
Abstract
New C-glycosides and α,β-unsaturated ketones incorporating the 4-hydroxy-3-methoxyphenyl (vanillin) moiety as inhibitors of carbonic anhydrase (CA, EC 4.2.1.1) isoforms have been investigated. The inhibition profile of these compounds is presented against four human CA (hCA) isozymes, comprising hCAs I and II (cytosolic, ubiquitous enzymes) and hCAs IX and XII (tumour associated isozymes). Docking analysis of the inhibitors within the active sites of these enzymes has been performed and is discussed, showing that the observed selectivity could be explained in terms of an alternative pocket out of the CA active site where some of these compounds may bind. Several derivatives were identified as selective inhibitors of the tumour-associated hCA IX and XII. Their discovery might be a step in the strategy for finding an effective non-sulfonamide CA inhibitor useful in therapy/diagnosis of hypoxic tumours or other pathologies in which CA isoforms are involved.
Graphical Abstract
1. Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are members of a great family of metalloproteins found in most organisms such as bacteria, archaea, fungi, protozoans, plants, algae, and vertebratesCitation1. CAs catalyze one of the most important physiological reactions: the reversible hydration of carbon dioxide with the formation of a proton and bicarbonateCitation1,Citation2. Up to now eight genetically distinct CA families are known, the α-, β-, γ-, δ-, ζ-, η-, θ-, and ι-CAs. Mammals possess only α-CAs, while many pathogenic organisms such as bacteria and fungi encode enzymes belonging to several families, among which α-, β-, γ- and ι-CAs. Most of these enzymes contain a metal ion (usually Zn2+) in their active site, which is coordinated by three histidine residues and a water molecule/hydroxide ion in the α-CA classCitation1,Citation2, whereas the recently described ι-CAs seem to be devoid of metal ions and perform the catalysis by a diverse mechanismCitation3.
In mammals, 16 different α-CA isoforms are known to date, with human CA isozymes hCA I and II (cytosolic forms) being the most widespread ones throughout the human bodyCitation1. The disregulated CA activity is linked with numerous pathological statesCitation1. In recent years, the connection between the overexpression of transmembrane isoforms hCA IX and XII with cancer progression has been investigated in detail and clarifiedCitation4. These isozymes are overexpressed in solid tumours as a consequence of the hypoxia-inducible factor-1 (HIF-1) signalling cascade, being less expressed in several normal tissuesCitation4,Citation5. Thus selective inhibition of tumour-associated isoforms hCA IX and XII (over the off-target hCA I and II) is a great promise for the use of CA inhibitors in the cancer therapy/diagnosisCitation4,Citation5.
The intake of protective factors for fortifying the natural bodýs defense capacity to reduce the risk of cancer is an approach called chemoprevention. Recently natural products containing phenols, have been recognised as cancer chemopreventive agentsCitation6–8. Ginger (Zingiber officinale) is one of the most widely consumed spices in the worldCitation6,Citation7 Ginger contains several phenolic compounds possessing antimicrobial and analgesic activity, such as 6-gingerol and 6-paradol that incorporate vanillin (4-hydroxy-3-methoxyphenyl) moiety in their molecules. Related 6-dehydroparadols have shown antitumor-promoting activity in mouse skin carcinogenesisCitation9. 6-Shogaol, another vanillin derivative found in ginger, suppresses the proliferation of non-small cell lung cancerCitation9. Phenol derivatives, including some natural products, have also been studied as CA inhibitors (CAIs)Citation10–13.
Several phenolic derivatives share a common moiety: an α,β-unsaturated carbonyl group, or enone. Incorporation of this functionality into a given molecular structure has been shown to increase antitumor activityCitation14. In that sense, the introduction of an enone moiety into a triterpenoid skeleton had enhanced the cytotoxic activity against human breast cancer cellsCitation15.
Our group has developed several selective CAIs by the attachment of C-glycosidic enones to carbohydratesCitation16. The use of carbohydrate moieties could induce the desired physicochemical properties such as water solubility, lower permeability in organs in which the enzyme is also present, etcCitation5. Although several pharmacophores have thoroughly been investigated in the design of sugar-based CAIsCitation17, there are still several functionalities that have been only poorly studied such as the vanillin moiety. To the best of our knowledge no alkyl nor aryl enones of vanillin have been studied so far as CAIsCitation18.
2. Materials and methods
2.1. Materials and apparatus
All starting materials and reagents were purchased from commercial suppliers. Reactions were monitored by TLC and TLC plates visualised with short wave UV fluorescence (k = 254 nm), sulphuric acid stain (5% H2SO4 in methanol). Silica gel flash chromatography was performed using silica gel 60 Å (230–400 mesh). All melting points are uncorrected. 1H and 13 C NMR spectra were recorded on a Bruker 600 (600 and 151 MHz, respectively). Chemical shifts were measured in ppm and coupling constants in Hz. High-resolution mass spectra were recorded using electrospray as the ionisation technique in positive ion or negative ion modes as stated. All MS analysis samples were prepared as solutions in methanol.
2.2. General procedure for the synthesis of compounds 1 and 2
A mixture of β-C-glucopyranosyl or β-C-galactopyranosyl ketone (1 mmol), 4-hydroxy-3-methoxybenzaldehyde (vanillin) (1.2 mmol), and L-proline (0.15 mmol)-TEA (0.3 mmol) in 3 ml of methanol was stirred at reflux for 48–96 h. The endpoint of the reaction was monitored by TLC. The solvent was evaporated under reduced pressure and the product was purified by column chromatography (eluant 8:2 DCM-MeOH) to afford pure material by 1H NMR and 13 C NMR spectroscopy. Yields 35–45%.
