
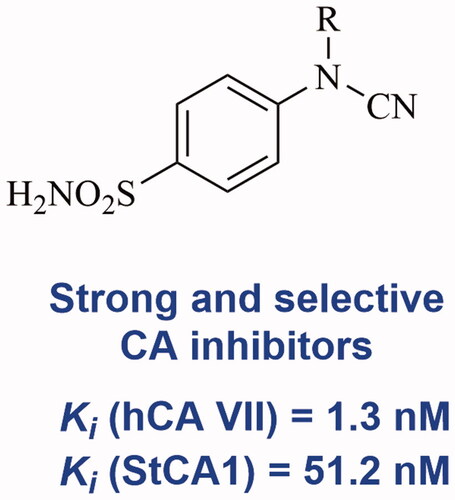
Abstract
A one-pot two-step protocol was developed for the synthesis of a series of novel 4-cyanamidobenzenesulfonamides from easily accessible methyl (4-sulfamoylphenyl)-carbamimidothioate. The new sulphonamides were investigated as inhibitors of the enzyme carbonic anhydrase (CA, EC 4.2.1.1), the human (h) cytosolic isoforms hCA I, II, VII, and XIII, as well as three bacterial enzymes belonging to the β-CA class, MscCA from Mammaliicoccus (Staphylococcus) sciuri and StCA1 and StCA2, from Salmonella enterica (serovar Typhimurium). The human isoforms were generally effectively inhibited by these compounds, with a clear structure-activity relationship privileging long aliphatic chains (C6, C7 and C18) as substituents at the cyanamide functionality. The bacterial CAs were also inhibited by these compounds, but not as effective as the hCAs. The most sensitive enzyme to these inhibitors was StCA1 (KIs of 50.7 − 91.1 nM) whereas SscCA was inhibited in the micromolar range (KIs of 0.86–9.59 µM).
Graphical abstract
Introduction
Sulphonamides are one of the crucial classes of bioactive compounds that played a pivotal role in the field of drug discovery and development due to their diversified pharmacological activities, with more than seventy FDA-approved medications containing one or more sulphonamide motifs in their structureCitation1. The best known and striking biological features of primary sulphonamide-containing compounds is their ability to inhibit the metalloenzyme carbonic anhydrase (CA, EC 4.2.1.1) by binding to the zinc ion within its catalytic siteCitation2. CAs are widespread in all types of organisms, in the three domains of life, Archaea, Bacteria and EukaryaCitation3. Presently, 15 different CA isoforms (CA I, II, III, IV, VA, VB, VI, VII, VIII, IX, X, XI, XII, XIII, and XIV) were described in humans/primates, 12 of which being able to catalyse the reversible interconversion of CO2 to HCO3− and a proton, whereas CA VIII, X, and XI are devoid of CO2 hydrase activityCitation3. This reaction plays fundamental roles in many physiological and pathological events, including respiration and CO2 transport, secretion of electrolytes, pH and other ions homeostasis, bone reabsorption, calcification, tumorigenesis, etc.Citation4. Therefore, the involvement of various CA isoform in such processes can be and is exploited for the development of different types of therapeutic agents, among which diuretics, anti-glaucoma, antiepileptic, anti-obesity and anti-tumour medicationsCitation5–7.
CAs are on the other hand widespread in bacteria, with at least four CA genetic families being present in these organisms, the α-, β-, γ-, and ι-class enzymes, of the eight CA classes described so farCitation8,Citation9. Many members of such CAs were discovered, isolated and characterised extensively, mainly in pathogenic bacteria, in the search of inhibitors acting as antibiotics with a diverse mechanism of action compared to the clinically used such agentsCitation8–11. Indeed, recent studies demonstrated that bacterial infections difficult to treat due to the drug resistance problems, such as those provoked by vancomycin-resistant Enterococci or Neisseria spp., can be managed by using CA inhibitors of the sulphonamide typeCitation9. Such promising results opened new directions in the design of CAIs selective for the various vertebrate isoforms, but also for compounds that might specifically target the bacterial over the host enzymesCitation2,Citation4,Citation8–11.
The cyanamide moiety is one of the bioactive motifs which is featured in a large number of biologically active molecules as well as some drugs, possessing a wide range of activities, such as antihistamineCitation12, anti-ischemiaCitation13, anti-tumourCitation14, anti-inflammatoryCitation15, and anti-apoptoticCitation16 effects. Without a slight doubt, expanding the database of carbonic anhydrase inhibitors by appending unique functionalities at aromatic ring periphery of arenesulphonamide scaffolds (mostly para position of well-studied benzenesulphonamide) is highly desirable for the development of isoform selective CAIs and has been the subject of large number of studies in recent years. Cyanamide (–NCN) moiety is not only one of the well-known pharmacophores but also a versatile building block in the synthesis of various cyclic and acyclic nitrogen-containing compounds. Therefore, synthesis and evaluation of CA inhibitory activity of hitherto unknown 4-cyanamidobenzenesulphonamide derivatives will not only help to expand the database of carbonic anhydrase inhibitors but also may provide useful intermediates which could subject to further manipulations for the preparation of novel and more complex sulphonamide-based CAIs.
