
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In continuation of our previous studies to optimise potent carbonic anhydrase inhibitors, two new series of isatin N-phenylacetamide based sulphonamides were synthesised and screened for their human (h) carbonic anhydrase (EC 4.2.1.1) inhibitory activities against four isoforms hCA I, hCA II, hCA IX and hCA XII. The indole-2,3-dione derivative 2h showed the most effective inhibition profile against hCAI and hCA II (KI = 45.10, 5.87 nM) compared to acetazolamide (AAZ) as standard inhibitor. Moreover, 2h showed appreciable inhibition activity against the tumour-associated hCA XII, similar to AAZ showing KI of 7.91 and 5.70 nM, respectively. The analogs 3c and 3d showed good cytotoxicity effects, and 3c revealed promising selectivity towards lung cell line A549. Molecular docking was carried out for 2h and 3c to predict their binding conformations and affinities towards the hCA I, II, IX and XII isoforms.
Graphical abstract

1. Introduction
The simple and fundamental interconversion of crucial ion species such as carbon dioxide and bicarbonate (CO2/HCO3−) occurs during biochemical processes in all living organisms. The CO2/HCO3- interconversion is catalysed by a superfamily of metalloenzymes called carbonic anhydrases (CAs, EC 4.2.1.1). The enzymes belonging to the α-CA family tightly coordinate a Zn2+ ion within their active sites, that interacts with a water molecule and histidine residues in a tetrahedral geometry of the metal ion. The zinc ion binds and activates a water molecule to catalyse the hydration reaction of carbon dioxide to bicarbonate and proton ions by a metal hydroxide nucleophilic mechanism. Consequently, CAs act among the major physiological buffering systemsCitation1–4.
Human CAs (hCAs) are represented by 15 isoforms which include the cytosolic (I, II, III, VII, XIII), mitochondrial (VA, VB), secreted (VI), and membrane associated (IV, IX, XII, XIV) in addition to the catalytic isoforms of carbonic anhydrase related proteins located in cytosol (VIII, X, XI)Citation5–8. Fluctuation in CA expression levels can lead to hypertension, glaucoma, epilepsy, neuropathic pain, cancer, etc. Accordingly, many hCA isoforms are considered as attractive therapeutic targets for many pharmacological applicationsCitation9,Citation10. Recently, CA inhibitors (CAIs) were adopted as a combination therapy for solid tumours upon acknowledging the overexpression of the membrane-associated isoenzymes hCA IX and hCA XII in that kind of tumours. Those isoenzymes were discovered to play pivotal role in microenvironment pH regulation thus being important in the survival, proliferation, metastasis and development of drug resistance of malignant cellsCitation11,Citation12.
Known CAIs are generally primary aromatic or heteroaromatic sulphonamides in which the sulphonamide moiety coordinates the Zn+2 ion in the enzyme active site with their terminal deprotonated nitrogen atom simulating the known non-selective CAI Acetazolamide (AAZ) I ()Citation13–15. Sulphonamide based CAIs have versatile pharmacological activities such as diureticCitation16, antiglaucomaCitation17, antiepilepticCitation18, antimicrobialCitation19, anti-inflammatoryCitation20, antioxidantCitation21 and anticancerCitation22–24. These activities made them attractive candidates for developing novel agents with varying biological activities through minor structural alterationsCitation9,Citation10.
Figure 1. Chemical structures of AAZ (I), Sunitinib (II) and reported CAIs incorporating sulphonamide moieties III-VIII.
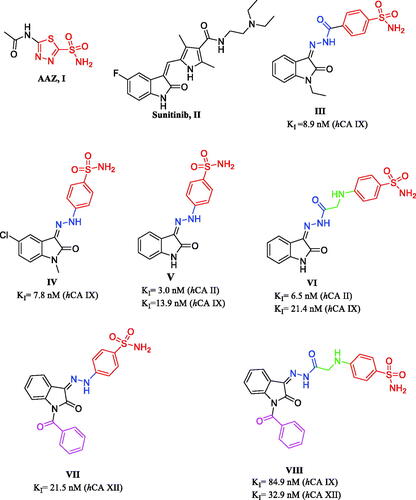
Moreover, isatin (indole-2,3-dione) is considered as a crucial core in many anticancer drugs in the market as sunitinib IICitation25. Furthermore, isatin-benzenesulfonamide conjugates were reported to have promising anticancer activity as well as carbonic anhydrase inhibitionCitation26–28. For example, 4-(2-(1-ethyl-2-oxoindolin-3-ylidene)hydrazine-1-carbonyl)benzenesulfonamide (III) revealed KI = 8.9 nM against hCA IXCitation26. Additionally, isatin hydrazinyl benzenesulfonamide derivative IV exhibited KI= 7.8 nM against hCA IX isoformCitation27. Based on this knowledge, the present study was designed to develop potent and selective isatin-based sulphonamide CAIs utilising the “tail approach”. This approach investigated the effect of adding different molecular fragments to the isatin scaffold on the binding interaction with the enzyme active site which in turn would modulate the potency and selectivityCitation29,Citation30.
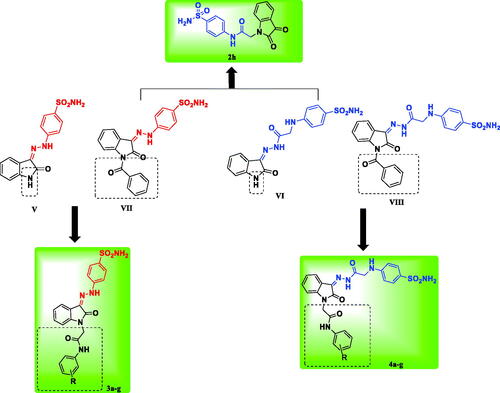
Previously reported unsubstituted isatin Schiff’s bases V and VI showed promising CA inhibitory activityCitation29 (), then the developed N-benzoyl candidates showed high potency and selectivity towards hCA XII (compounds VII, VIII)Citation30 (). Motivated by these results, we developed here new isatin-based sulphonamide candidates by incorporation of phenylacetamide moiety at the isatin nitrogen as a new structural modification. The attached phenylacetamide moiety was evaluated as unsubstituted as well as substituted by groups of diverse size, lipophilicity, and electronic property (such as Cl, Br, CH3, OCH3, SO2NH2) to investigate the interaction of the new adopted tail fragment with the CA active site. Additionally, the isatin scaffold was hybridised with the benzene sulphonamide through a hydrazinyl (compounds 3a–g), or hydrazinyl-2-oxoethylamino spacers (compounds 4a–g) in order to study the effect of this structure elongation along with the tail fragment on the CA inhibitory activity and selectivity (). All new synthesised candidates (2h, 3a–g, 4a–g) were screened for their CA inhibitory activity against four hCA isoforms (I, II, IX, and XII), and the most active ones were selected for further in vitro evaluation for their anti-proliferative activity. Furthermore, to rationalise their significant in vitro biological results, molecular docking simulation was carried out for the most active derivatives to study their binding pattern and affinity to the hCA II, IV, IX, and XII active sites. Furthermore, 2D-QSAR study was performed using all reported derivatives of our previous workCitation29,Citation30 in addition to the newly synthesised derivatives to obtain two validated models for the tumour associated isoforms hCA IX and XII elucidating the descriptors controlling the activity, therefore they can be used for prediction of activity of further isatin-based derivatives.
Figure 2. The rational of the newly synthesised isatin-based sulphonamide derivatives 2h, 3a-g and 4a-g.
2. Experimental
2.1. Chemistry
All chemicals were purchased from VWR International Merck, or Sigma-Aldrich, Germany. Melting points are uncorrected and were measured by open capillary tube method using Stuart SMP3 melting point apparatus. Elemental microanalyses were carried out at the Regional Centre for Mycology and Biotechnology, Al-Azhar University. Infra-red spectra were recorded on Shimadzu spectrometer IR Affinity-1 (FTIR- 8400S-Kyoto-Japan) and expressed in wave number (cm−1). 1H NMR and 13 C NMR Spectra were recorded on Bruker High Performance Digital FT-NMR Spectrometer Avance III 400 MHz, 13 C, 100 MHz NMR spectrometer. Chemical shifts were expressed in δ units and were related to that of the solvents. Mass Spectra were recorded using ISQLT Thermo Scientific Mass spectrometer at The Regional Centre for Mycology and Biotechnology, Al-Azhar University. All the reactions were monitored by TLC using silica gel F254 plates (Sigma-Aldrich), using chloroform: methanol 9.5:0.5 as eluting system and were visualised by UV-lamp. Compounds 1a–gCitation31,Citation32, 2a–fCitation33–35, and the reagents 4-hydrazinylbenzenesulfonamide hydrochlorideCitation36 and 4-(2-hydrazinyl-2-oxoethylamino)benzenesulfonamideCitation37 were prepared according to their reported methods.
2.1.1. General procedure for the synthesis of compounds 2a-h
A mixture of isatin (indole-2,3-dione) (0.29 g, 2 mmol) and potassium carbonate (0.82 g, 6 mmol) was stirred for 15 min. in DMF (5 ml), then an appropriate 2-chloro-N-phenylacetamide (1a-h) (2 mmol) was added. The reaction was stirred at room temperature for 4 h, then poured on ice water, the precipitated solid was filtered, washed with water and crystallised form methanol.
2.1.1.1. N-(2-Bromophenyl)-2–(2,3-dioxoindolin-1-yl)acetamide (2 g)
Yellow crystals, yield 80%, m.p. 221–222 °C; IR (KBr, ʋ cm−1): 3244 (NH), 3036 (CH aromatic), 2931 (CH aliphatic), 1736, 1670 (3 C=O), 1612 (NH bending), 1589, 1531 (C=C); 1H NMR (CDCl3, 400 MHz): δ 4.62 (s, 2H, NCH2), 7.17–7.20 (m, 3H, aromatic H), 7.38 (t, 1H, J = 7.64 Hz, aromatic H), 7.50 (d, J = 7.04 Hz, 1H, aromatic H), 7.63 (d, J = 6.60 Hz, 1H, aromatic H), 7.66–7.73 (m, 2H, aromatic H), 9.94 (s, 1H, NH, D2O exchangeable); 13 C NMR (CDCl3, 100 MHz): δ 44.7 (CH2), 110.8, 117.8, 122.1, 124.7, 125.8, 126.1, 128.5, 132.4, 134.6, 138.8, 149.9 (aromatic carbons), 158.3, 163.8, 181.9 (3 C=O); MS, m/z [%]: 361.10 [M++2, 3.27], 359.10 [M+, 3.87]; Anal. Calcd. for C16H11BrN2O3 (359.17): C, 53.50; H, 3.09; N, 7.80. Found: C, 53.74; H, 3.25; N, 8.07.
