
Abstract
Influenza viruses represent a major threat to human health and are responsible for seasonal epidemics, along with pandemics. Currently, few therapeutic options are available, with most drugs being at risk of the insurgence of resistant strains. Hence, novel approaches targeting less explored pathways are urgently needed. In this work, we assayed a library of nitrobenzoxadiazole derivatives against the influenza virus A/Puerto Rico/8/34 H1N1 (PR8) strain. We identified three promising 4-thioether substituted nitrobenzoxadiazoles (12, 17, and 25) that were able to inhibit viral replication at low micromolar concentrations in two different infected cell lines using a haemagglutination assay. We further assessed these molecules using an In-Cell Western assay, which confirmed their potency in the low micromolar range. Among the three molecules, 12 and 25 displayed the most favourable profile of activity and selectivity and were selected as hit compounds for future optimisation studies.
Graphical abstract
1. Introduction
Influenza is a respiratory infectious disease characterised by high morbidity and mortality especially in at-risk subjects. Influenza viruses (IVs) belong to the family of Orthomyxoviridae and are characterised by a segmented, single-stranded, negative-sense RNA genomeCitation1. Type A IVs infect a broad range of animals, including humans, while type B and C IVs are predominantly human pathogensCitation2. Type A and B IVs are both responsible for seasonal epidemics; in addition, type A IV has caused pandemics in the past leading to millions of deaths and hospitalisation. These include the 1918–1920 “Spanish” fluCitation3 and the 2009 (H1N1) pandemicCitation4,Citation5 which was the result of a triple reassortment of bird, swine, and human IVs that further combined with a Eurasian pig IVCitation6, leading to the term “swine flu”.
The therapeutic options currently available for IV infections include vaccination and antiviral drugs used to prevent and treat them, respectivelyCitation7,Citation8. To date, three classes of antivirals have been approved for clinical use: (i) the adamantanes, (ii) the inhibitors of the viral glycoprotein Neuraminidase (NA) and (iii) the compounds targeting the viral RNA-dependent RNA polymerase (RdRp), which represent the latest therapeutic approach to combat IVs. Adamantanes are the oldest and most affordable molecules to treat IVs and include amantadine and rimantadine ()Citation9. These compounds target the viral ion-channel Matrix (M2) protein required for virus uncoatingCitation10–12. However, both molecules possess potentially serious central nervous system side effects and nowadays most IV strains are resistant to these drugs. NA inhibitors impair the release of viral particles from infected host cells. Among them, the most used drugs are zanamivir and oseltamivir ()Citation13,Citation14 which have an improved safety profile compared to adamantanes. Nonetheless, the efficacy of these antivirals is often limited by the high antigenic variability of the virus,the huge circulation of different strains, and the development of resistance to the available drugs.
Figure 1. Most representative commercially available anti-IV drugs.
Given this situation, many lines of research have been directed towards the discovery of novel anti-influenza drugs. To this end, the RdRp has been identified as an interesting target for the inhibition of viral replication because it is an element conserved among different viral strains and is not subjected to great genetic variabilityCitation15. Drug discovery efforts finally led to two RdRp inhibitors recently approved for clinical use: baloxavir marboxil and favipiravir (). Baloxavir marboxil is a prodrug of baloxavir acid (), an inhibitor of the cap-dependent endonuclease activity of RdRp of both type A and B IVsCitation16. Favipiravir (), following activation to the corresponding ribofuranosyl-5′-triphosphate, acts as a purine nucleoside triphosphate analogue and it is thus recognised as an alternative substrate by the RdRpCitation17. Baloxavir marboxil has been licenced for the treatment and prevention of influenza in different countries, including JapanCitation18,Citation19, United States of AmericaCitation20,Citation21, and European UnionCitation22, while favipiravir is only licenced in JapanCitation23 for the emergency treatment of IV strains insensitive to current antivirals. These successful examples highlight the potential of RdRp as a drug target and, beyond marketed drugs, different groups have been researching novel inhibitors targeting either its enzymatic activity or its structural integrity.
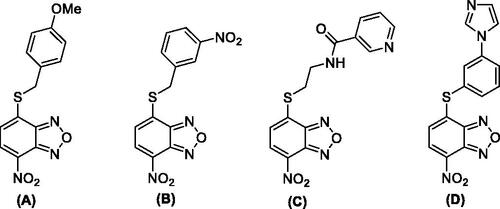
The enzyme RdRp is a heterotrimeric complex, consisting of three proteins: polymerase basic protein 1 (PB1), polymerase basic protein 2 (PB2), and polymerase acidic protein (PA). The three subunits form a multi-protein complex, with PB1 as the central core interacting with PB2 on one side and PA on the other side. Previous studies indicated that the PA/PB1 interaction is crucial for IVs replication and that it is rather conserved between the various viral strains. Consequently, the inhibition of protein–protein interactions (PPIs) represents an excellent approach for the development of new antivirals. This strategy is based on the dissociation of viral protein complexes using various types of inhibitors, including small molecules that are able to bind at the interface between the viral subunits PA and PB1. In order to identify novel PA/PB1 interaction inhibitors, Kessler and colleagues screened a large library of compounds through an ELISA-based assay. Then, a plaque reduction assay on MDCK cells infected with IV A/WSN/33 (H1N1) was used to determinate the IC50 of the best performing compoundsCitation24. This strategy led to the identification of some benzoxadiazole derivatives (A–D) endowed with anti-influenza A virus activity in the micromolar concentration range (). Nonetheless, these molecules displayed cytotoxic effects at relatively low concentrationsCitation24,Citation25. In addition, the mode of action of the compounds was not completely validated, thus raising doubts as to whether the observed effects are a direct consequence of PA/PB1 interaction disruption.
