-H, taut), 7.19–7.40 (m, 2H, C5,6-H), 7.43–7.53 (m, 1H, C7-H), 7.64–7.73 (m, 2H, 3-NO2-C6H4-C5,6-H), 8.23 (d, 1H, J = 7.00 Hz, 3-NO2-C6H4-C4-H), 8.42 (s, 1H, 3-NO2-C6H4-C2-H), 8.80 (s, 0.5H, NH, D2O exchangeable, taut). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 40.62 (morpholine C-3,5), 55.99 (morpholine C-2,6), 64.14 (C-1), 66.65 (C-4), 124.30, 125.68, 126.72, 128.11, 128.90, 129.67, 130.30, 130.88, 132.01, 132.62, 133.66, 133.85 (d, J = 238 Hz, C-F), 138.68, 139.90, 148.39 (ArCs). Anal. Calcd (%) for C21H17ClFN3O3S (445): C, 56.57, H, 3.84, N, 9.42. Found: C, 56.85, H, 3.99, N, 9.50.
2.1.8.3. 4-Chloro-3-(piperidin-1-yl)-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro [1]benzothieno[2,3-c]pyridine (8c)
Yield 68%, mp 241–243 °C, IR (KBr, cm−1): 3410 (NH), 3070 (C-H aromatic), 2935 (C-H aliphatic), 1608 (C=N), 1580 (C=C), 1527, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 0.86–0.89 (m, 2H, piperidine-C4-H), 1.11–1.22 (m, 4H, piperidine-C3,5-H), 1.54–1.63 (m, 4H, piperidine-C2,6-H), 2.97 (s, 1H, C1-H), 4.14 (s, 0.5H, C4-H, taut), 7.08–7.71 (m, 5H, C5,6,7-H and 3-NO2-C6H4-C5,6-H), 8.12 (d, 1H, J = 8.24 Hz, 3-NO2-C6H4-C4-H), 8.41 (s, 1H, 3-NO2-C6H4-C2-H), 9.01 (s, 0.5H, NH, D2O exchangeable, taut). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 14.33 (piperidine C-4), 22.62 (piperidine C-3,5), 25.92 (piperidine C-2,6), 44.11 (C-1), 54.20 (C-4), 127.92, 128.84 (d, J = 6 Hz, C-6), 129.11, 129.89, 130.88, 130.99, 132.05, 132.17, 132.51 (d, J = 300 Hz, C-F), 147.70, 148.70, 155.53, 155.34, 167.54, 169.05 (ArCs). MS: m/z (% relative abundance): 443 (M+.+2, 3.73%), 445 (M+., 8.18%) and 69 (100%). Anal. Calcd (%) for C22H19ClFN3O2S (443): C, 59.52, H, 4.31, N, 9.47. Found: C, 59.68, H, 4.40, N, 9.55.
2.1.8.4. 4-Cyano-3-(4-ethylpiperazin-1-yl)-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro [1]benzothieno[2,3-c]pyridine (8d)
Yield 63%, mp 224–226 °C, IR (KBr, cm−1): 3390 (NH), 3062 (C-H aromatic), 2823 (C-H aliphatic), 2210 (CN), 1593 (C=N), 1545 (C=C), 1531, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 1.00 (t, 3H, J = 7 Hz, CH3-CH2), 2.40 (q, 2H, J = 7 Hz, CH3-CH2), 2.70–2.95 (m, 4H, piperazine-C3,5-H), 3.01–3.21 (m, 4H, piperazine-C2,6-H), 3.94 (s, 0.5H, C4-H, taut), 4.07 (s, 1H, C1-H), 6.71 (t, 1H, J = 7.00 Hz, C6-H), 7.09 (d, 1H, J = 7.00 Hz, C5-H), 7.20–7.40 (m, 1H, 3-NO2-C6H4-C5-H), 7.50 (d, 1H, J = 8.00 Hz, 3-NO2-C6H4-C6-H), 7.69 (d, 1H, J = 8.00 Hz, 3-NO2-C6H4-C4-H), 7.85 (d, 1H, J = 7.00 Hz, C7-H), 8.20 (s, 1H, 3-NO2-C6H4-C2-H), 8.65 (s, 0.5H, NH, D2O exchangeable, taut). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 11.94 (CH3), 31.55 (CH2), 43.21 (piperazine-C-3,5), 49.34 (piperazine-C-2,6), 51.75 (C-4), 60.15 (C-1), 114.42, 120.31, 123.03, 125.86, 127.94, 128.80, 129.67, 131.06, 132.10, 132.27 (d, J = 242 Hz, C-F), 137.99, 139.21, 144.41, 149.26, 152.55, 158.97 (CN and ArCs). Anal. Calcd (%) for C24H22FN5O2S (463): C, 62.19, H, 4.78, N, 15.11. Found: C, 62.44, H, 4.89, N, 15.34.
2.1.8.5. 4-Cyano-3-(4-morpholino)-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro [1]benzothieno[2,3-c]pyridine (8e)
Yield 43%, mp 242–244 °C, IR (KBr, cm−1): 3356 (NH), 2843 (C-H aromatic), 2843 (C-H aliphatic), 2210 (CN), 1610 (C=N), 1593 (C=C), 1535, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 2.91–3.15 (m, 4H, morpholine-C3,5-H), 3.61–3.79 (m, 4H, morpholine-C2,6-H), 4.08 (s, 0.5H, C4-H, taut), 4.22 (s, 1H, C1-H), 6.65–6.90 (m, 1H, C6-H), 7.09 (d, 1H, J = 6.50 Hz, C5-H), 7.43 (d, 1H, J = 6.50 Hz, C7-H), 7.18–7.32 (m, 1H, 3-NO2-C6H4-C5-H), 7.68 (d, 1H, J = 8.00 Hz, 3-NO2-C6H4-C6-H), 7.84 (d, 1H, J = 8.00 Hz, 3-NO2-C6H4-C4-H), 8.19 (s, 1H, 3-NO2-C6H4-C2-H), 8.80 (s, 0.5H, NH, D2O exchangeable, taut). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 43.23 (morpholine-C-3,5), 48.43 (morpholine-C-2,6), 63.74 (C-4), 66.40 (C-1), 114.61, 116.93, 124.17, 124.37, 125.44, 128.24, 128.84 (d, J = 6 Hz, C-6), 130.36, 130.88, 131.56, 137.04 (d, J = 234 Hz, C-F), 138.13, 138.24, 147.56, 159.28, 160.35 (CN and ArCs). MS: m/z (% relative abundance): 436 (M+., 21.55%) and 328 (100.00%). Anal. Calcd (%) for C22H17FN4O3S (436): C, 60.64, H, 3.93, N, 12.84. Found: C, 60.38, H, 4.18, N, 13.12.
2.1.8.6. 4-Cyano-3-(piperidin-1-yl)-8-fluoro-1-(3-nitrophenyl)-1,2-dihydro [1]benzothieno[2,3-c]pyridine (8f)
Yield 50%, mp 280–282 °C, IR (KBr, cm−1): 3390 (NH), 3012 (C-H aromatic), 2947 (C-H aliphatic), 2210 (CN), 1593 (C=N), 1566 (C=C), 1531, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 0.89–1.10 (m, 2H, piperidine-C4-H), 1.20–1.60 (m, 4H, piperidine-C3,5-H), 2.50–2.90 (m, 4H, pipeirdine-C2,6-H), 3.81 (s, 0.5H, C4-H, taut), 3.92 (s, 1H, C1-H), 6.50–7.60 (m, 5H, C5,6,7-H and 3-NO2-C6H4-C5,6-H), 7.90–8.10 (m, 2H, 3-NO2-C6H4-C2,4-H), 8.35 (s, 0.5H, NH, D2O exchangeable, taut). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 22.54 (piperidine C-4), 26.01 (piperidine C-3,5), 44.03 (piperidine C-2,6), 48.44 (C-4), 55.30 (C-1), 122.74, 124.47, 125.34, 126.35, 128.25, 128.97, 130.36, 132.65 (d, J = 235 Hz, C-F), 133.62, 133.83, 135.91, 139.55, 141.04, 145.94, 147.53, 153.59 (CN and ArCs). Anal. Calcd (%) for C23H19FN4O2S (434): C, 63.58, H, 4.41, N, 12.89. Found: C, 63.71, H, 4.60, N, 13.10.