2.1.1.2. 2–(2,3-Dioxoindolin-1-yl)-N-(4-sulfamoylphenyl)acetamide (2 h)
Yellow orange crystals, yield 78%, m.p. 237–238 °C; IR (KBr, ʋ cm−1): 3325, 3267, 3205 (NH2, NH), 3067 (CH aromatic), 2935 (CH aliphatic), 1740, 1697 (3 C=O), 1612 (NH bending), 1597, 1543 (C=C), 1308, 1153 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 4.63 (s, 2H, NCH2), 7.16–7.18 (m, 2H, aromatic H), 7.28 (s, 2H, NH2, D2O exchangeable), 7.62–7.73 (m, 4H, aromatic H), 7.78 (d, J = 8.64 Hz, 2H, aromatic H), 10.59 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 43.7 (CH2), 111.5, 118.0, 119.6, 124.0, 125.0, 127.2, 138.8, 139.4, 141.7, 151.3 (aromatic carbons), 158.9, 166.0, 183.4 (C=O); MS, m/z [%]: 360.15 [M++1, 5.37], 359.20 [M+, 22.48]; Anal. Calcd. for C16H13N3O5S (359.36): C, 53.48; H, 3.65; N, 11.69. Found: C, 53.69; H, 3.80; N, 11.88.
2.1.2. General procedure for the synthesis of compounds 3a–g
A mixture of equimolar amounts of (2a–g) (1 mmol) and 4-hydrazinylbenzenesulfonamide hydrochloride (0.22 g, 1 mmol) in methanol (5 ml) was stirred at room temperature for 4 h. The obtained solid was filtered, washed with ethanol and recrystallized from ethanol.
2.1.2.1. 2–(2-Oxo-3–(2-(4-sulfamoylphenyl)hydrazono)indolin-1-yl)-N-phenylacetamide (3a)
Yellow crystals, yield 78%, m.p. 294–295 °C; IR (KBr, ʋ cm−1): 3364, 3329 (NH2), 3197, 3140 (2NH), 3059 (CH aromatic), 2974 (CH aliphatic), 1700, 1678 (2 C=O), 1605 (NH bending), 1570, 1551 (C=C), 1300, 1157 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 4.65, 4.70 (2 s, 2H, NCH2), 7.05–7.10, 7.13–7.19 (2 m, 3H, aromatic H), 7.25, 7.27 (2 s, 2H, NH2, D2O exchangeable), 7.31–7.36, 7.37–7.43 (2 m, 4H, aromatic H), 7.59, 7.68 (2d, J = 7.72 Hz, 2H, aromatic H), 7.62, 7.68 (2d, J = 7.72 Hz, 2H, aromatic H), 7.82, 7.84 (2d, J = 7.30 Hz, 2H, aromatic H), 10.34, 10.38 (2 s, 1H, NH, D2O exchangeable), 10.86, 12.68 (2 s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 43.1 (NCH2), 109.7, 114.5, 115.1, 119.4, 122.3, 123.1, 124.1, 127.6, 129.3, 129.7, 138.3, 139.0, 141.9, 145.6 (aromatic carbons), 161.7, 165.5 (C=O); MS, m/z [%]: 449 [M+, 40]; Anal. Calcd. for C22H19N5O4S (449.48): C, 58.79; H, 4.26; N, 15.58. Found: C, 59.03; H, 4.42; N, 15.79.
2.1.2.2. N-(4-Chlorophenyl)-2–(2-oxo-3–(2-(4-sulfamoylphenyl)hydrazono)indolin-1-yl)acetamide (3 b)
Yellow crystals, yield 75%, m.p. 297–298 °C; IR (KBr, ʋ cm−1): 3282 (NH2, 2 NH), 3067 (CH aromatic), 2932 (CH aliphatic), 1693, 1674 (2 C=O), 1601 (NH bending), 1570, 1539 (C=C), 1331, 1153 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 4.65, 4.70 (2 s, 2H, NCH2), 7.09–7.14, 7.16–7.20 (2 m, 4H, aromatic H), 7.24, 7.26 (2 s, 2H, NH2, D2O exchangeable), 7.61–7.64, 7.66–7.69 (2 m, 4H, aromatic H), 7.81–7.85 (m, 3H, aromatic H), 8.31 (d, J = 7.68 Hz, 1H, aromatic H), 10.49, 10.86 (2 s, 1H, NH, D2O exchangeable), 10.52, 12.67 (2 s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 43.0, 43.3 (NCH2), 109.7, 110.1, 114.5, 115.1, 116.0, 119.5, 120.6, 121.2, 121.3, 122.4, 123.2, 124.8, 127.6, 127.7, 128.0, 129.1, 129.2, 129.7, 131.1, 137.9, 138.0, 138.3, 141.8, 143.3, 145.6, 147.2 (aromatic carbons), 161.7, 164.7, 165.8, 166.0 (2 C=O); MS, m/z [%]: 486.15 [M++2, 13], 484.17 [M++1, 24]; Anal. Calcd. for C22H18ClN5O4S (483.93): C, 54.60; H, 3.75; N, 16.85. Found: C, 54.87; H, 3.92; N, 14.68.
2.1.2.3. N-(4-Bromophenyl)-2–(2-oxo-3–(2-(4-sulfamoylphenyl)hydrazono)indolin-1-yl)acetamide (3c)
Yellow crystals, yield 75%, m.p. 300–302 °C; IR (KBr, ʋ cm−1): 3333, 3267, 3155 (NH2, 2NH), 3059 (CH aromatic), 2931 (CH aliphatic), 1712, 1674 (2 C=O), 1605 (NH bending), 1570, 1539 (C=C), 1296, 1157 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 4.65, 4.70 (2 s, 2H, NCH2), 7.10 (d, J = 7.92 Hz, 1H, aromatic H), 7.15 (t, J = 7.34 Hz, 1H, aromatic H), 7.25 (s, 2H, NH2, D2O exchangeable), 7.40 (t, J = 7.68 Hz, 1H, aromatic H), 7.50 (d, J = 8.92 Hz, 2H, aromatic H), 7.56 (d, J = 8.92 Hz, 2H, aromatic H), 7.65 (d, J = 8.84 Hz, 2H, aromatic H), 7.84 (d, J = 8.72 Hz, 2H, aromatic H), 8.30 (d, 1H, J = 7.64 Hz, aromatic H), 10.49, 10.53 (2 s, 1H, NH, D2O exchangeable), 10.87, 12.67 (2 s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 43.14, 43.33 (NCH2), 109.7, 110.1, 114.5, 115.1, 115.6, 115.7, 116.0, 119.5, 120.6, 121.6, 121.7, 122.4, 123.1, 124.8, 127.7, 127.9, 129.1, 129.7, 131.2, 138.04, 138.3, 141.8, 143.3, 145.6, 147.2 (aromatic carbons), 161.7, 164.6, 165.8, 166.1 (2 C=O); MS, m/z [%]: 528.02 [M+, 25]; Anal. Calcd. for C22H18BrN5O4S (528.38): C, 50.01; H, 3.43; N, 13.25. Found: C, 50.12; H, 3.67; N, 13.49.
2.1.2.4. 2–(2-Oxo-3–(2-(4-sulfamoylphenyl)hydrazono)indolin-1-yl)-N-p-tolylacetamide (3d)
Yellow crystals, yield 73%, m.p. 298–299 °C; IR (KBr, ʋ cm−1): 3341, 3171 (NH2, 2NH), 3059 (CH aromatic), 2939 (CH aliphatic), 1713, 1682 (2 C=O), 1605 (NH bending), 1570, 1547 (C=C), 1296, 1157 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 2.26 (s, 3H, CH3), 4.63, 4.68 (2 s, 2H, NCH2), 7.09–7.19 (m, J = 8.08 Hz, 4H, aromatic H), 7.25, 7.27 (2 s, 2H, NH2, D2O exchangeable), 7.41 (t, J = 7.74 Hz, 1H, aromatic H), 7.47 (d, J = 8.28 Hz, 2H, aromatic H), 7.66 (d, 2H, J = 8.72 Hz, aromatic H), 7.84 (d, J = 8.60 Hz, 2H, aromatic H), 8.31 (d, J = 7.64 Hz, 1H, aromatic H), 10.27, 10.31 (2 s, 1H, NH, D2O exchangeable), 10.87, 12.68 (2 s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 20.9 (CH3), 43.0, 43.2 (NCH2), 109.7, 110.3, 114.4, 115.1, 116.0, 119.4, 119.6, 119.7, 120.5, 122.3, 123.1, 124.8, 127.6, 127.8, 129.1, 129.6, 131.1, 131.2, 132.9, 133.0, 136.5, 136.6, 138.0, 138.3, 141.9, 143.4, 145.6, 147.1 (aromatic H), 161.7, 164.6, 165.2, 165.6 (C=O); MS, m/z [%]: 463.51 [M+, 39]; Anal. Calcd. for C23H21N5O4S (463.51): C, 59.60; H, 4.57; N, 15.11. Found: C, 59.84; H, 4.73; N, 15.38.
