
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Pyrroles and its fused forms possess antimicrobial activities, they can easily interact with biomolecules of living systems. A series of substituted pyrroles, and its fused pyrimidines and triazines forms have been synthesised, all newly synthesised compound structures were confirmed by spectroscopic analysis. Generally, the compounds inhibited growth of some important human pathogens, the best effect was given by: 2a, 3c, 4d on Gram-positive bacteria and was higher on yeast (C. albicans), by 5c on Gram-negative bacteria and by 5a then 3c on filamentous fungi (A. fumigatus and F. oxysporum). Such results present good antibacterial and antifungal potential candidates to help overcome the global problem of antibiotic resistance and opportunistic infections outbreak. Compound 3c gave the best anti-phytopathogenic effect at a 50-fold lower concentration than Kocide 2000, introducing a safe commercial candidate for agricultural use. The effect of the compounds on DNA was monitored to detect the mode of action.
1. Introduction
Pathogenic bacteria are a major public health burden because they cause morbidity, mortality, and moreover increased healthcare costs. Moreover, the rapid evolution of resistance to antimicrobial agents puts an extra loadCitation1. Some of the most serious and resistant organisms are Staphylococcus aureus (S. aureus)Citation1 and Escherichia coli (E. coli) O157:H7 that belongs to the enterohemorrhagic E. coli (EHEC) classCitation2, and Pseudomonas aeruginosa (P. aeruginosa)Citation3 that represents the most common cause of nosocomial infections (OIs)Citation4.
Another major cause of OIs and the consequent fatality is the fungal pathogens. The most common opportunistic fungi include Candida species, Aspergillus species, and Fusarium speciesCitation5. Candida species, was a major cause of morbidity and mortality worldwide over the past few decadesCitation6. Aspergillus fumigatus (A. fumigatus) is a saprophyte that has become the most prevalent airborne fungal pathogenCitation7. A. fumigatus causes severe and usually fatal invasive aspergillosis in immuno-compromised hosts in developed countriesCitation7. The members of Fusarium oxysporum (F. oxysporum) complex, (FOSC) are soil borne pathogens that cause diseases to a broad range of important crops with a number of them having the ability to cause human diseases that can be fatalCitation8. Because fungi are eukaryotes, they have similarities with mammalian cells making it difficult to design a drug with selective toxicityCitation9. The first antifungal drug discovered was amphotericin BCitation10 followed by flucytosineCitation11, later on two imidazole derivatives, clotrimazole and miconazole, were extensively used in treatment of fungal infectionsCitation12–15. Triazole derivative, fluconazole, is an antifungal agent with low toxicity and high antifungal activityCitation16. Resistance of some fungi against imidazole is now becoming a serious clinical problemCitation17, several azole-related adverse drug effects (ADEs) are now considered such as hepatotoxicity and cardiac effects. In order to enrich the antifungal activity and/or diminish antifungal ADEs, new derivatives were synthesised containing other heterocycles instead of imidazole and 1,2,4-triazole, such as indole, 1,2,3-triazolesCitation14,Citation18–20. Voriconazole is a clinically important triazoles antifungal containing fluoropyrimidineCitation13,Citation20. Using quinazoline ring instead of a triazole ring results in albaconazole with higher in vitro activity than fluconazoleCitation21. Itraconazole with piperazinyl phenyl side chain showed remarkable in vitro antifungal activity against several pathogenic fungiCitation16, as shown in .
Scheme 1. Synthesis of targeted compounds 2–6.
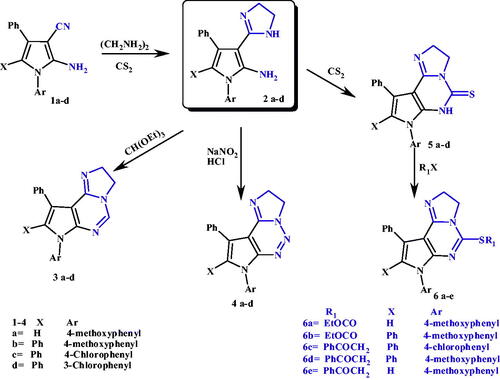
Figure 1. Azoles and triazoles as antimicrobial agents.
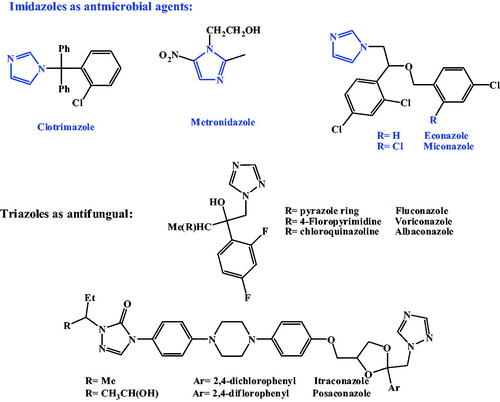
Pyrroles and its fused derivatives, are an important class of naturallyCitation22,Citation23 and syntheticallyCitation24–26 occurring compounds with a wide-range of biological activities; antibacterialCitation27–29, antifungalCitation30–33, antiviralCitation34–39, anticancerCitation40,Citation41 and anti-inflammatoryCitation42,Citation43. Pyrrolnitin and fludioxonil, are two naturally secreted pyrroles, reported to bear a broad spectrum antifungal activitiesCitation44–46. 7-deazapurine, naturally secreted pyrrolopyrimidines antibiotics, toyocamycin, tubercidin and sangivamycin, commonly have antibacterial, antifungal, anticancer, antiviral and anti-inflammatory activitiesCitation22,Citation47. Due to structural resemblance to purine, 7-deazapurines interfered with various cellular processes; toyocamycin united with tRNA, pyrrolopyrimidine inhibits tRNA aminoacylation. Sangivamycin has lately been revealed to inhibit protein kinasesCitation22, as revealed in .
Figure 2. pyrroles and pyrrolopyrimidines as bioactive compounds.
Health organisations, such as the WHO, CDC and FDA are considering the growing antimicrobial resistance a potential threat for human healthCitation48. Fungi also are showing a growing resistance with increased number of fatal systemic mycosesCitation9. That’s why the discovery of new effective antifungal and antibacterial drugs has become a high priority, and this is the aim of our study through preparation and testing of novel 3-imidazolinyl pyrroles, its fused form (namely; pyrimidnes and triazines).
2. Materials and methods
2.1. Synthesis of lead compounds
All commercial chemicals used as starting materials and reagents in this study were purchased from Merck (Darmstadt, Germany) and were of reagent grade. All melting points were uncorrected and measured using Electro-thermal IA 9100 apparatus (Shimadzu, Japan); IR spectra were recorded as potassium bromide pellets on a Perkin-Elmer 1650 spectrophotometer (USA), Faculty of Science, Cairo University, Cairo, Egypt. 1H-NMR spectra were determined on a Varian Mercury (300 MHz) spectrometer (Varian UK) and chemical shifts were expressed as ppm against TMS as internal reference (The Main Chemical Warfare Laboratories, Almaza, Cairo, Egypt). Mass spectra were recorded on 70 eV (EI Ms-QP 1000 EX, Shimadzu, Japan), Faculty of Science, Cairo University, Cairo, Egypt. Microanalyses were operated using Vario, Elmentar apparatus (Shimadzu, Japan), Organic Microanalysis Unit, Faculty of Science, Cairo University, Cairo, Egypt. Column Chromatography was performed on (Merck) Silica gel 60 (particle size 0.06-0.20 mm). All the listed compounds are new except compounds 1a-d was previously reportedCitation38,Citation49–51.
General procedure for the synthesis of compounds 2a-d:
To a mixture of 1a-d (5 mmol) and ethylenediamine (7.5 mL) carbon disulphide (0.7 mL) was added dropwise, and the reaction mixture was heated under reflux for 4 h. After cooling the reaction mixture was poured into cold water and the precipitate obtained was filtered off, washed with water, dried and crystallized from ethanol to yield compounds 2a-d
3–(4,5-Dihydro-1H-imidazol-2-yl)-1–(4-methoxyphenyl)-4-phenyl-pyrrol-2-amine (2a): Yield: 87%; m.p.: 187–189 °C; IR (KBr) υ (cm−1): 3347 (NH)), 3137 (NH2); MS (EI) m/z: 332 (M+, 34.24%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.02 (t, 2H, J = 8.1 Hz, CH2 imidazoline), 3.39 (t, 2H, J = 8.1 Hz, CH2 imidazoline), 3.45 (s, 3H, OCH3), 4.34 (s, 2H, NH2, D2O exchangeable), 6.72–8.47 (m, 10H, Ar-H), 9.64 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C20H20N4O (332.4): C, 72.29; H, 6.02; N, 16.87%. Found: C, 72.49; H, 6.31; N, 16.54%.
3–(4,5-Dihydro-1H-imidazol-2-yl)-1–(4-methoxyphenyl)-4,5-diphenyl-pyrrol-2-amine (2 b): Yield: 81%; m.p.: 251–253 °C; IR (KBr) υ (cm−1): 3290 (NH)), 3122 (NH2); MS (EI) m/z: 408 (M+, 67.19%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.37 (t, 2H, J = 8.4 Hz, CH2 imidazoline), 4.22 (s, 3H, OCH3), 4.57 (t, 2H, J = 8.4 Hz, CH2 imidazoline), 5.72 (s, 2H, NH2, D2O exchangeable), 7.01–8.37 (m, 14H, Ar-H), 8.84 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C26H24N4O (408.4): C, 76.47; H, 5.88; N, 13.73%. Found: C, 76.29; H, 6.03; N, 13.74%.
1–(4-Chlorophenyl)-3–(4,5-dihydro-1H-imidazol-2-yl)-4,5-diphenyl-pyrrol-2-amine (2c): Yield: 77%; m.p.: 210–212 °C; IR (KBr) υ (cm−1): 3403 (NH)), 3282 (NH2); MS (EI) m/z: 412 (M+, 19.2%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.53 (t, 2H, J = 8.2 Hz, CH2 imidazoline), 3.44 (t, 2H, J = 8.2 Hz, CH2 imidazoline), 5.20 (s, 2H, NH2, D2O exchangeable), 6.53–7.92 (m, 14H, Ar-H), 9.05 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C25H21ClN4 (412.14): C, 72.82; H, 5.10; N, 13.59%. Found: C, 73.08; H, 5.07; N, 13.66%.