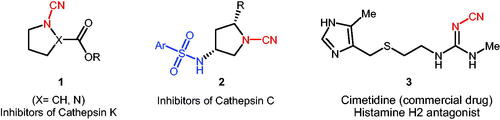
Additionally, there are several pathological processes which besides carbonic anhydrases other enzymes are also involved in their mechanism. An interesting example is bone resorption. Cathepsin K is considered the major cysteine protease expressed in osteoclasts and has been proposed to play a pivotal role in osteoclast-mediated bone resorption. Like all lysosomal enzymes, cathepsin K also require acidic pH for its optimum activity and it is well documented that CA II facilitates proton production, leading to the acidification of resorption lacunae, trigger cathepsin K activation, and ultimately to bone dissolution. Therefore, inhibition of either CA II or cathepsin K may prevent bone resorption. Similarly, CA VII and cathepsins (S, K) were recently validated as potential therapeutic targets in neuropathic pain, albeit more studies are needed to understand potential relationship between CA VII and cathepsin(s) in this syndrome. On one hand it is well known that (hetero)aryl sulphonamides are the widely used drug class for the inhibition of CAs, on the other hand, cyanamides are one of the main classes of cathepsin inhibitors. Therefore, synthesis of a novel panel of compounds possessing both sulphonamide and cyanamide moieties may help to develop effective multi-target drugs as well as unresisting the mechanism of various pathological processes. Importantly compounds containing electrophilic cyanamide warhead displayed significant inhibitory activities against human cathepsinsCitation17,Citation18, inhibiting the lysosomal cysteine protease cathepsin S (CatS)Citation19,Citation20 (Scheme 1).
Scheme 1. Selected examples of biologically active cyanamide derivatives of types 1–3.
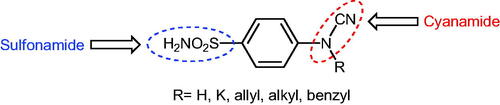
Scheme 2. Schematic presentation of compounds studied in this paper.
In this regard, in the next stage, we may study the inhibitory activity of the same set of compounds reported in this paper against cathepsin(s).
In light of the above-mentioned facts, we hypothesised that compounds possessing sulphonamide and cyanamide moieties might show efficient inhibitory activity against various human and bacterial CAs. Thus, in continuation of our interest in the development of selective CA inhibitors (CAIs)Citation21, herein, we present the synthesis of novel cyanamide-containing sulphonamides (Scheme 2) and evaluate their capability to inhibit various human and bacterial CAs.
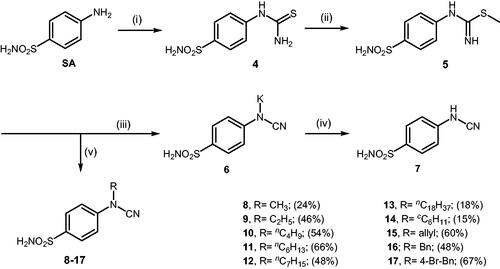
Scheme 3. Reagents and conditions: (i) KSCN, aq. 3.5 M HCl, reflux, 3 h, 31%; (ii) MeI, DMF, 40 °C, 2.5 h, 70%; (iii) K2CO3, DMF, 100 °C, 1.5 h, 89%; (iv) acetic acid (4 equiv.), DMF, RT, 10 min, 59%; (v) (a) K2CO3, DMF, 100 °C, 1.5 h; (b) RX (X = Br or I), DMF, 40–100 °C, 1–1.5 h.
Materials and methods
Chemistry
Reagents, starting materials and solvents were obtained from commercial sources and used as received. Thin-layer chromatography was performed on silica gel, spots were visualised with UV light (254 and 365 nm). NMR spectra were recorded on Bruker 300 spectrometer with chemical shifts values (δ) in ppm relative to TMS using the residual DMSO-d6 signal (1H 2.50; 13C 39.52). High-resolution mass spectra (HRMS) were recorded on a mass spectrometer with a Q-TOF micro mass analyser using the ESI technique.
Synthesis
4-Thioureidobenzenesulfonamide (4)
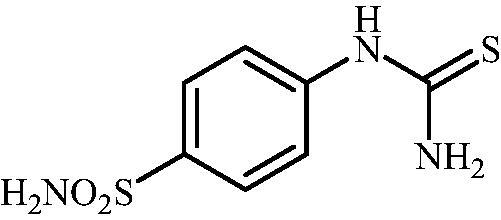
4-Aminobenzensulfonamide (30.00 g, 174.3 mmol) was dissolved in 180 ml of 3.5 M HCl at 70 °C. After cooling to room temperature, KSCN (16.94 g, 174.3 mmol) was added and the mixture was refluxed for 3 h. After cooling to room temperature, the reaction mixture was diluted with ice cooled water. Solids formed were filtered, washed with water, and air dried to afford 9 (12.49 g, 31%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 7.32 (s, 2H), 7.69 (d, 2H, J = 8.6 Hz), 7.77 (d, 2H, J = 8.6 Hz), 10.02 (s, 1H) ppm 13C NMR (75 MHz, DMSO-d6) δ = 122.8, 127.3, 139.8, 143.9, 182.8 ppm MS (ESI) [M + H]+: m/z 232.0.