2.3. General producer for the synthesis of compounds 3–7
A mixture of ketone (1 mmol) and 4-hydroxy-3-methoxybenzaldehyde (vanillin) (1 mmol) in 2 ml of anhydrous methanol has been added to the catalyst (see ). The reaction was stirred at the corresponding temperature until starting material was consumed as evidenced by TLC, followed by dilution with ice-cold water, acidification with cold 1 M HCl, and extraction with DCM. The solvent was evaporated under reduced pressure. The product was purified by column chromatography (eluant 9:1 DCM-MeOH) to afford pure material by 1H NMR and 13 C NMR spectroscopy. Yields 40–79%.
Table 1. Synthesis of vanillin enones 1–7.
1-(β-D-glucopyranosyl)-4–(4-hydroxy-3-methoxyphenyl)but-3-en-2-one (1). Sticky yellow solid. 1H NMR (600 MHz, DMSO) δ 9.62 (s, 1H, OH), 7.50 (d, 1H, J = 16.1 Hz, H-4), 7.31 (d, 1H, J = 2.0 Hz, ArH), 7.14 (dd, 1H, J = 8.2, 2.0 Hz, ArH), 6.82 (d, 1H, J = 1.6 Hz, ArH), 6.80 (d, 1H, J = 9.6 Hz, H-3), 5.03 (d, 1H, J = 5.7 Hz, OH), 4.91 (d, 1H, J = 4.6 Hz, OH), 4.85 (d, 1H, J = 4.8 Hz, OH), 4.34 (t, 1H, J = 5.7 Hz, OH), 3.83 (s, 3H, CH3O), 3.62 (dd, 1H, J = 9.2, 2.5 Hz, H-4′), 3.59 (m, 1H, H-6’b), 3.39 (dt, 1H, J = 11.2, 5.4 Hz, H-6’a), 3.17 (t, 1H, J = 7.7 Hz, H-1′), 3.06 (m, 2H, H-2′, H-5′), 2.95 (dt, 1H, J = 9.1, 4.8 Hz, H-1b), 2.91 (dd, 1H, J = 16.0, 2.6 Hz, H-3′), 2.77 (m, 1H, H-1a).13C NMR (151 MHz, DMSO) δ 198.23 (C-2), 149.81 (ArC), 148.39 (ArC), 143.28 (C-4), 126.43 (ArC), 124.32 (C-3), 123.77 (ArC), 116.07 (ArC), 111.80 (ArC), 81.14 (C-5′), 78.61 (C-1′), 76.38 (C-4′), 74.08 (C-3′), 70.79 (C-2′), 61.63 (C-6′), 56.15 (CH3O), 43.66 (C-1). HMRS m/z: calcd for C17H22O8, 354.1315; found, 352.1134.
1-(β-D-galactopyranosyl)-4–(4-hydroxy-3-methoxyphenyl)but-3-en-2-one (2). Sticky red solid. 1H NMR (600 MHz, DMSO) δ 9.04 (s, 1H, OH), 7.54 (d, 1H, J = 16.1 Hz, H-4), 6.97 (dd, 1H, J = 8.8, 1.9 Hz, ArH), 6.85 (dd, 1H, J = 8.2, 2.0 Hz, ArH), 6.82 (d, 1H, J = 8.1 Hz, ArH), 6.75 (d, 1H, J = 8.1 Hz, H-3), 5.42 (m, 1H, OH), 5.02 (d, 1H, J = 5.6 Hz, OH), 4.87 (d, 1H, J = 5.9 Hz, OH), 4.11 (q, 1H, J = 5.3 Hz, H-5′), 4.06 (m, 1H,H-2′), 3.93 (m, 1H, H-6’a), 3.81 (s, 1H, OH), 3.76 (s, 3H, CH3O), 3.58 (dd, 1H, J = 9.2, 3.0 Hz, H-1′), 3.44 (m, 1H, H-4′), 3.42 (m, 1H, H-6’b), 3.29 (dd, 1H, J = 9.3, 5.7 Hz, H-3′), 3.17 (d, 2H, J = 5.2 Hz, H-1a, H-1b).13C NMR (151 MHz, DMSO) δ 207.31 (C-2), 147.53 (ArC), 147.28 (ArC), 143.37 (C-4), 119.49 (ArC), 115.26 (C-3), 111.12 (ArC), 106.78 (ArC), 100.46 (ArC), 77.01 (C-5′), 76.43 (C-1′), 73.67 (C-4′), 70.35 (C-3′), 69.61 (C-2′), 69.25 (C-6′), 56.18 (CH3O), 46.69 (C-1). HMRS m/z: calcd for C17H22O8, 354.1315; found, 352.1130.
(E)-4–(4-Hydroxy-3-methoxyphenyl)but-3-en-2-one (3). White solid. Mp 122–122.5 °C. 1H NMR (600 MHz, CDCl3) δ 9.85 (sa, 1H, OH), 7.47 (d, 1H, J = 16.2 Hz,H-4), 7.10 (m, 3H, ArH), 6.95 (d, 1H, J = 8.2 Hz, ArH), 6.61 (d, 1H, J = 16.2 Hz, H-3), 3.95 (s, 3H, CH3O), 2.39 (s, 3H, CH3).13C NMR (151 MHz, CDCl3) δ 198.44 (C-2), 148.28 (ArC), 146.89 (ArC), 143.77 (C-4), 126.92 (ArC), 124.99 (C-3), 123.52 (ArC), 114.83 (ArC), 109.33(ArC), 55.97(CH3O), 27.31(CH3). HMRS m/z: calcd for C11H12O3, 192.0786; found, 192.0784.