2.1.9. 9-Fluoro-7-(3-nitrophenyl)-5,6,7,13-tetrahydro[1]benzothieno[3′,2′:4,5]-pyrido[2,3-b]quinoxaline (9)
An equimolar mixture of 7a (0.39 g, 0.001 mol), o-phenylenediamine (0.1 g, 0.001 mol), and triethylamine (2 drops) in dry benzene (25 ml) was heated under reflux for 5 h. After cooling, the separated solid was filtered, washed with ethanol, dried, and crystallised from acetonitrile.
Yield 83%, mp 160–162 °C, IR (KBr, cm−1): 3375–3321 (3NH), 3078 (C-H aromatic), 2924 (C-H aliphatic), 1635 (C=N), 1589 (C=C), 1527, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 4.06 (s, 1H, C7-H), 6.53–6.55 (m, 2H, C2,3-H) 6.65–6.68 (m, 2H, C1,4-H), 7.20–7.30 (m, 1H, C11-H), 7.35 (d, 1H, J = 8.16 Hz, C12-H), 7.60–7.91 (m, 2H, C10-H and 3-NO2-C6H4-C5-H), 8.16 (s, 2H, 2NH of quinoxaline, D2O exchangeable), 8.20–8.28 (m, 1H, 3-NO2-C6H4-C6-H), 8.33 (d, 1H, J = 8.44 Hz, 3-NO2-C6H4-C4-H), 8.39 (s, 1H, 3-NO2-C6H4-C2-H), 8.64 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, δ ppm: 57.21(C-7), 117.42, 117.75, 117.85 (d, J = 20 Hz, C-11), 119.25, 120.33, 124.68, 126.16, 126.50, 127.74, 128.98, 131.06, 132.64 (d, J = 317 Hz, C-F), 134.23, 136.50, 139.40, 142.10, 145.80, 149.77, 150.11 (ArCs). MS: m/z (% relative abundance): 430 (M+., 7.00%) and 65 (100%). Anal. Calcd (%) for C23H15FN4O2S (430): Calcd, C, 63.88; H, 3.96; N, 12.96. Found: C, 64.12; H, 4.10; N, 13.24.
2.1.10. 4-Cyano-8-fluoro-3((2-fluorophenyl) amino)-1–(3-nitrophenyl)-1,2-dihydro[1]benzothieno[2,3-c]pyridine (10)
Triethylamine (0.3 g, 0.003 mol) was added to a mixture of 7b (0.38 g, 0.001 mol) and 2-fluoroaniline (0.22 g, 0.002 mol) in absolute ethanol (25 ml), and the reaction mixture was heated under reflux for 8 h. The solvent was concentrated under reduced pressure, and the resulting solid product was filtered, dried, and crystallised from benzene.
Yield 81%, mp 240–242 °C, IR (KBr, cm−1): 3348, 3410 (2NH), 3050 (C-H aromatic), 2939 (C-H aliphatic), 2229 (CN), 1610 (C=N), 1593 (C=C), 1531, 1350 (NO2). 1H-NMR (DMSO-d6, 400 MHz, δ ppm): 4.06 (s, 0.5H, C4-H, taut), 4.20 (s, 1H, C1-H), 4.40 (s, 1H, NH, D2O exchangeable), 6.75–6.85 (m, 1H, C6-H), 6.89 (t, 1H, J = 7.50 Hz, 2-F-C6H4NH2-C4-H), 7.05–7.18 (m, 1H, C5-H), 7.22 (t, 1H, J = 7.50 Hz, 2-F-C6H3NH2-C5-H), 7.32–7.39 (m, 1H, 2-F-C6H4NH2-C6-H), 7.44 (d, 1H, J = 7.50 Hz, 2-F-C6H3NH2-C3-H), 7.68 (d, 1H, J = 8.40 Hz, C7-H), 7.71–7.78 (m, 1H, 3-NO2-C6H4-C5-H), 7.87 (d, 1H, J = 8.40 Hz, 3-NO2-C6H4-C6-H), 7.93–8.00 (m, 1H, 3-NO2-C6H4-C4-H), 8.31 (s, 1H, 3-NO2-C6H4-C2-H), 8.46 (s, 0.5H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz, δ ppm): 45.82 (C-4), 52.38 (C-1), 116.05, 124.09, 124.92, 125.53, 127.93, 128.71 (d, J = 24 Hz, C-6), 128.83, 128.88, 129.71, 130.01, 130.64, 130.90, 131.00, 131.57, 133.77 (d, J = 305 Hz, C-F), 135.30, 135.52, 136.61, 136.76, 140.59, 147.73, 151.32 (CN and ArCs). Anal. Calcd (%) for C24H14F2N4O2S (460): C, 62.60, H, 3.06, N, 12.17. Found: C, 62.47, H, 3.28, N, 12.06.
2.2. Anticancer activity
2.2.1. Measurement of cytotoxic activity
The cytotoxic activity of the twenty-seven newly synthesised compounds was evaluated using the NCI disease-oriented human cell lines screening assay. The cytotoxic assay was performed according to the protocol of the Drug evaluation branch, NCI, Bethesda, MD, USA. The new compounds were evaluated against a panel of 60 cancer cell lines derived from leukaemia, melanoma, lung, colon, CNS, ovarian, renal, prostate, and breast cancers at a single concentration of 10−5 M. The cultures were incubated for 48 h before performing the sulforhodamine (SRB) protein assay as described previouslyCitation30,Citation31. The compound with significant cytotoxic activity was further evaluated at five different concentrations ranging from 10−4 to 10−8 M. Cell viability and growth were estimated, and the results were reported as a percentage growth of the treated cells vs. untreated control cells. The molar concentration that produced a 50% decrease in cell growth (GI50) was calculated.
2.2.2. MTT cytotoxicity assay protocol
The MTT method for monitoring in vitro cytotoxicity was used with multiwell plates. Three human cell lines, namely prostate cancer cell line (PC-3), renal cancer cell line (UO-31), and breast cancer cell line (MCF-7) were obtained from the American Type Culture Collection ATCC, Manassas, VA, USA). The cells were trypsinized and seeded in 96 well plates with a density of 1.2 − 1.8 × 10,000 cells/well for 24 h at 37 °C. After treating the cells with five concentrations of compound 5c (0.01, 0.1, 1, 10, 100 µM), an MTT solution (Sigma Co., St. Louis, MO, USA) (5 mg/mL in phosphate buffer solution) was applied to each well and left for 48 h. The plates were cultivated in 5% CO2 for 4 h at 37 °C. Successively, the remaining formazan crystals were dissolved in 100 µl of DMSO, and the absorbance was determined with the ROBONIK P2000 spectrophotometer at a wavelength of 570 nm. The obtained values were analysed using Gen5 software (BioTek, UK). The proliferation percentage in each treated cell line was standardised based on their control wells. Both tests have been conducted in triplicate. The IC50 values were calculated using sigmoidal dose-response curve fitting models. For best results, cells in the log phase of growth were employed, and the final cell number did not exceed 106 cells/cm2. Each test included a blank containing a complete medium without cells.
