
Abstract
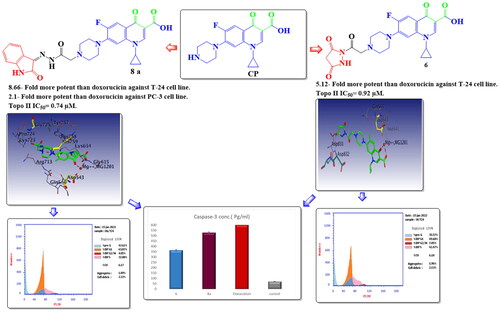
A series of novel ciprofloxacin (CP) derivatives substituted at the N-4 position with biologically active moieties were designed and synthesised. 14 compounds were 1.02- to 8.66-fold more potent than doxorubicin against T-24 cancer cells. Ten compounds were 1.2- to 7.1-fold more potent than doxorubicin against PC-3 cancer cells. The most potent compounds 6, 7a, 7b, 8a, 9a, and 10c showed significant Topo II inhibitory activity (83–90% at 100 μM concentration). Compounds 6, 8a, and 10c were 1.01- to 2.32-fold more potent than doxorubicin. Compounds 6 and 8a induced apoptosis in T-24 (16.8- and 20.1-fold, respectively compared to control). This evidence was supported by an increase in the level of apoptotic caspase-3 (5.23- and 7.6-fold, sequentially). Both compounds arrested the cell cycle in the S phase in T-24 cancer cells while in PC-3 cancer cells the two compounds arrested the cell cycle in the G1 phase. Molecular docking simulations of compounds 6 and 8a into the Topo II active site rationalised their remarkable Topo II inhibitory activity.
Graphical Abstract
Introduction
Cancer is a very dangerous and life-threatening disease, it is considered one of the most prevalent diseases in the worldCitation1. The defining characteristic of cancer is metastasis, the leading cause of death from cancerCitation1–3. Many antitumor agents are commercially available, but the emergence of acquired drug resistance with severe side effects of these clinically used anticancer drugs poses serious barriers to effective chemotherapyCitation4. Therefore, it is recommended to rationally develop new anticancer drugs with fewer side effects.
DNA topological problems arise from the intertwined nature of the DNA double helix structure, which causes tangles and supercoiling of the DNA duplex during the DNA replication and transcription. DNA supercoiling results in torsion that impair the function of DNA or RNA polymerases. Type II topoisomerase enzyme (Topo II) prevents and corrects these types of topological problems via transient double-stranded breaks, causing DNA metabolism to proceed, allowing the cell to efficiently replicate so enabling cellular division and vitalityCitation5–7. The role of divalent Mg2+ ions in Topo II-mediated reactions was recognised as an implication in enzyme-mediated DNA cleavage reactions. (2) participation in ATPase reactions and functions by providing the enzyme with magnesium–ATP substrateCitation8. Topo II enzyme inhibition leads to apoptosis and cell deathCitation9,Citation10, therefore, it is considered a valid strategy in cancer therapy. The presence of topoisomerase enzyme in both mammalian and bacterial cells makes it a pronounced target for antibacterial and anticancer drugsCitation10,Citation11. Recently, mammalian Topo II is considered a critical target for anticancer drug developmentCitation12–14.
The use of fluoroquinolone derivatives as anti-proliferative agents is of great interest to researchers, as they are less toxic, lower tumour resistance is exerted towards them, and they have less chance of developing secondary tumours. Moreover, they exhibited excellent pharmacological and pharmacokinetic profilesCitation15–17. Fluoroquinolones act by inhibiting the Topo II enzyme in both prokaryotic and eukaryotic cells, due to the similarities between the prokaryotic and eukaryotic topoisomerasesCitation18. A similar mechanism of action characterises several clinically important antitumor agents such as etoposide, doxorubicin, amsacrine, or mitoxantroneCitation19,Citation20. Recently, a great deal of work has been devoted to the antiproliferative activity of fluoroquinolones and several studies proved them as potent cytotoxic agentsCitation19,Citation21–23. Ciprofloxacin (CP), a broad-spectrum fluoroquinolone antibiotic, showed anti-proliferative activity against strains of human cancer cells. CP has been reported to pile up in urine and prostate tissues, therefore it is a privileged candidate for the treatment of bladder, and prostate cancersCitation24. CP, among fluoroquinolones, is distinguished by strong inhibition of Topo IICitation25. Additionally, it can induce the intrinsic apoptotic pathway by creating a double-stranded break in DNA or cell cycle arrest in the S/G2 phaseCitation26,Citation27. Thus, CP serves as a unique scaffold for the development of novel anticancer agents.
The SAR studies uncovered that fluorine atom, the 1-alkyl, and 1, 4-dihydro-4-oxo-quinoline-3-carboxylic acid skeleton are the fundamental pharmacophore lineaments for CP anticancer activityCitation19,Citation28–31.
Several studies have been conducted to determine the cytotoxic structural features of CP on eukaryotic cells. These studies changed the activity of fluoroquinolones from antibacterial to antitumor activityCitation32. Topo II inhibitory activity and pharmacokinetic features of CP are greatly affected by the modification at the piperazinyl N-4 position of the condensed CPCitation15. Hence, new fluoroquinolone analogues can be developed by structural modifications at the C-7 position of CP which diminished the zwitterion effect and greatly influenced the hydrophilicity nature leading to the improvement of antiproliferative activity of CPCitation33–38.
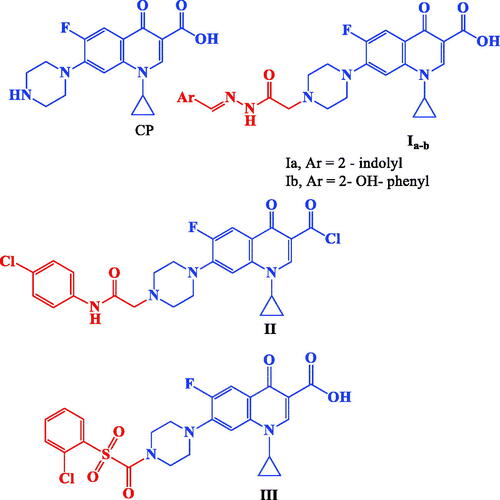
CP hybrids Ia,bCitation39, IICitation21, and IIICitation21 () incorporating N-acylarylhydrazone, arylacetamide, and aryl sulphonyl moieties, respectively at the N-4 position of piperazine revealed significant in vitro anti-proliferative activity.
Figure 1. Structures of CP and potent anticancer CP derivatives I–III.
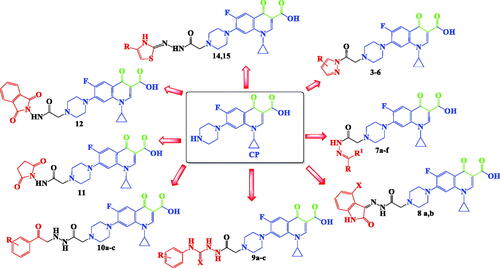
Inspired by all these findings, we have designed a series of novel CP derivatives with essential pharmacophoric features for Topo II inhibition. Our design strategy kept the two coplanar carbonyl groups at positions 3 and 4 of the CP scaffold which is a common structural motif of potent Topo II inhibitors such as merbaroneCitation40, vosaroxinCitation41, and A-65281Citation42 (). Moreover, this pharmacophore may guarantee the coordination with the Mg2+ ion that plays an essential role in promoting the DNA cleavage–rejoining activity of Topo II enzymeCitation8. The newly synthesised CP derivatives are featuring various biologically active moieties at the N-4 position of piperazine such as monocyclic heteroaryl scaffolds (pyrazole, pyrazolidine, pyrrolidine, or thiazolidine ring), benzo fused heteroaryl rings (indoline or isoindoline), N-acyl(alkyl or aryl) hydrazone, semicarbazide, thiosemicarbazide or arylacetamide. These moieties are well acknowledged for anti-proliferative activity via different mechanisms such as apoptosis induction, caspase activation, and DNA inter-chelationCitation21,Citation43–61. All these moieties are linked to the piperazine ring of CP via 2, 3, or 4 atoms spacers (). We have aimed to explore the impact of such variation at the N-4 position of piperazine of CP core with diverse lipophilic and electronic environments on the anticancer activity and to identify potent anti-proliferative agents. The synthesised CP hybrids were screened for their anti-proliferative activity in vitro against bladder T-24 and prostate PC-3 cancer cell lines. All derivatives were subjected to the determination of their half-maximal inhibitory concentration (IC50) values. The conversion of supercoiled plasmid DNA to relaxed DNA by recombinant Topo II was examined in the presence of each of the most potent compounds 6, 7a, 7b, 8a, 9a, and 10c. The most prominent Topo II inhibitors 6 and 8a were further investigated regarding their effects on cell cycle progression, induction of apoptosis, and level of active caspase-3 in the T-24 cell line.
Figure 2. Potent Topo II inhibitors.
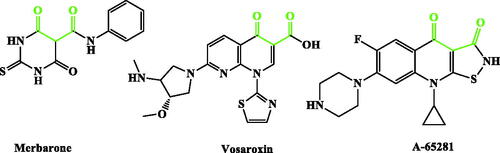
Figure 3. Design strategy for the synthesised CP hybrids.
Materials and methods
Chemistry
General
Melting points were determined on a Griffin apparatus and were uncorrected. Shimadzu IR 435 spectrophotometer (Shimadzu Corp., Kyoto, Japan), Faculty of Pharmacy, Cairo University, Cairo, Egypt, was used to record IR spectra, values were represented in cm−1. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded in ppm on the δ scale and coupling constants (J) were given in Hz on Bruker 400 MHz (Bruker Corp., Billerica, MA, USA) spectrophotometer, Faculty of Pharmacy, Cairo University, Cairo, Egypt. Tetra-methylsilane (TMS) was used as an internal standard. Progress of the reactions was monitored by TLC using pre-coated aluminium sheet silica gel MERCK 60F 254 and was visualised by a UV lamp.
Procedure for the preparation of 1-cyclopropyl-7-(4-(2-ethoxy-2-oxoethyl)piperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (1)
A mixture of ciprofloxacin (1.56 g, 0.005 mol), ethyl chloroacetate (0.61 g, 0.005 mol) and trimethylamine (10 g, 0.1 mol) in dimethylformamide (50 ml) was heated under reflux for 6 h. The reaction mixture was cooled, and the separated solid was filtered, dried, and crystallised from ethanol to give compound 1. MP 190–192 °C; yield 86%Citation39.
Procedure for the preparation of 1-cyclopropyl-6-fluoro-7-(4-(2-hydrazinyl-2-oxoethyl) piperazin-1-yl)-4-oxo-1,4-dihydroquinoline -3-carboxylic acid (2)
A mixture of ester derivative 1 (2.08 g, 0.005 mol) and hydrazine hydrate 99% (1.26 g, 0.025 mol) in absolute ethanol (5 ml) was heated under reflux for 6 h. The reaction mixture was cooled, and the separated solid was filtered, dried, and crystallised from ethanol to give compound 2. MP 226–228 °C (as reported); yield: 75%Citation39.
Procedure for the preparation of 1-cyclopropyl-6-fluoro-7-(4-(2-(3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)-2-oxoethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3)
A mixture of the hydrazinyl derivative 2 (0.50 g, 0.001 mol) and ethyl acetoacetate (0.13 g, 0.001 mol) in absolute ethanol (5 ml) was heated under reflux for 6 h. The reaction mixture was cooled, and the separated solid was filtered, dried, and crystallised from ethanol to give compound 3. MP 188–190 °C yield; 60%; IR (KBr) vmax: 3549 (OH), 3093 (C–H arom.), 2978 (C–H aliph.), 1730, 1720 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.18–1.20 (m, 2H, CH2 cyclopropyl), 1.30–1.35 (m, 2H, CH2 cyclopropyl), 2.47 (s, 3H, CH3C = N), 2.70–2.75 (m, 4H, 2CH2 piperazine), 3.30–3.40 (m, 4H, 2CH2 piperazine), 3.40–3.50 (m, 1H, CH cyclopropyl), 3.80 (s, 2H, CH2 pyrazolone), 4.30 (s, 2H, N-CH2CO), 7.54 (d, 1H, J = 8 Hz, ArH), 7.85 (d, 1H, J = 13.6 Hz, ArH), 8.60 (s, 1H, C2-H), 15.17 (s, 1H, COOH, D2O exchangeable) ppm; 13C NMR (DMSO-d6): δ 8.0, 14.6, 36.3, 49.8, 52.0, 58.6, 60.3, 107.1, 111.4, 119.0, 139.6, 145.5, 145.6, 148.4, 152.2, 154.7, 166.3, 170.3, 176.7–176.8 ppm. Anal. Calcd. for C23H24FN5O5 (469.47): C, 58.84; H, 5.15; N, 14.92. Found: C, 58.93; H, 5.19; N, 15.22.
