
Abstract
New polycyclic heterocycles were synthesised and evaluated as potential inhibitors of thymidine phosphorylase (TP). Inspired by the pharmacophoric pyrimidinedione core of the natural substrate, four series have been designed in order to interact with large empty pockets of the active site: pyrimidoquinoline-2,4-diones (series A), pyrimidinedione linked to a pyrroloquinoline-1,3-diones (series B and C), the polycyclic heterocycle has been replaced by a pyrimidopyridopyrrolidinetetraone (series D). In each series, the tricyclic nitrogen heterocyclic moiety has been synthesised by a one-pot multicomponent reaction. Compared to 7-DX used as control, 2d, 2l, 2p (series A), 28a (series D), and the open intermediate 30 showed modest to good activities. A kinetic study confirmed that the most active compounds 2d, 2p are competitive inhibitors. Molecular docking analysis confirmed the interaction of these new compounds at the active binding site of TP and highlighted a plausible specific interaction in a pocket that had not yet been explored.
1. Introduction
Angiogenesis – the formation of new blood vessels from pre-existing vasculature – has been validated as a target for several tumours and has been shown to promote tumour growth and metastasisCitation1. Given the complexity of this process, there is a need for new compounds targeting multiple pro-angiogenic factorsCitation2–5. In this context, thymidine phosphorylase (TP) seems to be an interesting and understudied therapeutic target.
The physiological role of TP is to catalyse the reversible phosphorolysis of thymidine into thymine and 2-deoxyribose-1-phosphate (2dR1P) that is metabolised into 2-deoxyribose (2dR) (). TP has been shown to be up-regulated in the hypoxic regions of many solid tumours (stomach, pancreas, kidney, oesophagus, breast, ovary, lung, colon, bladder, uterus, kidney …)Citation4,Citation6,Citation7. Usually, this over-expression is highly associated with tumour micro vessel level, infiltration, metastasis and also correlates with the aggressiveness and invasiveness of the cancerCitation6. Via 2dR, TP stimulates the secretion of several pro-angiogenic factors such as VEGF, MMP-1, IL-8 by both malignant cells and stromal cells located in the tumour microenvironmentCitation8,Citation9. Consequently, TP inhibitors seem to be promising agentsCitation10.
Figure 1. TP and 2dR1P roles in tumour development.
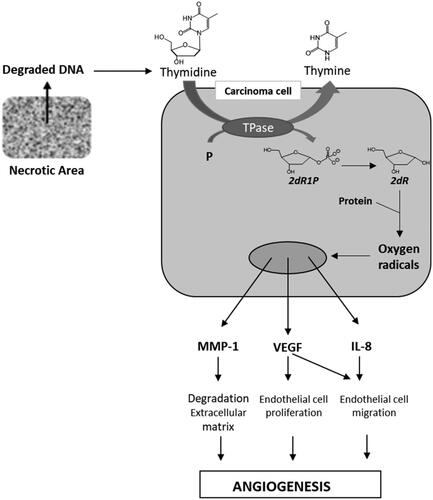
Various crystal structures of Escherichia coli TP and human TP (hTP) complexes with thymine or thymidine analogues have been publishedCitation11–16. Briefly, the binding of thymidine is ensured by three hydrogen bonds between the two carbonyls and the NH function with an arginine, a serine and a lysine. These studies revealed a large empty space (pocket 1) facing the C-5 and C-6 of the pyrimidinedione and another (pocket 2) close to the 1-position ().
Figure 2. Schematic representation of the pyridimidinedione binding site of hTP and chemical structures of 7-DX and TPI.
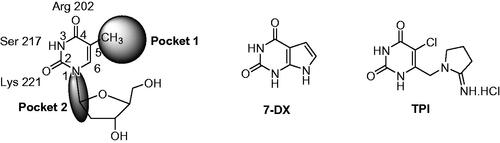
Various TP inhibitors have been reported in the literatureCitation8,Citation17,Citation18. For a recent review see Sajid et al.Citation19. In order to interact with the thymidine binding site, most of them are pyrimidine-2,4-dione derivatives. 7-Deazaxanthine (7-DX) and 5-chloro-6-[1-(2-iminopyrrolidinyl)methyl]uracil hydrochloride (TPI) have emerged ()Citation20,Citation21. Up to now, the most potent inhibitor is TPI (IC50 hTP = 0.035 µMCitation8), whose anti-angiogenic activity was attested through several in vitro and in vivo modelsCitation22–24. TPI has been co-crystallized with hTPCitation12. In combination with trifluridine, TPI has been recently approved (TAS-102, Lonsurf®) for the treatment of metastatic colorectal cancerCitation25. The role of TPI is dual, it prevents the degradation of trifluridine by TP and exerts anti-angiogenic effect.
Only a few polycyclic TP inhibitors have been reported (for a review see Bera et al.Citation8). Crystallographic studies data allowed us to design inhibitors in which the natural ligand feature is linked to a polycyclic aromatic nitrogen heterocycle either by ring annelation or via various linkers.
In the present study, we describe the synthesis and the biological in vitro evaluation as TP inhibitors of four series of new original aromatic derivatives. To elucidate the mechanism of enzyme inhibition of the hits, a brief kinetic study was attempted. In silico molecular docking studies have also been performed to explore the binding site and possible interactions mode of these new derivatives with TP.
2. Results and discussion
2.1. Chemistry
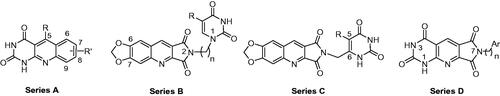
In a first time, we have designed three series of new heterocycles as potential TP inhibitors. The general structures are presented in . In series A, the pyrimidinedione was annelated to a quinoline giving a rigid tricyclic heterocycle. The substituents on the 5 to 9-positions could modulate the interaction with pocket 1. Compounds of series B and C were more flexible. Thus, a pyrimidinedione substituted by a methylenic chain either on the 1-position (series B) or on the 6-position (series C) was linked to the 2-position of a quinolopyrrolidinedione. In a second time, the results of a first docking study allowed us to design compounds of series D in which the quinolopyrrolidinedione has been replaced by a pyrimidopyridopyrrolidinetetraone.
Figure 3. General structures of the designed derivatives in series A–D.
Our strategies for synthesising the tricyclic heterocycles are based on one-pot multicomponent reactions, developed in our group, that involved an aniline, an aldehyde, and a 1,3-dicarbonyl derivativeCitation26–28.
2.1.1. Pyrimido[4,5-b]quinoline-2,4(1H, 3H)-diones (series A)
We have described a very simple and convenient one-pot reaction for the synthesis of pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones involving an aldehyde, an aniline, and barbituric acid 1 as the 1,3-dicarbonyl reagent ()Citation27. This straightforward method circumvents the preparation of unstable substituted 2-aminobenzaldehydes that limits the scope of previously described synthesesCitation29–31. The use of commercially available anilines allowed the facile syntheses of pyrimido[4,5-b]quinolinediones substituted on the 6 to 9-positions with electron donor or electron-withdrawing groups. Access to the 5-substituted derivatives is also possible starting from aliphatic or aromatic aldehydes. We have previously described the synthesis of compounds 2a–d, 2f–h, 2j–k, 2m, 2p–r which have been obtained in 25% to 88% yieldCitation27. According to this method, six new derivatives (2e, 2i, 2l, 2n, 2o, 2s) have been synthesised in modest-to-good yields ().
Table 1. Synthesis of the pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 2. 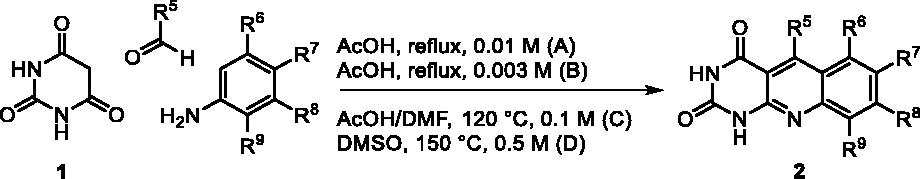
It is worth noting that the presence of two electron-withdrawing groups on anilines is allowed but provided compound 2l in low yield (17%). Tetra and pentacyclic derivatives have been obtained respectively using 1-naphthylamine and 1-aminoanthracene (compounds 2n and 2o). In the last case, it was necessary to dilute the reaction mixture (330 mL of AcOH/mmol instead of 100 mL) to avoid the precipitation of the poorly soluble dihydro intermediate. The angular structures of compounds 2n, and 2o have been unambiguously determined by NMR.
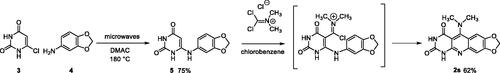
The preparation of compound 2s has required another reaction scheme. In the 1,3-dimethyl series, the literature described the synthesis of 5-dimethylaminopyrimidoquinolines using 6-arylamino-1,3-dimethyluracils as starting materials. When the synthesis was performed by nucleophilic substitution on an intermediate 5-thiomethyl heterocycle, the overall yields were very low due to the preparation of the thiomethylheterocycleCitation32. The use of phosgene iminium chloride (Viehe’s salt) furnished pyrimidoquinolines substituted either on the 5-position by a 5-dimethylamino group (in the presence of triethylamine) or by a chloride (without triethylamine)Citation33. The dimethylpyrimidoquinoline 2s has been prepared using this second procedure, so 6-arylaminouracil 5 was obtained by reaction of 6-chlorouracil 3 with 3,4-methylenedioxyaniline 4, under microwave irradiation, according to the method described by Fang et al.Citation34. Then, the reaction of the Viehe’s salt on 5 gave an iminium intermediate that cyclized into 2s (Scheme 1). Surprisingly, the reaction leads to the dimethylamino compound even in the absence of triethylamine. The low solubility of 2s, which precipitates in the reaction mixture, probably prevents the nucleophilic attack by chloride ions observed in other series.
Scheme 1. Synthesis of compound 2s.
2.1.2. 2H-pyrroloquinoline-1,3-diones/pyrimidinediones (series B and C)
Compounds of series B et C have been obtained by N2-alkylation of the 6,7-methylenedioxy-2H-pyrrolo[3,4-b]quinolines (10) using a pyrimidinedione substituted by a halogenoalkyl chain.
Although many publications have already reported the synthesis of N2-substituted 2H-pyrrolo[3,4-b]quinolines, only three have been covered regarding the N2-unsubstituted heterocycle. The reported methods are multistep syntheses. Starting from the corresponding 2-aminoacetophenones, the 9-methyl derivatives were obtained as a guanidinium salt in two steps and 38% overall yieldCitation35 and the 9-phenyl analogue in 5 steps and 12% overall yieldCitation36. The ultimate step of the third chemical scheme involved the condensation of urea on the quinoline-2,3-dicarboxylic diacid to give the unsubstituted heterocycleCitation37.
In a first time, we intended to apply this last method to the synthesis of the 6,7-methylenedioxy derivative 10 using a 6,7-methylenedioxyquinoline-2,3-dicarboxylate. In the literature, an analogous dimethyl ester has been already described either by reaction of the 2-aminobenzaldehyde with dimethyl acetylenedicarboxylate and subsequent acidic treatmentCitation38 or by reaction of the methylenedioxyaniline with the dimethyl acetylenedicarboxylate and subsequent treatment with the Vilsmeier reagentCitation39. In both cases, the yield of the second step was not specified.
We thought that it would be possible to prepare the diethyl ester 8 in a one-pot reaction. Indeed, 8 was obtained in 40% yield by refluxing in EtOH a mixture of aniline 4, diethyl acetylenedicarboxylate (7) and formaldehyde (6) (Scheme 2). Unfortunately, the condensation of 8 with urea afforded 10 in only 12% yield. Assays with the corresponding diacid were unsuccessful.
Scheme 2. Synthesis of compound 10.