2.2.3. In vitro cytotoxicity on WI-38 human cell line protocol
Cell Line cells were obtained from American Type Culture Collection and were cultured using DMEM (Invitrogen/Life Technologies) supplemented with 10% FBS (Hyclone), 10 µg/ml of insulin (Sigma), and 1% penicillin-streptomycin. All of the other chemicals and reagents were obtained from Sigma or Invitrogen. Plate cells were incubated (cells density 1.2–1.8 × 10,000 cells/well) in a volume of 100 µL complete growth medium and 100 ul of the tested compound per well in a 96-well plate for 24 h before the MTT assay. The cultures were removed from the incubator into a laminar flow hood or other sterile work area followed by the addition of reconstituted MTT in an amount equal to 10% of the culture medium volume, then incubated for an additional 2 h. After the incubation period, cultures were removed from the incubator, and the resulted formazan crystals were dissolved by adding an amount of MTT solubilisation solution [M-8910] equal to the original culture medium volume, then the absorbance at a wavelength of 570 nm was measured. Finally, the IC50 of the tested compound compared to the reference was calculated using GraphPad Prism software.
2.2.4. Cyp17 Enzyme inhibition in mice prostate cancer model
2.2.4.1. Animals and materials
Animals’ treatment protocol was approved by the Faculty of Pharmacy, Cairo University Animal Rights Committee (OC 2740). In all tests, adequate considerations were adopted to reduce the pain or discomfort of animals.
A total of 16 mice from the TRAMP/FVB background (Sprague-Dawley, male, average 45 days, average weight 120 gm, mice with prostate cancer) were taken and divided into 2 groups with 8 animals in each group. One group of mice was given a drug in 0.2 ml of DMSO and phosphate-buffered saline (1:10) ratio (5 mg/kg body weight, i.p., 5 days a week). The control group animals were treated with 0.2 ml of DMSO and phosphate-buffered saline (1:10) ratio. A total of eight mice, four animals from each group, were examined at 13 weeks of age, and the remaining animals from each group were examined at 20 weeks for prostate tumour development. Treatment was stopped 24 h before killing the animals.
2.2.4.2. ELISA assay for CYP17 enzyme
BioSource 2704060 96 assay kit is a sandwich enzyme immunoassay for in vitro quantitative measurement of CYP17 enzyme activity in mouse tissue homogenates, cell lysates, cell culture supernatant, and other biological fluidsCitation32. Tested tissues were rinsed in ice-cold PBS to remove excess blood and weighed before homogenisation. The tissues were minced and homogenised in fresh lysis buffer with a glass homogeniser. (catalog: IS007, different lysis buffer needs to be chosen based on subcellular location of the target protein) (w:v = 1:20–1:50, e.g. 1 ml lysis buffer is added in 20–50 mg tissue sample) The resulting suspension was sonicated with an ultrasonic cell disrupter till the solution was clarified. The homogenates were centrifuged for 5 min at 10,000 × g, and the supernatant was collected. An Aliquot was assayed and stored at ≤ −20 °C.
The assay sample and standard were incubated together with CYP-HRP conjugate in an appropriate number of microplates pre-coated with an antibody specific for the CYP17 enzyme for 1 h. After the incubation period, the wells were decanted and washed five times. The wells were then incubated with a substrate for the HRP enzyme. A blue-colored complex was produced as a result of an enzyme-substrate reaction. Finally, a stop solution (2N hydrochloric acid solution) was added to stop the reaction, turning the solution yellow. Colour intensity was measured spectrophotometrically at 450 nm in a microplate reader. A standard curve was plotted between the intensity of the colour vs. the concentration of standards. The CYP17 concentration in each sample was interpolated from this standard curve.
Assay procedures: Wells for a standard, blank, and sample were prepared. Dilutions of standard, blank, and sample (100 µL each) were added into the appropriate wells, covered with the plate sealer, then incubated for 1 h at 37 °C. The liquid was removed from each well without washing. Detection reagent A (100 µL) was added to each well, covered with the plate sealer, and incubated for 1 h at 37 °C. The solution was aspirated and washed three times with 350 µL of wash solution to each well. After the last wash, any remaining wash buffer was removed by aspirating or decanting. Detection reagent B (100 µL) was added to each well, covered with the plate sealer, and incubated for 30 min at 37 °C. The aspiration/washing process has been repeated a total of 5 times as before. Substrate solution (90 µL) was added to each well, covered with a plate sealer, and incubated for 10–20 min at 37 °C in the dark. The liquid turns blue by the addition of substrate solution. Stop solution (2 N hydrochloric acid solution, 50 µL) was added to each well till the liquid turns yellow. The absorbance was measured immediately at 450 nm.
2.2.5. Testosterone assay
The BioVendor mouse/rat testosterone ELISA assay is a competitive immunoassay for the measurement of testosterone in rat and mouse serum or plasma based on the principle of competitive bindingCitation33. An unknown amount of testosterone present in the sample (plasma or serum) and a known amount of testosterone conjugated to horseradish peroxidase compete for the binding sites of testosterone antiserum coated to the wells of a microplate. The sample was incubated with the microplate for 1 h, then washed four times. The HRP substrate solution was added followed by the stop solution. Colour change has been observed, and the optical density was measured at 450 nm.
Assay procedures: Sufficient number of microplate wells were prepared to accommodate standard and samples in duplicates. The standard, sample, and control (10 µL each) were dispensed with new disposable tips into appropriate wells. Incubation buffer 100 µL was dispensed into each well. Afterward, enzyme conjugate (50 µL) was added to each well and incubated for 60 min at room temperature on a microplate mixer. The content of the wells was discarded, and the wells were rinsed 4 times with diluted wash solution (300 µL per well). The substrate solution (200 µL) was added to each well and incubated in the dark without shaking for 30 min. The reaction was stopped by adding a stop solution (2 N hydrochloric acid solution, 50 µL) to each well. The absorbance of each well was determined at 450 nm.
2.2.6. Cell cycle analysis
The prostate PC-3 cells were placed in a six-well plate at 1 × 105 conc. of cells/well, then incubated for 24 h. The cells were treated with 2.34 µM of compound 5c or (0.1% DMSO) for 24 h. Thereafter, cells were collected and fixed for 12 h using ice-cold 70% ethanol at 4 °C. Then, ethanol was removed, and the cells were washed with cold Phosphate Buffer Saline (PBS) and incubated for 30 min at 37 °C in 0.5 ml of PBS. The cells were stained for 30 min with propidium iodide in the dark. The flow cytometer was used to detect DNA contentsCitation34.
2.2.7. Annexin V-FITC assay
PC-3 cells were placed in a 6-well plate at 1 × 105 conc. of cells per well, then incubated for 24 h. Afterward, the cells were treated with 2.34 µM of compound 5c or (0.1% DMSO) for 24 h, then harvested, washed with PBS, and stained with annexin V-FITC and PI in binding buffer (10 µM HEPES, 140 µM NaCl, and 2.5 µM CaCl2 at pH 7.4) for 15 min at room temperature in the dark, then analysed by the flow cytometerCitation35.