1–(3-Chlorophenyl)-3–(4,5-dihydro-1H-imidazol-2-yl)-4,5-diphenyl-pyrrol-2-amine (2d): Yield: 71%; m.p.: 205–207 °C; IR (KBr) υ (cm−1): 3337 (NH)), 3271 (NH2); MS (EI) m/z: 412 (M+, 22.52%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.53 (t, 2H, J = 8.4 Hz, CH2 imidazoline), 3.51 (t, 2H, J = 8.4 Hz, CH2 imidazoline), 5.87 (s, 2H, NH2, D2O exchangeable), 6.97–8.14 (m, 14H, Ar-H), 8.94 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C25H21ClN4 (412.14): C, 72.82; H, 5.10; N, 13.59%. Found: C, 72.94; H, 5.18; N, 13.34%.
General procedure for the synthesis of compounds 3a-d:
A mixture of 2a-d (5 mmol) and triethyl orthoformate (15 ml) was heated under reflux for 3 h. After cooling, the reaction mixture was poured into cold water. The solid precipitate was filtered off, washed with water, dried and recrystallized from dioxane to give 3a-d
7–(4-methoxyphenyl)-9-phenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidine (3a): Yield: 90%; m.p.: 138–140 °C; IR (KBr) υ (cm−1): 1596 (C = N); MS (EI) m/z: 342 (M+, 72.03%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.43 (t, 2H, J = 7.3 Hz, CH2 imidazoline), 3.97 (s, 3H, OCH3), 4.14 (t, 2H, J = 7.3 Hz, CH2 imidazoline), 6.91–7.34 (m, 11H, Ar-H); Anal. Calcd. for C21H18N4O (342.14): C, 73.68; H, 5.26; N, 16.37%. Found: C, 73.43; H, 5.11; N, 16.61%.
7–(4-methoxyphenyl)-8,9-diphenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidine (3 b): Yield: 88%; m.p.: 193–195 °C; IR (KBr) υ (cm−1): 1580 (C = N); MS (EI) m/z: 418 (M+, 8.99%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.55 (t, 2H, J = 8.6 Hz, CH2 imidazoline), 2.63 (t, 2H, J = 8.6 Hz, CH2 imidazoline), 4.38 (s, 3H, OCH3), 6.73–8.16 (m, 15H, Ar-H); Anal. Calcd. for C27H22N4O (418.17): C, 77.51; H, 5.26; N, 13.40%. Found: C, 77.41; H, 5.36; N, 13.62%.
7–(4-Chlorophenyl)-8,9-diphenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidine (3c): Yield: 82%; m.p.: 178–180 °C; IR (KBr) υ (cm−1): 1598 (C = N); MS (EI) m/z: 422 (M+, 10.37%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.36 (t, 2H, J = 7.4 Hz, CH2 imidazoline), 3.44 (t, 2H, J = 7.4 Hz, CH2 imidazoline), 6.61–7.82 (m, 15H, Ar-H); Anal. Calcd. for C26H19ClN4 (422.12): C, 73.93; H, 4.50; N, 13.27%. Found: C, 73.71; H, 4.44; N, 13.32%.
7–(3-Chlorophenyl)-8,9-diphenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidine (3d): Yield: 75%; m.p.: 138–140 °C; IR (KBr) υ (cm−1): 1603 (C = N); MS (EI) m/z: 422 (M+, 54.17%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.15 (t, 2H, J = 7.3 Hz, CH2 imidazoline), 3.99 (t, 2H, J = 7.3 Hz, CH2 imidazoline), 7.69–8.08 (m, 15H, Ar-H); Anal. Calcd. for C26H19ClN4 (422.12): C, 73.93; H, 4.50; N, 13.27%. Found: C, 74.04; H, 4.38; N, 13.02%.
General procedure for the synthesis of compounds 4a-d:
To a cold solution of 2a-d (5 mmol) in conc. hydrochloric acid (15 ml) and acetic acid (15 ml) a solution of sodium nitrite (2 g) in water (15 ml) was added portion-wise. After completion of the addition (30 min), the ice bath was removed and stirring was continued for 2 h. The solid product was filtered off and recrystallized from dioxane to afford 4a-d
7–(4-methoxyphenyl)-9-phenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e][1,2,3]triazine (4a): Yield: 85%; m.p.: 208–210 °C; IR (KBr) υ (cm−1): 1612 (C = N); MS (EI) m/z: 343 (M+, 18%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.18 (t, 2H, J = 8 Hz, CH2 imidazoline), 3.39 (t, 2H, J = 8 Hz, CH2 imidazoline), 4.55 (s, 3H, OCH3), 6.87–8.06 (m, 10H, Ar-H); Anal. Calcd. for C20H17N5O (343.14): C, 69.97; H, 4.96; N, 20.41%. Found: C, 70.04; H, 5.10; N, 20.23%.
7–(4-methoxyphenyl)-8,9-diphenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e][1,2,3]triazine (4 b): Yield: 86%; m.p.: 204–206 °C; IR (KBr) υ (cm−1): 1592(C = N); MS (EI) m/z: 419 (M+, 33.5%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.28 (t, 2H, J = 7.6 Hz, CH2 imidazoline), 3.75 (s, 3H, OCH3), 4.23(t, 2H, J = 7.6 Hz, CH2 imidazoline), 6.71–8.32 (m, 14H, Ar-H); Anal. Calcd. for C26H21N5O (419.17): C, 74.46; H, 5.01; N, 16.71%. Found: C, 74.33; H, 5.12; N, 16.56%.
7–(4-Chlorophenyl)-8,9-diphenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e][1,2,3]triazine (4c): Yield: 73%; m.p.: 159–161 °C; IR (KBr) υ (cm−1): 1595 (C = N); MS (EI) m/z: 423 (M+, 26.9%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.36 (t, 2H, J = 8.1 Hz, CH2 imidazoline), 3.43 (t, 2H, J = 8.1 Hz, CH2 imidazoline), 7.05–7.82 (m, 14H, Ar-H); Anal. Calcd. for C25H18ClN5 (423.13): C, 70.92; H, 4.26; N, 16.55%. Found: C, 70.78; H, 4.49; N, 16.30%.
7–(3-Chlorophenyl)-8,9-diphenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e][1,2,3]triazine (4d): Yield: 78%; m.p.: 183–185 °C; IR (KBr) υ (cm−1): 1607 (C = N); MS (EI) m/z: 423 (M+, 30.17%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.53 (t, 2H, J = 7.4 Hz, CH2 imidazoline), 3.43 (t, 2H, J = 7.4 Hz, CH2 imidazoline), 6.82–7.65 (m, 14H, Ar-H); Anal. Calcd. for C25H18ClN5 (423.13): C, 70.92; H, 4.26; N, 16.55%. Found: C, 70.76; H, 4.11; N, 16.60%.
General procedure for the synthesis of compounds 5a-d:
A mixture of 2a-d (5 mmol) and carbon disulphide (15 ml) in dry pyridine (50 ml) was heated under reflux for 10 h. The solid product formed on hot was filtered, washed with water, dried and crystallised from dioxane-water (2: 1) to afford 5a-d
7–(4-methoxyphenyl)-9-phenyl-6,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidine-5(3H)-thione (5a): Yield: 70%; m.p.: 210–212 °C; IR (KBr) υ (cm−1): 3214 (NH), 1606 (C = N), 1490, 1260, 1025, 800 (C = S); MS (EI) m/z: 374 (M+, 39.2%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.32 (t, 2H, J = 7.2 Hz, CH2 imidazoline), 4.36 (s, 3H, OCH3), 4.56 (t, 2H, J = 7.2 Hz, CH2 imidazoline), 7.10–8.45 (m, 10H, Ar-H), 8.88 (s, 1H, NH D2O exchangeable); Anal. Calcd. for C21H18N4OS (374.12): C, 67.38; H, 4.81; N, 14.97%. Found: C, 67.09; H, 4.91; N, 15.13%.
7–(4-methoxyphenyl)-8,9-diphenyl-6,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidine-5(3H)-thione (5 b): Yield: 71%; m.p.: 187–189 °C; IR (KBr) υ (cm−1): 3307 (NH), 1586 (C = N), 1492, 1280, 1010, 800 (C = S); MS (EI) m/z: 450 (M+, 76.28%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.53 (t, 2H, J = 7.2 Hz, CH2 imidazoline), 2.65 (t, 2H, J = 7.2 Hz, CH2 imidazoline), 3.56 (s, 3H, OCH3), 7.18–7.93 (m, 14H, Ar-H), 8.69 (s, 1H, NH D2O exchangeable); Anal. Calcd. for C27H22N4OS (450.15): C, 72.0; H, 4.89; N, 12.44%. Found: C, 72.13; H, 5.08; N, 12.28%.
7–(4-Chlorophenyl)-8,9-diphenyl-6,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidine-5(3H)-thione (5c): Yield: 69%; m.p.: 138–140 °C; IR (KBr) υ (cm−1): 3214 (NH), 1606 (C = N), 1488, 1273, 1017, 805 (C = S); MS (EI) m/z: 454 (M+, 71.9%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.69 (t, 2H, J = 8.1 Hz, CH2 imidazoline), 3.43 (t, 2H, J = 8.1 Hz, CH2 imidazoline), 7.01–8.13 (m, 14H, Ar-H), 12.17 (s, 1H, NH D2O exchangeable); Anal. Calcd. for C26H19ClN4S (454.1): C, 68.72; H, 4.19; N, 12.33%. Found: C, 68.78; H, 4.41; N, 12.20%.
7–(3-Chlorophenyl)-8,9-diphenyl-6,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidine-5(3H)-thione (5d): Yield: 65%; m.p.: 172–174 °C; IR (KBr) υ (cm−1): 3309 (NH), 1585 (C = N), 1479, 1283, 1020, 798 (C = S); MS (EI) m/z: 454 (M+, 44.19%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.48 (t, 2H, J = 7.8 Hz, CH2 imidazoline), 3.32 (t, 2H, J = 7.8 Hz, CH2 imidazoline), 6.92–7.61 (m, 14H, Ar-H), 8.65 (s, 1H, NH D2O exchangeable); Anal. Calcd. for C26H19ClN4S (454.1): C, 68.72; H, 4.19; N, 12.33%. Found: C, 68.80; H, 4.21; N, 12.25%.