Figure 2. Most relevant benzoxadiazole derivatives (A–D) previously reported as anti-IV A agentsCitation24,Citation25.
Previously, our group has developed several 7-nitro-2,1,3-benzoxadiazole (NBD) derivatives as glutathione-S-transferase P1-1 (GSTP1-1) inhibitorsCitation26–31. The GSTP1-1 is an isoenzyme of the GST superfamily, which is selectively overexpressed in tumour cells and associated with the inhibition of apoptosis through its direct interaction with the mitogen-activated protein kinase (MAPK) named c-Jun-N-terminal Kinase (JNK1), and the scaffolding protein TNFα-receptor-associated-factor 2 (TRAF2)Citation32–34. The prototype of our NBDs as GST inhibitors was the derivative 6-((7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)thio)hexan-1-ol (NBDHEX) 1, initially identified as a potent GSTP1-1 inhibitorCitation26 although suffering from poor water solubility and low target selectivity over the isoform GSTM2-2, which is widely expressed in many non-cancerous tissues.
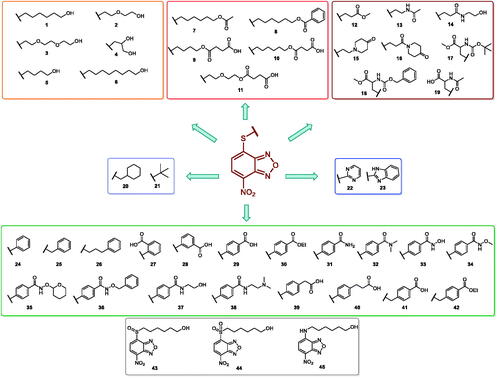
In an effort to obtain 1 derivatives with an improved pharmacological profile, we developed a library of more than 40 analogues of 1. These include alkyl or alkoxyalkyl derivatives (2–6) possessing a free primary or secondary hydroxyl function, which is also esterified in compounds 7–11. Compounds 12, 14, and 16–19 are propionic acid derivatives, while 13 and 15 possess an ethylamine side chain in which the nitrogen is either acetylated (13) or included in a piperidin-4-one ring (15). The library also includes compounds possessing small alkyl side chains (20, 21), heteroaromatic groups (22, 23), as well as aryl moieties variously functionalised at ortho, meta, and mainly para position (24–42). Finally, compounds 43–45 are analogues of 1 in which the sulphur in position 4 is oxidised to sulphinyl (43), sulphonyl (44) or replaced by an amino group (45)Citation27.
Since our GST inhibitors contain the same NBD central core of the compounds previously described as anti-IV molecules, we set out to evaluate derivatives 1–45 () as potential anti-IV agents to find derivatives possessing improved activity and/or selectivity and to draw preliminary structure–activity relationship (SAR) for this class of molecules.
Figure 3. Structures of the compounds (1–45) evaluated in the study.
2. Materials and methods
2.1. Chemistry
Unless otherwise specified, all chemicals used throughout this work were purchased from Sigma-Aldrich Srl (Milan, Italy). 1H-NMR spectra were recorded at 400 MHz on a Bruker AC 400 spectrometer (Bruker, Billerica, MA, USA); reporting chemical shifts in δ (ppm) units relative to the internal reference tetramethylsilane (Me4Si). All compounds were routinely checked by TLC and 1H-NMR. TLC was performed on aluminium-backed silica gel plates (Merck DC, Alufolien Kieselgel 60 F254, Kenilworth, NJ, USA) with spots visualised by UV light. Yields of all reactions refer to the purified products. All chemicals were of the highest purity. Mass spectra were recorded on an API-TOF Mariner by Perspective Biosystem; samples were injected by a Harvard pump using a flow rate of 5–10 µL/min, infused in the Electrospray system. Elemental analyses were performed by a PE 2400 (Perkin-Elmer, Waltham, MA) analyser and have been used to determine purity of the described compounds, which is >95%. Analytical results are within ±0.40% of the theoretical values.
Melting points were determined on a Buchi 530 melting point apparatus (Flawil, Switzerland) and are uncorrected.