(E)-1–(4-Hydroxy-3-methoxyphenyl)pent-1-en-3-one (4). Yellow solid. Mp 81–82 °C. 1H NMR (600 MHz, CDCl3) δ 7.51 (d, 1H, J = 16.1 Hz,H-1), 7.09 (m, 2H, ArH), 6.94 (d, 1H, J = 8.1 Hz, H-2), 6.62 (d, 1H, J = 16.1 Hz, ArH), 3.94 (s, 3H,CH3O), 2.70 (q, 2H, J = 7.3 Hz,H-4), 1.18 (t, 3H, J = 7.3 Hz,H-5).13C NMR (151 MHz, CDCl3) δ 201.10 (C-3), 148.15 (ArC), 146.88 (ArC), 142.57 (C-1), 127.07 (ArC), 123.81 (C-2), 123.34 (ArC), 114.83 (ArC), 109.47 (ArC), 55.97 (CH3O), 33.76 (C-4), 8.39 (C-5). HMRS m/z: calcd for C12H14O3, 206.0943; found, 206.0950.
(E)-1–(4-Hydroxy-3-methoxyphenyl)-5-methylhex-1-en-3-one (5). Sticky dark red solid. 1H NMR (600 MHz, CDCl3) δ 9.85 (d, 1H, J = 1.6 Hz, OH), 7.49 (d, 1H, J = 16.1 Hz, H-1), 7.12 (dd, 1H, J = 8.3, 1.8 Hz, ArH), 7.07 (d, 1H, J = 2.2 Hz, ArH), 6.95 (d, 1H, J = 8.1 Hz, H-2), 6.62 (d, 1H, J = 16.2 Hz, ArH), 3.95 (s, 3H, CH3O), 2.54 (d, 2H, J = 7.2 Hz, H-4), 2.25 (m, 1H, H-5), 1.00 (d, 6H, J = 6.6 Hz, H-6, H-7).13C NMR (151 MHz, CDCl3) δ 200.42 (C-3), 148.16 (ArC), 146.86 (ArC), 142.73 (C-1), 127.09 (ArC), 124.48 (C-2), 123.38 (ArC), 114.82 (ArC), 109.47 (ArC), 55.97 (CH3O), 49.69 (C-4), 25.39 (C-5), 22.74 (C-6,C-7). HMRS m/z: calcd for C14H18O3, 234.1256; found, 234.1260.
(E)-3–(4-Hydroxy-3-methoxyphenyl)-1-phenylprop-2-en-1-one (6). Sticky yellow solid. 1H NMR (600 MHz, CDCl3) δ 9.84 (sa, 1H, J = 1.4 Hz, OH), 7.77 (d, 1H, J = 15.6 Hz, H-3), 7.59 (m, 1H, ArH), 7.52 (d, 1H, J = 7.3 Hz, ArH), 7.44 (dd, 1H, J = 6.0, 1.7 Hz, ArH), 7.40 (d, 1H, J = 15.6 Hz,H-2), 7.28 (d, 1H, J = 1.4 Hz, ArH), 7.23 (dd, 1H, J = 8.3, 1.8 Hz, ArH), 7.15 (d, 1H, J = 1.8 Hz, ArH), 7.06 (dd, 1H, J = 8.5, 1.5 Hz, ArH), 6.98 (dd, 1H, J = 8.2, 1.4 Hz, ArH), 3.98 (s, 3H, J = 1.5 Hz,CH3O).13C NMR (151 MHz, CDCl3) δ 190.91 (C-1), 151.72 (ArC), 148.35 (ArC), 147.18 (C-3), 145.30 (C-2), 138.49 (ArC), 132.58 (ArC), 129.88 (ArC), 128.57 (ArC), 128.44 (ArC), 127.53 (ArC), 123.42 (ArC), 119.82 (C-3), 114.41 (ArC), 108.81 (ArC), 56.12 (CH3O). HMRS m/z: calcd for C16H14O3, 254.0943; found, 254.0948.
(E)-1–(4-Bromophenyl)-3–(4-hydroxy-3-methoxyphenyl)prop-2-en-1-one (7). Sticky yellow solid. 1H NMR (600 MHz, CDCl3) δ 9.85 (s, 1H, OH), 7.89 (d, 2H, J = 8.5 Hz, ArH), 7.77 (d, 1H, J = 15.6 Hz, H-3), 7.66 (d, 2H, J = 8.6 Hz, ArH), 7.33 (d, 1H, J = 15.6 Hz, H-2), 7.24 (dd, 1H, J = 8.2, 2.0 Hz, ArH), 7.14 (d, 1H, J = 1.9 Hz, ArH), 6.98 (d, 1H, J = 8.2 Hz, ArH), 3.99 (s, 3H, CH3O). 13 C NMR (151 MHz, CDCl3) δ 189.43 (C-1), 148.54 (ArC), 146.87 (ArC), 145.74 (C-3), 137.27 (ArC), 131.84 (2 x ArC), 129.94 (2 x ArC), 127.56 (ArC), 127.33 (ArC), 123.48 (ArC), 119.27 (C-2), 114.95 (ArC), 110.18 (ArC), 56.06 (CH3O). HMRS m/z: calcd for C16H13BrO3, 332.0048; found, 332.0061.
2.4. CA inhibition studies
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity as reported by KhalifahCitation19. Phenol red (at a concentration of 0.02 mM) has been used as an indicator, working at the absorbance máximum of 557 nm, with 20 mM Hepes (pH 7.5) as a buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled–deionized water and dilutions up to 0.01 nM were done thereafter with distilled–deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, and the Cheng-Prussoff equation as reported earlierCitation20 and represent the mean from at least three different determinations.
2.5. Computational methods
2.5.1. Preparation of the molecular systems
The simulations were based on X-ray crystal structures of human carbonic anhydrases (CA I: PDB ID 2FW4, CA II: PDB ID 3KS3, CA IX: PDB ID 6FE2, CA XII: PDB ID 1JCZ). These structures were selected from the Protein Data BankCitation21, based on resolution, validation parameters, and missing residues.
