
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Flavonoids are widely distributed in plants and constitute the most common polyphenolic phytoconstituents in the human diet. In this study, the in vitro inhibitory activity of 44 different flavonoids (1–44) against mushroom tyrosinase was studied, and an in silico study and type of inhibition for the most active compounds were evaluated too. Tyrosinase inhibitors block melanogenesis and take part in melanin production or distribution leading to pigmentation diseases. The in vitro study showed that quercetin was a competitive inhibitor (IC50=44.38 ± 0.13 µM) and achieved higher antityrosinase activity than the control inhibitor kojic acid. The in silico results highlight the importance of the flavonoid core with a hydroxyl at C7 as a strong contributor of interference with tyrosinase activity. According to the developed statistical model, the activity of molecules depends on hydroxylation at C3 and methylation at C8, C7, and C3 in the benzo-γ-pyrane ring of the flavonoids.
1. Introduction
Flavonoids are secondary plant metabolites that can be chemically divided into groups based on their substitutions. Flavonoid moieties can be modified by glycosylation, hydrogenation, hydroxylation, acetylation and methylation, as well as by malonylation and sulfatation. The chemical and biological potentials of flavonoids and their derivatives are connected with the position of diverse substitutions on the molecule and the saturation of double bonds in the structureCitation1. Thus, structure-activity relationship (SAR) studies can help predict the biological activities of compounds with related structures and help avoid off-target outcomes due to their toxicity and side effects. The substantial contribution of a treatment is based on selecting the right medicine for a given target. Available theoretical tools (e.g., molecular docking studies) can help predict potential inhibitory activity against enzymes, including tyrosinase, and virtual screening can be used to select compounds that target tyrosinase and to determine expected targets for well-known and newly discovered flavonoidsCitation2,Citation3. Tyrosinase is common in mammals, fungi, bacteria, and plants, and it plays a critical role in melanin biosynthesis. Tyrosinase consists of two identical H subunits as a catalytic component and two identical L subunits. The H subunit includes four helices that contain the catalytic binuclear copper-binding site. Each Cu2+ cofactor forms coordination bonds with three histidine residues (His61, His85, His94, and His259, His263, His296, respectively) located at the centre of two antiparallel α helix pairsCitation4,Citation5. His85 is covalently bound to Cys83 through a thioether bond, and these two residues are connected to each other via a threonine residue that forms a triad motif that is conserved among tyrosinases and is considered essential for catalytic activityCitation6,Citation7. The histidine ligands of the copper ions are stabilised via interactions with nearby residues such as Phe90 and Phe292 for catalytic activity; thus, interactions with the copper ions as well as their ligands and the nearby residues are required for effective inhibitionCitation8,Citation9. In humans, abnormal melanin production or distribution leads to pigmentation diseases, such as overtanning, freckles, age spots, and melasma. Disorderly melanin production plays a key role in melanotic melanoma, and inhibiting tyrosinase activity may reduce melanin content and be a useful process in skin-whitening compoundsCitation10,Citation11.