2.1.1. Synthesis of 4–(4-((7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)thio)butoxy)-4-oxobutanoic acid (9)
Succinic anhydride (196 mg, 1.96 mmol, 2.0 eq.) was added to a solution of 6-((7-nitrobenzo[c][1,2,5] oxadiazol-4-yl)thio)hexan-1-ol (5) (260 mg, 0.98 mmol, 1 eq.) and 4-(dimethylamino)pyridine (DMAP) (120 mg, 0.98 mmol, 1.0 eq.) in dry dichloromethane (DCM) (9 ml) and the resulting mixture was stirred at reflux for 4 h. Upon completion, the reaction mixture was poured into a solution of 2 N HCl (22 ml) and extracted with ethyl acetate (4 × 25 ml). The organic phase was then washed with saturated sodium chloride solution (25 ml), dried over anhydrous sodium sulphate, filtered, and concentrated under vacuum. Finally, the crude residue was purified by silica gel column chromatography eluting with the appropriate mixture CHCl3/MeOH 25:1 to provide the final compound as a yellow powder. Yield: 70%. M.p.: 90–93 °C. 1H-NMR (DMSO) δ 1.809–1.838 (m, 4H, –SCH2CH2CH2CH2O–), 2.478–2.509 (m, 4H, –OCOCH2CH2COOH), 3.408 (t, 2H, –SCH2CH2CH2CH2O–), 4.100 (t, 2H, –SCH2CH2CH2CH2O–), 7.534 (d, 1H, CH benzoxadiazole ring), 8.574 (d, 1H, CH benzoxadiazole ring), 12.126–12.266 (br s, 1H, COOH). MS (ESI), m/z: 368.1 [M – H]-. Anal. (C14H15N3O7S) Calcd. (%): C, 45.53; H, 4.09; -N, 11.38; S, 8.68. Found (%) C, 45.68; H, 4.11; N, 11.30; S, 8.64.
2.1.2. Synthesis of 4–(2–(2-((7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)thio)ethoxy)ethoxy)-4-oxobutanoic acid (11)
Succinic anhydride (277.8 mg, 2.77 mmol, 3.0 eq.) was added to a solution of 2-(2-((7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)thio)ethoxy)ethan-1-ol (264 mg, 0.93 mmol, 1 eq.) (2) and 4-(dimethylamino)pyridine (DMAP) (169.5 mg, 1.39 mmol, 1.5 eq.) in dry dichloromethane (DCM) (9 ml) and the resulting mixture was stirred at reflux for 4 h. Upon completion, the reaction mixture was poured into a solution of 2 N HCl (22 ml) and extracted with ethyl acetate (4 × 30 ml). The organic phase was then washed with saturated sodium chloride solution (30 ml), dried over anhydrous sodium sulphate, filtered, and concentrated under vacuum. Finally, the crude residue was purified by silica gel column chromatography eluting with the mixture CHCl3/MeOH 20:1 to provide the final compound. Yield: 51%. M.p.: 74–76 °C. 1H-NMR (DMSO) δ 2.461–2.509 (m, 4H, COCH2CH2COOH), 3.598 (t, 2H, –SCH2CH2OCH2CH2O–), 3.673 (t, 2H, –SCH2CH2OCH2CH2O–), 3.835 (t, 2H, –SCH2CH2OCH2CH2O–), 4.148 (t, 2H, –OCH2CH2O–), 7.583 (d, 1H, CH benzoxadiazole ring), 8.579 (d, 1H, CH benzoxadiazole ring), 12.170–12.2174 (br s, 1H, COOH). MS (ESI), m/z: 384.1 [M - H]-. Anal. (C14H15N3O8S) Calcd. (%): C, 43.64; H, 3.92; N, 10.90; S, 8.32. Found (%) C, 43.80; H, 3.94; N, 10.82; S, 8.27.
2.1.3. General procedure for the synthesis of compounds 19, 22, 23 example: preparation of 4-nitro-7-(pyrimidin-2-ylthio)benzo[c][1,2,5]oxadiazole (22)
Pyridine (0.69 ml, 8.62 mmol, 3.5 eq.) and 2-mercaptopyrimidine (275.9 mg, 2.46 mmol, 1.0 eq.) were added to a solution of 4-chloro-7-nitrobenzo[c][1,2,5]oxadiazole (491.8 mg, 2.46 mmol, 1.0 eq.) in a mixture (18 ml) EtOH:H2O (0.3:1 v/v). The mixture was stirred at room temperature for 16 h. Subsequently, the suspension was filtered, and the crude solid was washed on the filter with H2O. The compound obtained was first purified by trituration with CHCl3 and then by column chromatography on SiO2 gel eluting with a mixture CHCl3/MeOH (90:1). Yield: 76%. M.p.: 144–147 °C. 1H-NMR (DMSO): δ 7.416 (t, 1H, CH pyrimidine ring), 8.288 (d, 1H, CH benzoxadiazole ring), 8.683–8.696 (m, 2H, CH pyrimidine ring), 8.719 (d, 1H, CH benzoxadiazole ring). MS (ESI), m/z: 276.0 [M + H]+. Anal. (C10H5N5O3S) Calcd. (%): C, 43.64; H, 1.83; N, 25.44; S, 11.65. Found (%) C, 43.76; H, 1.85; N, 25.37; S, 11.59.
2.1.3.1. N-acetyl-S-(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)cysteine (19)
Yield: 78%. M.p.: 166–168 °C. 1H-NMR (DMSO): δ 1.857 (s, 3H, –NHCOCH3), 3.575–3.630 (dd, 1H, –SCHHCHNH–), 3.792–3.837 (dd, 1H, –SCHHCHNH–), 4.631–4.685 (m, 1H, CH2CHNH), 7.597 (d, 1H, CH benzoxadiazole ring), 8.540 (d, 1H, CHNHCOCH3), 8.601 (d, 1H, CH benzoxadiazole ring), 13.107–13.271 (br s, 1H, COOH). MS (ESI), m/z: 325.0 [M - H]-. Anal. (C11H10N4O6S) Calcd. (%): C, 40.49; H, 3.09; N, 17.17; S, 9.83. Found (%) C, 40.60; H, 3.11; N, 17.11; S, 9.76.
