
Abstract
In the current work, some 1,3,4-oxadiazole-naphthalene hybrids were designed and synthesised as VEGFR-2 inhibitors. The synthesised compounds were evaluated in vitro for their antiproliferative activity against two human cancer cell lines namely, HepG-2 and MCF-7. Compounds that exhibited promising cytotoxicity (5, 8, 15, 16, 17, and 18) were further evaluated for their VEGFR-2 inhibitory activities. Compound 5 showed good antiproliferative activity against both cell lines and inhibitory effect on VEGFR-2. Besides, it induced apoptosis by 22.86% compared to 0.51% in the control (HepG2) cells. This apoptotic effect was supported by a 5.61-fold increase in the level of caspase-3 compared to the control cells. Moreover, it arrested the HepG2 cell growth mostly at the Pre-G1 phase. Several in silico studies were performed including docking, ADMET, and toxicity studies to predict binding mode against VEGFR-2 and to anticipate pharmacokinetic, drug-likeness, and toxicity of the synthesised compounds.
1. Introduction
Although there are many advances in drug discovery for cancer control and treatment, still cancer is one of the most serious diseases responsible for a huge number of deathsCitation1. According to global statistics, cancer is a leading cause of death worldwide, accounting for nearly 10 million deaths in 2020Citation2. The cancer problem is localised not only in its widespread and metastasis but also in the lack of selectivity of anticancer drugs which leads to many side effectsCitation3. Moreover, the problem increases due to the continuous mutations in many targets of anticancer drugs which produce some sort of resistanceCitation4. Recently, medicinal chemists tried to develop new anticancer agents with high selectivity and can overcome the generated drug resistanceCitation5–7.
Vascular endothelial growth factor receptors (VEGFRs) are a group of tyrosine kinases that caught the attention of scientists for the discovery of new anticancer agentsCitation8,Citation9. VEGFRs group is subdivided into three categories. (i) VEGFR-1 is responsible for the control of angiogenesis in embryosCitation10. (ii) VEGFR-2 is the key element in tumour growth due to its crucial role in the formation of new vasculatures and angiogenesisCitation11. (iii) VEGFR-3 controls the process of lymphangiogenesisCitation12. VEGFR-2 took more interest due to its important role in tumour growthCitation13.
In embryonic vasculogenesis, VEGFR1 plays a critical function. VEGFR2 is a protein that controls embryonic and tumour angiogenesis. VEGFR2 has mostly overexpressed in tumour vasculature endothelial cells, with reduced expression in normal endothelial cellsCitation14. Overexpressed VEGFR-2 is found in a variety of malignancies, including hepatocellular carcinoma and breast cancerCitation15–17. Blocking VEGFR2 is a viable method for the identification of novel therapies for angiogenesis-dependent cancersCitation18. VEGFR2 is now the most significant target of antiangiogenic therapy. During development, VEGFR3 is found in all endothelium, but it is only found in the lymphatic endothelium in adultsCitation19. It is up-regulated in the microvasculature of tumours and woundsCitation20.
Till now, there are many drugs approved by the FDA targeting VEGFR-2 for the treatment of cancerCitation13. Sorafenib 1 was approved for the treatment of thyroid cancer, metastatic renal cell cancer, and hepatocellular carcinoma. It is associated with many adverse effects as diarrhoea, renal dysfunction, and cardiovascular problemsCitation21. Sunitinib II is an oral VEGFR-2 inhibitor that exhibits potent antiangiogenic and antitumor activities. It exhibits a high bioavailability and potency against VEGFR in the nanomolar range. Small-cell lung cancer, GI stromal tumours, breast cancer, acute myelogenous leukaemia, multiple endocrine neoplasia types 2A and 2B, and familial medullary thyroid carcinoma were all treated with sunitinibCitation22. Vorolanib III is a multi-target tyrosine kinase inhibitor that has successfully completed phase I studies. At a dose of 200 mg (once daily), it had an acceptable safety profile and a favourable therapeutic benefit for patients with advanced solid tumoursCitation23. Tivozanib IV is a powerful and highly selective orally accessible tyrosine kinase inhibitor with a long half-life (4 days) that targets VEGFR-1, VEGFR-2, and VEGF-3 at very low dosagesCitation24.
Four key pharmacophoric features of VEGFR-2 inhibitors have been identifiedCitation25–30. Each feature has its own binding area in VEGFR-2's active site. The hinge region is occupied by a flat heteroaromatic moiety that forms one hydrogen bond with Cys917Citation26. The second distinguishing feature is a linker moiety that sits between the hinge region and the DFG domainCitation31. The pharmacophore moiety, which occupies the DFG domain and forms two crucial hydrogen bonding interactions with Glu883 and Asp1044, is the third feature. At least one H-bond acceptor (HBA) and one H-bond donor (HBD) group (e.g. amide or urea) must be present in the pharmacophore moietyCitation32. The fourth feature is a terminal hydrophobic moiety that occupies the allosteric pocket, resulting in numerous hydrophobic interactionsCitation33 ().
Figure 1. Some reported VEGFR-2 inhibitors (I, II, III, and IV) and the target compounds (5–18) having the same pharmacophoric features.
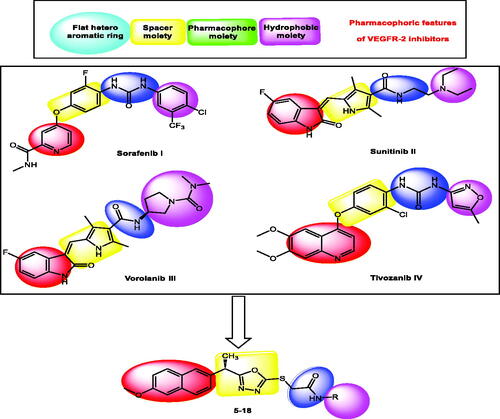
As an extension of our efforts to reach potent anti-VEGFR-2 agentsCitation29,Citation34–39, a new series of 1,3,4-oxadiazole-naphthalene hybrids was synthesised as a modified form of the reported VEGFR-2 inhibitors. The synthesised hybrids were designed to have the main features of VEGFR-2 inhibitors and evaluated to confirm their VEGFR-2 inhibitory activities.
1.1. Rationale of molecular design
The 1,3,4-oxadiazole moiety has various biological activities. The wide and potent activity of this moiety made it an important pharmacological scaffold for drug design especially in the field of cancer diseaseCitation5,Citation40,Citation41. Additionally, naphthalene is a main building block in many anticancer agentsCitation42,Citation43. The molecular hybridisation approach is one of the most interesting and efficient method for the design and discovery of new bioactive agentsCitation44,Citation45. Depending on this approach and in continuation of our activities in the discovery of VEGFR-2 inhibitors, we synthesised a new series 1,3,4-oxadiazole-naphthalene hybrids.
The 2-methoxynaphthalene moiety was used to occupy the hinge region of the VEGFR-2 binding site to validate the main pharmacophoric features of VEGFR-2 inhibitors. The naphthalene moiety's bicyclic structure is well-suited to the large size space of the hinge regionCitation46. Furthermore, the methoxy group serves as a hydrogen-bond acceptor, facilitating hydrogen bonding interactions in the hinge region. As a linker group, the 2-ethyl-1,3,4-oxadiazole moiety was used. In the linker region, the 1,3,4-oxadiazole moiety contains two nitrogen atoms that can form extra hydrogen bonds. As a pharmacophore moiety, we used an amide group to occupy the DFG region. Finally, different aromatic derivatives can occupy the allosteric hydrophobic region to investigate structure-activity relationships ().
2. Results and discussion
2.1. Chemistry
Different synthetic pathways were described in Scheme 1 for the preparation of final designed compounds. Firstly, the commercially available S(+)naproxen 1 ((S)-2–(6-methoxynaphthalen-2-yl)propanoic acid) was esterified using absolute ethanol and conc. H2SO4 to produce the ester form 2. Then, the ester derivative 2 was heated in ethanol and hydrazine hydrate to produce the corresponding hydrazide derivative 3.
Scheme 1. Synthesis of the target compounds 5–18. Reagents and conditions: (a) C2H5OH/H2SO4; (b) NH2NH2·H2O/C2H5OH; (c) CS2\KOH, EtOH; (d) RX/EtOH, NaOAc.