Methyl (4-sulfamoylphenyl)carbamimidothioate (5)

4-Thioureidobenzenesulfonamide (4) (300 mg, 1.3 mmol) was dissolved in DMF (4 ml) at room temperature. MeI (0.08 ml, 1.3 mmol) was added to the solution and the mixture was heated at 40 °C for 2.5 h. After cooling to room temperature, it was extracted with EtOAc (3 × 20 ml). The combined organic phases were washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl in water (1 × 20 ml) and dried over Na2SO4. Solvent removal in vacuo afforded 5 (223 mg, 70%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 2.37 (s, 3H), 6.63 (s, 2H), 6.94 (s, 2H), 7.22 (s, 2H), 7.71 (d, 2H, J = 8.4 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 14.2, 122.8, 127.7, 138.0, 153.9, 157.0 ppm HRMS (ESI) [M + H]+: m/z calcd for (C8H12N3O2S2) 246.0371. Found 246.0372.
Potassium cyano(4-sulfamoylphenyl)amide (6)
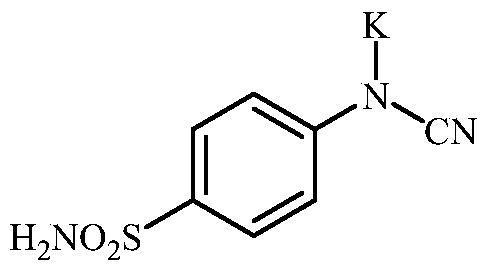
To a solution of methyl (4-sulfamoylphenyl) carbamimidothioate (5) (500 mg, 2.04 mmol) in DMF (8 ml) was added K2CO3 (564 mg, 4.08 mmol,) and the mixture was stirred at 100 °C for 1.5 h. The mixture was cooled to room temperature and precipitated K2CO3 was removed by filtration. To the solution EtOAc (80 ml) was added and precipitate formed was collected by filtration, washed with EtOAc (20 ml) and air dried to afford 6 (427 mg, 89%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 6.60 (d, 2H, J = 8.6 Hz), 6.85 (s, 2H), 7.29 (s, 1H), 7.38 (d, 2H, J = 8.6 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 118.0, 125.7, 127.9, 129.0, 160.9 ppm HRMS (ESI) [M – K]-: m/z calcd for (C7H6N3O2S) 196.0181. Found 196.0188.
4-Cyanamidobenzenesulfonamide (7)
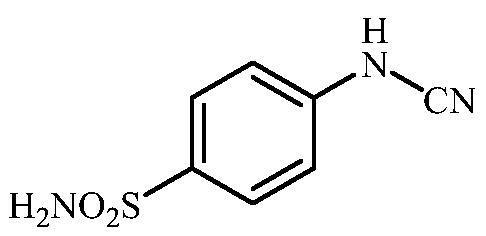
To a solution of methyl (4-sulfamoylphenyl)carbamimidothioate (5) (300 mg., 1.22 mmol) in DMF (4 ml) under stirring K2CO3 (338 mg, 2.44 mmol) was added in one portion. The mixture was stirred at 100 °C for 1.5 h. After cooling to room temperature, the mixture was neutralised by addition of acetic acid (0.28 ml, 4.88 mmol). It was extracted with EtOAc (3 × 20 ml). The combined organic phases were dried over Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel (DCM:MeOH, 95:5) to afford 7 (141 mg, 59%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 7.08 (dd, 2H, J = 6.7 and 1.9 Hz), 7.28 (s, 2H), 7.79 (dd, 2H, J = 6.7 and 1.9 Hz), 10.70 (br s, 1H) ppm 13C NMR (75 MHz, DMSO-d6) δ = 112.3, 115.9, 128.8, 139.0, 142.9 ppm HRMS (ESI) [M + H]+: m/z calcd for (C7H8N3O2S) 198.0337. Found 198.0333.
4-(N-Methylcyanamido)benzenesulfonamide (8)
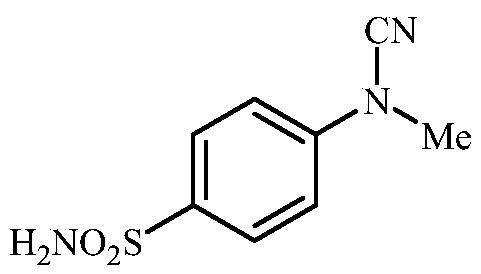
To a solution of methyl (4-sulfamoylphenyl)carbamimidothioate (5) (500 mg, 2.04 mmol) in DMF (8 ml) under stirring K2CO3 (564 mg, 4.08 mmol) was added in one portion The mixture was stirred at 100 °C for 1.5 h. After cooling to room temperature, MeI (0.127 ml, 2.04 mmol) was added and the mixture was stirred at 40 °C for 1 h. The reaction mixture was cooled to room temperature and extracted with EtOAc (3 × 20 ml). The combined organic phases were washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat.NH4Cl (1 × 20 ml) and dried over Na2SO4. The solvent was removed in vacuo and the residue was washed with EtOAc (2 × 10 ml) to afford 8 (103 mg, 24%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 3.42 (s, 3H), 7.32 (d, 2H, J = 8.8 Hz), 7.37 (s, 2H), 7.89 (d, 2H, J = 8.8 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 37.7, 114.1, 115.8, 128.5, 139.5, 144.2 ppm HRMS (ESI) [M]+: m/z calcd for (C8H9N3O2S) 211.0415. Found 211.0419.