2.1.3.2. 4-((1H-benzo[d]imidazol-2-yl)thio)-7-nitrobenzo[c][1,2,5]oxadiazole (23)
Yield: 81%. M.p.: 206–208 °C. 1H-NMR (DMSO): δ 7.136 − 7.417 (br m, 2H, CH benzimidazole ring), 7.430 − 7.716 (m, 3H, CH benzimidazole and benzoxadiazole rings), 8.582–8.633 (d, 1H, CH benzoxadiazole ring), 13.475 (br s, NH). MS (ESI), m/z: 314.0 [M + H]+. Anal. (C13H7N5O3S) Calcd. (%): C, 49.84; H, 2.25; N, 22.35; S, 10.23. Found (%) C, 49.90; H, 2.27; N, 22.28; S, 10.17.
2.2. Cell cultures
Madin-Darby canine kidney (MDCK) cells and human lung epithelial cells (A549) were grown in RPMI 1640 and DMEM medium, respectively, supplemented with 10% foetal bovine serum (FBS), 0.3 mg/mL glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin.
2.3. Cytotoxicity assay
The cytotoxicity of compounds was estimated on MDCK cells by using MTT [3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] (Sigma-Aldrich, St. Louis, MO, USA) assay. Briefly, cells were plated in a 96-well plate at a concentration of 2 × 104/well in RPMI-1640 without phenol red, supplemented with 10% FBS. After 24 h plating, the compounds dissolved in DMSO (concentrations range 1–30 µM) were added and cells incubated for the following 18 h. Then the medium was replaced with 50 μL of a 1 mg/ml solution of MTT and cells were incubated at 37 °C for 3 h. Following incubation, the remaining water insoluble formazan was solubilised in absolute isopropanol containing 0.1 N HCl. Absorbance of converted dye was measured in an ELISA plate reader at the wavelength of 570 nm. The cytotoxicity of the compounds was calculated as the percentage of reduction of the viable cells compared with the drug-free control cultureCitation35.
In parallel, cell viability was estimated by Trypan Blue (0.02%) exclusion in MDCK and A549 cells treated or not with the compounds (concentrations range 1–30 µM) and incubated at 37 °C for the following 24 h. Trypan Blue exclusion analysis was also used to calculate CC50 values for selected compounds. CC50 was defined as the drug concentration required for reducing the viability of A549 cells by 50%.
2.4. Virus production and infection
Influenza virus A/Puerto Rico/8/34 H1N1 (PR8 virus) was grown in the allantoic cavities of 10-day-old embryonated chicken eggs. After 48 h at 37 °C, the allantoic fluid was harvested and centrifuged at 5000 rpm for 30 min to remove cellular debris. Confluent monolayers of epithelial cells (A549 or MDCK) were challenged for 1 h at 37 °C with PR8 at a multiplicity of infection (m.o.i.) of 0.001 incubated for 1 h at 37 °C, washed with PBS, and then incubated with medium supplemented with 2% FCS. Mock infection was performed with the same dilution of allantoic fluid from uninfected eggsCitation36.
2.5. Haemagglutination (HAU) assay
MDCK cells plated at concentration of 2 × 105/mL were infected with PR8 (0.001 m.o.i.) and, after the adsorption period, washed with PBS and treated with different concentrations (0–30 μM) of each compound. After 24 h post infection (p.i.), viral production was quantified in the supernatants of infected cells by measuring the haemagglutinin units (HAU), using human type 0 Rh + erythrocytes. Compounds were dissolved in DMSO to prepare stock solutions and diluted in RPMI to final concentrations of 0–30 µM. Compounds were added after the adsorption period and maintained in the culture media until the end of the experiments. Control cells were treated with DMSO alone at the same concentration present in the test substance being evaluated. The highest DMSO concentration present in the culture medium was 0.2%. The concentration of compound required to inhibit viral replication of 50% (IC50) was determined by regression analysis using Microsoft Excel software, considering untreated infected cells as control (100%). Experiments were performed in triplicates and IC50 values reported as ± standard deviation (s. d.).
2.6. In-Cell Western (ICW) assay
The ICW assay was performed using the Odyssey Imaging System (LI-COR, Lincoln, NE)Citation37. A549 cells grown in 96-well plates (2 × 104 cells/well), either infected or mock-infected (Ctr) with PR8, were fixed with 4% formaldehyde, washed, permeabilised with 0.1% Triton X-100 and incubated with PBS containing Odyssey Blocking buffer (LI-COR Biosciences, Lincoln, NE). The cells were then stained at 4 °C overnight with mouse anti-NP (1:400; Santa Cruz Biotechnology) together with Cell Tag (1:2000; LI-COR Biosciences, Lincoln, NE) in PBS containing 5% Odyssey Blocking Buffer. Cells were then washed and stained with goat anti-mouse IRDyeTM 800 antibodies (1:3000; LI-COR Biosciences, Lincoln, NE). Protein expression was quantified using the Odyssey Imaging System. For statistical analysis, integrated intensities of fluorescence in wells were determined using software provided with the imager station (LI-COR). The relative amount of NP protein was obtained by normalising to the Cell Tag in all experiments. Data obtained from mock infected A549 cells treated with selected compounds at different concentrations and stained with Cell Tag were used to calculate the CC50 values.