Procedure for the preparation of 1-cyclopropyl-6-fluoro-7-(4-(2-(5-imino-3-oxopyrazolidin-1-yl)-2-oxoethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (4)
A mixture of hydrazinyl derivative 2 (0.50 g, 0.001 mol) and ethyl cyanoacetate (0.11 g, 0.001 mol) in glacial acetic acid (10 ml) was heated under reflux for 5 h. The reaction mixture was filtered while hot, the filtrate was left to cool, and the separated solid was filtered, washed with water (15 ml) dried and recrystallised from ethanol to give compound 4. MP 181–183 °C; yield 37%; IR (KBr) vmax: 3541 (OH), 3437, 3417 (2NH), 3059 (C–H arom.), 2870 (C–H aliph.), 1728, 1720 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.25–1.40 (m, 2H, CH2 cyclopropyl), 1.60–1.65 (m, 2H, CH2 cyclopropyl), 2.80–2.95 (m, 4H, 2CH2 piperazine), 3.36–3.41 (m, 4H, 2CH2 piperazine), 3.44 (s, 2H, NCH2O), 3.80–3.90 (m, 1H, CH cyclopropyl), 3.86 (s, 2H, CH2 pyrazalone) 7.57 (d, 1H, ArH), 7.90 (d, 1H, J = 8 Hz, ArH), 8.66 (s, 1H, C2-H), 9.68 (s, 1H, NH, D2O exchangeable), 9.75 (s, 1H, NH, D2O exchangeable), 15.19 (s, 1H, COOH D2O exchangeable) ppm; 13C NMR (DMSO-d6): δ 5.9, 12.5, 24.0, 34.2, 43.7, 47.5, 104.6, 109.3, 115.3, 116.8, 122.5, 137.5, 143.4, 146.2, 163.6, 164.2, 168.2, 169.5, 174.6 ppm. Anal. Calcd. for C22H23FN6O5 (470.45): C, 56.17; H, 4.93; N, 17.86. Found: C, 56.40; H, 4.81; N, 18.05.
Procedure for the preparation of 1-cyclopropyl-7-(4-(2-(3,5-dimethyl-1H-pyrazol-1-yl)-2-oxoethyl)piperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (5)
A mixture hydrazinyl derivative 2 (0.50 g, 0.001 mol) and acetylacetone (0.1 g, 0.001 mol) in absolute ethanol (5 ml) was heated under reflux for 6 h. The reaction mixture was cooled, and the separated solid was filtered, dried, and crystallised from ethanol to give compound 5. MP: 185–187 °C; yield: 60%; IR (KBr) vmax: 3444 (OH), 3055 (C–H arom.), 2912 (C–H aliph.), 1730, 1728, 1720 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.15–1.20 (m, 2H, CH2 cyclopropyl), 1.30–1.35 (m, 2H, CH2 cyclopropyl), 2.75–2.80 (m, 7H, 2CH2, CH3), 2.90 (s, 3H, CH3), 3.30–3.35 (m, 4H, 2CH2 piperazine), 3.40–3.50 (m, 1H, CH cyclopropyl), 3.80 (s, 2H, CH2CO), 7.55 (d, 1H, ArH), 7.88 (d, 1H, ArH), 7.90 (s, 1H, CH pyrazole), 8.63 (s, 1H, C2-H), 15.16 (s, 1H, COOH, D2O exchangeable) ppm; 13C NMR (DMSO-d6): δ 8.0, 14.6, 19.0, 36.3, 49.8, 52.0, 58.6, 106.8, 107.1, 111.2, 111.4, 118.9, 139.6, 145.6, 148.3, 152.2, 154.6, 166.3, 170.3, 176.7 ppm. Calcd. for C24H26FN5O4 (467.49): C, 61.66; H, 5.61; N, 14.98. Found: C, 61.92; H, 5.72; N, 15.17.
Procedure for the preparation of 1-cyclopropyl-7-(4-(2-(3,5-dioxopyrazolidin-1-yl)-2-oxoethyl)piperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6)
A mixture of hydrazinyl derivative 2 (0.20 g, 0.0005 mol) in sodium ethoxide (0.034 g atomic weight of sodium in 17 ml absolute ethanol), diethyl malonate (0.1 g, 0.0008 mol) was heated under reflux for 7 h. The reaction mixture was cooled, and the separated solid was filtered, washed with water (5 ml) dried, and crystallised from ethanol to give compound 6. MP: 140–142 °C; yield: 35%; IR IR (KBr) vmax: 3583 (OH), 3441 (NH), 3008 (C–H arom.), 2958 (C–H aliph.), 1680, 1660 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.15–1.30 (m, 4H, 2CH2 cyclopropyl), 2.70–2.80 (m, 4H, 2CH2 piperazine), 2.90 (s, 2H, CH2 pyrazolone), 3.20–3.25 (m, 4H, 2CH2 piperazine), 3.35 (s, 2H, CH2CO), 3.65 (s, 1H, NH, D2O exchangeable), 3.95–4.00 (m, 1H, CH cyclopropyl), 7.54 (d, 1H, ArH), 7.85 (d, 1H, ArH), 8.59 (s, 1H, C2-H) ppm; 13C NMR (DMSO-d6): δ 8.0, 14.6, 15.3, 36.3, 49.8, 60.3, 106.8, 117.6, 119.0, 130.1, 133.2, 148.3, 152.2, 154.7, 160.0, 166.3, 168.5, 170.3, 176.8 ppm. Anal. Calcd. for C22H22FN5O6 (471.44): C, 56.90; H, 4.70; N, 14.86. Found: C, 56.11; H, 4.68; N, 14.84.
General procedure for the preparation of 7-(4-(-1-(substituted)ethylidene)hydrazine carbonyl)methyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (7a–f)
A mixture of hydrazinyl derivative 2 (0.50 g, 0.001 mol), the appropriate ketone (0.001 mol), and glacial acetic acid (1 ml) in absolute ethanol (5 ml) was heated under reflux for 6 h. The reaction mixture was cooled, and the separated solid was filtered, dried, and recrystallised from ethanol to give compounds 7a–f.
7-(4-(2-(2-Cyclohexylidenehydrazinyl)-2-oxoethyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (7a)
M.P.: 256–258 °C; yield: 65%; IR (KBr) vmax: 3497 (OH), 3417 (NH), 3043 (C–H arom.), 2981, 2962 (C–H aliph.) cm−1; 1H NMR (DMSO-d6): δ 1.18–1.25 (m, 2H, CH2 cyclopropyl), 1.31–1.33 (m, 2H, CH2 cyclopropyl), 2.91–2.93 (m, 2H, CH2 cyclohexylidene) 3.24–3.25 (m, 4H, 2CH2 cyclohexylidene), 3.40–3.50 (m, 1H, CH cyclopropyl), 3.60–3.70 (m, 4H, 2CH2 cyclohexylidene), 3.82 (s, 2H, CH2CO) 7.55 (d, 1H, J = 8 Hz, ArH), 7.87 (d, 1H, J = 12 Hz, ArH), 8.13 (s, 1H, NH, D2O exchangeable), 8.65 (s, 1H, C2-H) ppm; 13C NMR (DMSO-d6): δ 8.0, 19.0, 34.2, 36.2, 45.8, 51.0, 51.1, 57.0, 77.0, 106.4, 107.2, 111.3, 118.7, 139.6, 146.1, 148.2, 166.4, 152.1, 154.6, 176.7 ppm. Anal. Calcd. for C25H30FN5O4 (483.54): C, 62.10; H, 6.25; N, 14.48. Found: C, 62.12; H, 6.28; N, 14.46.
7-(4-(2-(2-(1-(4-Chlorophenyl)ethylidene)hydrazinyl)-2-oxoethyl)-piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (7b)
M.P.: 178–180 °C; yield 75%; IR (KBr) vmax: 3444 (OH), 3298 (NH), 3078 (C–H arom.), 2924, 2827 (C–H aliph.), 1705 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.19–1.23 (m, 2H, CH2 cyclopropyl), 1.33–1.40 (m, 2H, CH2 cyclopropyl), 2.18 (s, 3H, CH3), 2.25–2.30 (m, 4H, 2CH2 piperazine), 2.70–2.90 (m, 4H, 2CH2 piperazine), 3.05 (s, 2H, CH2CO), 3.78–3.80 (m, 1H, CH cyclopropyl), 7.50–7.52 (m, 3H, ArH), 7.82–8.00 (m, 3H, ArH), 8.6 (s, 1H, C2-H), 10.42, 10.66 (2 s, 1H, NH, D2O exchangeable), 15.22 (s, 1H, COOH, D2O exchangeable) ppm. Anal. Calcd. for C27H27ClFN5O4 (539.99): C, 60.06; H, 5.04; N, 12.97. Found: C, 60.13; H, 5.18; N, 12.92.
7-(4-(2-(2-(1-(4-Aminophenyl)ethylidene)hydrazinyl)-2-oxoethyl)-piperazin-1-yl)-1-cyclopropyl -6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (7c)
M.P.: 173–175; yield 70%; IR (KBr) vmax: 3421 (OH), 3356, 3232 (NH, NH2), 3051 (C–H arom.), 2974 (C–H aliph.), 1730, 1728 (C = O) cm−1; 1HNMR (DMSO-d6): δ 1.19–1.22 (m, 2H, CH2 cyclopropyl), 1.26–1.33 (m, 2H, CH2 cyclopropyl), 1.84–1.87 (m, 1H, CH cyclopropyl), 2.80–2.93 (m, 4H, 2CH2 piperazine), 3.46–3.92 (m, 7H, 2CH2 piperazine, CH3), 3.87 (s, 2H, CH2CO), 4.20 (s, 2H, NH2, D2O exchangeable), 7.11 (s, 1H, NH, D2O exchangeable) 7.54 (d, 3H, ArH), 7.94 (d, 3H, ArH), 8.68 (s, 1H, C2-H), 11.23 (s, 1H, OH exchangeable by D2O) ppm. Anal. Calcd. for C27H29FN6O4 (520.56): C, 62.30; H, 5.62; N, 16.14. Found: C, 62.28; H, 5.57; N, 16.16.
1-Cyclopropyl-6-fluoro-7-(4-(2-(2-(1-(2-hydroxyphenyl)ethylidene)hydrazinyl)-2-oxoethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (7d)
M.P.: 166–168; yield 80%; IR (KBr) vmax; 3545 (OH), 3417 (NH), 3059 (C–H arom.), 2912 (C–H aliph.), 1728, 1705 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.18–1.22 (m, 2H, CH2 cyclopropyl), 1.32–1.33 (m, 2H, CH2 cyclopropyl), 2.53 (s, 3H, CH3), 2.75–2.80 (m, 4H, CH2 piperazine), 3.33–3.45 (m, 6H, 2CH2 piperazine, CH2CO), 3.81–3.90 (m, 1H, CH cyclopropyl), 6.95 (d, 2H, ArH), 7.40 (t, 1H, ArH), 7.55 (d, 1H, ArH), 7.76 (d, 1H, ArH), 7.89 (d, 1H, ArH), 8.64 (s, 1H, C2-H), 12.93 (s, 1H, NH, D2O exchangeable), 13.12 (s, 1H, OH, D2O exchangeable) 15.20 (s, 1H, COOH, D2O exchangeable) ppm. 13C NMR (DMSO-d6): δ 8.0, 15.3, 36.2, 49.8, 52.0, 60.3, 106.8, 107.1, 111.4, 117.6, 118.9, 119.5, 130.1, 133.2, 139.6, 145.5, 148.3, 152.1, 154.6, 160.0, 166.3, 168.6, 170.3, 176.7 ppm. Anal. Calcd. for C27H28FN5O5 (521.56): C, 62.18; H, 5.41; N, 13.43. Found: C, 61.96; H, 5.70; N, 13.62.
1-Cyclopropyl-6-fluoro-4-oxo-7-(4-(2-oxo-2-(2-(1-(thiophen-2-yl)ethylidene)hydrazinyl)ethyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (7e)
M.P.: 245–247; yield 85%; IR (KBr) vmax: 3441 (OH), 3271 (NH), 3082 (C–H arom.), 2947 (C–H aliph.), 1730, 1728 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.15–1.20 (m, 2H, CH2 cyclopropyl), 1.25–1.30 (m, 2H, CH2 cyclopropyl), 2.40 (s, 3H, CH3), 2.60–2.65 (m, 4H, 2CH2 piperazine), 2.70–2.80 (m, 4H, 2CH2 piperazine), 3.27 (s, 2H, CH2 CO), 3.85–3.90 (m, 1H, CH cyclopropyl), 7.05–7.15 (m, 2H, ArH), 7.50–7.70 (m, 2H, ArH), 7.95 (d, 1H, ArH), 8.65 (s, 1H, C2-H), 8.99, 9.67 (2 s, 1H, NH, D2O exchangeable), 15.19 (s, 1H, COOH, D2O exchangeable) ppm. 13C NMR (DMSO-d6): δ 8.0, 14.6, 36.3, 49.7, 49.9, 52.6, 106.8, 107.7, 111.2, 118.9, 119.0, 127.9, 128.5, 139.6, 143.5, 144.0, 145.6, 148.3, 154.6, 168.5, 171.5, 176.7 ppm. Anal. Calcd. for C25H26FN5O4S (511.57): C, 58.70; H, 5.12; N, 13.69. Found: C, 58.63; H, 5.15; N, 13.72.