4-(N-Ethylcyanamido)benzenesulfonamide (9)
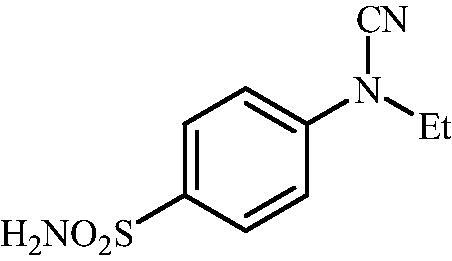
To a solution of methyl (4-sulfamoylphenyl)carbamimidothioate (5) (500 mg, 2.04 mmol) in DMF (8 ml) under stirring K2CO3 (564 mg, 4.08 mmol) was added in one portion The mixture was stirred at 100 °C for 1.5 h After cooling to room temperature, EtI (0.164 ml, 2.04 mmol) was added and the mixture was stirred at 100 °C for 1 h. The reaction mixture was cooled to room temperature and extracted with EtOAc (3 × 20 ml). The combined organic phases were washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat.NH4Cl (1 × 20 ml) and dried over Na2SO4. The solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (DCM:MeOH, 95:5) to afford 9 (211 mg, 46%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 1.36 (t, 3H, J = 7.0 Hz), 3.79 (q, 2H, J = 7.0 Hz), 7.35–7.38 (m, 4H), 7.89 (d, 2H, J = 8.2 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 13.3, 44.6, 113.0, 116.4, 128.6, 139.6, 143.5 ppm HRMS (ESI) [M - H]-: m/z calcd for (C9H10N3O2S) 224.0494. Found 224.0499.
4-(N-Butylcyanamido)benzenesulfonamide (10)

To a solution of methyl (4-sulfamoylphenyl)carbamimidothioate (5) (500 mg, 2.04 mmol) in DMF (8 ml) under stirring K2CO3 (564 mg, 4.08 mmol) was added in one portion The mixture was stirred at 100 °C for 1.5 h. After cooling to room temperature, nBuI (0.232 ml, 2.04 mmol) was added and the mixture was stirred at 100 °C for 1 h. The reaction mixture was cooled to room temperature and extracted with EtOAc (3 × 20 ml). The combined organic phases were washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat.NH4Cl (1 × 20 ml) and dried over Na2SO4. The solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (DCM:MeOH, 95:5) to afford 10 (279 mg, 54%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 0.96 (t, 3H, J = 7.1 Hz), 1.37–1.49 (m, 2H), 1.68–1.77 (m, 2H), 3.77 (t, 2H, J = 7.1 Hz), 7.37–7.39 (m, 4H), 7.88 (d, 2H, J = 8.7 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 14.5, 20.0, 29.8, 49.3, 113.4, 116.4, 128.6, 139.6, 143.6 ppm HRMS (ESI) [M]+: m/z calcd for (C11H15N3O2S) 253.0885. Found 253.0888.
4-(N-Hexylcyanamido)benzenesulfonamide (11)

To a solution of methyl (4-sulfamoylphenyl)carbamimidothioate (5) (500 mg, 2.04 mmol) in DMF (8 ml) under stirring K2CO3 (564 mg, 4.08 mmol) was added in one portion. The mixture was stirred at 100 °C for 1.5 h. After cooling to room temperature, 1-bromohexane (0.232 ml, 2.04 mmol) and KI (372 mg, 2.24 mmol) were added and the mixture was stirred at 100 °C for 1.5 h. The reaction mixture was cooled to room temperature and extracted with EtOAc (3 × 20 ml). The combined organic phases were washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat.NH4Cl (1 × 20 ml) and dried over Na2SO4. The solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (DCM:MeOH, 95:5) to afford 11 (379 mg, 66%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 0.89 (t, 3H, J = 6.5 Hz), 1.33–1.44 (m, 6H), 1.69–1.78 (m, 2H), 3.77 (t, 2H, J = 6.5 Hz), 7.37–7.39 (m, 4H), 7.86 (d, 2H, J = 7.9 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 14.8, 22.9, 26.3, 27.7, 31.7, 49.5, 113.4, 116.4, 128.6, 139.8, 143.5 ppm HRMS (ESI) [M - H]-: m/z calcd for (C13H18N3O2S) 280.1120. Found 280.1124.
4-(N-Heptylcyanamido)benzenesulfonamide (12)
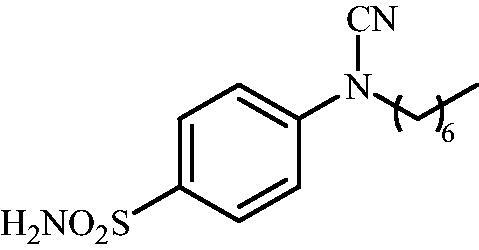
To a solution of methyl (4-sulfamoylphenyl)carbamimidothioate (5) (500 mg, 2.04 mmol) in DMF (8 ml) under stirring K2CO3 (564 mg, 4.08 mmol) was added in one portion The mixture was stirred at 100 °C for 1.5 h. After cooling to room temperature, 1-iodoheptane (0.335 ml, 2.04 mmol) was added and the mixture was stirred at 100 °C for 1.5 h. The reaction mixture was cooled to room temperature and extracted with EtOAc (3 × 20 ml). The combined organic phases were washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat.NH4Cl (1 × 20 ml) and dried over Na2SO4. The solvent was removed in vacuo. The semisolid residue was dissolved in EtOAc (5 ml) and precipitated by addition of hexane (50 ml). The precipitate formed was filtered, washed with hexane and air dried to afford 12 (290 mg, 48%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 0.89 (t, 3H, J = 6.5 Hz), 1.29 (br s. 5H), 1.37 (br s, 3H), 1.69–1.78 (m, 2H), 3.76 (t, 2H, J = 6.9 Hz), 7.36–7.39 (m, 4H), 7.88 (d, 2H, J = 8.6 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 14.9, 22.9, 26.6, 27.7, 29.2, 32.1, 49.5, 113.4, 116.4, 128.6, 139.6, 143.5 ppm HRMS (ESI) [M - H]-: m/z calcd for (C14H20N3O2S) 294.1276. Found 294.1285.