2.1.2.5. N-(4-Methoxyphenyl)-2–(2-oxo-3–(2-(4-sulfamoylphenyl)hydrazono)indolin-1-yl) acetamide (3e)
Yellow crystals, yield 70%, m.p. 294–295 °C; IR (KBr, ʋ cm−1): 3333, 3213, 3171 (NH2, 2NH), 3040 (CH aromatic), 2978, 2935 (CH aliphatic), 1713, 1678 (2 C=O), 1609 (NH bending), 1570, 1547 (C=C), 1300, 1157 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 3.72 (s, 3H, OCH3), 4.61, 4.62 (2 s, 2H, NCH2), 6.89 (d, J = 8.88 Hz, 2H, aromatic H), 7.09 (d, J = 7.88 Hz, 1H, aromatic H), 7.17 (t, J = 7.58 Hz, 1H, aromatic H), 7.25, 7.27 (2 s, 2H, NH2, D2O exchangeable), 7.41 (t, J = 7.74 Hz, 1H, aromatic H), 7.49 (d, J = 8.88 Hz, 2H, aromatic H), 7.65 (d, J = 8.80 Hz, 2H, aromatic H), 7.84 (d, J = 8.64 Hz, 2H, aromatic H), 8.30 (d, J = 7.64 Hz, 1H, aromatic H), 10.20, 10.21 (2 s, 1H, NH, D2O exchangeable), 10.86, 12.69 (2 s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 43.0, 43.2 (NCH2), 55.6 (OCH3), 109.7, 110.1, 114.4, 114.5, 115.1, 116.0, 119.5, 120.6, 121.2, 121.3, 122.4, 123.1, 124.8, 127.7, 127.9, 129.2, 129.7, 131.1, 131.3, 132.1, 132.2, 138.0, 138.3, 141.9, 143.4, 145.6, 147.2, 155.8, 155.9 (aromatic carbons), 161.7, 164.6, 165.0, 165.3 (C=O); MS, m/z [%]: 479.47 [M+, 26]; Anal. Calcd. for C23H21N5O5S (479.51): C, 57.61; H, 4.41; N, 14.61. Found: C, 57.49; H, 4.63; N, 14.88.
2.1.2.6. N-(2-Chlorophenyl)-2–(2-oxo-3–(2-(4-sulfamoylphenyl)hydrazono)indolin-1-yl) acetamide (3f)
Yellow crystals, yield 78%, m.p. 275–276 °C; IR (KBr, ʋ cm−1): 3363, 3333, 3136 (NH2, 2NH), 3059 (CH aromatic), 2974, 2935 (CH aliphatic), 1701 (2 C=O), 1601 (NH bending), 1570, 1512 (C=C), 1296, 1153 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 4.73, 4.78 (2 s, 2H, NCH2), 6.93 (d, J = 7.20 Hz, 1H, aromatic H), 7.13–7.23 (m, 2H, aromatic H), 7.27 (s, 2H, NH2, D2O exchangeable), 7.30–7.41 (m, 2H, aromatic H), 7.51 (d, J = 8.40 Hz, 1H, aromatic H), 7.62 (d, J = 8.80 Hz, 2H, aromatic H), 7.68 (t, J = 8.40 Hz, 1H, aromatic H), 7.82 (d, J = 8.80 Hz, 2H, aromatic H), 8.30 (d, J = 8.00 Hz, 1H, aromatic H), 9.99, 11.10 (2 s, 1H, NH, D2O exchangeable), 12.66 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 42.8 (NCH2), 110.1, 114.2, 114.4, 119.4, 120.6, 123.1, 126.6, 127.2, 127.6, 127.8, 129.1, 129.6, 130.0, 134.8, 138.3, 141.7, 145.6 (aromatic carbons), 161.6, 166.2 (2 C=O); MS, m/z [%]: 485.30 [M++2, 17.85], 483.30 [M+, 48.45]; Anal. Calcd. for C22H18ClN5O4S (483.93): C, 54.60; H, 3.75; N, 14.47. Found: C, 54.51; H, 3.89; N, 14.70.
2.1.2.7. N-(2-Bromophenyl)-2–(2-oxo-3–(2-(4-sulfamoylphenyl)hydrazono)indolin-1-yl) acetamide (3 g)
Yellow crystals, yield 73%, m.p. 279–280 °C; IR (KBr, ʋ cm−1): 3364, 3333, 3155 (NH2, 2NH), 3059 (CH aromatic), 2974, 2935 (CH aliphatic), 1697 (2 C=O), 1597 (NH bending), 1570, 1539 (C=C), 1300, 1153 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 4.71, 4.76 (2 s, 2H, NCH2), 7.10 (d, J = 7.84 Hz, 1H, aromatic H), 7.14–7.19 (m, 2H, aromatic H), 7.27, 7.25 (2 s, 2H, NH2, D2O exchangeable), 7.36–7.43 (m, 2H, aromatic H), 7.57–7.69 (m, 4H, aromatic H), 7.82 (d, J = 8.48 Hz, 2H, aromatic H), 8.31 (d, J = 7.60 Hz, 1H, aromatic H), 9.93, 10.87 (2 s, 1H, NH, D2O exchangeable), 9.97, 12.66 (2 s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 42.7, 43.0 (NCH2), 109.7, 110.2, 114.5, 115.1, 116.0, 118.6, 119.4, 120.6, 122.5, 123.2, 127.7, 127.9, 128.6, 129.2, 129.7, 131.2, 133.2, 136.2, 138.0, 138.3, 141.7, 143.1, 145.6, 147.2 (aromatic carbons), 161.6, 164.5, 166.1, 166.2 (C=O); MS, m/z [%]: 530 [M++2, 13.24], 528 [M+, 13.20]; Anal. Calcd. for C22H18BrN5O4S (528.38): C, 50.01; H, 3.43; N, 13.25. Found: C, 50.28; H, 3.61; N, 13.19.
2.1.3. General procedure for the synthesis of compounds 4a-g
A mixture of equimolar amounts of (2a-g) (1 mmol) and 4-((2-hydrazinyl-2-oxoethyl)amino)benzenesulfonamide (0.24 g, 1 mmol) was dissolved in methanol (5 ml) in the presence of glacial acetic acid (0.5 ml) and stirred at room temperature for 3 h. The obtained precipitate was filtered, washed with ethanol and recrystallized from ethanol.
2.1.3.1. 2–(2-Oxo-3–(2-(2–(4-sulfamoylphenylamino)acetyl)hydrazono)indolin-1-yl)-N-phenylacetamide (4a)
Yellow crystals, yield 80%, m.p. 272–273 °C; IR (KBr, ʋ cm−1): 3379, 3283, 3237, 3144 (NH2, 3NH), 3071 (CH aromatic), 2978, 2939 (CH aliphatic), 1720, 1693, 1663 (3 C=O), 1601 (NH bending), 1550, 1516 (C=C), 1312, 1150 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 4.38 (s, 2H, NHCH2), 4.64 (s, 2H, NCH2), 6.71–6.79 (m, 3H, aromatic H), 6.95 (s, 2H, NH2, D2O exchangeable), 7.06–7.15 (m, 3H, aromatic H), 7.32 (t, J = 7.90 Hz, 2H, aromatic H), 7.47 (t, J = 7.63 Hz, 1H, aromatic H), 7.54–7.59 (m, 4H, aromatic H), 8.13 (s, 1H, NH, D2O exchangeable), 10.35 (s, 1H, NH, D2O exchangeable), 11.41 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 43.2, 43.3 (2CH2), 110.2, 111.8, 115.2, 119.7, 123.8, 124.1, 126.3, 127.8, 133.1, 138.9, 139.0, 143.8, 144.9, 151.5 (aromatic carbons), 164.1, 165.2, 165.5 (3 C=O); MS, m/z [%]: 507.79 [M++1, 15]; Anal. Calcd. for C24H22N6O5S (506.53): C, 56.91; H, 4.38; N, 16.59. Found: C, 57.12; H, 4.60; N, 16.78.
2.1.3.2. N-(4-Chlorophenyl)-2–(2-oxo-3–(2-(2–(4-sulfamoylphenylamino)acetyl)hydrazono) indolin-1-yl)acetamide (4 b)
Yellow crystals, yield 78%, m.p. 270–271 °C; IR (KBr, ʋ cm−1): 3333, 3294, 3206, 3136 (NH2, 3NH), 3063–3032 (CH aromatic), 2978, 2947 (CH aliphatic), 1716, 1693, 1659 (3 C=O), 1612 (NH bending), 1554, 1512 (C=C), 1319, 1149 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 4.37 (s, 2H, NHCH2), 4.58, 4.64 (2 s, 2H, NCH2), 6.61 (d, J = 8.52 Hz, 2H, aromatic H), 6.72 (d, J = 8.48 Hz, 1H, aromatic H), 6.94, 6.95 (2 s, 2H, NH2, D2O exchangeable), 7.11, 7.20 (m, 2H, aromatic H), 7.38 (d, J = 8.76 Hz, 2H, aromatic H), 7.45 (d, J = 7.60 Hz, 1H, aromatic H), 7.51–7.63 (m, 3H, aromatic H), 7.67 (t, J = 7.74 Hz, 1H, aromatic H), 8.12 (s, 1H, NH, D2O exchangeable), 10.36, 10.49 (2 s, 1H, NH, D2O exchangeable), 11.37 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 43.6, 45.0 (2CH2), 111.4, 115.2, 117.9, 121.3, 121.6, 124.1, 125.0, 127.7, 129.2, 131.2, 137.7, 138.9, 144.7, 151.1, 158.9 (aromatic carbons), 165.7, 169.3, 183.4 (3 C=O); MS, m/z [%]: 542.94 [M++2, 32.72], 540.88 [M+, 40]; Anal. Calcd. for C24H21ClN6O5S (540.98): C, 53.28; H, 3.91; N, 15.53. Found: C, 53.61; H, 4.08; N, 15.79.