In this study, we investigated the tyrosinase inhibitory activity of 44 flavonoids, (7-methoxynaringenin (1), 7,4′-dimethoxynaringenin (2), butin (3), isookanin (4), hesperetin (5), hesperetin 7-O-rutinoside (hesperidin) (6), 5-hydroxyflavone (7), 5-hydroxy-2′-methoxyflavone (8), 5-hydroxy-2′,6′-dimethoxyflavone (9), zapotin (10), chrysin (11), apigenin (12), apigenin 7-O-glucoside (cosmosiin) (13), apigenin 8-C-glucoside (vitexin) (14), apigenin 8-C-glucosyl-2"-O-glucoside (15), acacetin (16), luteolin (17), luteolin 7-O-glucoside (cynaroside) (18), luteolin 7-O-sambubioside (19), luteolin-6-C-glucoside (isoorientin) (20), chrysoeriol (21), 5,7,3′-trihydroxy-4′-methoxyflavone-8-C-xylopyranoside-2"-O-glucoside (scleranthoside A) (22), 5,7,3′-trihydroxy-4′-acetoxyflavone-8-C- xylopyranoside-2"-O-glucoside (scleranthoside B) (23), 5,7-dihydroxy-3′-methoxy-4′-acetoxyflavone-8-C-xylopyranoside-2"-O-glucoside (24), 5,7-dihydroxy-3′-methoxy-4′-acetoxyflavone-8-C-xylopyranoside-2"-O-(4′"-acetoxy)-glucoside (scleranthoside D) (25), kaempferol (26), 8-methoxykaempferol (27), kumatakenin (28), kaempferol 3-O-glucoside (astragalin) (29), kaempferol 3-O-glucuronide (30), kaempferol 3-O-galactoside (hyperoside) (31), kaempferol 3-O-(6″-O-trans-p-coumaroyl)-glucoside (tiliroside) (32), ermanin (33), icariin (34), quercetin (35), quercetin 3-O-glucuronide (miquelianin) (36), quercetin 3-O-rutinoside (rutin) (37), quercetin 3-O-rutinoside-7-O-glucoside (38), 7-O-methoxyquercetin (rhamnetin) (39), 7-O-methoxyquercetin (isorhamnetin) (40), robinetin (41), myricetin (42), daidzein (43), and genistein (44), to find potentially active compounds. First, tyrosinase inhibition and enzyme kinetics were tested in an in vitro model to determine the types of inhibition for the compounds with the highest activity. Second, in an in silico model, docking studies were performed to describe SARs, which were also assessed using a statistical model. Thus, a combination of bioinformatics simulation and biological in vitro studies describe the functional mechanisms of the tested compounds.
2. Materials and methods
2.1. Chemical and reagents
Kojic acid (CAS number: 501-30-4), 3,4-dihydroxy-L-phenylalanine (L-DOPA) (CAS number: 59-92-7; purity ≥98%), and tyrosinase from mushrooms (CAS number: 9002-10-2) were purchased from Sigma–Aldrich Chemical Co. (St. Louis, MO, USA). Compounds 5 (CAS number: 69097-99-0), 11 (CAS number: 480-40-0), 12 (CAS number: 520-36-5), 14 (CAS number: 3681-93-4), 20 (CAS number: 4261-42-1), 21 (CAS number: 491-71-4), 26 (CAS number: 520-18-3), 35 (CAS number: 6151-25-3), 39 (CAS number: 90-19-7), 40 (CAS number: 480-19-3), 42 (CAS number: 529-44-2) and 44 (CAS number: 446-72-0) were obtained from Carl Roth (Karlsruhe, Germany), 16 (CAS number: 480-44-4), 34 (CAS number: 489-32-7), and 43 (CAS number: 552-66-9) were obtained from Cayman Chemical (Ann Arbour, MI, USA). Flavonoids 1, 2, 28, 33Citation12, 13Citation13, 15, 31, 38Citation14, 17Citation15, 18, 19Citation16, 7, 8, 9Citation17, 22, 23, 24, 25Citation18, 27, 32Citation19, 29, and 37Citation20 were isolated and identified in the Department of Pharmacognosy at the Medical University of Białystok. Compounds 3 (CAS number: 492-14-8), 4 (CAS number: 1036-49-3), and 10 (CAS number: 14813-19-5) were purchased from Biosynth Carbosynth (London, UK), 6 (CAS number: 520-26-3) and 41 (CAS number: 490-31-3) were purchased from Fluka Analysis (St. Louis, MO, USA), and 30 (CAS number: 22688-78-4) and 36 (CAS number: 22688-79-5) were obtained from Extrasynthese (Genay, France). All isolated compounds were >95% pure as measured by HPLC. Bioassay was performed on BioTek Instruments microplate spectrophotometer EPOCH 2 (Oxfordshire, UK).
2.2. Tyrosinase inhibition assay
The inhibitory effect on mushroom tyrosinase (TYase) was evaluated using a method reported in the literature with some modificationsCitation21. This assay was performed in PBS (100 mM, pH = 6.8 in 25 °C). Test samples 1–44 (80 µL) were preincubated with TYase solution (40 µL; 250 U/mL) at 25 °C for 10 min. Then, L-DOPA (80 µL; 0.19 mg/mL) was added, and after an additional 10 min at 25 °C, the absorbance was measured at 492 nm. A blank analysis was performed using PBS instead of a sample, and the positive control was conducted with kojic acid. The inhibitory effect was calculated as follows:
where B and C are the absorbances of the blank and the compounds, respectively. The compound concentration that inhibits 50% of tyrosinase activity (IC50) was calculated. All tests were performed in triplicate.