General procedure for the synthesis of compounds 6a-e:
A mixture of 5a-c (1 mmol), the appropriate halo derivative (ethyl chloroformate, ethyl chloroacetate, phenacyl bromide) (1 mmol) and sodium acetate (0.6 g) in ethanol (40 ml) was heated under reflux for 3 h. The reaction mixture was then cooled and poured into water. The solid precipitate was filtered off, washed with water, dried and recrystallized from ethanol to give 6a-e
O-ethyl S-7–(4-methoxyphenyl)-9-phenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e] pyrimidin-5-yl carbonothioate (6a): Yield: 72%; m.p.: 180–182 °C; IR (KBr) υ (cm−1): 1765 (C = O), 1606 (C = N); MS (EI) m/z: 446 (M+, 20.2%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 1.21 (t, 3H, J = 7.5 Hz, CH3*-CH2), 2.50 (t, 2H, J = 7.8 Hz, CH2 imidazoline), 3.39 (q, 2H, J = 7.5 Hz, CH2*-CH3), 3.54 (s, 3H, OCH3), 3.77 (t, 2H, J = 7.8 Hz, CH2 imidazoline), 6.98–7.54 (m, 10H, Ar-H); Anal. Calcd. for C24H22N4O3S (446.14): C, 64.57; H, 4.93; N, 12.56%. Found: C, 64.29; H, 4.90; N, 12.88%.
O-ethyl S-7–(4-methoxyphenyl)-8,9-diphenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidin-5-yl carbonothioate (6 b): Yield: 78%; m.p.: 163–165 °C; IR (KBr) υ (cm−1): 1720 (C = O), 1616 (C = N); MS (EI) m/z: 536 (M+, 13.42%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 1.24 (t, 3H, J = 7.4 Hz, H3*-CH2), 2.64 (s, 2H, CH2-S), 3.37 (t, 2H, J = 8.2 Hz, CH2 imidazoline), 3.96 (q, 2H, J = 7.4 Hz, CH2*-CH3), 4.32 (s, 3H, OCH3), 4.57 (t, 2H, J = 8.2 Hz, CH2 imidazoline), 7.01–8.86 (m, 14H, Ar-H); Anal. Calcd. for C31H28N4O3S (536.18): C, 69.40; H, 5.22; N, 10.45%. Found: C, 69.19; H, 5.40; N, 10.23%.
2–(7-(4-chlorophenyl)-8,9-diphenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidin-5-ylthio)-1-phenylethanone (6c): Yield: 68%; m.p.: 165–167 °C; IR (KBr) υ (cm−1): 1734 (C = O), 1599 (C = N); MS (EI) m/z: 572 (M+, 18.9%); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.92 (s, 2H, CH2-S), 4.16 (t, 2H, J = 7.5 Hz, CH2 imidazoline), 4.31 (t, 2H, J = 7.5 Hz, CH2 imidazoline), 6.69–7.43 (m, 19H, Ar-H); Anal. Calcd. for C34H25ClN4OS (572.14): C, 71.33; H, 4.37; N, 9.79%. Found: C, 71.57; H, 4.49; N, 9.52%.
2–(7-(4-methoxyphenyl)-8,9-diphenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidin-5-ylthio)-1-phenylethanone (6d): Yield: 73%; m.p.: 150–152 °C; IR (KBr) υ (cm−1): 1719 (C = O), 1594 (C = N); MS (EI) m/z: 568 (M+, 65.38); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.48 (t, 2H, J = 8 Hz, CH2 imidazoline), 3.35 (s, 3H, OCH3), 3.97 (t, 2H, J = 8 Hz, CH2 imidazoline), 4.08 (s, 2H, S-CH2-C = O), 6.72–7.60 (m, 19H, Ar-H); Anal. Calcd. for C35H28N4O2S (568.19): C, 73.94; H, 4.93; N, 9.86%. Found: C, 73.80; H, 5.02; N, 9.75%.
2–(7-(4-methoxyphenyl)-9-phenyl-3,7-dihydro-2H-imidazo[1,2-c]pyrrolo[3,2-e]pyrimidin-5-ylthio)-1-phenylethanone (6e): Yield: 80%; m.p.: 171–173 °C; IR (KBr) υ (cm−1): 1721 (C = O), 1587 (C = N); MS (EI) m/z: 492 (M+, 17.33); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.53 (t, 2H, J = 7.2 Hz, CH2 imidazoline), 3.35 (s, 3H, OCH3), 3.73 (t, 2H, J = 7.2 Hz, CH2 imidazoline), 4.09 (s, 2H, S-CH2-C = O), 6.580–7.51 (m, 15H, Ar-H); Anal. Calcd. for C29H24N4O2S (492.16): C, 70.73; H, 4.88; N, 11.38%. Found: C, 70.99; H, 4.70; N, 11.67%.
2.2. Biological method
2.2.1. Microbial strains media, reagents and chemicals
Bacterial strains used were two strains of Gram-negative bacteria, E. coli O157:H7 ATCC 51659 and P. aeruginosa ATCC 27853 and two strains of Gram-positive bacteria: S. aureus ATCC9144 and B. subtilis ATCC 6051. One strain of unicellular fungi (yeast) was used: Candida albicans (C. albicans) ATCC 90028 and also two multicellular filamentous fungi: F. oxysporum and A. fumigatus.
Antibiotics were purchased commercially. All media were prepared and sterilised by autoclaving at 121 °C for 15 min. Nutrient agar (Merck, Germany), nutrient broth, Trypticase soy broth and agar (Biolife, Italy) were used for bacteria. Sabouraud dextrose agar (SDA) and SD broth were used for C. albicans. Potato dextrose agar (PDA) medium was used for the filamentous fungal species (F. oxysporum and A. fumigatus). The pH value of the PDA and SDA is 5.6 ± 0.2 at 25 °C. The pH values of the nutrient agar, broth, and trypticase soy broth media are (6.9, and 7.3 ± 0.2) respectively. All chemical compounds to be tested were dissolved in DMSO. The PH of the tested compounds was neutral.
2.2.2. Determination of sensitivity of some Gram-positive, gram-negative bacteria and C. albicans to the synthesised compounds
The sensitivity was determined according to Kirby–Bauer disc diffusion methodCitation52,Citation53. Suspensions of bacterial and fungal standard strains were prepared from fresh slants in 3 ml sterile saline solution and the suspensions were adjusted to 0.5 McFarland (1–2 × 108 cfu/mL) then inoculated (5 drops) uniformly in nutrient agar plates for bacteria and SDA plates for C. albicans. Dried filter paper discs loaded with a standard amount (1000–2000 µg/mL) of the test compounds were applied to the surface of the plate. The plates were incubated at (37 °C for 24 h for bacteria) and (28 °C for 48 h for fungi) and sensitivity was determined by presence of zones of inhibition of microbial growth around the test compound disc. DMSO-loaded disc was used as a negative control. Ciprofloxacin disc was used as a standard antibacterial drug, and amphotericin B disc was used as a standard antifungal one. All experiments were conducted in triplicate and the mean values of the inhibition zones were calculated.
2.2.3. Determination of minimum inhibitory concentration (MIC) of the synthesised compounds against some Gram-positive, Gram-negative bacteria and C. albicans
The MICs of the tested compounds and control drugs against relevant bacterial and fungal strains were determined using the broth macrodilution method, according to guidelines outlined by the Clinical and Laboratory Standards InstituteCitation53–55.
Bacterial strains were grown aerobically overnight on nutrient agar at 37 °C, C. albicans was grown aerobically for 48 h. on SDA at 35 °C. Afterwards, suspensions of bacterial and fungal standard strains were prepared in 3 ml sterile saline solution and adjusted to 0.5 McFarland (1–2 × 108 cfu/mL). Tested compounds used to prepare stock solutions in DMSO at concentration of (1000 µg/mL) each. Further, test compounds were serially diluted in the corresponding culture medium (Trypticase soy broth and nutrient broth for antibacterial and SD broth for antifungal) in test tubes to achieve concentrations ranging from 50 to 400 µg/mL. Finally, the different strains were inoculated at 5 × 105 cfu/mL final concentration. The tubes were incubated aerobically at (37 °C for 24 h for bacteria) and (28 °C for 48 h for fungi) and the MICs were determined by visually assessing turbidity after the specified time. MIC was defined as the lowest concentration that resulted in complete lack of turbidity after the specified incubation period was conducted. For each tested compound, triplicate measurements were conducted. The mean MIC values, standard deviation (SD) and statistical differences were calculated using GraphPad Prism version 5 for Windows (GraphPad Inc., USA). DMSO was used as a negative control. Ciprofloxacin was used as standard antibacterial drug in bacteria experiments and amphotericin B was used as a standard antifungal one.
2.2.4. Molecular identification of the filamentous fungal species
2.2.4.1 DNA isolation of the filamentous fungal species
Molecular identification was carried out on the two filamentous fungal species used in this study. In summary, the genomic DNA was isolated by using a mortar and pestle to grind approximately 100 mg of frozen fungal mycelia with liquid nitrogen and mixing with 1 ml of 4 M guanidinium thiocyanate, 0.1 M sodium acetate pH 5.5, 10 mM ethylene-di-amine-tetra acetic acid (EDTA), 0.1 M 2-mercaptoethanol. Centrifugation of the extracts and the supernatants were loaded into a silica gel spin column (Wizard Plus SV Minipreps DNA Purification, Promega, USA). Finally, columns were washed with 70% ethanol, 10 mM sodium acetate pH 5.5 and DNA eluted with 50 µL of 20 mM Tris–HCl, pH 8.5.
2.2.4.2 Ribosomal DNA amplification and sequencing of the filamentous fungal species
PCR was carried out to amplify the ribosomal internal transcribed sequence (ITS) using primer pairs ITS1 (5′-CTTGGTC ATTTAGAGGAAGTAA-3′) and ITS4 (5′-TCCTCCGC TTATTGATATGC-3′)Citation56. Fungal DNA (1 µL) was amplified in a 20 µL reaction with 0.4 U Phusion DNA polymerase (Thermo Scientific) in the presence of 0.2 mM dNTPs, 3% dimethyl sulfoxide, 0.5 µM each primer and HF buffer (Thermo Scientific). Reaction consisted of an initial denaturation for 30 s at 98 °C, followed by 30 cycles of 10 s at 98 °C, 30 s at 55 °C and 30 s at 72 °C, and a final extension of 10 min at 72 °C. PCR products were separated by electrophoresis in a 1% agarose gel run for 75 min in buffer TAE (40 mM Tris, 20 mM sodium acetate, 1 mM EDTA, pH 7.2) and visualised using a UV transilluminator after ethidium bromide staining. The PCR products corresponding to ribosomal ITS, according to electrophoretic migration, were eluted from the gel using silica spin columns (DNA Clean and Concentrator, Zymo Research). Both strands of amplified ribosomal ITS DNA were sequenced using primers ITS1 and ITS4. The consensus sequence was used to search for homologous sequences using the BLAST search program at National Centre for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov).
