
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Human has used plants to treat many civilisation diseases for thousands of years. Examples include reserpine (hypertension therapy), digoxin (myocardial diseases), vinblastine and vincristine (cancers), and opioids (palliative treatment). Plants are a rich source of natural metabolites with multiple biological activities, and the use of modern approaches and tools allowed finally for more effective bioprospecting. The new phytochemicals are hyaluronidase (Hyal) inhibitors, which could serve as anti-cancer drugs, male contraceptives, and an antidote against venoms. In turn, tyrosinase inhibitors can be used in cosmetics/pharmaceuticals as whitening agents and to treat skin pigmentation disorders. However, the activity of these inhibitors is stricte dependent on their structure and the presence of the chemical groups, e.g. carbonyl or hydroxyl. This review aims to provide comprehensive and in-depth evidence related to the anti-tyrosinase and anti-Hyal activity of phytochemicals as well as confirming their efficiency and future perspectives.
Graphical Abstract
1. Introduction
Before the advancement of modern science, herbal medicines were widely used in ethnomedicine. Modern medicine primarily uses plant active ingredients that can be divided into primary and secondary metabolites. Secondary metabolites are defined as substances that are not directly necessary for the organism’s growth and development. It is believed that they play an important role in adapting plants to changing external conditions. Secondary metabolites are formed in various metabolic pathways, including amino acids and sugars intermediates, therefore, plants can synthesise many structurally diverse metabolites. It is estimated that plants produce at least 250,000 natural products, many of which have not yet been identified and characterised in structure and biological activityCitation1. The enormous diversity in the plant world (250,000–300,000 species) has provided many substances currently used in treatmentCitation2,Citation3. The isolation of morphine and codeine from Papaver somniferum L. ensured an effective pain treatmentCitation4. Obtaining digoxin from Digitalis purpurea L. allowed for an effective treatment of atrial fibrillation and heart failureCitation5. Salicylic acid, which is found, among others, in Salix alba L., after esterification forms acetylsalicylic acid, used in pain and thrombotic diseasesCitation6. The isolated artimizin from Artemisia annua L. is used in the treatment of a resistant form of malariaCitation7. Other compounds used in the treatment are capsaicin from Capsicum annuum L.Citation8, quinine from Cinchona L.Citation9, and inulinCitation10. Additionally, some compounds, after structure modification, have contributed to a new group of drugs. A good example is a phlorizin, which has served as the host structure for SGLT2 sodium-glucose co-transporter inhibitors, e.g. dapagliflozinCitation11. The development of new branches of science, such as genetics, biotechnology, and molecular biology, allowed for a better understanding of diseases’ pathophysiologyCitation12,Citation13. Hyals and tyrosinases are an underestimated group of enzymes commonly found in the world of organisms. Hyal, due to its participation in many biological processes, such as fertilisation, diffusion of toxins and microorganisms, inflammatory and allergic reactions, and cancer development, is perceived as a potential therapeutic targetCitation14. Besides, the inhibitors of tyrosinases and Hyals can be used in cosmetology to develop cosmetics or drugs used in dermatological diseases, such as eczema, acne, discolouration, and photoagingCitation15–17.
This study aims to systematise knowledge about the Hyal and tyrosinase inhibitors. We have hypothesised that there are the plant-based compounds and their chemically modified derivatives which inhibit the Hyal and tyrosinase.
2. Methods
To confirm our hypothesis we have searched for the different available databases (ScienceDirect, PubMed, Scopus, Web of Science, Google Scholar, and ClinicalTrials) in the regard to the relationship between the structure and activity of inhibitors, their action’s mechanism, and the prospects for their use in treatment. Search terms included “natural substances”, “plant substances”, “polyphenols”, “phenolic acids”, “chalcones”, “stilbenes”, “lignans”, “terpenes”, “alkaloids”, “glycosides”, “tyrosinase”, “tyrosinase inhibitors”, “hyaluronidase” (Hyal), “hyaluronidase inhibitors”, and “bacterial lyase”. The search equation was defined according to the formula [tyrosinase inhibitor OR tyrosinase OR Hyal inhibitor OR Hyal OR bacterial lyase inhibitor OR bacterial lyase] AND [natural substances OR plant substances OR polyphenols OR phenolic acids OR chalcones OR stilbenes OR lignans OR terpenes OR alkaloids OR glycosides]. Publications were searched from January 1990 to December 2021.
3. Types of inhibition
Inhibition is the process that slows down or completely stops a chemical reaction by a substance called an inhibitor. Depending on the way of binding of the inhibitor with the enzyme, one can distinguish reversible and irreversible inhibition ().
Table 1. Effect of the inhibitor on Km and Vmax.
3.1. Irreversible inhibition
An irreversible inhibitor is a compound with a structure similar to the substrate or product that forms a covalent bond with a group present in the active centre. In the case of irreversible inhibition, it is not necessary to maintain the inhibitor concentration at a sufficient level to ensure enzyme-substrate interaction. The complex formed does not dissociate, so the enzyme is inactive even when the inhibitor is absent. In contrast to irreversible inhibition, reversible inhibition is characterised by dissociating the enzyme-inhibitor complex. There are the following types of reversible inhibition: competent, incompetent, acompetent, and mixedCitation18.
3.1.1. Competent inhibition
The inhibitor shows a structural similarity to the substrate with which it competes for access to the enzyme’s active centre. The enzyme-inhibitor complex converts to an enzyme-substrate complex, and the inhibitor is displaced from the enzyme’s active site. The degree of inhibition of the enzyme depends on the concentration of the substrate. The higher substrate concentration relative to the inhibitor, the fewer enzyme molecules will bind to the inhibitor. Therefore, the inhibition of the reaction caused by a competent inhibitor can be reversed by increasing the substrate concentration.
3.1.2. Noncompetent inhibition
A noncompetent inhibitor usually bears no resemblance to the structure of the substrate. The inhibitor binds to the enzyme at a different site than the substrate. The inhibitor can attach to the free enzyme or the enzyme-substrate complex. Unlike competent inhibition, incompetent inhibition does not depend on the concentration of the substrate.
3.1.3. Accompetent inhibition
The inhibitor binds to the enzyme-substrate complex, forming an enzyme-inhibitor-substrate complex.
3.1.4. Mixed inhibition
It is a case of partially competent and incompetent inhibition. The inhibitor can bind to both the free enzyme and the ES complex, reducing the maximum rateCitation19.
4. Inhibitors of hyaluronidase
4.1. Hyaluronic acid and hyaluronidase
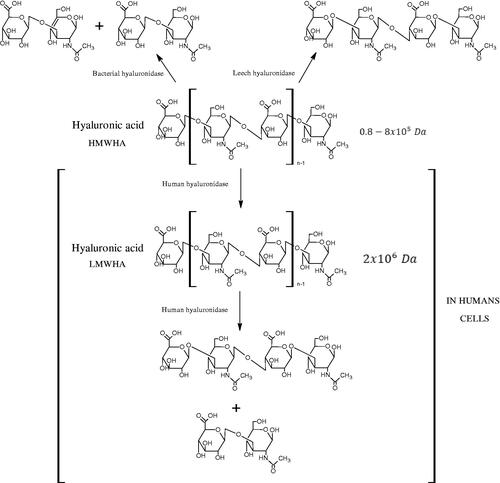
Hyal is responsible for the hydrolysis of hyaluronic acid (HA). HA is a linear, high molecular weight unsulfated polysaccharide composed of alternating N-acetyl D-glucosamine and D-glucuronic acid residues linked by glycosidic bonds. That compound is present both in prokaryotes and eukaryotes, commonly found in many tissues and body fluids, such as muscles, joints, synovial fluid, skin, and vitreous body. In addition to its structural functions, it is involved in wound healing, inflammation, and tumour development. Hyals are a group of enzymes commonly found in nature, e.g. they are an important component of the venom of bees, spiders, and snakesCitation20–22. Based on the structure and mechanism of action, there are three classes of Hyals (). The first class includes endo-β-N-acetylhexosaminidases (EC 3.2.1.35) present in mammals and in snake and hymenoptera venom. They hydrolyse β-1,4-glycosidic bonds in HA. The second class is endo-β-D-glucuronidases (EC 3.2.1.36), which hydrolyse the β-1,3-glycosidic linkages in the HA and are present in invertebrates. As a result of the activity of both enzymes, tetra, and hexasaccharide of HA are formed. The third class includes endo-β-N-acetylhexosaminidases (EC 4.2.2.1), present only in prokaryotes. Bacterial enzymes break β-1,4-glycosidic bonds in HA using a β-elimination reaction. As a result of their action, unsaturated di-, tetra-, and hexasaccharide are formed. There are five types of Hyals found in humans: HYAL-1, HYAL-2, HYAL-3, HYAL-4, and HYAL-5. HYAL-1 and HYAL-2 are found in most tissues and are involved in the circulation of HACitation23. HYAL-2 degrades long-chain hyaluronic acid (HMWHA) found in the extracellular matrix to short-chain hyaluronic acid (LMWHA), which after binding to the CD44 receptor, is transported into the cell. HYAL-1 is found inside the cell and is responsible for the degradation of LMWHA into tetra and hexasaccharideCitation24.
Figure 1. Classification of hyaluronidases (HMWHA: long-chain hyaluronic acid; LMWHA: short-chain hyaluronic acid).
Hyals are involved in the regulation of important physiological processes, such as fertilisation and skin ageing. Present in sperm, HYAL-5 plays a key role in fertilisation in mammals as it enables the sperm to connect to the egg cell by breaking down HA in the granule layer. The overactivity of collagenases, Hyal, and elastase leads to the formation of wrinklesCitation25.
Hyals are an important virulence factor for many bacterial species, such as Staphylococcus spp., Streptococcus spp., and Streptomyces spp. The decomposition of HA increases the viscosity of body fluids and reduces tissues’ integrity, facilitating the penetration of microorganisms and toxins into the skinCitation26.
Hyals are an important component of the venom of Hymenoptera, spiders, and snakes. Hyal, present in the venom, helps to distribute the toxins throughout the body. Several studies have shown Hyal to be involved in tumour growth, metastasis, and angiogenesisCitation27. However, the conducted studies do not provide unambiguous results showing that Hyals can function both as a suppressor and a promoter of carcinogenesis. HA promotes tumour metastasis, so the enzyme that breaks down hyaluronan (Hyal) inhibits their growth. On the other hand, low molecular weight HA stimulates angiogenesis, promoting tumour growth and metastasis formationCitation28.
4.2. Polyphenols as inhibitors of hyaluronidase: structure–activity relationships (SARS)
4.2.1. Flavonoids
Plant-based metabolites represent the chemically-different groups of compounds, of which many have been isolated for the first time from the plants used in the ethnomedicine of indigenous tribes. Those steps have allowed for their identification, biotechnological modification of their structure or to develop a synthesis path involving bacteria or fungi.
Phenolic compounds include a very numerous and important group of compounds commonly found in the world of plants. Polyphenols are plant secondary metabolites with very diverse chemical structures, containing at least two hydroxyl groups attached to an aromatic ring. Due to the number and way of connecting aromatic rings, we can distinguish phenolic acids, flavonoids, stilbenes, and lignansCitation29,Citation30. The presence of multiple hydroxyl groups gives phenolic compounds antioxidant propertiesCitation31. Phenolic compounds also show antitumor, anti-inflammatory, antiviral, antibacterial, antifungal, hepatoprotective, antiallergic, anticoagulant, and blood vessel sealing propertiesCitation32. Besides, many plant-derived polyphenols affect Hyal and other enzymes that regulate the metabolism of the extracellular matrixCitation33–36.
Inhibition of Hyal activity by polyphenolic compounds is related to, among others, the presence of hydroxyl groups (). Hertel et al. investigated the effect of flavonoids with a different number of hydroxyl groups on Hyals’ activity [flavones (apigenin and luteolin) and flavonols (kaempferol and quercetin) in concentration 0.1 mM]. In the case of bovine testicular Hyal (4 U/mL), the activity of the tested compounds was low (less than 20% for apigenin and luteolin). The inhibition of Hyal was only slightly enhanced by hydroxyl groups (quercetin or myricetin). Phenolic compounds with an additional hydroxyl group in the 3-position (quercetin ca. 90% inhibition or myricetin – ca. 70% inhibition) more strongly inhibited Streptococcus agalactiae hyaluronan lyase. Additionally, the introduction of a sugar moiety significantly reduces the test compounds’ activity (e.g. rutin). Aglycones were more potent inhibitors than their corresponding glycosides, what may probably result from the inhibitor’s difficult access to the Hyal active site in case of glycosidesCitation37.
Figure 2. Chemical groups of flavonoids involved in the inhibition of hyaluronidase.
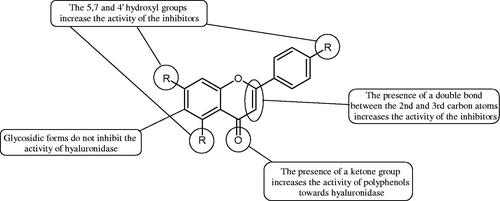
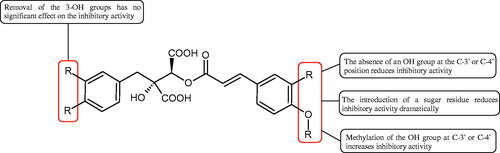
Another studyCitation38,Citation39 determined the effect of luteolin, apigenin, kaempferol, myricetin, quercetin, and morine on bovine testicular hyaluronidase (BTH). Flavonoids containing a double bond between the 2 and 3 carbon atoms showed a greater potency than flavonoids without the double bond. Additionally, a ketone group in the 4-position or hydroxyl group at 5,7, and 4′ positions may increase the inhibition of Hyal. In turn, methoxylation of the 4′-OH group in hesperitin and diosmetin reduces their activity. On the other hand, the presence of the 3-OH group did not affect the potency of Hyal inhibition. In addition, the presence of a catechol system in the B ring (3'4′-OH) may show a beneficial effect on the inhibitory activity of flavonoids. As in other studies, glycoside substituent presence completely reverses the inhibitory effect of flavonoids on BTH. The compounds possessing the malonyl group in the C-6 position of the sugar moiety, such as apigenin-7-O-(6′′-O-malonyl)glucoside (IC50 = 360 µM) or luteolin-7-O-(6′′-O-malonyl)glucoside (IC50 = 324 µM) show stronger inhibitory properties than compounds lacking this group, i.e. apigenin-7-O-rutinoside (IC50 > 1000 µM), naringenin7-O-rutinoside (IC50 > 1000 µM) and luteolin-7-O-glucoside (IC50 = 695 µM)Citation40. In another study, a relationship between the position of the sugar moiety and an activity against Hyal was found [apigenin 7-O-(3′’-O-acetyl)-glucuronide (200 µM, inhibition 10.0%) and apigenin 5-O-(3′’-O-acetyl)-glucuronide (200 µM, inhibition 13.9%)]Citation41.
Compounds containing a sugar residue in the C-3 position weakly block Hyal [kaempferol-3-O-α-L-rhamno pyranoside (18.26%), quercetin-3-O-α-L-rhamno pyranoside (6.88%), and quercetin-3-O-α-L-arabino pyranoside (13.81%)] ()Citation42.
Table 2. Structure of flavonoid glycosides with an anti-hyaluronidase activity.
Another possible mechanism involved in an inhibition of Hyals’ activity is based on the ability to associate compounds of low molecular weight. The acid function (hydroxyl, carboxyl, phosphate, or sulphate) is necessary to form an aggregate with multiple negative charges. More research is needed to determine the possibility of aggregating by low molecular weight flavonoids that can act as effective inhibitory units.
Bralley et al. determined antioxidants’ influence (phenolic acids, flavonoids, and condensed tannins) in Sorghum on Hyal activity. Amongst tested compounds, i.e. condensed tannin, apigenin, luteolin, kaempferol, quercetin, and rutin, only the four first were effective. Probably condensed tannins not only denature the enzyme but also interact with the hydrophobic channelCitation43. Tatemoto et al. investigated the effect of tannin, apigenin, and quercetin on Hyal activity and the fertilisation process in vitro. Tannins showed the greatest inhibitory properties at concentrations of 2–10 µg/mL. These data suggest that an adequate concentration of tannic acid prevents polyspermia by inhibiting sperm Hyal activity during IVF of porcine oocytes. However, the presence of apigenin or quercetin in the same concentrations as tannic acid could not prevent polyspermyCitation44.
The study by Zeng et al. determined how apigenin, luteolin, kempferol, quercetin, morin, naringenin, daidzein, and genistein bind to HAase. It was shown that those compounds interacted with the active centre of the enzyme through electrostatic forces, hydrophobic interactions, and hydrogen bonds. The binding of flavonoids caused changes in the active centre structure, resulting in inhibition of HAase ()Citation45.
Table 3. Structures of the active compounds.
4.2.2. Phenolic acids
Phenolic acids have demonstrated potent anti-Hyal properties. The impact of rosmarinic acid (IC50 = 24.3 µg/mL), protocatechuic acid (IC50 = 107.6 µg/mL), ferulic acid (IC50 = 396.1 µg/mL), and chlorogenic acid (IC50 = 162.4 µg/mL) on the activity of Hyal was noted.
Iwanaga et al. investigated the composition and effects of aqueous-acetone extracts from the aerial parts of Cimicifuga simplex and Cimicifuga japonica on HAase. The newly isolated fukiic acid derivatives (IC50 of compound 1. 255 µM; 2. 102 µM; 3. 173 µM; 4. 120 µM) inhibited Hyal more potently than rosmarinic acid (IC50 = 545 µM), caffeic acid (IC50 > 2000 µM), ferulic acid (IC50 > 2000 µM) and isoferulic acid (IC50 > 2000 µM). Based on the structure of compounds nr 2 and 4, the methoxy groups at the C-3′′ and C-4′′ positions may participate in the Hyal inhibitionCitation46.