2.3. Tyrosinase inhibition kinetic analysis
Based on IC50 values, the seven most active compounds (4, 17, 26, 32, 35, 41) were selected for kinetic analysis. The enzyme reaction kinetics of these compounds were measured by constructing Lineweaver-Burk plots of inverse velocities (1/V), contrary to the inverse of substrate concentration (1/S)Citation22,Citation23. Preincubations and measurement times were performed using the same protocol as described above. The enzyme concentration (250 U/mL) was kept constant in the presence of substrate solutions (L-DOPA) between 0.25 and 2 mM in all kinetic studies. The inhibitor concentrations for all test compounds were 0, 25, and 50 µM. The inhibitory types and inhibitory constant (Ki) values were described by 1/V versus inhibitor concentration plots (Dixon plot).
2.4. Molecular docking
The selected flavonoids were sketched in ACD/ChemSketch to build and generate their mol topology format. The compounds were modelled using LigPrep (2021–22, Schrödinger LLC, New York, NY) and MacroModel (2021–22, Schrödinger LLC, New York, NY) according to the OPLS4 forcefield parametersCitation24 and conjugate gradients method. The original configurations of the ligands were conserved, and their ionisation states at pH 7 were modelled. The mushroom tyrosinase structure (PDB ID: 2Y9XCitation5; resolution: 2.78 Å) was downloaded from the RCSB protein data bank (www.rcsb.org)Citation25 and prepared for docking using the Protein Preparation Wizard of Maestro (2021–22, Schrödinger LLC, New York, NY)Citation26. In this process, redundant molecules were removed, H atoms were added, partial charges were assigned, and ionisation, tautomeric states, and H bonds were set. The active site grids were generated using the Receptor Grid Generator panel of Maestro by setting the central coordinates as −0.62, 26.99, and −43.78, and the volume as 27.000 Å3. The ligand was docked to the active site using Glide (2021-2, Schrödinger LLC, New York, NY) in additional precision (XP) mode with 50 runs per ligand, and the results were visually evaluatedCitation27. Prior to docking of the ligands, the co-crystallized ligand in the PDB structure, tropolone, was removed and redocked to the active site, and the obtained binding mode was compared with the co-crystallized conformer by calculating the root-mean-square deviation (RMSD) to confirm the precision of the method. The predicted binding mode for tropolone was similar to that of the original conformer (RMSD: 1.33 Å).
2.5. Statistical analysis
Statistical analysis for IC50 calculations was performed using nonlinear regression using GraphPad Prism 9 (Trail, GraphPad Software, La Jolla, CA, USA). Data were collected as mean ± SD (n = 3), and the significance of differences was analysed using one-way analysis of variance (ANOVA). Statistical analysis of SARs was performed using Stata/IC 13.1 (StataCorp LP, TX 77845, USA).
3. Results and discussion
3.1. Inhibitory effects of tested compounds on tyrosinase
The effects of different concentrations of selected flavonoids () and kojic acid, a well-known inhibitor of L-DOPA oxidation by tyrosinase, were studied. The effects, expressed as IC50 values, are shown in .
Figure 1. Molecular structures of the tested compounds.
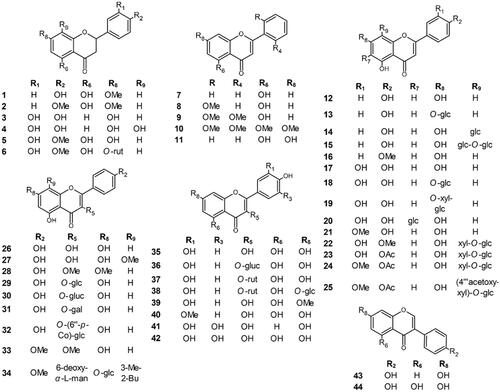
Table 1. Compounds 1–44 measured for anti-tyrosinase activity and their respective IC50 values.