7-(4-(2-(2-(1-(5-Chlorothiophen-2-yl)ethylidene)hydrazinyl)-2-oxoethyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (7f)
M.P.: 286–288; yield 85%; IR (KBr) vmax: 3456 (OH), 3275 (NH), 3059 (C–H arom.), 2924 (C–H aliph.), 1730, 1728 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.10–1.20 (m, 2H, CH2 cyclopropyl), 1.27–1.33 (m, 2H, CH2 cyclopropyl), 2.38 (s, 3H, CH3), 2.70–2.74 (m, 4H, 2CH2 piperazine), 3.30–3.42 (m, 6H, 2CH2 piperazine, CH2CO), 3.70–3.90 (m, 1H, CH cyclopropyl), 7.18–7.20 (m, 2H, ArH), 7.56 (d, 1H, ArH), 7.75 (d, 1H, ArH), 8.65 (s, 1H, C2-H), 10.30, 10.55 (2 s, NH, D2O exchangeable), 15.19 (s, 1H, COOH, D2O exchangeable) ppm; 13C NMR (DMSO-d6): δ 8.0, 14.6, 19.0, 36.3, 49.8, 56.4, 106.8, 107.1, 111.2, 111.4, 117.6, 119.0, 139.6, 145.5, 148.3, 152.2, 154.7, 166.3, 168.5, 170.3, 176.7 ppm. Anal. Calcd. for C25H25ClFN5O4S (546): C, 54.99; H, 4.61; N,12.83. Found: C, 54.92; H, 4.63; N,12.80.
General procedure for the preparation of 7-(4-((N'-(4-substituted-2-oxo-2,3-dihydro-1H-indol-3-ylidene)hydrazine carbonyl)methyl)piperazin-1-yl)1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (8a–b)
A mixture of hydrazinyl derivative 2 (0.50 g, 0.001 mol), substituted isatin (0.001 mol) and glacial acetic acid (1 ml) in absolute ethanol (5 ml) was heated under reflux for 7 h. The reaction mixture was cooled, and the separated solid was filtered, dried, and recrystallised from ethanol to give compounds 8a–b.
1-Cyclopropyl-6-fluoro-4-oxo-7-(4-(2-oxo-2-(2-(2-oxoindolin-3-ylidene)hydrazinyl)ethyl) piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (8a)
M.P.: 186–188; yield 85%; IR (KBr) vmax: 3441 (OH), 3240 (NH), 3051 (C–H arom.), 2974 (C–H aliph.), 1730, 1720, 1712 (C = O) cm−1. 1H NMR (DMSO-d6): δ 1.02–1.05 (m, 2H, CH2 cyclopropyl), 1.17–1.20 (m, 2H, CH2 cyclopropyl), 2.72–2.74 (m, 4H, 2CH2 piperazine), 3.29–3.31 (m, 4H, 2CH2 piperazine), 3.45 (s, 2H, CH2CO), 3.75–3.85 (m, 1H, CH cyclopropyl), 6.85–6.88 (m, 1H, ArH), 7.03–7.05 (m, 1H, ArH), 7.36–7.45 (m, 2H, ArH), 7.50 (d, 1H, ArH), 7.90 (d, 1H, ArH), 8.65 (s, 1H, C2-H), 10.96, 11.10 (2 s, 1H, NH, D2O exchangeable), 13.84 (s, 1H, NH, D2O exchangeable), 15.91 (s, 1H, COOH, D2O exchangeable) ppm. 13C NMR (DMSO-d6): δ 8.0, 14.6, 36.3, 49.8, 58.6, 60.3, 106.8, 111.5, 112.6, 118.2, 118.9, 123.2, 125.1, 128.6, 134.8, 138.8, 139.5, 145.6, 151.1, 152.1, 154.6, 166.3, 170.3, 176.7, 184.8 ppm. Anal. Calcd. for C27H25FN6O5 (532.52): C, 60.90; H, 4.73; N, 15.78. Found: C, 60.88; H, 4.71; N, 15.76.
7-(4-(2-(2-(4-Bromo-2-oxoindolin-3-ylidene)hydrazinyl)-2-oxoethyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (8b)
M.P.: 263–265; yield 70%; IR (KBr) vmax: 3498 (OH), 3200, 3174 (NH), 3078 (C–H arom.), 2916 (C–H aliph.), 1720, 1700, 1697 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.20–1.23 (m, 2H, CH2 cyclopropyl), 1.30–1.35 (m, 2H, CH2 cyclopropyl), 2.74–2.77 (m, 4H, 2CH2 piperazine), 3.34–3.35 (m, 4H, 2CH2 piperazine), 3.45 (s, 2H, CH2CO), 3.80–3.90 (m, 1H, CH cyclopropyl), 6.85–6.90 (m, 1H, ArH), 7.57–7.65 (m, 2H, ArH), 7.72 (d, 1H, ArH), 7.89 (d, 1H, J = 12 Hz, ArH), 8.60 (s, 1H, C2-H), 11.14, 11.26 (2 s, 1H, NH, D2O exchangeable), 13.83 (s, 1H, NH, D2O exchangeable), 15.21 (s, 1H, COOH, D2O exchangeable) ppm. 13C NMR (DMSO-d6): δ 8.0, 14.6, 49.8, 52.0, 58.6, 60.3, 106.9, 107.2, 111.2, 111.5, 114.7, 119.0, 120.0, 127.3, 139.6, 140.4, 145.5, 145.6, 148.4, 150.0, 159.4, 166.3, 170.3, 176.8, 183.6 ppm. Anal. Calcd. for C27H24BrFN6O5 (611.42): C, 53.04; H, 3.96; N, 13.75. Found: C, 57.13; H, 4.15; N, 14.18.
General procedure for the preparation of 7-(4-(substituted)phenyl)carbamoyl)amino)carbamoyl)methyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (9a-c)
A mixture of hydrazinyl derivative 2 (0.50 g, 0.001 mol), substituted phenyl isocyanate or phenyl isothiocyanate (0.001 mol), and glacial acetic acid (1 ml) in absolute ethanol (15 ml) was heated under reflux for 7 h. The reaction mixture was cooled, and the separated solid was filtered, dried, and recrystallised from ethanol to give compounds 9a–c.
7-(4-(2-(2-((4-Chloro-3-(trifluoromethyl)phenyl)carbamoyl)-hydrazinyl)-2-oxoethyl) piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (9a)
M.P.: 186–188; yield 65%; IR (KBr) vmax: 3437 (OH), 3414 (NH), 3055 (C–H arom.), 2908 (C–H aliph.), 1730, 1728 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.04–1.08 (m, 2H, CH2 cyclopropyl), 1.32–1.33 (m, 2H, CH2 cyclopropyl), 2.65 (s, 2H, CH2CO), 2.70–2.77 (m, 4H, 2CH2 piperazine), 3.30–3.45 (m, 4H, 2CH2 piperazine), 3.80–3.90 (m, 1H, CH cyclopropyl), 7.56–7.70 (m, 3H, ArH), 7.87–8.15 (m, 2H, ArH), 8.65 (s, 1H, C2-H), 9.30 (s, 1H, NH, D2O exchangeable), 9.50 (s, 1H, NH, D2O exchangeable), 10.10 (s, 1H, NH, D2O exchangeable), 15.07 (s, 1H, COOH, D2O exchangeable) ppm; 13C NMR (DMSO-d6): δ 8.3, 14.6, 49.8, 52.0, 60.3, 106.8, 106.9, 107.1, 111.2, 111.4, 118.9, 119.0, 139.6, 145.5, 145.6, 148.3, 152.2, 154.7, 166.3, 170.3, 176.7, 176.8 ppm. Anal. Calcd. for C27H25ClF4N6O5 (624.97): C, 51.89; H, 4.03; N, 13.45. Found: C, 52.17; H, 4.19; N, 13.71.
7-(4-(2-(2-((2-Chloro-6-methylphenyl)carbamoyl)hydrazinyl)-2-oxoethyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (9b)
M.P.: 285–287; yield 83%; IR (KBr) vmax: 3572 (OH), 3441 (NH), 3051 (C–H arom.), 2904 (C–H aliph.), 1730, 1728 (C = O) cm−1. 1H NMR (DMSO-d6): δ 1.15–1.25 (m, 4H, 2CH2 cyclopropyl), 2.20–2.25 (m, 11H, 4CH2 piperazine, CH3), 3.34 (s, 2H, CH2CO), 4.80–4.90 (m, 1H, CH cyclopropyl), 7.17–7.24 (m, 3H, ArH), 7.33–7.35 (m, 3H, ArH), 8.94 (brs, 3H, 3NH, D2O exchangeable). 13C NMR (DMSO-d6): δ 8.0, 18.8, 36.3, 44.1, 49.8, 49.9, 106.9, 107.8, 111.4, 119.0, 127.7, 129.2, 133.1, 135.6, 139.3, 145.4, 145.5, 148.2, 152.1, 154.4, 155.6, 166.3, 176.6, 176.7 ppm. Anal. Calcd. for C27H28ClFN6O5 (571): C, 56.79; H, 4.94; N, 14.72. Found: C, 57.05; H, 5.12; N, 14.98.
1-Cyclopropyl-6-fluoro-7-(4-(2-(2-((4-methoxyphenyl)carbamo-thioyl)hydrazinyl)-2-oxoethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (9c)
M.P.: 238–240; yield: 62%; IR (KBr) vmax: 3500 (OH), 3379 (NH), 3008 (C–H arom.), 2912 (C–H aliph.), 1712 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.17–1.20 (m, 2H, CH2 cyclopropyl), 1.33–1.35 (m, 2H, CH2 cyclopropyl), 3.40 (s, 2H, CH2CO), 3.49–3.50 (m, 4H, 2CH2 piperazine), 3.75 (s, 3H, CH3O), 3.83–3.84 (m, 1H, CH cyclopropyl), 4.14–4.15 (m, 4H, 2CH2 piperazine), 6.82 (d, 2H, J = 8.0 Hz, ArH), 7.19 (d, 2H, J = 8.0 Hz, ArH), 7.59 (d, 1H, J = 8.0 Hz, ArH), 7.90 (d, 1H, J = 13.20 Hz, ArH), 8.67 (s, 1H, C2-H), 9.34 (s, 2H, 2NH, D2O exchangeable) and 15.16 (s, 1H, COOH, D2O exchangeable); 13C NMR (DMSO-d6): δ 8.0, 36.3, 40.4, 47.7, 49.1, 55.6, 106.5, 107.1, 111.3, 111.5, 113.7, 118.8, 118.9, 127.8, 134.1, 145.1, 148.4, 151.9, 157.0, 166.5, 176.7, 182.2 ppm. Anal. Calcd. for C27H29FN6O5S (568.62): C, 57.03; H, 5.14; N, 14.78. Found: C, 57.21; H, 5.38; N, 15.02.
General procedure for the preparation of 1-cyclopropyl-6-fluoro-7-(4-((N'-(2-(substituted phenyl)-2-oxoethyl)hydrazine carbonyl)methyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (10a–c)
A mixture of hydrazinyl derivative 2 (0.50 g, 0.001 mol), anhydrous potassium carbonate (0.41 g, 0.003 mol) and substituted phenacyl bromide (0.001 mol) in dry benzene (8 ml) was heated under reflux for 24 h. The reaction mixture was filtered while hot. The residue was washed twice with water (20 ml), dried, and recrystallised from ethanol to give compound 10a–c.
1-Cyclopropyl-6-fluoro-4-oxo-7-(4-(2-oxo-2-(2-(2-oxo-2-phenyl-ethyl)hydrazinyl)ethyl) piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (10a)
M.P.: 183–185 °C; yield: 45%; (KBr) vmax: 3500 (OH), 3402 (NH), 3008 (C–H arom.), 2916 (C–H aliph.), 1732, 1700, 1697 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.11–1.18 (m, 2H, CH2 cyclopropyl), 1.30–1.32 (m, 2H, CH2 cyclopropyl), 2.75–2.79 (m, 2H, CH2 piperazine), 2.90–2.95 (m, 2H, CH2 piperazine), 3.40 (s, 2H,CH2CO), 3.45–3.50 (m, 2H, CH2 piperazine), 3.60–3.70 (m, 2H, CH2 piperazine), 3.80–3.90 (m, 1H, CH cyclopropyl), 4.00 (s, 2H, CH2CO), 5.61 (s, 1H, NH, D2O exchangeable), 7.37–8.05 (m, 7H, ArH), 8.56 (s, 1H, C2-H), 8.66 (s, 1H, NH, D2O exchangeable), ppm; 13C NMR (DMSO-d6): δ 7.9, 17.0, 26.0, 36.5, 46.2, 50.0, 102.0, 111.0, 116.5, 126.0, 127.4, 128.5, 129.4, 143.0, 144.5, 150.5, 161.0, 166.6, 174.5 ppm. Anal. Calcd. for C27H28FN5O5 (521.54): C, 62.18; H, 5.41; N, 13.43. Found: C, 61.96; H, 4.70; N, 13.72.