Oligomers composed of caffeic acid exhibited interesting anti-Hyal properties. Caffeic acid trimer isolated from Dracocephalum foetidum inhibited Hyal more strongly than disodium cromoglycate (IC50 = 220 µM and IC50 = 650 µM). Similar results were obtained by Aoshima’s team studying coffee acid oligomers isolated from Clinopodium gracile. It was appeared that coffee acid oligomers, such as clinopodic acid M, showed more potent inhibitory activity than rosmarinic acid (IC50 = 19 and 226 µM, respectively). The most active compounds possessed 3–(3,4-dihydroxyphenyl)-2-hydroxypropionic acid – danshensu grouping. The activity of other compounds with the structure of danshensu amounted to: clinopodic acid E (IC50 = 40 µM), clinopodic acid I (IC50 = 112 µM), clinopodic acid K(IC50 = 63 µM), clinopodic acid L(IC50 = 26 µM), clinopodic acid N(IC50 = 161 µM), clinopodic acid O(IC50 = 66 µM), clinopodic acid P(IC50 = 25 µM), clinopodic acid Q(IC50 = 165 µM), lithospermic acid (IC50 = 36 µM), salvianolic acid B (IC50 = 107 µM), and salvianolic acid A (IC50 = 206 µM). The IC50 values for compounds without the danshensu structure were equal 206 and 653 µM for clinopodic acid J and 8-epiblechnic acid, respectively. An interesting structure for further studies may be clinopodic acid E, which has instead of the structure of 2,3-dihydrobenzofuran, 1,4-benzodioxane. This compound is four times more potent than the analogue having the structure of 2,3-dihydrobenzofuran (clinopodic acid N). Additionally, as in other studies, a beneficial effect of the increase in oligomer mass on the inhibition of Hyal activity is seenCitation40,Citation47.
The structure of 1,4-benzodioxane is present in clinopodic acid C (IC50 = 80.1 µM) and clinopodic acid E (IC50 = 82.8 µM), isolated from the herbal drug takuran, which is produced from Lycopus lucidas. These compounds inhibited Hyal more strongly than rosmarinic acid (IC50 = 309 µM). It was found that an esterification of the carboxyl group associated with the structure of 1,4-benzodioxane decreased the activity of the compounds (lycopic acid A; IC50 = 134 µM; lycopic acid B; IC50 = 141 µM). Rosmarinic acid oligomers (trimer IC50 = 275 µM; tetramer IC50 = 183 µM) blocked Hyal more potent than rosmarinic acid (IC50 = 309 µM). Activity against hyalurinidase is also shown by seric acid A (IC50 = 119 µM), F (IC50 = 1330 µM), and G (IC50 = 1270 µM) isolated from the Oenanthe javanica root ( and ) ()Citation48.
Figure 3. Effect of alkyl chain length in phenolic acids on activity against hyaluronidase.
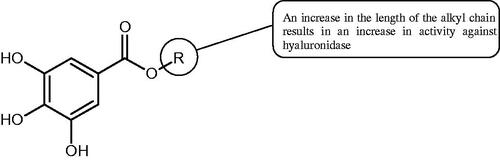
Figure 4. Potential groups engaged in an interaction oligomers phenolic acids-hyaluronidase.
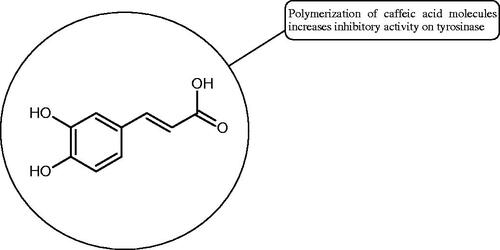
Table 4. Structure and activity of phenolic acids against hyaluronidase.
4.2.3. Tannins
Tannins are nitrogen-free plant substances of high molecular weight (500 − 3000), having numerous hydroxyl groups. Due to their chemical structure, they are divided into two groups: hydrolysing and non-hydrolysing (condensed) (). The first group is divided into galotannins (ester combinations of gallic acid and its derivatives) or elagotannins (ester combinations of ellagic acid). The second group is formed by the condensation of catechins (flavan-3-ol products). Tannins show the ability to form complexes with proteins, resulting in an astringent effect on the skin and mucous membranes. Tannins have been shown to have many properties, such as antibacterial, antiviral, anticancer, antioxidant, anti-inflammatory, and anti-hemorrhagicCitation49–51.
Figure 5. Potential groups engaged in an interaction fukiic acid derivative-hyaluronidase.
Sugimoto et al. investigated the effect of hydrolysing tannins isolated from an ethanolic extract of Eucalyptus globulus Labill. on Hyal activity (400 units/mL; from bovine testis Type IV-S). The following tannins were used in the study: pedunculagin (= 1.51 mM), tellimagrandin I (
= 0.9 mM), tellimagrandin II (
= 0.58 mM), heterophylliin A (
= 0.89 mM), 1,3-di-O-galloyl-4,6-hexahydroxydiphenoyl-β-D-glucose (
= 0.74 mM), 1,2,4-tri-O-galloyl-β-D-glucose (
= 1.57 mM), 1,2,3,6-tetra-O-galloyl-β-D-glucose (
= 0.35 mM), 1,2,4,6-tetra-O-galloyl-β-D-glucose (
= 0.68 mM), 1,2,3,4,6-penta-O-galloyl-β-D-glucose (
= 0.55 mM), ellagic acid (
= 4.66), gallic acid (
= 5.0 mM), and disodium cromoglycate (
= 0.45 mM) as a control. The activity of the tested compounds increases with the number of gallic acid residues attached to the sugar grouping (1,2,4-tri-O-galloyl-β-D-glucose −
= 1.57 mM, 1,2,4,6-tetra-O-galloyl-β-D-glucose −
= 0.68 mM, 1,2,3,4,6-penta-O-galloyl-β-D-glucose −
= 0.55 mM). Also, the localisation of gallic acid residues affects the inhibitory activity (1,2,3,6-tetra-O-galloyl-β-D-glucose −
= 0.35 mM vs. 1,2,4,6-tetra-O-galloyl-β-D-glucose −
= 0.68 mM). The inhibition level was similar for both ellagotannins and gallotannins. Another gallotannin, agrimoniin (
= 2.65 µM), also showed strong inhibition of Hyal activityCitation52.
In another studyCitation53, gallic acid esters with different n-alkanol chain lengths (from C-1 to C-12) were examined to determine their inhibitory activity against Hyal. With an increase of the alkyl chain length, the inhibitory activity was increased. Cromoglycan disodium (= 450 µM) was used as a positive control. Hexyl (
= 253 µM), heptyl (
= 112 µM), octyl (
= 106 µM), nonyl (
= 167 µM), and decyl (
= 580 µM) gallates inhibited Hyal. Next, the impact of hydroxyl groups in octyl gallate on an activity was checked. Octyl 3-hydroxybenzoate and octyl 4-hydroxybenzoate did not inhibit Hyal. Octyl 3,4-dihydroxybenzoate (
= 902 µM) blocked the enzyme to a small extent. The strongest inhibitor was octyl 3,5-dihydroxybenzoate (
= 113 µM). This shows the significance of the 3,5-OH grouping in gallic acid (). The type of inhibition was determined only for octyl gallate, which inhibited the enzyme in a competitive manner. The Ki value was estimated to be 45 µM ().
Figure 6. Chemical classification of tannins.
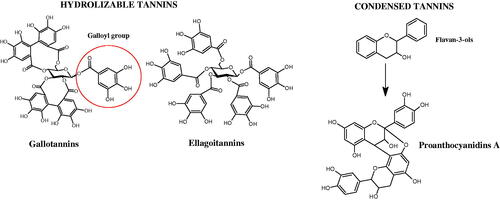
Table 5. Structures and activity of tannins and their esters with n-alkanol chain lengths against hyaluronidase.
Tokeshi et al. examined the effect of three tannins (tannic acid TA, gallic acid GA, and ellagic acid EA) on boar sperm Hyal. TA and EA strongly inhibited Hyal in the concentration range of 2–10 µMCitation54. The phlorotannins present in Eisenia bicyclis and Ecklonia kurome inhibited Hyal activity more strongly than standard substances such as disodium cromoglycate (IC50 = 270 µM), catechin (IC50 = 620 µM), and epigallocatechin gallate (IC50 = 190 µM). The IC50 values for phloroglucinol, phloroglucinol tetramer, eckol (trimer), phlorofucofuroeckol A (pentamer), dieckol, and 8,8′-bieckol (hexamers) were at the level of 280, 650, >800, 140, 120, and 40 µM, respectively. In the case of phlorofucofuroeckol A, dieckol, and 8,8′-bieckol an inhibition type (competitive inhibition) and the inhibition constant (Ki) values (130, 115, and 35 µM, respectively) were also established. Additionally, it was confirmed that the higher molecular weight of inhibitor the stronger inhibition is observed. This is probably associated with a stronger effect on the dimensional structure of the enzymeCitation55.
Procyanidin B1, procyanidin B3, epicatechin, and catechin exhibited a comparable inhibitory activity to disodium cromoglycate (51.1%) at the concentration of 250 µM (50.1, 28.9, 45.9, and 29.9%, respectively). At the concentration of 125 µM, epicatechin (30.3%), and procyanidin B1 (33.5%) showed a higher activity than DSCG (22.9%).
4.3. Non-polyphenols as inhibitors of hyaluronidase: structure–activity relationships (SARS)
4.3.1. Alkaloids
Natural alkaline nitrogen compounds are synthesised by plants, fungi, bacteria, and animals. Alkaloids, in minimal doses, have a strong physiological effect, especially on the central nervous system. Moreover, these compounds have antitumor, anaesthetic, antifungal, antibacterial, anti-inflammatory, and analgesic properties. Due to the heterocyclic ring system, we distinguish derivatives: pyridine and piperidine, tropane, quinoline, quinoline, indole, ergot, and purineCitation56. A study by Girish et al. determined the effect of aristolochic acid on the activity of purified Indian cobra venom hyaluronidase (NNH1) and the activity of whole venom Hyal. The tested compound inhibited NNH1 non-competitively. Besides, the venom’s administration with aristolochic acid to mice showed more than a twofold increase in survival time compared to mice injected with the venom alone. Lower survival was obtained by splitting the application of the inhibitor over time (10 min). Aristolochic acid did not bind to the enzyme’s active site but interacted with exposed tyrosine and tryptophan HAase residues. Aristolochic acid (50, 100, and 200 µM) inhibited NNH1 at the level of 100% for each concentration. Other alkaloids, ajmaline, and reserpine inhibited Hyal weaker than aristolochic acid (ajmaline 11, 26, 40%; reserpine 9, 23, 31%, respectively) ()Citation57.
Another studyCitation58 determined the effect of alkaloids isolated from the methanolic extract of Nelumbo nucifera Gaertn. flowers harvested at different stages of bloom (beginning of bloom, one-third in bloom, half in bloom, three-quarters in bloom, and full bloom). Samples flowering at half (52.69 mg per dried flower) had the highest alkaloid content (). Among the alkaloids, nornuciferin (= 22.5 µM), asymilobin (
= 11.7 µM), norarmepavin (
= 26.4 µM), coclaurin (
= 11.4 µM), and norjuzyfin (
= 24.3 µM) inhibited Hyal. The activity of alkaloids was more potent than that of the anti-allergic drug disodium cromoglycate (
= 64.8 µM). The N-methyl group reduces the ability of alkaloids to inhibit Hyal, e.g. nuciferine
> 100 µM < nornuciferine
= 22. µM or asimilobine
= 11.7 µM > N-methylasimilobine. On the other hand, demethylation of the hydroxyl groups increases their activity (asimilobine
= 11.7 µM > nornuciferine
= 22.5 µM). The observed structural relationships apply to both benzylisoquinoline alkaloids and apomorphine alkaloids ( and ).
Figure 7. Structure of benzylisoquinoline alkaloids and apomorphine alkaloids.
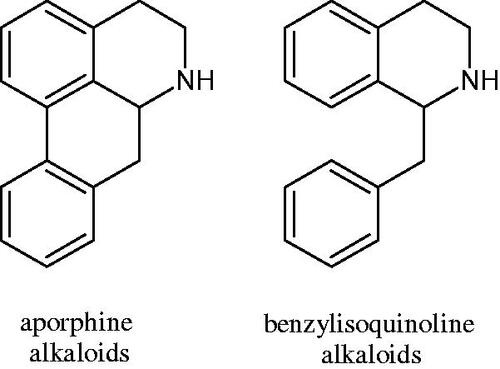
Table 6. Structures of the active alkaloids towards hyaluronidase.
Table 7. Activity of benzylisoquinoline alkaloids and apomorphine alkaloids against hyaluronidase.
4.3.2. L-ascorbic acid
Ascorbic acid (vitamin C – AA) is a compound commonly found in the world of plants and animals. Human is incapable of synthesising vitamin C, therefore, it must be supplied in the diet (parsley, red pepper, black currant, and Brussels sprouts). The ability of vitamin C to create an oxidative system (AA < => AA < => dehydroascorbic acid) determines its antioxidant properties. AA is the most important antioxidant of extracellular fluids in the human body. It is present in high concentrations in the eyeball and lymphocytes, protecting cells against reactive forms of oxygen and nitrogen. Besides its antioxidant activity, AA is involved in the absorption of non-heme iron; in the metabolism of fats, cholesterol, and bile; regeneration of vitamin E in the cell membrane; in the synthesis of collagen, accelerating the wound healing process. Additionally, the presence of AA in the skin may constitute a defense mechanism against an invasion of pathogenic bacteria. Despite the fact that AA is one of the most known biologically-active compound its anti-Hyal activity and SAR are still unknown in details. Several studies involving AA and AA derivatives have found to be able to inhibit HyalsCitation59–61.
In 2001, Li et al. first described a competitive type of AA inhibition on Hyal isolated from Streptococcus pneumoniae LHyal (hyaluronan lyase). The activity of AA towards Hyal is due to the structural similarity of vitamin C to glucuronic acid, being one of the basic building blocks of hyaluronan (HA) (β-1,4-glucuron-β-1,3-glucosamine). It was found that one AA molecule may bind to the enzyme’s active site. The AA carboxyl group provides a negative charge that directs the molecule to the positively charged enzyme gap. In the active centre of the enzyme, AA interacts with amino acids through hydrophobic (Trp-292), ionic (Arg-243, Arg-462, and Arg-466), and hydrogen (Tyr-408, Asn-290, and Asn-580) bondsCitation62.
In 2003, Okorukwu et al. confirmed the inhibitory effect of AA and AA analogs on the activity of bovine testicular Hyal (BTH – final concentration 3.5 Units/mL) and LHyal (Streptococcus zooepidemicus – final concentration 2.5 Units/mL). Gel permeation chromatography (GPC) was used in this study to evaluate the inhibitory activity. The AA and AA derivatives (D-iso-ascorbic acid and dehydroascorbic acid) blocked the hyaluronan lyase more strongly than BTH. D-Saccharic-1,4-lactone and saccharic acid inhibited LHyal without affecting the enzymatic activity of testicular Hyal. The introduction of a carboxyl group that gives the molecule a negative charge positively affects the inhibitory effect of AA derivatives. Hydrogenation of the double bond between the 2nd and 3rd carbon atoms decreases the activity of the compounds. Saccharic acid can be used to develop selective inhibitors of bacterial hyaluronan lyase ()Citation63.
Table 8. Structures of L-ascorbic acid and its derivatives with an anti-hyaluronidase activity.
In a study conducted by Botzki et al., a positive correlation was confirmed between the inhibition of Hyal activity and the increased hydrophobic interactions. L-Ascorbyl palmitate, through an increase in hydrophobic interactions with Phe343, His399, and Thr400 in the active centre, led to increased inhibition of hyaluronan lyase (competitive inhibition). A similar effect was achieved with BTH. The long alkyl chain interacts with a hydrophobic channel formed primarily by the amino acids Ala-84, Leu-91, Tyr-93, Tyr-220, and Leu-344Citation64.
The new LHyal inhibitors should have a larger ring system to favourably influence the hydrophobic bonding to the Trp-292 indole group and contain at least one negative charge group (carboxyl group), which brings the inhibitor to the cleft region (rich in positively charged arginine).
Spickenreither et al. examined the effect of 6-O-acylated AA derivatives on the Hyal activity of BTH and Streptococcus agalactiae strain 4755 (Sag Hyal 4755). All compounds showed more potent activity against bacterial lyase. Methylation of the endiol system reduces the activity of vitamin C analogs. On the other hand, 2 and 3 dibenzylated derivatives showed more potent inhibitory properties than AA. The increase in potency is due to additional hydrophobic interactions between the rings and the active centre. An increase in the length of the 6-O-acyl residue (13 b-j) results in increased inhibitory activity. The IC50 for octadecanoate was 0.9 and 39 µM for BTH and Sag Hyal 4755, respectively. Shortening of the aliphatic chain and adding phenyl, p-phenylene, or p-biphenyl groups leads to compounds with comparable inhibitory properties. Additionally, ether bonds in the synthesis of new inhibitors positively influence their activity. This is associated with the formation of additional hydrogen bonds in the active centre (; )Citation65.
Figure 8. Chemical groups of L-ascorbic acid involved in the inhibition of hyaluronidase.
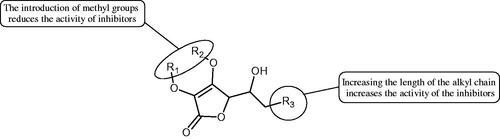
Figure 9. Chemical structures of saponins. A – triterpene saponins, B – steroid saponins, C – steroid alkaloids, and D – isoprene.
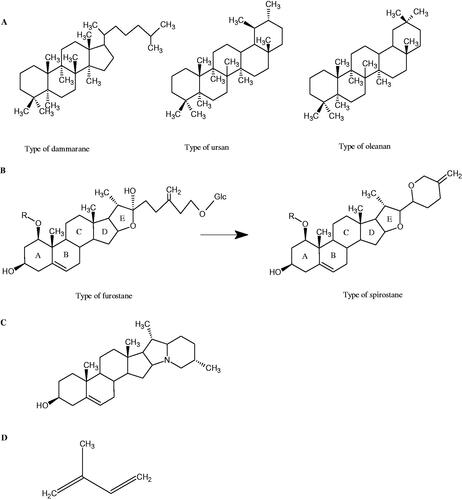
Table 9. Structures and activity of the vitamin C derivatives against hyaluronidase.Citation65
4.3.3. Glycosides
Glycosides are a group of the organic compounds consisting of sugar and an aglycone part. The bond between the sugar and the aglycone is called a glycosidic bond. The chemical nature of aglycones is very different, they can be alcohols, lactones, phenolic acids, thiols, etc. The sugar portion may consist of 1–12 monosaccharide, disaccharide, or oligosaccharide molecules. The glycosides widely occur in the plant world, especially in higher plants. Because of the chemical diversity of glycoside, the plant-based sources of glycosides are important in phytotherapyCitation66.
4.3.3.1. Cyanogenic glycosides
Cyanogenic glycosides are one of the glycosides groups that have an inhibitory effect on Hyal. In a study by Tanyildizi et al., it was checked how different doses of linamarine and amygdalin affect Hyal activity, motility, and morphology of bull sperm obtained from the Holstein bulls aged 2–3 years (). The samples were divided into 5 equal parts and mixed with linamarine at doses of 0.5, 0.75, 1, and 2 µM and with amygdalin at doses of 0.4, 0.8, 1, and 2 µM. Incubation of compounds with sperm resulted in a significant reduction (dose-dependent) of sperm motility and Hyal activity compared to the control group (isotonic saline solution). Both linamarine and amygdalin did not change sperm morphology. The authors concluded that the fertilisation ability of bull sperm could be inhibited by the over-consumption of plants rich in cyanogenic glycosidesCitation67.