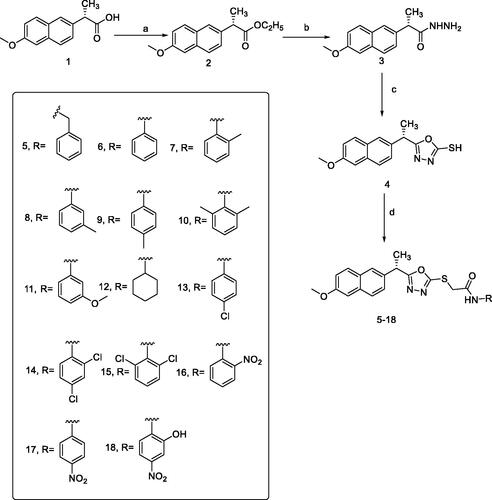
The synthesis of the key starting material 1,3,4-oxadiazolyl scaffold 4 was achieved by the reaction of acid hydrazide 3 with carbon disulphide in an alcoholic potassium hydroxide solution. Different chloroacetinilides were obtained by the treatment of aromatic amines with chloroacetyl chlorideCitation47. The potassium salt of 4 was allowed to react with the substituted chloroacetinilides. Unfortunately, the required products couldn't be separated from the reaction mixture with the desirable purity. The presence of more than one product for each reaction was attributed to the high reactivity and/or basicity of the potassium salt. Alternatively, a less basic condition was adopted and the mercapto-containing structure 4 was directly allowed to react with chloroacetinilides in the presence of sodium acetate. This alternative route successfully afforded the final products in satisfying yields and reasonable purities.
The spectral and elemental analytical data of this group of novel compounds confirmed their structures (see experimental section). In all cases, the characteristic thiol stretching band at 2624 cm−1 vanished in all IR spectra and was replaced by a higher frequency one distinctive band of the primary amide NH moiety. Furthermore, in all IR spectra, typical carbonyl stretching bands between 1655 and 1675 cm−1 were observed. These findings support the idea that the acetanilide moiety is linked to the oxadiazole nucleus via S-linkage. The later NH group revealed a broad singlet, corresponding to one proton around 10.4 ppm. As an example, the 1H NMR spectrum of compound 7 revealed a broad singlet signal, equivalent to one proton, at 10.36 ppm due to NH, and a multiplet signal, equivalent to ten protons, at 7.74–7.15 ppm due to aromatic protons. Finally, the ethyl linker between the naphthalene and oxadiazole rings produces a multiplet for one proton due (CH) and a doublet for three protons due to (CH3) at 4.53 and 1.78 ppm, respectively. Aliphatic (S-CH2) protons were detected as a singlet signal at 4.01 ppm, a singlet signal equivalent to three protons at 3.81 ppm due to OCH3, and a singlet signal of three protons at 1.95 ppm due to the benzylic methyl moiety. The 13C NMR spectrum of compound 7 revealed 24 signals.
The 1H NMR spectra of compounds 12, showed, in addition to naphthalene aromatic protons, broad singlet due to NH group at 8.12 ppm, the ethyl group appeared as multiplet and doublet signals at 4.57 and 1.66 ppm, a singlet for two protons at 4.94 ppm due S-CH2 group, another singlet for three protons at 3.83 ppm due to O-CH3. While the cyclohexyl side chain is represented by a multiplet signal between 1.66 and 1.04 ppm.
In general, mass spectra of compounds-containing halogen atoms showed the typical isotopic distribution patterns. The parent peak intensity of compounds 14 and 15 makes it easy to recognise the expected isotopic pattern of the two chlorine atoms; however, an anticipated isotopic ratio was observed with some fragments-containing chlorine; i.e. the anilinium fragment showed M+, (M + 2)+, and (M + 4)+ isotopic peaks as detailed in the experimental section.
2.2. Biological testing
2.2.1. In vitro cytotoxic activities
MTT assay was used to assess the cytotoxic activities of the synthesised compounds against MCF-7 (human breast cancer cell line) and HepG2 (human hepatocellular carcinoma cell line). And Sorafenib was used as a control drug (). When compared to sorafenib (IC50 = 10.8 and 10.2 µM against MCF-7 and HepG2 cells, respectively), the target compounds 5, 8, 15, 16, 17, and 18 showed promising cytotoxicity against the two cell lines with IC50 values ranging from 8.4 to 10.4 µM. Compound 5 demonstrated superior activity against MCF-7 and HepG2 cells, with IC50 values of 9.7 and 8.8 M, respectively. With IC50 values of 8.4 M, compound 15 demonstrated promising activity against HepG2 cells. Compounds 5, 8, 15, 16, 17, and 18 demonstrated excellent activity against HepG2 cells, with IC50 values of 8.8, 9.5, 8.4, 8.7, 9.2, and 8.7 µM, respectively. Compounds 6, 7, 10, 11, 12, 13, and 14 also demonstrated moderate activity against the tested cells, with IC50 values ranging from 10.4 M to 25.5 µM. Compound 9 on the other hand demonstrated weak cytotoxic activity against the two cell lines.
Table 1. The assessed compounds' in vitro cytotoxic activities (IC50) against MCF-7 and HepG2 cell lines.
2.2.2. VEGFR-2 inhibitory assay
The inhibitory effect of the most cytotoxic compounds 5, 8, 15, 16, 17, and 18 on VEGFR-2 was studied using sorafenib as a control drug. VEGFR-2 concentrations after inhibition by the aforementioned compounds against HepG2 cells were summarised in and .
Figure 2. The inhibitory effects of the compounds tested on VEGFR-2 in HepG2 cells compared to sorafenib. The data are presented as the mean ± SEM of three different experiments. *Significant from control group at p < 0.001 using unpaired t-test.
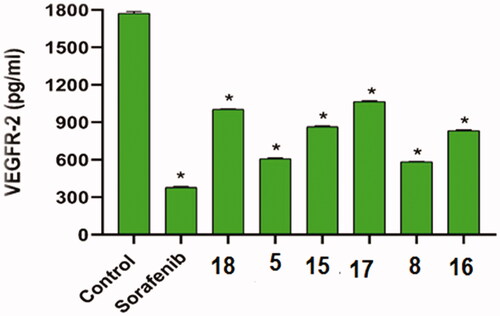
Table 2. The inhibitory effects of the assessed compounds on VEGFR-2 in HepG2 cells compared to sorafenib.
Compounds 5 and 8 inhibited VEGFR-2 well (concentrations of 610.4 and 583.7 pg/ml, respectively) when compared to sorafenib (378.7 pg/ml). Compounds 15 and 16 also had moderate effects (864.5 and 834.9 pg/ml, respectively). Compounds 17 and 18, on the other hand, had low effects (1067 and 1004 pg/ml, respectively).
2.2.3. In vitro cytotoxicity against normal cell
The cytotoxic effects of the most active compounds 5, 8, 15, 16, 17, and 18 against normal adult liver epithelial cells (Transformed Human Liver Epithelial-2, THLE-2 cells) were assessed in vitro (). The findings revealed that these compounds have low cytotoxicity against the normal THLE-2 cells with IC50 values of 33.7, 16.7, 34.9, 29.7, 22.8, and 28.6 µM, respectively. While IC50 value of sorafenib as a reference drug was 27.8 µM. The results showed that these synthesised compounds have low cytotoxicity against the normal cells in comparison to their cytotoxicity against cancer cell lines.
Table 3. In vitro cytotoxicity of the assessed compounds and sorafenib against normal adult liver epithelial cells (THLE-2 cells).
2.2.4. Structure–activity relationship (SAR)
The results of different biological analyses (cytotoxic activity and VEGFR-2 inhibitory assay) gave a valuable SAR. Comparing the cytotoxic activity of compounds 5 (incorporating benzyl moiety as a hydrophobic tail) and 6 (incorporating phenyl moiety as a hydrophobic tail), indicated that benzyl moiety enhances the cytotoxic activity. Modification of compound 6 by insertion of the methyl group at ortho-position (compound 7) gave a mild change in activity. While insertion of the methyl group at meta-position (compound 8) afforded a significant increase in the cytotoxic activity. On the other hand, the methyl substitution of phenyl moiety at para-position (compound 9) produced a dramatic decrease in the cytotoxic activity. Insertion of the methyl group at the two ortho-positions of phenyl ring (compound 10) did not produce a significant change in the cytotoxic activity. Comparing the cytotoxicity of compound 8 (with 3-methylphenyl moiety) and compound 11 (with 3-methoxyphenyl moiety), indicated that methyl moiety is more advantageous than the methoxy group. Insertion of the electron-withdrawing group at para-position (compound 13) gave a mild increase in the cytotoxic activity. Insertion of chloro atom at both 2 and 4 positions of phenyl ring (compound 14) did not produce a significant change in the cytotoxic activity. While insertion of the chloro atom at both 2 and 6 positions of the phenyl ring (compound 15) produced a significant change in the cytotoxic activity. Insertion of the nitro group at ortho-position of phenyl ring (compound 16) or para-position (compound 17) gave a dramatic increase in the biological activity. Modification of compound 17 by insertion of the hydroxyl group at position −2 of the phenyl ring to give compound 18 afforded a mild decrease in activity. Changing the phenyl ring of compound 6 by cyclohexyl moiety (compound 12), produced a dramatic decrease in the cytotoxic activity. This indicated that aromatic hydrophobic moiety is more efficient than not aromatic one.