2.1.3.3. N-(3-Bromophenyl)-2–(2-oxo-3–(2-(2–(4-sulfamoylphenylamino)acetyl)hydrazono) indolin-1-yl)acetamide (4c)
Orange yellow crystals, yield 78%, m.p. 260–262 °C; IR (KBr, ʋ cm−1): 3329, 3294, 3236, 3198 (NH2, 3NH), 3067, 3032 (CH aromatic), 2947, 2897 (CH aliphatic), 1725, 1697, 1650 (3 C=O), 1600 (NH bending), 1550, 1512 (C=C), 1315, 1149 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 4.36 (s, 2H, NHCH2), 4.58, 4.64 (2 s, 2H, NCH2), 6.61, 6.71 (2d, J = 8.44 Hz, 2H, aromatic H), 6.91, 7.11 (2d, J = 8.10 Hz, 1H, aromatic H), 6.94, 6.95 (2 s, 2H, NH2, D2O exchangeable),7.03, 7.17 (2t, J = 7.68 Hz, 1H, aromatic H), 7.45, 7.67 (2t, J = 7.70 Hz, 1H, aromatic H), 7.47–7.56 (m, 6H, aromatic H), 7.62 (d, J = 7.36 Hz, 1H, aromatic 8.12 (s, 1H, NH, D2O exchangeable), 10.37, 10.49 (2 s, 1H, NH, D2O exchangeable), 11.37 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 43.4, 43.7, 45.0 (2CH2), 111.5, 111.7, 115.7, 115.9, 117.9, 121.3, 121.6, 121.9, 122.2, 122.8, 124.0, 125.0, 126.3, 126.5, 127.7, 127.8, 131.2, 132.1, 138.1, 138.3, 138.8, 143.7, 144.7, 151.2, 151.4, 151.5, 158.9 (aromatic carbons), 165.5, 169.2, 183.3 (C=O); MS, m/z [%]: 583.20 [M+-2, 1.84]; Anal. Calcd. For C24H21BrN6O5S (585.43): C, 49.24; H, 3.62; N, 14.36. Found: C, 49.52; H, 3.83; N, 14.59.
2.1.3.4. 2–(2-Oxo-3–(2-(2–(4-sulfamoylphenylamino)acetyl)hydrazono)indolin-1-yl)-N-m-tolylacetamide (4d)
Yellow crystals, yield 75%, m.p. 273–274 °C; IR (KBr, ʋ cm−1): 3344, 3294 (NH2, 3NH), 3032 (CH aromatic), 2974, 2897 (CH aliphatic), 1717, 1697, 1659 (3 C=O), 1597 (NH bending), 1512 (C=C), 1315, 1145 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 2.26 (s, 3H, CH3), 4.37 (s, 2H, NHCH2), 4.62 (s, 2H, NCH2), 6.61, 6.71 (2d, J = 8.60 Hz, 2H, aromatic H), 6.94, 6.95 (2 s, 2H, NH2, D2O exchangeable), 7.10–7.14 (m, 4H, aromatic H), 7.44 (d, J = 8.04 Hz, 2H, aromatic H), 7.49 (d, J = 8.76 Hz, 2H, aromatic H), 7.55 (d, J = 8.72 Hz, 2H, aromatic H), 8.12 (s, 1H, NH, D2O exchangeable), 9.16 (s, 1H, NH, D2O exchangeable), 10.25 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 20.9 (CH3), 43.3, 45.0 (2CH2), 110.2, 111.6, 111.7, 115.2, 119.8, 122.8, 126.3, 127.8, 129.7, 131.2, 133.0, 136.5, 143.8, 144.9, 151.5 (aromatic carbons), 165.3, 169.2, 173.1 (3 C=O); MS, m/z [%]: 522.38 [M++2, 18], 520.41 [M+, 19]; Anal. Calcd. For C25H24N6O5S (520.56): C, 57.68; H, 4.65; N, 16.14. Found: C, 57.96; H, 4.82; N, 16.36.
2.1.3.5. N-(3-Methoxyphenyl)-2–(2-oxo-3–(2-(2–(4-sulfamoylphenylamino)acetyl) hydrazono)indolin-1-yl)acetamide (4e)
Yellow crystals, yield 70%, m.p. 278–279 °C; IR (KBr, ʋ cm−1): 3379, 3329, 3290, 3233 (NH2, 3NH), 3074 (CH aromatic), 2974, 2901 (CH aliphatic), 1720, 1693, 1659 (3 C=O), 1601 (NH bending), 1512 (C=C), 1311, 1149 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 3.72 (s, 3H, OCH3), 4.37 (s, 2H, NHCH2), 4.60 (s, 2H, NCH2), 6.70–6.73 (m, 2H, aromatic H), 6.89 (d, J = 9.00 Hz, 2H, aromatic H), 6.95, 6.97 (2 s, 2H, NH2, D2O exchangeable), 7.10–7.21 (m, 3H, aromatic H), 7.45–7.50 (m, 3H, aromatic H), 7.55 (d, J = 7.64 Hz, 2H, aromatic H), 8.12 (s, 1H, NH, D2O exchangeable), 10.19 (s, 1H, NH, D2O exchangeable), 11.40 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 43.1, 43.3 (2CH2), 55.6 (OCH3), 110.2, 111.8, 114.4, 115.2, 121.3, 122.8, 123.7, 126.3, 127.8, 132.1, 133.0, 143.8, 144.9, 151.4, 155.9 (aromatic carbons), 164.0, 164.5, 165.0 (3 C=O); MS, m/z [%]: 536.20 [M+, 40]; Anal. Calcd. For C25H24N6O6S (536.56): C, 55.96; H, 4.51; N, 15.66. Found: C, 56.13; H, 4.67; N, 15.89.
2.1.3.6. N-(2-Chlorophenyl)-2–(2-oxo-3–(2-(2–(4-sulfamoylphenylamino)acetyl)hydrazono) indolin-1-yl)acetamide (4f)
Yellow crystals, yield 65%, m.p. 206–208 °C; IR (KBr, ʋ cm−1): 3379, 3283, 3256 (NH2, 3NH), 3066 (CH aromatic), 2927, 2835 (CH aliphatic), 1740, 1720, 1674 (3 C=O), 1597 (NH bending), 1539, 1520 (C=C), 1312, 1150 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 3.69, 3.70 (2 s, 2H, NHCH2), 4.26, 4.32 (2s, 1H, NH, D2O exchangeable), 4.46, 4.71 (2 s, 2H, NCH2), 6.61 (d, J = 8.68 Hz, 2H, aromatic H), 6.70 (d, J = 8.72 Hz, 2H, aromatic H), 6.93 (s, 2H, NH2, D2O exchangeable), 7.08 (d, J = 7.68 Hz, 1H, aromatic H), 7.11 (d, J = 7.56 Hz, 1H, aromatic H), 7.23 (t, J = 6.96 Hz, 1H, aromatic H), 7.33 (t, J = 7.36 Hz, 1H, aromatic H), 7.42–7.46 (m, 1H, aromatic H), 7.51 (d, J = 8.72 Hz, 2H, aromatic H), 7.70 (d, J = 7.12 Hz, 1H, aromatic H), 9.15 (s, 1H, NH, D2O exchangeable), 9.96 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 45.03, 46.07 (2CH2), 111.64, 111.83, 122.84, 127.6, 127.8, 128.0, 128.3, 130.0, 131.2, 131.7, 132.1, 133.5, 134.8, 135.3, 138.1, 151.5 (aromatic carbons), 159.2, 169.2 (C=O); MS, m/z [%]: 541.10 [M++1, 1.63], 539.10 [M+-1, 2.34]; Anal. Calcd. For C24H21ClN6O5S (540.98): C, 53.28; H, 3.91; N, 15.53. Found: C, 53.44; H, 4.07; N, 15.76.
2.1.3.7. N-(2-Bromophenyl)-2–(2-oxo-3–(2-(2–(4-sulfamoylphenylamino)acetyl)hydrazono) indolin-1-yl)acetamide (4 g)
Buff crystals, yield 70%, m.p. 216–217 °C; IR (KBr, ʋ cm−1): 3379, 3345, 3283 (NH2, 3NH), 3062, 3032 (CH aromatic), 2920, 2850 (CH aliphatic), 1728, 1720, 1662 (3 C=O), 1601 (NH bending), 1520 (C=C), 1315, 1149 (SO2); 1H NMR (DMSO-d6, 400 MHz): δ 4.15, 4.16 (2 s, 2H, NHCH2), 4.62, 4.66 (2 s, 2H, NCH2), 6.33, 8.99 (2 s, 1H, NH, D2O exchangeable), 6.61 (d, J = 8.68 Hz, 2H, aromatic H), 6.69 (d, J = 8.72 Hz, 2H, aromatic H), 6.89, 6.93 (2 s, 2H, NH2, D2O exchangeable), 6.98, 8.36 (2d, J = 7.32 Hz, 1H, aromatic H), 7.12–7.20 (m, 2H, aromatic H), 7.38 (t, J = 7.62 Hz, 1H, aromatic H), 7.51 (d, J = 8.48 Hz, 2H, aromatic H), 7.61–7.70 (m, 2H, aromatic H), 9.21, 9.59 (2 s, 1H, NH, D2O exchangeable), 9.81, 10.02 (2 s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz): δ 42.85, 45.05 (2CH2), 111.5, 115.2, 118.8, 122.0, 125.1, 127.3, 127.6, 127.7, 128.6, 130.2, 131.2, 133.2, 136.4, 151.8 (aromatic carbons), 167.0, 173.9, 181.3 (3 C=O); MS, m/z [%]: 585.20 [M+, 0.90], 583.20 [M+-2, 1.38]; Anal. Calcd. For C24H21BrN6O5S (585.43): C, 49.24; H, 3.62; N, 14.36. Found: C, 49.45; H, 3.74; N, 14.63.
2.2. Carbonic anhydrase inhibitory activity
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activityCitation38. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for mainaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentration ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been use for determining the initial velocity. The un catalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together at room temperature (15 min) prior to assay, in order to allow for the formation of the E-I complex. Data from were obtained after 15 min incubation of enzyme and inhibitor, as for the sulphonamides reported earlierCitation39. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng-Prusoff equation, as reported earlierCitation40,Citation41 and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation42.
Table 1. Inhibition data of human CA isoforms hCA I, II, IX and XII with 2,3-dioxoindole sulfamoylphenyl acetamide 2h, 2-oxindole based benzenesulfonamides 3a-g, 4a-g and the standard inhibitor Acetazolamide (AAZ) using a stopped flow CO2 hydrase assay. 