7-(4-(2-(2-(2-(3-Bromophenyl)-2-oxoethyl)hydrazinyl)-2-oxo-ethyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (10b)
M.P.: 210–215 °C; yield: 62%; IR (KBr) vmax: 3500 (OH), 3240 (NH), 3020 (C–H arom.), 2920 (C–H aliph.), 1725 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.16–1.18 (m, 2H, CH2 cyclopropyl), 1.29–1.32 (m, 2H, CH2 cyclopropyl), 2.90–3.00 (m, 4H, 2CH2 piperazine), 3.22–3.24 (m, 4H, 2CH2 piperazine), 3.30 (s, 2H, CH2CO), 3.40–3.70 (m, 3H, CH cyclopropyl, CH2CO), 7.35–7.40 (m, 1H, ArH), 7.40–7.48 (m, 1H, ArH), 7.50 (d, 1H, J = 8 Hz, ArH), 7.81 (m, 5H, 3ArH, 2NH), and 8.62 (s, 1H, C2-H) ppm; 13C NMR (DMSO-d6): δ 8.0, 27.0, 36.2, 45.40, 50.6, 53.0, 106.5, 107.1, 111.1, 111.4, 118.7, 123.7, 124.3, 128.7, 129.3, 135.7, 139.6, 146.0, 148.2, 152.1, 154.6, 166.4, 167.2, 176.7 ppm. Anal. Calcd. for C27H27BrFN5O5 (600.45): C, 54.01; H, 4.53; N, 11.66. Found: C, 55.21; H, 4.82; N, 12.03.
1-Cyclopropyl-6-fluoro-7-(4-(2-(2-(2-(4-nitrophenyl)-2-oxoethyl)-hydrazinyl)-2-oxoethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (10c)
M.P.: 221–223 °C; yield: 55%; IR (KBr) vmax: 3540 (OH), 3402 (NH), 3008 (C–H arom.), 2950 (C–H aliph.), 1730 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.10–1.15 (m, 2H, CH2 cyclopropyl), 1.30–1.35 (m, 2H, CH2 cyclopropyl), 2.80–2.85 (m, 4H, 2CH2 piperazine), 3.20–3.25 (m, 4H, 2CH2 piperazine), 3.30 (s, 2H, CH2CO), 3.60 (s, 2H, CH2CO), 3.80–3.90 (m, 1H, CH cyclopropyl), 7.36 (s, 1H, NH, D2O exchangeable), 7.50–7.60 (m, 2H, ArH), 7.70 (d, 1H, ArH), 8.10–8.50 (m, 3H, ArH), 8.59 (s, 1H, NH, D2O exchangeable), and 8.66 (s, 1H, C2-H) ppm; 13C NMR (DMSO-d6): δ 8.0, 29.0, 31.5, 36.0, 45.5, 50.8, 106.5, 108.6, 111.2, 111.4, 119.3, 121.2, 128.2, 129.8, 131.4, 132.2, 139.5, 148.1, 152.1, 154.6, 166.6, 176.4 ppm. Anal. Calcd. for C27H27FN6O7 (566.55): C, 57.24; H, 4.80; N, 14.83. Found: C, 57.41; H, 4.98; N, 15.09.
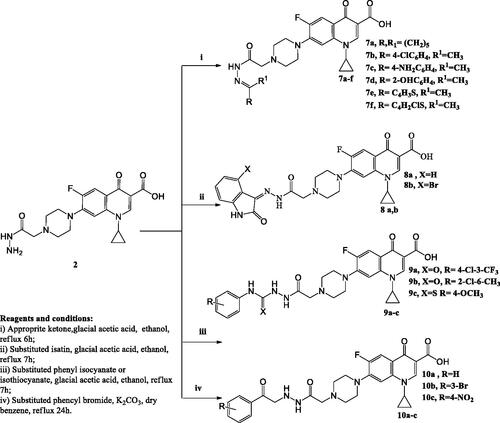
Procedure for the preparation of 1-cyclopropyl-7-(4-(2-((2,5-dioxopyrrolidin-1-yl)amino)-2-oxoethyl)piperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (11)
A mixture of hydrazinyl derivative 2 (0.50 g, 0.001 mol), succinic anhydride (0.10 g, 0.001 mol), and anhydrous sodium acetate (0.117 g, 0.0015 ml) in glacial acetic acid (10 ml) was heated under reflux for 5 h. The reaction mixture was concentrated to half its volume and allowed to cool, the separated solid was filtered, washed with cold ethanol, dried, and recrystallised from acetic acid to give compound 11. M.P.: 184–186 °C; yield: 55%; IR (KBr) vmax: 3441 (OH), 3224 (NH), 3016 (C–H arom.), 2947 (C–H aliph.), 1720, 1712, 1700 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.18–1.20 (m, 2H, CH2 cyclopropyl), 1.31–1.33 (m, 2H, CH2 cyclopropyl), 2.24 (s, 4H, 2CH2 pyrrolidine), 2.70–2.80 (m, 4H, 2CH2 piperazine), 3.10 (s, 2H, CH2CO), 3.30–3.40 (m, 4H, 2CH2 piperazine), 3.83–3.84 (m, 1H, CH cyclopropyl), 7.56 (d, 1H, J = 8 Hz, ArH), 7.90 (d, 1H, J = 13.6 Hz, ArH), 8.66 (s, 1H, C2-H), and 9.68 (s, 1H, NH, D2O exchangeable) ppm. 13C NMR (DMSO-d6): δ 8.0, 19.5, 20.8, 26.6, 28.7, 29.8, 106.6, 107.3, 111.2, 111.4, 119.0, 139.5, 145.5, 148.2, 152.1, 154.6, 168.4, 176.6 ppm. C23H24FN5O6 (485.46): C, 56.90; H, 4.98; N, 14.43. Found: C, 56.87; H, 4.95; N, 14.41.
Procedure for the preparation of 1-cyclopropyl-7-(4-(2-((1,3-dioxoisoindolin-2-yl)amino)-2-oxoethyl)piperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (12)
A mixture of compound 2 (0.50 g, 0.001 mol), phthalic anhydride (0.15 g, 0.001 mol), and anhydrous sodium acetate (0.117 g, 0.0015 ml) in glacial acetic acid (10 ml) was heated under reflux for 5 h. The reaction mixture was concentrated to half its volume and allowed to cool, the separated solid was filtered, washed with cold ethanol, dried, and recrystallised from acetic acid to give compound 12. M.P: 190–192 °C; yield: 50%; IR (KBr) vmax: 3441 (OH), 3224 (NH), 3012 (C–H arom.), 2897 (C–H aliph.), 1720, 1700 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.10–1.15 (m, 2H, CH2 cyclopropyl), 1.20–1.30 (m, 2H, CH2 cyclopropyl), 2.65–2.75 (m, 4H, 2CH2 piperazine), 3.11 (s, 2H, CH2CO), 3.25–3.35 (m, 4H, 2CH2 piperazine), 3.75–3.80 (m, 1H, CH cyclopropyl), 7.36–8.15 (m, 6H, ArH), 8.63 (s, 1H, C2-H), and 9.74 (s, 1H, NH, D2O exchangeable) ppm. Anal. Calcd. for C27H24FN5O6 (533.51): C, 60.78; H, 4.53; N, 13.13. Found: C, 60.92; H, 4.58; N, 13.11.
Procedure for the preparation of 7-(4-(2-(2-carbamothioylhydrazinyl)-2-oxoethyl) piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (13)
A mixture of hydrazinyl derivative 2 (0.50 g, 0.001 mol), potassium thiocyanate (0.194 g, 0.002 mol), and concentrated hydrochloric acid (1 ml) in absolute ethanol (15 ml) was heated under reflux for 6 h. The reaction mixture was cooled, and the separated solid was filtered, dried, and recrystallised from ethanol to give compound 13. M.P.: 188–200 °C; yield: 65%; IR (KBr) vmax: 3417 (OH), 3271, 3236 (NH, NH2), 3001 (C–H arom.), 2947 (C–H aliph.), 1747, 1728 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.19–1.21 (m, 2H, CH2 cyclopropyl), 1.34–1.35 (m, 2H, CH2 cyclopropyl), 3.30–3.50 (m, 8H, 4CH2 piperazine), 3.67 (s, 2H, CH2CO), 3.87–3.89 (m, 1H, CH cyclopropyl), 7.20 (s, 1H, NH, D2O exchangeable), 7.30 (s, 2H, NH2, D2O exchangeable), 7.40 (s, 1H, NH, D2O exchangeable), 7.63 (d, 1H, J = 8 Hz, ArH), 7.96 (d, 1H, J = 13 Hz, ArH), 8.68 (s, 1H, C2-H), and 15.20 (s, 1H, COOH, D2O exchangeable) ppm; 13C NMR (DMSO-d6): δ 8.0, 14.5, 17.0, 51.8, 56.1, 107.2, 130.0, 144.3, 148.4, 148.5, 152.0, 152.1, 154.5, 166.2, 166.3, 168.5, 176.7 ppm. Anal. Calcd. for C20H23FN6O4S (462.5): C, 51.94; H, 5.01; N, 18.17 Found: C, 51.91; H, 5.12; N, 18.22
Procedure for the preparation of 1-cyclopropyl-6-fluoro-4-oxo-7-(4-(2-oxo-2-(2-(4-oxothiazolidin-2-ylidene)hydrazinyl)ethyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (14)
A mixture of compound 13 (0.23 g, 0.0005 mol), ethyl chloroacetate (0.60 g, 0.005 mol) in absolute ethanol (15 ml) was heated under reflux for 6 h. The obtained precipitate was filtered, washed with water, dried, and recrystallised from ethanol to form compound 14. MP: 240–242 °C; yield: 70%; IR (KBr) vmax: 3417 (OH), 3271 (NH), 3020 (C–H arom.) 2947(C–H aliph.), 1720 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.18–1.20 (m, 2H, CH2 cyclopropyl), 1.34–1.36 (m, 2H, CH2 cyclopropyl), 2.60–2.80 (m, 4H, 2CH2 piperazine), 3.28 (s, 4H, 2CH2CO), 3.30–3.35 (m, 4H, 2CH2 piperazine), 3.51 (brs, 2H, 2NH, D2O exchangeable), 3.80–3.90 (m, 1H, CH cyclopropyl), 7.58 (d, 1H, J = 8.0 Hz, ArH), 7.90 (d, 1H, J = 13 Hz, ArH), 8.66 (s, 1H, C2-H), and 15.21 (s, 1H, COOH, D2O exchangeable) ppm. 13C NMR (DMSO-d6): δ 6.2, 21.4, 29.3, 44.6, 52.6, 60.5, 106.4, 111.2, 114.5, 121.0, 144.5, 144.6, 156.0, 165.5, 170.2, 178.1, 188.2 ppm. Anal. Calcd. for C22H23FN6O5S (502.52): C, 52.58; H, 4.61; N, 16.72. Found: C, 52.79; H, 4.75; N, 16.88.
Procedure for the preparation of 1-cyclopropyl-6-fluoro-4-oxo-7-(4-(2-oxo-2-(2-(4-phenylthiazol-2(3H)-ylidene)hydrazinyl)ethyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid (15)
A mixture of compound 13 (0.18 g, 0.0004 mol), phenacyl bromide (0.08 g, 0.0004 mol), and anhydrous sodium acetate (0.03 g, 0.0004 mol) in absolute ethanol (15 ml) was heated under reflux for 6 h. The obtained precipitate was filtered, washed with water, dried, and recrystallised from ethanol to form compound 15. M.P.: 256–258 °C; yield: 25%; IR (KBr) vmax: 3549 (OH), 3317 (NH), 3089 (C–H arom.) 2912 (C–H aliph.), 1728 (C = O) cm−1; 1H NMR (DMSO-d6): δ 1.19–1.20 (m, 2H, CH2 cyclopropyl), 1.33–1.35 (m, 2H, CH2 cyclopropyl), 2.21–2.23 (m, 4H, 2CH2 piperazine), 3.36–3.38 (m, 4H, CH2CO and 2NH), 3.70–3.75 (m, 4H, 2CH2 piperazine), 3.80–3.90 (m, 1H, CH cyclopropyl), 7.15–7.20 (m, 3H, ArH), 7.35 (d, 2H, ArH), 7.55 (d, 1H, J = 8.0 Hz, ArH), 7.85 (d, 1H, J = 13.0 Hz, ArH), 8.31 (s, 1H, thiazole-H), 8.62 (s, 1H, C2-H), and 14.80 (s, 1H, COOH, D2O exchangeable) ppm. 13C NMR (DMSO-d6): δ 8.0, 18.8, 36.3, 44.2, 49.9, 106.9, 107.1, 111.5, 119.1, 119.2, 127.2, 127.7, 129.3, 133.2, 135.6, 139.3, 145.5, 148.3, 152.1, 154.6, 155.7, 166.3, 176.7 ppm. Anal. Calcd. for C28H27FN6O4S (562.18): C, 59.78; H, 4.84; N, 14.94. Found: C, 59.50; H, 5.22; N, 14.84.