Table 10. Structures of the active cyanogenic glycosides towards hyaluronidase.
4.3.4. Terpene/terpenoids
Terpenes are composed of a varying number of isoprene units (). They are commonly found in the plant world in hydrocarbons or oxidised forms (with hydroxyl, carbonyl, or carboxyl groups). Depending on the number of isoprene residues, we distinguish monoterpenes (C10), diterpenes (C-20), sesquiterpenes (C-15), triterpenes (C-20), and tetraterpenes (C-40). Due to their highly diverse chemical structure, terpenes exhibit a variety of biological activities, such as antibacterial, antiviral, anticancer, anti-inflammatory, and sedative effectsCitation68.
4.3.4.1. Monoterpene/monoterpenoids
Morikawa et al. investigated the effect of methanolic extract of rhizome of Picrorhiza kurroa Royle ex Benth. on Hyal activity (Type IV-S from bovine testes). Seven new acylated iridoid glycosides (picrorhizaosides A–G) and six known iridoid glycosides were isolated from the extract. Among the isolates, picrorhizaosides D (= 43.4 µM), picrorhizaosides E (
= 35.8 µM), picrosides I (
= 60.7 µM), picrosides II (
= 22.3 µM), picrosides IV (
= 59.2 µM), and minecoside (
= 57.2 µM), showed similar or stronger Hyal inhibitory effects than the anti-allergic drugs disodium cromoglycate (
= 64.8 µM), ketotifen fumarate (
= 76.5 µM), and tranilast (
= 227 µM), but weaker than the alkaloids isolated from Nelumbo nucifera Gaertn., such as asimilobin (
= 11.7 µM) and coclaurin (
= 11.4 µM)Citation69.
4.3.4.2. Saponin
Saponins belong to the glycosides group composed of aglycone – sapogenin (sapogenol) and glycone-sugar. Depending on the type of sapogenin we distinguish triterpene saponins, steroidal saponins, and steroidal alkaloids (). These compounds reduce the surface tension of water solutions. They show anti-inflammatory, antibacterial, protozoal, antifungal, and antiviral activity, stimulate secretion of gastric juice, bile, and intestinal juice. They can affect cholesterol levelCitation70.
In a study by Zhou et al., they determined the effects of esculeoside A and its aglycone esculeogenin A on Hyal activity in vitro and in mice dermatitis model. Esculeoside A, a spirosolate-type glycoside, is identified as a significant component of ripe tomato fruit. The for esculeogenin A and esculeoside A was about 2 and 9 µM, respectively. Administration of esculeoside A at a dose of 10 mg/kg for four weeks to mice with dermatitis significantly reduced diseases symptomsCitation71. Esculeoside A is a competitive inhibitor of hyalurnidase (Ki = 11.0 µM)Citation72. Also, the other steroid alkaloids present in tomato juice showed beneficial inhibitory effects against Hyal. Administration of 10 mg/kg esculeoside B to mice with dermatitis for four weeks significantly reduced skin inflammation. In addition, it was found that esculeoside B administration significantly inhibited T-lymphocyte proliferation and decreased IL-4 productionCitation73.
More information about the effect of saponin structure on Hyal activity was provided by QSAR studies of ursolic and oleanolic acids, the results of which exhibited the higher activity of ursolic acid than oleanolic acid. In that experiment, the effect of the position of methyl groups at 29 and 30 carbon atoms was checked. Both geminal and vicinal positions had no significant impact on an inhibitor activity. For oleanolic acid, the activity increased when the methyl group was introduced at C-17 or C-16 and decreased when the methoxyl group was introduced at C-23. The 3-OH acetylation reduced the activity of the compounds. Carboxylation of C-30 increased the activity of the compounds, while esterification of the same carbon (C-30). In addition, the introduction of a sugar moiety into the 3-OH decreased their activity. In the case of ursolic acid, the activity decreased when the hydroxyl group was modified at C-3 (3-oxo, 3-hydroxyimino, and 3-acetylate derivatives) and at C-28. Replacement of the methyl group at C-23 with a carboxyl or hydroxy methylene group decreased the activity of the compounds. As in the case of oleanolic acid, the introduction of a sugar group caused a decrease in the inhibitory activity (; )Citation74.
Figure 10. Chemical groups of triterpenic acids involved in the inhibition of hyaluronidase.
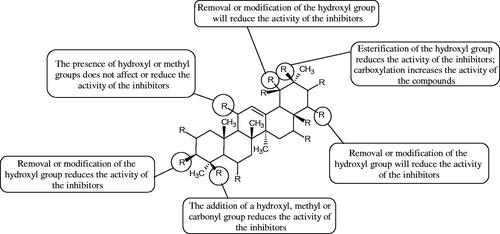
Figure 11. 3-O-β-D-glucuronopyranoside group.
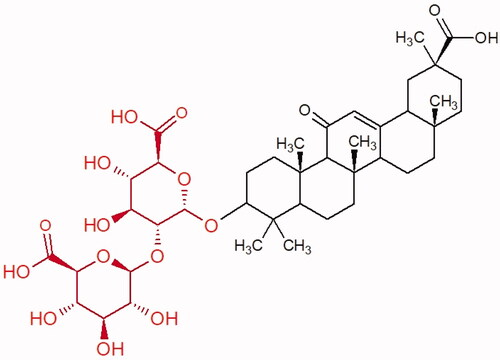
Table 11. Structures and an anti-hyaluronidase activity of oleanane-type saponinsCitation73.
Similar results were obtained in another experiment aimed at an investigation of the impact of the oleanic acid structure modification on anti-Hyal activity. Oxidation of the 3-OH group in oleanic acid led to a decrease of the activity about 4-fold (59.3–15.9%). A similar effect was obtained by esterification of the COOH group (12.7% inhibition). The activity of the compounds was evaluated at a concentration of 40 µg/mLCitation75.
In another studyCitation76, oleanane-type saponins, isoflavonoids, oxazoles, and glycosides (36 compounds) were isolated from the aerial part of Oxytropis lanata (Pall.) DC. The effect of saponins against Hyal (= 0.15–0.22 mM) was more potent than sodium cromoglycate, which was used as a positive control (
= 0.37 mM). On the basis of the structure analysis, it was appeared that all isolated saponins contain a 3-O-β-D-glucuronopyranoside grouping in the sugar moiety, which shows a potent inhibition of Hyal, as well as the compounds with a ketone group at C-22 more strongly blocked the enzyme (). Similar results were obtained when testing triterpene saponosides from the 3-O-β-D-glucuronopyranoside group, which blocked Hyal more strongly (2;
= 1.25 mM, 3:
= 0.68 mM, and 9:
= 0.82 mM) than rosmarinic acid used as a control (
= 1.36 mM)Citation77. The significance of the 3-O-β-D-glucuronopyranoside grouping was also noted by examining the triterpene saponosides camelliagenin A (IPS-1 and IPS-2) isolated from the methanolic root extract of Impatiens parviflora DC. A very interesting result has been obtained, because IPS-2 (
= 286.7 µg/mL) inhibited BTH Hyal activity more strongly than escin (
= 303.93 µg/mL). What is interesting, escin is recommended to be used as an anti-Hyal reference compound. The higher IC50 value was obtained for IPS-1; 368.1 µg/mL. Considering the structure of these compounds, a clear difference may be noticed significantly since the tested saponosides differed in their acetylation at C-16. The 16-O-acetylcameliagenin A derivative showed less activity. Therefore, the free OH group at position 16 may have a beneficial effect on Hyal inhibition, e.g. via participation in ions chelating in the reaction’s mediumCitation78.
Myose et al. demonstrated the effect of triterpene saponins isolated from methanolic extract of Camellia sinensis (L.) Kuntze seeds on Hyal activity. The isolated new saponin (Teaseedsaponin A-L) blocked Hyal more potently (= 19.3–55.6 µM) than rosmarinic acid (
= 240.1 µM)Citation79.
Another study examined how sugar moiety in saponins affects the activity of Hyals isolated from different bacterial species (Streptococcus agalactiae – Hyal B, Streptomyces hyalurolyticus – Hyal S, Streptococcus equisimilis – Hyal C) and bovine testes (BTH). Glycyrrhizin (1) and its aglycone glycyrrhetinic acid (2) were used as inhibitors. The tested compounds most potently blocked the activity of Hyal B [(1); = 0.440 mM, (2);
= 0.060 mM]. For other Hyals, the action was weak [Hyal S – (1); 1.020 mM, (2); 0.260 mM; Hyal C – (1); NA, (2); NA; BTH – (1); 1.300 mM, (2); 0.090 mM). Considering the results obtained, glycyrrhetinic acid inhibited the activity of tested enzymes weaker, which may prove the significance of the sugar moiety in the inhibition of Hyal activityCitation25.
Facino et al. investigated the inhibitory influence of saponins and sapogenins isolated from seeds of Aesculus hippocastanum L. (escin and escinol), leaves of Hedera helix L. (α-hederin, hederacoside C, oleanolic acid, and hederagenin), and rhizome of Ruscus aculeatus L. (ruscogenin). Of the Hedera helix L. components, only sapogenins inhibited Hyal in a dose-dependent manner. Hederagenin (= 280.4 µM) inhibited the enzyme at 100 µM (12.5%); 20.3% at 150 µM, 31% at 200 µM, 56.5% at 300 µM, and 74.2% at 500 µM (plateau). Oleanolic acid (
= 300.2 µM), 29.1% at 200 µM; 48.5% at 300 µM, and 67% (plateau) at 400 µM. Glycyrrhizic acid (positive control) blocked BTH Hyal significantly less (
= 550.2 µM). Hederacoside C and α-hederin showed no activity against Hyal. For Aesculus hippocastanurn L., escin had the highest activity (
= 149.9 µM), inhibiting Hyal starting at 50 µM (4.2%); at higher concentrations of 100 (27.4%), 150 (52.0%), 200 (79.2%), and 300 µM (93.6%). Escinol was much less active (
= 1.65 mM), and ruscogenin was utterly ineffective ()Citation80.
Table 12. Structures and an anti-hyaluronidase activity of the chosen saponins.
The olean-type saponins, spinasaponin A (lack of activity), spinasaponin A 28-O-glucoside (IC50 = 620 µM), udosaponin B (IC50 = 750 µM), and sandrosaponin IX (IC50 = 370 µM), isolated from the roots of Oenanthe javanica, showed moderate ability to inhibit Hyal. Esterification of the 28-COOH group with β-D-glucopranosyl increased the activity of the compoundsCitation81.
5. Inhibitors of tyrosinase
5.1. Tyrosine and tyrosinase
Tyrosinase is a key enzyme involved in melanogenesis. This enzyme belongs to the class of oxidoreductases (EC 1.14.18.1). It is responsible for the catalysis of tyrosine hydroxylation to L-DOPA and the oxidative conversion of L-DOPA to dopaquinone (). The active centre of tyrosinase consists of two copper atoms linked by a coordination bond with three histidine residues. Three different types of tyrosinase are involved in melanin production: oxy-tyrosinase, met-tyrosinase, and deoxy-tyrosinase. Oxy-tyrosinase and met-tyrosinase have Cu (II) copper atoms in their active centre, and deoxy-tyrosinase has two Cu (I) atoms. Deoxy-tyrosinase does not perform a catalytic function, but it is easily converted into the oxy form, which is the only form of the enzyme capable of transforming both monophenol and diphenol substrates. On the other hand, met-tyrosinase is formed during the reaction catalysed by the oxy form and is responsible only for reactions with diphenolic substrates ()Citation81–84.
Figure 12. A – reaction hydroxylation of monophenols to o-diphenols; reaction B – oxidation of o-diphenols to o-quinones.
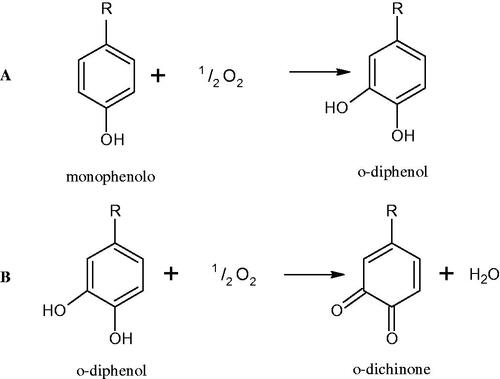
Figure 13. Mechanism of the tyrosinase action as monophenolase and diphenolase.
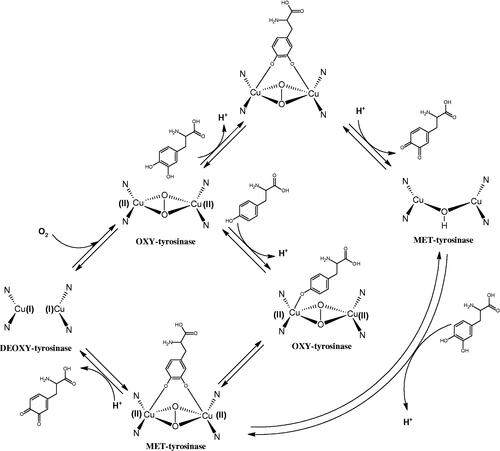
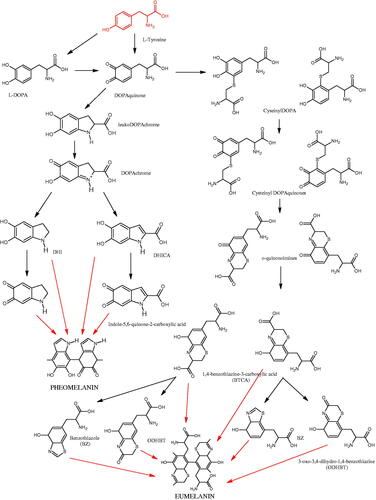
Tyrosinase is an enzyme widely distributed in nature, including in fungi and to a lesser extent in bacteria and algae. This enzyme is also found in plants and animals. In higher vertebrates, melatonin is produced in melanocytes present in the epidermis, hair follicles, the uveal membrane of the eye (choroid, ciliary body, and iris), the inner ear (cochlea), and the central nervous system (the arachnoid and internal filum terminale). Melanins are macromolecular polymeric pigments resulting from the oxidation and polymerisation of phenolic compounds (). Melanin synthesis takes place in vesicles called melanosomes and it is considered to be one of the most common pigments in nature. In mammals, melanosomal tyrosinase is involved in the formation of black-brown eumelanin and yellow-reddish phaeomelanin. Eumelanin has photoprotective properties, resulting from the ability to absorb ultraviolet radiation (UV) and neutralise free radicals and reactive oxygen species (ROS). On the other hand, pheomelanin has photosensitising properties and, under the influence of UV radiation, can participate in the generation of ROS. In mammals, melanin pigmentation performs many critical physiological tasks, such as adaptive colouration, protection of essential tissues against UV radiation, thermal control of the organism, regulation of vitamin D 3 biosynthesis. Abnormal tyrosinase activity is responsible for skin abnormalities such as vitiligo or freckles. Also, tyrosinase may play a role in carcinogenesis and neurodegenerative diseases such as Parkinson’s disease. Tyrosinase also contributes to the formation of brown colour in fruits and vegetables due to the reaction of dopaquinone with amino acids and proteins present in these foods. In most studies on the inhibition of tyrosinase activity, fungal tyrosinase was used due to its widespread availability. The enzyme isolated from the mushroom A. bisporus is very similar in structure to tyrosinase occurring in mammals, making it a suitable model for studying the process of melanogenesis. Since tyrosinase is a reasonably significant target in agriculture, food, medicine, and cosmetology, much attention has been paid to the development and screening of tyrosinase inhibitorsCitation85–87.
Figure 14. The pathway of melanin synthesis.
5.2. Polyphenols as inhibitors of tyrosinase: a structure–activity relationship
The positive effect of polyphenols on human health is mainly related to their antioxidant properties. The antioxidant activity of individual polyphenols depends on the number of hydroxyl groups and their location. It has been shown that the more hydroxyl groups in a molecule, the more potent antioxidant activity. Compounds with redox properties effectively prevent melanin biosynthesis due to their multidirectional mechanism of action. Tyrosinase inhibition by polyphenols is based on free radical scavenging properties and the ability to chelate copper in the tyrosinase active site.
5.2.1. Phenolic acids
Phenolic acids are one of the most common groups of organic compounds present in plants. They are made of a phenolic ring and a carboxylic acid residue. There are two subclasses of phenolic acids: derivatives of benzoic acid and cinnamic acid. Several studies have shown their inhibitory effect on tyrosinase activity and that activity was related to the number and position of hydroxyl groupsCitation88. Considering the structure of phenolic acids, three mechanisms of tyrosinase inhibition can be distinguished. The first is related to the chelation of copper ions in the active centre. The second mechanism is associated with the disturbance of the enzyme’s tertiary structure through hydrogen bond formation. The third mechanism involves constructing hydrogen bonds between the hydroxyl groups of phenolic acids and the carbonyl oxygen of the Tyr98 ORF378 protein, preventing the interaction between tyrosinase and ORF378. Consequently, ORF378 cannot serve as a Cu (II) ion transporter to the enzyme’s active siteCitation89,Citation90.
5.2.1.1. Hydroxybenzoic acids
Kubo et al. investigated the effect of anisic acid and its derivatives on L-DOPA oxidation by tyrosinase. As the concentration of anisic acid increased, the enzymatic activity decreased sharply but was not completely inhibited ( = 0.60 mM). The inhibition of fungal tyrosinase by anisic acid is a reversible reaction in which the tested acid is a non-competitive inhibitor (
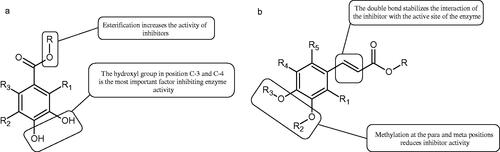
=0.603 mM). The modification of the alkyl chain in anisic acid influenced the activity and type of inhibition of the tested compounds. P-ethoxybenzoic acid showed a kind of incompetent inhibition, p-propoxybenzoic acid was of mixed, and p-butoxybenzoic acid was of competitive. With the increase in the alkyl chain’s length, the inhibitory activity of the tested compounds decreasedCitation91,Citation92.