2.3. Anti-proliferative activity
Cancer cell lines were obtained from Nawah Scientific Inc. (Mokatam, Cairo, Egypt). Cells were maintained in DMEM media supplemented with 100 mg/mL of streptomycin, 100 units/mL of penicillin and 10% of heat-inactivated foetal bovine serum in humidified, 5%(v/v) CO2 atmosphere at 37 °C.
Cell viability assay was assessed by SRB assay. Aliquots of 100 μL cells suspension (5x103cells) were in 96 well plates and incubated in complete media for 24 h. Cells were treated with another aliqot of 100 μL media containing drugs at various concentrations. After 72 h of drug exposure, cells were fixed by replacing media with 150 μL of 10%TCA and incubated at 4 °C for 1 h. The TCA solution was removed, and the cells were washed 5 times with distilled water. Aliquots of 70 μL SRB solution (0.4%w/v) were added and incubated in a dark place at room temperature for 10 min. Plates were washed 3times with 1% acetic acid and allowed to air-dry overnight. Then, 150 μL of TRIS (10 mM) was added to dissolve protein-bound SRB stain; the absorbance was measured at 540 nm using a BMGLABTECH® -FLUO star Omega microplate reader (Ortenberg, Germany)Citation43,Citation44. All experiments were performed in triplicate wells for each condition. IC50 values were calculated from concentration-response curves by Sigma Plot software, version 12.0 (System Software, San Jose, CA, USA), using an E-max model equation. Also selectivity index was determined.
2.4. Molecular docking
Molecular Operating Environment MOE® 2014.0901 was used as the computational software throughout the docking study. The best quality chains of the four PDB files (3W6H, 3HS4, 3IAI and 1JD0 for hCA I, II, IX and XII, respectively) were selected where the unnecessary ions, ligands and water molecules were removed in a radius of 4.0 Å. The 3D protein structures were protonated using their corresponding pH and temperature conditions. The selected compounds 2h and 3c were constructed then deprotonated in-silico using amber:10EHT forcefield followed by energy minimisation at a gradient of 0.01 RMSD. On the fly-docking protocol was implemented at the same forcefield using triangle matcher, London dG and GBVI/WSA dG as placement and rescoring function 1 and 2, respectively.
For accurate molecular docking protocol, validation was done by self-docking the co-crystallised ligands to get the lowest root mean square deviation (RMSD) and the highest energy score (S).
2.5. 2D-QSAR study
The compounds were constructed, and energy minimised using MOE® 2014.0901 MMFF94x forcefield at the default parameters with RMS gradient 0.1 Kcal/mol/A2 then divided into training and testing sets for model generation and validation, respectively. For hCA IX, 34 compounds were used as the training set and 5 as test set of compounds while for hCA XII, 34 and 6 compounds were used as the training and test set, respectively. A variety of 413 1D and 2D descriptors were calculated using Chemopy® descriptors from ChemDes® open-source webserver. Thereafter, descriptors with constant values across the training set or equal to zero were excluded. RapidMiner® Studio 9.10 program was used for descriptors filtration through the forward selection optimisation algorithm against the dependent variable log KI then the best descriptors were transferred back to MOE® for a linear model generation using the partial least square (PLS) method.
3. Results and discussion
3.1. Chemistry
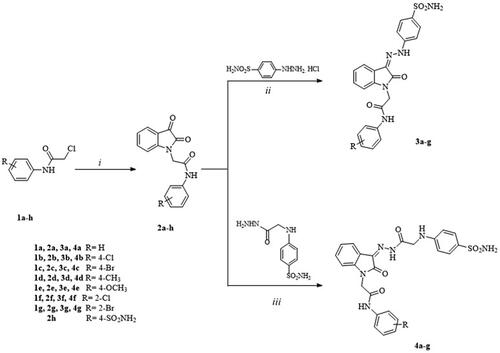
Scheme 1 illustrates the synthesis of the target 2-oxindole benzenesulfonamide analogs 3a-g and 4a-g. Known 2-chloro-N-[(un)sub)]phenylacetamides (1a-h) were prepared according to the reported method by alkylation of the appropriate substituted aniline derivatives with chloroacetylchlorideCitation31,Citation32. The reported key intermediates (2a-f)Citation33–35 and the new ones (2g, h) were prepared according to the conventional method by the reaction of the commercially available isatin (indole-2,3-dione) with the corresponding 2-chloro-N-phenacetamide (1a-h) in dry N, N-dimethyl formamide (DMF) in the presence of potassium carbonate. Then, 2a-g were condensed with the prepared 4-hydrazinylbenzenesulfonamide hydrochlorideCitation36 at room temperature to give new 4-[2–(2-oxindolin-3-ylidene)hydrazinyl]benzenesulfonamide (3a-g). Similarly, the condensation of 2a-g with the prepared 4–(2-hydrazinyl-2-oxoethylamino)benzenesulfonamideCitation37 in methanol at room temperature afforded 4–(2-oxindolin-3-ylidene)hydrazinyl-2-oxoethylamino benzenesulfonamide derivatives (4a-g). All new derivatives were confirmed by spectral and elemental analyses as detailed in the experimental part.
Scheme 1. Synthetic pathway for the target compounds 1–4. Reagents and reaction conditions. (i) Glacial acetic acid, sodium acetate, 0 °C. (ii) MeOH, r.t. (iii) MeOH, glacial acetic acid, r.t.
It is noteworthy that all target compounds 3a-g and 4a-g contain an exocyclic C=N imine bond which results in their presence as E/Z geometrical isomers. However, it was reported that E/Z ratio is dependent on each isomer stability and NMR solventCitation28,Citation45,Citation46. This fact was observed in many compounds like 3a,b,f,g and 4b,c,f,g which were found in a mixture of E and Z isomers as observed from their 1H NMR spectra that revealed additional signals due to both isomers. For instance, 1H NMR spectrum of compound 3a had two singlets attributed to the methylene protons at 4.65 ppm (E-isomer) and 4.70 ppm (Z-isomer) with integration ratio 3.23:3.42, respectively, whereas compounds 3c,d,e showed both isomeric forms with integration ratio (6:1) for Z–isomer. On the other hand, compounds 4a,d,e were isolated mainly as Z isomers. (see the experimental section).
3.2. Carbonic anhydrase inhibitory activity
The new candidates 2,3-dioxoindole sulfamoylphenyl acetamide 2h, 2-oxoindole derivatives (hydrazinyl benzenesulfonamide (3a-g) and hydrazinyl 2-oxoethylamino benzenesulfonamide congeners (4a-g) were evaluated for their ability to inhibit four physiologically relevant hCA isoforms (cytosolic isoforms; hCA I, hCA II and the transmembrane, tumour-associated isoforms hCA IX, hCA XII) through a stopped flow CO2 hydrase assayCitation38–42. Their potency as KI (nM) were compared to the standard CA inhibitor acetazolamide (AAZ) and these results were summarised in (). A brief study of SAR revealed the followings:
Regarding the ability of the tested compounds to inhibit hCA I, all tested compounds except 2h revealed relatively lower activity than other isoforms. It was noticed that 2,3-dioxoindole 2h showed an extremely high potency (KI = 45.1 nM) compared to AAZ (KI = 250 nM) and to other tested compounds that showed inhibitory activity ranged from 656 to 9318 nM. Derivatives linked to hydrazinyl benzenesulfonamide (3a-g) were more active (KIs = 656-3971 nM) than their analogs linked to hydrazinyl 2-oxoethylamino benzenesulfonamide congeners (4a-g) (KIs = 2353-9318 nM). Also, in hydrazinyl benzenesulfonamides (3a-g) substituted phenylacetamide moiety revealed activity better than unsubstituted congener, the order of activity was (4-CH3> 4-Br> 4-OCH3> 4- Cl> 2-Br> 2-Cl > H), thus, the inductive effect of substituents at the phenyl ring has no absolute impact on the activity. On the other hand, in hydrazinyl 2-oxoethylamino benzenesulfonamide congeners (4a-g) the order of activity was 4-Br> 2-Cl > H> 2-Br> 4-Cl> 4-CH3> 4-OCH3), that the presence of electron withdrawing groups is more favourable for activity than electron donating moieties.
Activity towards hCA II isoform was better with KI ranged from 5.87 to 4586 nM; like hCA I, compound 2h was also the most potent one with KI = 5.87 nM. Derivatives linked to hydrazinyl benzenesulfonamides (3a-g) were more active (KIs = 56.4-804 nM) than their analogs linked to hydrazinyl 2-oxoethylamino benzenesulfonamide congeners (4a-g) (KI = 458-4586 nM). In both series (3a-g) and (4a-g) it was obvious that substitution of phenylacetamide with 4-Br or 4-CH3 was preferable for activity.
All the tested compounds except compounds 2h, and 4e-g represented a reasonable selectivity towards tumour isoform hCA IX compared to hCA I isoform. The hydrazinyl benzenesulfonamide (3a-g) were more active (KIs = 56.9–673 nM) than their analogs linked to hydrazinyl 2-oxoethylamino benzenesulfonamide congeners (4a-g) (KIs = 574–7336 nM). The 4-bromophenylacetamide derivative (3c) was the most active one. Similar to hCA II activity, in both series (3a-g) and (4a-g) it was obvious that substitution of phenylacetamide with 4-Br or 4-CH3 was preferable for activity.
Regarding the hCA XII inhibition, all the tested compounds except compounds 2h, revealed very weak selectivity towards hCA XII. Compound 2h was relatively the most potent one with KI = 7.91 nM. Hydrazinyl benzenesulfonamide (3a-g) were more active than their analogs linked to hydrazinyl 2-oxoethylamino benzenesulfonamide congeners (4a-g). In hydrazinyl benzenesulfonamide (3a-g) it was obvious that substitution of phenylacetamide with 4-Br or 4-Cl was preferable for activity, while in hydrazinyl 2-oxoethylamino benzenesulfonamide congeners (4a-g) substitution at ortho position of phenylacetamide with Br or Cl was more favoured.