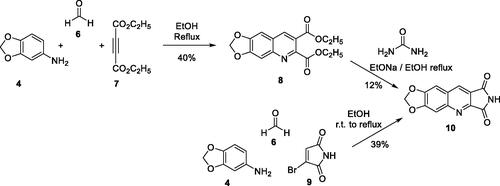
Finally, we have synthesised 10 in 39% yield according to an adjustment of our three-component one-pot method replacing the cyclic 1,3-diketone by bromomaleimide (9) which is more easily accessible than the hydroxymaleimide (Scheme 2)Citation28. With this example, we have shown that the scope of the one-pot reaction could be extended to 1,3-diketone analogues such as a cyclic β-halogenoketone.
It is well known that pyrimidinediones could be alkylated on the N1 and/or on the N3. As an example, the reaction of thymine with the ω-dibromobutane and the ω-dibromohexane lead to a mixture of N1-substituted derivatives and N1,N1′-dimers. With the dibromopentyl chain in the same conditions, only the N1,N3-disubstituted pyrimidinedione has been isolatedCitation40. In several papers, the N1-alkylation was achieved in two steps via the formation of the 2,4-O-disilyl pyrimidine in order to avoid the formation of side productsCitation41–46. We have prepared compounds 12a,b and 15 according to the described procedure but without isolation of the unstable disilyl intermediates (Scheme 3).
Scheme 3. Synthesis of compounds 13a, 13b, 16a, 16b, and 17.
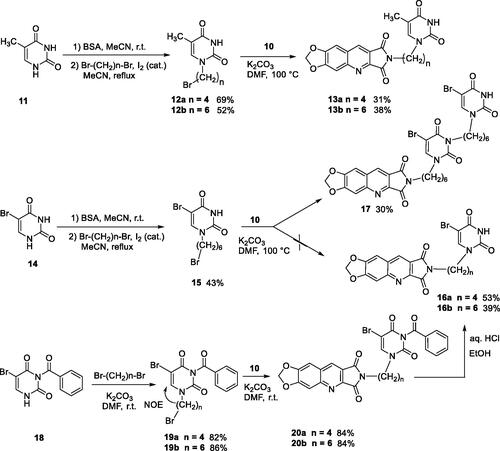
Final compounds 13a and 13b were obtained by alkylation of 10 by 12a and 12b in DMF in the presence of K2CO3. Unfortunately, in the bromo-uracil series, it was not possible to isolate compound 16b which reacted again with another molecule of 15 to give 17 (Scheme 3).
Access to the 5′-bromo derivatives required the use of a benzoyl protecting group on the N3 position of 5-bromouracil (14)Citation47. After alkylation of the N3-benzoylbromouracil (18)Citation48, the reaction of the resulting compounds 19a,b with 10 and subsequent deprotection of the pyrimidinedione in acidic medium afforded compounds 16a and 16b (Scheme 3). A NOE NMR experiment realised on compound 19a confirmed the N1-alkylation of the pyrimidinedione from bromouracil (14).
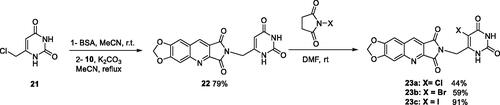
For the synthesis of series C, it was necessary to protect the 6-chloromethyluracil (21) by bis(trimethylsilyl)acetamide (BSA) before the condensation with 10. The reaction of the resulting 5-unsubstituted compound 22 with N-halogenosuccinimides lead to the 5′-halogeno compounds 23a–c (Scheme 4).
Scheme 4. Synthesis of compounds 22 and 23a–c.
Biological results and molecular modelling have prompted us to synthesise the N2-benzyl substituted compound 24 () in which the pyrimidinedione moiety of derivative 22 is replaced by a phenyl nucleus. Thus, the reaction of benzyl bromide with 10 in the presence of K2CO3 in DMF furnished 24 in 65% yield.
2.1.3. Pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6,8(3H,7H)-tetraones (series D)
Derivatives of series D have been synthesised from the unsubstituted tricyclic heterocycle 27 (Scheme 5). We have previously described easy access to this heterocyclic skeleton via a new synthon, the 4-formyl-3-hydroxy-2,5-dioxo-2,5-dihydro-1H-pyrrole (26)Citation28. The functionalization of 27 was achieved by reaction with two equivalents of the appropriate amine in refluxed DMF, and cyclisation of the resulting diamide using two equivalents of p-toluenesulfonic acid (PTSA)Citation28. Four new derivatives 28c, 28e, 28f, and 28h have been synthesised for the present study.
Scheme 5. Synthesis of compounds 28a–h and 30.
In an attempt to prepare 28i, the reaction of the 6-aminomethyluracil hydrochloride (29)Citation49,Citation50 with 27 afforded only the diamide 30 which could not be cyclized into 28i whatever the acidic conditions used (PTSA, TFA, HCl, H3PO4, B(OH)3) (Scheme 5).
2.2. In vitro thymidine phosphorylase inhibiting activity and molecular docking study
Inhibitory activities of the new heterocyclic derivatives in series A-D have been tested in vitro against recombinant E. coli TP. The adopted protocol is a modification of the original method developed by KhanCitation51. TP activity and inhibition assays were based on the phosphorolysis of thymidine to thymine, this conversion results in a significant spectrophotometric decrease. At 290 nm and pH 7.4, thymidine has a higher extinction coefficient than thymine (Δε = −480 M−1 cm−1). The absorbance at 290 nm was measured every 2 min for 30 min in 96-well plates at 25 °C.
A preliminary screening has been carried out at 50 µM or at the maximum concentration allowed by the solubility of the tested compound in the incubation medium (aqueous buffer containing 2.5% of DMSO). Results are given in percentage of inhibition (average of three measurements). For the most promising compounds (inhibition > 30% and solubility > 50 µM), IC50 values have been determined (). 7-DX, which presents the activity of the same order of magnitude (IC50 E. coli TP = 40 µMCitation8) was used as a reference.
Table 2. Escherichia coli TP inhibition of series A derivatives.
Table 3. Escherichia coli TP inhibition of series B and C derivatives.
Table 4. Escherichia coli TP inhibition of series D derivatives.
To help understand structure–activity relationships, the geometry optimised structures of the synthesised compounds were docked into the active site of hTPCitation12 using GOLD 5.1. In order to validate the docking protocol, an X-ray crystal of TP complexed with TPI was retrieved, and the ligand was redocked into the active site of the enzyme. The predicted binding mode and interaction pattern of TPI generated in our study were found to be in accordance with the reported crystallographic study with an RMS deviation of 0.654 ÅCitation12.
Finally, a brief kinetic study was performed to elucidate the mechanism of inhibition of the two most interesting derivatives.
2.2.1 Series A
Series A has been designed as a conformationally constrained series in which the pyrimidinedione moiety should interact with the thymine binding site and the other nuclei with pocket 1 facing the C-5 and C-6 of the thymine (). In order to modulate the interactions and/or the solubility, various substituents have been introduced on the 5 to 9-positions.
Seven derivatives exhibited inhibiting activity towards E. coli TP (compounds 2c, 2d, 2e, 2i, 2l, 2m, 2p), three of them (compounds 2d, 2l, 2p) are interesting in terms of activity and solubility as compared to 7-DX (). This preliminary enzyme inhibition study suggested the following structure-activity relationships for series A compounds:
A methyl on the benzenic ring was unfavourable except for the 9-methyl derivative 2c that was weakly active.
Monosubstitution on the 8-position by an ether group seemed to be interesting in the case of a methoxy group (compound 2d) but the activity decreased with the substituent size (compounds 2e and 2f). The replacement of the oxygen by methylene seemed to be possible (compare compounds 2e–2i). Compounds 2g and 2h were sparingly soluble; consequently, it is not possible to conclude about the effect of the presence of two ether groups.
The presence of an electron-withdrawing substituent on the 8-position such as a chlorine atom (compound 2j) or a trifluoromethyl group (compound 2k) was unfavourable. However, the 7,8-dichloro derivatives 2l exhibited an IC50 in the same order of magnitude as the 8-methoxy derivative 2d.
The enzyme seemed to accept the presence of an additional angular benzene ring in compound 2m. It is not possible to conclude concerning the geometrical isomer 2n or the homologue 2o which were less soluble.
The introduction of methyl on the 5-position of the 8-methoxy derivative 2d was possible (compound 2p). In the case of compound 2g, the 5-substituted derivatives remained inactive whatever the substituent sizes (compounds 2q, 2r, 2s).
In this series, results were found to be optimum for the two 8-methoxypyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 2d and 2p, and the 7,8-dichloro derivative 2l.
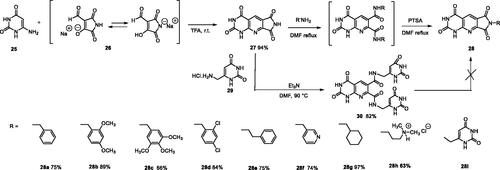
As expected, preliminary docking study showed that the pyrimidinedione moiety of the series A derivatives interacted with the active site of hTP in a similar binding mode as the 5-chlorouracil fragment of TPI, that is, by establishing hydrogen bonds with arginine 202, serine 217 and lysine 221 of the α domain and histidine 116 of the α/β domain. As an example, docking of compound 2d superposed on TPI (in green) is presented in . Results were the same for the other active derivatives 2p and 2l (Supporting information, Figure S1).
Figure 4. Docking superposition in the hTP active site of compounds 2d (in blue), 2e (in pink), 2i (in orange), and TPI (in green).
The less marked activity of compounds 2c and 2e was also supported by the modelling results that highlighted two major binding modes corresponding in both 180° vertical and 180° horizontal rotations of the tricyclic moieties (Supporting information, Figure S2 for compound 2c as an example). It is worth noting that several substituents on the 8-position seemed to be located in the same area of the active site as showed in : methoxy group of compound 2d, benzyl of compound 2i, and phenoxy for one of the conformations of compound 2e.
2.2.2. Series B and C
Series B and C have been designed as more flexible series than series A with the aim of interactions of the pyrimidinedione in the thymidine binding site of TP and of the methylenedioxypyrroloquinoline moiety in pocket 1 (see ).
In terms of enzymatic inhibition, series B and C did not present really interesting activities in comparison with 7-DX. The unsubstituted derivative 22 and the 5-bromopyrimidinediones 16a, 16b, or 23b seemed to the more interesting but, unfortunately, they are the less soluble. The spacer size has no influence. In the case of a methylene linker (series C), the presence of a halogen on the 5-position was not favourable. More interestingly, the pyrimidinedione did not seem to be essential, it can be replaced by a phenyl nucleus (compound 24) ().
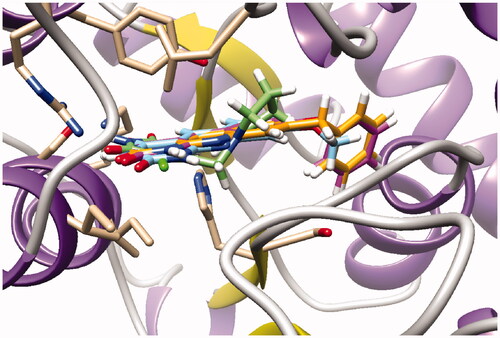
Surprisingly, docking study showed that in both series, the dioxolane of the pyrroloquinoline moiety is positioned in the same place as the pyrimidinedione of TPI suggesting interactions of the methylenedioxy group with the active site; the pyrimidinedione moiety interacting with a large pocket which has never been explored (superposition of compound 16b and TPI on ).
Figure 5. Docking superposition in the hTP active site of TPI (in green), compounds 16b (dark green cloud), and 23b (white cloud).
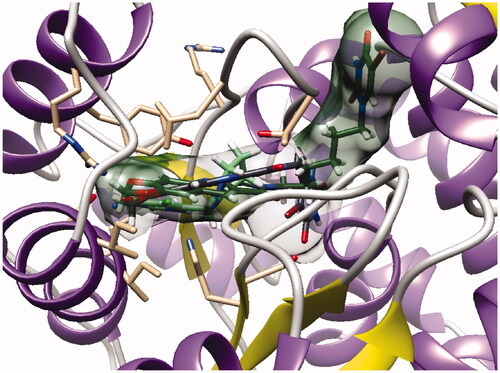
However, as presented in for compound 16b (dark green cloud) and compound 23b (white cloud), two differences between series B and C were noticed. On the one hand, the tricyclic moieties were not superposed (180° vertical rotation). On the other hand, the aliphatic chains and pyrimidinediones of the B-series molecules were positioned in the upper part of the pockets, whereas pyrimidinediones of series C derivatives interacted with the lower part of this pocket. Interestingly, the phenyl nucleus of compound 2i (series A) was also located in this white cloud (Supporting information, Figure S3).