Although the rendered activity is accumulative of an entire molecule, a structure-activity relationship was recognised by examining the effect of different substitutions: the presence or absence of hydroxyl groups or their methylation/acetylation/glycosylation in all carbons of the B-ring, as well as in carbons 3, 5, 6, 7, and 8 of the benzo-γ-pyrane ring, on the potential inhibitory activity.
Some flavonoids (4, 17, 35) exhibited IC50 values that were comparable to the positive control, kojic acid (IC50=49.48 ± 0.23 µM). Compound 4 (IC50=58.43 ± 0.38 µM), tested in this study for the first time, was one of the most effective inhibitors of tyrosinase, along with compound 17 (IC50=58.88 ± 0.78 µM) and compound 35 (IC50=44.38 ± 0.13 µM). The distinction in position C3 in 35 and 36 (IC50=220.10 ± 1.14 µM) or 37 (IC50=141.67 ± 1.30 µM) is only a difference in the glycosylation of a hydroxyl group. However, this small difference in their structure causes immense differences in their inhibitory potentials. It is presumed that substitutions at C3 led to steric hindrances that prevent molecules from binding to enzymes. However, the most typical locations for C-glycosyl radicals are the C6 and C8 positions at the A-ring. It seems that aglycone 4 (IC50=58.43 ± 0.38 µM) is a more effective tyrosinase inhibitor than it glycoside: 20 (IC50=132.55 ± 2.32 µM). The exact location of the glycosidic residue (carbon 6 or 8) does not affect activity. Also, disaccharides linked with the flavonoid core in C8 (22, 23, 24, 25) exhibit weak inhibitory potential with IC50 values that are higher than those of the maximum tested concentration. When the antityrosinase activity of methylated 26, 27, and 28 molecules with IC50 values 65.11 ± 1.09 µM, 290.46 ± 1.19 µM, and >500 µM, respectively, was compared, the presence of methyl groups likely prevents appropriate interactions with the enzyme active siteCitation28. The same SAR pattern was observed with compounds 4 (without methylation), 1 (one methyl group at C7), and 2 (two methyl groups at C7, C4’), with IC50 values of 58.43 ± 0.38 µM, 441.92 ± 1.91 µM, and >500 µM, respectively. The links of the B-ring in the C-ring allow flavone and isoflavone activity to be compared. Isoflavones (3-B-ring) with IC50 values >500 µM (43 and 44) exhibit no inhibitory potential. Additionally, the confrontation of the activity of 35 (IC50=44.38 ± 0.13 µM) with 42 (IC50=100.33 ± 1.86 µM) leads to the conclusion that the additional hydroxyl group in C5' reduces phenol inhibitory potency. It was not possible to determine the IC50 values of flavonoids 2, 21, 22, 23, 25, 28, 40, 43 and 44 up to 500 µM, which was the highest tested concentration. Thus, based on the in vitro study, molecules containing more unsubstituted hydroxyl groups in C3, C5, and C7, as well as in C3' and C4', generally achieve highly potent activity against tyrosinase, this finding is consistent with previous studyCitation11. Similar conclusions of SAR were presented using inhibitors of xanthine oxidase and elastase and scavengers of superoxide radicalsCitation29,Citation30.
3.2. Kinetic analysis of tyrosinase inhibition
Kinetic analysis of compound-induced inhibition was performed to determine the type of inhibition of the most active constituents (i.e., IC50 values below 100 µM). Lineweaver-Burk plots () in double-reciprocal form and Dixon plots () were used to determine the inhibition types and evaluate the dissociation constants for the inhibitors (Ki) (). The inhibitory type of isookanin (4) and robinetin (41) was tested for the first time in this study.
Figure 2. Lineweaver–Burk plots for inhibition of tyrosinase in the presence of compounds: 4 (A), 17 (B), 26 (C), 32 (D), 35 (E), and 41 (F). The concentrations of the compounds were 0.00, 25, and 50 µM. The substrate L-DOPA concentrations were 0.25, 0.50, 1, and 2 mM.