4-(N-Octadecylcyanamido)benzenesulfonamide (13)

To a solution of methyl (4-sulfamoylphenyl)carbamimidothioate (5) (500 mg, 2.04 mmol) in DMF (8 ml) under stirring K2CO3 (564 mg, 4.08 mmol) was added in one portion The mixture was stirred at 100 °C for 1.5 h. After cooling to room temperature, 1-bromooctadecane (0.335 ml, 2.04 mmol) and KI (680 mg, 2.24 mmol) were added and the mixture was stirred at 100 °C for 1.5 h. The reaction mixture was cooled to room temperature and extracted with EtOAc (3 × 20 ml). The combined organic phases were washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat.NH4Cl (1 × 20 ml) and dried over Na2SO4. The solvent was removed in vacuo. Residual yellowish solids were washed with cold EtOAc (15 ml) to afford 13 (165 mg, 18%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 0.88 (t, 3H, J = 6.7 Hz), 1.26 (br s, 30H), 1.68–1.77 (m, 2H), 3.75 (t, 2H, J = 6.7 Hz), 7.35–7.38 (m, 4H), 7.88 (d, 2H, J = 8.6 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 14.9, 23.1, 26.7, 27.8, 29.5, 29.7, 30.0 (br), 32.3, 49.5, 113.4, 116.4, 128.6, 139.6, 143.5 ppm HRMS (ESI) [M - H]-: m/z calcd for (C25H42N3O2S) 448.2998. Found 448.2994.
4-(N-Cyclohexylcyanamido)benzenesulfonamide (14)
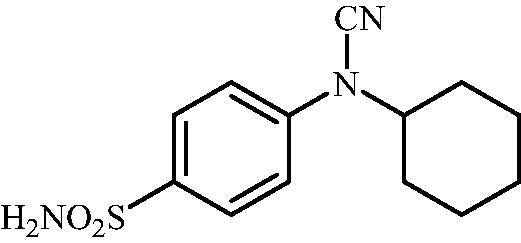
To a solution of methyl (4-sulfamoylphenyl)carbamimidothioate (5) (500 mg, 2.04 mmol) in DMF (8 ml) under stirring K2CO3 (564 mg, 4.08 mmol) was added in one portion The mixture was stirred at 100 °C for 1.5 h. After cooling to room temperature, bromocyclohexane (0.252 ml, 2.04 mmol) and KI (372 mg, 2.24 mmol) were added and the mixture was stirred at 100 °C for 1.5 h. The reaction mixture was cooled to room temperature and extracted with EtOAc (3 × 20 ml). The combined organic phases were washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat.NH4Cl (1 × 20 ml) and dried over Na2SO4. The solvent was removed in vacuo. The residue was purified by column chromatography on silica gel (DCM:MeOH, 95:5) to afford 14 (85 mg, 15%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 1.19–1.27 (m, 1H), 1.41–1.55 (m, 4H), 1.68 (d, 1H, J = 12.1 Hz), 1.83 (s, 2H), 2.02 (s, 2H), 3.99 (s, 1H), 7.38 (s, 2H), 7.43 (d, 2H, J = 8.6 Hz), 7.88 (d, 2H, J = 8.6 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 25.4, 25.6, 31.5, 56.8, 111.9, 117.1, 128.6, 139.8, 143.3 ppm HRMS (ESI) [M - H]-: m/z calcd for (C13H16N3O2S) 278.0963. Found 278.0969.
4-(N-Allylcyanamido)benzenesulfonamide (15)
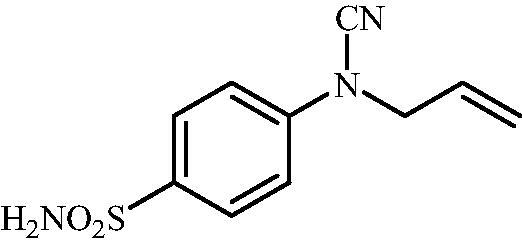
To a solution of methyl (4-sulfamoylphenyl)carbamimidothioate (5) (500 mg, 2.04 mmol) in DMF (8 ml) under stirring K2CO3 (564 mg, 4.08 mmol) was added in one portion The mixture was stirred at 100 °C for 1.5 h. After cooling to room temperature, allyl bromide (0.176 ml, 2.04 mmol) was added and the mixture was stirred at 100 °C for 1.5 h. The reaction mixture was cooled to room temperature and extracted with EtOAc (3 × 20 ml). The combined organic phases were washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat.NH4Cl (1 × 20 ml) and dried over Na2SO4. The solvent was removed in vacuo. The semisolid residue was dissolved in EtOAc (5 ml) and precipitated by addition of hexane (50 ml). The precipitate formed was filtered, washed with hexane and air dried to afford 15 (290 mg, 60%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 4.46 (d, 2H, J = 4.9 Hz), 5.36 (s, 1H), 5.41 (d, 1H, J = 7.8 Hz), 5.94–6.06 (m, 1H), 7.36 (d, 2H, J = 8.6 Hz), 7.38 (s, 2H), 7.88 (d, 2H, J = 8.6 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 52.0, 113.4, 116.5, 120.5, 128.5, 131.6, 139.7, 143.3 ppm HRMS (ESI) [M - H]-: m/z calcd for (C10H10N3O2S) 236.0494. Found 236.0489.