Finally, from our previous studies to develop isatin-based CA derivativesCitation29,Citation30 we can find in the current study that the presence of substituted phenylacetamide increase the potency specially for hCA I and shifted the selectivity mainly towards hCA IX comparing to substituted benzoyl moiety in the previous studyCitation30 that reflects the ability of these compounds to have promising anticancer activity. Also, in our current study increasing the length of the N-substitution from N-benzoyl to N-phenylacetamido group leading to decrease in potency mainly against all isoforms and slightly shifted the selectivity in case of 2-substituted phenylacetamide analogs.
3.3. Anti-proliferative activity
As CA IX is overexpressed in MCF-7 breast cancer cell lineCitation47 and in A549 lung cancer cellsCitation48, therefore, compounds 2h, 3c and 3d eliciting the best CA IX inhibition were selected for further evaluation for their anti-proliferative activity against these cancer cell lines. Moreover, they were tested for their cytotoxicity against normal human skin fibroblast cell line HSF to confirm their safety, the results are illustrated in and Figures S1–S6. Compound 3d showed good cytotoxic activity against MCF-7 and A549 cell lines with IC50 = 0.869 and 5.12 μM, respectively. On the other hand, 3c exerted only cytotoxic activity against A549 cell line with IC50 = 5.90 μM. Furthermore, 3c showed promising safety margin towards normal cells through its selectivity towards A549 cells in contrast to doxorubicin and 3d which were unselectively cytotoxic to both normal and cancer cell lines. Unfortunately, compound 2h found to have no cytotoxic activity against both cell lines, thus, its anti-proliferative activity is not correlated to its CA IX inhibition, however, it can be considered as an adjuvant with an anti-cancer agent to enhance its cytotoxic effect due to its CA IX inhibition activity similar to acetazolamide.
Table 2. Cytotoxic activity and selectivity index of compounds 2h, 3c, and 3d on cancer and normal cell lines
3.4. Molecular docking study
Molecular docking was done for 2h and 3c to relate their in vitro biological results with their possible conformations for binding to the four CA isoforms. Hence, the crystal structure of each isoform co-crystallised with AAZ was downloaded from the Protein Data Bank (PDB) using PDB ID: 3W6H, 3HS4, 3IAI and 1JD0 for isoforms hCA I, II, IX and XII, respectivelyCitation49. Molecular docking method was validated using the co-crystallised AAZ by self-docking till getting the same interaction pattern with each isoform binding site residues as presented in Supplementary Figure S10a-d. Considering the importance of sulphonamide moiety deprotonation to coordinate with the positively charged zinc ionCitation50, the tested compounds sulphonamide group were in-silico deprotonated and energy minimised before docking.
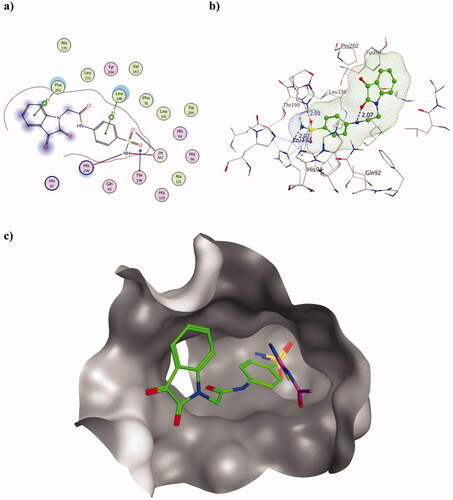
The molecular docking of the most active derivative 2h augmented the achieved in vitro KI values that surpassed AAZ using both hCA isoforms I and II. Considering hCA I (PDB ID 3W6H), it revealed the formation of the two characteristic co-ordinations with zinc ion using the deprotonated nitrogen of the sulphonamide moiety () which translated into KI value of 45.1 and 250.0 nM for 2h and AAZ, respectively. The achieved co-ordinations were distanced by 2.01 (zinc- nitrogen) and 2.65 Å (zinc – oxygen) from zinc which are consistent with many crystallographic findings (). Moreover, the sulphonamide negatively charged deprotonated nitrogen was able to interact with the basic His200, a unique hCA I isoform zinc ligandCitation51. This interaction could support the conformation of the sulphonamide moiety to approach zinc and stabilised the outcome interaction. Furthermore, two arene-H bonds were formed with Leu198, and Pro202 by the two phenyl groups beside the sulphonamide oxygen adeptness to accept the H-bond from Thr199. Another explanation of the resulted lower KI value of 2h than AAZ was that the capability of 2h to block most of the hCAI active site entrance by its isatin moiety ().
Figure 3. Molecular docking data of 2h on hCAI isoform using PDB ID 3W6H as 2D (a) and 3D (b) presentations showing 2h as green ball and stick model with the formed H-bonds and arene-H were shown in blue and black dotted lines, respectively with their distance in Å highlighting the interaction site. c) The overlaid 3D presentation of 2h (green) and AAZ (magenta) inside the CA I active site.
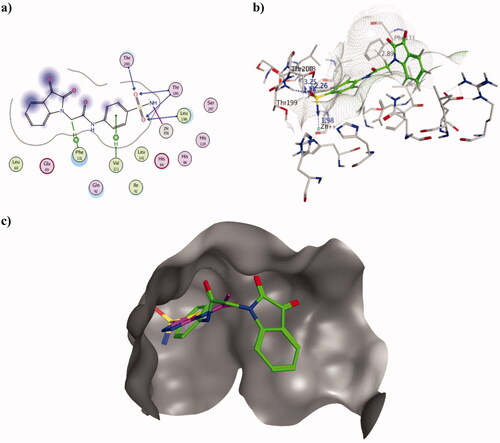
Similarly, 2h resulted in better Ki inhibitory value of hCA II isoform than AAZ exhibiting 5.87 and 12.0 nM, respectively. The binding mode of 2h inside the active site of PDB ID 3HS4 managed to explain the achieved in vitro results. Its sulphonamide moiety interacted with zinc, Thr199, Thr200 and Leu198 forming four H-bonds ranging 2.26–3.2 Å and zinc co-ordination at 1.98 Å. In addition, two arene-H interactions were formed with Val121 and Phe131 using phenyl moiety of sulphonamide terminus and acetamide linkage, respectively (). Similar to hCAI, 2h oriented in a conformation that blocked the entrance of hCA II active site by its isatin moiety (). The binding energy of 2h was better than AAZ calculated as −10.11 and −7.69 Kcal/mol, respectively which complemented the binding conformations and achieved KI values.
Figure 4. Molecular docking data of 2h on hCA II isoform using PDB ID 3HS4 as 2D (a) and 3D (b) presentations showing 2h as green ball and stick model with the formed H-bonds and arene-H were shown in blue and black dotted lines, respectively with their distance in Å highlighting the interaction site. c) The overlaid 3D presentation of 2h (green) and AAZ (magenta) inside the hCA II active site.
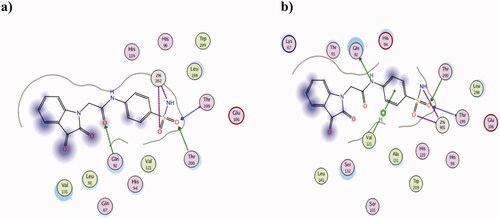
Regarding the tumour-associated hCA IX isoform, 2h molecular docking data using PDB ID 3IAI showed its interaction with zinc in resemblance to other isoforms with the sulphonamide oxygen and deprotonated nitrogen suggesting good inhibitory potential (). Moreover, the oxygen atoms of sulphonamide group and acetamide linkage acted as acceptors to three H-bonds with Thr199, Thr200 and Gln92, respectively. Despite its in silico interaction pattern, its binding energy score was not as favourable as AAZ giving −8.75 and −10.81 Kcal/mol, respectively which explained the superiority of the achieved AAZ Ki value. Nonetheless, 2h showed better KIvalue on the other tested tumour-associated hCA XII isoform comparing to AAZ showing 7.91 and 5.7 nM, respectively. Its binding mode using hCA XII PDB 1JD0 supported the achieved KI value in which it interacted with zinc, Thr199, Thr200, Gln92 and Val131 (). However, AAZ surpassed 2h binding energy score giving −9.12 Kcal/mol comparing to −8.09 Kcal/mol of 2h which was translated into a slight difference in their KI values.
Figure 5. The 2D presentation of 2h molecular docking study interacting with the tumour-associated isoforms hCA IX (a) and XII (b) using PDB ID 3IAI and 1JD0, respectively.
The following most promising derivative 3c showed equal sub-nanomolar KI values on hCA II and IX giving 56.4 and 56.9 nM, respectively compared to AAZ with KI 12.0 and 25.0 nM for both isoforms, respectively. Its molecular docking investigation using PDB ID 3HS4 and 3IAI for hCA II and hCA IX, respectively explained the achieved KI values (Figure S11(a,b)). Zinc ion co-ordination was detected in both isoforms yet, the number of interactions of 3c with the active site residues were more at hCA IX than hCA II. This was translated into half the inhibitory activity in case of hCA IX compared to almost one-fifth the inhibition in case of hCA II in comparison to AAZ. Furthermore, the superiority of AAZ was clarified by its type of interactions with the crucial active site residues of hCA II and hCA IX (Figure S11(c,d)) together with its favourable binding energy −11.85 and −10.98 Kcal/mol compared to 3c with −8.45 and −9.98 Kcal/mol for both isoforms, respectively. In contrast, 3c did not show as promising inhibitory activity on the other two isoforms I and XII which were elucidated by its molecular docking data (Figure S12(a,b)).