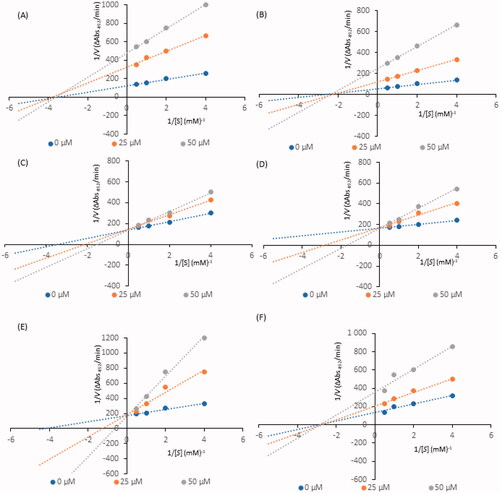
Figure 3. Dixon plots for inhibition of tyrosinase in the presence of compounds: 4 (A), 17 (B), 26 (C), 32 (D), 35 (E), and 41 (F). The concentrations of the compounds were 0.00, 25, and 50 µM. The substrate L-DOPA concentrations were 0.25, 0.50, 1, and 2 mM.
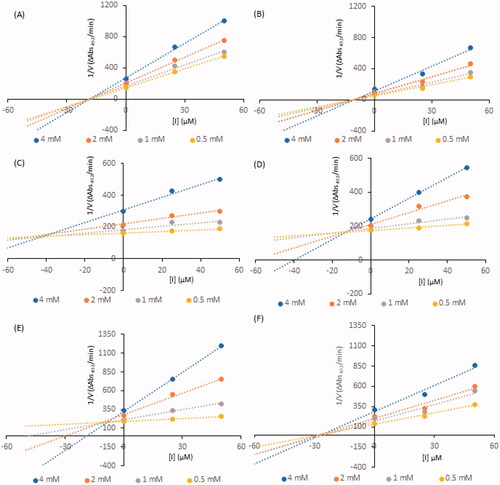
Table 2. Kinetic analysis of active compounds on tyrosinase.
As shown in , the straight lines intersected at the same point on the x-axis, implying that the Km value remained constant, while the maximum reaction rate (Vmax) decreased with increasing concentrations of tested inhibitors (0, 25, and 50 µM). Thus, isookanin (4), luteolin (17) and robinetin (41) likely caused non-competitive tyrosinase inhibition. Compounds 4, 17 and 41 produce allosteric regulation, which is a specific type of enzyme inhibition that is characterised by an inhibitor binding to an allosteric site, resulting in decreased enzyme efficacy. The luteolin type of inhibition was consistent with previous studiesCitation31. The inhibition constant Ki was obtained from Dixon plots () as 18.64 ± 0.53 µM, 11.32 ± 0.77 µM and 27.42 ± 0.62 µM for compounds 4, 17, and 41, respectively.
In contrast, kaempferol (26), tiliroside (32) and quercetin (35) exhibit tyrosinase inhibition that are similarCitation11,Citation32,Citation33. As shown in , all straight lines crossed at the same point on the y-axis, which indicated that Vmax remained constant. As the x-axis intercepted increased with increasing inhibitor concentrations, Km increased because a higher concentration of substrate is required to overcome the inhibitory effects of a competitor. Thus, compounds 26, 32, 35 can bind to the active site and prevent binding to the real substrate. Based on Dixon plots (), Ki for compounds 26, 32, 35 were calculated to be 40.20 ± 0.46 µM, 9.83 ± 0.57 µM, and 8.21 ± 0.86 µM, respectively. The inhibition constant for quercetin (35) was the lowest, suggesting that the inhibitory effect of this compound is leading among all tested flavonoid structures. This result is consistent with the tyrosinase inhibitory activity test (IC50=44.38 ± 0.13 µM).
3.3. Molecular docking studies
Active compounds were generally predicted to show high affinity to the active site, as represented by their docking scores (). Compounds 26 and 41 were particularly noteworthy based on their docking scores, which were based on a set of profitable interactions with the enzyme and its cofactors.
Table 3. Docking scores of the active flavonoids.
Isookanin’s (4) binding mode lacked interactions with Cu2+ and its ligands (), which resulted in a moderate docking score. This result was most likely due to the lack of aromaticity (i.e., a double bond between C2 and C3) of its flavonoid core, which, in the case of other compounds, was able to reach deeper into the copper zone and make π-stacks with the key histidine residues. The lack of aromaticity in isookanin’s flavonoid core also cancelled out the compound’s planarity and led to bending, which may have made it sterically difficult to approach the copper ions.