2.2.5. Cell cycle analysis
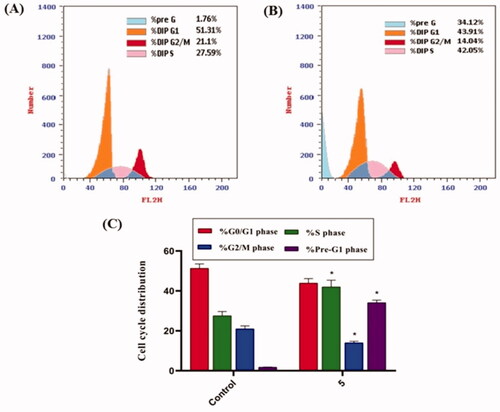
Because compound 5 effectively inhibited the growth of HepG2 cells, it was assumed that this inhibitory effect was due to its ability to obstruct cell cycle progression. As a result, the cell cycle process was investigated after HepG2 cells were exposed to compound 5 at a concentration of 8.8 µM (IC50 value of compound 5). As a control, HepG2 cells were not treated with compound 5. According to flow cytometry data, the percentage of cells arrested in the S phase increased from 27.59% (in control cells) to 42.05% (in compound 5 treated cells). Furthermore, the percentage of HepG2 cells increased from 1.76 to 34.12% during the Pre-G1 phase. In contrast, the percentage of HepG2 cells decreased slightly during the G0/G1 phase, from 51.31 to 43.91%. These findings revealed that compound 5 primarily inhibited HepG2 cell growth during the Pre-G1 phase ().
Figure 3. Flow cytometric analysis of cell cycle phases after compound 5 treatment of HepG2 cells. (A) The representative histogram depicts the cell cycle distribution of control (HepG2) cells; (B) the representative histogram depicts the cell cycle distribution of compound 5-treated cells. (C) A column graph depicts the percentage of cells in each phase of the cell cycle in both control (HepG2) and compound 5 treated cells. The percentages are given as the mean SEM of three different experiments. Using unpaired t-tests, *p < 0.05 indicates statistically significant differences from the untreated control (HepG2) group.
2.2.6. Apoptosis analysis
The apoptosis induced by compound 5 was quantified using an Annexin-V/propidium iodide (PI) staining assay. Compound 5 was applied to HepG2 cells at a concentration of 8.8 µM. As shown in and , compound 5 had a significantly higher apoptotic effect in HepG2 cells (22.86%) than in control cells (0.51%).
Figure 4. In HepG2 cells, compound 5 caused apoptosis. (A) Control, (B) Compound 5, and (C) represent the graphical representation of the percent of apoptotic and necrotic cells in control (HepG2) cells and compound 5 treated cells. The percentages are given as the mean SEM of three different experiments. Using unpaired t-tests, *p < 0.05 indicates statistically significant differences from the untreated control (HepG2) group.
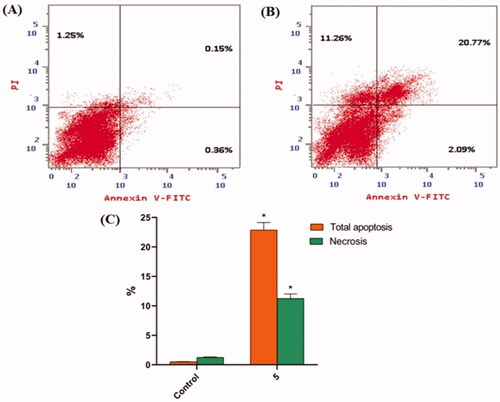
Table 4. Effect of compound 5 on the cell death process in HepG2 cells.
Percentages equal mean ± SEM of three experiments. *p < 0.05 indicates a statistically significant difference from the corresponding control (HepG2) group using unpaired t-tests.
2.2.7. Caspase-3 determination
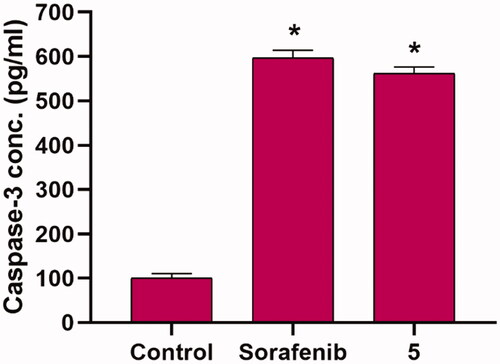
To test the effect of the synthesised compounds on caspase-3 levels, the most promising member 5, at a concentration of 8.8 µM, was applied to the most sensitive cells (HepG2) for 24 h. The results showed that compound 5 significantly increased the level of caspase-3 (5.61-fold) when compared to control cells ().
Figure 5. Effects of compound 5 on Caspase 3 level in HepG2 cells. Values are reported as mean ± SEM of three different experiments. *p < 0.001 indicates statistically significant differences from the untreated control (HepG2) group using unpaired t-test.
2.3. In silico studies
2.3.1. Docking studies
Docking studies were performed in this study to gain a better understanding of the binding modes of the synthesised compounds into the VEGFR-2 binding site (PDB ID: 2OH4). Sorafenib was used as a control drug. displays the binding free energies (ΔG). Asp1044 and Glu883 have been identified as key amino acids involved in the binding of VEGFR-2 inhibitorsCitation33,Citation48.
Table 5. Results of docking scores.
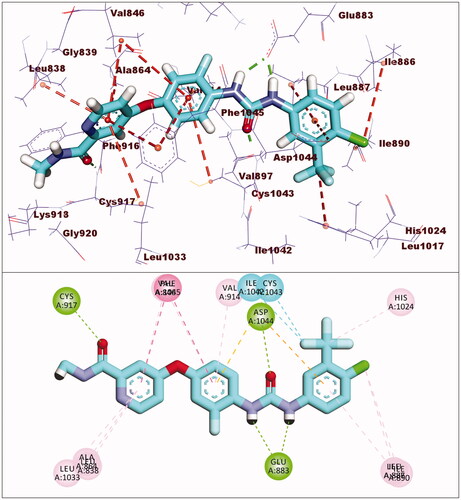
Sorafenib had a binding affinity of −22.46 kcal/mol. Three hydrogen bonding interactions occurred between the urea moiety and Glu883 and Asp1044. Four hydrophobic interactions were formed by the central phenyl group with Val914, Val846, Cys1043, and Phe1045. The hinge region was occupied by the N-methylpicolinamide moiety, which formed one hydrogen bond with Cys917 and five hydrophobic interactions with Val846, Leu838, Leu1033, Phe1045, and Ala864. The allosteric binding pocket was occupied by the terminal 1-chloro-2-(trifluoromethyl)benzene moiety, which formed five hydrophobic interactions with His1024, Ile890, Ile886, and Leu887. It also had one electrostatic interaction with Asp1044 ().
Figure 6. Interaction of sorafenib with VEGFR-2.
The findings revealed that the synthesised compounds have a binding mode similar to that of sorafenib. summarises the generated ΔG (binding energies) against VEGFR-2.
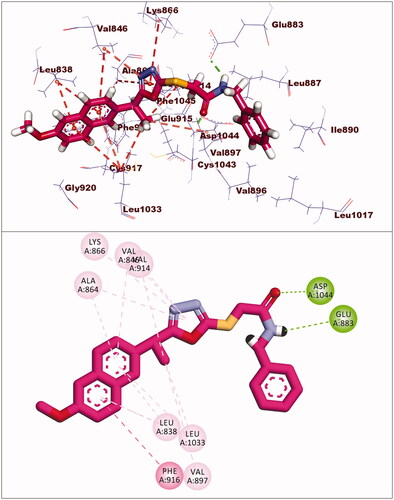
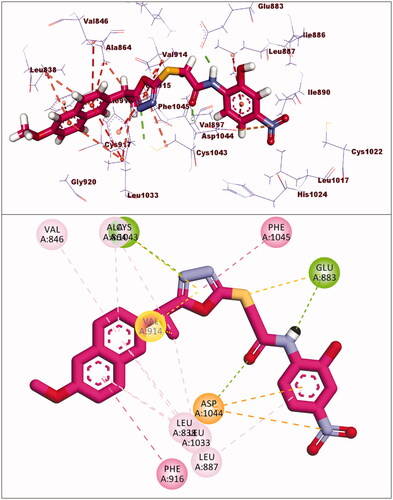
The docking score for compound 5 was −23.66 kcal/mol. Through its amide group, it formed two hydrogen bonds with Glu883 and Asp1044. The spacer moiety (2-ethyl-1,3,4-oxadiazole) interacted hydrophobically with Lys886, Val914, Val846, Val864, Val897, and Leu1033. Many hydrophobic interactions in the hinge region occurred between the 2-methoxynaphthalene moiety and different amino acid residues including Leu1033, Leu838, Val846, Val864, and Phe916. Furthermore, the hydrophobic group was positioned in the allosteric pocket, in close proximity to Ile890 and Leu887 ().