3. Results
3.1. Chemistry
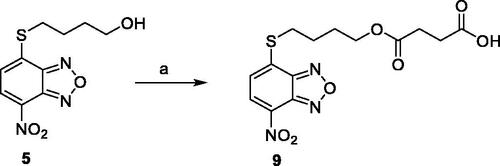
Compounds 1Citation26, 2–3Citation38, 4–6Citation27, 7–8Citation39, 10Citation40, 12Citation27,13–18Citation27, 20–21Citation27, 24–43Citation27 have been prepared as described in previous reports. Compound 9 was prepared through a reaction between succinic anhydride and 5 in dry dichloromethane (DCM) in the presence of 4-(dimethylamino)pyridine (DMAP) under reflux conditions (Scheme 1).
Scheme 1. Compound 9 preparation. Reagents and conditions: (a) succinic anhydride, DMAP, dry DCM, reflux.
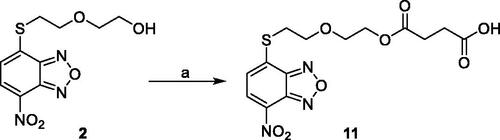
Compound 11 was prepared through a reaction between succinic anhydride and 2 in dry DCM in the presence of DMAP under reflux conditions (Scheme 2).
Scheme 2. Reagents and conditions: (a) succinic anhydride, DMAP, dry DCM, reflux.
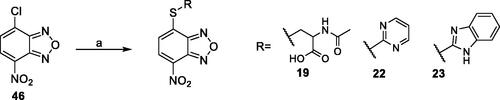
The derivatives 19, 22, and 23 were prepared through a reaction between the commercially available 4-chloro-7-nitrobenzo[c][1,2,5]oxadiazole (46) and the proper commercial thiol in a mixture EtOH:H2O (0.3:1 v/v) in the presence of pyridine at room temperature (Scheme 3).
Scheme 3. Preparation of compounds 19, 22, and 23. Reagents and conditions: (a) appropriate thiol (R-SH), pyridine, EtOH:H2O (0.3:1 v/v), rt.
3.2. Cytotoxicity and antiviral activity of NBD derivatives in MDCK cells
In a first set of experiments, we assessed the potential cytotoxicity of the compounds. We plated MDCK at a concentration of 2 × 104/well and, after 24 h, the cells were treated with various concentrations (range 1–30 µM) of each compound and incubated for the following 24 h. The cytotoxicity of each compound was assessed through a MTT assay, as described in the Materials and Methods section. In parallel, we estimated cell viability in the presence of each compound at the tested concentrations by Trypan Blue exclusion. Microscopic examination and Trypan Blue exclusion demonstrated that most compounds exerted a toxic effect at all tested concentrations and were therefore excluded for the antiviral activity screening (). Notably, compounds 8, 10, 11, 14, 19, 21, 23, 27, 28, 30, 31, 33, 37, 38, 39, 42, 43, 44, and 45 were not toxic at any tested concentration, while compounds 9, 12, 17, 25, 40, and 41 were toxic only at higher concentrations (starting from 20–30 μM).
Table 1. Antiviral activity and cytotoxicity of NBD derivatives 1–45. 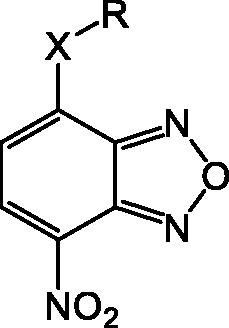
Based on cytotoxicity studies, MDCK cells were infected with influenza A/PR8 H1N1 virus strain and, after viral adsorption, cells were treated with different concentrations (ranging from 0.25 to 30 μM) of each compound. After 24 h post infection (p.i.), we quantified viral production through a haemagglutination assay (HAU) on the supernatants of infected cells. Compounds 9, 11, 12, 17, 19, 25, 40, and 41 were effective against IV ( and Figure S1), as demonstrated by their IC50 (the concentration of compound required to inhibit viral replication of 50%). Compounds 6, 8, 14, 21, 23, 27, 28, 30, 31, 32, 33, 37, 38, 39, 43, 44, and 45 did not inhibit viral replication up to the maximum tested dose (30 µM) and are indicated as inactive in .
3.3. Antiviral activity of compounds 12, 17 and 25 in A549 cells
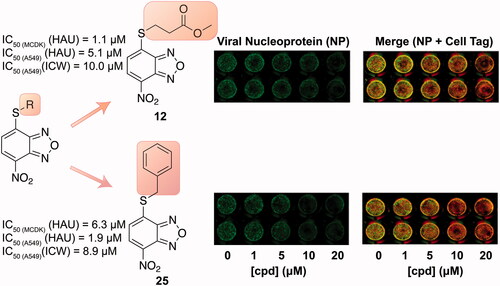
Among the active molecules, 12, 17 and 25 were the only ones to display IC50 values below 10 µM, thus being the most effective compounds against IV. Therefore, we decided to evaluate their antiviral activity on a different cell line highly permissive to IVs, the human lung epithelial A549 cells. Viral replication measured by HAU assay demonstrated that all three compounds were effective against IV. In particular, the IC50 for 12 was 5.1 μM, while the CC50 calculated by counting the number of dead cells stained with Trypan Blue compared to the live cells was 54.5 μM. Hence, the Selectivity Index (SI = CC50/IC50) for compound 12 is equal to 11, suggesting that the molecule is active at concentrations that are far below its cytotoxicity threshold. The IC50 values for 17 and 25 were 11.7 and 1.9 μM, respectively, with the SIs equal to 2 and 6, respectively.