2.2.3. Series D
The B and C series modelling results led us to design the D-series in which the methylenedioxyphenyl rings were replaced by a pyrimidinedione with the aim to optimise interactions with the thymidine binding site of the natural ligand. Furthermore, various substituents have been introduced on the imide nitrogen in an attempt to explore the lower part of the “new” pocket.
In a physicochemical point of view, series D compounds are more soluble than the previous series. However, they were uninteresting in terms of enzymatic inhibition activity except for the N-benzyl derivative 28a ().
As expected, the tricyclic moiety of compound 28a was well superposed with compound 2i, the benzyl analog in series A. The benzyl nuclei were positioned in the same area but not in the same position ().
Figure 6. Docking superposition in the hTP active site of compounds 2i (in orange), 28a (in purple), and 30 (in blue).
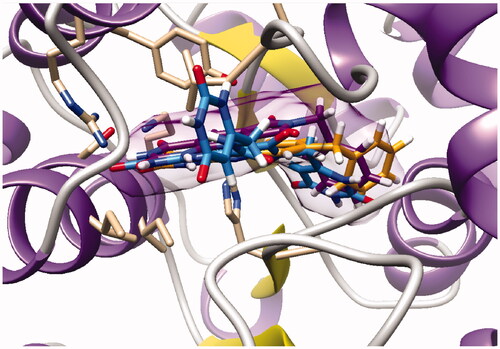
Surprisingly, the flexible open intermediate derivative 30 was as active as compound 28a. Interestingly, one 6-pyrimidinomethylamino arm was positioned in the same area as previously described compounds (), the other one was located at the entry of the pocket.
2.3. Mechanism of enzyme inhibition
The synthesised compounds have been designed as TP inhibitors interacting with the thymidine fixation site. However, TP being a two substrates enzyme, it was necessary to verify that new inhibitors do not bind to the phosphate site. This study has been realised with one of the most active and/or the most soluble compounds in each series, that is, compounds 2p, 23b, 28a, and 30. E. coli TP activity was determined in the presence of a saturating concentration of thymidine (equivalent to three times the value of the Michaelis-Menten constant [km]Citation52) and variable amounts of phosphate (2–30 mM). Tested compounds have been used at a concentration corresponding to the IC50. For each tested derivative, phosphate concentration did not have any significant influence on the percentage of inhibition, indicating that the selected inhibitors did not bind to the phosphate site.
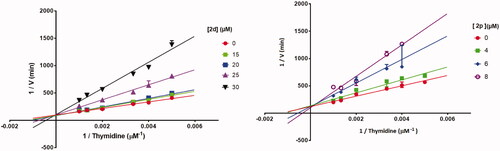
To explore whether 2d and 2p, the most active compounds which showed the same activities as 7-DX (TP CI50: 26–28 µM), acted as a competitive, uncompetitive, or mixed TP inhibitor, a brief kinetic study was performed according to previously described methodCitation51. The percentage of TP inhibition was determined for different concentrations of the inhibitors and thymidine. Lineweaver-Burk plots showed that all straight lines converged at the same point on the positive side of the y-axis (). Consequently, 2d and 2p exhibited competitive inhibition kinetic on TP with thymidine as substrate. This was further supported by the fact that the Km values increased in the presence of 2d and 2p, while the Vmax values did not change significantly as the inhibitor concentration increased.
Figure 7. Lineweaver–Burk plots of E. coli TP inhibition by 2d and 2p, in the presence of variable concentrations of thymidine demonstrating competitive-type enzyme inhibition. Results are presented as means ± SD; SD denoted by error bars (experiments carried out in triplicate).
2.4. Cell proliferation assay (MTT)
The two most active compounds against TP (compounds 2d and 2p) were evaluated for growth-inhibiting properties in two cells lines namely human umbilical vein endothelial cells (HUVEC) and epidermal carcinoma cells (A431) which are well known to overexpress epidermal growth factor receptor (EGFR), a pathway involved in the cell proliferation and angiogenesis.
The inhibitory effect on cell proliferation was assessed using MTT assayCitation53 after 72 h of treatment through dose-response assays performed in the 100 µM to 0.1 µM concentration range. The results were expressed as percentages of growth inhibition.
Compound 2d exhibited better activity against A431 cell line (57% at 50 µM) than against HUVEC cell line (56% at 100 µM) but antiproliferative effect remains low. Compound 2p can be considerate as less cytotoxic against the two cell lines (50% and 31% at 100 µM against respectively A431 and HUVEC cells).
3. Conclusion
In conclusion, a small library of 38 derivatives has been synthesised and evaluated for its TP inhibiting activity. Around the pharmacophoric pyrimidinedione core of the natural substrate thymidine, four series have been designed in order to interact with wide empty pockets of the active site.
The natural ligand has been annealed to a quinoline in series A (pyrimidoquinoline-2,4-diones), substituted via a methylenic chain by a quinolopyrrolidinedione in the more flexible series B and C, and the polycyclic heterocycle has been replaced by a pyrimidopyridopyrrolidinetetraone in series D. The tricyclic moieties of these new polycyclic nitrogen heterocycles have been synthesised by one-pot multicomponent reactions that involved an aniline, an aldehyde, and a 1,3-dicarbonyl derivative or an analogue.
The biological evaluation identified several structurally distinct TP inhibitors in series A and D. Among them, compounds 2d, 2l, 2p, 28a, and surprisingly the open intermediate 30 showed a modest to good TP inhibition (IC50 values ranging from 26 to 87 µM) when compared to 7-DX used as a positive control. The two most active compounds 2d and 2p were shown to interact with the thymidine fixation site and to exhibit a competitive mode of inhibition towards TP.
Molecular docking analysis confirmed the interaction of these newly synthesised compounds at the active binding site of TP. Moreover, docking studies highlighted a plausible specific interaction in a wide pocket that had not been yet explored.
For the first time, our study showed that it is possible to inhibit TP with tricyclic heterocycles. It is worth noting that the active compounds have in common a pyrido[2,3-d]pyrimidinedione nucleus and possess a chain interacting with the same part of this pocket. Interestingly, the open intermediate 30 becomes a starting point for a novel series that will be further exploited in order to improve the activity against TP.
4. Experimental section
4.1. Chemistry
General procedures: Commercial reagents were used as received without further purification. Microwave irradiation reaction was performed with an InitiatorTM 2.0 device, Biotage. Reactions were followed with thin-layer chromatography (TLC) (using 0.20 mm silica or alumina gel 60 F254 aluminium plates, Merck) and visualisation was achieved with UV light (254 and 365 nm). Purifications were achieved through recrystallization or flash chromatography (using 40–63 µM silica, Merck). 1H and 13C NMR spectra were recorded on a Bruker AC 300 or 400 spectrometer by using DMSO-d6 as the solvent and internal standard. Chemical shifts are reported in ppm and coupling constants (J) are given in Hertz. Spin multiplicities are reported as follows: s = singlet, d = doublet, dd = doublet of doublet, m = multiplet, q = quadruplet, quint = quintuplet, t = triplet. Melting points were measured on a Stuart SMP3 melting point apparatus and are uncorrected. IR spectra were obtained on Perkin-Elmer 1600 spectrophotometer. Elemental analyses were performed at the CNRS Analysis Laboratory, Gif-sur-Yvette, France.
4.1.1. Pyrimido[4,5-b]quinoline-2,4 (1H, 3H) -diones (series A)
4.1.1.1. General procedure for the synthesis of compounds 2
A suspension of the requisite aniline (1.00 mmol, 1.0 eq.), barbituric acid 1 (128 mg, 1.00 mmol, 1.0 eq.), and paraformaldehyde (30.0 mg, 1.00 mmol, 1.0 eq.) was refluxed in AcOH (100 mL, method A; 300 mL, method B) or heated at 120 °C in a 1:1 mixture of AcOH/DMF (10 mL, method C). Reaction times and yields are given in .
4.1.1.2. 8-Phenoxypyrimido[4,5-b]quinoline-2,4(1H,3H)-dione (2e)
Method C using 3-phenoxyaniline. After removal of the solvent under vacuum, the solid was boiled in H2O (10 mL) for 1 h, filtered and washed successively with H2O, EtOH and Et2O. Recrystallization from DMF afforded 2e as a yellowish solid. mp: 334–336 °C (decomposition). 1H NMR (DMSO-d6, 400 MHz) δ: 11.61 (s, 1H), 11.46 (s, 1H), 8.95 (s, 1H), 8.16 (d, J = 9.0 Hz, 1H), 7.5 (t, J = 7.8 Hz, 2H), 7.34–7.28 (m, 2H), 7.22 (d, J = 7.8 Hz, 2H) 6.98 (d, J = 2.0 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 162.2, 161.6, 154.6, 151.1, 150.7, 150.7, 138.7, 132.1, 130.4, 125.2, 120.9, 120.5, 118.2, 110.4, 109.5 ppm. IR ν: 3136, 3057, 3041, 2904, 2839, 1733, 1678, 1613, 1591, 1509, 1487, 1464, 1425, 1403, 1377, 1340, 1287, 1264, 1222, 1127, 970, 840, 817, 790, 775, 749, 707, 684 cm−1. Anal. Calcd. for C17H11N3O3 · 0.25 H2O (309.79): C, 65.91; H, 3.74; N, 13.56. Found: C, 65.93; H, 3.87; N, 13.77%.
4.1.1.3. 8-Benzylpyrimido[4,5-b]quinoline-2,4(1H,3H)-dione (2i)
Method C using 3-benzylaniline. Work up was the same as used for 2e. Recrystallization from DMF afforded 2i as a yellowish solid. mp: 297–299 °C (decomposition). 1H NMR (300 MHz, DMSO-d6) δ: 11.65 (s, 1H), 11.47 (s, 1H), 8.92 (s, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.64 (s, 1H), 7.41 (dd, J = 8.4 Hz, 1.3, 1H), 7.35–7.28 (m, 4H), 7.26–7.18 (m, 1H), 4.16 (s, 2H) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 162.3, 150.7, 150.2, 149.8, 147.2, 140.2, 138.7, 129.8, 129.0, 128.6, 126.8, 126.3, 125.7, 123.1, 110.5, 41.3 ppm. IR ν: 3135, 3045, 2909, 2833, 1731, 1703, 1679, 1611, 1580, 1513, 1490, 1451, 1397, 1344, 1315, 1285, 1268, 1027, 829, 789, 749, 705, 677 cm−1. Anal. Calcd. for C18H13N3O2 · 0.25 H2O (307.81): C, 70.23; H, 4.42; N, 13.65. Found: C, 70.00; H, 4.54; N, 13.72%.
4.1.1.4. 7,8-Dichloropyrimido[4,5-b]quinoline-2,4(1H,3H)-dione (2l)
Method A using 3,4-dichloroaniline. The suspension was filtered and washed successively with H2O and Et2O to afford pure 2l as a yellow solid. mp: > 360 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.97 (s, 1H), 11.88 (s, 1H), 11.61 (s, 1H), 9.04 (s, 1H), 8.54 (s, 1H), 8.09 (s, 1H) 1.91 (s, 3H) ppm. IR ν: 3179, 3053, 3024, 2837, 1690, 1655, 1624, 1578, 1560, 1481, 1450, 1418, 1369, 1335, 1265, 1182, 1126, 1034 cm−1. Anal. Calcd. for C11H5Cl2N3O2 · CH3COOH (342.13): C, 45.64; H, 2.65; N, 12.28. Found: C, 45.40; H, 2.39; N, 12.23%.