2.2.5. In vitro antifungal activity of the synthetic compounds against two filamentous fungal species
Poisoned food techniqueCitation57 was used to measure the antifungal activity of the newly synthesised compounds on two different pathogenic filamentous fungal species (F. oxysporum and A. fumigatus) that cause human and plant diseases as well. In summary, three different concentrations (1, 10, 50) µg/mL for each compound were used separately to supplement the agar medium, then from each of fungal species a (0.5 cm) disc of the active mycelial growth, eight-day-old fungal culture, was placed in the centre of each prepared plate. Finally, plates were incubated at 26 °C ± 2 for 8 days. The antifungal effect of synthetic compound was evaluated by measuring the radial growth of fungal growth in each inoculated plateCitation58 and calculating the inhibitory growth rate:
“where R is the radial growth of fungal hyphae on the control plate and r is the radial growth of fungal hyphae on the plate supplemented with the synthetic compound”. Itraconazole (Johnson & Johnson, New Jersey, USA) and Kocide 2000 (Certis USA, Columbia, MD) were used as positive control. The experiments were repeated three times. The mean values of Inhibitory growth rate (%), standard deviation (SD) and statistical differences were calculated using GraphPad Prism version 5 for Windows (GraphPad Inc., USA).
2.2.6. The effect of the tested compounds on some selected species nucleic acid
2.2.6.1 The effect on the bacterial nucleic acid
DNA damage analysis was performed on P. aeruginosa ATCC 27853 according toCitation59 as a representative of the bacterial strains used in this study. Total DNA was isolated from P. aeruginosa cells after three hours treatment with each the MIC value of each tested compound individually. Isolation of P. aeruginosa DNA and polymerase chain reaction (PCR) amplification of the 16S–23SrDNA intergenic transcribed spacer Isolated from each microcosm were cultivated on LB agar for 24 h at 30 W C, then the total nucleic acids were extracted by means of the InstaGene™ Matrix (bio-Rad Laboratories) according to the protocol described by the manufacturer. The corresponding DNA encoding 16S–23S rDNA intergenic transcribed spacer (ITS) was amplified from genomic DNA using forward primer Paer1, 5′-TCCAAA CAATCGAAAGC-3′ and reverse primer Paer2, 5′- CCGAAAACTTGCGCTTGAAC-3′. All of the amplifications were performed in a 100 PTC- thermal cycler, as previously reported (Tyler et al. 1995)Citation60. The reaction mixture (50 μL) included 1x reaction buffer (5 μL), 0.2 mM of each deoxynucleoside triphosphate, 1 μM of forward and reverse primer, 1 μL of the DNA template, 1.5 mM of MgCl2, and 2.5 U of Taq DNA polymerase. Each PCR reaction consisted of 30 amplification cycles, with an initial denaturation of 2 min at 94 °C followed successively by denaturation (30 s) at 94 °C, annealing (30 s) at 60 °C, and extension (1 min), ending with a 10 min extension at 72 °C and subsequent cooling to 4 °C. The molecular weight standard used was DNA Hyper Ladder II (Promega, USA). The PCR products were loaded onto a 1% agarose gel for electrophoresis followed by ethidium bromide stainingCitation61.
2.2.6.2 The effect on the filamentous fungal nucleic acid
DNA damage study on F. oxysporum was performed according to the protocol ofCitation58,Citation62,Citation63. DNA amplified by PCR was treated with our synthetic compounds at (1 mM) and incubated for 1 h. DNA without the compound treatment served as a control. After treatment DNA was separated by electrophoresis using a 1% agarose gel at 75 V for 30 min. The gel was stained with ethidium bromide and then subjected to UV irradiation to visualise the DNA bands.
3. Results and discussion
3.1. Chemistry
N-aryl-2-amino-3-cyanopyrroles 1a-d were obtained by reaction of α-hydroxy ketones (namely; benzoin or phenacyl bromide) with different aryl amine and malonodinitrile according to a procedure defined in the literatureCitation50,Citation64,Citation65. Reaction of nitriles, carboxylic acids, esters, ortho-esters or hydroxyamides with ethylenediamine, using carbon disulphide as solvent, is a well-known method for preparation of the corresponding 2-substituted imidazolines, as used here for the preparation of compounds 3–(2-dihydroimidazolyl)pyrrole-2-amines 2a-dCitation66–69. The structures of these compounds were confirmed by spectral analysis; IR spectra showed disappearance of CN absorption bands and appearance of absorption frequency for NH at 3403–3290 cm−1. 1H-NMR spectra revealed the appearance of two triplet peaks for two methylene groups of imidazoline ring in the range 2.53–4.57 ppm and distinctive D2O exchangeable peak of NH at 8.84–9.64 ppm.
On the other hand, the 3–(2-dihydroimidazolyl)pyrrole-2-amines (2a-d) were converted to the corresponding pyrrolo[3,2-e]imidazo[1,2-c]pyrimidines (3a-d, 5a-d) via condensation with triethylorthoformate (TEOF) or carbon disulphide (CS2), respectivelyCitation70–72. Structures of pyrrolo[3,2-e]imidazo[1,2-c]pyrimidine 3a-d were confirmed by disappearance of both NH and NH2 absorption bands in IR spectra and also their peaks in1H-NMR spectra were disappeared and in Mass spectra molecular ion peaks were recorded exactly at the molecular weight of each compound. Also structures of pyrrolo[3,2-e]imidazo[1,2-c]pyrimidine-5-thiones 5a-d were confirmed by disappearance of NH2 absorption bands in IR spectra, but NH absorption bands were still present, and appearance of four distinctive absorption bands for C = S group at 1492–798 cm−1. 1H-NMR spectra also revealed disappearance of NH2 peaks and presence of NH peaks.
Formation of pyrrolo[3,2-e]imidazo[1,2-c]1,2,3-triazines (4a-d) were obtained via reaction of 2 a-d with sodium nitrite (NaNO2)Citation73–75, Confirmation of structures of compounds 4a-d was lied on disappearance of both NH and NH2 absorption bands or peaks in both IR and 1H-NMR spectra. Alkylation of thione derivatives 5a-d with appropriate halo derivative (namely; ethyl chloroformate, ethyl chloroacetate, or phenacyl bromide) afforded alkylated thio derivatives 6a-e. The structures of these compounds were confirmed by spectral analysis; IR spectra showed disappearance of both NH and C = S absorption bands and appearance of absorption frequency for C = O at 1765–1719 cm−1. 1H-NMR spectra revealed the disappearance of NH peaks and appearance of new peaks in a reasonable correspondence with excess aliphatic part in the compounds. The mass spectra showed molecular ion peaks at m/z values as it should be for each derivative, as revealed in Scheme 1.
3.2. Anti-microbial activities
3.2.1. Sensitivity of some gram-positive, gram-negative bacteria and C. albicans to the tested compounds:
The newly synthesised compounds were tested for antimicrobial activity by the disc diffusion method. Results are shown in .
Table 1. The in vitro antimicrobial activity of the synthesised compounds and the control drugs (1000 µg/mL) against some Gram-positive, Gram-negative bacteria and C. albicans.
To discuss the former results, the results indicated good antimicrobial activities for all tested compounds. Among these, compounds 2a, 3c and 4d were found to possess significant activity against Gram-positive bacteria and C. albicans when compared to standard antibacterial and antifungal drugs with compound 3c as the most potent one. Their mean zones inhibition (MZI) were: 2a (22, 24, and 24 mm), 3c with the highest MZI (25, 24, and 23 mm), 4d (21, 23, and 28 mm) against S. aureus, B. subtilis and C. albicans, respectively. Where 2a, 3c and 4d shown higher activity over standard drug Ciprofloxacin (19 and 18 mm) against S. aureus, and B. subtilis. All the three compounds 2a, 3c and 4d gave better anticandidal effect with higher MZI values than the standard drug Amphotericin B (22 mm) and the most potent was 4d. For E. coli, the best effective compounds were 5 b and 5c with MZI (23 and 24 mm), respectively. While for P. aeruginosa, the best effective compounds were 5a and 5c with MZI (24 and 23 mm), respectively.
3.2.2. MIC Of the synthesised compounds against some Gram-positive, Gram-negative bacteria and C. albicans
To further evaluate antimicrobial potential, the compounds that gave the highest MZI against the tested microorganisms were selected to get their MIC determined against the corresponding genus. Results are shown in .
Table 2. The MIC (µg/mL) of the synthesised compounds and a control drugs using the broth macrodilution method.
Results showed that all the tested compounds against both S. aureus and B. subtilis (2a, 3c and 4d) showed a higher antibacterial effect than the standard drug, with their MIC values (30, 30, 35 µg/mL), respectively against S. aureus and (33, 31, 33 µg/mL), respectively, against B. subtilis, all of them are less than the MIC values of ciprofloxacin (45 and 40 µg/mL) against S. aureus and B. subtilis, respectively. Compound 3c is considered the best antimicrobial compound against both of the tested Gram-positive bacteria with a slight difference from compounds 2a and 4d. As for E. coli, 5 b has an MIC (25 µg/mL) that’s equal to ciprofloxacin (25 µg/mL) 5c has almost a lower one (20 µg/mL), which means better antibacterial effect. Compound 5c is considered to have the best antimicrobial effect against both Gram-negative bacterial strains because with P. aeruginosa, it had the same MIC value as ciprofloxacin (30 µg/mL), while compound 5a had a higher one (40 µg/mL).