Figure 4. Predicted binding mode of compounds 4 (A), 17 (B), 26 (C), 32 (D), 35 (E), and 41 (F) in the tyrosinase active site. Compounds are represented as colour stick balls, interacting tyrosinase residues as grey sticks, Cu2+ as orange spheres, their histidine ligands as wireframes, and binding interactions as colour dashed lines. The distances between Cu2+ and interacting atoms are shown in Å.
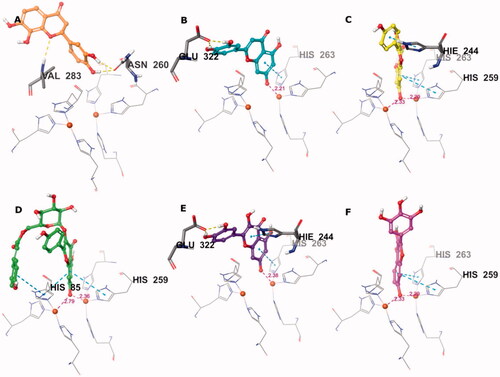
Conversely, compounds 17, 26, 32, 35, and 41 were found to be suitable chelators of Cu2+ due to the common flavonoid core and the ionisable OH substituent at the 7th position (). The ionised hydroxyl was effective even for double Cu2+ engagement in the case of 26, 32 and 41, which were the best scoring compounds (see ). Aromaticity of the flavonoid core enabled π stacking with the histidine residues serving as copper ligands, as well as His244. The hydroxyl groups on ring B of 17 and 35 formed H bonds with Glu322. The hydroxyphenyl attached to the glucopyranosyl moiety of 32 made additional π stacking with His85. Ring B of 17, 26, 35, and 41 was observed to face active site entry and disposed to solvent molecules (). With three hydroxyls on ring B, 41 was the most advantageous compound in this situation, which contributed to its docking score.
Figure 5. Binding interactions of compounds 4 (A), 17 (B), 26 (C), 32 (D), 35 (E), and 41 (F) with the tyrosinase active site residues. Binding interactions are shown as coloured lines, and compound moieties exposed to solvent are highlighted with grey shades.
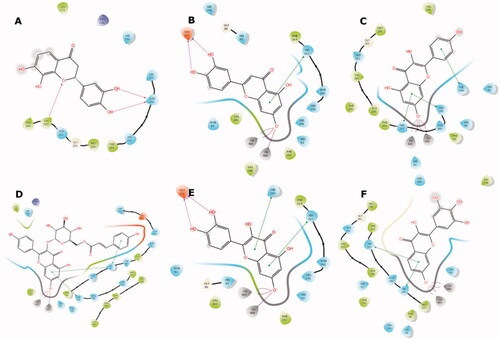
These results highlight the importance of the flavonoid core with a free hydroxyl at C-7 as a key component for copper chelation and for interactions with the key residues of the tyrosinase active site. The exceptional binding mode of 4 in the catalytic site with respect to its activity can be explained by more effective binding to a possible allosteric site.
3.4. Statistical modelling approach
To create a statistical model, 26 attributes () that were consistent with all tested flavonoid compounds and their variables (IC50 values) were identified.
Table 4. Attributes definitions used to for the statistical analysis.
shows the multivariate linear regression model for the ln IC50. Statistical analysis explained more than 50% of the variability of the IC50 variable (adjusted R2=50.88%) with p= 0.0002. All 11 independent variables in this approach yield p< 0.2, while five of them (A4, A10, A18, A21 and A22) have a statistically significant influence on the dependent variable (IC50).
Table 5. The multivariate linear regression model for the ln IC50.
Based on these results, the presence of methoxy substitution at C3'(A4) increases the mean IC50 value by approximately 4.15 times, and increasing this value (by approximately 2.47 times) relates to methoxy substitution at C7 (A18). Methylation at C8 (A22) caused an increase in the IC50 value, as shown by lnIC50 (1.452888), which indicates that, in this case, the mean IC50 increases by approximately 4.28 times. Conversely, the presence of hydroxy substitution at C3 (A10) and hydroxy substitution at C8 (A21) reduced the mean IC50 by approximately 2.83 and 5.11 times, respectively. Overall, statistical modelling suggests that flavonoids with higher antityrosinase activity possess an OH group at C3 and at C8, while methylation of the hydroxyl group at carbon 3 and at carbon 8 notably reduces the inhibitory activity of the molecules.