4.1.1.5. Benzo[h]pyrimido[4,5-b]quinoline-8,10(9H,11H)-dione) (2n)
Method A using 1-naphthylamine. Work up was the same as used for 2l to afford pure 2n as a pink solid. mp: 326–328 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.88 (s, 1H), 11.57 (s, 1H), 9.08 (d, J = 8.0 Hz, 1H), 9.00 (s, 1H), 8.05 (d, J = 8.0 Hz, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.84–7.74 (m, 2H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 162.4, 150.7, 150.1, 148.5, 137.9, 134.8, 129.8, 129.4, 128.2, 127.1, 126.0, 125.9, 124.7, 122.4, 110.4 ppm. IR ν: 3063, 3038, 3022, 3007, 2841, 1688, 1645, 1618, 1601, 1578, 1559, 1541, 1508, 1473, 1437, 1419, 1394, 1327, 1281, 1271, 1225, 1213, 1196, 1146 cm−1. Anal. Calcd. for C15H9N3O2 · 1.25 H2O (285.77): C, 63.04; H, 4.06; N, 14.70. Found: C, 62.93; H, 3.58; N, 14.97%.
4.1.1.6. Naphto[2,3-h]pyrimido[4,5-b]quinoline-2,4(1H,3H)-dione (2o)
Method B using 1-aminoanthracene. Work up was the same as used for 2e. Recrystallization from DMF afforded 2o as a green-brownish solid. mp: > 360 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.96 (s, 1H), 11.58 (s, 1H), 9.66 (s, 1H), 8.93 (s, 1H), 8.62 (s, 1H), 8.25 (d, J = 8.0 Hz, 1H), 8.21 (d, J = 8.0 Hz, 1H), 7.91 (q, J = 8.0 Hz, 2H), 7.70 (m, 2H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 162.8, 151.1, 150.9, 150.5, 137.6, 133.7, 132.4, 131.7, 129.4, 128.4, 128.3, 127.9, 127.2, 127.0, 126.8, 125.9, 125.2, 122.9, 110.8 ppm. IR ν: 3042, 3028, 3011, 2992, 2974, 1713, 1682, 1609, 1585, 1566, 1541, 1506, 1489, 1454, 1393, 1337, 1277, 1206, 1038 cm−1. Anal. Calcd. for C19H11N3O2 · 0.75 H2O (326.82): C, 69.82; H, 3.86; N, 12.86. Found: C, 69.98; H, 3.38; N, 12.95%.
4.1.1.7. 6-(3,4-Methylenedioxyphenylamino)pyrimidine-2,4(1H,3H)-dione (5)
A suspension of 6-chloropyrimidinedione (3) (146 mg, 1.00 mmol, 1.0 eq.) and methylenedioxyaniline (4) (548 mg, 4.00 mmol, 4.0 eq.) in DMAC. After evaporation of the solvent under reduced pressure, Et2O was added. The resulting suspension was filtered and washed with Et2O and MeOH to give crude 5 (185 mg, 75% yield) which was used without further purification in the next step. An analytical sample was obtained by recrystallization of a small amount from MeOH. mp: 305 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.40 (s, 1H), 10.16 (s, 1H), 8.01 (s, 1H), 6.91 (d, J = 9.0 Hz, 1H), 6.83 (d, J = 2.0 Hz, 1H), 6.67 (dd, J = 9.0 and 2.0 Hz, 1H), 6.04 (s, 2H), 4.46 (s, 1H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 164.7, 153.7, 151.3, 148.2, 145.4, 131.9, 117.9, 108.9, 106.3, 101.9, 75.6 ppm. IR ν: 3298, 2899, 1732, 1625, 1490, 1395, 1354, 1326, 1288, 1248, 1226, 1199, 1102, 1040, 990, 928, 813, 728, 757, 649, 600, 547, 531 cm−1. Anal. Calcd. for C11H9N3O4 · 0.25 H2O (251.71): C, 52.49; H, 3.80; N, 16.69. Found: C, 52.16; H, 3.67; N, 16.82%.
4.1.1.8. 5-Dimethylamino-7,8-methylenedioxypyrimido[4,5-b]quinoline-2,4(1H,3H)-dione (2s)
To a suspension of 5 (247 mg, 1.00 mmol, 1.0 eq.) in anhydrous chlorobenzene (11 mL) was added Viehe’s salt (195 mg, 1.20 mmol, 1.2 eq.). The reaction mixture was refluxed for 12 h. The resulting solid was isolated by filtration, washed with H2O and recrystallized from DMF to give 2s as a yellow solid (186 mg, 62% yield). mp: 348 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.15 (s, 1H), 10.94 (s, 1H), 7.44 (s, 1H), 7.05 (s, 1H), 6.20 (s, 2H), 3.06 (s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 161.4, 159.3, 152.6, 151.3, 150.8, 148.8, 146.4, 119.2, 104.3, 102.7, 102.0, 101.8, 44.5 ppm. IR ν: 3166, 3053, 2922, 1732, 1691, 1670, 1614, 1577, 1523, 1468, 1433, 1309, 1254, 1231, 1045, 801, 573, 535.cm−1. Anal. Calcd. for C14H12N4O4 · 0.5 H2O (309.28): C, 54.37; H, 4.24; N, 18.12. Found: C, 54.07; H, 3.88; N, 17.94%.
4.1.2. 2H-Pyrroloquinoline-1,3-diones/pyrimidinediones (series B and C)
4.1.2.1. Diethyl 6,7-methylenedioxyquinoline-2,3-dicarboxylate (8)
To a solution of aniline 4 (137 mg, 1.00 mmol, 1.0 eq.) in EtOH (10 mL) was added 37% aqueous formaldehyde solution (0.24 mL, 3.00 mmol, 3.0 eq.) and ethyl acetylene dicarboxylate (7) (0.16 mL, 1.00 mmol, 1.0 eq.). The reaction mixture was refluxed for 1 h. After evaporation of the solvent under vacuum, the residue was purified by flash column chromatography, using DMC as an eluant to afford 8 as a yellow powder (127 mg, 40% yield). mp: 132.5–133.5 °C. 1H NMR (400 MHz, DMSO-d6) δ: 9.23 (s, 1H), 7.43 (s, 1H), 7.07 (s, 1H), 6.17 (s, 2H), 4.56 (q, J = 7.0 Hz, 2H), 4.45 (q, J = 7.0 Hz, 2H), 1.45 (t, J = 7.0 Hz, 3H), 1.42 (t, J = 7.0 Hz, 3H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 167.2, 164.6, 152.7, 149.4, 149.0, 148.0, 140.7, 120.5, 117.9, 106.1, 102.5, 100.7, 62.3, 61.2, 14.2, 14.1 ppm. IR ν: 2983, 2968, 2928, 1757, 1723, 1573, 1502, 1467, 1368, 1344, 1315, 1278, 1198, 1150, 1104, 1049, 1035, 1020, 941, 851, 760, 742, 669, 637 cm−1. Anal. Calcd. for C16H15NO6 (317.29): C, 60.57; H, 4.77; N, 4.41. Found: C, 60.61; H, 4.72; N, 4.21%.
4.1.2.2. 6,7-Methylenedioxypyrrolo[3,4-b]quinoline-1,3-dione (10)
Method A: To a solution of Na (23 mg, 1.00 mmol, 2.8 eq.) in EtOH (0.5 mL) were added 8 (115 mg, 0.36 mmol, 1.0 eq.) and urea (34 mg, 0.56 mmol, 1.6 eq.). The reaction mixture was heated under reflux for 1.5 h. The resulting brown solid was isolated by filtration, washed with a 1:1 AcOH/H2O mixture (5 mL) to afford 10 as a yellow solid (10 mg, 12% yield). Method B: 3,4-Methylenedioxyaniline (4) (411 mg, 3.00 mmol, 1.0 eq.), paraformaldehyde (270 mg, 9.00 mmol, 3.0 eq.) and 3-bromomaleimide 9 (528 mg, 3.00 mmol, 1.0 eq.) were stirred in EtOH (30 mL) at r.t. for 1 night. The reaction mixture was then heated at 60 °C for 6 h. The resulting suspension was filtered off and the solid was washed with EtOH and H2O to afford crude 10 (283 mg, 39%) which was used without further purification. A small amount was recrystallized from DMF to give an analytical sample (yellow solid). mp: 366–368 °C (decomposition). 1H NMR (400 MHz, DMSO-d6) δ: 11.50 (s, 1H), 9.01 (s, 1H), 7.88 (s, 1H), 7.57 (s, 1H), 6.35 (s, 2H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 170.2, 169.3, 153.6, 151.2, 151.0, 141.3, 134.6, 123.2, 119.0, 106.3, 103.8, 98.7 ppm. IR ν: 3449, 2956, 2723, 1771, 1720, 1623, 1573, 1471, 1411, 1330, 1265, 1100, 1030, 935, 872, 790, 748 cm−1. Anal. Calcd. for C12H6N2O4 · 0.25 H2O (246.69): C, 58.42; H, 2.66; N, 11.36. Found: C, 58.35; H, 2.49; N, 11.35%.
4.1.2.3. General method for preparation of compounds 12 and 15
To a suspension of thymine (11) or 5-bromouracile (14) (4.00 mmol, 1.0 eq.) in MeCN (6 mL) was added BSA (2.5 mL, 10.00 mmol, 2.5 eq.) at r.t. When the reaction mixture became clear (15 min), the ω-dibromoalkyl derivative (6.00 mmol, 1.5 eq.) and I2 (cat. amount) were added. The mixture was refluxed for 2 h and then either maintained at r.t. for 1 night (12a,b) or refluxed for 2 days (15). After elimination of the solvent under vacuum, the residue was stirred with water (10 mL), isolated by filtration, and then purified by flash column chromatography.
4.1.2.4. 1-(4-Bromobutyl)-5-methylpyrimidine-2,4(1H,3H)-dione (12a)
Chromatography solvents: DCM/MeOH, 100:0 to 97:3. White solid, 720 mg, 69% yield. mp: 143 °C. 1H NMR (300 MHz, DMSO-d6) δ: 11.23 (s, 1H), 7.54 (s, 1H), 3.65 (t, J = 7.0 Hz, 2H), 3.55 (t, J = 7.0 Hz, 2H), 1.75 (m, 7H) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 164.5, 151.5, 141.8, 109.2, 46.8, 35.0, 29.6, 27.8, 12.4 ppm. IR ν: 3161, 3033, 2929, 2858, 2834, 1692, 1673, 1474, 1426, 1356, 1270, 1222, 872, 765, 692, 560 cm−1. Anal. Calcd. for C9H13BrN2O2 (261.11): C, 41.40; H, 5.02; N, 10.73. Found: C, 41.59; H, 4.86; N, 10.73%.
4.1.2.5. 1-(6-Bromohexyl)-5-methylpyrimidine-2,4(1H,3H)-dione (12b)
Chromatography solvents: DCM/MeOH, 100:0 to 97:3. White solid, 600 mg, 52% yield. mp: 118 °C. 1H NMR (300 MHz, DMSO-d6) δ: 11.20 (s, 1H), 7.53 (s, 1H), 3.60 (t, J = 7.0 Hz, 2H), 3.52 (t, J = 7.0 Hz, 2H), 1.79 (quint, J = 7.0 Hz, 2H), 1.75 (s, 3H), 1.56 (quint, J = 7.0 Hz, 2H), 1.40 (quint, J = 7.0 Hz, 2H), 1.26 (quint, J = 7.0 Hz, 2H) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 164.7, 151.3, 141.9, 108.8, 47.5, 35.5, 32.5, 28.7, 27.6, 25.4, 12.4 ppm. IR ν: 3166, 3036, 2929, 1702, 1647, 1473, 1425, 1356, 1270, 1221, 1185, 1113, 1066, 927, 911, 871, 784, 764, 692, 560 cm1. Anal. Calcd. for C11H17BrN2O2 (289.17): C, 45.69; H, 5.93; N, 9.69. Found: C, 45.67; H, 5.68; N, 9.77%.