Figure 7. Interaction of compound 5 with VEGFR-2.
The binding energy of compound 8 was −24.98 kcal/mol. Two hydrogen bonds were formed between the pharmacophore (amide) group and Glu883 and Asp1044. Six hydrophobic interactions were formed by the 2-ethyl-1,3,4-oxadiazole moiety with Val914, Cys1043, Val864, and Leu1033. Furthermore, it formed an additional hydrogen bond with Cys1043. The 2-methoxynaphthalene moiety formed seven hydrophobic bonds, which were Leu1033, Leu838, Val846, Val864, and Phe916. In the allosteric pocket, the m-tolyl group formed numerous hydrophobic bonds with Leu1017, His1024, Val897, and Leu887. It also had an electrostatic interaction with Asp1044 ().
Figure 8. Interaction of compound 8 with VEGFR-2.
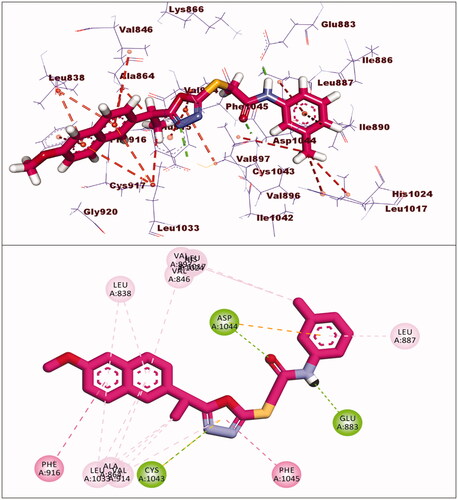
Compounds 17 and 18 exhibited binding energies of −23.78 and −23.40 kcal/mol, respectively. The pharmacophore groups in each compound formed two hydrogen bonds with Glu883 and Asp1044. The 2-ethyl-1,3,4-oxadiazole moiety in each compound formed many hydrophobic interactions in the linker region with extra hydrogen bonding with Cys1043. The 2-methoxynaphthalene moiety of compounds 17 and 18 formed four and seven hydrophobic bonds, respectively. The p-nitrophenyl of compound 17 and 2-hydroxy-4-nitrophenyl of compound 18 were oriented into the allosteric binding pocket-forming two electrostatic interactions with Asp1044 and one hydrophobic interaction with Leu887 ().
Figure 9. Interaction of compound 17 with VEGFR-2.
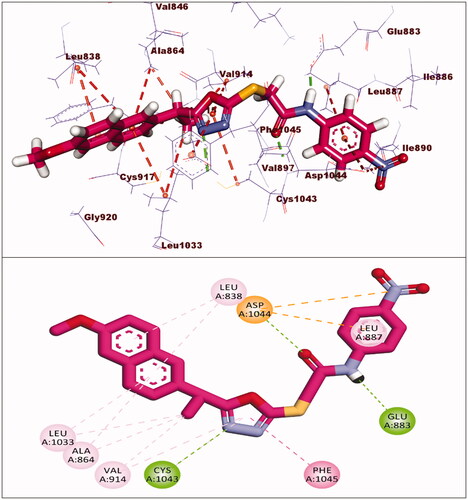
Figure 10. Predicted binding mode of compound 18 with the active site of VEGFR-2.
2.3.2. In silico ADMET studies
Discovery studio 4.0 was used to investigate the pharmacokinetic properties (ADMET studies) of the synthesised compounds. As a control molecule, sorafenib was used. summarises the ADMET parameter values.
Table 6. Predicted ADMET for the designed compounds and reference drug.
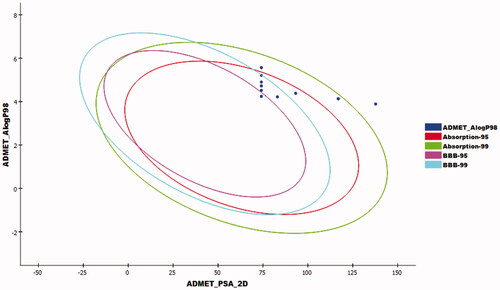
Compounds 14–18 exhibited a very low level of BBB penetration, while compounds 5–13 had medium to high levels. The aqueous solubility of the synthesised compounds ranged from very low to very low. For absorption parameters, compounds 5–13 showed good levels, while compounds 14–17 were expected to have moderate absorption levels. Compound 18 showed poor absorption. In addition, all the tested compounds were anticipated to have a non-inhibitory effect against CYP2D6, and plasma protein binding ability of more than 90% ().
Figure 11. The expected ADMET studies.
2.3.3. In silico toxicity studies
The synthesised compounds were examined in silico to reach the expected toxicity profile using Discovery Studio softwareCitation49,Citation50. As a control molecule, sorafenib was used. The toxicity studies include seven models: FDA rodent carcinogenicity, carcinogenic potency TD50, and carcinogenic potency TD50Citation51, rat maximum tolerated doseCitation52,Citation53, rat oral LD50Citation54, rat chronic LOAELCitation55,Citation56, ocular irritancyCitation57, and skin irritancyCitation57 ().
Table 7. Toxicity properties of the synthesised compounds.
Except for compound 7, all compounds were predicted to be non-carcinogenic. TD50 values for compounds 6, 7, 8, 16, and 18 were 20.304, 23.695, 18.535, 22.826, and 21.184 mg/kg body weight/day, respectively, when compared to sorafenib (TD50 = 17.535 mg/kg body weight/day). Except for compound 18 (rat maximum tolerated dose = 0.132 g/kg body weight), all compounds had lower rat maximum tolerated doses (ranging from 0.042 to 0.053 g/kg body weight) than sorafenib. The rat chronic LOAEL ranged from 0.012 to 0.041 g/kg body weight, which was higher than sorafenib (0.004 g/kg body weight). Furthermore, all compounds were predicted to be non-irritants in skin irritancy models and mild irritants in ocular irritancy models.
3. Conclusion
Fourteen 1,3,4-oxadiazole-naphthalene hybrids were synthesised and tested for their in vitro antiproliferative activity against two human cancer cell lines MCF-7 and HepG-2. Compounds 5, 8, 15, 16, 17, and 18 exhibited promising cytotoxicity ranging from 8.4 to 10.4 µM, comparing to sorafenib (IC50 = 10.8 µM and 10.2 µM against MCF-7 and HepG2 cells, respectively). Compounds 5, 8, 15, 16, 17, and 18 showed VEGFR-2 inhibitory effects with concentrations of 610.4, 583.7, 864.5, 834.9, 1067, and 1004 pg/ml, respectively. SAR study revealed that substitution at the hydrophobic tail with electron-withdrawing is more beneficial than substitution with the electron-donating group for cytotoxicity. Compound 5, the most active counterpart, induced a significant increase in apoptosis (22.86% compared to 0.51% in the control) and arrested the HepG2 cell growth mostly at the Pre-G1 phase. Additionally, compound 5 exerted a significant increase in the level of caspase-3 (5.61-fold). Docking studies revealed that the new derivatives have the same binding mode of sorafenib against the VEGFR-2 active site. ADMET and toxicity studies appeared that the new members have a high degree of drug-likeness profile.
4. Experimental
4.1. Chemistry
4.1.1. General
Reagents, solvent, and apparatus used in chemical synthesis were shown in Supplementary Data. Compounds 2, 3, and 4 were prepared according to the reported proceduresCitation58,Citation59.
4.1.2. General procedure for synthesis of compounds (5–18)
The appropriate chloroacetanilide (0.7 mmol) was added to a suspension of 5-(1-(6-methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazole-2-thiol 4 (0.2 g, 0.7 mmol) and anhydrous sodium acetate (0.1 g, 1.1 mmol) in absolute ethanol (30 ml). For 2–4 h, the reaction mixture was heated under reflux. The precipitate was collected and recrystallized from absolute ethanol after cooling to room temperature to provide the desired products (5–18).
4.1.2.1. N-Benzyl-2-{[5-(1-(6-methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}acetamide (5)
Yellow solid (0.25 g–83%); mp = 192–194 °C; IR (KBr) cm−1: 3295 (NH), 3082 (CH aromatic), 2925 (CH aliphatic), 1675 (C=O amide), 1628 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:8.76 (brs, 1H, NH), 7.78–7.12 (m, 11H, Ar-H), 4.56–4.54 (m, 1H, CH3-CH), 4.22 (s, 2H, S-CH2), 4.04 (s, 2H, Ph-CH2), 3.83 (s, 3H, O-CH3), 1.66 (d, J = 4.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 170.48, 166.12, 162.66, 157.48, 139.15, 135.71, 133.93, 129.78, 128.77, 128.53, 128.0, 127.79, 127.27, 126.77, 126.26, 119.65, 106.16, 55.33, 42.71, 37.11, 36.21, 19.77; MS (m/z) 433; Anal. Calc. for: (C24H23N3O3S, Mwt = 433): C, 66.49; H, 5.35; N, 9.69; Found: C, 66.56; H, 5.41; N, 9.76%.