3.5. 2D-QSAR study
As a useful manipulation of our current and previously achieved in vitro hCA inhibitory data of benzenesulfonamide derivatives as hCA inhibitors, () Citation29,Citation30, the tumour associated hCA IX and XII data were employed to build two 2D Quantitative Structure-Activity Relationship (2D-QSAR) models given their scaffold similarity, substitution variation and the unified in vitro assay method, these QSAR models were used to correlate the achieved inhibitory activity with their structural features for future structural optimisation. The collective biology data showed wide biology spectrum ranges from KI = 14 to 7336 nM and 8 to 6025 nM for hCA IX and hCAXII, respectively. Different mathematical forms of the dependent variable were tried such as KI, log KI and PkI where log KI were found to be linear with approximate r2 value= 0.95 (Figures S13,14). Linear regression method was used for descriptors selection and optimisation by RapidMiner® Studio then the model was generated using Partial Least Square (PLS) method by MOE® program. In PLS method, the results of the fit are presented as the root mean square error (RMSE) which is the square root of the MSE function at the chosen descriptors value and the correlation coefficient (r2) which is equal to 1-MSE/YVAR where YVAR is the sample variance of the experimental valuesCitation52,Citation53. The calculated 1D and 2D descriptors by Chemopy®Citation54 included different constitutional, topological, charge-related, thermodynamic and atom/bond-type descriptors properties of the compounds. An exclusion of descriptors with constant values amongst the training set or equal to zero was done before descriptors selection using the forward linear regression algorithm with leave 10 out validation method of RapidMiner® Studio program. The selected descriptors co-linearity of both models was evaluated using correlation matrices to prevent model over-fitting.
Figure 6. Chemical structures of the reported benzenesulfonamide derivatives as hCA inhibitors included in the 2D-QSAR training set Citation29,Citation30.
3.5.1. 2D-QSAR model of hCA IX
For the tumour associated hCA IX, the 40 compounds were divided into training N = 34 and test n = 5 sets and their descriptors were calculated and filtered to 147 after excluding descriptors with fixed values. The selected linear descriptors values of the training set were presented in Table S1. The descriptors governed the activity were found to be the distance and adjacency matrix of the heavy atoms (BCUT_PEOE-0)Citation55, the partial equalisation of orbital electronegativities (PEOE_VSA-3)Citation52, fractional positive polar van der Waals surface area (QVSA_FPPOS)Citation56, partition coefficient of subdivided surface area (SLogP_VSA 4)Citation57 and wiener polarity number (wienerPol)Citation58 according to the model presented in EquationEquation (1)(1) . The generated model showed regression coefficient r2= 0.862 and RMSE= 0.269.
(1)
Examining the coefficient values of the independent variables displayed in EquationEquation (1)(1) , it revealed that the distance and adjacency of the heavy atoms had the main proportional impact on the dependent inhibitory activity of hCA IX. Furthermore, electronegativity and polarity of the compound had appreciable effect on the predicted inhibitory action.
This model was capable of predicting the inhibitory activity of the most potent analogue 7a amongst all in-vitro tested derivatives of benzenesulfonamide with very small difference in log KI value of −0.11. The predicted log KI of 7a was 1.26 (KI= 18.2 nM) compared to the actual value 1.146 (KI= 13.9 nM).
The generated QSAR model was validated using leave one out (LOO) cross validation method which involved developing a number of models with one example excluded at a time and externally validated using the test set compounds. The achieved r2(LOO) = 0.797 with a corresponding RMSE= 0.328 while test set predictive dependent variables showed r2(validation) = 0.766. The predictive values of the dependent variables and their corresponding graphical presentation of both LOO and external validation were presented in and . As shown in , the model managed to predict the inhibitory activity of the test set of compounds that were compatible with the actual experimental values with small residuals. Moreover, the obtained Z-score during cross validation remained less than 2.4 suggesting compounds fitting to the generated model with no outliers. Additionally, Fisher-value test (F) and standard deviation (s2) were calculated for the model to confirm its statistical reliability and significanceCitation59. This model revealed F = 33.039 and s2 = 0.089 with the higher value of F relative to s2 was an additional support of the model validation.
Figure 7. Graphical presentation between the experimental and predicative log Ki of hCAIX training set (LOO) validation (a) and test set (b) where the corresponding squared linear coefficient r2 were displayed.
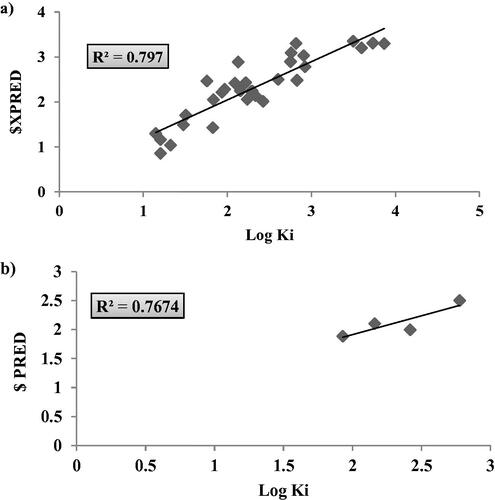
Table 3. The validation data of hCAIX 2D-QSAR model as calculated from LOO method and test set external validation.
3.5.2. 2D-QSAR model of hCA XII
Similarly, the achieved hCA XII inhibitory data was used to construct a 2D-QSAR model to correlate with their physicochemical and topological properties. The compounds were classified into 34 and 6 as training and testing set, respectively where the descriptors selection was done in a similar way to hCA IX model complying with the usual linear regression method. The four descriptors governed the activity were depicted in EquationEquation (2)(1) and Table S2 were the fractional positive polar van der Waals surface area (PEOE_VSA_FPPOS)Citation56, fractional hydrophobic van der Waals surface area (PEOE_VSA_ FHYD)Citation56, partition coefficient of subdivided surface area (SLogP_VSA 1)Citation57 and the total positive partial charge (Q_PC+)Citation56. The estimated model showed regression coefficient r2= 0.699 and RMSE= 0.408.
(2)
According to the shown coefficient value of each independent variable in EquationEquation (2)(2) , it was suggested that both van der Waals hydrophobic and polar fractional surface area had the main influence on the inhibitory activity of hCA XII in direct and inverse proportional, respectively. Similar to hCA IX 2D-QSAR model, both fractional positive polar van der Waals surface area and partition coefficient of subdivided surface area had an appreciable effect on the inhibition of hCA XII.
The obtained model managed to predict the inhibitory activity of the most potent hCAXII inhibitor 2 h amongst the tested analogues with difference of 0.051. The model suggested a log KI of 0.954 (KI= 8.99 nM) compared to the actual log KI of 0.903 (KI= 7.91 nM).
Resembling the hCAIX inhibitors 2D-QSAR model validation parameters, the predicative log KI values were presented in . This model displayed r2 (LOO) = 0.62 with RMSE= 0.46 however, its external validation using the test set revealed more promising r2(validation) = 0.841 () accompanied with F = 16.27 and s2= 0.197.
Figure 8. Graphical presentation between the experimental and predicative log KI of hCAXII training set (LOO) validation (a) and test set (b) where the corresponding squared linear coefficient r2 were displayed.
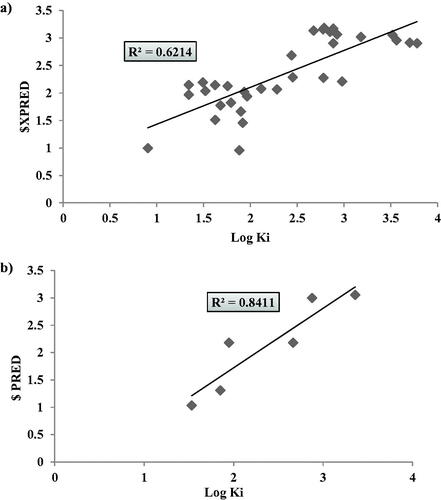
Table 4. The validation data of hCAXII 2D-QSAR model as calculated from LOO method and test set external validation.
Considering the formerly mentioned internal and external validation and statistical analysis of the calculated descriptors of both hCA IX and XII 2D-QSAR models, it was concluded that they showed sufficient predictive capability of the inhibitory activity with minimal error.
4. Conclusion
From our current study we can conclude that isatin N-phenylacetamide based sulphonamides could be considered as promising scaffold for future research to develop potent and selective carbonic anhydrase inhibitors along with anti-proliferative activity. It was observed that 2,3-dioxoindole 2h, hydrazinyl benzenesulfonamides 3c and 3d were relatively the most potent ones. Derivative 2h showed promising potency against all tested isoforms with the best inhibitory activity towards hCAI and hCA II (KI = 45.10 and 5.87 nM, respectively) more than that of acetazolamide (AZZ) (250 and 12 nM, respectively). Moreover, compounds 3c and 3d showed good cytotoxicity where compound 3c revealed a promising selectivity towards human lung adenocarcinoma A549 (SI= 16.94). Docking simulation of the most active ones 2h and 3c predicted their binding pattern and binding affinity to the hCA II, IV, IX and XII active sites and rationalising their selectivity based on their docking binding patterns and scores. Furthermore, 2D-QSAR developed models using training set of the published data of our previous work of benzenesulfonamide derivatives as hCA IX and XII inhibitors showed sufficient predictive capability of the inhibitory activity with minimal error.
Supplemental Material
Download PDF (2.8 MB)Disclosure statement
No potential conflict of interest was reported by all authors. CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- Boone CD, Pinard M, McKenna R, Silverman D. Catalytic mechanism of α-class carbonic anhydrases: CO2 hydration and proton transfer. Subcell Biochem 2014;75:31–52.
- McKenna R, Supuran CT. Carbonic anhydrase inhibitors drug design. Subcell Biochem 2014;75:291–323.
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72.
- Luca LD, Mancuso F, Ferro S, et al. Inhibitory effects and structural insights for a novel series of coumarin-based compounds that selectively target human CA IX and CA XII carbonic anhydrases. Eur J Med Chem 2018;143:276–82.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- Awadallah FM, El-Waei TA, Hanna MM, et al. Synthesis, carbonic anhydrase inhibition and cytotoxic activity of novel chromone-based sulfonamide derivatives. Eur J Med Chem 2015;96:425–35.
- Awadallah FM, Bua S, Mahmoud WR, et al. Inhibition studies on a panel of human carbonic anhydrases with N1-substituted secondary sulfonamides incorporating thiazolinone or imidazolone-indole tails. J Enzyme Inhib Med Chem 2018;33:629–38.