From the former results, the relatively low effect of the tested compounds on P. aeruginosa in comparison to their effect on the other tested strains () is due to the fact that it is remarkably capable of resisting antibiotics and its strains have high levels of both intrinsic and acquired resistance mechanisms to most antibiotics. Moreover, P. aeruginosa has adaptive antibiotic resistance mechanisms like biofilm- mediated resistance and formation of multidrug-toleranceCitation3. It can also be noticed from the results ( and ) that the antibacterial activity of the tested compounds against Gram-positive strains in general was higher than that against Gram-negative ones. This result can be explained by the fact that Gram-negative bacteria are more resistant than Gram-positive ones because of their special structureCitation76 that differs in the composition of the cell wallCitation77,Citation78. Gram-negative bacteria have an outer membrane (OM), while Gram-positive bacteria do not and the OM of former is responsible for making them resistant to many antibiotics, because most antibiotics have to pass through it to enter the cellCitation79.
As for the fungus C. albicans; all the tested compounds possessed very significant antifungal activities that are much higher than the standard drugs as the MIC values of compounds (2a, 3c and 4d) were (15, 30 and 12 µg/mL), respectively, while that of amphotericin B was (56 µg/mL). Compound 4d had the best antifungal effect with an MIC that’s almost fivefold lower than that of amphotericin B, with a slight difference from compound 2a, MIC almost four-fold the amphotericin B one. The results indicated that compounds 2a, 3c and 4d have a very good activity against both Gram-positive bacteria and candida but that the anti-candida effect is higher than the antibacterial one, these compounds exceeded one of the most active antifungal agent, amphotericin B, against most Candida speciesCitation6.
3.2.3. Identification of the filamentous fungal species
Accurate identification of both used species was confirmed by sequence analysis of the nuclear ribosomal ITS region as F. oxysporum and A. fumigatus.
3.2.4. In vitro antifungal activity of the synthetic compounds against two filamentous fungal species
We used the poison food technique to analyse the in vitro inhibitory effect of the tested compounds at different concentrations (1, 10 and 50) µg/mL compared to two chemical fungicides: Kocide 2000 2500 µg/mL and itraconazole 6.25 µg/mL. Results are shown in and .
Table 3. Mean of inhibitory growth rate (%) ±SD of a pathogenic F. oxysporum with the tested compounds: All control plates have 100 ± 0 growth rate on the 8th day of cultivation.
Table 4. Mean of inhibitory growth rate (%) ±SD of a pathogenic A. fumigatus with tested compounds: All control plates have 100 ± 0 growth rate on the 8th day of cultivation.
First: in comparison to Kocide 2000 (Cu(OH)2) that is used against plant pathogens, the obtained results clearly indicated that a number of our synthetic compounds markedly exhibited higher antifungal activity than the control drug. The mycelial growth of pathogenic F. oxysporum and A. fumigatus was inhibited to various extents our newly synthesised compounds.
A concentration of 50 µg/mL of the compounds 3a-d, 4a,d, 5a,b,d and 6a,b,d,e induced the highest levels of inhibition rate (%) of mycelial growth of both of the tested pathogenic species F. oxysporum and A. fumigatus compared to the chemical fungicide (Kocide 2000) after incubation period of 8 days. The values against F. oxysporum were in range between (73.0 ± 1.0 to 89.9 ± 1.5) exceeded these of the chemical fungicide Kocide 2000 (70.5 ± 0.5). On testing the other strain, A. fumigatus, the values of the chemical compounds (3a-d, 4a,d, 5a,b,d and 6a,b,d,e) were in range between (73.0 ± 1.0 to 89.9 ± 1.5) compared to the chemical fungicide value (72.5 ± 1.0 Kocide 2000). The compound 3c is showing the highest inhibition rate (%) of the mycelial growth of both pathogenic species (F. oxysporum and A. fumigatus) with the value (89.9 ± 1.5).
To Discuss the former results; The antifungal effect shown by the tested compounds was not only higher than that of the commercially well-known fungicide Kocide 2000, but also at a lower concentration than its recommended concentration (2500 µg/mL). That is why trying to find new antifungal agents that can be used on plants is a major concern. So that finding makes the synthesised compounds very promising candidates for commercial use as they produce the desired antifungal effect at a much lower concentration that can produce a safer alternative on edible plants but this needs further investigation. This result is highly important given the fact that F. oxysporum causes a global economic loss by infecting important plantsCitation80.
Second: in comparison to itraconazole (triazole derivative) as the control drug for treatment of the human mycelial diseases, the results () showed that some compounds had a very good effect at different concentrations compared to itraconazole at 6.25 µg/mL. The compounds 3 b, 3c, 5a, 5 b, 6 b and 6e displayed good antifungal activity against the tested pathogenic species F. oxysporum and A. fumigatus. The values of mycelial growth inhibition rate (%) at a concentration of 1 µg/mL against both F. oxysporum and against A. fumigatus were in range between (82.31 ± 2.0 to 88.21 ± 0.4), in comparison to itraconazole at a concentration of (6.25 µg/mL) the values were (93.5 ± 0.5 and 92.2 ± 5.0), respectively. Compound 5a had the highest activity with growth inhibition rate % (88.21 ± 0.4) at 1 µg/mL against both F. oxysporum and A. fumigatus.
To understand the former results; this result is very promising as it means that the antifungal effect of our tested compounds against the filamentous fungal human pathogens was highly equivalent to that of itraconazole and can be produced at even a lower concentration (1 µg/mL). Knowing that itraconazole is considered as one of the very few fungicides for human use that still can overcome the resistance of both of the species used in this study. If we look at the current treatments of A. fumigatus we can find Amphotericin B as an effective antifungal, yet with significant side effects. Other effective drugs are echinocandins (cyclic hexapeptide antibiotics bearing pyrrolidine moiety) lack the inhibition ability against A. fumigatus, so they are only used for salvage therapy of invasive aspergillosis. Likewise, the most recommended drugs for both treatment and prophylaxis of aspergillosis include triazole antifungals: itraconazole, voriconazole, and posaconazole ()Citation81 and again, unfortunately, A. fumigatus isolates are becoming globally more resistant to them to a variable extentCitation82. The great importance of the triazoles containing drugs against A. fumigatus comes from the fact that they are the only drug that can be given orally for long term therapy. The increasing resistance documented of course compromises such clinical progressCitation15. Our results also come in agreement with Dabur et.al.Citation83 that discovered naturally occurring antifungal pyrrole derivatives from Datura leaves that was effective against pathogenic A. fumigatusCitation83.
It can be concluded from our results against the tested filamentous fungi that our newly synthesised compounds possess a very good and broad-spectrum antifungal activity against both the yeast and filamentous fungi, such a characteristic makes the compounds own an important feature of the ideal antifungal agent according to Mazu et.al.Citation9.
3.2.5. The effect of the tested compounds on some selected species nucleic acid
To analyse the effect of the tested compounds on the pathogenic filamentous fungal species nucleic acid, we set up a gel electrophoresis analysis.
3.2.5.1 The effect on bacterial nucleic acid
P. aeruginosa was selected as a representative of the bacteria for this experiment for the following reasons: The Gram-negative bacteria are more resistant than Gram-positive ones, out of the former class comes P. aeruginosa as a major cause of the most challenging issue regarding nosocomial infections due to resistance to antibioticsCitation76. P. aeruginosa resistance is multifactorial and remarkable against many of the currently used antibiotics making them ineffective. The effect on the DNA of P. aeruginosa ATCC 27853 is shown in .
Figure 3. Agarose gel electrophoresis showing the effect of the tested compounds on the nucleic acid of P. aeruginosa. From right to left: lane 1marker (M), lane 2 (negative control), lane 3 (positive control) lane 4 to 19 (fungal DNA treated with the synthetic compounds.
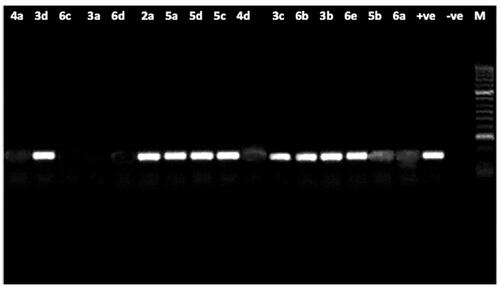
The results showed intact DNA bands with the untreated DNA (positive control), where no significant damage occurred. The same was shown with nine of tested compounds, treated DNA showed intact bands like the untreated DNA, which means that those compounds have no effect on DNA, maybe because of a developed mechanism of resistance, and also they could be acting through any other mechanism. In contrast, compounds 3a, 4a, 4d, 5 b and 6a,c, d showed substantial alteration in electrophoretic migration with compounds 3a, 6c showing the highest effect, which suggests that the antimicrobial effect of them occurs by a direct chemical damage to DNA.
To Discuss the former results; Comparing the MIC values of the compounds against P. aeruginosa ( and ) with their results on the bacterial DNA () showed that, first: some compounds (namely; 3a, 4a, 4d, 5 b and 6a,c, d) could exert their antibacterial effect on P. aeruginosa through damaging its DNA, and second: as the compounds have closely related MIC values, yet very different effect on its DNA, this leads to the conclusion that the effect on the bacterial DNA is mostly not the main mode of antibacterial action of all the examined compounds and there must be other targets inside the P. aeruginosa bacterial cell that get affected by those compounds. It can also be concluded that P. aeruginosa developed a resistance mechanism such as; an efflux pump, modification of target or deactivation of the chemical compound, that protected its DNA from compounds revealed no effect on it.
The effect shown by the tested compounds on the DNA of P. aeruginosa comes in agreement with the well-known effect of many antibiotics on the bacterial DNA and the possible mechanism could be related to any of the several mechanisms exhibited by them. For example, there is a recently reported mechanism of DNA acting antimicrobials active on Gram-positive bacteria through inhibiting the DNA polymerase IIICCitation84, and another possible mechanism could be through affecting the DNA primaseCitation85. Usually, the bacterial DNA damage caused by antibiotics can be exerted either by direct chemical modification inducing breakage in DNA, like metronidazole (imidazole derivative) or through affecting the enzymes associated with its replication. To the second class belong: quinolones, coumermycins and novobiocin. The quinolones selectively inhibit DNA gyrase (topoisomerase II) leading to chromosomal fragmentation. Quinolones include nalidixic acid and the newer second-generation fluoroquinolones (FQ): norfloxacin and ciprofloxacin. FQ are much more effectiveCitation86 and are able to inhibit all Gram-negative organisms, including Pseudomonas speciesCitation87 with ciprofloxacin coming as the current first choice against themCitation86. The coumermycins and novobiocin also inhibit DNA gyrase but with a mechanism different from quinolonesCitation88,Citation89. FQs are among the most widely used antimicrobials on the marketCitation90, yet the increasing resistance to it urged the development of novel topoisomerase inhibitorsCitation91.