4.1.2.2. 2-{[5-(1-(6-Methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}-N-phenylacetamide (6)
Light yellow solid (0.23 g–80%) mp = 187–189 °C; IR (KBr) cm−1: 3300 (NH), 3075 (CH aromatic), 2922 (CH aliphatic), 1685 (C=O amide), 1633 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:10.76 (brs, 1H, NH), 7.76–7.04 (m, 11H, Ar-H), 4.55–4.54 (m, 1H, CH3-CH), 4.22 (s, 2H, S-CH2), 3.83 (s, 3H, O-CH3), 1.70 (d, J = 8.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 170.10, 166.57, 160.70, 158.27, 135.85, 133.98, 129.86, 129.75, 129.41, 129.36, 127.93, 127.70, 126.60, 126.32, 126.09, 119.65, 106.15, 55.82, 42.33, 37.11, 19.77; MS (m/z) 419; Anal. Calc. for: (C23H21N3O3S, Mwt = 419): C, 65.85; H, 5.05; N, 10.02; Found: C, 65.93; H, 5.09; N, 10.09%.
4.1.2.3. 2-{[5-(1-(6-Methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}-N-(o-tolyl)acetamide (7)
Light yellow solid (0.23 g–77%) mp = 190–192 °C; IR (KBr) cm−1: 3293 (NH), 3089 (CH aromatic), 2954 (CH aliphatic), 1685 (C=O amide), 1638 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:10.36 (brs, 1H, NH), 7.74–7.15 (m, 10H, Ar-H), 4.53–4.51 (m, 1H, CH3-CH), 4.01 (s, 2H, S-CH2), 3.81 (s, 3H, O-CH3), 1.95 (s, 3H, Ph-CH3), 1.78 (d, J = 4.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 171.77, 164.09, 157.53, 147.73, 136.45, 134.69, 133.66, 131.67, 131.23, 129.67, 129.62, 129.16, 128.74, 126.95, 125.82, 123.23, 122.12, 119.08, 106.31, 55.80, 44.0, 33.18, 18.64, 17.39; MS (m/z) 433; Anal. Calc. for: (C24H23N3O3S, Mwt = 433): C, 66.49; H, 5.35; N, 9.69; Found: C, 66.57; H, 5.43; N, 9.77%.
4.1.2.4. 2-{[5-(1-(6-Methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}-N-(m-tolyl) acetamide (8)
Light yellow solid (0.24 g–82%) mp = 193–195 °C; IR (KBr) cm−1: 3288 (NH), 3010 (CH aromatic), 2925 (CH aliphatic), 1682 (C=O amide), 1638 (C=N) 1H NMR (400 MHz, DMSO-d6) δ:10.36 (brs, 1H, NH), 7.75–7.09 (m, 10H, Ar-H), 4.96–4.93 (m, 1H, CH3-CH), 4.08 (s, 2H, S-CH2), 3.82 (s, 3H, O-CH3), 2.28 (s, 3H, Ph-CH3), 1.62 (d, J = 8.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 172.60, 169.99, 160.92, 157.47, 139.16, 136.99, 135.32, 133.54, 133.49, 129.77, 129.20, 128.89, 128.81, 128.32, 127.07, 126.64, 125.76, 119.26, 106.15, 55.82, 43.60, 33.17, 21.05, 18.88; MS (m/z) 433; Anal. Calc. for: (C24H23N3O3S, Mwt = 433): C, 66.49; H, 5.35; N, 9.69; Found: C, 66.53; H, 5.39; N, 9.73%.
4.1.2.5. 2-{[5-(1-(6-Methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}-N-(p-tolyl)acetamide (9)
Light yellow solid (0.24 g–82%) mp = 198–200 °C; IR (KBr) cm−1: 3309 (NH), 3122 (CH aromatic), 2900 (CH aliphatic), 1665 (C=O amide), 1633 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:10.49 (brs, 1H, NH), 7.83–7.01 (m, 10H, Ar-H), 4.53–4.48 (m, 1H, CH3-CH), 4.05 (s, 2H, S-CH2), 3.85 (s, 3H, O-CH3), 2.30 (s, 3H, Ph-CH3), 1.78 (d, J = 12.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 172.22, 165.94, 158.70, 157.67, 137.51, 134.05, 133.02, 130.57, 129.17, 128.45, 128.14, 127.43, 126.41, 126.01, 123.65, 119.80, 106.96, 55.92, 44.43, 34.04, 20.81, 18.37; MS (m/z) 433; Anal. Calc. for: (C24H23N3O3S, Mwt = 433): C, 66.49; H, 5.35; N, 9.69; Found: C, 66.58; H, 5.46; N, 9.73%.
4.1.2.6. N-(2,6-Dimethylphenyl)-2-{[5-(1-(6-methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}acetamide (10)
Light yellow solid (0.26 g–85%) mp = 206–208 °C; IR (KBr) cm−1: 3295 (NH), 3082 (CH aromatic), 2925 (CH aliphatic), 1675 (C=O amide), 1628 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:9.65 (brs, 1H, NH), 7.77–7.01 (m, 10H, Ar-H), 4.58–4.56 (m, 1H, CH3-CH), 4.22 (s, 2H, S-CH2), 3.83 (s, 3H, O-CH3), 2.05 (s, 6H, Ph-2CH3), 1.67 (d, J = 4.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 171.53, 166.33, 161.83, 153.10, 137.83, 134.04, 133.02, 130.18, 129.17, 128.45, 128.14, 127.75, 127.42, 126.01, 123.96, 119.47, 106.42, 55.92, 44.75, 33.33, 22.54, 18.76; MS (m/z) 447; Anal. Calc. for: (C25H25N3O3S, Mwt = 447): C, 67.09; H, 5.63; N, 9.39; Found: C, 67.16; H, 5.69; N, 9.47%.
4.1.2.7. 2-{[5-(1-(6-Methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}-N-(3-methoxyphenyl)acetamide (11)
Brownish yellow solid (0.25 g–82%) mp = 209–212 °C; IR (KBr) cm−1: 3295 (NH), 3080 (CH aromatic), 2900 (CH aliphatic), 1688 (C=O amide), 1631 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:10.37 (brs, 1H, NH), 7.76–7.06 (m, 10H, Ar-H), 4.56–4.54 (m, 1H, CH3-CH), 4.21 (s, 2H, S-CH2), 3.83 (s, 3H, O-CH3), 3.68 (s, 3H, O-CH3), 1.66 (d, J = 8.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 170.47, 165.32, 163.93, 158.81, 157.77, 140.44, 135.40, 133.93, 129.92, 129.48, 128.68, 127.71, 126.45, 126.18, 119.26, 112.01, 109.56, 106.35, 105.59, 55.74, 55.60, 42.88, 36.91, 19.71; MS (m/z) 449; Anal. Calc. for: (C24H23N3O4S, Mwt = 449): C, 64.13; H, 5.16; N, 9.35; Found: C, 64.15; H, 5.21; N, 9.39%.
4.1.2.8. N-Cyclohexyl-2-{[5-(1-(6-methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}acetamide (12)
Yellow solid (0.23 g–78%) mp = 188–191 °C; IR (KBr) cm−1: 3261 (NH), 3071 (CH aromatic), 2944 (CH aliphatic), 1681 (C=O amide), 1630 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:8.12 (brs, 1H, NH), 7.78–7.13 (m, 6H, Ar-H), 4.57–4.49 (m, 1H, CH3-CH), 4.94 (s, 2H, S-CH2), 3.83 (s, 3H, O-CH3), 1.66–1.04 (m, 14H, CH-CH3 and cyclohexyl 11H). 13C NMR (101 MHz, DMSO) δ: 170.15, 165.34, 163.72, 157.87, 135.87, 129.75, 128.88, 127.93, 126.44, 126.12, 126.0, 119.66, 106.15, 105.59, 55.78, 48.68, 36.81, 36.39, 32.50, 25.53, 24.85, 19.69; MS (m/z) 425; Anal. Calc. for: (C23H27N3O3S, Mwt = 425): C, 64.92; H, 6.40; N, 9.87; Found: C, 64.99; H, 6.47; N, 9.95%.