- Ghorab MM, Alsaid MS, Soliman AM, Al-Mishari AA. Benzo[g]quinazolin-based scaffold derivatives as dual EGFR/HER2 inhibitors. J Enzyme Inhib Med Chem 2018;33:67–73.
- Ghorab MM, Alsaid MS, Soliman AM, Ragab FA. VEGFR-2 inhibitors and apoptosis inducers: synthesis and molecular design of new benzo[g]quinazolin bearing benzenesulfonamide moiety. J Enzyme Inhib Med Chem 2017;32:893–907.
- Mboge MY, McKenna R, Frost SC. Advances in anti-cancer drug development targeting carbonic anhydrase IX and XII. Top Anti-Cancer Res 2015;5:3–42.
- Supuran CT. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs 2018;27:963–70.
- Krasavin M, Korsakov M, Dorogov M, et al. Probing the 'bipolar' nature of the carbonic anhydrase active site: aromatic sulfonamides containing 1,3-oxazol-5-yl moiety as picomolar inhibitors of cytosolic CA I and CA II isoforms. Eur J Med Chem 2015;101:334–3478.
- Ibrahim HS, Allam HA, Mahmoud WR, et al. Dual-tail arylsulfone-based benzenesulfonamides differently match the hydrophobic and hydrophilic halves of human carbonic anhydrases active sites: Selective inhibitors for the tumor-associated hCA IX isoform. Eur J Med Chem 2018;152:1–9.
- Chiaramonte N, Romanelli MN, Teodori E, Supuran CT. Amino acids as building blocks for carbonic anhydrase inhibitors. Metabolites 2018;8:36–57.
- Supuran CT. Diuretics: from classical carbonic anhydrase inhibitors to novel applications of the sulfonamides. Curr Pharm Des 2008;14:641–8.
- Borras J, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406.
- Supuran CT. Acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6.
- Ghorab MM, Soliman AM, Alsaid MS, Askar AA. Synthesis, antimicrobial activity and docking study of some novel 4-(4, 4-dimethyl-2, 6- dioxocyclohexylidene) methylamino derivatives carrying biologically active sulfonamide moiety. Arab J Chem 2020;13:545–56.
- Borne RF, Peden RL, Waters I, et al. Anti-inflammatory activity of para-substituted N-benzenesulfonyl derivatives of anthranilic acid. J Pharm Sci 1974;63:615–7.
- Soliman AM, Karam HM, Mekkawy MH, Ghorab MM. Antioxidant activity of novel quinazolinones bearing sulfonamide: potential radiomodulatory effects on liver tissues via NF-kB/PON1 pathway. Eur J Med Chem 2020;197:112333.
- Soliman AM, Alqahtani AS, Ghorab M. Novel sulphonamide benzoquinazolinones as dual EGFR/HER2 inhibitors, apoptosis inducers and radiosensitizers. J Enzyme Inhib Med Chem 2019;34:1030–40.
- Ghorab MM, Alsaid MS, El-Gaby MS, et al. Biological evaluation of some new N-(2,6-dimethoxypyrimidinyl) thioureido benzenesulfonamide derivatives as potential antimicrobial and anticancer agents. Eur J Med Chem 2016;124:299–310.
- Ghorab MM, Ragab FA, Heiba HI, Soliman AM. Anticancer and radiosensitizing evaluation of some new sulfonamide derivatives bearing pyridone, thiophene, and hydrazone moieties. Res Chem Intermed 2017;43:4657–81.
- Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol 2007;25:884–96.
- Abo-Ashour MF, Eldehna WM, Nocentini A, et al. Novel hydrazido benzenesulfonamides-isatin conjugates: Synthesis, carbonic anhydrase inhibitory activity and molecular modeling studies. Eur J Med Chem 2018;157:28–36.
- Abo-Ashour MF, Eldehna WM, Nocentini A, et al. 3-Hydrazinoisatin-based benzenesulfonamides as novel carbonic anhydrase inhibitors endowed with anticancer activity: Synthesis, in vitro biological evaluation and in silico insights. Eur J Med Chem 2019;184:111768.
- Eldehna WM, Abo-Ashour MF, Nocentini A, et al. Enhancement of the tail hydrophobic interactions within the carbonic anhydrase IX active site via structural extension: Design and synthesis of novel N-substituted isatins-SLC-0111 hybrids as carbonic anhydrase inhibitors and antitumor agents. Eur J Med Chem 2019;162:147–60.
- George RF, Said MF, Bua S, Supuran CT. Synthesis and selective inhibitory effects of some 2-oxindole benzenesulfonamide conjugates on human carbonic anhydrase isoforms CA I, CA II, CA IX and CAXII. Bioorg Chem 2020;95:103514.
- George RF, Bua S, Supuran CT, Awadallah FM. Synthesis of some N-aroyl-2-oxindole benzenesulfonamide conjugates with carbonic anhydrase inhibitory activity. Bioorg Chem 2020;96:103635.
- Kumar R, Kaur M, Bahia MS, Silakari O. Synthesis, cytotoxic study and docking based multidrug resistance modulator potential analysis of 2-(9-oxoacridin-10(9H)-yl)-N-phenyl acetamides. Eur J Med Chem 2014;80:83–91.
- Monforte AM, Ferro S, Luca LD, et al. Design and synthesis of N1-aryl-benzimidazoles 2-substituted as novel HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem 2014;22:1459–67.
- Wang Y, Chan FY, Sun N, Lui HK, et al. Structure-based Design, Synthesis, and Biological Evaluation of Isatin Derivatives as Potential Glycosyltransferase Inhibitors. Chem Biol Drug Des 2014;84:685–96.
- Akgul O, Tarikogullari AH, Kose FA, Ballar P. Synthesis and cytotoxic activity of some 2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)acetamide derivatives. Turkish J Chem 2013;37:204–12.
- Firoozpour L, Gao L, Moghimi S, et al. Efficient synthesis, biological evaluation, and docking study of isatin based derivatives as caspase inhibitors. J Enzyme Inhib Med Chem 2020;35:1674–84.
- Soliman R. Preparation and antidiabetic activity of some sulfonylurea derivatives of 3,5-disubstituted pyrazoles. J Med Chem 1979;22:321–5.
- Wani TV, Bua S, Khude PS, et al. Evaluation of sulphonamide derivatives acting as inhibitors of human carbonic anhydrase isoforms I, II and Mycobacterium tuberculosis β-class enzyme Rv3273. J Enzyme Inhib Med Chem 2018;33:962–71.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stopflow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Nocentini A, Trallori E, Singh S, et al. 4-Hydroxy-3-nitro-5-ureido-benzenesulfonamides selectively target the tumor-associated carbonic anhydrase isoforms IX and XII showing hypoxia-enhanced antiproliferative profiles. J Med Chem 2018;61:10860–74.
- Entezari Heravi Y, Bua S, Nocentini A, et al. Inhibition of Malassezia globosa carbonic anhydrase with phenols. Bioorg Med Chem 2017;25:2577–82.
- Nocentini A, Carta F, Tanc M, et al. Deciphering the mechanism of human carbonic anhydrases inhibition with sulfocoumarins: computational and experimental studies. Chemistry 2018;24:7840–4.
- Nocentini A, Lucidi A, Perut F, et al. α,γ-Diketocarboxylic acids and their esters act as carbonic anhydrase IX and XII selective inhibitors . ACS Med Chem Lett 2019;10:661–5.
- Skehan P, Storeng R, Scudiero D, et al. 82(13):, New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107–12.
- Allam RM, Al-Abd AM, Khedr A, et al. Abdel-Naim AB. Fingolimod interrupts the cross talk between estrogen metabolism and sphingolipid metabolism within prostate cancer cells. Toxicol Lett 2018;291:77–85.
- Eldehna WM, Fares M, Ceruso M, et al. Amido/ureidosubstituted benzenesulfonamides-isatin conjugates as low nanomolar/subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform XII . Eur J Med Chem 2016;110:259–66.
- Smirnov AS, Nikolaev DN, Gurzhiy VV, Smirnov SN, Suslonov VS, et al. Conformational stabilization of isatin Schiff bases-biologically active chemical probes. RSC Adv 2017;7:10070–3.
- Chia SK, Wykoff CC, Watson PH, et al. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J Clin Oncol 2001;19:3660–8.
- İlie M, Mazure NM, Hofman V, et al. High levels of carbonic anhydrase IX in tumour tissue and plasma are biomarkers of poor prognostic in patients with non-small cell lung cancer. Br J Cancer 2010;102:1627–35.
- Available from: https://www.rcsb.org
- Kanamori K, Roberts JD. Nitrogen-15 nuclear magnetic resonance study of benzenesulfonamide and cyanate binding to carbonic anhydrase. Biochemistry 1983;22:2658–64.
- Briganti F, Mangani S, Orioli P, et al. Carbonic anhydrase activators: X-ray crystallographic and spectroscopic investigations for the interaction of isozymes I and II with histamine. Biochemistry 1997;36:10384–92.
- Helland IS. On the structure of partial least squares regression. Comm Statist B, Simulation Comput 1988;17:581–607.
- Geladi P, Kowalski R. Partial least squares regression: a tutorial. Analytica Chimica Acta 1986;185:1–17.
- Chemopy Descriptor Calculator. http://www.scbdd.com/chemopy_desc/index/ (6/10/2021).
- Pearlman RS, Smith KM. Novel software tools for chemical diversity. Persp Drug Disc Des 1998;9:339–53.
- Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity - a rapid access to atomic charges. Tetrahedron 1980;36:3219–28.
- Wildman SA, Crippen GM. Prediction of physiochemical parameters by atomic contributions. J Chem Inf Comput Sci 1999;39:868–73.
- Balaban AT. Five new topological indices for the branching of tree-like graphs. Theoretica Chimica Acta 1979;53:355–75.
- Gramatica P, On the Development and Validation of QSAR Models. In: Computational toxicology: Volume II, Methods in molecular biology. Reisfeld MA. Springer Science + Business Media, LLC; 2013, p. 499–526.