Biological activity
Cell culture protocol
Human bladder cancer (T-24) and human prostate carcinoma (PC-3) cell lines were obtained from American Type Culture Collection (ATCC; Manassas, VA USA). Cells were maintained in Dulbecco’s modified Eagle medium with 10% foetal bovine serum at 37 °C and 5% CO2. All the operations were carried out under strict aseptic conditions. The culture medium was removed to a centrifuge tube containing 9.0 ml complete culture medium and spanned at approximately 125 × g for 5 to 7 min. The cell layer was rinsed with 0.25% (w/v) trypsin- 0.53 µM EDTA solution by which all traces of serum-containing inhibitor were removed. Trypsin EDTA solution 2.0 to 3.0 ml was added to a flask and cells were monitored under an inverted microscope until the cell layer was dispersed (usually within 5–15 min). A complete growth medium was added (6.0–8.0 ml), and cells were aspirated by gentle pipetting. The cell pellet was suspended with the recommended complete medium and dispensed into a 75 cm2 culture flask. The culture vessel containing the complete growth medium was placed in the incubator for 24 h to at 37 °C. The cells were treated with different concentrations (0.39, 1.60, 6.25, 25, and 100 μg/ml) of each of the test compounds or doxorubicin, followed by incubation for 48 h at 37 °C, then the plates were examined under the inverted microscope and finally the MTT assay was carried out.
Cell viability assay
Anti-cancer activity of the newly synthesised compounds was evaluated in vitro against both T-24 and PC-3 cell lines according to the MTT methodCitation62. Cells were seeded into 96-well plates (flat bottom) at a density of 10 000 cells/well for 24 h. The vial of MTT to be used was reconstituted with 3 ml of medium or balanced salt solution without phenol red and serum. then, a reconstituted MTT vial was added in an amount equal to 10% of the culture medium volume. Cultures were incubated for 2–4 h depending on the cell type and maximum cell density and removed from the incubator and the formazan crystals were dissolved via the addition of an amount of MTT solubilisation solution (M-8910) equal to the original culture medium volume. ROBONIK P2000 spectrophotometer at a wavelength of 570 nm was used to measure the colour intensity spectrophotometrically. The survival curve of both T-24 and PC-3 cells was obtained by plotting the percentage of surviving cells against the drug after each compound. The IC50 value for each test compound and the reference drug doxorubicin was calculated.
In vitro DNA topo II-mediated relaxation assay
DNA Topo II inhibitory activities of the targeted compounds were measured as follows. A mixture of 100 ng of supercoiled pBR322 plasmid DNA (Fermentas, USA) and 2 units of human DNA Topo II (USB Corp., USA) was incubated with and without the prepared compounds in the assay buffer (10 µM Tris-HCl pH 7.9) containing 50 µM NaCl, 4 µM MgCl2, 1 µM EDTA, 1 µM ATP, and 15 mg/ml bovine serum albumin for 30 min at 37 °C. The reaction in a final volume of 10 µl was terminated by the addition of 4 µl of 6 µM EDTA. DNA samples were then electrophoresed on 1% agarose gel containing 0.5 µg/ml ethidium bromide in gel and buffer at high voltage (100–250 v) until the dye front has migrated about 4–6 cm down the gel. with a running buffer of TAE Tris-acetate– EDTA (TAE). Gels were stained for 15 min in water. DNA bands were visualised by transillumination with UV light and quantitated using AlphaI-maker TM (Alpha Innotech Corporation).
Cell cycle analysis of compounds 6 and 8a
The cell cycle was done using a propidium iodide flow cytometry kit (Abcam, ab139418) according to the manufacturer’s instructions. T-24 and PC-3 cells were treated with IC50 of compounds 6 and 8a for 24 h. After treatment, the cells were washed twice with ice-cold phosphate buffer saline, then centrifuged and fixed using ice-cold 66% (v/v) ethanol, washed with phosphate buffer saline re-suspended with 0.10 mg/ml, and stained with 200 µl propidium iodide. Cells were analysed by flow cytometry. The cell cycle distributions were calculated using cell–quest software (Bectopn Dickinson).
Apoptosis determination
Apoptosis was evaluated using Annexin V fluorescein isothiocyanate (FITC) and propidium iodide (PI) using the Annexin V-FITC/PI apoptosis detection kit (Biovision, Mountain View, CA # K101-25). According to the manufacturer’s instructions after staining the cells with annexin V fluorescein (FITC) isothiocyanate. Briefly, 1–5 × 105 cells were exposed to compounds 6 and 8a at their IC50 concentrations for 24 h. Cells were centrifuged and washed once with serum-containing media followed by resuspension in 500 µl of binding buffer. Then 5 µl Annexin V-FITC and 5 µl of (PI 50 mg/ml) were added and incubated for 5 min at room temperature in the dark. Analyses were performed using a flow cytometer (EX = 488 nm: Em = 530 nm) using a FITC signal detector and PI staining by a phycoerythrin emission signal detector.
Measurement of the effect of compounds 6 and 8a on the level of caspase-3 protein (a marker of apoptosis)
The level of the apoptotic marker caspase-3 was measured using The Invitrogen Caspase-3 (active) Human ELISA Kit. The procedure of the used kit was done according to the manufacturer’s instructions.
Molecular docking study
Molecular docking studies were performed using the Molecular Operating Environment (MOE, 2014.0901) software. All minimisations were performed using MOE with MMFF94x force field, and the partial and formal charges were calculated automatically. The X-ray crystallographic structures of topoisomerase IIα co-crystallised with DNACitation63 were downloaded from the protein data bankCitation64. To prepare the enzyme for the docking study, first DNA chains and water molecules were removed, and then the protonate 3D protocol in MOE with default options was used. For the docking protocol, the triangle matcher placement method and London dG scoring function were used. Validation of the docking results was carried out by docking the reference topoisomerase inhibitor merbarone and comparing the results with a previously reported study. The MOE-validated setup was used to predict the binding interactions and affinity of the synthesised compounds at the active site.
Results and discussion
Chemistry
The synthetic approaches designed in this study for the synthesis of the target compounds are elucidated in Schemes 1–3. The primary starting compound 1, was prepared by reacting CP with ethyl chloroacetate in dimethylformamide in the presence of trimethylamineCitation39. Compound 1 was reacted with hydrazine hydrate to afford compound 2Citation39.
Compound 3 was prepared by reacting hydrazinyl derivative 2 with ethyl acetoacetate in absolute ethanol. The 1H NMR spectrum of this compound displayed the disappearance of the signal corresponding to NH2 of the parent compound 2 and the appearance of two singlet signals at δ 2.47 and 3.80 ppm corresponding to CH3C=N and CH2 pyrazolone protons, respectively.
CP derivative 4 was prepared by reacting compound 2 with ethyl cyanoacetate in glacial acetic acid, the 1H NMR spectrum displayed a singlet signal at δ 3.86 ppm expressing the CH2 pyrazolone protons. Compound 5 was obtained via refluxing CP hydrazinyl derivative 2 with acetylacetone in ethanol. The 1H NMR spectrum displayed the disappearance of the signal corresponding to NH2 of the parent compound 2 in addition to the presence of one singlet signal at δ 7.90 ppm corresponding to CH of pyrazole.
Compound 6 was obtained by reacting compound 2 with diethyl malonate in the presence of sodium ethoxide. The 1H NMR spectrum of this compound revealed the appearance of a singlet signal at δ 2.90 ppm due to the CH2 of pyrazolone.
In Scheme 2, the hydrazones 7a–f were obtained by reacting compound 2 with the appropriate ketone in ethanol in the presence of glacial acetic acid. The 1H NMR and 13C NMR spectra of these compounds displayed the presence of signals corresponding to different alkyl or aryl groups which were not present in compound 2.
Scheme 1. The synthetic path and reagents for the preparation of the target compounds 1–6.
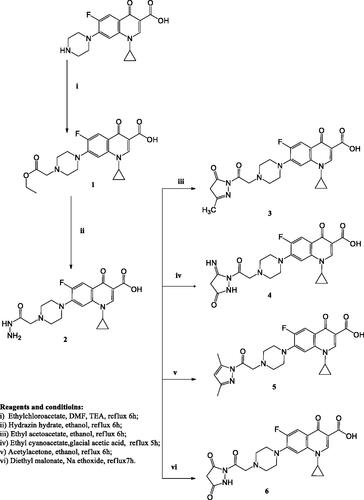
Scheme 2. The synthetic path and reagents for the preparation of the target compounds 7–10.
Refluxing compound 2 with the suitable isatin in absolute ethanol in the presence of glacial acetic acid afforded compounds 8a–b. The 1H NMR and 13C NMR spectra of these compounds revealed the presence of different signals of indoline moieties.
Compounds 9a–c were prepared via reacting compound 2 with the appropriate phenyl isocyanates or phenyl isothiocyanate in absolute ethanol in the presence of glacial acetic acid. The 1H NMR and 13C NMR spectra of these derivatives displayed the characteristic signals corresponding to different aryl moieties. Further structural evidence stemmed from the 1H NMR spectra that showed the exchangeable singlet signals corresponding to NH protons.
CP derivatives 10a–c were prepared via reacting compound 2 with the suitable phenacyl bromide in dry benzene in the presence of potassium carbonate. The 1H NMR and 13C NMR spectra of these derivatives showed the appearance of new signals corresponding to the added phenyl ring in addition to singlet signals that appeared at δ 3.30–3.40 ppm due to CH2CO protons.
In Scheme 3, the reaction of compound 2 with succinic anhydride or phthalic anhydride in glacial acetic acid in the presence of anhydrous sodium acetate yielded compounds 11 and 12, respectively. The 1H NMR and 13C NMR spectra of these compounds showed the appearance of new signals corresponding to pyrrolidine and isoindoline moieties.
Scheme 3. The synthetic path and reagents for the preparation of the target compounds 11–15.
Reacting compound 2 with potassium thiocyanate and concentrated hydrochloric acid in absolute ethanol afforded compound 13. 13C NMR spectrum revealed the appearance of (C = S) carbon at δ 176.7 ppm. Compound 14 was prepared via reacting derivative 13 with ethyl chloroacetate in ethanol. The 1H NMR spectrum of compound 14 revealed the disappearance of the exchangeable signal of NH2 protons.
Finally, compound 15 was prepared by refluxing compound 13 with phenacyl bromide in absolute ethanol in the presence of anhydrous sodium acetate. The 1H NMR and 13C NMR spectra showed the expected signals corresponding to the phenyl group, which were not present in the starting compound 14. Additionally, the 1H NMR spectrum revealed the appearance of a single signal corresponding to CH of the thiazole ring at δ 8.31 ppm.
Biological activity
Growth inhibition against human tumour cell lines
In this study, all the newly synthesised CP derivatives were subjected to anticancer activity evaluation against bladder (T-24) and prostate cancer (PC-3) cell lines. The compounds were evaluated for their activity using 5 doses determinations (100 μg/ml, 25 μg/ml, 6.25 μg/ml, 1.60 μg/ml, and 0.39 μg/ml). Their half-maximal inhibitory concentration (IC50) values were measured. Doxorubicin was chosen as a reference anticancer drugCitation65.
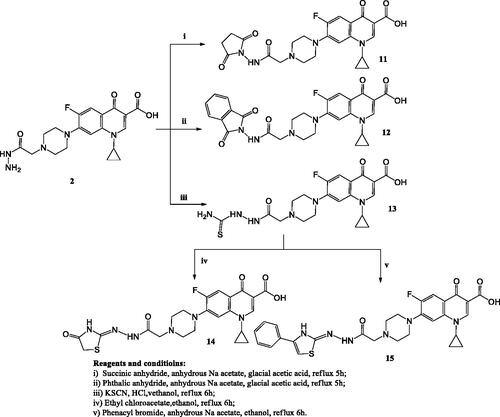
The test compounds showed anticancer activity against the T-24 cell line with IC50 values ranging from 3.36–366 μM. They exhibited IC50 values against the PC-3 cell line in the range of 3.25–159.24 μM ().
Table 1. The half-maximal inhibitory concentration (IC50) of CP derivatives and doxorubicin after treatment for 24 h on both the prostate cancer cell line (PC-3) and bladder cancer cell line (T-24).
Compounds 4–6, 7a–c, 7e, 8a, 9a–b, 10b–c, 14, and 15 showed 1.02- to 8.66-fold more potent anti-proliferative activity than the reference standard doxorubicin against T-24 cell line (.
Figure 4. Graphical representation for the half-maximal inhibitory concentration (IC50) of CP derivatives and doxorubicin after treatment for 24 h on bladder cancer cell line.