4.1.2.6. 5-Bromo-1-(6-bromohexyl)pyrimidine-2,4(1H,3H)-dione (15)
Chromatography solvents: DCM/AcOEt, 90:10. White solid, 610 mg, 43% yield. mp: 151 °C. 1H NMR (300 MHz, DMSO-d6) δ: 11.73 (s, 1H), 8.24 (s, 1H), 3.65 (t, J = 7.0, 2H), 3.52 (t, J = 7.0, 2H), 1.79 (quint, J = 7.0, 2H), 1.58 (quint, J = 7.0, 2H), 1.39 (quint, J = 7.0, 2H), 1.26 (quint, J = 7.0, 2H) ppm. 13C NMR (100 MHz, DMSO-d6): δ: 160.1, 150.7, 145.8, 94.9, 48.2, 35.5, 32.5, 28.6, 25.6, 25.3 ppm. IR (KBr) ν: 3151, 3028, 2932, 2854, 1693, 1620, 1460, 1430, 1357, 1336, 1261, 1047, 749, 635, 560 cm−1. Anal. Calcd. for C10H14Br2N2O2 (354.04): C, 33.92; H, 3.99; N, 7.91. Found C, 34.31; H, 4.03; N, 7.85%.
4.1.2.7. 2-[4-(2,4-Dioxo-5-methyl-3,4-dihydropyrimidine-1(2H)-yl)butyl]-6,7-methylenedioxy-2H-pyrrolo[3,4-b]quinoline-1,3-dione (13a)
A suspension of imide 10 (242 mg, 1.00 mmol, 1.0 eq.) and K2CO3 (166 mg, 1.20 mmol, 1.2 eq.) in anhydrous DMF (9 mL) was stirred for 0.5 h and then a solution of 12a (390 mg, 1.50 mmol, 1.5 eq.) in anhydrous DMF (1 mL). was added. The reaction mixture was heated at 100 °C for 1 h. After evaporation of the solvent under vacuum, H2O (10 mL) was added to give a suspension which was filtered, washed with H2O, and purified by column chromatography (DCM/MeOH, 98:2 to 90:10) to afford 13a as a yellow solid (130 mg, 31% yield). mp: 302 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.19 (s, 1H), 9.05 (s, 1H), 7.89 (s, 1H), 7.57 (s, 1H), 7.52 (s, 1H), 6.36 (s, 2H), 5.76 (CH2Cl2), 3.62 (m, 4H), 1.72 (s, 3H), 1.62 (m, 4H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 168.8, 168.1, 164.7, 153.8, 151.4, 151.3, 151.2, 141.9, 141.1, 133.9, 122.3, 118.9, 108.9, 106.3, 103.8, 98.7, 55.4 (CH2Cl2), 47.0, 37.6, 26.3, 25.3, 12.4 ppm. IR ν: 3062, 3039, 2991, 2817, 1770, 1761, 1690, 1625, 1496, 1467, 1434, 1397, 1364, 1268, 1233, 1180, 1104, 1032, 936, 913, 857, 803, 761, 747, 708, 569 cm−1. Anal. Calcd. for C21H18N4O6 · 0.5 CH2Cl2 (464.86): C, 54.72; H, 4.06; N, 11.87. Found: C, 54.84; H, 4.11; N, 11.76%.
4.1.2.8. 2-[4-(2,4-Dioxo-5-methyl-3,4-dihydropyrimidine-1(2H)-yl)hexyl]-6,7-methylenedioxy-2H-pyrrolo[3,4-b]quinoline-1,3-dione (13b)
Starting from 12b (432 mg, 1.50 mmol, 1.5 eq.), 13b was prepared in the same procedure and work-up as described for 13a. Yellow solid, 170 mg, 38% yield. mp: 262 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.18 (s, 1H), 9.02 (s, 1H), 7.86 (s, 1H), 7.54 (s, 1H), 7.51 (s, 1H), 6.37 (s, 2H), 3.58 (m, 4H), 1.73 (s, 3H), 1.57 (m, 4H), 1.30 (m, 4H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 168.7, 168.1, 164.7, 153.8, 151.3, 151.2, 141.8, 141.1, 133.8, 122.2, 118.9, 108.8, 106.3, 103.8, 98.6, 47.5, 37.9, 28.8, 28.2, 26.3, 25.9, 12.4 ppm. IR ν: 3042, 2936, 2854, 2817, 1768, 1712, 1666, 1623, 1498, 1463, 1429, 1395, 1351, 1310, 1267, 1232, 1181, 1033, 941, 914, 885, 864, 810, 793, 760, 566 cm−1. Anal. Calcd. for C23H22N4O6 · 0.25 H2O (454.95): C, 60.72; H, 4.99; N, 12.31. Found: C, 60.73; H, 4.82; N, 12.39%.
4.1.2.9. 2-[4-[5-Bromo-3-[4-(5-bromo-2,4-dioxo-3,4-dihydropyrimidine-1(2H)-yl)hexyl]-2,4-dioxo-3,4-dihydropyrimidine-1(2H)-yl]hexyl]-6,7-méthylènedioxy-2H-pyrrolo[3,4-b]quinoline-1,3-dione (17)
To a suspension of imide 10 (200 mg, 0.82 mmol, 1.0 eq.) and K2CO3 (136 mg, 0.924 mmol, 1.2 eq.) in anhydrous DMF (8 mL) was added, drop by drop, a solution of 15 (400 mg, 1.23 mmol, 1.5 eq.) in anhydrous DMF (12 mL). The mixture was heated at 100 °C for 6.5 h. After removing the solvent under vacuum, the residue was stirred with water (10 mL) and a aqueous 1 N HCl solution was added until a pH = 3 was reached. The resulting solid was isolated by filtration, washed with H2O and EtOH and recrystallized from MeOH to afford 17 as a yellow solid (194 mg, 30% yield). mp: 144–148 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.72 (s, 1H), 9.05 (s, 1H), 8.28 (s, 1H), 8.21 (s, 1H), 7.89 (s, 1H), 7.57 (s, 1H), 6.36 (s, 2H), 3.69 (m, 8H), 1.54 (m, 8H), 1.30 (m, 8H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 168.8, 168.1, 160.1, 159.2, 153.8, 151.3, 151.2, 150.8, 150.7, 150.6, 145.7, 144.3, 141.1, 133.8, 122.2, 118.9, 106.3, 103.8, 98.7, 94.9, 94.4, 49.4, 48.2, 42.1, 37.9, 28.7, 28.6, 28.2, 27.2, 26.2, 25.8, 25.7 ppm. IR ν: 3186, 3057, 2934, 2858, 1768, 1709, 1655, 1460, 1343, 1262, 1228, 1178, 1032, 939, 865, 803, 760, 620, 574 cm−1. Anal. Calcd. for C32H32Br2N6O8 · 0.5 H2O (797.45): C, 48.20; H, 4.17; N, 10.54. Found: C, 48.03; H, 3.94; N, 10.61%.
4.1.2.10. 3-Benzoyl-5-bromo-1-(4-bromobutyl)pyrimidine-2,4(1H,3H)-dione (19a)
1,4-Dibromobutane (3.24 mL, 24.00 mmol) was added to a suspension of 18Citation48 (885 mg, 3.00 mmol) and K2CO3 (1.656 g, 12.00 mmol, 4.0 eq.) in anhydrous DMF (40 mL). The mixture was stirred at r.t. for 2 h. After removing the solvent under vacuum, the residue was stirred with water (10 mL), isolated by filtration, and purified by flash column chromatography using DCM as a solvent to give 19a as a white solid (1.058 g, 82% yield). mp: 129 °C. 1H NMR (300 MHz, DMSO-d6) δ: 8.51 (s, 1H), 8.04 (d, J = 8.0 Hz, 2H), 7.81 (t, J = 8.0 Hz, 1H), 7.61 (t, J = 8.0 Hz, 2H), 3.79 (t, J = 6.0 Hz, 2H), 3.58 (t, J = 6.0 Hz, 2H), 1.81 (m, 4H) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 167.9, 158.5, 149.2, 143.9, 135.6, 130.9, 130.5, 129.4, 96.1, 48.5, 32.7, 29.2, 27.7 ppm. IR ν: 3081, 2961, 1751, 1702, 1655, 1619, 1598, 1427, 1243, 1179, 1091, 985, 937, 924, 798, 782, 761, 743, 708, 686, 657, 563, 546 cm−1. Anal. Calcd. for C15H14Br2N2O3 (430.09): C, 41.89; H, 3.28; N, 6.51. Found: C, 41.73; H, 3.21; N, 6.47%.
4.1.2.11. 3-Benzoyl-5-bromo-1-(4-bromohexyl)pyrimidine-2,4(1H,3H)-dione (19b)
Starting from 1,6-dibromohexane (7.23 mL, 24.00 mmol), 19b was prepared in the same procedure, work-up and purification as described for 19a. White solid (1.18 g, 86% yield). mp: 89 °C. 1H NMR (300 MHz, DMSO-d6) δ: 8.51 (s, 1H), 8.01 (d, J = 8.0 Hz, 2H), 7.80 (t, J = 8.0 Hz, 1H), 7.61 (t, J = 8.0 Hz, 2H), 3.74 (t, J = 7.0 Hz, 2H), 3.52 (t, J = 7.0 Hz, 2H), 1.80 (quint, J = 7.0 Hz, 2H), 1.65 (quint, J = 7.0 Hz, 2H), 1.41 (quint, J = 7.0 Hz, 2H), 1.30 (quint, J = 7.0 Hz, 2H) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 169.2, 158.9, 149.5, 146.8, 136.2, 131.2, 131.0, 130.0, 94.5, 48.9, 35.6, 32.5, 28.5, 27.5, 25.2 ppm. IR ν: 2939, 1745, 1699, 1662, 1619, 1599, 1427, 1352, 1333, 1255, 1189, 986, 708, 686, 659, 559 cm−1. Anal. Calcd. for C17H18Br2N2O3 (458.14): C, 44.57; H, 3.96; N, 6.11. Found: C, 44.46; H, 3.86; N, 6.04%.
4.1.2.12. 2-[4-(3-Benzoyl-5-bromo-2,4-dioxo-3,4-dihydropyrimidine-1(2H)-yl)butyl]-6,7-methylenedioxy-2H-pyrrolo[3,4-b]quinoline-1,3-dione (20a)
To a suspension of 10 (242 mg, 1.00 mmol, 1.0 eq.) and K2CO3 (166 mg, 1.20 mmol, 1.2 eq.) in anhydrous DMF (10 mL) was added 19a (645 mg, 1.50 mmol, 1.5 eq.). The mixture was stirred at r.t. for 0.5 h. After elimination of the solvent by evaporation under vacuum, the residue was purified by flash column chromatography (DCM/EtOAc, 95:5 to 80:20) to afford 20a (496 mg, 84% yield). mp: 257 °C. 1H NMR (400 MHz, DMSO-d6) δ: 9.07 (s, 1H), 8.48 (s, 1H), 8.01 (d, J = 9.0 Hz, 2H), 7.91 (s, 1H), 7.79 (t, J = 9 Hz, 1H), 7.60 (m, 3H), 6.37 (s, 2H), 3.78 (t, J = 6.0 Hz, 2H), 3.63 (t, J = 6.0 Hz, 2H), 1.68 (m, 4H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 169.1, 168.8, 168.1, 158.9,153.8, 151.3, 151.2, 149.5, 146.8, 141.1, 136.2, 133.8, 131.1, 131.0, 130.0, 122.2, 118.9, 106.3, 103.8, 98.6, 94.5, 48.6, 37.6, 26.1, 25.3 ppm. IR ν: 2920, 1742, 1704, 1659, 1618, 1455, 1426, 1399, 1330, 1277, 1259, 1230, 1177, 1085, 1032, 940, 912, 867, 802, 758, 712, 658, 572 cm−1. Anal. Calcd. for C27H19BrN4O7 (591.37): C, 54.84; H, 3.24; N, 9.47. Found: C, 55.01; H, 3.52; N, 9.08%.