The preparation of proteins was done with ChimaeraCitation22. Water molecules and other ligands were removed. All Asp and Glu residues were considered to have a negative charge and all the Arg and Lys residues were considered to have a positive charge. Hydrogens were added following the hydrogen-bonding pattern. Ligand structures were built with AvogadroCitation23 and then optimised using the PM6 semiempirical method, implemented in OpenMOPACCitation24. The solvent effect was considered using the COSMO implicit modelCitation25, with a dielectric constant value of 78.4, corresponding to an aqueous medium. The geometry optimisation termination criteria were set to 0.1 kcal/mol/Å gradient norm requirement.
2.5.2. Molecular docking
Molecular docking was carried out to find and score protein-ligand binding poses on carbonic anhydrase structures with Smina with Vina as scoring functionCitation26. Protein and ligand PDBQT files were prepared with AutoDockTools softwareCitation27. To unify the box set and simplify the analysis of results, chains A of 2FW4, 6FE2, 1JCZ structures were aligned to 3KS3 using PymolCitation28. A docking box with size 27.75 Å × 27.00 Å × 28.50 Å was centred on the catalytic binding site. The ligands were docked using a flexible-ligand/rigid-receptor approach. The exhaustiveness value was increased to 20.
In order to have alternative poses, DOCK 6.8Citation29 was also used with grid score and flexible ligands and applying a subsequent minimisation. The spheres selected where the grid is calculated were the ones that fit inside the box used with Smina. Other parameters were set to their default values.
The post-docking analysis included visualisation of the ligand-receptor complexes with Pymol to analyse the potential interactions with the amino acid.
2.5.3. Binding energy estimated by semiempirical method
In order to obtain greater precision in the characterisation of the interaction of ligands with carbonic anhydrase isozymes, a calculation scheme based on the semi-empirical PM6Citation30 was adopted, employing OpenMOPAC Citation24. It was decided to implement a quantum method in view of the presence of Zn2+ in the carbonic anhydrase, which makes it somewhat difficult to deal with molecular mechanical force fields, in particular, to estimate binding energy.
Due to the considerable size of the system, the MOZYMECitation31,Citation32 approach was used, which has already been used to improve docking scoring functions or estimate binding energiesCitation33–35. The solvent effect was also considered using the COSMO implicit model.
A molecular optimisation of all the poses obtained by docking was performed, both on the complexes, as well as on the ligands and the carbonic anhydrase separately. Then, binding enthalpies were calculated as the heat of formation differences (ΔHbind = ΔfHcomplex - ΔfHligand - ΔfHCA).
2.5.4. Molecular dynamics
MD simulations were performed on poses resulting from molecular docking for CA I and CAII. The system, CAs, and ligands have a positive net charge, so chloride anions were added as counterions with the Leap module to achieve electroneutrality. The neutralised ligand/CA complexes were immersed in a box of TIP3P waters which extended up to 15 Å from the solute. CAs were described using the Amber14SB force fieldCitation36. The ligand was described using the Generalised Amber Force FieldCitation37 with charges derived from RESP, which were calculated with the Antechamber module. Leap and Antechamber are included in the package AmberTools 20.0Citation38. Zn2+ cation, neighbouring residues and a water molecule were modelled with MCPB.pyCitation39, also included in AmberTools 20.0.
All MD simulations were run using the NAMD 2.13 softwareCitation40. The van der Waals interaction cut-off distances were set at 12 Å and long-range electrostatic forces were computed using the particle mesh Ewald summation method with a grid size set to 1.0 Å. The 1–4 contributions were multiplied by a factor of 0.83 to match the AMBER force field requirements. The system was subjected to 100000 minimisation steps, heating from 0 to 300 K in 30 ps, and 10 ns of equilibration/production simulation. This trajectory extension was chosen to achieve a balance between the number of simulations to run with respect to sample at equilibrium conditions, and also considering that the CAs quickly reach the equilibrium condition. For all equilibration/production simulations, constant temperature (300 K) was maintained using Langevin dynamics with a damping coefficient of 5 ps−1, while pressure was kept constant at 1 atm through the Nosé-Hoover Langevin piston method with a decay period of 200 fs and a damping time constant of 100 fs. A time step of 1 fs was used along with molecular mechanics. Bonds involving hydrogen atoms of waters were constrained using the SHAKE algorithm. RMSD values were depicted to determine the convergence and stability of simulations. (See Electronic Supplementary Material, and )
2.5.5. Binding energy estimated by MM/GBSA
Binding free energies of ligands with CAs were computed using the MM-GB/SA method, where the binding free energy is calculated as the difference between the bound and unbound states of protein and ligandCitation41,Citation42.
The solvation free energy was calculated using the generalised Born (GB) modelCitation43 implemented in MMPBSA.py moduleCitation44, igb = 5 as the selected model. The hydrophobic contribution was determined using the solvent-accessible surface area. The CAs–ligands binding free energies were calculated using a single trajectory (for ligand, protein, and complex) based on 500 snapshots taken from the last 5 ns portion of the MD simulation trajectories. Entropies were estimated using quasi-harmonic approximation.
To obtain the detailed representation of interactions, free energy decomposition analysis was employed to decompose the total binding free energies into ligand–amino acid pairs. These calculations were performed using a pairwise energy decomposition scheme (idecomp option 3) also with the MMPBSA.py module.
3. Results and discussion
3.1. Chemistry and CA inhibition
The enones 1–7 () have been prepared by aldol condensation of vanillin with the appropriate alkyl, aryl, or glycosidic methyl ketones (Scheme 1). Knoevenagel reaction of D-glucose or D-galactose with pentane-2,4-dione in the presence of aqueous sodium bicarbonate at 90 °C afforded the β-D-glycosyl-propan-2-ones in 50% and 54% yields, respectively. It should be noted that higher yields were described in the literatureCitation45, but the products had not been purified as in our report.