4.1.2.9. N-(4-Chlorophenyl)-2-{[5-(1-(6-methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}acetamide (13)
Brownish yellow solid (0.23 g–78%) mp = 214–216 °C; IR (KBr) cm−1: 3321 (NH), 3088 (CH aromatic), 2954 (CH aliphatic), 1687 (C=O amide), 1644 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:10.49 (brs, 1H, NH), 7.75–7.11 (m, 10H, Ar-H), 4.56–4.54 (m, 1H, CH3-CH), 4.21 (s, 2H, S-CH2), 3.83 (s, 3H, O-CH3), 1.65 (d, J = 8.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 173.20, 164.66, 157.86, 134.0, 131.78, 131.08, 130.50, 129.77, 129.32, 129.21, 127.91, 127.20, 126.90, 126.34, 126.07, 121.33, 119.44, 106.39, 55.69, 43.51, 37.17, 19.41; MS (m/z) 453; Anal. Calc. for: (C23H20ClN3O3S, Mwt = 453): C, 60.86; H, 4.44; N, 9.26; Found: C, 60.94; H, 4.52; N, 9.31%.
4.1.2.10. N-(2,4-Dichlorophenyl)-2-{[5-(1-(6-methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}acetamide (14)
Brownish yellow solid (0.24 g–72%) mp = 226–228 °C; IR (KBr) cm−1: 3298 (NH), 3068 (CH aromatic), 2914 (CH aliphatic), 1688 (C=O amide), 1635 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:10.58 (brs, 1H, NH), 8.36 (s, 1H, Ph-H3), 7.71–7.12 (m, 8H, Ar-H), 4.56–4.54 (m, 1H, CH3-CH), 4.24 (s, 2H, S-CH2), 3.82 (s, 3H, O-CH3), 1.69 (d, J = 8.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 170.47, 163.57, 159.60, 157.48, 152.58, 147.60, 141.82, 136.99, 134.43, 133.54, 131.76, 131.37, 129.59, 128.32, 127.04, 125.75, 118.76, 116.20, 106.15, 55.83, 44.09, 34.55, 18.88; MS (m/z) 487; Anal. Calc. for: (C23H19Cl2N3O3S, Mwt = 487): C, 56.56; H, 3.92; N, 8.60; Found: C, 56.61; H, 3.97; N, 8.64%.
4.1.2.11. N-(2,6-Dichlorophenyl)-2-{[5-(1-(6-methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}acetamide (15)
Brownish yellow solid (0.23 g–68%) mp = 223–225 °C; IR (KBr) cm−1: 3311 (NH), 3082 (CH aromatic), 2923 (CH aliphatic), 1687 (C=O amide), 1630 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:10.57 (brs, 1H, NH), 7.71–7.10 (m, 9H, Ar-H), 4.62–4.57 (m, 1H, CH3-CH), 4.34 (s, 2H, S-CH2), 3.79 (s, 3H, O-CH3), 1.38 (d, J = 12.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 170.28, 160.10, 157.38, 137.44, 134.26, 133.75, 132.52, 129.79, 129.54, 128.84, 128.77, 127.13, 126.83, 125.88, 125.37, 119.24, 106.17, 55.46, 43.61, 33.0, 18.77; MS (m/z) 487; Anal. Calc. for: (C23H19Cl2N3O3S, Mwt = 487): C, 56.56; H, 3.92; N, 8.60; Found: C, 56.59; H, 3.97; N, 8.64%.
4.1.2.12. 2-{[5-(1-(6-Methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}-N-(2-nitrophenyl) acetamide (16)
Yellow solid (0.19 g–60%) mp = 228–230 °C; IR (KBr) cm−1: 3333 (NH), 3069 (CH aromatic), 2974 (CH aliphatic), 1688 (C=O amide), 1628 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:10.22 (brs, 1H, NH), 8.0–7.11 (m, 10H, Ar-H), 4.54–4.52 (m, 1H, CH3-CH), 4.25 (s, 2H, S-CH2), 3.82 (s, 3H, O-CH3), 1.56 (d, J = 8.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 170.50, 166.33, 162.86, 159.0, 157.28, 153.81, 146.16, 134.04, 133.33, 131.92, 130.58, 129.48, 127.12, 126.40, 123.96, 118.76, 116.31, 106.95, 55.54, 43.41, 33.64, 18.77; MS (m/z) 464; Anal. Calc. for: (C23H20N4O5S, Mwt = 464): C, 59.47; H, 4.34; N, 12.06; Found: C, 59.54; H, 4.41; N, 12.11%.
4.1.2.13. 2-{[5-(1-(6-Methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}-N-(4-nitrophenyl) acetamide (17)
Yellow solid (0.20 g–62%) mp = 232–234 °C; IR (KBr) cm−1: 3298 (NH), 3060 (CH aromatic), 2931 (CH aliphatic), 1671 (C=O amide), 1632 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:10.37 (brs, 1H, NH), 8.0 (d, J = 8.0 Hz, 2H, Ph-H3,H5), 7.76–7.10 (m, 8H, Ar-H), 4.52–4.48 (m, 1H, CH3-CH), 4.27 (s, 2H, S-CH2), 3.84 (s, 3H, O-CH3), 1.64 (d, J = 8 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 173.26, 167.36, 163.58, 159.0, 154.83, 150.35, 137.12, 134.04, 133.02, 130.19, 129.16, 128.45, 126.41, 125.71, 123.97, 119.08, 106.56, 55.53, 43.27, 33.65, 18.76; MS (m/z) 464; Anal. Calc. for: (C23H20N4O5S, Mwt = 464): C, 59.47; H, 4.34; N, 12.06; Found: C, 59.56; H, 4.40; N, 12.11%.
4.1.2.14. N-(2-Hydroxy-4-nitrophenyl)-2-{[5-(1-(6-methoxynaphthalen-2-yl)ethyl)-1,3,4-oxadiazol-2-yl]thio}acetamide (18)
Yellow solid (0.20 g–62%) mp = 237–240 °C; IR (KBr) cm−1: 3435 (OH), 3300 (NH), 3075 (CH aromatic), 2922 (CH aliphatic), 1685 (C=O amide), 1633 (C=N); 1H NMR (400 MHz, DMSO-d6) δ:10.41 (brs, 1H, OH), 10.29 (brs, 1H, NH), 8.29–7.14 (m, 9H, Ar-H), 4.27–4.25 (m, 1H, CH3-CH), 4.24 (s, 2H, S-CH2), 3.83 (s, 3H, O-CH3), 1.66 (d, J = 12.0 Hz, 3H, CH-CH3). 13C NMR (101 MHz, DMSO) δ: 172.24, 166.33, 162.16, 160.11, 153.49, 147.20, 134.36, 133.02, 132.63, 129.87, 129.16, 128.46, 128.13, 127.74, 127.43, 126.41, 123.97, 119.48, 106.57, 55.53, 43.72, 33.33, 18.76; MS (m/z) 480; Anal. Calc. for: (C23H20N4O6S, Mwt = 480): C, 57.49; H, 4.20; N, 11.66; Found: C, 57.57; H, 4.28; N, 11.72%.
4.2. Biological testing
4.2.1. Cell culture
The MCF-7, HepG2, and THLE-2 cell lines were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). MCF-7 and HepG2 cells were grown in a medium that contains Roswell Park Memorial Institute (RPMI) 1640 with L-glutamine, 10% foetal bovine serum (FBS), streptomycin (100 μg/ml) and penicillin (100 U/ml). While THLE-2 cells were cultivated in Bronchial Epithelial Growth Media (BEGM) with 10% FBS, 5 ng/ml EGF, and 70 ng/ml phosphoethanolamine. All cells were maintained in a humidified incubator and 5% (v/v) CO2 atmosphere at 37 °CCitation60.
4.2.2. In vitro cytotoxic activity
The MCF-7, HepG2, and THLE-2 cells were tested for cytotoxicity using the 3–(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assayCitation61–65. The ability of living cells to reduce the yellow product MTT to a blue product, formazan, via a reduction reaction that occurs in the mitochondria is used to assess cell population growth. In this assay, 5000 cells/well were plated in a 96-well plate and allowed to grow for 24 h before being treated with over-mentioned suitable media containing increased concentrations of tested compounds (0, 0.1, 1, 10, 100, and 1000 µM). Each experiment was conducted in triplicate. After removing the media, 100 µL of MTT was added to each well and incubated for 4 h. Following that, 100 µL of dimethyl sulfoxide (DMSO) solution was added to solubilise the resulting formazan product, and absorbance at 570 nm was measured using an ELISA microplate reader (Epoc-2 C micro-plate reader, Bio Tek, VT, USA). The IC50 values [the concentration required to inhibit cell viability by 50%] were calculated.
4.2.3. Assessment of VEGFR-2 level
In vitro VEGFR-2 concentration was evaluated using Enzyme-Linked Immunosorbent Assay (ELISA) kit (Cat. NO. EK0544) (AVIVA System Biology, USA) according to manufacturer instructionsCitation66.