4.1.2.13. 2-[6-(3-Benzoyl-5-bromo-2,4-dioxo-3,4-dihydropyrimidine-1(2H)-yl)hexyl]-6,7-methylenedioxy-2H-pyrrolo[3,4-b]quinoline-1,3-dione (20b)
Starting from 19b (596 mg, 1.30 mmol, 1.3 eq.), 20b was prepared in the same procedure, work-up and purification as described for 20a. Yellow solid (520 mg, 84% yield). mp: 196 °C. 1H NMR (400 MHz, DMSO-d6) δ: 9.05 (s, 1H), 8.48 (s, 1H), 8.01 (d, J = 6.0 Hz, 2H), 7.90 (s, 1H), 7.78 (t, J = 6 Hz, 1H), 7.59 (m, 3H), 6.36 (s, 2H), 3.74 (t, J = 6.0 Hz, 2H), 3.59 (t, J = 6.0 Hz, 2H), 1.62 (m, 4H), 1.32 (m, 4H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 169.1, 168.8, 168.1, 158.7,153.8, 151.3, 151.2, 149.4, 146.7, 141.1, 136.1, 133.8, 131.2, 131.0, 130.0, 122.2, 118.9, 106.3, 103.8, 98.7, 94.4, 48.9, 37.8, 28.6, 28.2, 26.2, 25.7 ppm. IR ν: 2933, 1746, 1704, 1663, 1621, 1598, 1498, 1458, 1425, 1396, 1334, 1318, 1260, 1230, 1176, 1033, 973, 943, 865, 799, 769, 757, 657, 572 cm−1. Anal. Calcd. for C29H23BrN4O7. 0.25 H2O (623.92): C, 55.83; H, 3.80; N, 8.98. Found: C, 55.71; H, 3.84; N, 8.79%.
4.1.2.14. 2-[4-(5-Bromo-2,4-dioxo-3,4-dihydropyrimidine-1(2H)-yl)butyl]-6,7-methylenedioxy-2H-pyrrolo[3,4-b]quinoline-1,3-dione (16a)
To a suspension of 19a (387 mg, 0.65 mmol, 1.0 eq.) in EtOH (6 mL) was added an aqueous 5 N HCl solution (4 mL). The mixture was refluxed for 2 days. After cooling at r.t., the solid was collected by filtration, washed with H2O. Recrystallization from DMF afforded 16a as a yellow solid (168 mg, 53% yield). mp: 276 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.72 (s, 1H), 9.06 (s, 1H), 8.21 (s, 1H), 7.89 (s, 1H), 7.58 (s, 1H), 6.37 (s, 2H), 3.69 (t, J = 6.0 Hz, 2H), 3.63 (t, J = 6.0 Hz, 2H), 1.63 (m, 4H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 168.8, 168.1, 160.1, 153.8, 151.3, 151.2, 150.8, 145.8, 141.1, 133.9, 122.3, 118.9, 106.3, 103.8, 98.7, 95.0, 47.8, 37.6, 26.2, 25.2 ppm. IR ν: 3449, 3042, 2953, 2792, 1769, 1709, 1623, 1497, 1465, 1397, 1349, 1266, 1232, 1181, 1108, 1034, 938, 913, 868, 803, 749, 619, 576, 566 cm−1. Anal. Calcd. for C20H15BrN4O6 · 0.5 H2O (496.27): C, 48.40; H, 3.25; N, 11.29. Found: C, 48.17; H, 3.41; N, 11.64%.
4.1.2.15. 2-[6-(5-Bromo-2,4-dioxo-3,4-dihydropyrimidine-1(2H)-yl)hexyl]-6,7-methylenedioxy-2H-pyrrolo[3,4-b]quinoline-1,3-dione (16b)
Starting from 20b (520 mg, 0.84 mmol, 1.0 eq.), 16b was prepared in the same procedure, work-up and purification as described for 16a. Yellow solid (168 mg, 39% yield). mp: 282–283 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.72 (s, 1H), 9.04 (s, 1H), 8.22 (s, 1H), 7.89 (s, 1H), 7.57 (s, 1H), 6.36 (s, 2H), 3.64 (t, J = 6.0 Hz, 2H), 3.57 (t, J = 6.0 Hz, 2H), 1.59 (m, 4H), 1.30 (m, 4H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 168.8, 168.1, 160.1, 153.8, 151.3, 151.2, 150.7, 145.7, 141.1, 133.8, 122.2, 118.9, 106.3, 103.8, 98.7, 94.6, 48.2, 37.9, 28.7, 28.2, 26.3, 25.8 ppm. IR ν: 3043, 2989, 2934, 2788, 1770, 1714, 1691, 1622, 1495, 1468, 1438, 1392, 1349, 1335, 1269, 1231, 1146, 1181, 1037, 941, 868, 743, 617 cm−1. Anal. Calcd. for C22H19BrN4O6 (515.31): C, 51.28; H, 3.72; N, 10.87. Found: C, 51.04; H, 3.66; N, 10.87%.
4.1.2.16. 2-[(2,6-Dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)methyl]-6,7-methylenedioxy-1H-pyrrolo[3,4-b]quinoline-1,3(2H)-dione (22)
To a suspension 21 (482 mg, 3.00 mmol, 1.5 eq.) in anhydrous MeCN (10 mL) was added BSA (1.5 mL, 6.00 mmol, 3.0 eq.) at r.t. When the reaction mixture became clear (15 min), a suspension of imide 10 (484 mg, 2.00 mmol, 1.0 eq.) and K2CO3 (415 mg, 3.00 mmol, 1.5 eq.) in anhydrous MeCN (10 mL) was added and the mixture was refluxed for 5 days. Then, another amount of 21 (241 mg, 1.50 mmol, 0.75 eq.) and BSA (0.85 mL, 3.48 mmol, 1.74 eq.) in MeCN (5 mL) were added and the reflux was maintained for 2 more days. After evaporation of the solvent under vacuum, the residue was stirred with H2O (10 mL) and an aqueous 1 N HCl solution was added until a pH = 3 was reached. The resulting solid was isolated by filtration, washed with H2O and EtOH. Recrystallization from DMF afforded 22 as a yellow solid (579 mg, 79% yield). mp: 407–409 °C (decomposition). 1H NMR (400 MHz, DMSO-d6) δ: 11.07 (s, 1H), 11.03 (s, 1H), 9.10 (s, 1H), 7.89 (s, 1H), 7.60 (s, 1H), 6.37 (s, 2H), 5.55 (s, 1H), 4.51 (s, 2H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 167.8, 167.0, 163.8, 153.4, 151.3, 150.9, 150.8, 150.7, 140.8, 133.8, 122.1, 118.5, 105.9, 103.4, 98.2, 97.1, 37.4 ppm. IR ν: 3185, 3106, 3056, 3001, 2817, 1772, 1702, 1650, 1494, 1455, 1414, 1392, 1341, 1322, 1307, 1264, 1230, 1177, 1104, 1030, 1014, 930, 866, 847, 828, 794, 752 cm−1. Anal. Calcd. for C17H10N4O6 · 0.5 H2O (375.29): C, 54.41; H, 2.95; N, 14.93. Found: C, 54.10; H, 3.05; N, 14.94%.
4.1.2.17. 2-[(5-Chloro-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)methyl]-6,7-methylenedioxy-1H-pyrrolo[3,4-b]quinoline-1,3(2H)-dione (23a)
To a suspension of 22 (183 mg, 0.50 mmol) in anhydrous DMF (5 mL), NCS (93.5 mg, 0.70 mmol, 1.4 eq.) was added and the mixture was stirred overnight at r.t. Then, the reaction was cooled at 0 °C and H2O (5 mL) was added. The resulting precipitate was isolated by filtration, washed successively with H2O and EtOH. Recrystallization from DMF afforded 23b as a yellow solid (88 mg, 44% yield). mp: 352–354 °C (decomposition). 1H NMR (400 MHz, DMSO-d6) δ: 11.65 (s, 1H), 11.33 (s, 1H), 9.12 (s, 1H), 7.91 (s, 1H), 7.61 (s, 1H), 6.38 (s, 2H), 4.77 (s, 2H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 167.7, 166.9, 159.4, 153.5, 151.1, 150.7, 149.9, 146.7, 140.8, 133.8, 122.1, 118.4, 105.9, 104.3, 103.4, 98.1, 36.9 ppm. IR ν: 3256, 3180, 3143, 3051, 3005, 2808, 1773, 1702, 1668, 1618, 1501, 1462, 1420, 1386, 1344, 1316, 1259, 1224, 1105, 1035, 925, 892, 876, 796, 762, 750 cm−1. Anal. Calcd. for C17H9ClN4O6 · 0.5 H2O (409.74): C, 49.83; H, 2.46; N, 13.67. Found: C, 49.87; H, 2.22; N, 13.48%.
4.1.2.18. 2-[(5-Bromo-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)methyl]-6,7-methylenedioxy-1H-pyrrolo[3,4-b]quinoline-1,3(2H)-dione (23b)
To a suspension of 22 (293 mg, 0.80 mmol) in anhydrous DMF (8 mL), NBS (214 mg, 1.20 mmol, 1.5 eq.) was added and the mixture was stirred overnight at r.t. Work up and purification were the same as used for 23a to give 23b as a yellow solid (210 mg, 59% yield). mp: 370–372 °C (decomposition). 1H NMR (400 MHz, DMSO-d6) δ: 11.64 (s, 1H), 11.37 (s, 1H), 9.14 (s, 1H), 7.92 (s, 1H), 7.63 (s, 1H), 6.39 (s, 2H), 4.74 (s, 2H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 168.3, 167.0, 160.2, 154.0, 151.6, 151.2, 150.7, 148.8, 141.3, 134.4, 122.7, 118.9, 106.4, 103.9, 98.6, 94.8, 39.7 ppm. IR ν: 3002, 2957, 2912, 2820, 1786, 1707, 1650, 1619, 1492, 1457, 1427, 1385, 1340, 1314, 1263, 1233, 1032, 930, 873, 861, 802, 757, 743, 733, 684, 662 cm−1. Anal. Calcd. for C17H9BrN4O6 · 0.75 H2O (458.69): C, 44.51; H, 2.31; N, 12.21. Found: C, 44.64; H, 2.22; N, 12.00%.
4.1.2.19. 2-[(5-Iodo-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)methyl]-6,7-methylenedioxy-1H-pyrrolo[3,4-b]quinoline-1,3(2H)-dione (23c)
To a suspension of 22 (147 mg, 0.40 mmol) in anhydrous DMF (4 mL), NIS (135 mg, 0.60 mmol, 1.5 eq.) was added and the mixture was stirred overnight at r.t. Work up was the same as used for 23a to give pure crude 23c as a white solid (180 mg, 91% yield). mp: 365–367 °C (decomposition). 1H NMR (400 MHz, DMSO-d6) δ: 11.49 (s, 1H), 11.27 (s, 1H), 9.12 (s, 1H), 7.92 (s, 1H), 7.62 (s, 1H), 6.38 (s, 2H), 4.70 (s, 2H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 167.9, 167.1, 161.2, 153.5, 151.1, 150.8, 150.7, 150.6, 140.8, 134.0, 122.3, 118.4, 105.9, 103.4, 98.1, 70.4, 43.5 ppm. IR ν: 3040, 2948, 2912, 2814, 1785, 1725, 1703, 1649, 1614, 1492, 1458, 1437, 1387, 1338, 1314, 1264, 1233, 1032, 931, 873, 862, 803, 760, 743, 733 cm−1. Anal. Calcd. for C17H9IN4O6 (492.18): C, 41.49; H, 1.84; N, 11.38. Found: C, 41.10; H, 1.51; N, 11.08%.