Chen et al. obtained similar results when testing p-alkoxybenzoic acid derivatives. The tested compounds behave as reversible tyrosinase inhibitors in the presence of L-DOPA substrate (λ 475 nm; spectrophotometric method, 6680 U/mg). Among them, p-hydroxybenzoic acid (= 1.3 mM;
= 0.73 mM) is a competitive inhibitor, p-methoxybenzoic acid (
=0.42 mM;
= 0.43/0.43 mM) is non-competitive, p-ethoxybenzoic acid (
=1.1 mM;
= 1.46/0.84 mM) is of the mixed type, and the others show a type of non-competitive inhibition (p-propoxybenzoic acid, p-butoxybenzoic acid, p-pentoxybenzoic acid, and p-hexyloxybenzoic acid). Additionally, it was appeared that increasing the chain length above two carbon atoms changed the type of braking from competitive to non-competitiveCitation93. (22)
Another studyCitation94 determined the effect of methoxylation of hydroxyl groups on the tested acids’ activity and their esters. Protocatechuic acid methyl ester, protocatechuic acid, vanillic acid methyl ester, vanillic acid, isovanillic acid methyl ester, isovanillic acid, veratric acid methyl ester, and veratric acid were used in the study. Only protocatechuic acid and its methyl ester inhibited the enzyme (60.1 and 75.4% inhibition; =0.42 µmol/mL and 0.28 µmol/mL). The hydroxyl group at the para and meta position is an important part of the structure of inhibitors. Protocatechuic acid methyl ester inhibited the enzyme activity more strongly than protocatechuic acid, which may result from the esterification of the carboxyl group.
Kubo et al. provided more information about the impact of an esterification on the activity of gallic acid. It was appeared that, apart from gallic acid (4.5 mM), the of all esters was almost comparable (<0.5 mM). Based on the above observation, it can be concluded that gallic acid esters with an increase in the number of carbon atoms of the alkyl chain (>C10) may be more challenging to incorporate into the protein pocket to reduce the rate of oxidation by the enzyme. Hence, gallates with a longer alkyl chain (>C-10) become inhibitors but not substrates, which indicates that gallates with a longer alkyl group (>C-10) can be expected as more suitable inhibitors. However, tetradecanoyl gallate (C-14) and hexadecanyl gallate (C-16) are sparingly soluble in water (; Tables 1S and )Citation95.
Table 13. An anti-tyrosinase activity of hydroxybenzoic and hydroxycinnaminic acids and their derivatives (NR-not reported; a-L-DOPA; b-L-tyrosin).
5.2.1.2. Hydroxycinnaminic acids
Shi et al. investigated the effect of cinnamic acid and its derivatives on the activity of fungal tyrosinase. Cinnamic acid (=2.10 mM), 4-methoxycinnamic acid (
=0.42 mM), and 4-hydroxycinnamic acid (
=0.52 mM) strongly inhibited the conversion of diphenol to dichinone. Cinnamic acid and 4-methoxycinnamic acid showed a non-competitive type of inhibition (Ki = 1.994 and 0.458 mM), interacting with a different site of the enzyme than the active site. In contrast, 4-hydroxycinnamic acid competitively inhibited the enzyme (Ki = 0.244), which may result from the similar structure of the tested compound to the tyrosinase substrate. One of the tested compounds, i.e. 2-hydroxycinnamic acid did not inhibit the enzyme activity, probably because of the presence of the 2-OH group causing spherical hindrancesCitation96.
Another studyCitation97 assessed the influence of methoxylation of cinnamic acid and its derivatives (cinnamic acid, 2-hydroxycinnamic acid, 3-hydroxycinnamic acid, 4-hydroxycinnamic acid, 3,4-dihydroxycinnamic acid, 2-methoxycinnamic acid, 3-methoxycinnamic acid, and 4-methoxycinnamic acid; concentrations from 0.1 to 3 mmol/L) on the action of tyrosinase (3130 U/mg) in the presence of L-tyrosine or L-DOPA as a substrate. Cinnamic acid (=2.1 mM), 2-hydroxycinnamic acid (
=0.5 mM), and the O-methyl (
=0.42 mM) forms exhibited inhibitory properties compared to 3-hydroxycinnamic acid, 4-hydroxycinnamic acid, and 3,4-dihydroxycinnamic acid, which turned out to be substrates of tyrosinase. All inhibitors showed competitive inhibition. The inhibition constants are about the same for the oxidation of L-tyrosine and L-DOPA, indicating that the inhibitors bind to the same form of the enzyme. In the next studyCitation98, the inhibitory effect of caffeic acid and ferulic acid on tyrosinase activity isolated from murine B16 melanoma cells (90 U) was analysed. Both ferulic acid (27.4% non-toxic conc.) and caffeic acid (24.4% non-toxic conc.) effectively inhibited melanin production in B16 melanoma cells. Ferulic acid was reducing tyrosinase activity by binding directly to the enzyme, whereas no binding was observed between caffeic acid and tyrosinase.
One of the important structural elements regulating the enzyme’s activity is a type of bonds present in the inhibitor’s structure. Some compounds, such as p-coumaric acid (=115.6 µM; % inhibition 74.4) and isoferulic acid (
=114.9 µM; % inhibition 77.8) lose their tyrosinase inhibitory properties after saturation of the double bond (dihydro-p-coumaric acid
=1000 µM; 74.4% inhibition and dihydroisoferulic acid
=195.7 µM; 60.6% inhibition). None of the compounds tested were more effective than kojic acid (
=51.6 µM) or arbutin (
=210.5 µM). All compounds tested show a non-competitive type of inhibition. The results indicate that the reduction of double bonds weakens the inhibitory activity of the test compounds. The C = C binding is necessary for the proper interaction of the inhibitor with the active siteCitation99.
In another study, the type of inhibition and the effect of caffeic acid, p-coumaric acid, and rosmarinic acid on monophenolase and diphenalase activities were determined. The most active compound was p-coumaric acid (L-tyr, IC50 =0.3 µM; L-DOPA, IC50 =0.62 µM), which inhibited the enzyme noncompetitively (L-tyr, Ki=0.033 µM; L-DOPA, Ki=2.2 µM) followed by caffeic acid (L-tyr, IC50 =1.50 µM; L-DOPA, IC50 =2.30 µM) and rosmarinic acid (L-tyr, IC50 =4.14 µM; L-DOPA, IC50 =8.59 µM). These compounds blocked the enzyme more potently than kojic acid (L-tyr, IC50 =33.45 µM; L-DOPA, IC50 =38.98 µM) (; Tables 2S and )Citation100.
Figure 15. a) Potential groups engaged in an interaction hydroxybenzoic acid-tyrosinase and b) Potential groups engaged in an interaction hydroxycinnamic acid-tyrosinase.
5.2.2. Flavonoids
Flavonoids belong to a group of compounds commonly found in the plant world. These compounds differ from each other by the presence of a double bond between the second and third carbon atom, a ketone group at position 4, and the position of the B ring. Additionally, individual flavonoids differ from hydroxyl, methyl, isoprenoid, and methoxy groups arranged in different rings. Many flavonoids have tyrosinase inhibitory activityCitation101.
5.2.2.1. Hydroxyl groups
The distribution and number of hydroxyl groups in flavonoid molecules significantly affect their activity. Chrysin (flavone), having no hydroxyl groups in the B-ring, does not block the activity of tyrosinase. The presence of the hydroxyl group in the C-4′ position of apigenin (=c. 40 µM) significantly increased its activity compared to chrysin. The additional hydroxyl group in the C-3′ position of luteolin (
=c. 186 µM) decreased its activity compared to apigenin. Galagin (
=c. 10 µM) without hydroxyl groups on the B-ring blocks tyrosinase activity more strongly than chrysin. Addition of the hydroxyl group at the C-4′ or C-3′, C-4′ position reduces the activity of kaempferol (
=c. 73 µM) and quercetin (
=c. 30 µM) compared to galagin. Flavonols, due to the additional hydroxyl group at the C-3 position, block tyrosinase much more strongly than flavonesCitation102.
Kim et al. provided more information regarding the influence of the position and number of hydroxyl groups on their inhibitory activity. Natural and synthetic flavones and flavonols were used in the study. L-tyrosine was used as a substrate for fungal tyrosinase. It appears that the hydroxyl groups at the C-7 (ring A) and C-4′ (ring B) positions may increase the inhibitory activity of flavonoids. In this study, 3′,4′,7,8-tetrahydroxy flavone showed the strongest inhibitory properties (=0.07 µM). Two catechol groups in ring A and B are probably responsible for the inhibition. Lack of the fourth hydroxyl group in 4′′,7,8-trihydroxy flavone (
=12.95 µM) and 3′′,4′′,7-trihydroxy flavone (
=24.1 µM) weakens their activity. The 5-OH group also has a significant influence on the activity of the inhibitors, e.g. 2′,5,7-trihydroxyflavone (
=12.95 µM) and 3′,4′,5,7-tetrahydroxy flavone (
=12.95 µM) block tyrosinase more strongly than 2′,7-dihydroxy flavone, and 3′,4′,7-trihydroxy flavone. Also, the hydroxyl group in the C-3 position, which divides flavonoids into flavones and flavonols, influences on the activity with a weaker inhibition for flavones. Additionally, an increase in the number of hydroxyl groups in flavonoids reduces their inhibitory activity against tyrosinase (3′,4′,5,6,7-pentahydroxy flavonol
=314.23 µM). The location of the hydroxyl groups plays a more important role than their number. Summarising the increase in the number of hydroxyl groups on the other side of the flavonoids (C-7, C-8, C-2′, C-3′, and C-4′) increases compounds’ activityCitation103. In contrast, an increase in the number of hydroxyl groups on the ketone side (C-5, C-6, C-5′, C-6′) reduces the activity of inhibitors, which is related to the disturbance of the interaction with tyrosinase. Isoflavonoids are characterised by a linked ring B at the third carbon atom. The location and number of hydroxyl groups in the A-ring of isoflavonoids can strongly affect both the inhibitory power and inhibition type. 6,7,4′-trihydroxyisoflavone, daidzein, glycitin, daidzin and genistin showed strong monophenolase inhibitory activity but weak diphenolase inhibitory activity. 4′,6,7-trihydroxyisoflavone (
=9 µM) shows the strongest properties. Presence of a single hydroxyl group in the C-7 position (4′,7-trihydroxyisoflavone
=203 µM; 6-methoxy-7,4′-dihydroxyisoflavone
=218 µM) or no hydroxyl groups (4′-hydroxyisoflavone-7-O-glucoside
=267 µM) in ring A significantly reduces the activity of the compounds. The presence of the hydroxyl groups in the C-7 and C-8 position can influence the type of inhibition, shifting from reversible to irreversible typeCitation104. Similar results were obtained in case of anthocyanins and an assessment of the impact of the number and distribution of hydroxyl groups in the B ring on tyrosinase activity. The following compounds were investigated: pelargonidin (
=66 µM), cyanidin (
=27.1 µM), and delphinidin (
=57.4 µM) in the presence of kojic acid (
=34.8 µM) as a positive control. The substrate for the reaction was L-DOPA. These results indicate that the structure with two hydroxyl groups in ring B has the greatest inhibitory effectCitation105.
Another study examined the effect of isoflavonoids isolated from the roots of Pueraria lobata, such as daidzein and formononetin. These compounds have appeared to be of weak inhibitors with the IC50 values for daidzein L-tyr. 350 µM; L-DOPA 350 µM and for formononetin L-tyr. IC50 >350 µM; L-DOPA IC50 >350 µM. A significant increase in an activity was obtained by introducing OH groups into the B ring at the C-3′ position and methylation of the 4′-OH group - calycosin (L-tyr. IC50 =7.02 µM; L-DOPA IC50 =1.45 µM). This compound is more active than kojic acid (L-tyr. IC50 =12.10 µM; L-DOPA IC50 =9.14 µM). It is seen that the presence of a hydroxyl group at the C-3′ position and a methoxyl group at the C-4′ position of the isoflavone backbone plays a major role in the anti-tyrosine activityCitation106. The inhibitory activity of calycosin against tyrosinase (monophenolase) was also confirmed by Kim et al. with the IC50 equal of 38.4 µM. In turn, the IC50 value for kojic acid and arbutin was 51.5 and 120.9 µM, respectivelyCitation107. However, calycosin was more toxic than standards (LD50 =120 µM vs. LD50 >200 µM for kojic acid and arbutin). The results of another study conducted by Kim et al. have also shown an inhibitory activity of calycosin with the IC50 =30.8 µM and for kojic acid IC50 =50.1 µMCitation108.
The isoflavonoids isolated from the stem of Maackia fouriei have also exhibited their anti-tyrosinase activity. Methylation of the 4′-OH group in formononetin, texasin, and odoratin, resulted in impaired anti-tyrosinase properties. The presence of 5-OH group in genistein (IC50=33 µM) and tectorigenin (IC50=20 µM) favourably affects the activity of isoflavonoids in comparison to daidzein (IC50=41 µM) without 5-OH group. An interesting compound isolated from M. fouriei is the bishomoflavonoid derivative, mircoin (IC50=5 µM). This compound inhibited the enzyme competently. Due to its strong inhibition, further structure-activity studies are needed for this compound. In this study, kojic acid (IC50=45 µM) was used as a positive controlCitation109.
In another study, the effect of OH groups at the C-6, C-7 and C-4′ positions on isoflavonoid activity was investigated. 6,7,4′ -Trihydroxyisoflavone inhibited tyrosinase competently (IC50=9 µM; Ki value of 5.72–6.24 µM). Methylation of the 6-OH group in glycitein (IC50=264 µM), the absence of the 6-OH group in daidzein (IC50=237 µM), or the presence of an OH group at the 5-OH position in genistein (IC50=822 µM), decreased the anti-tyrosinase activity ()Citation110.
Table 14. Structure and activity of flavonoids with an anti-tyrosinase activity.
Similar effects of isoflavonoids on tyrosinase were noted by studying extracts of Otholobium pubescens (Pior.) J.W. Grimes. In this study, L-tyrosine as substrate and β-arbutin (IC50 = 1830 µM) as a control were used. Daidzein and its aglycone, genistein showed no effect (daidzein, IC50 =1580 µM; genistein, IC50 =7660 µM)Citation111.
The discrepancies in the study are due to the variation in experimental conditions such as temperature, pH, and concentrations of enzyme and substrates used. Flavonols are the most potent tyrosinase inhibitors among flavonoids. This is due to the similarity of flavonols to the structure of kojic acid − 3-hydroxy-4-keto moiety ( and ).
Figure 16. Potential groups engaged in an interaction flavonoid-tyrosinase.
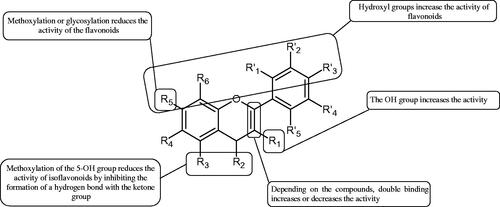
Figure 17. Structure relationship between flavanol (a) and kojic acid (b) and mode of copper chelation by 3-hydroxy-4-keto group in flavanol (c), and kojic acid (d).
5.2.2.2. Methoxylation
Methoxylation of the hydroxyl groups in the flavonoid molecules reduces their activity. Methoxylated flavones such as 5,6,7,4′-tetramethylscutellarein (5.21% inhibition), 5,7,4′-trimethylscutellarein (3.23% inhibition), and ladanein (4.24% inhibition) showed a ten-fold decrease in the inhibitory activity than kojic acid (% inhibition 80.02). It was proved that methoxylation of isoflavonoid also decreases the inhibition e.g. 2′-hydroxygenistein (37.3% inhibition) inhibited tyrosinase stronger than its methylated forms, such as 5-O-methyl-2′-hydroxygenistein (25.8% inhibition) and 7-O-methyl-2′-hydroxygenistein (31.2% inhibition). The weaker activity of 5-O-methyl-2′-hydroxygenistein may be related to the disturbance of a hydrogen bond formation between 5-OH and carbonyl oxygen (C-4). Kojic acid (=11.3 µM) was used as a control and L-DOPA as a substrateCitation112.
More information about the effect of hydroxyl groups and methylation on flavonoid activity was provided by studying derivatives having a methoxy group at position 3, a hydroxy group at position 5, and oxidised aromatic carbons at C4′ and C7 (). The most potent inhibitors 1 (IC50 =6.71 µM), 2 (IC50 =13.20 µM), and 3 (IC50 =17.66 µM) have three hydroxy groups at the C-3′, C-4′ and C-5′ positions in the B ring. Compound 1, which contains an additional methoxy group at C6 and a hydroxy group at C7, was the most active. Comparing the IC50 of 1 vs. 2 and 5 (IC50 =73.03 µM) vs. 6 (IC50 =103.56 µM), it seems that compounds containing a methoxy group at position C6 are more active than those that are unsubstituted at this position ()Citation113.
Table 15. Effect of the position and number of hydroxyl groups on the activity of flavonoids towards tyrosinase.
5.2.2.3. Double bond
The double bond between the second and third carbon atoms is preferred for flavonoids to maintain a flat molecular structure. Naringenin (=c. 555 µM) showed less inhibitory activity than apigenin (
=c. 40 µM). Dihydromyricetin (
=c. 37 µM) showed greater inhibitory activity than myricitin (
=c. 85 µM), while taxifolin (
=c. 800 µM) showed less inhibitory activity than quercetin (
=c. 30 µM). These results suggested that C2 = C3 binding affects the inhibitory properties of the flavonoidsCitation102.
5.2.2.4. Glycosides
Flavonoids occur mainly in the form of 3- and 7-glycosides. Some studies have revealed that a sugar moiety can modify flavonoids’ activity, e.g. 3-O-glycosides, hyperin ( not detected) and rutin (
=c. 4571 µM) show weaker tyrosinase inhibition than aglycone – quercetin (
=c. 30 µM). Similarly, 7-O-glycosides, baicalin (
=c. 215 µM) and naringin (
=c. 1900 µM) also inhibited tyrosinase weaker. A clear evidence was provided when monoglycosides, diglycosides and acylated monoglycosides towards tyrosinase inhibition were tested. Monoglycosides such as luteolin-7-O-glucoside (27.35% inhibition,
=74 µM), kaempferol-3-O-glucoside (24.2% inhibition,
=74 µM), and isorhamnetin-3-O-glucoside (24.22% inhibition,
=70 µM) showed stronger inhibition tyrosinases in a comparison to diglycosides such as kaempferol-3-O-rutinoside (16.05% inhibition,
=56 µM), isorhamnetin-3-O-rutinoside (13.13% inhibition,
=53 µM), and rutin (12.65% inhibition,
=55 µM). It is suggested that a presence of acyl groups on sugar residues of monoglycosides kaempferol-3-O-(6′′-pCm)-glucoside (14.69% inhibition,
=11 µM), quercetin-3-O-(6′′-pCm) -glucoside (21.86% inhibition,
=55 µM), isorhamnetin-3-O-(6′′-OAc)-glucoside (23.31% inhibition,
=64 µM), isorhamnetin-7-O-(6′′-pCm)-glucoside (21.10% inhibition,
=53 µM), apigenin-7-O-(6′′-pCm)-glucoside (17.66% inhibition,
=58 µM), apigenin-7-O-(3′′,6′′-di-pCm)-glucoside (20.69% inhibition,
=46 µM), chrysoeriol-7-O-(3′′,6′′-di-pCm)-glucoside (15.59% inhibition,
=44 µM) promotes an inhibitory effect compared to mono and diglycosides. The increase in the size of the flavonoids may prevent the flavonoids from entering the active site of tyrosinase (Tables 3S and )Citation102.