4-(N-Benzylcyanamido)benzenesulfonamide (16)

To a solution of methyl (4-sulfamoylphenyl)carbamimidothioate (5) (500 mg, 2.04 mmol) in DMF (8 ml) under stirring K2CO3 (564 mg, 4.08 mmol) was added in one portion The mixture was stirred at 100 °C for 1.5 h. After cooling to room temperature, benzyl bromide (0.236 ml, 2.04 mmol) was added and the mixture was stirred at 100 °C for 1.5 h. The reaction mixture was cooled to room temperature and water was added until precipitate was formed. The precipitate was filtered, washed with water (10 ml) and Et2O (10 ml) and air dried to afford 16 (282 mg, 48%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 5.06 (s, 2H), 7.35–7.44 (m, 9H), 7.86 (d, 2H, J = 8.4 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 52.5, 113.2, 116.2, 128.1, 128.3, 128.8, 129.4, 135.0, 139.3, 142.8 ppm HRMS (ESI) [M - H]-: m/z calcd for (C14H12N3O2S) 286.0650. Found 286.0649.
4-(N-(4-Bromobenzyl)cyanamido)benzenesulfonamide (17)
To a solution of methyl (4-sulfamoylphenyl)carbamimidothioate (5) (500 mg, 2.04 mmol) in DMF (8 ml) under stirring K2CO3 (564 mg, 4.08 mmol) was added in one portion The mixture was stirred at 100 °C for 1.5 h. After cooling to room temperature, 4-bromobenzyl bromide (659 mg, 2.04 mmol) was added and the mixture was stirred at 100 °C for 1.5 h. The reaction mixture was cooled to room temperature and water was added until precipitate was formed. The precipitate was filtered, washed with water (10 ml) and Et2O (10 ml) and air dried to afford 17 (501 mg, 67%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 5.06 (s, 2H), 7.36–7.42 (m, 6H), 7.65 (d, 2H, J = 8.0 Hz), 7.87 (d, 2H, J = 8.0 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 52.3, 113.5, 116.7, 122.6, 128.6, 131.1, 132.8, 134.9, 139.9, 143.2 ppm HRMS (ESI) [M - H]-: m/z calcd for (C14H11N3O2SBr) 363.9755. Found 363.9758.
CA inhibitory assay
An applied photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activityCitation22. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer for α-CAs or 20 mM TRIS (pH 8.4) as buffer for β-CAs, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10 − 100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5 − 10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled – deionised water, and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 6 h at room temperature prior to assay in order to allow for the formation of the E – I complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3 and the Cheng – Prusoff equation, as reported earlierCitation23–25 and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation26–28 and their concentrations in the assay system ranged between 8–15 nM.
Results and discussion
Chemistry
The drug design of the new CAIs reported here considered the benzenesulphonamide scaffold, which has been widely employed earlier for derivatization reactions, mainly by using the tail approach, as it allows a facile chemistry and generally high yields of new productsCitation2–4. The general synthetic route of the target compounds is shown in Scheme 3. In order to bypass the need for toxic cyanogen bromide, a multi-step procedure was designed which involves the initial formation of methyl (4-sulfamoylphenyl) carbamimidothioate 5 via a two-step procedure through thioureation of sulphanilamide SA (4-amino-benzenesulfonamide) using KSCN and subsequent S-methylation of the resulted thiourea 4 with MeI. The elimination of methyl sulphide from carbamimidothioate 5 by treatment with K2CO3 afforded potassium cyano(4-sulphamoylphenyl)amide 6 in excellent yield which by treatment with acid was converted into the corresponding secondary cyanamide 7. The target 4-(N-alkyl/benzyl-cyanamido)benzenesulfonamide derivatives 8–17 were synthesised by reaction of in situ generated potassium cyano(4-sulfamoylphenyl)amide 6 with selected alkyl/benzyl halides (iodides or bromides) under catalyst- and additive-free conditions in DMF at elevated temperatures (Scheme 3).