4.1.2.20. 6,7-Methylenedioxy-2-benzylpyrrolo[3,4-b]quinoline-1,3-dione (24)
A suspension of imide 10 (121 mg, 0.50 mmol) and K2CO3 (207 mg, 1.50 mmol, 3.0 eq.) in anhydrous DMF (7 mL) was stirred at r.t. After solubilisation of 10, benzyl bromide (0.293 mL, 2.50 mmol, 5.0 eq.) was added. After 5 min, K2CO3 was removed by filtration, and H2O (40 mL) was added. The resulting precipitate was isolated by filtration and solubilised in DCM. The organic layer was dried over anhydrous Na2SO4, filtrated, and concentrated under reduced pressure. The residual solid was washed 4 times with Et2O to afford pure 24 as a yellowish solid (108 mg, 65% yield). mp: 191–193 °C. 1H NMR (300 MHz, DMSO-d6) δ: 9.05 (s, 1H), 7.85 (s, 1H,), 7.54 (s, 1H), 7.42–7.23 (m, 5H), 6.35 (s, 2H), 4.79 (s, 2H) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 168.1, 167.4, 153.4, 150.9, 150.8, 140.8, 136.5, 133.3, 128.6, 127.5, 121.7, 118.5, 105.9, 103.4, 98.2, 41.0 ppm. IR ν: 3107, 3048, 2914, 1765, 1704, 1617, 1495, 1454, 1427, 1390, 1343, 1316, 1258, 1228, 1175, 1033, 938, 886, 860, 796, 757, 747, 703, 696 cm−1. Anal. Calcd. for C19H12N2O4 · 0.25 H2O (336.81): C, 67.75; H, 3.74; N, 8.32. Found: C, 67.46; H, 3.71; N, 8.06%.
4.1.3. 1 H-Pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6,8(3H,7H)-tetraones (series D)
4.1.3.1. General method for preparation of compounds 28
To a suspension of 27Citation28 (70.0 mg, 0.30 mmol, 1.0 eq.) in DMF (444 µL), the requisite amine (0.60 mmol, 2.0 eq.) was added. The reaction mixture was refluxed for a time T1. To the resulting solution, PTSA (115 mg, 0.60 mmol, 2.0 eq.) was added and reflux was maintained during a time T2. The resulting precipitate was filtered off and washed to give a crude pure compound.
4.1.3.2. 1H-7-(3,4,5-Trimethoxybenzyl)-pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6,8 (3H,7H)-tetraone (28c)
T1 = 1 h, T2 = 17 h. Another amount of PTSA (0.5 eq.) was added and the reaction was heated at 120 °C for additional 24 h. The resulting precipitate was washed with DMF and H2O to afford pure 28c as a yellow solid (81 mg, 66% yield). mp: 316–318 °C (decomposition). 1H NMR (400 MHz, DMSO-d6) δ: 12.50 (s, 1H), 11.88 (s, 1H), 8.51 (s, 1H), 6.64 (s, 2H), 4.73 (s, 2H), 3.73 (s, 6H), 3.61 (s, 3H) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 165.3, 165.2, 161.5, 157.0, 156.1, 152.9, 150.0, 136.8, 131.9, 131.5, 121.3, 112.7, 104.9, 60.0, 55.9, 41.5 ppm. IR ν: 3439, 3182, 3075, 2838, 1743, 1707, 1676, 1614, 1589, 1550, 1511, 1458, 1424, 1394, 1382, 1346, 1331, 1281, 1242, 1191, 1127, 1105, 1038, 995, 966, 942, 835, 812, 795, 777, 752, 733, 697, 667 cm−1. Anal. Calcd. for C19H16N4O7 · H2O (430.37): C, 53.02; H, 4.22; N, 13.02. Found: C, 53.09; H, 4.25; N, 13.23%.
4.1.3.3. 7-(Pyridin-3-ylmethyl)-1H-pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6,8(3H,7H)-tetraone (28f)
T1 = 24 h, T2 = 24 h, washing solvent: H2O. 28f: white solid (72 mg, 74% yield). mp: >360 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.50 (s, 1H), 11.87 (s, 1H), 8.60 (s, 1H), 8.55–8.44 (m, 2H), 7.77 (d, J = 7.9 Hz, 1H), 7.36 (dd, J = 7.9, 4.8 Hz, 1H), 4.85 (s, 2H) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 165.2, 165.1, 161.5, 157.0, 156.1, 150.0, 149.0, 148.7, 135.6, 131.9, 131.5, 123.6, 121.3, 112.7, 38.9 ppm. IR ν: 3232, 3101, 3030, 2905, 2795, 1777, 1715, 1606, 1584, 1478, 1462, 1424, 1381, 1356, 1343, 1306, 1287, 1248, 1187, 1112, 1098, 1066, 1043, 985, 964, 932, 853, 837, 818, 795, 748, 723, 678 cm−1. Anal. Calcd. for C15H9N5O4 · 0.5 H2O (332.27): C, 54.22; H, 3.03; N, 21.08. Found: C, 54.18; H, 3.00; N, 20.99%.
4.1.3.4. 7-Phenylethyl-1H-pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6,8(3H,7H)-tetraone (28e)
T1 = 5 h, T2 = 17 h, washing solvent: H2O. 28e: white solid (76 mg, 75% yield). mp: 359–361 °C. 1H NMR (300 MHz, DMSO-d6) δ: 12.47 (s, 1H), 11.85 (s, 1H), 8.46 (s, 1H), 7.15–7.32 (m, 5H), 3.84 (t, J = 6.9 Hz, 2H), 2.92 (t, J = 6.9 Hz, 2H) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 165.1, 165.0, 161.5, 157.0, 155.8, 150.0, 138.2, 131.3, 128.7, 128.5, 126.5, 121.0, 112.8, 39.0, 33.6 ppm. IR ν: 3604, 3478, 3164, 3028, 3055, 2817, 1778, 1740, 1697, 1674, 1604, 1540, 1497, 1466, 1455, 1437, 1398, 1383, 1338, 1287, 1241, 1188, 1150, 1116, 1107, 1040, 1028, 987, 905, 866, 807 cm−1. Anal. Calcd. for C17H12N4O4 · 0.75 H2O (349.81): C, 58.37; H, 3.89; N, 16.02. Found: C, 58.48; H, 3.89; N, 16.13%.
4.1.3.5. 7-(3-(Dimethylamino)propyl)-1H-pyrrolo[3′,4′:5,6]pyrido [2,3-d]pyrimidine-2,4,6,8 (3H,7H)-tetraone, hydrochloride (28h)
T1 = 2 h, T2 = overnight, washing solvent: EtOH. The solid was then stirred in EtOH (10 mL) with bubbling HCl gas during few seconds and the resulting precipitate was isolated by filtration, washed with EtOH and Et2O to afford pure 28h as a white solid (67 mg, 63% yield). mp: 309–311 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.47 (s, 1H), 11.85 (s, 1H), 10.29 (s, 1H), 8.50 (s, 1H), 3.69 (t, J = 6.5 Hz, 2H), 3.15–3.05 (m, 2H), 2.71 (s, 6H), 2.08–1.96 (m, 2H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 165.3, 165.2, 161.4, 156.9, 156.1, 149.9, 131.2, 121.2, 112.5, 54.1, 42.0, 35.1, 23.0 ppm. IR ν: 3184, 3134, 3034, 2910, 2694, 1785, 1713, 1680, 1607, 1527, 1435, 1396, 1366, 1355, 1285, 1185, 1114, 1000, 886, 778, 768, 742, 692 cm−1. Anal. Calcd. for C14H16ClN5O4 · 0.75 H2O (367.27): C, 45.78; H, 4.80; N, 19.07. Found: C, 46.18; H, 4.49; N, 18.83%.
4.1.3.6. N6,N7-Bis((2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl) methyl)-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6,7-dicarboxamide (30)
To a suspension of 27 (232 mg, 1.00 mmol) in DMF (10 mL), Et3N (0.697 mL, 5.00 mmol, 5 eq.) and 29Citation49,Citation50 (533 mg, 3.00 mmol, 3 eq.) were added and the mixture was heated at 90 °C overnight. Once cooled at r.t., the reaction mixture was poured on Et2O (10 mL) and maintained at 4 °C for 2 h. To a solution of the resulting solid in H2O (12 mL), a 1 N HCl solution was added until a pH < 3 was reached. The solid was filtrated, washed successively with H2O and EtOH to afford pure 30 as a white solid (410 mg, 82% yield). mp: 259–261 °C (decomposition). 1H NMR (400 MHz, DMSO-d6) δ: 12.10 (s, 1H), 11.70 (s, 1H), 10.97 (s, 2H), 10.90 (s, 1H), 10.82 (s, 1H), 9.10 (t, J = 5.8 Hz, 1H), 8.96 (t, J = 6.1 Hz, 1H), 8.58 (s, 1H), 5.53 (s, 1H), 5.48 (s, 1H), 4.18–4.07 (m, 4H) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 166.0, 165.3, 164.0, 161.6, 156.9, 153.4, 153.3, 152.4, 151.4, 151.3, 150.3, 136.8, 124.6, 109.8, 96.8, 96.6, 39.3, 38.7 ppm. IR ν: 3563, 3489, 3247, 3118, 2988, 2819, 1689, 1657, 1609, 1583, 1527, 1502, 1419, 1357, 1321, 1284, 1233, 1020, 841, 819, 766, 743, 703 cm−1. Anal. Calcd. for C19H15N9O8 · 3.5 H2O (560.43): C, 40.72; H, 3.96; N, 22.49. Found: C, 40.97; H, 3.73; N, 22.02%.
4.2. Biologicals
A spectrophotometric assay method was adopted to evaluate in vitro E. coli TP inhibiting activity of all the synthesised compounds. The conversion of thymidine to thymine was recorded at 290 nm. Thymidine was dissolved in buffer (3 mM). DMSO stock solutions of the tested compounds were diluted in buffer, the incubation medium contained 2.5% of DMSO. All experiments were conducted in triplicate.
4.2.1. In vitro thymidine phosphorylase essay
Initially, all synthesised compounds were tested against E. coli TP at 50 µM or at the maximum concentration allowed by their solubility.
The method reported by Khan et al.Citation51 has been adapted to a screening in 96-well plates (200 µL well, UV-Star® 96-well microplate, Greiner). Each well contained 160 µL of 50 mM KH2PO4 buffer (pH 7.4), 0.006 U of E. coli TP (T2807-1KU, Sigma Aldrich), and 20 µL of the tested compound solution. The reaction was initiated by the addition of 20 µL thymidine (i.e. a 300 µM final concentration corresponding to the KmCitation52). The decrease in absorbance due to conversion of thymidine to thymine was followed during 30 min (PowerWave HT Microplate Reader, Biotek) at 25 °C, with one measure every 2 min and a 30 s shaking before each measure. Data were processed using Biotek KC4 software and the results were expressed in percentage of inhibition at 50 µM or at the maximum concentration allowed by the solubility of the tested compound (). For the most active and soluble compounds 2d, 2l, 2p, 28a, and 30 (inhibition > 30% and solubility > 50 µM), IC50 values have been determined.
4.2.2. Phosphate competition study
For this study, the reaction mixture was composed of 160 µL of 10 mM Tris buffer (pH 7.4), 150 mM NaCl, 2 mM EDTA, 0.006 U of E. coli TP, different concentrations of KH2PO4 (2, 5, 10 and 30 mM). Compounds were tested at a concentration close to their IC50. The reaction was initiated by the addition of 20 µL of thymidine (3 mM and 10 mM solutions).
4.2.3. Enzyme inhibition kinetic study
The TP inhibiting activity at differents concentrations of compound 2d (0, 15, 20, 30 µM) and 2p (0, 4, 6, 8 µM) was evaluated in the presence of different concentrations of thymidine (200, 250, 300, 500, 750, 1000 mM) in KH2PO4 buffer (pH 7.4).
4.2.4. In vitro anti-proliferative assay
Cell viability was determined on HUVEC and A431 cell lines using 3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) assayCitation53 after treatment with compounds 2d and 2p. A total of 5 × 103 cells/well (A431 cell line) or 104 cells/well (HUVEC cell line) were plated in a 96-well plate and treated, in duplicates, with compounds 2d and 2p at five concentrations (0.1 µM, 1 μM, 10 μM, 50 μM, 100 μM) for 72 h. Absorbance was read at 570 nm using Labsystem Multiskan MS microplate reader. The results were expressed as percentages of growth inhibition compared to positive control cell growth (100%).
4.3. Molecular docking study
Molecular modelling studies were carried out with the GOLD software (version 5.1). The ligands were constructed using the standard fragments of the Sybyl library 6.9.1. Their geometry was optimised using the Tripos force field by assigning partial loads calculated by the Gasteiger–Hückel method to a gradient of 0.0001 Kcal/mol/Å.