3. Results and discussion
3.1. Chemistry
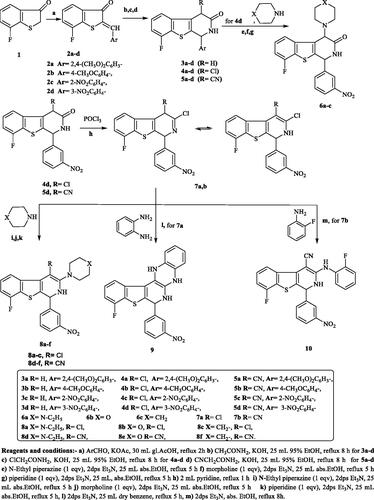
The synthetic pathways of the target compounds are illustrated in Scheme 1. Herein, the arylidene derivatives 2a–d are the key intermediates for the synthesis of the designed [1]benzothieno[2,3-c]pyridine derivatives 3a–d, 4a–d, and 5a–d. Compounds 2a–d were prepared by condensation of 7-fluoro[1]benzothiophen-3(2H)-one (1) with some aromatic aldehydes in glacial acetic acid and anhydrous potassium acetate as a catalyst, following previously reported procedureCitation36. After that, cyclisation of the produced α,β-unsaturated ketones 2a–d with acetamide, 2-chloroacetamide, or 2-cyanoacetamide in the presence of potassium hydroxide as a catalyst afforded the target compounds 3a–d, 4a–d, and 5a–d, respectively, according to a reported procedureCitation37. Previous studies in this field showed that compounds containing 3-nitrophenyl moiety at the 4-position of 1,2-dihydro[1]bezothienopyridine ring expressed promising anticancer activityCitation29. On this basis, compounds 4d and 5d were chosen in the preparation of other new bezothienopyridine derivatives hoping to obtain compounds potentially active as anticancer agents. The reaction of 4d with several secondary amines in the presence of triethylamine afforded 6a–c. On the other hand, the preparation of 7a,b has been accomplished via chlorination of 4d or 5d with excess phosphorus oxychloride in pyridine, adopting the reported procedureCitation38. Furthermore, amination of 7a,b with a number of secondary amines afforded 3-substitutedamino-4-chloro[1]benzothieno[2,3-c]pyridines 8a–c and 3-substituted amino-4-cyano-[1]benzothieno[2,3-c]pyridine derivatives 8d–f. Moreover, the reaction of 7a with o-phenylenediamine in the presence of triethylamine produced the quinoxaline derivative 9. Finally, heating of 7b with o-fluoroaniline in absolute ethanol and triethylamine as a catalyst produced 4-cyano-8-fluoro-3[(2-fluorophenyl) amino][1]benzothieno[2,3-c]pyridine 10 (Scheme 1).
Scheme 1. The synthetic pathways and reagents for the preparation of the target compounds 2a–d, 3a–d, 4a–d, 5a–d, 6a–c, 7a,b, 8a–f, 9, and 10. Reagents and conditions: (a) ArCHO, KOAc, 30 mL gl.AcOH, reflux 2h, (b) CH3CONH2, KOH, 25 mL 95% EtOH, reflux 8 h for 3a–d, (c) ClCH2CONH2, KOH, 25 mL 95% EtOH, reflux 8 h for 4a–d, (d) CNCH2CONH2, KOH, 25 mL 95% EtOH, reflux 8 h for 5a–d, (e) N-Ethyl piperazine (1 eqv), 2dps Et3N, 25 mL abs.EtOH, reflux 5 h, (f) morpholine (1 eqv), 2dps Et3N, 25 mL abs.EtOH, reflux 5 h, (g) piperidine (1 eqv), 2dps Et3N, 25 mL, abs.EtOH, reflux 5 h, (h) 2 mL pyridine, reflux 1 h, (i) N-Ethyl piperazine (1 eqv), 2dps Et3N, 25 mL abs.EtOH, reflux 5 h, (j) morpholine (1 eqv), 2dps Et3N, 25 mL abs.EtOH, reflux 5 h, (k) piperidine (1 eqv), 2dps Et3N, 25 mL abs.EtOH, reflux 5 h, (l) 2dps Et3N, 25 mL dry benzene, reflux 5 h, and (m) 2dps Et3N, abs. EtOH, reflux 8 h.
1H-NMR spectra of compounds 2a,c,d revealed the appearance of benzylidene proton (=CH) in the range of 8.26–8.70 ppm.
IR spectra of the target derivatives 3a–d, 4a–d, and 5a–d, showed tautomeric OH, NH and C=O bands. Compounds 5a–d displayed sharp bands at 2206–2210 cm−1, confirming the presence of CN function. 1H-NMR spectra of 3a–d, 4a–d, and 5a–d, demonstrated the disappearance of benzylidene proton signal. On the other hand, they revealed a singlet signal in a range of 3.78–4.75 ppm corresponding to C1-H proton and a D2O exchangeable singlet signal attributable to NH proton. In addition, 1H-NMR spectra of compounds 3a–d showed a singlet signal at about 3.00–3.93 ppm corresponding to CH2 protons, while the spectra of compounds 4a–d and 5a–d revealed a singlet signal at about 3.84–4.21 ppm corresponding to C4-H. Finally, tautomeric OH singlet signal integrated for half proton was observed in compounds 3d, 4a, and 5a, indicating the presence of two tautomers. 13C-NMR spectra of the target compounds revealed two signals in the aliphatic region corresponding to carbons at the position 1 and 4. The characteristic C=O signal was displayed in the range of 177.52–192.27 ppm. Mass spectra of compounds 3d, 4d, and 5d were consistent with their structures. Compound 4d showed M+ and M+ +2 peaks confirming the presence of the Cl atom.
Further, IR spectra of 6a–c showed NH str. and C=O str. bands. The 1H-NMR spectrum of 6a displayed triplet and quartette signals confirming the ethyl group of N-ethylpiperazine. Moreover, the spectrum showed two multiplets corresponding to the piperazine ring protons. However, the 1H-NMR spectrum of 6b revealed two multiplet signals of the morpholine ring protons. On the other hand, compound 6c showed three multiplet signals at about 1.50–1.57, 1.58–1.70, and 2.90–3.07 ppm corresponding to piperidine-C4-H, piperidine-C3,5-H, and piperidine-C2,6-H protons, respectively. Further structural evidence steamed from the 13C-NMR spectra of 6a, 6b, and 6c, which were consistent with the exact structure of these compounds. The 13C-NMR spectrum of 6a illustrated six signals of aliphatic carbons corresponding to ethyl group carbons, ethylpiperazine-C-3,5, ethylpiperazine-C-2,6, C-1, and C-4. Also, the 13C-NMR spectrum of 6b showed four signals in the aliphatic region, attributable to morpholine-C-3,5, morpholine-C-2,6, C-1, and C-4. However, the 13C-NMR spectrum of 6c demonstrated five signals in the aliphatic area for piperidine-C-4, piperidine-C-3,5, C-1, piperidine-C-2,6, and C-4. All compounds displayed a carbonyl carbon. Moreover, the mass spectrum of compound 6b demonstrated its molecular ion peak with the absence of M+.+2 peak of its chloro precursor 4d.
IR spectrum of the chloro derivatives 7a and 7b lacked the C=O str. band of the precursors 4d and 5d. On the other hand, they displayed NH absorption bands. Their 1H-NMR spectra demonstrated a singlet signal at 4.06 corresponding to C1-H. In addition, compounds 7a and 7b exhibited two singlets, each integrated for half proton, attributed to C4-H tautomer, as well as a D2O exchangeable signal of NH proton. Their 13C-NMR spectra showed two signals corresponding to the aliphatic carbons at C-1 and C-4. In addition, the aromatic carbons were in full correspondence to the exact structures. Mass spectra of compound 7a showed M+., M+.+2, and M+.+4 isotopes, confirming the presence of two Cl atoms. On the other hand, the presence of one chlorine atom in 7b was approved by the presence of M+. and M+.+2 peaks.