Scheme 1. Preparation of vanillin enones 1–7 discussed in the paper
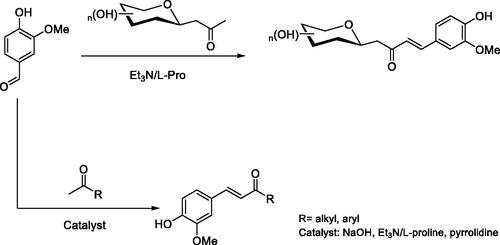
Figure 1. Structure of compounds 1–7.
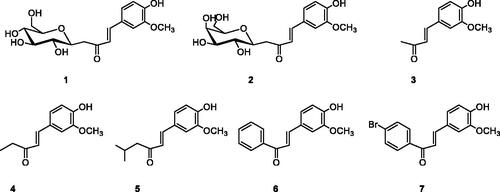
C-glycosides 1–2 have been prepared by reaction of β-C-glucosyl or β-C-galactosylketone with vanillin in the presence of L-proline/Et3N with moderate yields (). Next, we studied the aldol condensation of aliphatic or aromatic ketones and vanillin with different catalysts. The best reaction conditions found in the synthesis of the α,β-unsaturated ketones are shown in . Enones 1–7 were successfully purified by flash chromatography. The 1H NMR, 13 C NMR, 2 D COSY and HSQC were in full agreement with their structures (see Supplementary information). The large coupling constant (J ≈ 16 Hz) between the two olefinic protons, was consistent with the E configuration of the double bond. In enones, 1–2 the large coupling constant (J = 9.4 Hz) between H-10 and H-20 indicated a diaxial relationship and thus confirmed the β-configuration.
The inhibitory activity of compounds 1–7 against cytosolic isoforms hCA I and II as well as the membrane-associated isoforms hCA IX and XII was assayed by using a stopped-flow assay method and the results are shown in . A number of structure-activity relationships were identified in this study and are summarised as follows.
Table 2. Inhibition of hCA I, II, IX, XII with vanillin enones 1–7.Table Footnotea,b
3.1.1. Off-target CA isozymes
C-glycosides 1 and 2 were micromolar inhibitors of hCA I. A similar trend was found for the aryl derivatives 6 and 7 and the methyl one 3. The alkyl derivatives 4 and 5 were very poor inhibitors of hCA I. It is of great interest to relate the behaviour of these compounds towards hCA I and it can be concluded that attachment of a voluminous scaffold such as the carbohydrate one, to the enone functionality, does not lead to a decrease in the inhibitory potency of these compounds against hCA I.
Vanillin derivatives showed an interesting inhibition profile against hCA II. It should be noted that the C-glycosidic derivatives 1-2 and the alkyl enones 3-5 were very poor inhibitors of hCA II. On the other hand, the aromatic derivatives 6-7 were inhibitors acting in the micromolar range. However, being a ubiquitous, housekeeping isoform, this may not be a valuable property in another context if compounds targeting other isoforms (such as hCA IX and XII) should possess activity against hCA II. However, it is interesting to note that compounds 1-5 showed poor inhibition against hCA II while retaining effective inhibition against hCA IX and XII.
3.1.2. Cancer-associated CA isozymes
The tumour-associated target isoform hCA IX was inhibited in the submicromolar range by all enones except compound 7, which was a poor inhibitor of this isozyme. The best inhibitor was the C-galactoside 2, which weakly inhibited hCA II too.
The second tumour-associated isoform, hCA XII, was the most inhibited isoform by all vanillin derivatives. The phenyl enone 6 was the most active compound in the series.
Selectivity for inhibiting the tumour-associated isoforms (hCA IX and XII) over the widespread cytosolic forms (hCA I and II) is a key issue when designing CAIs. As can be in several compounds showed better activity profiles against hCA IX and XII over I and II which is highly desirable when only the tumour-associated isoforms would be targeted. It was observed that enones 1-5, which showed very good inhibition of isoforms IX and XII were also shown to be highly selective. The most effective hCA IX inhibitor 2 showed an excellent selectivity ratio over hCA II. Only aryl enones 6-7 showed almost no selectivity and are not useful in the design of selective inhibitors.
3.2. Molecular docking
It was observed, from the poses generated using molecular docking, that the various ligands presented different binding patterns for the analysed CA isozymes. Thus, in addition to ligand poses within the catalytic active site, the location of some poses indicates that the inhibitors might bind at an adjacent pocket, at the entrance of the active site, . This site was previously described for hCA II as an alternative site for inhibitor interaction, and only one carboxylic acid derivative was observed to bind in it by means of X-ray crystallographyCitation51. The hCA I active site is aligned among others by residues Asp1, Gly3, Tyr4, Asp5, Asn8, His61, Lys167, Ser228, and Met238. In the case of hCA II, the site is integrated, in addition to the previously reported residues by Gly6, Tyr7, Gly8, Asn11, His64, Phe231, Asn232, and Glu239, by Trp5, Gly63, and Lys170Citation51.
Figure 2. Binding poses of 7 to four CA isozymes obtained through molecular docking using Smina program. The ligand poses can be seen inside the catalytic site. In addition, some poses show the inhibitor bound in the adjacent pocket (oriented to the left) to the entrance at the active site cavity in the case of isoforms CA I, II, and XII.