Table 16. An anti-tyrosinase activity of flavonoids (UE-unable to establish; c-with respect to L-tyrosine; d-with respect to both L-tyrosine and L-DOPA; NR-not reported; NT-not tested).
Comparing baicalein (IC50 =290 µM) with chrysin (no activity), the other hydroxyl group at the C-6 position of baicalein results in more potent tyrosinase inhibition. Comparing the inhibitory potency of baicalein (aglycone) with its glycosides, oroxin B (no activity) and oroxin A (IC50 =500 µM), a decrease in an inhibitory activity was noted. Glycosylation of the hydroxyl group at C7 was negatively correlated with the inhibitory activity of flavonoids. In addition, the activity of glycosides was influenced by the type of sugar moiety. The presence of the β-D-gentiobiosyl group reduced the inhibitory activity stronger than β-D-glucopyranosyl. This effect is due to spherical collapsesCitation114.
5.2.3. Lignans
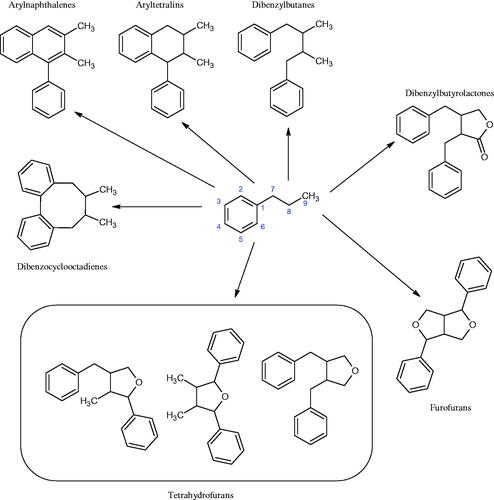
Lignans are phenylpropanoid dimers belonging to the group of plant phytoestrogens (). These compounds are widespread in seeds (lentils), vegetables (garlic and asparagus), and fruits (pears and plums), however, the richest source is linseed and whole cereal grains. They are part of the cell wall and can be released by intestinal bacteria. Due to the similar structure to oestrogens, lignans compete for oestrogen receptors. In oestrogen deficiency, lignans gently complement their action, and when there is an excess of them, they reduce their activity because they have a much weaker oestrogenic effect. As a result, they help maintain the hormonal balance in the body and reduce the risk of various hormone-dependent diseases. Besides, these compounds protect against osteoporosis, lower LDL cholesterol, inhibit bacteria and fungi’ growth, and lower blood glucose levels. The most important compounds in this group are sesamine, sesaminol, sesamoline, pinoresinol, secoisolaricresinol, matairesinol, schizandrin, and schizandrolCitation115–120.
Figure 18. Chemical classification of lignans.
Eight lignans were isolated from the methanolic extract of Vitex negundo L., i.e. negundin A, negundin B, 6-hydroxy-4-(4-hydroxy-3-methoxy)-3-hydroxymethyl-7-methoxy-3,4-dihydro-2-naphthalodehydrate, vitrofolal E, (+)-lyoniresinol, (+)-lyoniresinol-3α-O-β-D-glucoside, (+)-(-)-pinoresinol, and (+)-diasyringaresinol. The lactam ring present in negundin A caused moderately strong (=10.06 µM) inhibition of tyrosinase as compared to kojic acid (
=16.67 µM). Negundin B, with the -CH2OH group in the C-2 position and the C = C bond between C-1 and C-2, showed stronger (
=6.72 µM) inhibition of tyrosinase compared to kojic acid (
=16.67 µM). Compound 3, in which the CH2OH group in the C-2 position was replaced with an aldehyde group, blocked tyrosinase to a lesser extent (
=7.81 µM) than negundin B. Removal of the CH2OH group in the C-3 position and introduction of the C = C bond between C3 and C-4 reduced vitrofolal E’s strength (
=9.76 µM). The strongest inhibitor was (+)-lyoniresinol, in which both positions C-2 and C-3 contain the CH2OH group (
=3.21 µM). Glycosylation of (+)-lyoniresinol at position C-3 rendered inactive. The presence of a sugar residue hinders the interaction between the enzyme’s active site and the inhibitor. (+)-(-)-Pinoresinol showed moderate inhibition (
=15.13 µM). The introduction of -OCH3 groups in the 5′ and 3′ positions in (+)-(-)-pinoresinol, lead to the formation (+)-diasyringaresinol which strongly inhibits tyrosinase (
=5.61 µM). The compound (+)-lyoniresinol could be used as a potential lead molecule in bioprospectingCitation121. Also, other lignans exhibited an anti-tyrosinase activity, e.g. 5,5-dimethoxylaryresinol-4-O-β-d-glucopyranoside and eleutheroside
showed significant inhibition with the
value of 42.1 and 28 µM, respectivelyCitation122.
Lignan glycosides showed a moderate inhibitory effect on tyrosinase (4–5 times less than kojic acid) in the presence of L-DOPA as a substrate (4-O-lariciresinol-glucoside − 17.74% inhibition and 4′-O-lariciresinol-glucoside − 12.27% inhibition). When compared to lignan diglucoside (11.06% inhibition), they showed a lower activity, probably due to the complete absence of free hydroxyl groups ()Citation112.
Table 17. An anti-tyrosinase activity of lignans.
5.2.4. Flavonolignans
Phytochemicals composed of part flavonoid and part phenylpropanoid, which are commonly found in nature. The richest source of these compounds is Silybum marianum from the Asteraceae family. They show hepatoprotective, anticancer, and anti-inflammatory effectsCitation123.
For example, isosilybin A (IC50 =2.1 µM) was more effective than its three mother compounds 3′-O-methyltaxifolin (IC50 =51.2 µM), dihydrokaempferol (IC50 =73.6 µM), and taxifolin (IC50 =23.0 µM). Analysing the structure of the mother compounds, deletion or methylation of the 3′-OH group 2.5–3 times reduces the activity of the compounds. Silychristin A (IC50 =3.2 µM; IC50 =28.8 µM) and silychristin B (IC50 =4.5 µM; IC50 =44.9 µM) having a double bond between C-2 and C-3 more potently inhibited tyrosinase activity than 2,3-dihydrosilychristin (IC50 =7.6 µM; IC50 =35.9 µM) having no double bond. The isolated compounds showed a mixed type of inhibition (Ki: L-tyr, 0.7–4.7 µM; L-DOPA, 8.5–36.7 µM). The mother compounds inhibited the enzyme in a competent manner ()Citation124.
Table 18. An anti-tyrosinase activity of flavonolignans.
5.2.5. Stilbenes
These compounds belong to phytoalexins, low molecular weight cell components with antibacterial and antifungal properties. Besides, they show other biological properties such as antioxidant, anti-inflammatory, and antiproliferative effects. A characteristic feature of their structure is the presence of a 1,2-diphenylethylene core. More than 400 natural stilbenes have been discovered, but due to the low abundance of the critical enzyme stilbene synthase, they are not widely distributed in nature. The primary source of stilbenes in the human diet are grapes, red wine, and peanuts. The most famous representative of this group is resveratrolCitation125.
5.2.5.1. Hydroxyl groups
Many naturally occurring stilbenes exhibit tyrosinase inhibitory activity, that is related to the characteristic elements of their structure. The inhibitory properties are due to the number and distribution of oxygen atoms attached to the aromatic rings. Dioxyl stilbene, pinosylvin, showed weak inhibitory properties (=46 µM), while resveratrol, a stilbene representative with three hydroxyl groups, inhibited tyrosinase even more strongly than kojic acid. However, when compared to oxyresveratrol, representative of tetroxyl stilbenes, that compound showed a nine-fold increase in the inhibition than resveratrol (
=1.5 vs. 14.4 µM)Citation126. To better understand, the structure–activity relationship in a model hydroxystilbene-tyrosinase new derivatives of trans-stilbene were synthesisedCitation127. Monohydroxy trans stilbenes showed no inhibitory effect on tyrosinase, only after attachment of another hydroxyl group to the aromatic ring resulted in an increase of inhibition. The braking force depended on the position of the hydroxyl groups to each other, e.g. 3,3′-dihydroxy-transstilbene (26.3% inhibition
>200 µM) has a more substantial inhibitory effect than 2,3-dihydroxy-trans-stilbene (4.4% inhibition
>200 µM) and 3,4-dihydroxy-trans-stilbene (9.5% inhibition
>200 µM) or 3,5-dihydroxy-trans-stilbene (18.6% inhibition
>200 µM). The 3,3′,4-trihydroxy-trans-stilbene (87.7% inhibition
=74.3 µM) and 3,3′,4,4′-tetrahydroxy-trans-stilbene (98.3% inhibition
=29.1 µM) showed more potent activity against the enzyme than 3,3′-dihydroxy-trans-stilbene. 3,3,4,4′-tetrahydroxy-trans-stilbene (
=29.1 µM) inhibited the tyrosinase activity almost completely. It is seen that an increase of the inhibitory power of the hydroxystilbenes is correlated with an increase of the number of hydroxyl groups. O-methylation decreased the action of the stilbenes.
5.2.5.2. Stilbene glycosides
Other studies have examined the difference in an action between stilbene glycosides and their aglycones. Several hydroxystilbenes were isolated from the methanolic extract of Veratrum patulum L. (=100 µM), including piceid, the aglycone of which is resveratrol. The inhibitory activity of piceid was 6.9 and 8.2 (L-DOPA and L-tyrosine) times lower than that of resveratrol (phenylthiourea was used as a positive control). The tested compounds showed a more significant effect on the monophenolase activity than on the diphenolase activityCitation128. Kim et al. who studied the impact of mulberroside A (isolated from the ethanolic Morus alba L. root extract) enzymatic biotransformation to oxyresveratrol and their anti-tyrosinase activity. The inhibitory activity of oxyresveratrol was approximately 110-fold higher than that of mulberroside A (
= 0.49 and 53.6 µM, respectively). Kojic acid and arbutin were selected as controls in the study. Inhibition of tyrosinase activity by oxyresveratrol (L-tyrosine) was 43-fold and 1503-fold higher than that of kojic acid and arbutin (
=21.1 and 736.5 µM, respectively). Tyrosinase was almost completely inhibited by oxyresveratrol, mulberroside A, and kojic at concentrations of 2.5, 500, and 250 µM, respectively. Arbutin weakly inhibited tyrosinase (about 85%, 3000 µM). On the basis of the kinetic parameters, it has been shown that mulberroside A is a competitive inhibitor of fungal tyrosinase with L-tyrosine and L-DOPA as a substrate, oxyresveratrol showed mixed inhibition and non-competitive inhibition to L-tyrosine and L-DOPA as the substrate, respectively. Tyrosinase catalyses two different reactions: the hydroxylation of monophenols to o-diphenols (monophenolase activity) and the oxidation of o-diphenols to o-quinones (diphenolase activity). Considering the
(L-tyrosine 0.49; L-DOPA 11.9) and Ki (L-Tyrosine 1.093, 0.521; L-DOPA 1.272) values, oxyresveratrol had a more significant impact on the activity of monophenolase than on the diphenolase activityCitation129.
Isolated from the water-methanol extract of the rhizome of Rheum officinale Baill galloyl glucosides of resveratrol, e.g. 3,4′, 5-trihydroxystilbene-4′-O-β-D- (2′′-O-galloyl) glucopyranoside (A) and 3,4′, 5-trihydroxystilbene-4′-O-β-D- (6″ -O-Galloyl) glucopyranoside (B) inhibit the activity of tyrosinase. These compounds showed a competitive type of inhibition and blocked the enzyme stronger than kojic acid. The compounds inhibited the conversion of L-tyrosine to L-DOPA more strongly than L-DOPA to DOPA quinone. Inhibitory effect for compounds A (=6.71 µM) and B (
=14.7 µM) was higher than for kojic acid (
=28.9 µM), when L-tyrosine was used as a substrate. Inhibitory effect of compound B (
=82.3 µM) was significantly less than that of kojic acid (
=23 µM),when L-DOPA was used as a substrate. In the case of compound A the
value was comparable to that of kojic acid (
=24.6 µM)Citation130.
The obtained results indicate that the deglycosylation of stilbenes has a positive influence on their activity and indicates that the aglycones are more active. This is probably related to particle size because as larger compounds, glycosides have restricted an access to the active site of the enzyme.
5.2.5.3. Isoprenyl chain
The inhibition of tyrosinase may also be stimulated by the compounds with isoprenyl chain in their structure, e.g. 4-[(2′′E)-7′′-hydroxy-3′’,7′′-dimethyloct-2′′-enyl] − 2′,3,4′,5-tetrahydroxy-trans-stilbene (compound A) and chlorophorin, both isolated from Chlorophora excels (Welw.) Benth. core, showed a different inhibitory activity dependent on the presence of the isoprenyl chain. The values for compound A and chlorophorin were equal 96 and 1.3 µM, respectively (kojic acid,
=20 µM). Attaching a water molecule to the geranyl chain in compound A reduced its tyrosinase activity. This is probably due to reducing the chain’s interaction and the hydrophobic protein pocket close to the active siteCitation131. The other investigations have shown that the presence of a prenyl chain in a compound having a 4-substituted resorcinol backbone increased the inhibitory activity compared to that of oxyresveratrol (
=0.66 and 0.98 µM, respectively).
In another study, the impact of chain length, functional groups (polarity), and cyclisation of isoprenyl chains on the anti-tyrosinase activity was investigated. An increase in chain length from one isoprene unit to two resulted in a 4-fold increase in activity (IC50 = 15.87 µM, IC50 = 60.14 µM). In turn, the introduction of hydroxyl groups or cyclisation of the isoprenyl chain caused a drastic decrease in anti-tyrosinase activity. Similar results of isoprenyl chain influence were observed in stilbene derivatives isolated from Angelica keiskei roots.
5.2.5.4. Double bond
Another compound inhibiting tyrosinase is gnetol, a tetrahydroxystilbene isolated from Gnetum gnemon L. It was appeared that gnetol is approximately 30 times more potent than kojic acid. Additionally, gnetol inhibited tyrosinase much more than dihydrognetol (100% and 20%, respectively). The double bond is crucial for the activity. The double bond’s role in the stilbene backbone in inhibiting tyrosinase examined the cis-olefin structure. The cis isomer of 3,3′-dihydroxystilbene (% inhibition c. 1) compared to the trans isomer (26% inhibition) shows no inhibitory effect. The saturation of the double bond in the oxyresveratrol (=0.98 µM) significantly reduces the activity (
=58 µM). However, dihydrooxyresveratrol showed eight times more inhibitory effect on the activity of fungal tyrosinase than oxyresveratrol (
=1.6 and 12.7 µM, respectively). The higher activity of dihydrooxyresveratrol, compared to oxyresveratrol, was probably due to its dibenzyl structure, which provided greater flexibility, and thus allowed for a more effective interaction of phenolic groups with the enzyme ()Citation132.
Figure 19. Chemical structure of cis- and trans-isomer of stilbenes.
Similar results of the effect of double bond saturation were obtained by studying dihydrostilbene derivatives isolated from the 80% ethanolic extract of the Dendrobium loddigesii Rolfe stem. It was appeared that 3,4,5-trihydroxy-3′,4′-dihyroxyhydrostilbene; 3,5-dihydroxy-3′,4′-dihyroxyhydrostilbene and their methoxy derivatives did not inhibit tyrosinase activity. The exception to this rule is 3,5-dihydroxy-3′-dihyroxyhydrostilbene (IC50 = 37.90 µM). Attachment of dioxolane (aphyllals C) to the B ring of 3,5-dihyroxyhydrostilbene results in a marked increase in an activity (IC50 =152.56 µM). Methylation of the 3-OH group in aphyllals C abolishes the compound’s activity. The above examples indicate that methylation of the 3-OH group inactivates the compound. Kojic acid was used as a positive control (IC50 =8.02 µM). A noteworthy compound for (Q)SAR studies is 1,3-benzodioxol derivative (benzene ring linked to dioxolane). Due to the ambiguity of the results, further studies on the effect of a double bond in stilbenes are needed.
5.2.5.5. Stilbene oligomers
The next group with an anti-tyrosinase activity is oligomers of stilbenes. The following resveratrol oligomers: ε-viniferin as a dimer; vaticanol A, vaticanol G, and α-viniferin as trimers; vaticanol B, vaticanol C and (-)-hopeaphenol as tetramers were tested towards tyrosinase from murine B16 melanoma cells and L-DOPA as the substrate and kojic acid as a control (=119.7 µM; c. 49.3% inhibition). Resveratrol (
=10.8 µM) at a concentration of 100 µM inhibited the activity of tyrosinase at the level of 98%. However, the oligomers have appeared to be weak inhibitors, e.g. dimer-ε-viniferin at a concentration of 100 µM showed approx. 24.6% inhibition, the trimmers, and tetrameters showed an inhibition level of less than 8%. It may be suggested that the inhibitory potency of the resveratrol oligomers decreases with increasing molecular weight (; Tables 4S and )Citation133.
Figure 20. Potential groups engaged in an interaction stilbene-tyrosinase.
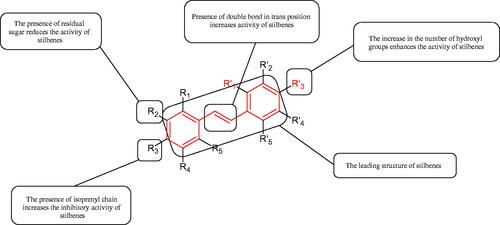
Table 19. An anti-tyrosinase activity of stilbenes.