The 3-substituted amino derivatives 8a–f revealed an NH absorption band. Besides, compounds 8d–f showed the band of CN function.1H-NMR spectra of compounds 8a and 8d showed triplet and quartette signals of the ethyl group protons of N-ethylpiperazine. In addition, two multiplets of the piperazine-C3,5-H and piperazine-C2,6-H were observed. Besides, two singlet signals integrated as half proton each were attributed to C4-H and the D2O exchangeable singlet signal of NH. 1H-NMR spectra of compounds 8b and 8e demonstrated two multiplets corresponding to morpholine-C3,5-H and morpholine-C2,6-H, in addition to three singlet signals for C1-H, C4-H, and a D2O exchangeable singlet signal of NH. The signals for C4-H and NH were integrated for half proton, each attributed to the presence of two tautomers. On the other hand, the 1H-NMR of 8c and 8f showed three multiplets of piperidine-C4-H, piperidine-C3,5-H, and piperidine-C2,6-H. Besides that, the spectra showed two singlets; for C1-H and for C4-H (integrated for half proton) in addition to the NH D2O exchangeable singlet signal. 13C-NMR spectra of compounds 8a and 8d illustrated the carbons of the ethyl group of ethylpiperazine as well as the piperazine ring carbons in the aliphatic region. The number of the other carbons was consistent with the proposed structure. Further, 13C-NMR spectra of compounds 8b and 8e demonstrated signals of morpholine C-3,5 morpholine C-2,6. The position and the total number of the signals were consistent with the proposed structures. The piperidine ring in compounds 8c and 8f exhibited three signals in the aliphatic region, which were attributed to piperidine C-4, piperidine C-3,5, and piperidine C-2,6. Other carbons in these compounds were observed in the expected chemical shift. The mass spectra of compounds 8c and 8e confirmed the presence of their molecular ion peaks. Furthermore, 8c showed the M+.+2 peak, indicating the existence of one chlorine atom.
The 1H-NMR spectrum of the quinoxaline derivative 9 revealed D2O exchangeable signals corresponding to two NH protons of the quinoxaline ring and pyridine-NH proton. Also, the 13C-NMR spectrum showed C-7 at 57.21 ppm, and the total number of the signals was consistent with the proposed structure. Moreover, the mass spectrum demonstrated the molecular ion peak with the absence of a peak corresponding to M+.+2, confirming the absence of the Cl atom of the precursor, which ascertains ring closure to the corresponding quinoxaline.
On the other hand, the IR spectrum of compound 10 showed 2NH absorption bands and the CN band at 2229 cm−1. Its 1H-NMR spectrum demonstrated two singlet signals corresponding to C1-H and C4-H (integrated for half proton), in addition to two D2O exchangeable signals (integrated for half proton) for 2NH protons. The 13C-NMR spectrum of 10 demonstrated two signals in the aliphatic region for C-4 and C-1, which ensures the existence of the compound in two tautomeric forms.
3.2. Cytotoxic activity
3.2.1. In vitro anticancer activity against a panel of 60 human tumour cell lines
Herein, twenty-seven newly synthesised compounds; 2a,c, 3a–d, 4a–d, 5a–d, 6a–c, 7a,b, 8a–f, 9, and 10 were evaluated by National Cancer Institute (NCI, USA) at a single dose (10−5 M) against 60 different human cell lines, representing leukaemia, melanoma, lung, colon, CNS, ovary, kidney, prostate and breast cancer. The growth inhibition percentages for the tested compounds were obtained from a single dose screening, and the mean growth inhibition percent of the treated cells compared to the untreated control cells was calculated for each compound (). The results revealed that compounds 5a–c exhibited prominent anticancer activity against almost all human cancer cell lines, with mean growth inhibition ranging from 46.88 to 52.52%.
Table 1. Growth inhibition (%) obtained from a single dose (10−5 M) of the tested compounds 2a,c, 3a–d, and 4a–c.
Table 2. Growth inhibition (%) obtained from a single dose (10−5 M) of the tested compounds 4d, 5a–d, 6a–c, and 7a.
Table 3. Growth inhibition (%) obtained from a single dose (10−5 M) of the tested compounds 7b, 8a–f, 9, and 10.
Table 4. Mean growth inhibition (%) for the tested compounds 2a,c, 3a–d, 4a–d, 5a–d, 6a–c, 7a,b 8a–f, 9, and 10.
3.2.2. Determination of GI50, IC50, TGI, and LC50 on full NCI-60 cell panel
Compound 5c was further selected by National Cancer Institute (NCI, USA) to be evaluated against 60 cell lines at 5 dose concentration levels (0.01, 0.1, 1, 10, and 100 µM). The results were exhibited in terms of four response parameters: median growth inhibition (GI50, the compound’s concentration that causes a 50% decrease in net cell growth), half-maximal inhibitory concentration (IC50, the concentration of drug which exhibited 50% cell viability) , total growth inhibition (TGI, the compound’s concentration leading to total inhibition of cell growth) and median lethal concentration (LC50, the compound’s concentration causing a net 50% loss of initial cells at the end of the incubation period) as well as mean graph midpoints (MG-MID) to obtain an average activity parameter overall tested cell lines (). The in-vitro screening revealed that compound 5c exhibited potent broad-spectrum anticancer activity against almost all human cancer cell lines showing GI50 range between 4 nM to 37 µM with MG-MID GI50, LC50, and TGI values of 0.21, 0.03, and 0.01 µM, respectively. Moreover, compound 5c exerted GI50 at a submicromolar concentration (<1 µM) in 13 tested cancer cell lines.
Table 5. GI50, IC50, LC50, and TGI of compound 5c on 60 cancer cell lines.
3.2.3. In vitro MTT cytotoxicity assay
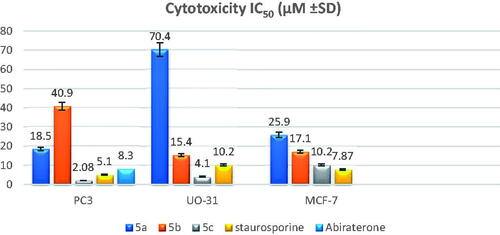
Compounds 5a–c were subjected to MTT cytotoxicity assays based on the results of one dose screening, particularly against prostate cancer (PC-3), renal cancer (UO-31), and breast cancer (MCF-7) cell lines, and their IC50 was determined using staurosporine as the reference drug. The mean estimations of three-fold experiments are represented in (). Compound 5c showed higher potency than the reference drug staurosporine against the PC-3 and UO-31 cell lines, but it exhibited half potency against the MCF-7 cell line. On the other hand, compounds 5a,b generally showed moderate activity against the three cancer cell lines, except for compound 5a which exhibited weak activity against the UO-31 cell line, compared to the reference drug. Furthermore, compound 5c demonstrated fourfold the potency of the abiraterone reference drug against the PC-3 cell line (IC50 8.3 M).
Figure 3. Graphical representation of IC50 of the tested compounds 5a–c compared to staurosporine as a reference standard.
3.2.4. In-vitro cytotoxicity on WI-38 human cell line
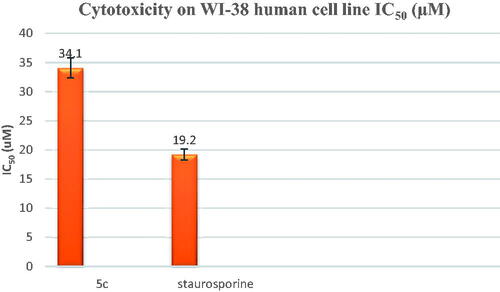
Compound 5c was further evaluated against the WI-38 human cell line (Normal cell composed of fibroblasts and derived from lung tissue of a 3-month-gestation aborted female fetus). The tested compound 5c showed low cellular cytotoxicity with an IC50 of 34.1 µM compared to the reference drug staurosporine (IC50= 19.2 µM), as shown in .