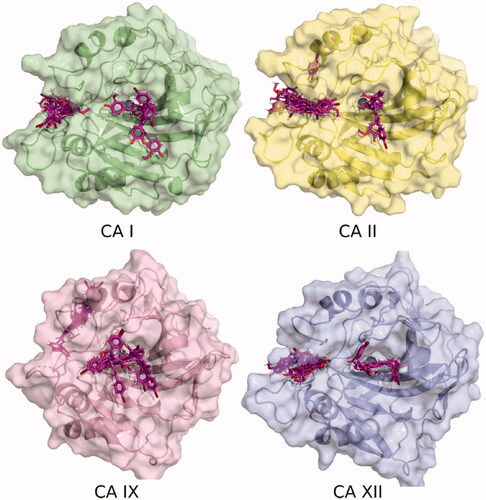
The hCA XII pocket is characterised by the presence of residues Lys1, Lys166, and Arg238, which are bulkier than the equivalent residues in hCA I and hCA II. In the case of hCA IX (PDB Id 6FE2), this pocket is absent and some docking poses are located close to this, on the rear face of the structure, as shown in .
3.2.1. Calculated binding enthalpies
In order to obtain more precise energies of the interaction with CAs, semi-empirical calculations were performed on the obtained docking poses. The binding enthalpies calculated with PM6 and the MOZYME approach are listed in . The energies were classified with a Python script according to the location of ligands, at the catalytic site or in the adjacent pocket, reporting the most stable energy value for each category when it was observed. In most cases, it was found that the poses in the alternative pocket have a lower (more favourable) binding enthalpy with respect to the location within the catalytic site. The exception to this observation is the CA IX isoform, where poses located at the catalytic site were more favourable than the out of the active site bindingCitation51.
3.2.2. Molecular dynamics and binding free energies
Although the ligand structures and nearby residues are optimised when applying the semiempirical methodology, this represents a "snapshot" of the enzyme-inhibitor complex. To analyse the "film" of the system, molecular dynamics simulations and subsequent estimation of the binding energy on the hCA I and hCA II isozymes were thereafter performed. The calculated binding free energies including the entropy as quasi-harmonic approximation are shown in . It was observed in most cases that the interaction in the alternative pocket was more stable than poses placing the inhibitor inside the catalytic cavity with the methoxyphenol derivative close to the Zn2+ cation, this situation being more favourable than the poses localising the inhibitor inside the catalytic cavity with the outward-facing methoxyphenol.
Table 3. Binding free energies (kcal mol−1) of ligands with hCA I and hCA II calculated with MM-GBSA.
A correlation was observed for the compounds that presented a more favourable calculated binding energy with respect to those that showed better inhibitory capacity in the experimental assays (compounds 3, 6, and 7 with isoform hCA I, and compounds 6 and 7 with isoform hCA II). In the case of compounds 1 and 2, the correlation was not good, probably due to the difficulty in modelling the carbohydrate fragment, which is structurally very flexible. It should be noted that entropy had to be included in the calculations despite the increased computational cost. Consequently, the entropic term was often neglected. However, from preliminary tests that were performed (data not shown) it was found that the relative order of stabilities between the possible binding sites was altered when only the enthalpic term was considered.
In order to analyse which residues could interact with the compounds, a per-residue decomposition of MM-GBSA binding energy was performed from molecular dynamics trajectories. shows this analysis for ligands located in the alternative pocket of CA I and CA II. In the case of the CA I pocket, there is a pronounced interaction with the residues Tyr7, Asp8, Gly63, His64, Lys170, Met241, and His243.
Figure 3. Per-residue decomposition of binding energy (only the enthalpy term considered) of ligand-bound hCA I and II, located in the pocket adjacent to the catalytic site. These calculations were performed with the MMPBSA.py module and plotted using Python's Matplotlib library. The residues shown were those that evidenced at least one interaction of < 0.3 kcal mol−1 with a ligand.
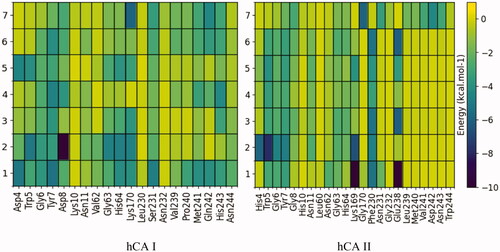
While the interaction in the CA II pocket occurs mainly with the residues Gly6, Tyr7, Gly63, and His64. It should be noted that 7, in contrast to the other compounds, also interacts with the residues Leu239, Met240, Val241, Asp242, and Asn243.
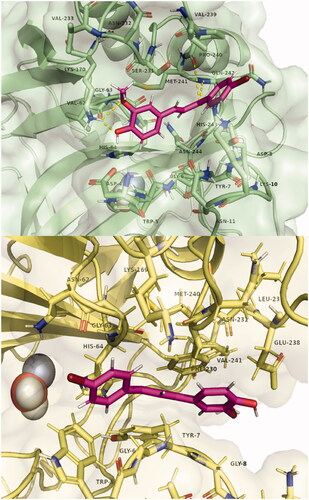
shows the three-dimensional structures of compound 7 in the alternative CA I and CA II pocket. A possible explanation that we put forward initially is the possibility that this alternative pocket competes with the binding within the catalytic site. Thus, those compounds that fit within this alternative pocket do not block the catalytic site and therefore do not inhibit CA by the canonical inhibition mechanism. Along these lines, the size of this pocket is reduced in the order: CA II, CA I, CA XII, and is absent in CA IX. Precisely in this last isoform, all compounds except 7, produced inhibition. However, computationally estimated binding energies do not support this hypothesis. From these results, the experimentally observed inhibition differences could be correlated with the interaction of the compounds with this alternative binding pocket at the entrance of the active site. Further studies, both computational and experimental, are warranted to fully reveal the mode of interaction and inhibition of these compounds with these CA isoforms.
Figure 4. Poses of compound 7 bound in the adjacent pocket to the active site entrance of hCA I (above) and hCA II (below). Hydrogen bonds with residues Lys170, Pro 240, and Gln242 are highlighted in yellow. Images were generated with Pymol. The selected structures of all ligands with CAs are presented in the Supplementary Material.