4. Conclusions
Results from the literature that describe the effect of flavonoids on mushroom tyrosinase activity are dispersed and sometimes contradictory. This dissidence may relate to various experimental conditions (e.g., concentration of enzyme or substrate L-DOPA, wavelength), which led to different ranges of reported inhibitory activity. Furthermore, generally few flavonoids were included in such investigations, which yields restricted structure-activity relationships information. The statistical model developed in this study provides accurate SAR data in a comprehensive format. Many flavonoids were tested under the same conditions to obtain values that are as close to those in the real world as possible. In summary, a comparison of IC50 values, kinetic reactions, molecular docking scores and statistical data allowed us to identify the characteristics of flavonoid structures that facilitate tyrosinase inhibition: the presence of hydroxyls at C3 and C7, O- and C-glycosylation, methylation and acetylation of OH groups. As in many other cases reported in the literatureCitation34, the natural products represent a gold mine for interesting biological activities which can be translated to potential biomedical applications.
Author contributions
Study design: K.J, and M.T.; Biological evaluation: K.J.; Data Interpretation: K.J.; Computational studies: S.S., and D.S.; Statistical analysis: R.M., and K.J.; Supervised experiments: M.T., and D.S.; Wrote the original paper: K.J., S.S., and R.M.; Manuscript formal analysis: M.T., and C.T.S.
Acknowledgement
The authors express thanks to J.W. Strawa and A.M. Juszczak from the Medical University of Białystok for isolation of compounds to in vitro studies.
Disclosure statement
No potential conflict of interest was reported by the authors. CT Supuran is Editor-in-Chief of Journal of Enzyme Inhibition and Medicinal Chemistry and he was not involved in the assessment, peer review or decision making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Correction Statement
This article was originally published with errors, which have now been corrected in the online version. Please see Correction (10.1080/14756366.2022.2024999).
References
- Jakimiuk K, Wink M, Tomczyk M. Flavonoids of the Caryophyllaceae. Phytochem Rev 2021;20:1–40.
- García-Sosa AR, Maran U. Improving the use of ranking in virtual screening against HIV-1 integrase with triangular numbers and including ligand profiling with antitargets. J Chem Inf Model 2014;54:3172–85.
- Glisic S, Sencanski M, Perovic V, et al. Arginase flavonoid anti-leishmanial in silico inhibitors flagged against anti-targets. Molecules 2016;21:589–14.
- Flurkey WH, Inlow JK. Proteolytic processing of polyphenol oxidase from plants and fungi. J Inorg Biochem 2008;102:2160–70.
- Ismaya WT, Rozeboom HJ, Weijn A, et al. Crystal structure of Agaricus bisporus mushroom tyrosinase: identity of the tetramer subunits and interaction with tropolone. Biochem 2011;50:5477–86.
- Klabunde T, Eicken C, Sacchettini JC, Krebs B. Crystal structure of a plant catechol oxidase containing a dicopper center. Nat Struct Mo. Biol 1998;5:1084–90.
- Zolghadri S, Bahrami A, Hassan Khan MT, et al. A comprehensive review on tyrosinase inhibitors. J Enzyme Inhib Med Chem 2019;34:279–309.
- Hazes B, Magnus K, Bonaventura C, et al. Crystal structure of deoxygenated Limulus polyphemus subunit II hemocyanin at 2.18 A resolution: clues for a mechanism for allosteric regulation. Protein Sci 1993;2:597–619.
- Ferro S, Deri B, Germanò MP, et al. Targeting tyrosinase: development and structural insights of novel inhibitors bearing arylpiperidine and arylpiperazine fragments. J Med Chem 2018;61:3908–17.
- Lai X, Wichers HJ, Soler-Lopez M, Dijkstra BW. Structure and function of human tyrosinase and tyrosinase-related proteins. Chem–An Eur J 2018;24:47–55.
- Şöhretoğlu D, Sari S, Barut B, Özel A. Tyrosinase inhibition by some flavonoids: inhibitory activity, mechanism by in vitro and in silico studies. Bioorg Chem 2018;81:168–74.