Figure 4. In-vitro cytotoxicity (IC50) of compound 5c and staurosporine on WI-38 human cell line.
3.2.5. Cyp17 enzyme inhibition assay
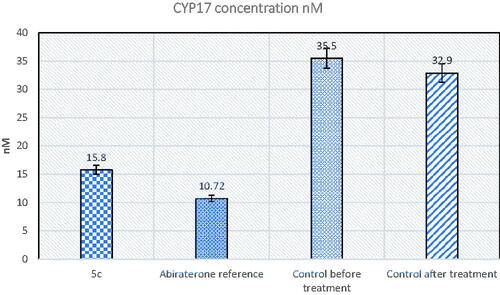
MTT cytotoxicity assay showed that compound 5c had high cytotoxic activity against PC-3 cancer cell line and was selected by NCI for further screening at a five-dose concentration level since it was the most active of the other tested derivatives. On this basis, the effect of compound 5c was further evaluated as a CYP17 enzyme inhibitor in mice with prostate cancer in comparison to the abiraterone reference drug. Afterward, in vitro ELISA quantitative measurement of CYP17 enzyme activity in mice tissue was carried out. The results showed that compound 5c was able to decrease the enzyme concentration to 15.80 nM, which was almost comparable to the reference drug ().
Figure 5. The effect of compound 5c and abiraterone reference on CYP17 enzyme compared to control.
3.2.6. Testosterone assay
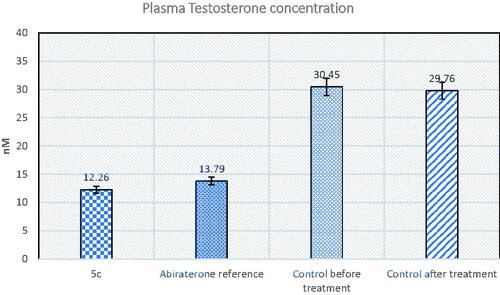
Furthermore, compound 5c was tested for its ability to inhibit testosterone production in serum mice prostate cancer models. The plasma concentration of testosterone in the samples of each group was quantified using testosterone Biovendor rat ELISA assay. Testosterone inhibitory activity was compared to abiraterone as a reference standard. The results revealed that compound 5c had 1.1 folds the potency of abiraterone ().
Figure 6. The effect of compound 5c and abiraterone reference on plasma testosterone.
3.2.7. Cell cycle analysis
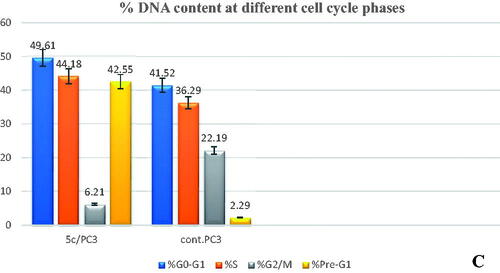
To investigate the mechanism of the antiproliferative activity of the most active compound on the cell cycle progression, the prostate cancer PC-3 cells were treated with compound 5c for 24 h then analysed. The results indicated that compound 5c increased the accumulation of cells at both G0/G1-phase (49.61%) and S-phase (44.18%) compared to 41.52 and 36.29% of the control cells, respectively (). In addition, compound 5c significantly suppressed cell accumulations in the G2/M-phase from 22.19 to 6.21%. Furthermore, compound 5c showed a marked increase in cells in Pre-G1-phase by 15.58-fold from 2.29 to 42.55%, thereby indicating the induction of apoptosis.
Figure 7. (A) Cell cycle analysis of PC-3 treated with DMSO only. (B) Cell cycle analysis of PC-3 after treatment with 5c (2.34 µM). (C) Graphical representation of effect of compound 5c on cell cycle profile of PC-3 cells.
3.2.8. Annexin V-FITC apoptosis determination
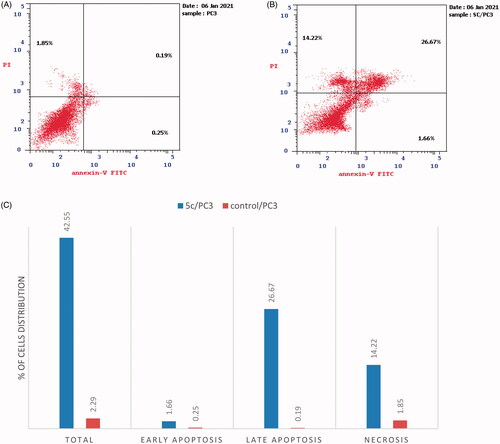
Apoptosis has significant effects on both carcinogenesis and cancer treatment. A large number of synthetic and natural compounds have been reported to be effective against several cancer diseases through the induction of apoptosis in their target cellsCitation39. Herein, the apoptotic potential of 5c was further studied using Annexin V-FITC assay in prostate cancer PC-3 cell line. The results expressed in () showed a significant increase in the early and late apoptotic cells by 6.64- and 140.36-fold, respectively, in addition to the elevation in the percentage of necrosis by 7.68-fold with a total increase by 18.58-fold as compared to the control cells.
Figure 8. (A) Apoptosis and necrosis in prostate cancer PC-3 cell line. (B) The effect of compound 5c (2.34 µM) on apoptosis and necrosis in prostate cancer PC-3 cells. (C) Graphical representation of the effect on apoptosis and necrosis of 5c on PC3 cell line in comparison with control cells.
3.2.9. In silico physicochemical properties, ADMET profiles, and drug-likeness data of 5c compared to abiraterone
Novel molecules with appropriate pharmacokinetic or pharmacodynamic features are considered potential drug candidates. The efficacy of newly synthesised compounds is determined by their biological activity as well as their physicochemical, pharmacokinetic, and drug-likeness properties. Swiss ADME Online software (www.SwissADME.ch) has been used to measure in silico ADME profile of the compound 5c in comparison with abiraterone as a reference drug. The Boiled-Egg chartCitation40 showed that 5c is expected to be highly absorbable by the gastrointestinal tract, similar to the standard abiraterone, due to its location in the human intestinal absorption (HIA) area. Also, the target compound 5c was characterised by a lack of BBB permeability, unlike abiraterone, implying that it will not reach the CNS (). Furthermore, the metabolism of compound 5c is postulated to inhibit four of the five main cytochrome P-450 (CYP) isoforms (CYP2C9, CYP1A 2, CYP2D6, CYP2C19, and CYP3A4) in the liver, indicating that 5c should be administered in a time interval with any other medications to avoid any potential drug-drug interactions ().
Figure 9. (A) The boiled-egg chart of the compound 5c and the reference abiraterone. (B) The bioavailability radar chart for compound 5c and abiraterone.
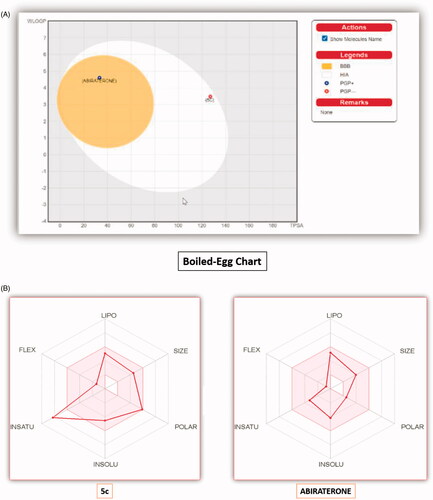
Table 6. The in silico predicted ADME profiles for compound 5c and abiraterone.