A question may arise about the effect that the compounds may have on the human DNA, but since the protein importance in the bacterial DNA replication is different from the eukaryotic counterparts, chromosome replication represents a promising therapeutic targetCitation90,Citation91 without such a fear on human safetyCitation92. There are many examples about the selectivity of the DNA acting antibacterial agents based on the structure difference of the targeted enzymes between bacteria and human, like quinolones which do not interfere with the human topoisomerase enzymesCitation93, bacterial primase inhibitorsCitation85 and Pol IIIC inhibitorsCitation84.
3.2.5.2. The effect on the filamentous fungal nucleic acid
The effect on the DNA of the pathogenic fungus F. oxysporum is shown in ().
Figure 4. Agarose gel electrophoresis showing the effect of the tested compounds on the nucleic acid of the pathogenic fungus F. oxysporum. From right to left: lane 1marker (M), lane 2 (positive control), lane 3 (negative control) lane 4 to 19 (fungal DNA treated with the synthetic compounds.
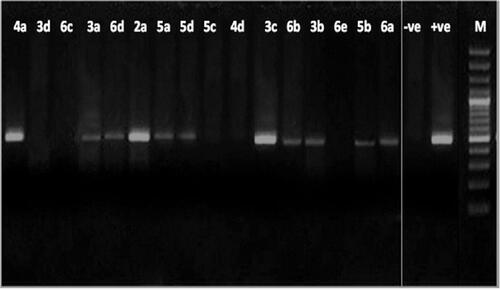
The results showed intact DNA bands with the untreated DNA (positive control), where no significant damage occurred. DNA treated with compounds 2a, 3c and 4a also showed intact bands like the untreated DNA, which means that those compounds have no effect on DNA and must be acting through any other mechanism. In contrast, the remaining tested compounds showed substantial alteration in electrophoretic migration with compounds 3d, 4d, 5c, 6c and 6e showing the highest effect.
The former results indicated that; most of the compounds (thirteen out of sixteen examined compounds) could be exerting their antifungal effect by direct chemical damage to the DNA or at least it is one of the target sites. Comparing the MIC values of the compounds against F. oxysporum () with their results on the fungal DNA () shows that two compounds (2a and 3c), which showed the (lowest and highest) antifungal activity, respectively, are showing the same effect on DNA which is no damage, such an observation can suggest that the effect on DNA is not the MAIN mode of action of at least these two compounds or that there may be a strong resistance mechanism inside the fungal cell that leads to decreasing their effect on DNA like efflux pump, change in the target site, deactivation of the compound, or decreased permeability Citation76.
Comparing showed that the effect of the examined compounds on the DNA of the filamentous fungi is of much wider range than that on P. aeruginosa (13 compounds are effective () in comparison to only seven (). Also, from the same comparison, it can be shown that there is an agreement between the MIC values and the effect on the DNA in case of F. oxysporum of more compounds () than what’s seen in case of P. aeruginosa () which means that still the effect on DNA is more common in fungi and it could have a bigger role in the antifungal effect than the antibacterial one of the tested compounds.
If the antimicrobial drug is affecting the DNA of the filamentous fungi, which are eukaryotes, must it affect the human one too? actually the answer is: not necessarily, because the literatures show that there are important differences between mammalian and fungal cell types and such differences have already enabled researchers to develop some drugs like 5-fluorocytosine (5FC), that interferes with pyrimidine metabolism, RNA, DNA, and protein synthesis of fungi, yet it’s safe for human use due to the enzyme differences. Topoisomerases I (TOP) inhibitors represent another successful example and the reason is that it affects the enzymes of C. albicans more than the human ones like aminocatechol A-3253Citation5.
Several antifungals target fungal DNA or RNA like a natural product called Yatakemycin that acts by sequence-specific DNA alkylation. Icofungipen is another example, it is an orally administered, synthetic derivative of the naturally occurring β-amino acid cispentacin, blocks isoleucyl-tRNA synthetase, which leads to the inhibition of protein synthesis and growth of fungal cellsCitation94. Other products that give their antifungal activity by interfering with the fungal cell nucleic acid biosynthesis are some natural products; sampangine, meridine, onychine and berberineCitation5. Of course in order to be completely sure that our compounds are safe for human use further investigations on their toxicity profile will be needed in future work.
Some possible effects of pyrrole could also be on the synthesis of proteins; for example: the antibacterial properties of some halogenated pyrroles were due to their ability to affect the synthesis of macromolecules in Gram-positive and Gram-negative bacteriaCitation95. Also, A pyrrole derivative exhibited its antifungal activity against A. fumigatus by inhibiting the expression of two important proteins at the time of spore germinationCitation83. Other pyrroles affected proteins but not at the level of synthesis as protein inhibitors like toyocamycin and sangivamycin, two pyrrole derivatives that are reported as inhibitors of protein kinase C and/or protein kinase ACitation50 and also several other pyrrole derivatives were found to be inhibitors of several types of protein kinasesCitation27. There are other antifungal agents reported to produce their effect through acting on some proteins, Sordarins are one example that acts on the yeast protein synthesis elongation cycle. Elongation Factor-3 is required by the 40S ribosomal subunit of yeast but not by human cells so the inhibition happens with no effect on the protein synthesis in human. Also some antibiotics with antifungal activity (Enactins and neoenactins) affect the protein post-translational modification by inhibiting the protein-N-myristoyltransferase (NMT). NMT has different substrate specificities in fungi and mammalian cells, making it a good target for designing antifungal agents against fungi that requires it like Cryptococcus neoformans (C. neoformans). Another inhibitors of NMT, the analogs of 2-bromotetradecanoic acid, were active against filamentous fungi such as A. niger and also on yeasts like S. cerevisiae, C. albicans and C. neoformansCitation5.
We tried to find out a possible mechanism that is responsible for the DNA damage inside the filamentous fungi that was produced by our compounds in this study; an interesting finding was about the reactive oxygen species (ROS). ROS are molecules with unstable highly reactive free radicals, they get produced during normal cell routine or as a response to exposure to drugs then when they reach high levels they can damage the cellular DNACitation96.
Such a finding was assumed to be the possible mechanism because literatures have a lot of examples of antifungals that cause increased ROS in fungal cells, for example,Citation97,Citation98 showed some antifungals that lead to intracellular production of ROS in filamentous fungi such as A. fumigatus. Also, miconazole use was reported to generate ROS in C. albicansCitation14,Citation98, Finally, there is fluconazole (FLC) that leads to generation of ROS in C. albicans neoformansCitation96.
Pyrroles bearing an imidazoline group at C-3,were prepared and examined as an investigation for new anti-microbial agents, especially as an anti-candidaCitation99. The anti-microbial activities of these compounds indicates; higher anti-candida activity over the Amphotericin B, one of the most active antifungal agent against most Candida speciesCitation6. Compounds 2a, 3c and 4d have a very good activity against both Gram-positive bacteria and candida but that the anti-candida effect is higher than the antibacterial one, owning these activities to the pyrrole moiety.
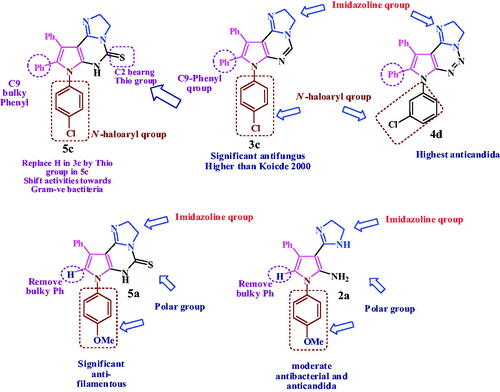
The highest active compound for both Gram-positive and filamentous fungi as plant pathogen is; pyrrolopyrimidine 3c bearing imidazoline, phenyl at C9 and N-haloaryl. Replace the pyrimidine with triazine ring as in 4d shift the activities against fungi, with enhancing the antibacterial and anti-candida activities. Replace the C9-bulky phenyl in 3c with H and N-haloaryl with hydrophilic N-methoxyphenyl group mask the bacterial and yeast activity, but enhance the antifungal activity as in thio derivative 5a (highest activity against filamentous as human pathogen). Another thio derivative, bearing N-haloaryl and phenyl at C9, compound 5c shown the highest activity against Gram-negative bacteria. Three fused ring exhibit a predominant activities over the free pyrrole bearing imidazoline at C2 as in compound 2a which show moderate activity against Gram positive and yeast, as shown in .
Figure 5. Relation between chemical structure and microbial activity of compounds (2a, 3c, 4d, 5a and 5c).
Conclusion
In this study, we have presented novel 3-imidazolyl pyrroles, and their fused form (pyrimidines and triazines) that can act as both antibacterial and antifungal agents. Compounds 2a, 3c, 4d and 5c showed a very good activity against Gram-positive bacteria in comparison to ciprofloxacin with higher MZI and lower MIC values. Compound 5c was the best against Gram-negative bacteria with MZI value comparable to that of ciprofloxacin, an equal MIC against P. aeruginosa ATCC 27853 and an even lower one against E. coli O157:H7 ATCC 51659. Against C. albicans, the effect of compounds 2a, 3c and 4d exceeded one of the most active antifungal agents, amphotericin B, giving higher MZI values; moreover, their MIC values showed several fold decrease than amphotericin B up to fivefold. The effect of 3 b, 3c, 5a, 5 b, 6 b and 6e against the filamentous fungal human pathogens was highly equivalent to that of itraconazole at even a lower concentration (1 µg/mL) with compound 5a showing the highest inhibition rate % (88.21 ± 0.4). The outcomes of this study present some potential candidates that might participate to find a solution to the continuous huge problem of antibiotic resistance both bacterial and fungal and the high need to develop new treatment options for opportunistic infections. In addition, a lot of our synthetic compounds showed potential protection of plants from filamentous fungal pathogens, with a much less toxicity profile that may have a very good agricultural economic impact as they exhibited the desired antifungal activity at a much lower concentration (50 µg/mL) than Kocide 2000 (2500 µg/mL), and compound 3c was the best candidate with the highest inhibition rate (%) (89.9 ± 1.5). We also propose a possible target of these derivatives inside the bacterial and fungal cells, which is the DNA of the filamentous fungi and bacteria. Yet, further investigation about the exact mode of action is required.