Compounds 7a–b, 7d, 8a, 9b–c, 10b–c, 14, and 15 exhibited 1.2- to 7.1-fold more potent anti-proliferative activity than doxorubicin against PC-3 cells line ().
Figure 5. Graphical representation for half-maximal inhibitory concentration (IC50) of the target compounds and doxorubicin after treatment for 24 h on the prostate cancer cell line.
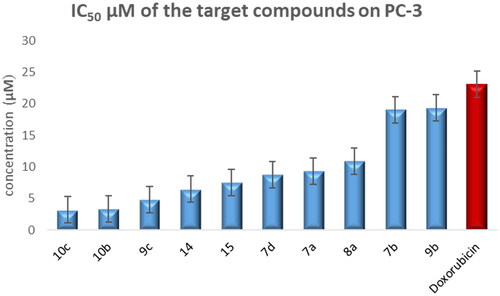
Regarding the activity towards bladder cancer, compounds 6, 7a, 7b, 8a, 9a, and 10c were the most potent among the synthesised CP derivatives.
It is worth mentioning that the T-24 bladder cancer cell line was more sensitive to the synthesised CP derivatives than the PC-3 prostate cancer cell line.
The Structure-activity correlation of the newly synthesised CP derivatives revealed that modification at the piperazinyl N-4 position of the CP scaffold resulted in variable potency. CP derivatives bearing pyrazole ring through acetyl spacer (4–6) showed potent anticancer activity. They showed more potency against bladder cancer cell line, compound 6 incorporating pyrazolidine-3,5-dione moiety was the most prominent among them. Regarding CP hydrazones 7a–e, compound 7a incorporating cyclohexylidene moiety showed marked potency against bladder and prostate cancer cell lines. Further analysis of these compounds revealed that hydrazones carrying substituted phenyl ring (7b–d) were more potent than their counterparts having thiophene ring (7e,f). An interesting phenomenon is that CP hydrazone 7d featuring ortho hydroxyl group on the phenyl ring showed the most potent anti-proliferative activity among CP aryl hydrazones against prostate cancer. CP derivative 8a having the acylhydrazone scaffold with a benzene ring carrying a nearby NH proved to marked anti-proliferative activity against both cell lines. It was clear that the introduction of a bromine atom on the indoline scaffold (8b) had a bad impact on the anticancer activity. CP semicarbazide and thiosemicarbazide derivatives 9a–c possessed potent activity. It was noticed that compound 9a featuring the 4-chloro-3-trifluoromethylphenyl moiety group was the most potent. Reviewing compounds 10a–c, revealed that the substitution of phenyl moiety with electron withdrawing group in 10b and 10c significantly improved the anticancer activity against the two cell lines. The incorporation of monocyclic pyrrolidine or pyrrolidine fused with benzene in CP derivatives 11 and 12 resulted in lower anticancer activity. It is worth mentioning that, grafting thiazole moiety in derivatives 14 and 15 highly improved the antiproliferative activity.
Recombinant topoisomerase II inhibitory activities of compounds (6, 7a, 7b, 8a, 9a, and 10c)
The conversion of supercoiled plasmid DNA to relaxed DNA by recombinant Topo II was examined in the presence of each of the most potent compounds 6, 7a, 7b, 8a, 9a, and 10c for measuring their Topo II inhibitory activities. Well-known Topo II inhibitor doxorubicin was used as a positive control. The reaction products of Topo II relaxation assays were analysed by electrophoretic mobility and developed in ethidium bromide in the presence of UV light. The inhibitory activities were evaluated at 4 concentrations of 100, 10, 1, and 0.1 μM for all compounds as shown in (). Topo II inhibitory activities of the tested compounds are summarised in ().
Figure 6. Recombinant Topo II inhibitory activities of compounds 6, 7a, 7b, 8a, 9a, and 10c at 100, 10, 1, 0.1 µM. Lane P: pBR322 DNA only; lane N: pBR322 DNA + Topo II; lanes (6, 7a, 7 b, 8a, 9a and 10c): pBR322 DNA + Topo II + compounds 6, 7a, 7 b, 8a, 9a and 10c.
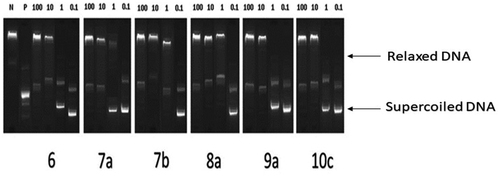
Table 2. Recombinant Topo II inhibitory activities of compounds (6, 7a, 7b, 8a, 9a, and 10c).
All test compounds showed significant Topo II inhibitory activity in the range of 83–90% at 100 μM concentration. While at 0.1 μM concentration, the range of Topo II inhibition was 13.7–32.5% (). The IC50 value of each compound was calculated. Compounds 6, 8a, and 10c were 1.01- to 2.32-fold more potent than doxorubicin ().
Table 3. Topo II IC50 results of compounds (6, 7a, 7b, 8a, 9a, and 10c) compared to doxorubicin.
Cell cycle analysis and detection of apoptosis
The most prominent compounds 6 and 8a that showed Topo II IC50 values in the sub-micro-molar range were further investigated in terms of their effects on apoptosis induction and cell cycle progression in both T-24 and PC-3 cell lines.
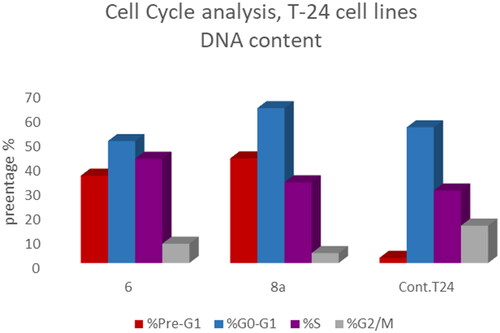
T-24 and PC-3 cells were treated with compounds 6 and 8a at their IC50 values for 24 h and their effects on the normal cell cycle profile and induction of apoptosis were recorded. Treatment of T-24 and PC-3 cells with compounds 6 and 8a resulted in an interference with the normal distribution of the cell cycle. In T-24 cell lines, both compounds 6 and 8a induced a significant increase in the percentage of cells at pre-G1 by 16.7- and 20.1-fold, respectively when compared to the control. Also, they showed an increase in the percentage of cells in the S phase by 1.4- and 1.11-fold, sequentially compared to the control (). The accumulation of cells in the pre-G1 phase indicated that CP derivatives 6 and 8a induced tumour cell death and cytotoxicity via apoptosis. While the accumulation of the cells in the S phase might result from the cell cycle arrest of T-24 cells in the S phase.
Figure 7. Effect of compound 6 (5.68 μM) on DNA-ploidy flow cytometric analysis of T-24 cells after 24 h.
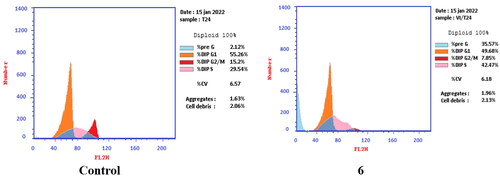
Figure 8. Effect of compound 8a (3.36 μM) on DNA-ploidy flow cytometric analysis of T-24 cells after 24 h.
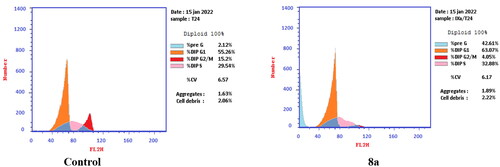
Figure 9. Bar presentation showing effects of compounds 6 (5.68 μM) and 8a (3.36 μM) on DNA-ploidy flow cytometric analysis of T-24 cells after 24 h.
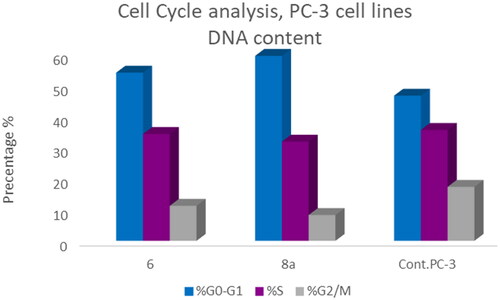
In PC-3 cell lines, both compounds 6 and 8a induced an increase in the percentage of cells at the G1 phase by 1.16- and 1.27-fold, respectively when compared to the control (). This was confirmed by a concomitant decrease in the percentage of cells in the G2/M phase, where both compounds 6 and 8a induced a decrease in the percentage of cells at the G2/M phase by 1.5- and 20.1-fold, respectively. Compounds 6 and 8a induced G1-phase cell cycle arrest of PC-3 cells at their IC50 concentrations. Topoisomerase inhibitors cause DNA damage which is related to G1/S and S phase arrest. Cell cycle arrest prevents the replication of damaged DNA. In PC-3 cell line compounds 6 and 8a probably arrested the cell cycle in the G1 phase by inhibition of Cyclin D–Cdk4–Cdks complex, however in the T-24 cell line, they might arrest the cell cycle in the S phase by the inhibition of both Cyclin D–Cdk4–Cdks complex and cyclins E1–E2/Cdk2, which promote G1/S transitionCitation66–69.
Figure 10. Effect of compound 6 (92.16 μM) on DNA ploidy flow cytometric analysis of PC-3 cells after 24 h.
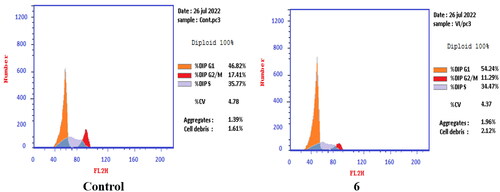
Figure 11. Effect of compound 8a (10.95 μM) on DNA-ploidy flow cytometric analysis of PC-3 cells after 24 h.
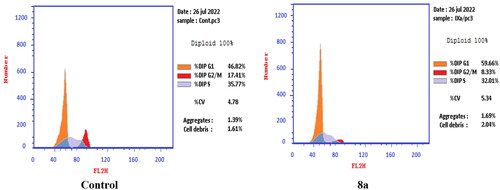
Figure 12. Bar presentation showing effects of compounds 6 (92.16 μM) and 8a (10.95 μM) on DNA-ploidy flow cytometric analysis of PC-3 cells after 24 h.
Similar effects regarding cell cycle arrest at different phases by CP derivatives have been reported, which approved that CP is a promising framework for anticancer drug design and discovery. In 2018, two CP derivatives induced a significant increase in the percentage of cells at pre-G1and G2/M phases by 13.9-, 8.8-fold, and 2.05-, 2.06-fold, respectively compared to the negative control in the UO-31 cell lineCitation39.
In 2021, Abdel-Rahman et al. reported that a Mannich base CP derivative induced an increase in the percentage of cells at pre-G1and G2/M phases by 17.9- and 4.1-fold, respectively, in the OVCAR-3 cell line compared to the negative controlCitation70.
Apoptosis determination by annexin V-FITC assay
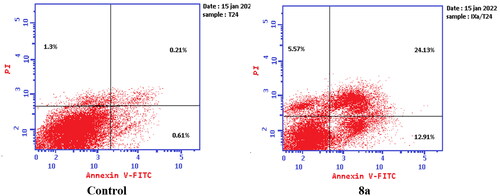
To verify the ability of compounds 6 and 8a to induce apoptosis, a flow cytometry assay was performed using propidium iodide (PI), and immunofluorescent markers of the protein annexin-V. Dual staining for annexin-V and with PI provides the distinction between viable cells, early apoptotic cells, late apoptotic cells, and necrotic cellsCitation71–73. Treatment of T-24 cells with compounds 6 and 8a at their IC50 concentrations resulted in a decrease in the percentage of viable cells. The results showed that compound 6 induced both early and late apoptosis with 16.27- and 83.38-fold more than the control, respectively, and induced total apoptosis and necrosis with 16.77- and 6.25-fold more than the control, respectively (). While compound 8a induced both early and late apoptosis with 21.16- and 114.90-fold more than the control, respectively, and induced total apoptosis and necrosis with 20.1- and 4.28-fold more than the control, sequentially ().
Figure 13. Representative dot plots of T-24 cells treated with 6 (5.68 μM) for 24 h and analysed by flow cytometry after double staining of the cells with annexin-V FITC and PI.
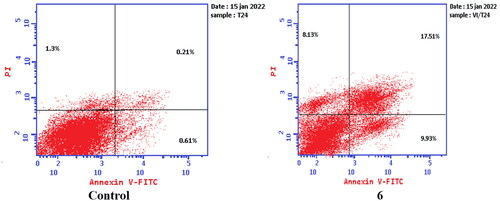
Figure 14. Representative dot plots of T-24 cells treated with 8a (3.36 μM) for 24 h and analysed by flow cytometry after double staining of the cells with annexin-V FITC and PI.