Finally, we tested these compounds following a recently described method for viral titrationCitation37,Citation41, the In Cell Western (ICW) assay. This approach is based on the evaluation of the viral nucleoprotein (NP) expression revealed by labelling cells with anti-NP antibodies. Briefly, infected confluent monolayers of A549, treated with increasing concentrations (1–20 μM) of the three compounds, were fixed and permeabilised (see Materials and Methods section), then incubated with anti-NP antibodies and with Cell Tag (a non-specific cell stain used to evaluate the integrity of cell monolayer). The fluorescence intensity of NP (green) and Cell Tag (red) was measured by Odyssey Imaging System. In parallel, mock infected A549 cells were treated with compounds 12, 17, and 25 and stained with Cell Tag to evaluate their toxicity and to calculate the CC50 using the ICW assay. As reported in (merge panel), the green fluorescence intensity released by viral NP labelled with specific antibodies indicated that NP expression was dose-dependently decreased in cells treated with the compounds compared to untreated ones. This is confirmed by the plots on the right panel of Figure 4, showing that the intesity ration between NP and Cell Tag decreases as compounds concentrations increase. The IC50 values obtained by ICW were in line with what already observed with the previous approach (IC50 for 12 = 10 μM, IC50 for 17 = 9.6 μM, and IC50 for 25 = 8.9 μM). Moreover, SI values calculated using the ICW IC50s and CC50s () confirmed the selectivity of the three molecules.
Figure 4. ICW assay on A549 cells infected with PR8 and treated with NBD derivatives 12, 17 and 25 (concentration range 1–20 μM). Left panel: The integrity of cell monolayer was revealed by Cell Tag on the 680 nm channel (red); viral NP expression on the 800 nm channel (green); merged images show the overlapping between viral protein and infected cells (yellow). Right panel. Fluorescence intensities determined by the Odyssey software and the ratios betwen NP and Cell Tag signal were calculated and averaged for duplicate wells. The values are shown as a function of compounds concentration. Error bars indicate s. d. The percentage (%) of fluorescence intensity was calculated respect to untreated infected cells (considered as 100%).
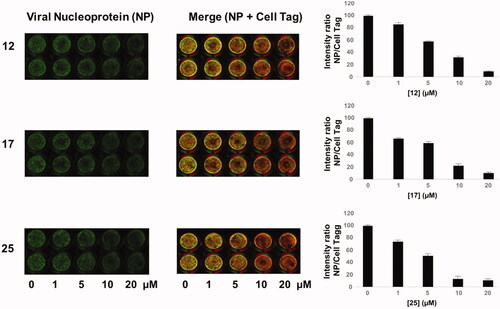
Table 2. Antiviral activity and selectivity of compounds 12, 17, and 25 in A549 cells as determined by both HAU and ICW assays.
4. Discussion
Over the past few years, many efforts have been made to obtain novel drugs for the treatment of infections caused by IVs. These endeavours resulted in the recent antivirals baloxavir marboxil and favipiravir both targeting IV RdRp with different modes of action. In an effort to find new anti-IV agents, we assessed our in-house library of NBD derivatives since they possess the same core of previously reported molecules endowed with anti-IV activity, although those compounds were cytotoxic at relative low concentrations and still necessitate validation of their mode of actionCitation24,Citation25. We evaluated our compound library in two different cell lines infected by PR8, a strain of H1N1 influenza A virus.
The initial assay devoted to preliminary assessing the cytotoxicity of the compounds revealed that 25 molecules were not toxic in the concentration range we tested (1–30 µM) or toxic only at higher concentrations (≥20 µM) (). All 4-thioether-NBD derivatives possessing an alkyl/alkoxy alcohol (1 − 6) side chain were cytotoxic at all tested concentrations. Esterification of 1 with an acetyl group (7) did not change the toxicity profile; similarly, acylated or alkylated ethylamine derivatives 13 and 15 were cytotoxic.
Conversely, compound 8, the benzoic ester of 1, was not toxic at any tested concentration, but did not display any antiviral activity. Notably, esterification of 1, 2, and 5 with succinic acid yielding compounds 10, 9, and 11, respectively, abolished the cytotoxicity and resulted in mild anti-IV activity. Among the propionic acid derivatives, only two compounds were cytotoxic, namely the piperidin-4-one amide derivative 16 and compound 18, bearing a Cbz-protected amino group in C2. Among the non-toxic molecules, the methyl propionate derivatives 12 and 17 displayed the best antiviral activity, while 19, bearing a free carboxylic acid, showed mild antiviral properties and the hydroxyethyl amide derivative 14 was inactive. Compounds 20 and 21 were toxic and inactive, respectively, suggesting that purely alkyl side chains may be detrimental for antiviral activity. Similarly, compounds bearing an aromatic moiety directly linked to the sulphur were either toxic or substantially inactive. Moreover, compounds 29 and 39, bearing a benzoic or phenylacetic moiety were toxic and inactive, respectively. Remarkably, benzyl derivatives 25, 41 and 42 were not cytotoxic and possessed good anti-IV activity, suggesting that the presence of a methylene spacer between the sulphur and the side-chain aromatic group is essential for activity and selectivity. Finally, 1 analogue in which the sulphur at position 4 was replaced by a 4-sulphinyl (43), 4-sulphonyl (44) or 4-amino (45) were not toxic, but did not possess any antiviral activity at the tested concentrations. These data suggest that the presence of a reduced sulphur is pivotal for antiviral activity.