Carbonic anhydrase inhibition
The cyanamido-benzenesulfonamides 6–17 reported here have been tested as inhibitors of three human (h) CA isoforms, the cytosolic hCA I, II, VII and XIII, as well as the bacterial β-CAs from Mammaliicoccus (Staphylococcus) sciuriCitation26 and Salmonella enterica (serovar Typhimurium), StCA1 and StCA2Citation27. It should be mentioned that the first bacterial enzyme, SscCA, was originally reported by us as Staphylococcus aureus β-CA, SauCA based on a genomic sequence annotated in the data bases in 2017Citation26. A recent reanalysis of that sequence revealed that the original annotation was erroneous, and that the sequence encodes a β-class CA from another Staphylococcaceae family member, i.e. Staphylococcus sciuri, which is a Gram-positive, oxidase-positive, coagulase-negative member of these infectious bacteria known to provoke disease in humans and animals (it was originally isolated from the squirrel)Citation29. In 2020, Madhaiyan et al. renamed the species as belonging to a new genus, as Mammaliicoccus sciuriCitation30. In fact, the taxonomy of the Staphylococcaceae family is rather complex, and as mentioned earlier, many genome annotations were inexact or were overlapping between various genetically similar speciesCitation30. Thus, we report here that the enzyme previously known as SauCA is in fact MscCACitation31.
The following should be noted regarding data of , where the inhibition data of these enzymes are presented:
Table 1. Inhibition data of human CA isoforms CA I, II, VII, and XIII and bacterial β-CA isoforms SscCA, from Mammaliicoccus (Staphylococcus) sciuri, and StCA1 and StCA2, from Salmonella enterica (serovar Typhimurium), with compounds 6–17 using acetazolamide (AAZ) as standard drug. 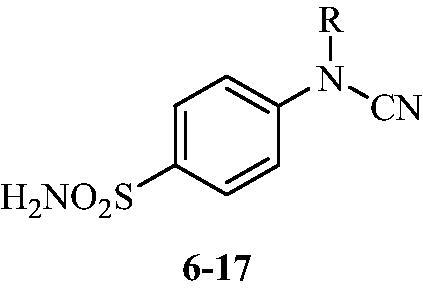
hCA I was effectively inhibited by cyanamido-benzenesulphonamides 6–17 reported here with KIs ranging between 9.3 and 889 nM (acetazolamide, the standard CAI is a medium potency inhibitor with a KI of 250 nM). The most effective hCA I inhibitors in the series were 10–14, KIs of 9.3–50.6 nM, all of which incorporate rather long aliphatic R moieties at the cyanamide functionality. The optimal substitution seems to be an n-hexyl group, in 11, which is the most effective hCA I inhibitor reported here. Smaller aliphatic R moieties, unsaturated or aromatic ones lead to a decrease of the hCA I inhibitory potency.
hCA II is also effectively inhibited by these sulphonamides, with KIs ranging between 5.3 and 148 nM (). Derivatives 11–13 were the most effective inhibitors, with KIs of 5.3–9.5 nM, even better than AAZ (KI of 12 nM). Again the presence of long aliphatic chains (C6, C7 and C18) induced these effective inhibitory effects, whereas shorter, unsaturated or aromatic/benzylic R groups reduce (in some cases only slightly) the potency.
hCA VII, the mainly brain expressed cytosolic isoform was the most susceptible to inhibition with the newly prepared sulphonamides, which showed KIs ranging between 1.3 and 30.6 nM (). Thus, all substitution patterns present in these compounds lead to highly effective CAIs, with the optimal substitutions being those present in 10–17. Thus, for this isoform, apart the long aliphatic groups, the unsaturated, benzylic and substituted benzyl moieties afforded highly effective inhibitors.
MscCA was on the other hand poorly inhibited by the cyanamido-benzenesulphonamides 6–17 reported here, with KIs in the micromolar range, more precisely 860–9592 nM (). The best inhibitors were the unsubstituted cyanamides 6 and 7, which like acetazolamide, are medium potency bacterial CA inhibitors.
StCA1 was the best inhibited bacterial CA among the three such enzymes investigated here with cyanamido-benzenesulphonamides 6–17. Indeed, these compounds showed KIs ranging between 50.7 and 91.1 nM. Thus, all of them show a quite flat structure-activity relationship, acting as effective (but not highly potent) CAIs.
The second S. enterica (serovar Typhimurium) CA isoform StCA2 was on the other hand less sensitive to inhibition with these new sulphonamides compared to StCA1, and the KIs were in the range of 92.4–791 nM. In this case, the best inhibitors were the ones with small R groups (H or potassium), compounds 6 and 7, whereas the increase of the R moiety led to a decrease of the inhibitory power ().
Conclusions
A one-pot two-step protocol was developed for the synthesis of novel 4-cyanamidobenzenesulfonamide derivatives from easily accessible methyl (4-sulfamoylphenyl)carbamimidothioate under catalyst- and additive-free conditions. The small series of prepared compounds was investigated for the inhibition of human and bacterial CAs belonging to the α- and β-CA classes. Several highly effective hCA I, II, and VII inhiibtors were detected, whereas hCA XIII was less inhibited by most such compounds. The structure-activity relationship for the inhibition of the human isoforms is rather straightforward, with long aliphatic chains (C6, C7 and C18) at the cyanamide functionality inducing the most effective inhibitory effects. Among the three bacterial CAs investigated here for their inhibition with cyanamidobenzenesulfonamides, StCA1 was the most sensitive to these inhibitors, followed by StCA2, whereas MscCA was inhibited only in the micromolar range. Although these compounds do not show selectivity for the inhibition of bacterial versus human CAs, they may be considered as interesting starting points for the development of novel pharmacological agents belonging to this class of enzyme inhibitors.