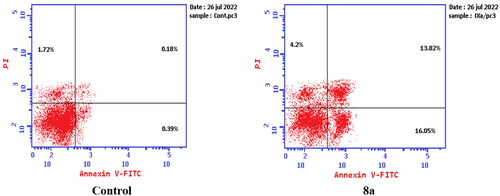
While treatment of PC-3 cells with compounds 6 and 8a at their IC50 concentrations resulted in a decrease in the percentage of viable cells. The results showed that compound 6 induced both early and late apoptosis with 54.69- and 31.72-fold more than the control, respectively, and induced total apoptosis and necrosis with 13.02- and 1.62-fold more than the control, respectively (). While compound 8a induced both early and late apoptosis with 41.15- and 76.77-fold more than the control, respectively, and induced total apoptosis and necrosis with 14.87- and 2.44-fold more than the control, sequentially ().
Figure 15. Representative dot plots of PC-3 cells treated with 6 (92.16 μM) for 24 h and analysed by flow cytometry after double staining of the cells with annexin-V FITC and PI.
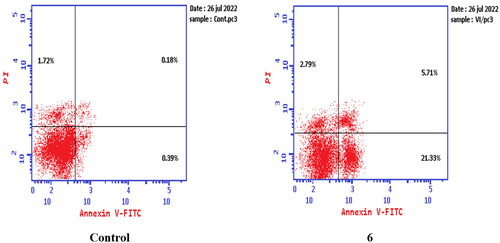
Figure 16. Representative dot plots of PC-3 cells treated with 8a (10.95 μM) for 24 h and analysed by flow cytometry after double staining of the cells with annexin-V FITC and PI.
Effect of compounds 6 and 8a on the level of active caspase-3 (key executioner of apoptosis)
Caspase-3 (a key effector enzyme) plays an important role in apoptosis since its activation leads to catalysing specific enzymes responsible for DNA fragmentation, which leads to cell deathCitation74–77. Apoptosis induction in T-24 cells by compounds 6 and 8a was investigated via caspase 3, compared to doxorubicin as a reference drug.
Treatment of T-24 cells with compounds 6 and 8a at concentrations 5.68 and 3.36 μM, sequentially enhanced the level of active caspase-3 compared to the control (5.23- and 7.6-fold) (, ).
Figure 17. Graphical representation for active caspase-3 assays of compounds 6 and 8a compared to doxorubicin as a positive control.
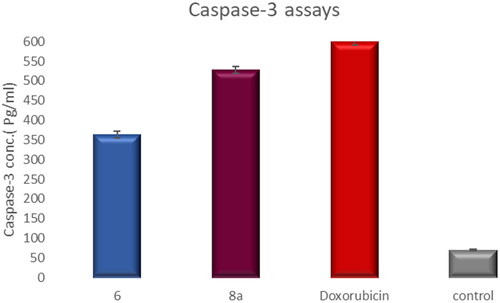
Table 4. Active caspase-3 assay results.
Molecular docking study
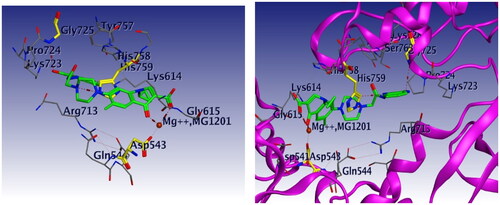
Docking studies were carried out for compounds 6 and 8a which showed potent activity in the Topo II enzyme inhibition assay. We used topoisomerase IIα co-crystallised with DNA (PDB ID: 4FM9)Citation63 for molecular docking of compounds 6, 8a, and merbarone. Docking of merbarone in the DNA binding site was carried out for validation of the molecular docking and it was compared with a previously reported studyCitation78. Interestingly, the test compounds exerted favourable interactions with the Topo II enzyme active site, with almost the same binding pattern as merbarone. Merbarone showed coordinate bond interactions with Mg2+ and H-bond interaction with amino acid Asp 543 (). Compound 6 interacted in a similar pattern, the oxygen atoms of both coplanar carbonyl groups at positions 3 and 4 formed coordinate bond interactions with Mg2+ and H-bond interaction with amino acid Asp 543. Moreover, compound 6 interacted as an H-bond acceptor through the carbonyl group of its pyrazolidine moiety with amino acid Asp 831(). Regarding compound 8a, it interacted with the Topo II enzyme through coordinate bonding with the magnesium ion Mg2+ via the oxygen atoms of both carbonyl groups at positions 3 and 4 and H-bond interaction with amino acid Asp 543. Additionally, it interacted as an H-bond acceptor with amino acids His 759 and Gly 725 through the N atom of piperazine and carbonyl group of indoline moiety, respectively. It exerted arene cation interaction with Arg 713 ().
Figure 18. 3D interaction of merbarone with DNA binding site of topoisomerase IIα. Red dashed lines represent coordinate bond interactions with Mg2+. Red tiny, dashed lines are hydrogen bonding interactions with amino acid Asp 543. Mg2+ is shown as a nonbonded sphere (crimson red). Residues that are involved in hydrogen bonding are shown in the stick presentation.
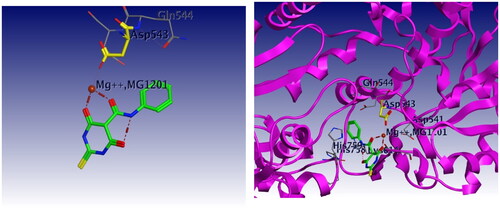
Figure 19. 3D interaction of compound 6 with DNA binding site of topoisomerase IIα. Red dashed lines represent coordinate bond interactions with Mg2+. Red tiny, dashed lines are hydrogen bonding interactions with amino acid Asp 543, and Asp 831. Mg2+ is shown as a nonbonded sphere (crimson red). Residues that are involved in hydrogen bonding are shown in the stick presentation.
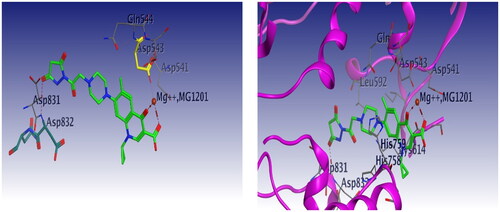
Figure 20. 3D interaction of compound 8a with DNA binding site of topoisomerase IIα. Red dashed lines represent coordinate bond interactions with Mg2+. Red tiny, dashed lines are hydrogen bonding interactions with amino acid Asp 543, acids His 759 and Gly 725. Mg2+ is shown as a nonbonded sphere (crimson red). Residues that are involved in hydrogen bonding are shown in the stick presentation.
Conclusion
In summary, a series of novel CP derivatives were designed and synthesised. They were evaluated for their anti-proliferative activity against bladder T-24 and prostate PC-3 cancer cell lines. 14 compounds exhibited potent antiproliferative activity against the T-24 cell line, with IC50 values between 3.36 and 28.55 µM, which was 1.02- to 8.66-fold more potent than the reference drug doxorubicin. 10 compounds proved their potency against the PC-3 cell line with IC50 values between 3.24 and 19.33 µM, which was 1.2- to 7.1-fold more potent than doxorubicin. The most potent compounds 6, 7a, 7b, 8a, 9a, and 10c showed significant Topo II inhibitory activity. Compounds 6, 8a, and 10c were 1.01- to 2.32-fold more potent than doxorubicin. The most prominent compounds 6 and 8a were further investigated regarding their effects on cell cycle progression, induction of apoptosis, and level of active caspase-3 in the T-24 and PC-3 cell lines. Both compounds induced apoptosis in T-24 cells (16.8- and 20.1-fold, respectively compared to control). This evidence was supported by an increase in the level of apoptotic caspase-3 (5.23- and 7.6-fold). Compounds 6 and 8a arrested the cell cycle in the S phase. Regarding the induction of apoptosis in PC-3 cell lines, both compounds 6 and 8a induced G1-phase cell cycle arrest at their IC50 concentrations. This was confirmed by the increase in the percentage of cells in the G1 phase (1.16- and 1.27-fold, respectively), and a concomitant decrease in the percentage of cells in G2/M phase, (1.5- and 20.1-fold, respectively). The treatment of PC-3 cells with compounds 6 and 8a at their IC50 concentrations resulted in a decrease in the percentage of viable cells. The results showed that both compounds 6 and 8a induced both early and late apoptosis. The molecular docking study of these compounds with Topo II protein revealed more favourable binding modes compared to merbarone, explaining their remarkable Topo II inhibitory potency. CP derivatives are promising leads for further studying, designing, and synthesis of potent anti-proliferative candidates.
Supplemental Material
Download PDF (1.2 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- World Health Organization. Cancer [Internet]. Geneva (Switzerland): WHO; 2021 [cited 2022 Feb 3]. Available from: https://www.who.int/news-room/fact-sheets/detail/cancer.
- Norman A. On the origin of cancer foci. Cancer. 1952;5(3):581–582.
- Biller LH, Schrag D. Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA. 2021;325(7):669–685.
- Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull. 2017;7(3):339–348.
- Nitiss JL, Kiianitsa K, Sun Y, Nitiss KC, Maizels N. Topoisomerase assays. Curr Protoc. 2021;1(10):e250.
- Lee JH, Berger JM. Cell cycle-dependent control and roles of DNA topoisomerase II. Genes. 2019;10(11):859.
- Baguley BC, Drummond CJ, Chen YY, Finlay GJ. DNA-binding anticancer drugs: one target, two actions. Molecules. 2021;26(3):552.
- Sissi C, Palumbo M. Effects of magnesium and related divalent metal ions in topoisomerase structure and function. Nucleic Acids Res. 2009;37(3):702–711.
- Lehninger A, Nelson DL, Cox MM. Biochemistry principles. 7th ed. New York: W.H. Freeman; 2017. p. 1328.
- Delgado JL, Hsieh C-M, Chan N-L, Hiasa H. Topoisomerases as anticancer targets. Biochem J. 2018;475(2):373–398.
- Hevener KE, Verstak TA, Lutat KE, Riggsbee DL, Mooney JW. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm Sin B. 2018;8(6):844–861.
- Buzun K, Bielawska A, Bielawski K, Gornowicz A. DNA topoisomerases as molecular targets for anticancer drugs. J Enzyme Inhib Med Chem. 2020;35(1):1781–1799.
- Skok Z, Zidar N, Kikelj D, Ilas J. Dual inhibitors of human DNA topoisomerase II and other cancer- related targets. J Med Chem. 2020;63(3):884–904.
- Johnson-Arbor K, Dubey R. Doxorubicin. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022. PMID: 29083582.
- Abdel-Aal MAA, Abdel-Aziz SA, Shaykoon MSA, Abuo-Rahma GEDA. Towards anticancer fluoroquinolones: a review article. Arch Pharm. 2019;352(7):e1800376.
- Ezelarab HAA, Abbas SH, Hassan HA, Abuo-Rahma GEDA. Recent updates of fluoroquinolones as antibacterial agents. Arch Pharm Chem Life Sci. 2018;351(9):1800141.
- Yadav V, Talwar P. Repositioning of fluoroquinolones from antibiotic to anti-cancer agents: an underestimated truth. Biomed Pharmacother. 2019;111:934–946.
- Kathiravan MK, Khilare MM, Nikoomanesh K, Aparna S, Jain KS. Topoisomerase as target for antibacterial and anticancer drug discovery. J Enzyme Inhib Med Chem. 2013;28(3):419–435.
- Idowu T, Schweizer F. Ubiquitous nature of fluoroquinolones: the oscillation between antibacterial and anticancer activities. Antibiotics. 2017;6(4):26.
- Gouvea LR, Garcia LS, Lachter DR, Nunes PR, de Castro Pereira F, Silveira-Lacerda EP, Louro SRW, Barbeira PJS, Teixeira LR. Atypical fluoroquinolone gold(III) chelates as potential anticancer agents: relevance of DNA and protein interactions for their mechanism of action. Eur J Med Chem. 2012;55:67–73.
- Samir M, Ramadan M, Hamed M, Osman M, Abou-Rahma G. Recent strategies in design of antitumor and antibacterial fluoroquinolones. J Adv Biomed Pharm Sci. 2021;4(3):134–151.
- Bisacchi GS, Hale MR. A “double-edged” scaffold: antitumor power within the antibacterial quinolone. Curr Med Chem. 2016;23(6):520–577.
- Wang L-L, Battini N, Rammohan RY, Bheemanaboina S-L, Zhang CHZ. Design and synthesis of aminothiazolyl norfloxacin analogues as potential antimicrobial agents and their biological evaluation. Eur J Med Chem. 2019;167:105–123.
- Kloskowski T, Szeliski K, Fekner Z, Rasmus M, Paweł D, Wolska A. Ciprofloxacin and levofloxacin as potential drugs in genitourinary cancer treatment – the effect of dose-response on 2d and 3d cell cultures. Int J Mol Sci. 2021;22(21):11970.
- Gupta P, Gao HL, Ashar YV, Karadkhelkar NM, Yoganathan S, Chen ZS. Ciprofloxacin enhances the chemosensitivity of cancer cells to ABCB1 substrates. Int J Mol Sci. 2019;20(2):268.
- Herold C, Ocker M, Ganslmayer M, Gerauer H, Hahn EG, Schuppan D. Ciprofloxacin induces apoptosis and inhibits proliferation of human colorectal carcinoma cells. Br J Cancer. 2002;86(3):443–448.