5.2.6. Chalcones
Chalcones belong to the group of unsaturated aromatic ketones and are thought to be the precursors for the synthesis of flavonoids. The core of chalcones consists of two aromatic rings linked by a three-carbon α, β-unsaturated carbonyl system (1,3-diphenyl-2-propen-1-one) (). Chalcones can exist in trans (E) and cis (Z) forms, but cis isomers are unstable due to spherical effects. Chalcones are characterised by a broad spectrum of biological properties, including anticancer, antioxidant, antidiabetic, anti-inflammatory, antibacterial, and antiviral activitiesCitation133.
Chalcones have shown an anti-tyrosinase activity dependent on their structure and concentration. Khatib et al. studied the effect of catechol and resorcinol groupings in the A and B rings of chalcones. Two substrates for tyrosinase, L-tyrosine (first step – L-tyrosine to L-DOPA) and L-DOPA (second step – L-DOPA to o-quinone), were used. All of the compounds tested showed greater activity in blocking the first step than the second step. 3,4,2′,4′- Hydroxychalcone, with a resorcinol moiety (at the 2′ and 4′ positions) in the A ring and catechol in the B ring inhibited the first step more potently (=29.3 µM) than the second step (
>100 µM). 2,4,3′,4′- Hydroxychalcone, with the opposite structure to 3,4,2′,4′- hydroxychalcone, inhibited tyrosinase about 146.5-fold more potently (
=0.2 µM – stage 1, and
=7.5 µM stage 2). The compound 3,5,2′,4′- hydroxychalcone, in which the OH groups of the B ring are located at positions 3 and 5 while maintaining the identical position of the catechol group as in 3,4,2′,4′- hydroxychalcone, blocked the enzyme weaker (
=31.7 µM for step 1,
>1000 µM for step 2). Compound 2,4,2′,4′- hydroxychalcone, made up of two resorcinol groups in rings A and B, blocked tyrosinase activity most strongly (
=0.02 µM for stage 1 and up to 90 µM for Stage 2). Additionally, 2,4,3′,4′- hydroxychalcone is 7.5 times more active than trans-stilbene, with the same catechol and resorcinol arrangement. Moreover, the position of OH groups in the A and B ring affects the mechanism of chalcone inhibition. Compounds with a catechol group showed the ability to chelate copper ions, while 2,4,2′,4′- hydroxychalcone (resorcinol structure) did not chelate copper ions. The catechol group in ring A acted as a chelator of copper ions, while the catechol in ring B is oxidised to o-quinone. However, catechol groups in the A or B ring had no significant impact on tyrosinase inhibition. The compound with two resorcinol moieties has the most potent effect on tyrosinase activityCitation134.
A promising group of tyrosinase inhibitors may be 2′,4′,6′-trihydroxychalcone derivativesCitation135. It was confirmed, that the absence of OH groups at the 4′ or 6′ position in 2′,4′,6′-trihydroxychalcones results in loss of an inhibitory activity (less than 20% inhibition at 400 µM concentration). Comparing the 2′,4′,6′-trihydroxychalcone activity to kojic acid, chalcone has exhibited a 10-fold weaker inhibition (=120 and 12 µM, respectively). Methoxylation of hydroxyl groups at the 4′ and 6′ position of compounds: 2,2′,3,4′,6′-trihydroxychalcones, 2′,3,4,4′,5,6′-trihydroxychalcones, and 2′,3,4,4′,6′-trihydroxychalcones also cause loss of their activity. 2′,2,4,4′,6′-Trihydroxychalcones (
=1 µM) were found to exhibit the highest activity, even better than 2,2′,4,4′-tetrahydroxychalcone (
=5 µM) and kojic acid (
=12 µM). In contrast, methoxylation of the 6′-OH group in 2′,2,4,4′,6′-trihydroxychalcones impairs activity (
=3.1 µM).
Nguyen et al. provided more information about the influence of the position and number of OH groups on the activity of chalcones. It was appeared that a presence one or two hydroxyl groups in the A ring do not strength the inhibition, e.g. inhibition for 4-hydroxychalcone, 2-hydroxychalcone, and 2,4-dihydroxychalcone was about 14% at 50 µM, respectively. However, the changes had been observed after the introduction of the 4′-OH group in the B ring, which resulted in the activity’s increase. The inhibition at 50 µM for 4′-hydroxychalcone, 2,4,4′-trihydroxychalcone was 71% and 67%, respectively. Additionally, the presence of the 2-OH group in the A ring drastically reduces the activity of 2,4′-dihydroxychalcone (10% inhibition at 50 µM concentration). The weakening effect of the 4′-OH group on tyrosinase activity is due to conformational changes. These changes are due to the formation of hydrogen bonds between 2-OH and the carbonyl group. The importance of the arrangement of hydroxyl groups is related to the structure of chalcones. Ring A is associated with a carbonyl carbon atom, while ring B is related to a vinyl carbon atom. This is in contrast to stilbenes, in which the rings are bound to identical carbon atoms ()Citation136.
Figure 21. a) A basic structure of chalcones (1,3-diphenyl-2-propen-1-one); b) The difference in structure between stilbenes and chalcones.
The anti-tyrosinase properties have also been confirmed in the group of the chalcone glycosides, e.g. licuraside, isoliquiritin, and the aglycone licochalcone A isolated from two species of Glycyrrhiza (Glycyrrhiza uralensis Fisch and Glycyrrhiza inflate Bat., respectively). The for licuraside, isoliquiritin, and licochalcone A, in the presence of L-tyrosine, were of 72, 38, and 25.8 µM, respectively. The compounds inhibited enzyme competitive (L-tyrosine). The inhibitory effect of chalcones on diphenolase activity (L-DOPA as substrate) was much lower. The difference in inhibition between monophenolase and diphenolase activity is related to the structure of the chalcones, those ones which inhibit monophenolase more strongly show a similarity to L-tyrosine ()Citation134.
Figure 22. a) Similarity in the structure of L-tyrosine and 4-hydroxychalcones; b) Similarity in structure of L-DOPA and flavonol.
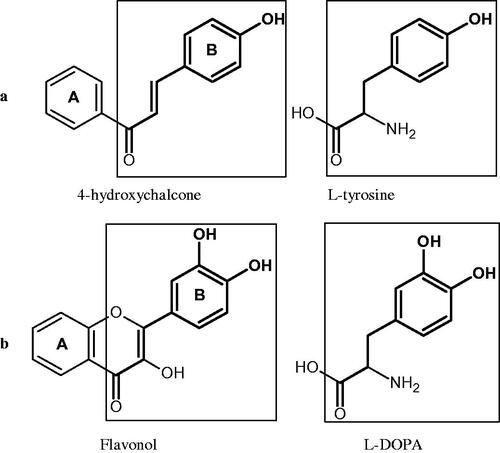
The 4-OH group in the B ring of chalcones affects the potency of the inhibitor (similarity to the tyrosine backbone). Licochalcone A is a more potent inhibitor than licurad and isoliquiritin because it has a free 4-OH group in the B ring and has less steric hindrance. The sugar residue at the 4′-OH position hinders access to the enzyme’s active centre, resulting in reduced inhibitory activity. In addition, the 3,3- dimethylpropylene group at position 5 (ring B) of licochalcone A disrupts the quaternary structure of tyrosinase, inhibiting the enzymeCitation134.
In the case of diphenolase activity, for which the substrate is L-DOPA, the presence of both 3′-OH and 4′-OH groups in the B ring is necessary in the inhibitor’s structure in order to resemble L-DOPA. The absence of the 3′-OH group in the B ring of chalcones resulted in the lack of diphenolase inhibitory activity of tyrosinaseCitation137. The tyrosinase activity may be also regulated via the introduction of alkyl chains into chalcone molecules. It has been proved that prenylated chalcone, curaridine, blocked the enzyme activity very strongly (=0.6 µM) compared to kojic acid (
=20.5 µM). The important elements of the studied compound are the 2′-OH and 4′-OH groups in the B ring, and 4-OH, and the prenyl chain at C-5 (lavandulyl) in the A ringCitation138. On the other hand, the addition of two isoprenyl groups at the C-5 and C-3′ positions to 4,4′,6-trihydroxychalcone abolishes tyrosinase inhibition. This is probably due to steric hindrance (; Tables 5S and )Citation134.
Figure 23. Potential groups engaged in an interaction chalcone-tyrosinase.
Table 20. An anti-tyrosinase activity of chalcones.
5.2.7. Phenylpropanoid sucrose esters (PSEs)
Phenylpropanoid sucrose esters (PSEs) are composed of a sucrose core linked to one or more phenylpropanoid residues (Ph-CH = CH-CO-) via an ester bond. PSEs include substituted/unsubstituted caffeic, coumaric, ferulic, cinnamic, and sinapic acids. PSEs have been isolated from various species of medicinal plants in the families Arecaceae, Boraginaceae, Brassicaceae, Caryophyllaceae, Liliaceae, Melanthiaceae, Polygonaceae, Poaceae, Polygalaceae, Rutaceae, and Rosaceae. These compounds exhibit anti-inflammatory, antioxidant, hypoglycaemic, and anticancer activities ()Citation139–141.
Figure 24. Structure of Phenlpropanoid Sucrose Esters – PSEs (R: phenylpropanoid residues).
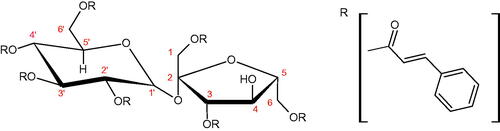
The PSEs isolated from Persicaria orientalis (L.) Spach showed low to medium tyrosinase inhibitory abilities [hydropiperoside (L-tyr, IC50 =27.1 µM; L-DOPA, IC50 =166.15 µM), vanicoside A (L-tyr, IC50 =37.29 µM; L-DOPA, IC50 =135.91 µM), vanicoside B (L-tyr, IC50 =62. 0 µM; L-DOPA, IC50 =113.13 µM), vanicoside C (L-tyr, IC50 =39.0 µM; L-DOPA, IC50 =91.38 µM), and vanicoside E (L-tyr, IC50 =45.23 µM; L-DOPA, IC50 =189.96 µM)]. In this study, kojic acid (L-tyr, IC50 =14.15 µM; L-DOPA, IC50 =181.40 µM) was used as a positive controlCitation142. Cho et al. investigated the inhibitory effect of PSEs isolated from the Oryza sativa roots on tyrosinase activity. The most active compounds were 3,6-diferuloyl-3′,6′-diacetylsucrose (IC50 =47.33 µM) and smilaside A (IC50 =45.13 µM). The IC50 for 3-feruloyl-4′,6′-diacetyl sucrose, 3-feruloyl-6′-acetylsucrose 3,6-diferuloylsucrose was >400 µM. None of the isolated compounds inhibited the enzyme more strongly than the positive control, kojic acid (IC50 =28.60 µM)Citation143.
Summarising results of the above studies, they indicate the significance of the presence of a feruloyl group at C-6, an acetyl group at C-6′, and another acetyl group at C-3′/C-4′ in inhibiting of the enzyme. Additionally, it seems that the introduction of additional feruloyl groups in the fructose moiety increases the activity of the compounds ().
Table 21. Structure and an anti-tyrosinase activity of phenylpropanoid sucrose esters.
5.3. Coumarin
Coumarins are derivatives of α-pyrone, condensed with benzene. Benzo-α-pyrone is usually substituted at C-7, less often at C-5, C-6, and C-8 positions with a hydroxyl group to which methyl groups or sugar moieties may be attached. A furan or pyran ring may be condensed with the benzo-α-pyrone structure ().
Figure 25. Structure of α-piron (a); benzo-α-piron (b).
Coumarins isolated from Euphorbia lathyris seeds showed weak inhibitory properties against tyrosinase. The exception was esculetin, whose IC50 was 43 µM (kojic acid; IC50 =10 µM) lCitation144. Different results were obtained for the constituents present in the leaf extract from M. alba L. Scopoletin showed the strongest anti-tyrosinase properties (IC50 =0.2 µM), while esculetin and scopoline appeared to be less potent with the IC50 equal 6.9 and 15.9 µM, respectively. All inhibitors blocked the enzyme competentlyCitation145.
Coumarin glycosides present in the Morus nigra roots showed weak inhibitory properties against tyrosinase, probably the presence of a sugar residue reduced the inhibitory activityCitation146.
Some coumarins present in Rhododendron collettianum inhibited tyrosinase more strongly than kojic acid (IC50 =16.67 µM). 8′-Epi-cleomiscosin A (IC50 =1.33 µM) blocked the enzyme activity most strongly. Cleomiscosin A (IC50 =18.69 µM) differs from 8′-epi-cleomiscosin A by the position of the proton at position 8. Due to the change in stereochemistry of the single proton, the inhibitory activity of the compounds changes drastically. This may be due to stereochemically favourable binding conditions at the enzyme active site. Aquillochin (IC50 =15.69 µM) and 5,6,7-trimethoxycoumarin (IC50 =8.65 µM) also inhibited the enzyme more strongly than caffeic acid. This study also showed a negative impact of the sugar residue on an anti-tyrosinase activity (8-O-β-D-glucopyranosyl-6-hydroxy-2-methyl-4H-1-benzopyrane-4-one; IC50 =256.97 µM)Citation147.
More information on the structure–activity relationship was provided by examining semi-synthetic/synthetic coumarin derivatives. In a study conducted by Matos et al., the effect of 3-phenylcoumarin and 3-thiophenylcoumarin derivatives on tyrosinase activity was investigated. L-DOPA was used as a substrate for the enzyme. The results showed that some synthesised derivatives exhibited an inhibitory activity against mashroom tyrosinase. The two most active compounds (5,7-dihydroxy-3–(3-thiophenyl)coumarin and 3–(4′-bromophenyl)-5,7-dihydroxycoumarin) showed tyrosinase inhibitory activity (IC50=0.19 and 1.05 µM, respectively), higher than kojic acid (IC50=17.90 µM). The presence of two hydroxyl groups at the C-5 and C-7 positions of the coumarin scaffold improved the inhibitory activity. The presence of the resorcinol grouping enhanced the ability to chelate copper ionsCitation148.
In subsequent studies, the effect of the position of hydroxyl, methoxyl, ethoxyl, and bromine groups in 3-phenylcoumarin derivatives on the activity against tyrosinase was examined. 3-Phenyl-6-hydroxy-8-bromocoumarin (IC50 =215 µM) inhibited tyrosinase more strongly than kojic acid (IC50 =420 µM). In addition, the introduction of more hydroxyl groups in the coumarin grouping increased the inhibitory activity. Compared to 6-hydroxy-8-bromocoumarin (IC50 =302 µM), one more hydroxyl group was introduced in the 3-phenyl-6-hydroxy-8-bromocoumarin, which improved the inhibitory activity about 1.5 times. A bromine substituent and a C-4′ hydroxyl group at the C-6 positions increase the inhibitory activity. In turn, methoxy and ethoxy derivatives weakly inhibited the enzyme. It seems that brominated hydroxycoumarin derivatives may be promising inhibitors of tyrosinaseCitation149.
In the study of Asthan et al., the effect of the position of the hydroxyl group in benzo-α-pyrone on tyrosinase activity was checked. The position of the hydroxyl group at C-6 and C-7 causes the molecule to behave as a weak substrate for the enzyme. This is related to the interaction of the hydroxyl group with the copper ion in the enzyme’s active centre, which means that the compounds with the OH group in the pyrone ring cannot be substrates for tyrosinase. Among investigated compounds, only 3-hydroxycoumarin inhibited the enzyme activity. The studies indicate the possibility of application of 3-hydroxycoumarin structure as a new class of tyrosinase inhibitorsCitation150.
In another study, thiosemicarbothioamide derivatives, such as 2–(1-(coumarin-3-yl)ethylidene)hydrazinecarbothioamide inhibited tyrosinase stronger than kojic acid with the IC50 values 3.44 and 23.0 µM, respectively. The compound blocked the enzyme irreversiblyCitation151.
Ashraf et al. synthesised several umbelliferone derivatives and studied their effects on tyrosinase. Compounds 4e and 4c, having 2,4-dihydroxy and 3,4-dihydroxyphenyl group, inhibited fungal tyrosinase most potently with the IC50 values 8.96 and 118.48 µM, respectively. The other umbelliferone derivatives showed weak activity against mashroom tyrosinase compared to kojic acid (IC50 = 16.69 µM). On the other hand, umbelliferone derivatives obtained in this study inhibited tyrosinase more potently than umbelliferone (IC50 = 420 µM) (; )Citation152.
Figure 26. Potential groups engaged in an interaction coumarin-tyrosinase.
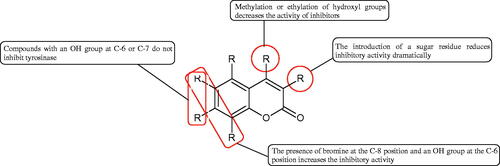
Table 22. Structure and activity of coumarin against tyrosinase.
5.4. Tannins
Another group of natural compounds with tyrosinase inhibitory potential is tannins. The flavan-3-ol derivatives isolated from Potrntilla anserina L. rhizome showed interesting inhibitory properties against tyrosinase. In both cases, the compounds inhibited diphenolases more strongly than monophenolases. Flavan-3-ol dimers (potenserin C – L-tyr IC50 = 16.53 µM, L-DOPA IC50 = 2.63 µM; gallocatechin-(4′→O → 7)-epigallocatechin-L-tyr IC50 = 18. 55 µM, L-DOPA IC50 = 6.62 µM; bis-6,8′-catechinylmethane – L-tyr IC50 = 30.56 µM, L-DOPA IC50 = 12.26 µM; bis-8,8′-catechinylmethane – L-tyr IC50 = 33. 89 µM, L-DOPA IC50 = 15.11 µM; catechin (4α → 8) catechin – L-tyr IC50 = 39.09 µM, L-DOPA IC50 = 25.49 µM; catechin (4α → 8) epicatechin – L-tyr IC50 = 37.22 µM, L-DOPA IC50 = 21.76 µM) showed similar or stronger inhibitory properties than kojic acid (L-tyr IC50 = 48.55 µM, L-DOPA IC50 = 21.00 µM). The presence of an additional OH group at the C-3′ position of catechins (L-tyr IC50 = 65.26 µM, L-DOPA IC50 = 42.71 µM) enhanced the anti-tyrosinase properties (gallocatechin – L-tyr IC50 = 41.96 µM, L-DOPA IC50 = 30.11 µM). In turn, methylation of the 3′-OH and 5′-OH groups in (2 R, 3S)- 3′, 5′-dimethoxy gallocatechin decreases the inhibitor activityCitation153. A similar result was obtained by testing proanthocyanidin oligomers (OPC) from red wine (Vitis vinifera) for tyrosinase activity at a concentration of 1 mM. Procyanidin dimers as procyanidin B-3 (69.6% inhibition) and B-4 (69.4% inhibition), which possess (+)-catechin, more strongly blocked the enzyme than procyanidin B-1 (16.7% inhibition) and B-2 (26.0% inhibition). The IC50 values for PB3 and PB4 were 545 and 726 µM, respectively. The trimeric OPCs showed an inhibition rate of 25.3–31.1% at 1 mM, meaning no significant differences between the trimeric proanthocyanidinsCitation154.