4. Conclusions
In conclusion, a small series of enones 1–7 have been prepared by aldol condensation of methyl ketones, including C-glycosylated ones with vanillin, being obtained in moderate yields. The compounds have been investigated as inhibitors against four isozymes of CA comprising the cytosolic, ubiquitous isozymes hCA I and II as well as the transmembrane, tumour-associated isoforms hCA IX and XII which are validated antitumor targets. In this study, C-glycosyl and alkyl enones derivated from vanillin, have been identified as selective inhibitors of hCA IX and XII. Molecular docking studies, quantum semiempirical calculations, and molecular dynamics simulations have been carried out in order to understand the inhibition profile of the compounds 1–7 with the different CA isozymes. A good correlation was found between the calculated binding free energies and the experimental inhibitory activity. An alternative pocket, already discovered for the binding of a carboxylic acid derivative several years agoCitation51, close to the active site entrance, is proposed as the site of interaction with these CAIs and could be useful in the design of potent and selective inhibitors. Further studies, both computational and experimental, are needed to fully validate the mode of interaction and inhibition of these compounds with these as well as other CA isoforms of pharmacological interest.
Supplemental Material
Download PDF (1.6 MB)5. Disclosure statement
CT Supuran is Editor-in-Chief of Journal of Enzyme Inhibition and Medicinal Chemistry and he was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.b) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.c) Mishra CB, Tiwari M, Supuran CT. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev 2020;40:2485–565.d) Supuran CT. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin Drug Discov 2020;15:671–86.
- a) Supuran CT. Emerging role of carbonic anhydrase inhibitors. Clin Sci 2021;135:1233–49.b) Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74.c) Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32.d) Supuran CT, Capasso C. Antibacterial carbonic anhydrase inhibitors: an update on the recent literature. Expert Opin Ther Pat 2020;30:963–82.
- a) Hirakawa Y, Senda M, Fukuda KY, et al. Characterization of a novel type of carbonic anhydrase that acts without metal cofactors. BMC Biol 2021;19:105.b) Del Prete S, Nocentini A, Supuran CT, Capasso C. Bacterial ι-carbonic anhydrase: a new active class of carbonic anhydrase identified in the genome of the Gram-negative bacterium Burkholderia territorii. J Enzyme Inhib Med Chem 2020;35:1060–8.c) Nocentini A, Supuran CT, Capasso C. An overview on the recently discovered iota-carbonic anhydrases. J Enzyme Inhib Med Chem 2021;36:1988–95.
- a) Pastorek J, Pastoreková S, Callebaut I, et al. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene 1994;9:2877–88.b) Türeci O, Sahin U, Vollmar E, et al. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc Natl Acad Sci USA 1998;95:7608–13.c) Svastová E, Hulíková A, Rafajová M, et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 2004;577:439–45.d) Supuran CT. Experimental carbonic anhydrase inhibitors for the treatment of hypoxic tumors. J Exp Pharmacol 2020;12:603–17.e) Supuran CT. Experimental carbonic anhydrase inhibitors for the treatment of hypoxic tumors. J Exp Pharmacol 2020;12:603–17.
- a) Nocentini A, Angeli A, Carta F, et al. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J Enzyme Inhib Med Chem 2021;36:561–80.b) Angeli A, Carta F, Nocentini A, et al. Carbonic anhydrase inhibitors targeting metabolism and tumor microenvironment. Metabolites 2020;10:412.c) McDonald PC, Chia S, Bedard PL, et al. A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am J Clin Oncol 2020;43:484–90.d) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.e) Chafe SC, Vizeacoumar FS, Venkateswaran G, et al. Genome-wide synthetic lethal screen unveils novel CAIX-NFS1/xCT axis as a targetable vulnerability in hypoxic solid tumors. Sci Adv 2021;7:eabj0364.
- Anand P, Thomas SG, Kunnumakkara AB, et al. Biological activities of curcumin and its analogues (Congeners) made by man and mother nature. Biochem Pharmacol 2008;76:1590–611.
- Surh YJ, Park KK, Chun KS, et al. Anti-tumor-promoting activities of selected pungent phenolic substances present in ginger. J Environ Pathol Toxicol Oncol 1999;18:131–9.
- Atanasov AG, Zotchev SB, Dirsch VM, et al. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov 2021;20:200–16.
- Kim MO, Lee M-H, Oi N, et al. [6]-Shogaol inhibits growth and induces apoptosis of non-small cell lung cancer cells by directly regulating Akt1/2. Carcinogenesis 2014;35:683–91.
- a) Karioti A, Ceruso M, Carta F, et al. New natural product carbonic anhydrase inhibitors incorporating phenol moieties. Bioorg Med Chem 2015;23:7219–25.b) D'Ambrosio K, Carradori S, Cesa S, et al. Catechols: a new class of carbonic anhydrase inhibitors. Chem Commun 2020;56:13033–6.
- a) Alissa SA, Alghulikah HA, ALOthman ZA, et al. Inhibition survey with phenolic compounds against the δ- and η-class carbonic anhydrases from the marine diatom thalassiosira weissflogii and protozoan Plasmodium falciparum. J Enzyme Inhib Med Chem 2020;35:377–82.b) Nocentini A, Bua S, Del Prete S, et al. Natural polyphenols selectively inhibit β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa: activity and modeling studies. ChemMedChem 2018;13:816–23.c) Karioti A, Carta F, Supuran CT. Phenols and polyphenols as carbonic anhydrase inhibitors. Molecules 2016;21:1649.
- a) Cau Y, Mori M, Supuran CT, Botta M. Mycobacterial carbonic anhydrase inhibition with phenolic acids and esters: kinetic and computational investigations. Org Biomol Chem 2016;14:8322–30.b) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.c) Supuran CT. Novel carbonic anhydrase inhibitors. Future Med Chem 2021.