4.2.4. Cell cycle analysis using flow cytometry
The effect of compound 5 on the cell cycle phases was investigated using propidium iodide staining and flow cytometric analysis according to the cell cycle kit (PN C03551). Briefly, HepG2 cells were allowed to grow in 25 cm3 flask, then treated with compound 5 for 48 h. After that, the cells were harvested and cell fixation was performed, cells were centrifuged at 2000 rpm for 5 min then, the supernatant was aspirated. The pellet of fixed cells was resuspended in a 0.5 ml cell cycle kit, vortexed, and incubated at 25 °C for 15 min. Finally, DNA was stained with 50 µg/ml propidium iodide for 30 min. Flow cytometric analysis of cell cycle performed on a COULTER® EPICS® XL™ Flow Cytometer (USA)Citation67–70.
4.2.5. Flow cytometric analysis for detection of apoptosis
To assess the effect of compound 5 on cell apoptosis, Annexin V–FITC Kit was used according to the kit protocol (PN IM3546), followed by flow cytometric analysis. In brief, HepG2 cells were allowed to grow in 25 cm3 flask, after that treated for 48 h. then, washed in phosphate-buffered saline and suspended to 5 × 105–5 × 106 cells/mL in 1X binding buffer. Then we added to 100 µL of the cell suspensions, 5 µL of dissolved PI, and 1 µL of annexin VFITC solution and incubated in the dark for 15 min. Next to that, we added 400 µL of ice-cold 1X binding buffer and mixed gently. Apoptotic cells were determined by flow cytometric analysis on a COULTER® EPICS® XL™ Flow Cytometer (USA)Citation67,Citation71,Citation72.
4.2.6. Caspase-3 determination
The effect of compound 5 on the Caspase-3 level was assessed using an ELISA kit (Catalog # KHO1091) according to manufacturer instructions.
4.3. In silico studies
4.3.1. Docking studies
Docking studies were performed on VEGFR-2 [PDB ID: PDB ID: 2OH4, resolution: 2.05] using the reported procedureCitation45,Citation73–76 as described in Supplementary Data.
4.3.2. ADMET studies
The ADMET descriptors were calculated using Discovery Studio 4.0 in accordance with the reported methodCitation73,Citation74,Citation77 (Supplementary Data).
4.3.3. Toxicity studies
The toxicity potential of the synthesised compounds was predicted using the Discovery studio 4.0 software, as reported in Supplementary DataCitation78.
Supplemental Material
Download PDF (12.4 MB)Acknowledgements
The authors extend their appreciation to the Research center at AlMaarefa University for funding this work under TUMA project number "TUMA-2021-4"
Disclosure statement
No potential conflict of interest was reported by the authors.
Correction Statement
This article was originally published with errors, which have now been corrected in the online version. Please see Correction (10.1080/14756366.2022.2024999).
Additional information
Funding
References
- Baselga J, Bhardwaj N, Cantley LC, et al. AACR cancer progress report 2015. Clin Cancer Res 2015;21:S1–S128.
- WHO, Cancer. https://www.who.int/health-topics/cancer#tab=tab_1. (Accessed May 2020 2020).
- Thurston DE. Chemistry and pharmacology of anticancer drugs. 2nd ed. London: CRC Press, Tylor and Francis; 2006.
- Nikolaou M, Pavlopoulou A, Georgakilas AG, Kyrodimos E. The challenge of drug resistance in cancer treatment: a current overview. Clin Exp Metastasis 2018;35:309–18.
- El-Zahabi MA, Sakr H, El-Adl K, et al. Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents. Bioorg Chem 2020;104:104218.
- Nasser AA, Eissa IH, Oun MR, et al. Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFRWT and EGFRT790M. Org Biomol Chem 2020;18:7608–34.
- Eldehna WM, Abo-Ashour MF, Nocentini A, et al. Novel 4/3-((4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylidene)amino) benzenesulfonamides: synthesis, carbonic anhydrase inhibitory activity, anticancer activity and molecular modelling studies. Eur J Med Chem 2017;139:250–62.
- El-Adl K, El-Helby A-GA, Sakr H, et al. Design, synthesis, molecular docking and anticancer evaluations of 5-benzylidenethiazolidine-2,4-dione derivatives targeting VEGFR-2 enzyme. Bioorg Chem 2020;102:104059.
- Eissa IH, El-Helby A-GA, Mahdy HA, et al. Discovery of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors: design, synthesis, and anti-proliferative evaluation. Bioorg Chem 2020;105:104380.
- Shibuya M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis. Angiogenesis 2006;9:225–30.
- Takahashi S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biol Pharma Bull 2011;34:1785–8.
- Flister MJ, Wilber A, Hall KL, et al. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1. J Am Soc Hematol 2010;115:418–29.
- El‐Adl K, Ibrahim MK, Khedr F, et al. N‐Substituted‐4‐phenylphthalazin‐1‐amine‐derived VEGFR‐2 inhibitors: design, synthesis, molecular docking, and anticancer evaluation studies. Archiv Pharma 2021;354:2000219.
- Padro T, Bieker R, Ruiz S, et al. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia. Leukemia 2002;16:1302–10.
- Yang C, Qin S. Apatinib targets both tumor and endothelial cells in hepatocellular carcinoma. Cancer Med 2018;7:4570–83.
- Guo S, Colbert LS, Fuller M, et al. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim Biophys Acta 2010;1806:108–21.
- Martins SF, Garcia EA, Luz MAM, et al. Clinicopathological correlation and prognostic significance of VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3 expression in colorectal cancer. Cancer Genomics Proteomics 2013;10:55–67.
- Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal 2007;19:2003–12.
- Kaipainen A, Korhonen J, Mustonen T, et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci USA 1995;92:3566–70.
- Tammela T, Zarkada G, Wallgard E, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008;454:656–60.
- Peng F-W, Liu D-K, Zhang Q-W, et al. VEGFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012–2016). Expert Opin Ther Pat 2017;27:987–1004.
- Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol 2007;25:884–96.
- Song Y, Wang J, Ren X, et al. Vorolanib, an oral VEGFR/PDGFR dual tyrosine kinase inhibitor for treatment of patients with advanced solid tumors: an open-label, phase I dose escalation and dose expansion trial. Chin J Cancer Res 2021;33:103–14.
- Jamil MO, Hathaway A, Mehta A. Tivozanib: status of development. Curr Oncol Rep 2015;17:24.
- Xie Q-Q, Xie H-Z, Ren J-X, et al. Pharmacophore modeling studies of type I and type II kinase inhibitors of Tie2. J Mol Graph Model 2009;27:751–8.
- Lee K, Jeong K-W, Lee Y, et al. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur J Med Chem 2010;45:5420–7.
- Eskander RN, Tewari KS. Incorporation of anti-angiogenesis therapy in the management of advanced ovarian carcinoma-mechanistics, review of phase III randomized clinical trials, and regulatory implications. Gynecol Oncol 2014;132:496–505.
- Eissa IH, Ibrahim MK, Metwaly AM, et al. Design, molecular docking, in vitro, and in vivo studies of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors with potential activity against hepatocellular carcinoma. Bioorg Chem 2021;107:104532.
- El-Metwally SA, Abou-El-Regal MM, Eissa IH, et al. Discovery of thieno[2,3-d]pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg Chem 2021;112:104947.
- Alsaif NA, Dahab MA, Alanazi MM, et al. New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: design, molecular modeling, and synthesis. Bioorg Chem 2021;110:104807.
- Machado VA, Peixoto D, Costa R, et al. Synthesis, antiangiogenesis evaluation and molecular docking studies of 1-aryl-3-[(thieno[3,2-b]pyridin-7-ylthio)phenyl]ureas: discovery of a new substitution pattern for type II VEGFR-2 Tyr kinase inhibitors. Bioorg Med Chem 2015;23:6497–509.
- Wang Z, Wang N, Han S, et al. Dietary compound isoliquiritigenin inhibits breast cancer neoangiogenesis via VEGF/VEGFR-2 signaling pathway. PLOS One 2013;8:e68566.
- Dietrich J, Hulme C, Hurley LH. The design, synthesis, and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: a structural analysis of the binding interactions of Gleevec, Nexavar, and BIRB-796. Bioorg Med Chem 2010;18:5738–48.
- Mahdy HA, Ibrahim MK, Metwaly AM, et al. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4(3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorg Chem 2020;94:103422.
- El‐Helby AGA, Sakr H, Eissa IH, et al. Benzoxazole/benzothiazole‐derived VEGFR‐2 inhibitors: design, synthesis, molecular docking, and anticancer evaluations. Archiv Pharma 2019;352:1900178.
- El‐Helby AGA, Sakr H, Eissa IH, et al. Design, synthesis, molecular docking, and anticancer activity of benzoxazole derivatives as VEGFR‐2 inhibitors. Archiv Pharm 2019;352:1900113.