The procyanidin epicatechin-(4β→8, 2β→O → 7)-epicatechin-(4β→8)-epicatechin isolated from Guioa villosa and procyanidin B1 have shown a minimal effect on tyrosinase activityCitation155,Citation156.
Another study examined the effect of tannins isolated from the methanolic extract of Ecklonia stolonifera. The extract contained five phlorotannins: phloroglucinol, eckstolonol, eckol, phlorofucofuroeckol A, and dieckol. Particularly noteworthy was dieckol (IC50 =2.16 µg/mL), which blocked the enzyme more strongly than kojic acid (IC50 =6.32 µg/mL) and arbutin (IC50 =112.0 µg/mL). Phloroglucinol and eckstolonol showed the competent type of inhibition. Eckol, phlorofucofuroeckol A, and dieckol inhibited the enzyme incompetently. The isolated phlorotannin derivatives owe their properties to the presence of a resorcinol group, which can chelate copper in the active tyrosinase centerCitation157.
Shoji et al. examined the effect of procyanidin polymerisation on tyrosinase activity. All oligomer fractions (1mer to 7mer) strongly inhibited tyrosinase activity, as did kojic acid. The IC50 values were similar for all groups: monomer, 74 µM; dimer, 235 µM; trimmer, 140 µM; tetramer, 149 µM; pentamer, 184 µM; hexamer, 127 µM; and heptamer 103 µM. No correlation was observed between the degree of procyanidin polymerisation and tyrosinase inhibition. Nevertheless, these observations suggest that procyanidins are effective inhibitors of tyrosinase ()Citation158.
Table 23. Structure and activity of tannin against tyrosinase.
5.5. Terpenes
A very promising group of tyrosinase inhibitors are terpenes, for which that activity was proved by Lin et al. who examined the effect of terpenes isolated from the Eucalyptus globulus Labill leaves. Among few tested terpenes, the sesquiterpene, (-)-globulol (IC50 =9.79 µM) showed the strongest inhibition, followed by the isoiphionane sesquiterpene derivatives, 5-β-11-dihydroxy-iphionan-4-one (IC50 =10.08 µM) and 3-β-11-dihydroxyisoiphion-4-one (IC50 =14.17 µM). They have appeared to be more effective inhibitors than kojic acid (IC50 =17.32 µM)Citation159. Isolated from the Aquilaria genus prezizane-type sesquiterpenes, agarosanol A–F weakly inhibited tyrosinase at a concentration of 100 µM (less than 30%) ()Citation160.
Table 24. Structure and an anti-tyrosinase activity of terpenes.
6. Conclusion and future perspectives
Despite the fact that the plant-derived substances have always been a mainstay in medical treatment, the use of plants as a source of new drugs is still poorly studied. Of the 250,000–500,000 plant species, only a small number have been adequately studied. Besides the possibility of a direct use of plant materials in medicine, isolated substances can be used as a source of the structures for the synthesis of new drugs.
Commercially available Hyal and tyrosinase inhibitors are characterised by a range of adverse properties. Tyrosinase overactivity can be reduced by inhibitors such as hydroquinone, arbutin, vitamin C, azelaic acid, kojic acid, and ellagic acid. However, those inhibitors have many side effects, e.g. hydroquinone, the most commonly used inhibitor, shows mutagenic and irritating effects (dermatitis and irritation), while arbutin, a prodrug of hydroquinone, is a chemically unstable compound. Kojic acid is carcinogenic, L-AA is quickly degraded, ellagic acid, due to its poor solubility, shows a low bioavailability. Therefore, it is necessary to search for new safe inhibitorsCitation161–165.
One of the Hyal inhibitors available for treatment is escin (aescin). Unfortunately, due to its physicochemical properties, the compound is absorbed orally only to a small extentCitation166.
From this review, it is clear that currently used tyrosinase and Hyal inhibitors are not without their drawbacks, therefore, there is a great need to search for the new inhibitors with more valuable pharmacological properties. In the further bioprospecting, the following criteria should be considered:
Examine the effect of extracts and newly isolated compounds on cell cultures to determine cytotoxicity. Some compounds, e.g. alkaloids or terpenes, may exhibit toxic effects. Therefore, early determination of their toxicity is necessary. In addition, the study of new inhibitors using cell cultures will allow us to initially determine the metabolic pathways and mode of absorption (active or passive transport) of the tested compounds.
To better understand the utility of new substances in inhibiting Hyal or tyrosinase, more studies using animal models should be planned to determine the safety and efficacy of the tested inhibitors.
Most of the work was done with tyrosinase isolated from the fungus Agaricus bisporus. Human tyrosinase is membrane-bound, while fungal tyrosinase is a cytosolic enzyme. Furthermore, the human enzyme is a monomer that undergoes an intensive glycation process, unlike the fungal tyrosinase, which is a tetramer. Knowledge about the effects of tyrosinase inhibitors on the human enzyme is limited, thus, it is necessary to determine the suitability of fungal tyrosinase inhibitors relative to the human enzyme.
The search for selective Hyal inhibitors is also essential. The availability of selective inhibitors that target one type of Hyal without affecting another Hyal is important because different isoforms of Hyal regulate physiological processes. Additionally, the search for selective Hyal inhibitors will allow for a better understanding of the role of Hyals in both physiological and pathological processes.
A significant problem when comparing the results of studies is the deficiencies in analysing enzymatic reaction kinetic. Information on Km, Vmax, and Cmax of the enzymatic reaction is often missing in studies.
Many studies, especially on Hyal, focus only on determining the properties of anti-Hyal extracts. A more careful analysis of the active ingredients is needed.
Disclosure statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Additional information
Funding
References
- Stafford A, Morris P, Fowler MW. Plant cell biotechnology: a perspective. Enzyme Microb Technol 1986;8:578–87.
- Veeresham C. Natural products derived from plants as a source of drugs. J Adv Pharm Technol Res 2012;3:200–1.
- Shakya AK. Medicinal plants: future source of new drugs. Int J Herb Med 2016;4:59–64.
- Stranska I, Skalicky M, Novak J, et al. Analysis of selected poppy (Papaver somniferum L.) cultivars: pharmaceutically important alkaloids. Ind Crop Prod 2013;41:120–6.
- Li Y, Gao Z, Piao C, et al. A stable and efficient Agrobacterium tumefaciens-mediated genetic transformation of the medicinal plant Digitalis purpurea L. Appl Biochem Biotechnol 2014;172:1807–17.
- Shara M, Stohs SJ. Efficacy and safety of white willow bark (Salix alba) extracts. Phytother Res 2015;29:1112–6.
- Meshnick SR. Artemisinin: mechanisms of action, resistance and toxicity. Int J Parasitol 2002;32:1655–60.
- Sanati S, Razavi BM, Hosseinzadeh H. A review of the effects of Capsicum annuum L. and its constituent, capsaicin, in metabolic syndrome. Iranian J Basic Med Sci 2018;21:439–48.
- Júnior WSF, Cruz MP, dos Santos LL, Medeiros MFT. Use and importance of quina (Cinchona spp.) and ipeca (Carapichea ipecacuanha (Brot.) L. Andersson): Plants for medicinal use from the 16th century to the present. J Herb Med 2012;2:103–12.
- Shoaib M, Shehzad A, Omar M, et al. Inulin: properties, health benefits and food applications. Carbohydr Polym 2016;147:444–54.
- Osorio H, Bautista R, Rios A, et al. Effect of phlorizin on SGLT2 expression in the kidney of diabetic rats. J Nephrol 2010;23:541–6.
- Wink M. Modes of action of herbal medicines and plant secondary metabolites. Medicines (Basel) 2015;2:251–86.
- Maffei ME, Gertsch J, Appendino G. Plant volatiles: production, function and pharmacology. Nat Prod Rep 2011;28:1359–80.
- Khan N, Niazi ZR, Akhtar A, et al. Hyaluronidases: a therapeutic enzyme. Protein Peptide Lett 2018;25:663–76.
- Zaidi KU, Ali AS, Ali SA, et al. Microbial tyrosinases: promising enzymes for pharmaceutical, food bioprocessing, and environmental industry. Biochem Res Int 2014;2014:1–16.
- Jung H. Hyaluronidase: an overview of its properties, applications, and side effects. Arch Plastic Surg 2020;47:297–300.
- Buhren BA, Schrumpf H, Hoff NP, et al. Hyaluronidase: from clinical applications to molecular and cellular mechanisms. Eur J Med Res 2016;21:1–7.
- Saboury AA, Enzyme inhibition and activation: a general theory. J Iranian Chem Soc 2009;6:219–29.
- Sharma R. Enzyme inhibition: mechanisms and scope. In: Enzyme Inhibition and Bioapplications 2012;3–36.
- Weber GC, Buhren BA, Schrumpf H, et al. Clinical applications of hyaluronidase. Adv Exp Med Biol 2019;1148:255–77.
- Lee A, Grummer SE, Kriegel D, et al. Hyaluronidase. Dermatol Surg 2010;36:1071–7.
- McAtee CO, Barycki JJ, Simpson MA. Emerging roles for hyaluronidase in cancer metastasis and therapy. Adv Cancer Res 2014;123:1–34.
- Krupkova O, Greutert H, Boos N, et al. Expression and activity of hyaluronidases HYAL-1, HYAL-2 and HYAL-3 in the human intervertebral disc. Eur Spine J 2020;29:605–15.
- Papakonstantinou E, Roth M, Karakiulakis G. Hyaluronic acid: a key molecule in skin aging. Dermato-endocrinology 2012;4:253–8.
- Marei WF, Salavati M, Fouladi-Nashta AA. Critical role of hyaluronidase-2 during preimplantation embryo development. MHR: Basic Sci Reprod Med 2013;19:590–9.
- Wang Z, Guo C, Xu Y, et al. Two novel functions of hyaluronidase from Streptococcus agalactiae are enhanced intracellular survival and inhibition of proinflammatory cytokine expression. Infect Immun 2014;82:2615–25.
- Wittkowski KM. Hyaluronidase and hyaluronan in insect venom allergy. Int Arch Allergy Immunol 2011;156:205–11.
- Wiezel GA, dos Santos PK, Cordeiro FA, et al. Identification of hyaluronidase and phospholipase B in Lachesis muta rhombeata venom. Toxicon 2015;107:359–68.
- Panche AN, Diwan AD, Chandra SR. Flavonoids: an overview. J Nutr Sci 2016;5:
- Harborne JB, Marby H, and, Marby TJ, The flavonoids. Berlin, Germany: Springer; 2013.
- Maleki SJ, Crespo JF, Cabanillas B. Anti-inflammatory effects of flavonoids. Food Chem 2019;299:125124.
- Buer CS, Imin N, Djordjevic MA. Flavonoids: new roles for old molecules. J Integr Plant Biol 2010;52:98–111.
- Agati G, Azzarello E, Pollastri S, et al. Flavonoids as antioxidants in plants: location and functional significance. Plant Sci 2012;196:67–76.
- Wiciński M, Gębalski J, Mazurek E, et al. The influence of polyphenol compounds on human gastrointestinal tract microbiota. Nutrients 2020;12:350.
- Mandal SM, Chakraborty D, Dey S. Phenolic acids act as signaling molecules in plant-microbe symbioses. Plant Signal Behav 2010;5:359–68.
- Karak P. Biological activities of flavonoids: an overview. Int J Pharm Sci Res 2019;10:1567–74.
- Hertel W, Peschel G, Ozegowski JH, et al. Inhibitory effects of triterpenes and flavonoids on the enzymatic activity of hyaluronic acid-splitting enzymes. Arch Pharm Int J Pharm Med Chem 2006;339:313–8.
- Kuppusamy UR, Das NP. Inhibitory effects of flavonoids on several venom hyaluronidases. Experientia 1991;47:1196–200.
- Kuppusamy UR, Khoo HE, Das NP. Structure-activity studies of flavonoids as inhibitors of hyaluronidase. Biochem Pharmacol 1990;40:397–401.
- Aoshima H, Miyase T, Warashina T. Caffeic acid oligomers with hyaluronidase inhibitory activity from Clinopodium gracile. Chem Pharm Bull 2012;60:499–507.
- Kubínová R, Gazdová M, Hanáková Z, et al. New diterpenoid glucoside and flavonoids from Plectranthus scutellarioides (L.) R. Br. South Afr J Bot 2019;120:286–90.
- Karakaya S, Süntar I, Yakinci OF, et al. In vivo bioactivity assessment on Epilobium species: a particular focus on Epilobium angustifolium and its components on enzymes connected with the healing process. J Ethnopharmacol 2020;262:113207.
- Bralley E, Greenspan P, Hargrove JL, et al. Inhibition of hyaluronidase activity by select sorghum brans. J Med Food 2008;11:307–12.
- Tatemoto H, Tokeshi I, Nakamura S, et al. Inhibition of boar sperm hyaluronidase activity by tannic acid reduces polyspermy during in vitro fertilization of porcine oocytes. Zygote 2006;14:275–85.
- Zeng HJ, Ma J, Yang R, et al. Molecular interactions of flavonoids to hyaluronidase: insights from spectroscopic and molecular modeling studies. J Fluoresc 2015;25:941–59.
- Iwanaga A, Kusano G, Warashina T, Miyase T. Phenolic constituents of the aerial parts of Cimicifuga simplex and Cimicifuga japonica. Journal of Natural Products 2010;73:609–12.
- Iwanaga A, Kusano G, Warashina T, Miyase T. Hyaluronidase inhibitors from “Cimicifugae Rhizoma”(a mixture of the rhizomes of Cimicifuga dahurica and C. heracleifolia). J Nat Prod 2010;73:573–8.
- Murata T, Katagiri T, Osaka M, et al. Hyaluronidase and degranulation inhibitors from the edible roots of Oenanthe javanica including seric acids F and G that were obtained by heating. Biosci Biotechnol Biochem 2021;85:369–77.
- Hassanpour S, MaheriSis N, Eshratkhah B. Plants and secondary metabolites (Tannins): a review. Int J For Soil Eros 2011;1:47–53.
- Piluzza G, Sulas L, Bullitta S. Tannins in forage plants and their role in animal husbandry and environmental sustainability: a review. Grass Forage Sci 2014;69:32–48.
- Sieniawska E, B, T. Tannins In pharmacognosy. Cambridge (MA): Academic Press; 2017:199–232.
- Sugimoto K, Nakagawa K, Hayashi S, et al. Hydrolyzable tannins as antioxidants in the leaf extract of Eucalyptus globulus possessing tyrosinase and hyaluronidase inhibitory activities. Food Sci Technol Res 2009;15:331–6.
- Barla F, Higashijima H, Funai S, et al. Inhibitive effects of alkyl gallates on hyaluronidase and collagenase. Biosci Biotechnol Biochem 2009;73:2335–7.
- Tokeshi I, Yoshimoto T, Muto N, et al. Antihyaluronidase action of ellagic acid effectively prevents polyspermy as a result of suppression of the acrosome reaction induced by sperm-zona interaction during in vitro fertilization of porcine oocytes. J Reprod Dev 2007;53:755–64.
- Shibata T, Fujimoto K, Nagayama K, et al. Inhibitory activity of brown algal phlorotannins against hyaluronidase. Int J Food Sci Technol 2002;37:703–9.
- Kurek J. Introductory chapter: alkaloids-their importance in nature and for human life. London: IntechOpen; 2019.
- Girish KS, Kemparaju K. Inhibition of Naja naja venom hyaluronidase: role in the management of poisonous bite. Life Sci 2006;78:1433–40.
- Morikawa T, Okugawa S, Manse Y, et al. Quantitative determination of principal aporphine and benzylisoquinoline alkaloids due to blooming state in lotus flower (flower buds of nelumbo Nucifera) and their hyaluronidase inhibitory activity. Nat Prod Commun 2019;14:1934578X1985783.
- Shenoy N, Creagan E, Witzig T, et al. Ascorbic acid in cancer treatment: let the phoenix fly. Cancer Cell 2018;34:700–6.
- Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta 2012;826:443–57.
- Smirnoff N. Ascorbic acid metabolism and functions: a comparison of plants and mammals. Free Radic Biol Med 2018;122:116–29.
- Li S, Taylor KB, Kelly SJ, et al. Vitamin C inhibits the enzymatic activity of streptococcus pneumoniae hyaluronate lyase. J Biol Chem 2001;276:15125–30.
- Okorukwu ON, Vercruysse KP. Effects of ascorbic acid and analogs on the activity of testicular hyaluronidase and hyaluronan lyase on hyaluronan. J Enzyme Inhibit Med Chem 2003;18:377–82.
- Botzki A, Rigden DJ, Braun S, et al. L-Ascorbic acid 6-hexadecanoate, a potent hyaluronidase inhibitor: X-Ray structure and molecular modeling of enzyme-inhibitor complexes. J Biol Chem 2004;279:45990–7.
- Spickenreither M, Braun S, Bernhardt G, et al. Novel 6-O-acylated vitamin C derivatives as hyaluronidase inhibitors with selectivity for bacterial lyases. Bioorg Med Chem Lett 2006;16:5313–6.
- Kytidou K, Artola M, Overkleeft HS, et al. Plant glycosides and glycosidases: a treasure-trove for therapeutics. Front Plant Sci 2020;11:357.
- Tanyildizi S, Bozkurt T. In vitro effects of linamarin, amygdalin and gossypol acetic acid on hyaluronidase activity, sperm motility and morphological abnormality in bull sperm. Turk J Vet Anim Sci 2004;28:819–82.
- Cox-Georgian D, Ramadoss N, Dona C, et al. Therapeutic and medicinal uses of terpenes. In: Medicinal plants. Cham, Switzerland: Springer; 2019:333–359.
- Morikawa T, Nakanishi Y, Inoue N, et al. Acylated iridoid glycosides with hyaluronidase inhibitory activity from the rhizomes of picrorhiza kurroa royle ex benth. Phytochemistry 2020;169:112185.
- Juang YP, Liang PH. Biological and pharmacological effects of synthetic saponins. Molecules 2020;25:4974.
- Zhou JR, Kanda Y, Tanaka A, et al. Anti-hyaluronidase activity in vitro and amelioration of mouse experimental dermatitis by tomato saponin, Esculeoside A. J Agric Food Chem 2016;64:403–8.