- Isidorov V, Szoka L, Nazaruk J. Cytotoxicity of white birch bud extracts: perspectives for therapy of tumours. PLoS One 2018;13:e0201949–10.
- Nazaruk J, Jakoniuk P. Flavonoid composition and antimicrobial activity of Cirsium rivulare (Jacq.) all flowers. J Ethnopharmacol 2005;102:208–12.
- Tomczyk M, Gudej J. Quercetin and kaempferol glycosides from Ficaria verna flowers and their structure studied by 2D NMR spectroscopy. Pol J Chem 2002;76:1601–5.
- Strawa J, Wajs-Bonikowska A, Jakimiuk K, et al. Phytochemical examination of woolly burdock Arctium tomentosum leaves and flower heads. Chem Nat Compd 2020;56:345–7.
- Juszczak AM, Czarnomysy R, Strawa JW, et al. In vitro anticancer potential of Jasione montana and its main components against human amelanotic melanoma cells. Int J Mol Sci 2021;22:1–25.
- Strawa J, Galanty A, Jakimiuk K, Grabowska K, Podolak I, Tomczyk M. Cytotoxic effect on human melanoma cell lines and tyrosinase inhibition of Hottonia palustris. Poster session presented at: 69th International Congress and Annual Meeting of the Society for Medicinal Plant and Natural Product Research; 5-9 September 2021; Bonn, Germany; Virtual Conference.
- Jakimiuk K, Strawa JW, Granica S, Tomczyk M. New flavone C-glycosides from Scleranthus perennis and their anti-collagenase activity. Molecules 2021;26:5631–10.
- Tomczyk M. Secondary metabolites from Potentilla recta L. and Drymocallis rupestris (L.) Soják (syn. Potentilla rupestris L.) (Rosaceae). Biochem Syst Ecol 2011;39:893–6.
- Gudej J, Tomczyk M. Polyphenolic compounds from flowers of Ficaria verna Huds. Acta Pol Pharm 1999;56:475–6.
- Ciganović P, Jakimiuk K, Tomczyk M, Zovko-Končić M. Glycerolic licorice extracts as active cosmeceutical ingredients: extraction optimization, chemical characterization, and biological activity. Antioxidants 2019;8:445–14.
- Yoshino M, Murakami K. A graphical method for determining inhibition constants. J Enzyme Inhib Med Chem 2009;24:1288–90.
- Butt ARS, Abbasi MA, Rehman AA, et al. Synthesis and structure-activity relationship of tyrosinase inhibiting novel bi-heterocyclic acetamides: mechanistic insights through enzyme inhibition, kinetics and computational studies. Bioorg Chem 2019;86:459–72.
- Lu C, Wu C, Ghoreishi D, et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput 2021;17:4291–300.
- Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res 2000;28:3908–17.
- Sastry GM, Adzhigirey M, Day T, et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 2013;27:221–34.
- Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 2006;49:6177–96.
- Jacob V, Hagai T, Soliman K. Structure-activity relationships of flavonoids. Curr Org Chem 2011;15:2641–57.
- Cos P, Ying L, Calomme M, et al. Structure-activity relationship and classification of flavonoids as inhibitors of xanthine oxidase and superoxide scavengers. J Nat Prod 1998;61:71–6.
- Jakimiuk K, Gesek J, Atanasov AG, Tomczyk M. Flavonoids as inhibitors of human neutrophil elastase. J Enzyme Inhib Med Chem 2021;36:1016–28.
- Zhang L, Zhao X, Tao G-J, et al. Investigating the inhibitory activity and mechanism differences between norartocarpetin and luteolin for tyrosinase: a combinatory kinetic study and computational simulation analysis. Food Chem 2017;223:40–8.
- Fan M, Zhang G, Hu X, et al. Quercetin as a tyrosinase inhibitor: inhibitory activity, conformational change and mechanism. Food Res Int 2017;100:226–33.
- Lu Y, Chen J, Wei D, et al. Tyrosinase inhibitory effect and inhibitory mechanism of tiliroside from raspberry. J Enzyme Inhib Med Chem 2009;24:1154–60.
- Atanasov AG, Zotchev SB, Dirsch VM, Supuran CT. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov 2021;20:200–16.