The oral bioavailability of the compound 5c and abiraterone is demonstrated in the radar chart (). It includes six important oral bioavailability parameters; SIZE (size), POLAR (polarity), INSATU (saturation), LIPO (lipophilicity), INSOLU (solubility), and FLEX (flexibility). The radar chart's pink area represents the ideal range for each value of the six parameters, and the red lines indicate the computed physicochemical features of the compound being studied. The measured physicochemical properties for 5c were located in the ideal pink area for the parameters, except for the INSATU parameter, which demonstrated a violation.
The physicochemical properties of 5c are shown in . The molecular weight of 5c is < 500 Da, proposing its easy diffusion and absorption via the cell membrane. Also, 5c is supposed to possess a strong membrane permeability, as it fulfils the ideal log p-values. Furthermore, compound 5c was displayed ideal H-bond acceptors (5) and H-bond donors (1) that enhance water solubility and allow the molecule to transmit with passive diffusion via the aqueous pores of biological membranes. Moreover, 5c includes four rotatable bonds, every single bond attached to a heavy atom, which proposes reasonable molecular flexibility. On the other hand, it showed moderate TPSA generated by the molecule's polar atoms. NRB and TPSA have revealed that 5c has interesting oral bioavailability. In general, compound 5c demonstrated almost comparable physicochemical properties to abiraterone ().
Table 7. In silico physicochemical properties for compound 5c and abiraterone.
The SwissADME Web-tool illustrated that the studied compound 5c compiled almost all rules of drug-likeness created by the leading pharmaceutical companies; Lipinski's (Pfizer),Citation41 Ghose's (Amgen)Citation42, Veber's (GSK)Citation43, Egan's (Pharmacia)Citation44, and Muegge's (Bayer)Citation45 filters. One of the most important identifications of drug-like compounds is Lipinski and Veber rules. Lipinski's rules are concerned with identifying compounds that have absorption and permeability issues, whereas Veber's rules indicate the topological polar surface area and molecular flexibility, both of which are important in determining oral bioavailability. The investigated compound 5c was completely aligned with both Lipinski and Veber’s rules. From the point of view of medicinal chemistry, compound 5c lacks PAINS (Pan Assay Interference Structures) alertsCitation46 (), which emphasises the absence of interference of 5c in any protein test, supposing that the results obtained from in vitro bioassays should be robust.
Table 8. The drug-likeness of compound 5c and the reference abiraterone drug.
3.3. Structure-activity relationship study
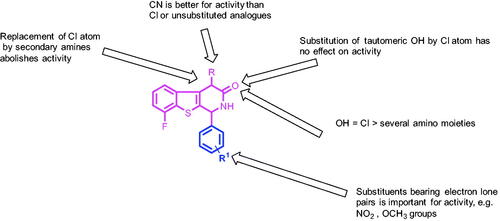
SAR studies of the tested compounds revealed that compounds 5a–c with a CN group at C-4 were of particular interest since they demonstrated potent anticancer activity with growth inhibition percentages ranging from 18.98 to 109.11% when compared to the unsubstituted and chloro substituted analogs 3a–d and 4a–d, respectively. The replacement of the Cl substituent at C-4 in compound 4d with secondary amines (compounds 6a–c) was found to be unfavourable for cytotoxicity. Furthermore, the substitution of 3-OH in 4d and 5d by a Cl atom (compounds 7a,b) does not affect activity. However, replacing Cl in compounds 7a,b with certain amino compounds in compounds 8a–f, 9, and 10 resulted in a decrease in cytotoxic activity ().
Figure 10. SAR study of the synthesised benzothienopyridine derivatives.
4. Conclusions
Several [1]benzothieno[2,3-c]pyridine derivatives were synthesised and selected by NCI, USA, for one-dose anticancer screening. Among these, compounds 5a–c, possessing CN group at C-4, demonstrated potent anticancer activity with GI50% ranging from 18.98 to 109.11%. Five dose screening of 5c exerted broad-spectrum anticancer activity with a GI50 range of 4 nM-37 µM. The IC50 of compounds 5a–c was determined against three cancer cell lines using in vitro MTT cytotoxicity assay. The results revealed that compound 5c was the most active derivative against prostate cancer PC3 cell line, with an IC50 of 2.08 µM, almost double the activity of staurosporine (IC50 = 5.10 µM) and quadruple the activity of abiraterone (IC50 = 8.3 µM) reference drugs. In addition, compound 5c showed low cellular cytotoxicity on the WI-38 human cell line with an IC50 of 34.1 µM when compared to staurosporine (IC50= 19.2 µM). Furthermore, compound 5c inhibited the CYP17 enzyme in mice prostate cancer models at concentrations ranging from 35.50 to 15.80 nM and was nearly equipotent to abiraterone in decreasing plasma testosterone levels in prostate cancer-treated mice. Moreover, compound 5c induced a significant disruption in the cell cycle profile besides marked induction of apoptosis. It also exhibited significant physicochemical properties and drug-likeness via analysing ADME parameters and drug-likeness data. On this basis, [1]benzothieno[2,3-c]pyridines have proven to be an attractive chemical scaffold with potential cytotoxic properties, and compound 5c is a potentially active orally absorbed CYP17 inhibitor with lower cytotoxicity on normal cells than steroidal derivatives, such as abiraterone.
| Abbreviations | ||
| CYP17 | = | cytochrome P450 17α-hydroxylase/C17,20-lyase |
| ADME | = | absorption, distribution, metabolism, and excretion |
| NCI-USA | = | National Cancer Institute-United States of America |
| PC-3 | = | prostate cancer cell line |
| UO-31 | = | renal cancer cell line |
| MCF-7 | = | breast cancer cell line |
| PSA | = | prostate specific antigen |
| DRE | = | digital rectal exam |
| PBH | = | prostatic hyperplasia |
| ADT | = | androgen deprivation therapy |
| AR | = | androgen receptors |
| m-CRPC | = | metastatic castrate-resistant prostate cancer |
| ELISA | = | enzyme-linked immunoassay |
| MTT | = | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide |
| CNS | = | central nervous system |
Supplemental Material
Download PDF (9.7 MB)Acknowledgements
The authors are grateful to all members of the National Cancer Institute, USA, for carrying out the anticancer screening. The authors thank E. Rashwan, Head of the Confirmatory Diagnostic Unit VACSERA-EGYPT, for carrying out in-vivo CYP17 ELISA assay and in-vivo plasma testosterone assay in mice prostate cancer models.
Disclosure statement
The authors declare that they have no competing interests.
References
- Kaninjing ET, Dagne G, Atawodi SE, Alabi A. Modifiable risk factors implicated in prostate cancer mortality and morbidity among west African men. J Health Dispar 2019;5:1–13.
- Rafiemanesh H, Mehtarpour M, Khani F, et al. Epidemiology, incidence and mortality of lung cancer and their relationship with the development index in the world. J Thorac Dis 2016;6:1094–102.
- Carter HB, Coffey DS. The prostate: an increasing medical problem. Prostate 1990;16:39–48.
- Smith R, Andrews K, Brooks D, et al. Cancer screening in the United States, 2019: a review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J Clin 2019;69:184–210.
- Halpern JA, Oromendia C, Shoag JE, et al. Use of digital rectal examination as an adjunct to prostate specific antigen in the detection of clinically significant prostate cancer. J Urol 2018;199:947–53.