- Zhou, J. R., Kimura, S., Nohara, T., & Yokomizo, K. (2018). Competitive inhibition of mammalian hyaluronidase by tomato saponin, esculeoside A. Nat Prod Commun 2018;13. DOI:10.1177/1934578X1601101226
- Zhou JR, Urata J, Shiraishi T, et al. Tomato juice saponin, esculeoside B ameliorates mice experimental dermatitis. Funct Foods Health Dis 2018;8:228–41.
- Abdullah NH, Thomas NF, Sivasothy Y, et al. Hyaluronidase inhibitory activity of pentacylic triterpenoids from Prismatomeris tetrandra (Roxb.) K. Schum: isolation, synthesis and QSAR study. Int J Mol Sci 2016;17:143.
- He H, Li H, Akanji T, et al. Synthesis and biological evaluations of oleanolic acid indole derivatives as hyaluronidase inhibitors with enhanced skin permeability. J Enzyme Inhibit Med Chem 2021;36:1664–77.
- Buyankhishig B, Murata T, Suganuma K, et al. Hyaluronidase inhibitory saponins and a trypanocidal isoflavonoid from the aerial parts of Oxytropis lanata. Fitoterapia 2020;145:104608.
- Murata T, Suzuki A, Mafune N, et al. Triterpene saponins from Clethra barbinervis and their hyaluronidase inhibitory activities. Chem Pharm Bull 2012;c12-00566.
- Grabowska K, Wróbel D, Żmudzki P, et al. Anti-inflammatory activity of saponins from roots of Impatiens parviflora DC. Nat Prod Res 2020;34:1581–5.
- Myose M, Warashina T, Miyase T. Triterpene saponins with hyaluronidase inhibitory activity from the seeds of Camellia sinensis. Chem Pharm Bull (Tokyo) 2012;60:612–23.
- Facino RM, Carini M, Stefani R, et al. Anti-elastase and anti-hyaluronidase activities of saponins and sapogenins from Hedera helix, Aesculus hippocastanum, and Ruscus aculeatus: factors contributing to their efficacy in the treatment of venous insufficiency. Archiv Der Pharm 1995;328:720–4.
- Casanola-Martin GM, Le-Thi-Thu H, Marrero-Ponce Y, et al. Tyrosinase enzyme: 1. An overview on a pharmacological target. Curr Top Med Chem 2014;14:1494–501.
- Zaidi KU, Ali AS, Ali SA. Purification and characterization of melanogenic enzyme tyrosinase from button mushroom. Enzyme Res 2014;2014:120739.
- Lai X, Wichers HJ, Soler-Lopez M, et al. Structure and function of human tyrosinase and tyrosinase-related proteins. Chem A Eur J 2018;24:47–55.
- Bae-Harboe YSC, Park HY. Tyrosinase: a central regulatory protein for cutaneous pigmentation. J Invest Dermatol 2012;132:2678–80.
- Mann T, Gerwat W, Batzer J, et al. Inhibition of human tyrosinase requires molecular motifs distinctively different from mushroom tyrosinase. J Invest Dermatol 2018;138:1601–16.
- Goldfeder M, Kanteev M, Adir N, et al. Influencing the monophenolase/diphenolase activity ratio in tyrosinase. Biochim Biophys Acta 2013;1834:629–33.
- Ismaya WT, Rozeboom HJ, Weijn A, et al. Crystal structure of Agaricus bisporus mushroom tyrosinase: identity of the tetramer subunits and interaction with tropolone. Biochemistry 2011;50:5477–86.
- Ghasemzadeh A, Ghasemzadeh N. Flavonoids and phenolic acids: role and biochemical activity in plants and human. J Med Plants Res 2011;5:6697–703.
- Girish KS, Kemparaju K, Nagaraju S, et al. Hyaluronidase inhibitors: a biological and therapeutic perspective. Curr Med Chem 2009;16:2261–88.
- Goncalves S, Romano A, Inhibitory properties of phenolic compounds against enzymes linked with human diseases. Phenolic compounds-biological activity. London: IntechOpen; 2017:99–118.
- Kubo I, Kinst-Hori I. Tyrosinase inhibitory activity of the olive oil flavor compounds. J Agric Food Chem 1999;47:4574–8.
- Kubo I, Chen QX, Nihei KI, et al. Tyrosinase inhibition kinetics of anisic acid. Zeitschr Naturforschung C 2003;58:713–8.
- Chen QX, Song KK, Qiu L, et al. Inhibitory effects on mushroom tyrosinase by p-alkoxybenzoic acids. Food Chem 2005;91:269–74.
- Miyazawa M, Oshima T, Koshio K, et al. Tyrosinase inhibitor from black rice bran. J Agric Food Chem 2003;51:6953–6.
- Kubo I, Kinst-Hori I, Kubo Y, et al. Molecular design of antibrowning agents. J Agric Food Chem 2000;48:1393–9.
- Shi Y, Chen Q, Wang Q, et al. Inhibitory effects of cinnamic acid and its derivatives on the diphenolase activity of mushroom (Agaricus bisporus) tyrosinase. Food Chem 2005;92:707–12.
- Garcia-Jimenez A, García-Molina F, Teruel-Puche JA, et al. Catalysis and inhibition of tyrosinase in the presence of cinnamic acid and some of its derivatives. Int J Biol Macromol 2018;119:548–54.
- Maruyama H, Kawakami F, Lwin T-T, et al. Biochemical characterization of ferulic acid and caffeic acid which effectively inhibit melanin synthesis via different mechanisms in B16 melanoma cells. Biol Pharm Bull 2018;41:806–10.
- Lee HS, Shin KH, Ryu GS, et al. Synthesis of small molecule-peptide conjugates as potential whitening agents. Bull Korean Chem Soc 2012;3:3004–8.
- Crespo MI, Chabán MF, Lanza PA, et al. Inhibitory effects of compounds isolated from Lepechinia meyenii on tyrosinase. Food Chem Toxicol 2019;125:383–91.
- Samanta A, Das G, Das SK. Roles of flavonoids in plants. Carbon 2011;100:12–35.
- Fan M, Ding H, Zhang G, et al. Relationships of dietary flavonoid structure with its tyrosinase inhibitory activity and affinity. LWT 2019;107:25–34.
- Kim D, Park J, Kim J, et al. Flavonoids as mushroom tyrosinase inhibitors: a fluorescence quenching study. J Agric Food Chem 2006;54:935–41.
- Chang TS. Two potent suicide substrates of mushroom tyrosinase: 7,8,4'-trihydroxyisoflavone and 5,7,8,4'-tetrahydroxyisoflavone. J Agric Food Chem 2007;55:2010–5.
- Tsuda T, Osawa T. Inhibition of tyrosinase activity by the anthocyanin pigments isolated from Phaseolus vulgaris L. food science and technology international. Tokyo 1997;3:82–3.
- Wagle A, Seong SH, Jung HA, Choi JS. Identifying an isoflavone from the root of Pueraria lobata as a potent tyrosinase inhibitor. Food Chem 2019;276:383–9.
- Kim JH, Kim MR, Lee ES, Lee CH. Inhibitory effects of calycosin isolated from the root of Astragalus membranaceus on melanin biosynthesis. Biol Pharm Bull 2009;32:264–8.
- Kim AJ, Choi JN, Kim J, et al. Metabolomics-based optimal koji fermentation for tyrosinase inhibition supplemented with Astragalus Radix. Biosci Biotechnol Biochem 2012;76:863–9.
- Kim JM, Ko RK, Jung DS, et al. Tyrosinase inhibitory constituents from the stems of Maackia fauriei. Phytother Res 2010;24:70–5.
- Chang TS, Ding HY, Lin HC, et al. Identifying 6,7,4'-trihydroxyisoflavone as a potent tyrosinase inhibitor. Biosci Biotechnol Biochem 2005;69:1999–2001.
- Zuo G, Wang Z, Guillen Quispe YN, et al. S. S. Target guided isolation of potential tyrosinase inhibitors from Otholobium pubescens (Poir.) JW Grimes by ultrafiltration, high-speed countercurrent chromatography and preparative HPLC. Ind Crop Prod 2019;134:195–205.
- Karioti A, Protopappa A, Megoulas N, et al. Identification of tyrosinase inhibitors from Marrubium velutinum and Marrubium cylleneum. Bioorg Med Chem 2007;15:2708–14.
- Santi MD, Bouzidi C, Gorod NS, et al. In vitro biological evaluation and molecular docking studies of natural and semisynthetic flavones from Gardenia oudiepe (Rubiaceae) as tyrosinase inhibitors. Bioorg Chem 2019;82:241–5.
- Yin XS, Zhang XQ, Yin JT, et al. Screening and identification of potential tyrosinase inhibitors from Semen Oroxyli extract by ultrafiltration LC-MS and in silico molecular docking. J Chromatogr Sci 2019;57:838–46.
- Zhang J, Chen J, Liang Z, et al. New lignans and their biological activities. Chem Biodiv 2014;11:1–54.
- Peterson J, Dwyer J, Adlercreutz H, et al. Dietary lignans: physiology and potential for cardiovascular disease risk reduction. Nutr Rev 2010;68:571–603.
- Landete JM. Plant and mammalian lignans: a review of source, intake, metabolism, intestinal bacteria and health. Food Res Int 2012;46:410–24.
- Dar AA, Arumugam N. Lignans of sesame: purification methods, biological activities and biosynthesis–a review. Bioorg Chem 2013;50:1–10.
- Malik A, Hassan Khan MT, Ali Shah AH, et al. Tyrosinase inhibitory lignans from the methanol extract of the roots of Vitex negundo Linn and their structure–activity relationship. Phytomedicine 2016;13:255–60.
- Magid AA, Abdellah A, Pecher V, et al. Flavonol glycosides and lignans from the leaves of Opilia amentacea. Phytochem Lett 2017;21:84–9.
- Begum SA, Sahai M, Ray AB. Non-conventional lignans: coumarinolignans, flavonolignans, and stilbenolignans. Fortschr Chem Org Naturst 2010;93:1–70.
- Kim JY, Kim JY, Jenis J, et al. Tyrosinase inhibitory study of flavonolignans from the seeds of Silybum marianum (Milk thistle). Bioorg Med Chem 2019;27:2499–507.
- Chong J, Poutaraud A, Hugueney P. Metabolism and roles of stilbenes in plants. Plant Sci 2009;177:143–55.
- Likhitwitayawuid K. Stilbenes with tyrosinase inhibitory activity. Curr Sci 2008;94:44–52.
- Ohguchi K, Tanaka T, Kido T, et al. Effects of hydroxystilbene derivatives on tyrosinase activity. Biochem Biophys Res Commun 2003;307:861–3.
- Kim DH, Kim JH, Baek SH, et al. Enhancement of tyrosinase inhibition of the extract of Veratrum patulum using cellulase. Biotechnol Bioeng 2004;87:849–54.
- Kim JK, Kim M, Cho SG, et al. Biotransformation of mulberroside A from Morus alba results in enhancement of tyrosinase inhibition. J Ind Microbiol Biotechnol 2010;37:631–7.
- Iida K, Hase K, Shimomura K, et al. Potent inhibitors of tyrosinase activity and melanin biosynthesis from Rheum officinale. Planta Medica 1995;61:425–8.
- Shimizu K, Yasutake S, Kondo R. A new stilbene with tyrosinase inhibitory activity from Chlorophora excelsa. Chem Pharm Bull 2003;51:318–9.
- Ohguchi K, Tanaka T, Ito T, et al. Inhibitory effects of resveratrol derivatives from dipterocarpaceae plants on tyrosinase activity. Biosci Biotechnol Biochem 2003;67:1587–9.
- Rammohan A, Reddy JS, Sravya G, et al. Chalcone synthesis, properties and medicinal applications: a review. Environ Chem Lett 2020;18:433–58.
- Khatib S, Nerya O, Musa R, et al. Chalcones as potent tyrosinase inhibitors: the importance of a 2,4-substituted resorcinol moiety. Bioorg Med Chem 2005;13:433–41.
- Jun N, Hong G, Jun K. Synthesis and evaluation of 2',4',6'-trihydroxychalcones as a new class of tyrosinase inhibitors. Bioorg Med Chem 2007;15:2396–402.
- Nguyen NT, Nguyen MHK, Nguyen HX, et al. Tyrosinase inhibitors from the wood of Artocarpus heterophyllus. J Nat Prod 2012;75:1951–5.
- Fu B, Li H, Wang X, et al. Isolation and identification of flavonoids in licorice and a study of their inhibitory effects on tyrosinase. J Agric Food Chem 2005;53:7408–14.
- Kim SJ, Son KH, Chang HW, et al. Tyrosinase inhibitory prenylated flavonoids from Sophora flavescens. Biol Pharm Bull 2003;26:1348–50.
- Panda P, Appalashetti M, Judeh ZMA. Phenylpropanoid sucrose esters: plant-derived natural products as potential leads for new therapeutics. Curr Med Chem 2011;18:3234–51.
- Kiem PV, Kiem PV, Nhiem NX, et al. & KNew phenylpropanoid esters of sucrose from Polygonum hydropiper and their antioxidant activity. Arch Pharm Res 2008;31:1477–82.
- Deng R, Li W, Berhow MA, et al. Phenolic sucrose esters: evolution, regulation, biosynthesis, and biological functions. Plant Mol Biol 2021;1–15.
- Masum MN, Choodej S, Yamauchi K, Mitsunaga T. Isolation of phenylpropanoid sucrose esters from the roots of Persicaria orientalis and their potential as inhibitors of melanogenesis. Med Chem Res 2019;28:623–32.
- Cho JG, Cha BJ, Seo WD, et al. Feruloyl sucrose esters from Oryza sativa roots and their tyrosinase inhibition activity. Chem Nat Compound 2015;51:1094–8.
- Masamoto Y, Ando H, Murata Y, et al. Mushroom tyrosinase inhibitory activity of esculetin isolated from seeds of Euphorbia lathyris L. Biosci Biotechnol Biochem 2003;67:631–4.
- Li HX, Heo M, Go Y, et al. Coumarin and moracin derivatives from mulberry leaves (Morus alba L.) with soluble epoxide hydrolase inhibitory activity. Molecules 2020;25:3967.
- Zheng ZP, Cheng KW, Zhu Q, et al. Tyrosinase inhibitory constituents from the roots of Morus nigra: a structure-activity relationship study. J Agric Food Chem 2010;58:5368–73.
- Ahmad VU, Ullah F, Hussain J, et al. Tyrosinase inhibitors from Rhododendron collettianum and their structure-activity relationship (SAR) studies. Chem Pharm Bull 2004;52:1458–61.
- Matos MJ, Varela C, Vilar S, et al. Design and discovery of tyrosinase inhibitors based on a coumarin scaffold. RSC Adv 2015;5:94227–35.
- Matos MJ, Santana L, Uriarte E, et al. New halogenated phenylcoumarins as tyrosinase inhibitors. Bioorg Med Chem Lett 2011;21:3342–5.
- Asthana S, Zucca P, Vargiu AV, et al. Structure-activity relationship study of hydroxycoumarins and mushroom tyrosinase. J Agric Food Chem 2015;63:7236–44.
- Liu J, Wu F, Chen L, et al. Biological evaluation of coumarin derivatives as mushroom tyrosinase inhibitors. Food Chem 2012;135:2872–8.
- Ashraf Z, Rafiq M, Seo SY, et al. Design, synthesis and bioevaluation of novel umbelliferone analogues as potential mushroom tyrosinase inhibitors. J Enzyme Inhibit Med Chem 2015;30:874–83.
- Yang D, Wang L, Zhai J, et al. Characterization of antioxidant, α-glucosidase and tyrosinase inhibitors from the rhizomes of Potentilla anserina L. and their structure-activity relationship. Food Chem 2021;336:127714.
- Fujimaki T, Mori S, Horikawa M, Fukui Y. Isolation of proanthocyanidins from red wine, and their inhibitory effects on melanin synthesis in vitro. Food Chem 2018;248:61–9.
- Momtaz S, Mapunya BM, Houghton PJ, et al. Tyrosinase inhibition by extracts and constituents of Sideroxylon inerme L. stem bark, used in South Africa for skin lightening. J Ethnopharmacol 2008;119:507–12.
- Magid AA, Voutquenne-Nazabadioko L, Bontemps G, et al. Tyrosinase inhibitors and sesquiterpene diglycosides from Guioa villosa. Planta Med 2008;74:55–60.
- Kang HS, Kim HR, Byun DS, et al. Tyrosinase inhibitors isolated from the edible brown alga Ecklonia stolonifera. Arch Pharm Res 2004;27:1226–32.
- Shoji T, Masumoto S, Moriichi N, et al. Procyanidin trimers to pentamers fractionated from apple inhibit melanogenesis in B16 mouse melanoma cells. J Agric Food Chem 2005;53:6105–11.
- Lin QM, Wang Y, Yu JH, et al. Tyrosinase inhibitors from the leaves of Eucalyptus globulus. Fitoterapia 2019;139:104418.
- Yang L, Yang YL, Dong WH, et al. Sesquiterpenoids and 2-(2-phenylethyl)chromones respectively acting as α-glucosidase and tyrosinase inhibitors from agarwood of an Aquilaria plant. J Enzyme Inhib Med Chem 2019;34:853–62.
- Zolghadri S, Bahrami A, Hassan Khan MT, et al. A comprehensive review on tyrosinase inhibitors. J Enzyme Inhibit Med Chem 2019;34:279–309.
- Loizzo MR, Tundis R, Menichini F. Natural and synthetic tyrosinase inhibitors as antibrowning agents: an update. Comprehens Rev Food Sci Food Saf 2012;11:378–98.
- Batubara I, Darusman LK, Mitsunaga T, et al. Potency of Indonesian medicinal plants as tyrosinase inhibitor and antioxidant agent. J Biol Sci 2010;10:138–44.
- Bonesi M, Xiao J, Tundis R, et al. Advances in the tyrosinase inhibitors from plant source. Curr Med Chem 2019;26:3279–99.
- Scotti L, Singla RK, Ishiki HM, et al. Recent advancement in natural hyaluronidase inhibitors. Curr Top Med Chem 2016;16:2525–31.
- Gallelli L. Escin: a review of its anti-edematous, anti-inflammatory, and venotonic properties. Drug Design Dev Ther 201913:3425–37.
- Scotti L, Singla RK, Ishiki HM, et al. Recent advancement in natural hyaluronidase inhibitors. Curr Top Med Chem 2016;16:2525–31.
- Gallelli, L. Escin: A review of its anti-edematous, anti-inflammatory, and venotonic properties. Drug Des Devel Ther 2019;13:3425.