- a) Innocenti A, Öztürk Sarikaya SB, Gülçin I, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of mammalian isoforms I-XIV with a series of natural product polyphenols and phenolic acids. Bioorg Med Chem 2010;18:2159–64.b) Taslimi P, Gülçin İ, Öztaşkın N, et al. The effects of some bromophenols on human carbonic anhydrase isoenzymes. J Enzyme Inhib Med Chem 2016;31:603–7.c) Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: inhibition of mammalian isoforms I-XIV with a series of substituted phenols including paracetamol and salicylic acid. Bioorg Med Chem 2008;16:7424–8.d) Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: interactions of phenols with the 12 catalytically active mammalian isoforms (CA I-XIV). Bioorg Med Chem Lett 2008;18:1583–7.
- An B, Zhang S, Yan J, et al. Synthesis, in vitro and in vivo evaluation of new hybrids of millepachine and phenstatin as potent tubulin polymerization inhibitors. Org Biomol Chem 2017;15:852–62.
- Heller L, Schwarz S, Perl V, et al. Incorporation of a Michael acceptor enhances the antitumor activity of triterpenoic acids. Eur J Med Chem 2015;101:391–9.
- Colinas PA. Novel glycomimetics: anomeric and N-glycosyl sulfonamides. Curr Org Chem 2012;16:1670–6.
- Winum JY, Colinas PA, Supuran CT. Glycosidic carbonic anhydrase IX inhibitors: a sweet approach against cancer. Bioorg Med Chem 2013;21:1419–26.
- Maresca A, Akyuz G, Osman SM, et al. Inhibition of mammalian carbonic anhydrase isoforms I-XIV with a series of phenolic acid esters. Bioorg Med Chem 2015;23:7181–8.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. J Biol Chem 1971;246:2561–73.
- a) Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996;31:1001–10.b) Eldehna WM, Abo-Ashour MF, Nocentini A, et al. Enhancement of the tail hydrophobic interactions within the carbonic anhydrase IX active site via structural extension: design and synthesis of novel N-substituted isatins-SLC-0111 hybrids as carbonic anhydrase inhibitors and antitumor agents. Eur J Med Chem 2019;162:147–60.c) Fares M, Eldehna WM, Bua S, et al. Discovery of potent dual-tailed benzenesulfonamide inhibitors of human carbonic anhydrases implicated in glaucoma and in vivo profiling of their intraocular pressure-lowering action. J Med Chem 2020;63:3317–26.d) Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun (Camb) 2012;48:1868–70.
- Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res 2000;28:235–42.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12.
- Hanwell MD, Curtis DE, Lonie DC, et al. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 2012;4:17.
- Stewart J, MOPAC2012.
- Klamt A, Schüürmann G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc Perkin Trans 2 1993;5:799–805.
- Koes DR, Baumgartner MP, Camacho CJ. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model 2013;53:1893–904.
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91.
- Delano LW. The PyMOL Molecular Graphics System. http://www.pymol.org. 2002. Available from: https://ci.nii.ac.jp/naid/10020095229/ [last accessed 31 May 2019].
- Allen WJ, Balius TE, Mukherjee S, et al. DOCK 6: impact of new features and current docking performance. J Comput Chem 2015;36:1132–56.
- Stewart JJP. Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model 2007;13:1173–213.
- Stewart JJP. Calculation of the geometry of a small protein using semiempirical methods. J Mol Struct 1997;401:195–205.
- Stewart JJP. Application of the PM6 method to modeling proteins. J Mol Model 2009;15:765–805.
- Vasilyev V, Bliznyuk A. Application of semiempirical quantum chemical methods as a scoring function in docking. Theor Chem Acc 2004;112:313–7.
- Bikadi Z, Hazai E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J Cheminform 2009;1:15.
- Nikitina E, Sulimov V, Zayets V, Zaitseva N. Semiempirical calculations of binding enthalpy for protein-ligand complexes. Int J Quantum Chem 2004;97:747–63.
- Maier JA, Martinez C, Kasavajhala K, et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 2015;11:3696–713.
- Wang J, Wolf RM, Caldwell JW, et al. Development and testing of a general Amber force field. J Comput Chem 2004;25:1157–74.
- Case Ross C Walker DA, Darden Junmei Wang T. Amber 2016 Reference Manual Principal Contributors to the Current Codes. Available from: http://ambermd.org/contributors.html [last accessed 20 April 2021].
- Li P, Merz KM. MCPB.py: a python based metal center parameter builder. J Chem Inf Model 2016;56:599–604.
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem 2005;26:1781–802.
- Srinivasan J, Thomas E, Cheatham I, et al. Continuum solvent studies of the stability of DNA. RNA Phosphoramidate DNA Helices. J Biomol Struct Dyn. 1998;16:671–82.
- Vorobjev YN, Hermans J. ES/IS: estimation of conformational free energy by combining dynamics simulations with explicit solvent with an implicit solvent continuum model. Biophys Chem 1999;78:195–205.
- Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc 1990;112:6127–9.
- Miller BR, McGee TD, Swails JM, et al. MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput 2012;8:3314–21.
- Rodrigues F, Canac Y, Lubineau A. A convenient, one-step, synthesis of β-C-glycosidic ketones in aqueous media. Chem Commun 2000;20:2049–50.
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62.
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44.
- Temperini C, Innocenti A, Scozzafava A, et al. The coumarin-binding site in carbonic anhydrase accommodates structurally diverse inhibitors: the antiepileptic lacosamide as an example and lead molecule for novel classes of carbonic anhydrase inhibitors. J Med Chem 2010;53:850–4.
- Maresca A, Supuran CT. Coumarins incorporating hydroxy- and chloro-moieties selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones i and II. Bioorg Med Chem Lett 2010;20:4511–4.
- Maresca A, Scozzafava A, Supuran CT. 7,8-disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8.
- D'Ambrosio K, Carradori S, Monti SM, et al. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem Commun (Camb) 2015;51:302–5.