Disclosure of interest
Authors would like to report no conflict of interest.
Supplemental Material
Download PDF (1.4 MB)References
- Lee CR, Lee JH, Park M, et al. Biology of Acinetobacter baumannii: Pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front Cell Infect Microbiol 2017;7:55. (MAR).
- Tadesse DA, Zhao S, Tong E, et al. Antimicrobial drug resistance in Escherichia coli from humans. Emerg Infect Dis 2012;18:741–9.
- Pang Z, Raudonis R, Glick BR, et al. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol Adv 2019;37:177–92.
- Barbier F, Andremont A, Wolff M, Bouadma L. Hospital-acquired pneumonia and ventilator-associated pneumonia: recent advances in epidemiology and management. Curr Opin Pulm Med 2013;19:216–28. https://journals.lww.com/co-pulmonarymedicine/Fulltext/2013/05000/Hospital_acquired_pneumonia_and.6.aspx.
- Mazu TK, Bricker BA, Flores-Rozas H., Ablordeppey SY. The Mechanistic Targets of Antifungal Agents: An Overview. Mini-Reviews Med Chem 2016;16:555–78. doi:https://doi.org/http://dx.doi.org/10.2174/1389557516666160118112103
- Amran F, Aziz MN, Ibrahim HM, et al. In vitro antifungal susceptibilities of Candida isolates from patients with invasive candidiasis in Kuala Lumpur Hospital, Malaysia. J Med Microbiol 2011;60:1312–6.
- Frisvad JC, Larsen TO. Extrolites of aspergillus fumigatus and other pathogenic species in aspergillus section fumigati. Front Microbiol 2015;6:1485–14.
- O'Donnell K, Gueidan C, Sink S, et al. A two-locus DNA sequence database for typing plant and human pathogens within the Fusarium oxysporum species complex. Fungal Genet Biol 2009;46:936–48.
- Mazu TK, Bricker BA, Ablordeppey HF-R, et al. The mechanistic targets of antifungal agents: an overview. Mini Rev Med Chem 2016;16:555–78. doi:https://doi.org/http://dx.doi.org/10.2174/1389557516666160118112103
- Dutcher JD. The discovery and development of amphotericin B. Dis Chest 1968;54:Suppl 1:296–8.
- Titsworth E, Grunberg E. Chemotherapeutic activity of 5-fluorocytosine and amphotericin B against Candida albicans in mice. Antimicrob Agents Chemother 1973;4:306–8.
- Mert S, Kasımoğulları R, İça T, et al. Synthesis, structure-activity relationships, and in vitro antibacterial and antifungal activity evaluations of novel pyrazole carboxylic and dicarboxylic acid derivatives. Eur J Med Chem 2014;78:86–96.
- Sharma V, Chitranshi N, Agarwal AK. Significance and biological importance of pyrimidine in the microbial world. Int J Med Chem 2014;2014:31–202784.
- Francois I, Cammue B, Borgers M, et al. Azoles: Mode of antifungal action and resistance development. Effect of miconazole on endogenous reactive oxygen species production in Candida albicans. Antiinfect Agents Med Chem 2006;5:3–13.
- Verweij PE, Chowdhary A, Melchers WJG, Meis JF. Azole resistance in Aspergillus fumigatus : can we retain the clinical use of mold-active antifungal azoles ? 2016;62: 362–8.
- Sadeghpour H, Khabnadideh S, Zomorodian K, et al. Design, synthesis, & biological activity of new triazole & nitro-triazole derivatives as antifungal agents. Molecules 2017;22:1150–11.
- Writers AM. Long-term use of systemic azole antifungals can result in hepatotoxicity and other serious adverse effects. Drugs Ther Perspect 2020;36:112–5.
- Landbouwcatalogus C, Mode of action of the phenylpyrrole fungicide fenpiclonil in Fusarium Sulphureum. 1994.
- Arif T, Bhosale JD, Kumar N, et al. Natural products-antifungal agents derived from plants. J Asian Nat Prod Res 2009;11:621–38.
- Peyton LR, Gallagher S, Hashemzadeh M. Triazole antifungals: a review. Drugs Today (Barc) 2015;51:705–18.
- Bartroli J, Turmo E, Algueró M, et al. New azole antifungals. 3. Synthesis and antifungal activity of 3-substituted-4(3H)-quinazolinones. J Med Chem 1998;41:1869–82.
- Shuai H, Myronovskyi M, Nadmid S, Luzhetskyy A. Identification of a biosynthetic gene cluster responsible for the production of a new pyrrolopyrimidine natural product—huimycin. Biomolecules 2020;10:1074–10.
- Pathania S, Rawal RK. Pyrrolopyrimidines: An update on recent advancements in their medicinal attributes. Eur J Med Chem 2018;157:503–26.
- Dı L, Beleta J, Ca A, et al. New pyrrolopyrimidin-6-yl benzenesulfonamides: potent A2B adenosine receptor antagonists. Bioorg Med Chem Lett 2006;16:3642–5.
- Ojha RP, Sanyal NK. Biological activity of an antibiotic puromycin — a theoretical study. J Biosci 1990;15:417–25.
- Fatahala SS, Khedr MA, Mohamed MS. Synthesis and structure activity relationship of some indole derivatives as potential anti-inflammatory agents. Acta Chim Slov 2017;64:865–76.
- Mohamed MSS, Fathallah SSS. Pyrroles and Fused Pyrroles : Synthesis and Therapeutic Activities. Mini Rev Org Chem 2014;6:477–507.
- Tari LW, Trzoss M, Bensen DC, et al. Pyrrolopyrimidine inhibitors of DNA gyrase B (GyrB) and topoisomerase IV (ParE). Part I: Structure guided discovery and optimization of dual targeting agents with potent, broad-spectrum enzymatic activity. Bioorg Med Chem Lett 2013;23:1529–36.
- Acs G, Reich E. Tubercidin and Related Pyrrolopyrimidine Antibiotics. Antibiotics 1967;(1956):494–8.
- Kaur R, Manjal SK, Rawal RK, Kumar K. Recent synthetic and medicinal perspectives of pyrroles: An overview. J Pharm Chem Chem Sci 2017;1:17–32. https://www.alliedacademies.org/articles/recent-synthetic-and-medicinal-perspectives-of-pyrroles-an-overview-9135.html.
- Varshney H, Ahmad A, Rauf A, et al. Synthesis and antimicrobial evaluation of fatty chain substituted 2,5-dimethyl pyrrole and 1,3-benzoxazin-4-one derivatives. J Saudi Chem Soc 2017;21:S394–S402.
- Swapna M, Premakumari C, Reddy N, et al. Synthesis, antimicrobial and cytotoxic activities of sulfonamidomethane linked heterocycles. Chem Pharm Bull 2013;61:722–30.
- Raimondi MV, Cascioferro S, Schillaci D, Petruso S. Synthesis and antimicrobial activity of new bromine-rich pyrrole derivatives related to monodeoxypyoluteorin. Eur J Med Chem 2006;41:1439–45.
- Carbone A, Parrino B, Barraja P, et al. Synthesis and antiproliferative activity of 2,5-bis(3'-indolyl)pyrroles, analogues of the marine alkaloid nortopsentin. Mar Drugs 2013;11:643–54.
- Zhang S, Tan X, Liang C, Zhang W. Design, synthesis, and antifungal evaluation of novel coumarin‐pyrrole hybrids. J Heterocycl Chem 2020;23:1–9.
- Tokarenko A, Lišková B, Smoleń S, et al. Synthesis and cytotoxic and antiviral profiling of pyrrolo- and furo-fused 7-deazapurine ribonucleosides. J Med Chem 2018;61:9347–59.
- Khan AT, Lal M, Bagdi PR, et al. Synthesis of tetra-substituted pyrroles, a potential phosphodiesterase 4B inhibitor, through nickel(II) chloride hexahydrate catalyzed one-pot four-component reaction. Tetrahedron Lett 2012;53:4145–50.
- Mohamed MS, Kamel R, Abd El-hameed RH. Evaluation of the anti-inflammatory activity of some pyrrolo[2,3-d]pyrimidine derivatives. Med Chem Res 2013;22:2244–52.
- Balaraman S, Nayak N, Subbiah M, Elango KP. Synthesis and antiviral study of novel 4-(2-(6-amino-4-oxo-4,5-dihydro-1H-pyrrolo[2,3-d]pyrimidin-3-yl)ethyl)benzamide derivatives. Med Chem Res 2018;27:2538–46.
- Adel M, Serya RAT, Lasheen DS, Abouzid KAM. Pyrrolopyrimidine, a multifaceted scaffold in cancer targeted therapy. Drug Res (Stuttg) 2018;68:485–98.
- Fatahala S, Shalaby E, Kassab S, et al. A promising anti-cancer and anti-oxidant agents based on the pyrrole and fused pyrrole: synthesis, docking studies and biological evaluation. Anticancer Agents Med Chem 2015;15:517–26.
- Mohame MS, Mostafa AG, El-hameed RHA, et al. Evaluation of the anti-inflammatory activity of novel synthesized pyrrole, pyrrolopyrimidine and spiropyrrolopyrimidine derivatives. Pharmacophore 2012;3:44–54. http://www.pharmacophorejournal.com.
- Sarg MT, Koraa MM, Bayoumi AH, Gilil SE. Synthesis of pyrroles and condensed pyrroles as anti-inflammatory agents with multiple activities and their molecular docking study. Open J Med Chem 2015;5:49–96.
- Zhang SG, Liang CG, Sun YQ, et al. Design, synthesis and antifungal activities of novel pyrrole- and pyrazole-substituted coumarin derivatives. Mol Divers 2019;23:915–25.
- Bhardwaj V, Gumber D, Abbot V, et al. Pyrrole: a resourceful small molecule in key medicinal hetero-aromatics. RSC Adv 2015;5:15233–66.
- Pawar S, Chaudhari A, Prabha R, et al. Microbial pyrrolnitrin: Natural metabolite with immense practical utility. Biomolecules 2019;9:443.
- Gilson P, Josa-Prado F, Beauvineau C, et al. Identification of pyrrolopyrimidine derivative PP-13 as a novel microtubule-destabilizing agent with promising anticancer properties. Sci Rep 2017;7:10209–14.