- Yadav V, Varshney P, Sultana S, Yadav J, Saini N. Moxifloxacin and ciprofloxacin induces S-phase arrest and augments apoptotic effects of cisplatin in human pancreatic cancer cells via ERK activation. BMC Cancer. 2015;15(1):1–15.
- Liu H, Huang J, Wang J, Wang M, Liu M, Wang B, Guo H, Lu Y. Synthesis, antimycobacterial and antibacterial evaluation of l-[(1R, 2S)-2-fluorocyclopropyl]fluoroquinolone derivatives containing an oxime functional moiety. Eur J Med Chem. 2014;86:628–638.
- Mohammadhosseini N, Alipanahi Z, Alipour E, Emami S, Faramarzi MA, Samadi N, Khoshnevis N, Shafiee A, Foroumadi A. Synthesis and antibacterial activity of novel levofloxacin derivatives containing a substituted thienylethyl moiety. DARU. 2012;20(1):1.
- Gao F, Zhang X, Wang T, Xiao J. Quinolone hybrids and their anti-cancer activities: an overview. Eur J Med Chem. 2019;165:59–79.
- Zhang GF, Zhang S, Pan B, Liu X, Feng LS. 4-Quinolone derivatives and their activities against Gram positive pathogens. Eur J Med Chem. 2018;143:710–723.
- Sissi C, Palumbo M. The quinolone family: from antibacterial to anticancer agents. Curr Med Chem Anticancer Agents. 2003;3(6):439–450.
- Suaifan GARY, Mohammed AAM. Fluoroquinolones structural and medicinal developments (2013–2018): where are we now? Bioorg Med Chem. 2019;27(14):3005–3060.
- Liu J, Ren Z, Fan L, Wei J, Tang X, Xu X, Yang D. Design, synthesis, biological evaluation, structure-activity relationship, and toxicity of clinafloxacin-azole conjugates as novel antitubercular agents. Bioorg Med Chem. 2019;27(1):175–187.
- Feng L, Lv K, Liu M, Wang S, Zhao J, You X, Li S, Cao J, Guo H. Synthesis and in vitro antibacterial activity of gemifloxacin derivatives containing a substituted benzyloxime moiety. Eur J Med Chem. 2012;55:125–136.
- Zhang YB, Feng LS, You XF, Guo Q, Guo HY, Liu ML. Synthesis and in vitro antibacterial activity of 7-(3-alkoxyimino-4-methyl-4-methylaminopiperidin-1-yl)-fluoroquinolone derivatives. Arch Pharm. 2010;343(3):143–151.
- Abuo-Rahma GDAA, Sarhan HA, Gad GFM. Design, synthesis, antibacterial activity and physicochemical parameters of novel N-4-piperazinyl derivatives of norfloxacin. Bioorg Med Chem. 2009;17(11):3879–3886.
- Alovero FL, Pan XS, Morris JE, Manzo RH, Fisher LM. Engineering the specificity of antibacterial fluoroquinolones: benzenesulfonamide modifications at C-7 of ciprofloxacin change its primary target in Streptococcus pneumoniae from topoisomerase IV to gyrase. Antimicrob Agents Chemother. 2000;44(2):320–325.
- Kassab AE, Gedawy EM. Novel ciprofloxacin hybrids using biology oriented drug synthesis (BIODS) approach: anticancer activity, effects on cell cycle profile, caspase-3 mediated apoptosis, topoisomerase II inhibition, and antibacterial activity. Eur J Med Chem. 2018;150:403–418.
- Khélifa T, Beck WT. Merbarone, a catalytic inhibitor of DNA topoisomerase II, induces apoptosis in CEM cells through activation of ICE/CED-3-like protease. Mol Pharmacol. 1999;55(3):548–556.
- Hotinski AK, Lewis ID, Ross DM. Vosaroxin is a novel topoisomerase-II inhibitor with efficacy in relapsed and refractory acute myeloid leukaemia. Expert Opin Pharmacother. 2015;16(9):1395–1402.
- Kohlbrenner WE, Wideburg N, Weigl D, Saldivar A, Chu DTW. Induction of calf thymus topoisomerase II-mediated DNA breakage by the antibacterial isothiazoloquinolones A-65281 and A-65282. Antimicrob Agents Chemother. 1992;36(1):81–86.
- Chauhan S, Paliwal S, Chauhan R. Anticancer activity of pyrazole via different biological mechanisms. Synth Commun. 2014;44(10):1333–1374.
- Luzina EL, Popov AV. Synthesis and anticancer activity evaluation of 3,4-mono- and bicyclosubstituted N-(het)aryl trifluoromethyl succinimides. J Fluor Chem. 2014;168:121–127.
- Milosevic NP, Kojic V, Curcic J, Jakimov D, Milic N, Banjac N, Uscumlic G, Kaliszan R. Evaluation of in silico pharmacokinetic properties and in vitro cytotoxic activity of selected newly synthesized N-succinimide derivatives. J Pharm Biomed Anal. 2017;137:252–257.
- Tan A, Yaglioglu AS, Kishali NH, Sahin E, Kara Y. Evaluation of cytotoxic potentials of some isoindole-1,3-dione derivatives on HeLa, C6 and A549 cancer cell lines. Med Chem. 2020;16(1):69–77.
- Firoozpour L, Gao L, Moghimi S, Pasalar P, Davoodi J, Wang M-W, Rezaei Z, Dadgar A, Yahyavi H, Amanlou M, et al. Efficient synthesis, biological evaluation, and docking study of isatin based derivatives as caspase inhibitors. J Enzyme Inhib Med Chem. 2020;35(1):1674–1684.
- Ma L, Wang H, Wang J, Liu L, Zhang S, Bu M. Novel steroidal 5α,8α-endoperoxide derivatives with semicarbazone/thiosemicarbazone side-chain as apoptotic inducers through an intrinsic apoptosis pathway: design, synthesis and biological studies. Molecules. 2020;25(5):1209.
- Muğlu H. Synthesis, characterization, and antioxidant activity of some new N4-aryl substituted-5-methoxyisatin-β-thiosemicarbazone derivatives. Res Chem Intermed. 2020;46(4):2083–2098.
- El Majzoub R, Fayyad-Kazan M, Nasr El Dine A, Makki R, Hamade E, Grée R, Hachem A, Talhouk R, Fayyad-Kazan H, Badran B, et al. A thiosemicarbazone derivative induces triple negative breast cancer cell apoptosis: possible role of miRNA-125a-5p and miRNA-181a-5p. Genes Genomics. 2019;41(12):1431–1443.
- Hsu DC, Roth HS, West DC, Botham RC, Novotny CJ, Schmid SC, Hergenrother PJ. Parallel synthesis and biological evaluation of 837 analogues of procaspase-activating compound 1 (PAC-1). ACS Comb Sci. 2012;14(1):44–50.
- Liang Z, Zhang D, Ai J, Chen L, Wang H, Kong X, Zheng M, Liu H, Luo C, Geng M, et al. Identification and synthesis of N′-(2-oxoindolin-3-ylidene)hydrazide derivatives against c-Met kinase. Bioorg Med Chem Lett. 2011;21(12):3749–3754.
- Hantgan RR, Stahle MC. Integrin priming dynamics: mechanisms of integrin antagonist-promoted αIIbβ3:PAC-1 molecular recognition. Biochemistry. 2009;48(35):8355–8365.
- Kasiotis KM, Tzanetou EN, Haroutounian SA. Pyrazoles as potential anti-angiogenesis agents: a contemporary overview. Front Chem. 2014;2:78–77.
- Awadallah FM, Piazza GA, Gary BD, Keeton AB, Canzoneri JC. Synthesis of some dihydropyrimidine-based compounds bearing pyrazoline moiety and evaluation of their antiproliferative activity. Eur J Med Chem. 2013;70:273–279.
- Rathore P, Yaseen S, Ovais S, Bashir R, Yaseen R, Hameed AD, Samim M, Gupta R, Hussain F, Javed K, et al. Synthesis and evaluation of some new pyrazoline substituted benzenesulfonylureas as potential antiproliferative agents. Bioorg Med Chem Lett. 2014;24(7):1685–1691.
- George RF, Fouad MA, Gomaa IEO. Synthesis and cytotoxic activities of some pyrazoline derivatives bearing phenyl pyridazine core as new apoptosis inducers. Eur J Med Chem. 2016;112:48–59.
- Fahmy HH, Khalifa NM, Ismail MMF, El-Sahrawy HM, Nossier ES. Biological validation of novel polysubstituted pyrazole candidates with in vitro anticancer activities. Molecules. 2016;21(3):271.
- Best J, Schotten C, Lohmann G, Gerken G, Dechêne A. Tivantinib for the treatment of hepatocellular carcinoma. Expert Opin Pharmacother. 2017;18(7):727–733.
- Devi KS, Subramani P, Parthiban S, Sundaraganesan N. One-pot synthesis, spectroscopic characterizations, quantum chemical calculations, docking and cytotoxicity of 1-((dibenzylamino)methyl)pyrrolidine-2,5-dione. J Mol Struct. 2020;1203:127403.
- Han SH, Kim S, De U, Mishra NK, Park J, Sharma S, Kwak JH, Han S, Kim HS, Kim IS, et al. Synthesis of succinimide-containing chromones, naphthoquinones, and xanthones under Rh(III) catalysis: evaluation of anticancer activity. J Org Chem. 2016;81(24):12416–12425.
- Mitra I, Mukherjee S, Reddy BVP, Dasgupta S, Bose KJC, Mukherjee S, Linert W, Moi SC. Benzimidazole based Pt(II) complexes with better normal cell viability than cisplatin: synthesis, substitution behavior, cytotoxicity, DNA binding and DFT study. RSC Adv. 2016;6(80):76600–76613.
- Wendorff TJ, Schmidt BH, Heslop P, Austin CA, Berger JM. The structure of DNA-bound human topoisomerase II alpha: conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J Mol Biol. 2012;424(3–4):109–124.
- Protein Data Bank. Available from: http://www.rcsb.org/pdb.
- Vincenzi B, Frezza AM, Santini D, Tonini G. New therapies in soft tissue sarcoma. Expert Opin Emerg Drugs. 2010;15(2):237–248.
- Poli A, Mongiorgi S, Cocco L, Follo MY. Protein kinase C involvement in cell cycle modulation. Biochem Soc Trans. 2014;42(5):1471–1476.
- Chen Y-J, Dominguez-Brauer C, Wang Z, Asara JM, Costa RH, Tyner AL, Lau LF, Raychaudhuri P. A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J Biol Chem. 2009;284(44):30695–30707.
- Black AR, Black JD. Protein kinase C signaling and cell cycle regulation. Front Immunol. 2012;3:423.
- Ding L, Cao J, Lin W, Chen H, Xiong X, Ao H, Yu M, Lin J, Cui Q. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int J Mol Sci. 2020;21(6):1960.
- Abdel-Rahman IM, Mustafa M, Mohamed SA, Yahia R, Abdel-Aziz M, Abuo-Rahma GE-DA, Hayallah AM. Novel Mannich bases of ciprofloxacin with improved physicochemical properties, antibacterial, anticancer activities and caspase-3 mediated apoptosis. Bioorg Chem. 2021;107:104629.
- D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019;43(6):582–592.
- Obeng E. Apoptosis (programmed cell death) and its signals - a review. Braz J Biol. 2021;81(4):1133–1143.
- Majtnerová P, Roušar T. An overview of apoptosis assays detecting DNA fragmentation. Mol Biol Rep. 2018;45(5):1469–1478.
- Araya LE, Soni IV, Hardy JA, Julien O. Deorphanizing caspase-3 and caspase-9 substrates in and out of apoptosis with deep substrate profiling. ACS Chem Biol. 2021;16(11):2280–2296.
- Mai F-Y, He P, Ye J-Z, Xu L-H, Ouyang D-Y, Li C-G, Zeng Q-Z, Zeng C-Y, Zhang C-C, He X-H, et al. Caspase-3-mediated GSDME activation contributes to cisplatin- and doxorubicin-induced secondary necrosis in mouse macrophages. Cell Prolif. 2019;52(5):e12663.
- Jeelani R, Chatzicharalampous C, Kohan-Ghadr H-R, Bai D, Morris RT, Sliskovic I, Awonuga A, Abu-Soud HM. Hypochlorous acid reversibly inhibits caspase-3: a potential regulator of apoptosis. Free Radic Res. 2020;54(1):43–56.
- Beroske L, Van den Wyngaert T, Stroobants S, Van der Veken P, Elvas F. Molecular imaging of apoptosis: the case of caspase-3 radiotracers. Int J Mol Sci. 2021;22(8):3948.
- Baviskar AT, Amrutkar SM, Trivedi N, Chaudhary V, Nayak A, Guchhait SK, Banerjee UC, Bharatam PV, Kundu CN. Switch in site of inhibition: a strategy for structure-based discovery of human topoisomerase IIα catalytic inhibitors. ACS Med Chem Lett. 2015;6(4):481–485.