Overall, 10 compounds of the library possessed antiviral activity, all of them being 4-thioether derivatives of NBD. The active molecules can be divided into three subgroups: (i) the emisuccinic esters 9, 10, 11; (ii) the propionic acid derivatives 12, 17, and 19; (iii) the 4-benzylthio derivatives 25, 41, 42. In group (i), it is apparent that increasing the length of the alkyl chain reduces the antiviral activity as demonstrated by the increased IC50 value of 10 compared to 9. In addition, the introduction of polar atoms, such as oxygen, is detrimental for compound activity as indicated by the higher IC50 value of 11 compared to 9. In group (ii), methylation of the carboxylic acid function of N-α protected cysteine residues decreases the IC50 value at least six-fold (compare 17 with 19), and the subsequent removal of the protected amino group at α position leads to a further increase of antiviral potency (compare 12 with 17) suggesting that a small methyl propionate side chain is important for compound activity. In group (iii), the introduction of both carboxy (41) and carbethoxy (42) groups determine a decrease in antiviral activity compared to 25, indicating that substitutions with polar moieties at para position are unfavourable for the 4-benzylthio-NBD derivatives series. Overall, the present data suggest that compact, hydrophobic substitutions are favourable for antiviral activity, alternatively benzyl derivatives are promising anti-IV compounds, although different substitutions in the benzene ring need to be explored.
Given their promising activity in MCDK cells, we assessed compounds 12, 17, and 25 also in the A549 cell line. The HAU and ICW assays performed on these cells infected with PR8 confirmed the antiviral activity and selectivity of the three molecules. Overall, compound 25 resulted as the most active in A549 cells, while 12 was the best one in MCDK cells and displayed the best selectivity profile in both cell lines ().
Despite their specific mode of action has not been clarified yet, the results of this study demonstrate that some 4-thio-NBD derivatives are effective against IV and shed light on structure–activity relationship that could be exploited for the design of new molecules. Indeed, compounds 12 and 25 with their simple structures amenable for modifications represent promising hit compounds for future medicinal chemistry optimisation studies.
In conclusion, we hope that the present study might drive the search to the identification and characterisation of new potential compounds for fighting IV.
ienz_a_1982932_sm7185.pdf
Download PDF (183.2 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Shaw ML, Palese P, Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, eds. Fields virology. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2013: 1671–1740.
- Chen R, Holmes EC. The evolutionary dynamics of human influenza B virus. J Mol Evol 2008;66:655–63.
- Tumpey TM, Belser JA. Resurrected pandemic influenza viruses. Annu Rev Microbiol 2009;63:79–98.
- Neumann G, Noda T, Kawaoka Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009;459:931–9.
- Fraser C, The WHO Rapid Pandemic Assessment Collaboration, Donnelly CA, Cauchemez S, et al. Pandemic potential of a strain of influenza a (H1N1): early findings. Science 2009;324:1557–61.
- Trifonov V, Khiabanian H, Rabadan R. Geographic dependence, surveillance, and origins of the 2009 influenza a (H1N1) virus. N Engl J Med 2009;361:115–9.
- Das K. Antivirals targeting influenza a virus. J Med Chem 2012;55:6263–77.
- Syrjänen RK, Jokinen J, Ziegler T, et al. Effectiveness of pandemic and seasonal influenza vaccines in preventing laboratory-confirmed influenza in adults: a clinical cohort study during epidemic seasons 2009–2010 and 2010–2011 in Finland. PLOS One 2014;9:e108538.
- Davies WL, Grunert RR, Haff RF, et al. Antiviral activity of 1-adamantanamine (amantadine). Science 1964;144:862–3.
- Cady SD, Schmidt-Rohr K, Wang J, et al. Structure of the amantadine binding site of influenza m2 proton channels in lipid bilayers. Nature 2010;463:689–92.
- Pielak RM, Chou JJ. Flu channel drug resistance: a tale of two sites. Protein Cell 2010;1:246–58.
- Jing X, Ma C, Ohigashi Y, et al. Functional studies indicate amantadine binds to the pore of the influenza a virus m2 proton-selective ion channel. Proc Natl Acad Sci USA 2008;105:10967–72.
- von Itzstein M, Wu WY, Kok GB, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993;363:418–23.
- Kim CU, Lew W, Williams MA, et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: Design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J Am Chem Soc 1997;119:681–90.
- Stevaert A, Naesens L. The influenza virus polymerase complex: an update on its structure, functions, and significance for antiviral drug design. Med Res Rev 2016;36:1127–73.
- Noshi T, Kitano M, Taniguchi K, et al. In vitro characterization of baloxavir acid, a first-in-class cap-dependent endonuclease inhibitor of the influenza virus polymerase PA subunit. Antiviral Res 2018;160:109–17.
- Shiraki K, Daikoku T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol Ther 2020;209:107512.