- Chiu PKF, Roobol MJ, Teoh JY, et al. Prostate health index (PHI) and prostate-specific antigen (PSA) predictive models for prostate cancer in the Chinese population and the role of digital rectal examination-estimated prostate volume. Int Urol Nephrol 2016;48:1631–7.
- Saini S. PSA and beyond: alternative prostate cancer biomarkers. Cell Oncol 2016;39:97–106.
- Chan JM, Stampfer MJ, Giovannucci EL. What causes prostate cancer? A brief summary of the epidemiology. Sem Cancer Biol 1998;8:363–273.
- Nam RK, Cheung P, Herschorn S, et al. Incidence of complications other than urinary incontinence or erectile dysfunction after radical prostatectomy or radiotherapy for prostate cancer: a population-based cohort study. Lancet Oncol 2021;15:223–31.
- Sooriakumaran P, Nyberg T, Akre O, et al. Comparative effectiveness of radical prostatectomy and radiotherapy in prostate cancer: observational study of mortality outcomes. BMJ 2014;348:g1502–13.
- Boevé L, Hulshof M, Vis A, et al. Effect on survival of androgen deprivation therapy alone compared to androgen deprivation therapy combined with concurrent radiation therapy to the prostate in patients with primary bone metastatic prostate cancer in a prospective randomised clinical trial: data from the HORRAD trial. Eur. Urol 2019;75:410–8.
- Rusthoven CG, Jones BL, Flaig TW, et al. Improved survival with prostate radiation in addition to androgen deprivation therapy for men with newly diagnosed metastatic prostate cancer. J Clin Oncol 2016;34:2835–42.
- Sun M, Choueiri TK, Hamnvik OPR, et al. Comparison of gonadotropin-releasing hormone agonists and orchiectomy: effects of androgen-deprivation therapy. JAMA Oncol 2016;2:500–7.
- Shim M, Bang W, Oh C, et al. Effectiveness of three different luteinizing hormone-releasing hormone agonists in the chemical castration of patients with prostate cancer: goserelin versus triptorelin versus leuprolide. Investig Clin Urol 2019;60:244–50.
- Ahmed A, Ali S, Sarkar FH. Advances in androgen receptor targeted therapy for prostate cancer. J Cell Physiol 2014;229:271–6.
- Teo M, Rathkopf D, Kantoff P. Treatment of advanced prostate cancer. Annu Rev Med 2019;70:479–99.
- Gillessen S, Attard G, Beer T, et al. Re: Silke Gillessen, Gerhardt Attard, Tomasz M. Beer, et al. Management of Patients with Advanced Prostate Cancer: Report of the Advanced Prostate Cancer Consensus Conference 2019. Eur Urol. 2020;77: 508-47. Eur Urol 2020;78:e201–547.
- Alex AB, Pal SK, Agarwal N. CYP17 inhibitors in prostate cancer: latest evidence and clinical potential. Ther Adv Med Oncol 2016;8:267–75.
- Fizazi K, Tran N, Fein L, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. NEJM 2017;377:352–60.
- Njar VC, Brodie AM. Discovery and development of Galeterone (TOK-001 or VN/124-1) for the treatment of all stages of prostate cancer. J Med Chem 2015;58:2077–87.
- Bastos DA, Antonarakis ES. Galeterone for the treatment of advanced prostate cancer: the evidence to date. Drug Des Devel Ther 2016;10:2289–97.
- Dellis A, Papatsoris A. Phase I and II therapies targeting the androgen receptor for the treatment of castration resistant prostate cancer. Expert Opin Invest Drugs 2016;25:697–707.
- Smith MR, Saad F, Chowdhury S, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. NEJM 2018;378:1408–18.
- Matsunaga N, Kaku T, Itoh F, et al. C17,20-lyase inhibitors I. Structure-based de novo design and SAR study of C17,20-lyase inhibitors. Bioorg Med Chem 2004;12:2251–73.
- Ideyama Y, Kudoh M, Tanimoto K, et al. Novel nonsteroidal inhibitor of cytochrome P45017α (17α‐hydroxylase/C17-20 lyase), YM116, decreased prostatic weights by reducing serum concentrations of testosterone and adrenal androgens in rats. Prostate 1998;37:10–8.
- Wang M, Fang Y, Gu S, et al. Discovery of novel 1,2,3,4-tetrahydrobenzo[4,5]thieno[2,3-c]pyridine derivatives as potent and selective CYP17 inhibitors. Eur J Med Chem 2017;132:157–72.
- Auchus R, Sharifi N. Sex hormones and prostate cancer. Annu Rev Med 2020;71:33–45.
- Roviello G, Sigala S, Danesi R, et al. Incidence and relative risk of adverse events of special interest in patients with castration resistant prostate cancer treated with CYP-17 inhibitors: a meta-analysis of published trials. Crit Rev Oncol Hematol 2016;101:12–20.
- Mouineer A, Zaher A, El-Malah A, Sobh EA. Design, synthesis, antitumor activity, cell cycle analysis and ELISA assay for cyclin dependant kinase-2 of a new (4-aryl-6-fluoro-4H-benzo[4,5]thieno[3,2-b]pyran) derivatives. Mediterr 2017;6:165–79.
- Boyd MR. The NCI human tumor cell line (60-Cell) screen. In: Anticancer drug development guide. Totowa (NJ): Humana Press; 2004:41–61.
- Boyd MR. In vitro anticancer drug discovery screen. In: Anticancer drug development guide. Totowa (NJ): Humana Press; 1997:23–42.
- Li J, Li Y, Son C, et al. 4-Pregnene-3-one-20β-carboxaldehyde: a potent inhibitor of 17α-hydroxylase/C17, 20-lyase and of 5α-reductase. J Steroid Biochem Mol Biol 1992;3:313–20.
- Shrivastav TG, Basu A, Kariya KP. One step enzyme linked immunosorbent assay for direct estimation of serum testosterone. J Immunoass Immunoch 2003;2:205–17.
- Turky A, Bayoumi AH, Ghiaty A, et al. Design, synthesis, and antitumor activity of novel compounds based on 1,2,4-triazolophthalazine scaffold: apoptosis-inductive and PCAF-inhibitory effects. Bioorg Chem 2020;101:104019.
- Andree H, Reutelingsperger C, Hauptmann R, et al. Binding of vascular anticoagulant alpha (VAC alpha) to planar phospholipid bilayers. J Biol Chem 1990;265:4923–8.
- Dalgliesh CE, Mann FG. The comparative reactivity of the carbonyl groups in the thionaphthenquinones. Part II. The influence of substituent groups in the thionaphthenquinones. J Chem Soc 1945;5:893–909.
- Eisa MA. Synthesis of some new fused heterocyclic rings derived from 3-benzofuranone. AREJ 2006;17:76–85.
- Wang H, Wen K, Wang L, et al. Large-scale solvent-free chlorination of hydroxy-pyrimidines, -pyridines, -pyrazines and -amides using equimolar POCl3. Molecules 2012;17:4533–44.
- Joshi A, Haque N, Lateef A, et al. Apoptosis and its role in physiology. Int J Livestock Res 2017;7:33–45.
- Daina A, Zoete V. A boiled-Egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016;11:1117–21.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2012;64:4–17.
- Ghose AK, Viswanadhan VN, Wendoloski JJ. A. knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem 1999;1:55–68.
- Veber DF, Johnson SR, Cheng HY, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002;45:2615–23.
- Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem 2000;43:3867–77.
- Muegge I, Heald SL, Brittelli D. Simple selection criteria for drug-like chemical matter. J Med Chem 2001;44:1841–6.
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 2010;53:2719–40.