- Alanazi MM, Mahdy HA, Alsaif NA, et al. New bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: design, synthesis, in silico studies, and anticancer evaluation. Bioorg Chem 2021;112:104949.
- Alanazi MM, Eissa IH, Alsaif NA, et al. Design, synthesis, docking, ADMET studies, and anticancer evaluation of new 3-methylquinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers. J Enzyme Inhib Med Chem 2021;36:1760–82.
- Parmar DR, Soni JY, Guduru R, et al. Discovery of new anticancer thiourea-azetidine hybrids: design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorg Chem 2021;115:105206.
- Abadi AH, Eissa AAH, Hassan GS. Synthesis of novel 1,3,4-trisubstituted pyrazole derivatives and their evaluation as antitumor and antiangiogenic agents. Chem Pharm Bull 2003;51:838–44.
- Vaidya A, Pathak D, Shah K. 1,3,4-oxadiazole and its derivatives: a review on recent progress in anticancer activities. Chem Biol Drug Des 2021;97:572–91.
- Wang G, Peng Z, Zhang J, et al. Synthesis, biological evaluation and molecular docking studies of aminochalcone derivatives as potential anticancer agents by targeting tubulin colchicine binding site. Bioorg Chem 2018;78:332–40.
- Wang G, Liu W, Gong Z, et al. Synthesis, biological evaluation, and molecular modelling of new naphthalene-chalcone derivatives as potential anticancer agents on MCF-7 breast cancer cells by targeting tubulin colchicine binding site. J Enzyme Inhib Med Chem 2020;35:139–44.
- Ibrahim M, Taghour M, Metwaly A, et al. Design, synthesis, molecular modeling and anti-proliferative evaluation of novel quinoxaline derivatives as potential DNA intercalators and topoisomerase II inhibitors. Eur J Med Chem 2018;155:117–34.
- Ibrahim MK, Eissa IH, Abdallah AE, et al. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of novel quinoxaline derivatives as potential PPARγ and SUR agonists. Bioorg Med Chem 2017;25:1496–513.
- Hou T, Zhu L, Chen L, Xu X. Mapping the binding site of a large set of quinazoline type EGF-R inhibitors using molecular field analyses and molecular docking studies. J Chem Inform Comput Sci 2003;43:273–87.
- Bhinge SD, Chature V, Sonawane LV. Synthesis of some novel 1,3,4-thiadiazole derivatives and biological screening for anti-microbial, antifungal and anthelmintic activity. Pharma Chem J 2015;49:367–72.
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2006;2:358–64.
- Xia X, Maliski EG, Gallant P, Rogers D. Classification of kinase inhibitors using a Bayesian model. J Med Chem 2004;47:4463–70.
- BIOVIA, QSAR, ADMET and Predictive Toxicology.https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html. (Accessed May 2020).
- Gold L S, Slone T H, Bernstein L J E H P, Summary of carcinogenic potency and positivity for 492 rodent carcinogens in the carcinogenic potency database, (1989);79:259-272.
- Goodman G, Wilson R. Comparison of the dependence of the TD50 on maximum tolerated dose for mutagens and nonmutagens. Risk Anal 1992;12:525–33.
- Haseman J, Seilkop S J F, Toxicology A, An examination of the association between maximum-tolerated dose and carcinogenicity in 326 long-term studies in rats and mice, (1992);19(2):207-213.
- Diaza RG, Manganelli S, Esposito A, et al. Comparison of in silico tools for evaluating rat oral acute toxicity. SAR QSAR Environ Res 2015;26:1–27.
- Pizzo F, Benfenati E. In silico models for repeated-dose toxicity (RDT): prediction of the no observed adverse effect level (NOAEL) and lowest observed adverse effect level (LOAEL) for drugs. In: In silico methods for predicting drug toxicity. Switzerland AG: Springer; 2016:163–76.
- Venkatapathy R, Moudgal CJ, Bruce RM. Assessment of the oral rat chronic lowest observed adverse effect level model in TOPKAT, a QSAR software package for toxicity prediction. J Chem Inform Comput Sci 2004;44:1623–9.
- Wilhelmus KR. The Draize eye test. Surv Ophthalmol 2001;45:493–515.
- Han Mİ, Bekçi H, Uba AI, et al. Synthesis, molecular modeling, in vivo study, and anticancer activity of 1,2,4‐triazole containing hydrazide–hydrazones derived from (S)‐naproxen. Arch Pharm 2019;352:1800365.
- El-Husseiny WM, Magda A-A, Abdel-Aziz NI, et al. Structural alterations based on naproxen scaffold: synthesis, evaluation of antitumor activity and COX-2 inhibition, and molecular docking. Eur J Med Chem 2018;158:134–43.,
- Sefried S, Häring H-U, Weigert C, Eckstein SS. Suitability of hepatocyte cell lines HepG2, AML12 and THLE-2 for investigation of insulin signalling and hepatokine gene expression. Open Biol 2018;8:180147.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986;89:271–7.
- Thabrew MI, Hughes RD, McFarlane IG. Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay. J Pharma Pharmacol 2011;49:1132–5.
- Al-Rashood ST, Hamed AR, Hassan GS, et al. Antitumor properties of certain spirooxindoles towards hepatocellular carcinoma endowed with antioxidant activity. J Enzyme Inhib Med Chem 2020;35:831–9.
- Ismail A, Doghish AS, Elsadek BE, et al. Hydroxycitric acid potentiates the cytotoxic effect of tamoxifen in MCF-7 breast cancer cells through inhibition of ATP citrate lyase. Steroids 2020;160:108656.
- Sharma K, Suresh P, Mullangi R, Srinivas N. Quantitation of VEGFR2 (vascular endothelial growth factor receptor) inhibitors-review of assay methodologies and perspectives. Biomed Chromatogr 2015;29:803–34.
- El-Mahdy HA, El-Husseiny AA, Kandil YI, El-Din AMG. Diltiazem potentiates the cytotoxicity of gemcitabine and 5-fluorouracil in PANC-1 human pancreatic cancer cells through inhibition of P-glycoprotein. Life Sci 2020;262:118518.
- Kassab AE, Gedawy EM, Hamed MI, et al. Design, synthesis, anticancer evaluation, and molecular modelling studies of novel tolmetin derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. J Enzyme Inhib Med Chem 2021;36:922–39.
- Eldehna WM, Hassan GS, Al-Rashood ST, et al. Synthesis and in vitro anticancer activity of certain novel 1-(2-methyl-6-arylpyridin-3-yl)-3-phenylureas as apoptosis-inducing agents. J Enzyme Inhib Med Chem 2019;34:322–32.,
- Al-Warhi T, Abo-Ashour MF, Almahli H, et al. Novel [(N-alkyl-3-indolylmethylene)hydrazono]oxindoles arrest cell cycle and induce cell apoptosis by inhibiting CDK2 and Bcl-2: synthesis, biological evaluation and in silico studies. J Enzyme Inhib Med Chem 2020;35:1300–9.
- Sabt A, Abdelhafez OM, El-Haggar RS, et al. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: synthesis, in vitro biological evaluation, and QSAR studies. J Enzyme Inhib Med Chem 2018;33:1095–107.
- Al-Sanea MM, Al-Ansary GH, Elsayed ZM, et al. Development of 3-methyl/3-(morpholinomethyl)benzofuran derivatives as novel antitumor agents towards non-small cell lung cancer cells. J Enzyme Inhib Med Chem 2021;36:987–99.
- El-Zahabi MA, Elbendary ER, Bamanie FH, et al. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of phthalimide-sulfonylurea hybrids as PPARγ and SUR agonists. Bioorg Chem 2019;91:103115.
- Ibrahim MK, Eissa IH, Alesawy MS, et al. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of quinazolin-4(3H)-one derivatives as potential PPARγ and SUR agonists. Bioorg Med Chem 2017;25:4723–44.
- El-Gamal KM, El-Morsy AM, Saad AM, et al. Synthesis, docking, QSAR, ADMET and antimicrobial evaluation of new quinoline-3-carbonitrile derivatives as potential DNA-gyrase inhibitors. J Mol Struct 2018;1166:15–33.
- El-Naggar AM, Eissa IH, Belal A, El-Sayed AA. Design, eco-friendly synthesis, molecular modeling and anticancer evaluation of thiazol-5(4H)-ones as potential tubulin polymerization inhibitors targeting the colchicine binding site. RSC Adv 2020;10:2791–811.
- El-Demerdash A, Metwaly AM, Hassan A, et al. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against SARS-CoV-2 (COVID-19). Biomolecules 2021;11:460.
- Eissa IH, Dahab MA, Ibrahim MK, et al. Design and discovery of new antiproliferative 1,2,4-triazin-3(2H)-ones as tubulin polymerization inhibitors targeting colchicine binding site. Bioorg Chem 2021;112:104965.