- Shionogi. Shionogi announces supplemental new drug application for XOFLUZA® in Japan for the post-exposure prophylaxis of influenza virus infection was approved [Internet]. ; 2020, November 27 [cited 2021 Jul 27]. Available from: https://www.shionogi.com/content/dam/shionogi/global/news/pdf/2020/11/e-201127.pdf
- Pharmaceutical Evaluation Division, Pharmaceutical Safety and Environmental Health Bureau – Ministry of Health, Labour and Welfare. Report on the deliberation results [Internet]. 2018, February 8 [cited 2021 ]. Available from: https://www.pmda.go.jp/files/000225380.pdf
- U. S. Food & Drug Administration. FDA approves new drug to treat influenza [Internet]. 2018, October 24 [cited 2021 Jul 27]. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-treat-influenza
- U. S. Food & Drug Administration. FDA expands approval of influenza treatment to post-exposure prevention [Internet]. 2020, November 23 [cited 2021 Jul 27]. Available from: https://www.fda.gov/news-events/press-announcements/fda-expands-approval-influenza-treatment-post-exposure-prevention
- European Medicines Agency. Xofluza [Internet].2021, January 22 [cited 2021 Jul 27]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/xofluza
- Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau – Ministry of Health, Labour and Welfare. Report on the deliberation results [Internet]. 2014, March 4 [cited 2021 Jul 27]. Available from: https://www.pmda.go.jp/files/000210319.pdf
- Kessler U, Castagnolo D, Pagano M, et al. Discovery and synthesis of novel benzofurazan derivatives as inhibitors of influenza a virus. Bioorg Med Chem Lett 2013;23:5575–7.
- Pagano M, Castagnolo D, Bernardini M, et al. The fight against the influenza a virus H1N1: Synthesis, molecular modeling, and biological evaluation of benzofurazan derivatives as viral rna polymerase inhibitors. ChemMedChem 2014;9:129–50.
- Ricci G, De Maria F, Antonini G, et al. 7-nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione s-transferases. Mechanism of action of potential anticancer drugs. J Biol Chem 2005;280:26397–405.
- Rotili D, De Luca A, Tarantino D, et al. Synthesis and structure-activity relationship of new cytotoxic agents targeting human glutathione-s-transferases. Eur J Med Chem 2015;89:156–71.
- Graziani G, Artuso S, De Luca A, et al. A new water soluble mapk activator exerts antitumor activity in melanoma cells resistant to the braf inhibitor vemurafenib. Biochem Pharmacol 2015;95:16–27.
- Sciarretta F, Fulci C, Palumbo C, et al. Effects of glutathione transferase-targeting nitrobenzoxadiazole compounds in relation to pd-l1 status in human melanoma cells. Chemotherapy 2019;64:138–45.
- De Luca A, Carpanese D, Rapanotti MC, et al. The nitrobenzoxadiazole derivative mc3181 blocks melanoma invasion and metastasis. Oncotarget 2017;8:15520–38.
- Palumbo C, De Luca A, Rosato N, et al. C-jun n-terminal kinase activation by nitrobenzoxadiazoles leads to late-stage autophagy inhibition. J Transl Med 2016;14:37.
- Tew KD, Monks A, Barone L, et al. Glutathione-associated enzymes in the human cell lines of the national cancer institute drug screening program. Mol Pharmacol 1996;50:149–59.
- Ruiz-Gómez MJ, Souviron A, Martínez-Morillo M, Gil L. P-glycoprotein, glutathione and glutathione s-transferase increase in a colon carcinoma cell line by colchicine. J Physiol Biochem 2000;56:307–12.
- Inoue T, Ishida T, Sugio K, et al. Glutathione s transferase pi is a powerful indicator in chemotherapy of human lung squamous-cell carcinoma. Respiration 1995;62:223–7.
- Checconi P, Sgarbanti R, Celestino I, et al. The environmental pollutant cadmium promotes influenza virus replication in mdck cells by altering their redox state. Int J Mol Sci 2013;14:4148–62.
- Fioravanti R, Celestino I, Costi R, et al. Effects of polyphenol compounds on influenza a virus replication and definition of their mechanism of action. Bioorg Med Chem 2012;20:5046–52.
- De Angelis M, Casciaro B, Genovese A, et al. Temporin G, an amphibian antimicrobial peptide against influenza and parainfluenza respiratory viruses: insights into biological activity and mechanism of action. Faseb J 2021;35:e21358.
- De Luca A, Rotili D, Carpanese D, et al. A novel orally active water-soluble inhibitor of human glutathione transferase exerts a potent and selective antitumor activity against human melanoma xenografts. Oncotarget 2015;6:4126–43.
- Fulci C, Rotili D, De Luca A, et al. A new nitrobenzoxadiazole-based gstp1-1 inhibitor with a previously unheard of mechanism of action and high stability. J Enzyme Inhib Med Chem 2017;32:240–7.
- Di Paolo V, Fulci C, Rotili D, et al. Characterization of water-soluble esters of nitrobenzoxadiazole-based gstp1-1 inhibitors for cancer treatment. Biochem Pharmacol 2020;178:114060.
- Marcocci ME, Amatore D, Villa S, et al. The amphibian antimicrobial peptide temporin b inhibits in vitro herpes simplex virus 1 infection. Antimicrob Agents Chemother 2018;62:e02367–02317.