- Bello-Vieda NJ, Pastrana HF, Garavito MF, et al. Antibacterial activities of azole complexes combined with silver nanoparticles. Molecules 2018;23:1–17.
- Mohamed MS, El-Domany RA, Abd El-Hameed RH. Synthesis of certain pyrrole derivatives as antimicrobial agents. Acta Pharm 2009;59:145–58.
- Mohamed MS, Abd El-Hameed RH, Sayed AI. Synthesis Strategies and Biological Value of Pyrrole and Pyrrolopyrimidine. J Adv Pharm Res 2017;1:1–24.
- Fatahala SS, Mahgub S, Taha H, et al. Synthesis and evaluation of novel spiro derivatives for pyrrolopyrimidines as anti-hyperglycemia promising compounds. J Enzyme Inhib Med Chem 2018;33:809–17.
- Jorgensen JH, Turnidge JD, Susceptibility Test Methods: Dilution and Disk Diffusion Methods*. In: Manual of clinical microbiology, 11th ed. Wayne, PA : American Society of Microbiology; 2015:1253–1273.
- Clinical ALSI. Document M27-A4. Ref Method Broth Dilution Antifung Suscetibility Test Yeasts 2012.
- CLSI. M07-A10: Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically.; 2015.
- Clinical and Laboratory Standards Institute. Performance standards for antimicrobial disk susceptibility tests: approved standard – 11th ed. Vol 32. Wayne, PA ; 2012. doi:M02-A11
- Gardes M, Bruns T. ITS Primers with enhanced specificity for Basidiomycetes - Application to the identification of mycorrhizae and rusts. Mol Ecol 1993;2:113–8.
- Al-Burtamani SKS, Fatope MO, Marwah RG, et al. Chemical composition, antibacterial and antifungal activities of the essential oil of Haplophyllum tuberculatum from Oman. J Ethnopharmacol 2005;96:107–12.
- Abdel-Hafez SII, Nafady NA, Abdel-Rahim IR, et al. Assessment of protein silver nanoparticles toxicity against pathogenic Alternaria solani. 3 Biotech 2016;6:199.
- Babii C, Bahrin LG, Neagu AN, et al. Antibacterial activity and proposed action mechanism of a new class of synthetic tricyclic flavonoids. J Appl Microbiol 2016;120:630–7.
- Tyler SD, Strathdee CA, Rozee KR, Johnson WM. Oligonucleotide primers designed to differentiate pathogenic pseudomonads on the basis of the sequencing of genes coding for 16S-23S rRNA internal transcribed spacers. Clin Diagn Lab Immunol 1995;2:448–53.
- Sambrook J, Fritsch EF, Maniatis T, Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory Press, Cold Spring Harbour; 1989.
- Vahdati AR, Sadeghi B. A study on the assessment of DNA strand-breaking activity by silver and silica nanoparticles. J Nanostructure Chem 2013;3:7.
- Eftekhari-Sis B, Vahdati-Khajeh S. Ultrasound-assisted green synthesis of pyrroles and pyridazines in water via three-component condensation reactions of arylglyoxals. Curr Chem Lett 2013;2:85–92.
- Traxler PM, Furet P, Mett H, Buchdunger E, et al. 4-(Phenylamino)pyrrolopyrimidines: potent and selective, ATP site directed inhibitors of the EGF-receptor protein tyrosine kinase. J Med Chem 1996;716:2285–92.
- Mohamed MS, Ali SA, Abdelaziz DHA, Fathallah SS. Synthesis and evaluation of novel pyrroles and pyrrolopyrimidines as anti-hyperglycemic agents. Biomed Res Int 2014;2014:249780–13.
- Pathan MY, Paike VV, Pachmase PR, et al. Microwave-assisted facile synthesis of 2-substituted 2-imidazolines. ARKIVOC 2007;2006:205–10.
- Sun M, Wei HT, Li D, et al. Mild and efficient one-pot synthesis of 2-imidazolines from nitriles using sodium hydrosulfide as catalyst. Synth Commun 2008;38:3151–8.
- Moghadam M, Mohammadpoor-Baltork I, Mirkhani V, et al. Rapid and efficient synthesis of imidazolines and bisimidazolines under microwave and ultrasonic irradiation. Monatshefte Fur Chemie 2007;138:579–83.
- Levesque G, Gressier JC, Proust M. 4,5-Dihydroimidazole from dithiocarboxylic esters, thiocarboxamides or nitriles. Synthesis (Stuttg) 1981;12:963–5. http://library1.nida.ac.th/termpaper6/sd/2554/19755.pdf.
- Mohamed MS, Kamel R, Fatahala SS. New condensed pyrroles of potential biological interest syntheses and structure-activity relationship studies . Eur J Med Chem 2011;46:3022–9.
- Mohamed MS, Kamel R, Fathallah SS. Synthesis of new pyrroles of potential anti-inflammatory activity. Arch Pharm (Weinheim) 2011;344:830–9.
- Fatahala SSSS, Hasabelnaby S, Goudah A, et al. Pyrrole and fused pyrrole compounds with bioactivity against inflammatory mediators. Molecules 2017;22:1–18.
- Karrouchi K, Radi S, Ramli Y, et al. Synthesis and pharmacological activities of pyrazole derivatives: a review. Molecules 2018;23:134.
- Voskoboynik OY, Kolomoets OS, Berest GG, et al. Preparation and biological properties of 2-thio-containing pyrimidines and their condensed analogs. Chem Heterocycl Compd (N Y) 2017;53:256–72.
- Farghaly AR, El-Kashef H. Synthesis of imidazo[1,2-c]pyrazolo [4,3-e]pyrimidines derived from indole and related heterocycles. Monatshefte Fur Chemie 2006;137:1195–202.
- Jubeh B, Breijyeh Z, Karaman R. Resistance of gram-positive bacteria to current antibacterial agents and overcoming approaches. Molecules 2020;25:2888.
- Coico R. Gram staining basic protocol commonly used techniques. Curr Protoc Microbiol 2005;(1):3–4.
- Moyes RB, Reynolds J, Breakwell DP. Differential staining of bacteria: Gram stain. Curr Protoc Microbiol 2009;(SUPPL. 15):1–8.
- Silhavy TJ, Kahne D, Walker S. The Bacterial Cell Envelope1 T. J. Silhavy, D. Kahne and S. Walker,. Cold Spring Harb Perspect Biol. 2010;2:1–16. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2857177/pdf/cshperspect-PRK-a000414.pdf.
- Donnell KO, Sutton DA, Rinaldi MG, et al. Genetic diversity of human pathogenic members of the Fusarium oxysporum complex inferred from multilocus DNA sequence data and amplified fragment length polymorphism analyses : evidence for the recent dispersion of a geographically widespread clonal linea. J Clin Microbiol 2004;42:5109–20.
- Srivastava V, Singla RK, Dubey AK. Emerging virulence, drug resistance and future anti-fungal drugs for candida emerging virulence, drug resistance and future anti-fungal drugs for candida pathogens. Curr Top Med Chem 2018;8:1–20.
- Chowdhary A, Kathuria S, Xu J, Meis JF. Emergence of azole-resistant Aspergillus fumigatus strains due to agricultural azole use creates an increasing threat to human health. PLoS Pathog 2013;9:e1003633–7.
- Dabur R, Ali M, Singh H, et al. A novel antifungal pyrrole derivative from Datura metel leaves. Pharmazie 2004;59:568–70.
- Xu WC, Silverman MH, Yu XY, et al. Discovery and development of DNA polymerase IIIC inhibitors to treat Gram-positive infections. Bioorganic Med Chem 2019;27:3209–17.
- Ilic S, Cohen S, Singh M, et al. DnaG primase—A target for the development of novel antibacterial agents. Antibiotics 2018;7:72.
- Pham TDM, Ziora ZM, Blaskovich MAT. Quinolone antibiotics. Medchemcomm 2019;10:1719–39.
- Patrick GL. An introduction to medicinal chemistry. Biochem Education 2003;4(4):82.
- Hugo WB, Russell AD. Pharmaceutical microbiology. Ciba Foundation Symposium 102, vol. 102; 1984:204–221.
- Harris M. Pharmaceutical microbiology. J Med Educ 1964;39:xix.
- Sanyal G, Doig P. Bacterial DNA replication enzymes as targets for antibacterial drug discovery. Expert Opin Drug Discov 2012;7:327–39.
- van Eijk E, Wittekoek B, Kuijper EJ, Smits WK. DNA replication proteins as potential targets for antimicrobials in drug-resistant bacterial pathogens. J Antimicrob Chemother 2017;72:1275–84.
- Trojanowski D, Kołodziej M, Hołówka J, et al. Watching antibiotics in action: Exploiting time-lapse microfluidic microscopy as a tool for target-drug interaction studies in Mycobacterium. Antimicrob Agents Chemother 2019;63:e00739-19.
- Drlica K, Hiasa H, Kerns R, et al. Quinolones: action and resistance updated. Curr Top Med Chem 2009;9:981–98.
- Gubbins PO, Anaissie EJ, CHAPTER 7 - Antifungal therapy. In: Anaissie, EJ, McGinnis MR, Pfaller MA, eds. Clinical mycology. 2nd ed. Edinburgh: Churchill Livingstone; 2009:161–195. doi:
- Cascioferro S, Raimondi MV, Cusimano MG, et al. Pharmaceutical potential of synthetic and natural pyrrolomycins. Molecules 2015;20:21658–71.
- Dbouk NH, Covington MB, Nguyen K, Chandrasekaran S. Increase of reactive oxygen species contributes to growth inhibition by fluconazole in Cryptococcus neoformans. BMC Microbiol 2019;19:1–10.
- Shekhova E, Kniemeyer O, Brakhage AA. Crossm induction of mitochondrial reactive oxygen species production by amphotericin B as a mode of action against Aspergillus fumigatus. Antimicrob Agents Chemother 2017;61:1–14.
- Ferreira GF, Baltazar LDM, Ribeiro J, et al. The role of oxidative and nitrosative bursts caused by azoles and amphotericin B against the fungal pathogen Cryptococcus gattii. J Antimicrob Chemther 2013;68:1801–11.
- Özdemir A, Altintop MD, Sever B, et al. A new series of pyrrole-based chalcones: Synthesis and evaluation of antimicrobial activity, cytotoxicity, and genotoxicity. Molecules 2017;22:1–16.