The conformations obtained for each compound were classified by a consensus scoring function and then the conformation groups were observed visually to evaluate their consistency and their complementarity with the active site. Only the most representative conformations of each group were selected.
In order to check the validity of the results, the TPI reference inhibitor was relocated to the hTP binding site (PDB code: 1UOU) following the same protocol as for the inhibitors. The best solution has an RMS on heavy atoms of 0.654 Å with respect to the 1UOU crystallographic structure. Results were visualised by Chimaera and Maestro.
Supplementary data includes docking superposition of compounds 2d, 2p, and 2l in the hTP active site; two docked conformations of compound 2c in the hTP active site; docking superposition of compounds 2i, and 23b in the hTP active site; NMR spectra of the final compounds.
Supplemental Material
Download PDF (2.3 MB)Acknowledgement
The authors gratefully thank Prof. Marc Lecouvey’s team for the anti-proliferative assay.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Battaglin F, Puccini A, Intini R, et al. The role of tumor angiogenesis as a therapeutic target in colorectal cancer. Expert Rev Anticancer Ther 2018;18:251–66.
- Folkman J, Merler E, Abernathy C, Williams G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med 1971;133:275–88.
- Weidner N, Semple JP, Welch WR, Folkman J. Tumor Angiogenesis and metastasis-correlation in invasive breast carcinoma. N Engl J Med 1991;324:1–8.
- Akiyama SI, Furukawa T, Sumizawa T, et al. The role of thymidine phosphorylase, an angiogenic enzyme, in tumor progression. Cancer Sci 2005;95:11851–7.
- Jayson GC, Kerbel R, Ellis M, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. The Lancet 2016;388:518–29.
- Bronckaers A, Gago F, Balzarini J, Liekens S. The dual role of thymidine phosphorylase in cancer development and chemotherapy. Med Chem Rev 2009;29:903–53.
- Elamin YY, Rafee S, Osman N, et al. Thymidine phosphorylase in cancer; enemy or friend? Cancer Microenviron 2016;9:33–43.
- Bera H, Chigurupati S. Recent discovery of non-nucleobase thymidine phosphorylase inhibitors targeting cancer. Eur J Med Chem 2016;124:992–1003.
- Brown N, Jones A, Fujiyama C, et al. Cooperative stimulation of vascular endothelial growth factor expression by hypoxia and reactive oxygen species: the effect of targeting vascular endothelial growth factor and oxidative stress in an orthotopic xenograft model of bladder carcinoma. Cancer Res 2000;60:6298–302.
- Bijnsdorp IV, Capriotti F, Kruyt FAE, et al. Thymidine phosphorylase in cancer cells stimulates human endothelial cell migration and invasion by the secretion of angiogenic factors. Br J Cancer 2011;104:1185–92.
- Walter MR, Cook WJ, Cole LB, et al. Three-dimensional structure of thymidine phosphorylase from Escherichia coli at 2.8 A resolution. J Biol Chem 1990;265:14016–22.
- Norman RA, Barry ST, Bate M, et al. Crystal structure of human thymidine phosphorylase in complex with a small molecule inhibitor. Structure 2004;12:75–84.
- El Omari K, Bronckaers A, Liekens S, et al. Structural basis for non-competitive product inhibition in human thymidine phosphorylase: implications for drug design. Biochem J 2006;399:199–204.
- Mitsiki E, Papageorgiou AC, Iyer S, et al. Structures of native human thymidine phosphorylase and in complex with 5-iodouracil. Biochem Biophys Res Commun 2009;386:666–70.
- Timofeev VI, Abramchik YA, Fateev IV, et al. Three-dimensional structure of thymidine phosphorylase from E. coli in complex with 3'-azido-2'-fluoro-2',3'-dideoxyuridine. Crystallogr Rep 2013;58:842–53.
- Timofeev V, Abramchik Y, Zhukhlistova N, et al. 3′‐Azidothymidine in the active site of Escherichia coli thymidine phosphorylase: the peculiarity of the binding on the basis of X‐ray study. Acta Crystallogr Sect D 2014;70:1155–65.
- Pérez-Pérez MJ, Priego EM, Hernandez AN, et al. Thymidine phosphorylase inhibitors: recent developments and potential therapeutic applications. Mini Rev Med Chem 2005;5:1113–23.
- Nencka R. Anti-angiogenesis drug discovery and development. Amsterdam (The Netherlands): Wiley; 2011:116–47.
- Sajid MA, Khan ZA, Shahzad SA, et al. Recent advances in thymidine phosphorylase inhibitors: syntheses and prospective medicinal applications. Turk J Chem 2017;41:1–28.
- Balzarini J, Gamboa AE, Esnouf R, et al. 7-Deazaxanthine, a novel prototype inhibitor of thymidine phosphorylase. FEBS Lett 1998;438:91–5.
- Fukushima M, Suzuki N, Emura T, et al. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2'-deoxyribonucleosides. Biochem Pharmacol 2000;59:1227–36.
- Matsuhita S, Nitanda T, Furukawa T, et al. The effect of a thymidine phosphorylase inhibitor on angiogenesis and apoptosis in tumors. Cancer Res 1999;50:1911–6.
- Takao S, Akiyama SI, Nakajo A, et al. Suppression of metastasis by thymidine phosphorylase inhibitor. Cancer Res 2000;60:5345–8.
- Yano S, Kazuno H, Sato T, et al. Synthesis and evaluation of 6-methylene-bridged uracil derivatives. Part 2: optimization of inhibitors of human thymidine phosphorylase and their selectivity with uridine phosphorylase. Bioorg Med Chem 2004;12:3443–50.
- Raedler LA. Lonsurf (trifluridine plus tipiracil): a new oral treatment approved for patients with metastatic colorectal cancer. Am Health Drug Benefits 2016; 9:97–100.
- Tratrat C, Giorgi-Renault S, Husson HP. A multicomponent reaction for the one-pot synthesis of 4-aza-2,3-didehydropodophyllotoxin and derivatives. Org Lett 2002;4:3187–9.
- Aknin K, Desbène-Finck S, Helissey P, Giorgi-Renault S. A new synthetic approach to functionalize pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones via a three-component one-pot reaction. Mol Divers 2010;14:123–30.
- Tourteau A, Merlet E, Bontemps A, et al. Easy access to 1H‐pyrrolo[3′,4′:5,6]pyrido[2,3‐d]pyrimidine‐ 2,4,6,8(3H,7H)‐tetraone and selectively N7‐substituted analogues through key synthons. Eur J Org Chem 2015;32:7028–35.
- Conrad M, Reinbach H. Condensationen von barbitursäure und aldehyden. Ber Dtsch Chem Ges 1901;34:1339–44.
- Tröger J, Cohaus C. Über mittels 6‐Amino‐3‐methoxybenzaldehyd ausgeführte Chinolinsynthesen und über ein aus diesen Aldehyden entstehendes Kondensationsprodukt. J Prakt Chem 1927;117:97–116.
- King FE, King TJ. New potential chemotherapeutic agents. Part VI. Derivatives of 1: 3-diaza-acridine. J Chem Soc 1947;726–34.
- Tominaga Y, Okuda H, Kohra S, Mazume H. Synthesis of pyrimidine derivatives using N‐bis(methylthio)methylenecyanamide. J Heterocycl Chem 1991;28:1245–55.
- Kokel B. The reaction of N,N‐dimethyldichloromethyleniminium chloride (phosgeniminium chloride) with 6‐N‐arylaminouracils. A new and convenient “one pot” synthesis of l,3‐dimethyl‐5‐ dimethylaminopyrimido[4,5‐b]quinoline‐(1H,3H)‐2,4‐ diones, 1,3‐dimethyl‐5‐chloropyrimido[4,5‐b]quinoline‐(1H,3H)‐2,4‐diones and 3‐ methyl‐10‐alkyl‐5‐chloropyrimido[4,5‐b]quinoline‐(3H,10H)‐2,4‐diones (3‐methyl‐ 10‐alkyl‐5‐chloro‐5‐deazaflavins). J Heterocycl Chem 1994;31:845–55.
- Fang WP, Cheng YT, Cheng YR, Chern YJ. Synthesis of substituted uracils by the reactions of halouracils with selenium, sulfur, oxygen and nitrogen nucleophiles under focused microwave irradiation. Tetrahedron 2005;61:3107–13.
- Kurasawa Y, Takada A. A convenient synthesis of pyridazino [4,5-b] quinolines and pyrrolo [3,4-b] quinolines. Chem Pharm Bull 1980;28:3457–65.
- Gal’pern MG, Kudrevich SV, Novozhilova IG. Synthesis and spectroscopic properties of soluble aza analogs of phthalocyanine and naphthalocyanine. Chem Heterocycl Compd 1993;29:49–54.
- Brown FK, Brown PJ, Bickett DM, et al. Matrix metalloproteinase inhibitors containing a (carboxyalkyl)amino zinc ligand: modification of the P1 and P2' residues. J Med Chem 1994;37:674–88.
- Hendrickson JB, Rees R, Templeton JF. A new general heterocycle synthesis; use of acetylenedicarboxylic esters. J Am Chem Soc 1964;86:107–11.
- Peel MR, Sternbach DD. The synthesis and evaluation of flexible analogues of the topoisomerase I inhibitor, camptothecin. Bioorg Med Chem Lett 1994;4:2753–8.
- Itahara T. NMR and UV Study of 1,1′-(α,ω-Alkanediyl)bis[thymine] and 1,1′-(α,ω- Alkanediyl)bis[uracil]. Bull Chem Soc Jpn 1997;70:2239–47.
- Browne DT, Eisinger J, Leonard NJ. Synthetic spectroscopic models related to coenzymes and base pairs. II. Evidence for intramolecular base-base interactions in dinucleotide analogs. J Am Chem Soc 1968;90:7302–23.
- Leonard NJ, McCredie RS, Logue MW, Cundall RL. Synthetic spectroscopic models related to coenzymes and base pairs. XI. Solid state ultraviolet irradiation of 1,1'-trimethylenebisthymine and photosensitized irradiation of 1,1'-polymethylenebisthymines. J Am Chem Soc 1973;95:2320–4.
- Tjoeng FS, Kraas E, Breitmaier E, Jung G. Einfache darstellung von 2‐amino‐ω‐ (uracil‐l‐yl und thymin‐1‐yl)‐n‐alkansäuren. Chem Ber 1976;109:2615–21.
- Decout JL, Lhomme J. Synthetic models related to DNA-intercalating molecules a study of the ring-ring stacking interactions between 8-alkoxypsoralen and thymine. Tetrahedron Lett 1981;22:1247–50.
- Nowick JS, Chen JS, Noronha G. Molecular recognition in micelles: the roles of hydrogen bonding and hydrophobicity in adenine-thymine base-pairing in SDS micelles. J Am Chem Soc 1993;115:7636–44.
- Nawrot B, Michalak O, Olejniczak S, et al. Alkylation of thymine with 1,2-dibromoethane. Tetrahedron 2001;57:3979–85.
- Allan AL, Gladstone PL, Price ML, et al. Synthesis and evaluation of multisubstrate bicyclic pyrimidine nucleoside inhibitors of human thymidine phosphorylase. J Med Chem 2006;49:7807–15.
- Russ P, Schelling P, Scapozza L, et al. Synthesis and biological evaluation of 5-substituted derivatives of the potent antiherpes agent (north)-methanocarbathymine. J Med Chem 2003;46:5045–54.
- Wade JJ. Synthesis of imidazo[1,5‐c]pyrimidine derivatives. J Heterocyclic Chem 1986;23:981–7.
- Yano S, Kazuno H, Suzuki N, et al. Synthesis and evaluation of 6-methylene-bridged uracil derivatives. Part 1: discovery of novel orally active inhibitors of human thymidine phosphorylase. Bioorg Med Chem 2004;12:3431–41.
- Khan KM, Ambreen N, Hussain S, et al. Schiff bases of 3-formylchromone as thymidine phosphorylase inhibitors. Bioorg Med Chem 2009;17:2983–8.
- Panova NG, Alexeev CS, Kuzmichov AS, et al. Substrate specificity of Escherichia coli thymidine phosphorylase. Biochemistry 2007;72:21–8.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.