Disclosure statement
No potential conflict of interest was reported by all author(s) except CTS. CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- (a) Scott KA, Njardarson JT. Analysis of US FDA-approved drugs containing sulfur atoms. Top Curr Chem. 2018;376(1):5; (b) Carta F, Supuran CT, Scozzafava A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med Chem. 2014;6(10):1149–1165; (c) Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem. 2018;33(1):485–495.
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7(2):168–181; (b) Supuran CT. Emerging role of carbonic anhydrase inhibitors. Clin Sci. 2021;135(10):1233–1249; (c) Supuran CT. Novel carbonic anhydrase inhibitors. Future Med Chem. 2021;13(22):1935–1937.
- (a) Aspatwar A, Tolvanen MEE, Barker H, Syrjänen L, Valanne S, Purmonen S, Waheed A, Sly WS, Parkkila S. Carbonic anhydrases in metazoan model organisms: molecules, mechanisms, and physiology. Physiol Rev. 2022;102(3):1327–1383; (b) Aspatwar A, Syrjänen L, Parkkila S. Roles of carbonic anhydrases and carbonic anhydrase related proteins in Zebrafish. IJMS. 2022;23(8):4342.
- (a) Mishra CB, Tiwari M, Supuran CT. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev. 2020;40(6):2485–2565; (b) Alterio V, Di Fiore A, D'Ambrosio K, Supuran CT, De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev. 2012;112(8):4421–4468; (c) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov. 2017;12(1):61–88.
- (a) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem. 2016;31(3):345–360; (b) Supuran CT. Carbonic anhydrases as drug targets-an overview. Curr Top Med Chem. 2007;7(9):825–833; (c) Nocentini A, Angeli A, Carta F, Winum J-Y, Zalubovskis R, Carradori S, Capasso C, Donald WA, Supuran CT. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J Enzyme Inhib Med Chem. 2021;36(1):561–580.
- (a) Angeli A, Carta F, Nocentini A, Winum J-Y, Zalubovskis R, Akdemir A, Onnis V, Eldehna WM, Capasso C, Simone GD, et al. Carbonic anhydrase inhibitors targeting metabolism and tumor microenvironment. Metabolites. 2020;10(10):412; (b) Angeli A, Carta F, Nocentini A, Winum J-Y, Zalubovskis R, Onnis V, Eldehna WM, Capasso C, Carradori S, Donald WA, et al. Response to perspectives on the classical enzyme carbonic anhydrase and the search for inhibitors. Biophys J. 2021;120(1):178–181; (c) McDonald PC, Chafe SC, Supuran CT, Dedhar S. Cancer therapeutic targeting of hypoxia induced carbonic anhydrase IX: from bench to bedside. Cancers (Basel). 2022;14(14):3297; (d) Supuran CT. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs. 2018;27(12):963–970.
- (a) Nerella SG, Singh P, Arifuddin M, Supuran CT. Anticancer carbonic anhydrase inhibitors: a patent and literature update 2018-2022. Expert Opin Ther Pat. 2022;32(8):833–847; (b) Supuran CT. Carbonic anhydrase inhibitors: an update on experimental agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs. 2021;30(12):1197–1208; (c) Mincione F, Nocentini A, Supuran CT. Advances in the discovery of novel agents for the treatment of glaucoma. Expert Opin Drug Discov. 2021;16(10):1209–1225; (d) Supuran CT. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin Drug Discov. 2020;15(6):671–686.
- (a) Amedei A, Capasso C, Nannini G, Supuran CT. Microbiota, bacterial carbonic anhydrases, and modulators of their activity: links to human diseases? Mediators Inflamm. 2021;2021:6926082; (b) Nocentini A, Supuran CT, Capasso C. An overview on the recently discovered iota-carbonic anhydrases. J Enzyme Inhib Med Chem. 2021;36(1):1988–1995; (c) Campestre C, De Luca V, Carradori S, Grande R, Carginale V, Scaloni A, Supuran CT, Capasso C. Carbonic anhydrases: new perspectives on protein functional role and inhibition in Helicobacter pylori. Front Microbiol. 2021; 12:629163; (d) Del Prete S, Nocentini A, Supuran CT, Capasso C. Bacterial ι-carbonic anhydrase: a new active class of carbonic anhydrase identified in the genome of the Gram-negative bacterium Burkholderia territorii. J Enzyme Inhib Med Chem. 2020;35(1):1060–1068.
- (a) De Luca V, Carginale V, Supuran CT, Capasso C. The gram-negative bacterium Escherichia coli as a model for testing the effect of carbonic anhydrase inhibition on bacterial growth. J Enzyme Inhib Med Chem. 2022;37(1):2092–2098; (b) Supuran CT, Capasso C. Antibacterial carbonic anhydrase inhibitors: an update on the recent literature. Expert Opin Ther Pat. 2020;30(12):963–982; (c) Giovannuzzi S, Hewitt CS, Nocentini A, Capasso C, Costantino G, Flaherty DP, Supuran CT. Inhibition studies of bacterial α-carbonic anhydrases with phenols. J Enzyme Inhib Med Chem. 2022;37(1):666–671.
- (a) Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin Ther Pat. 2018;28(10):745–754; (b) Flaherty DP, Seleem MN, Supuran CT. Bacterial carbonic anhydrases: underexploited antibacterial therapeutic targets. Future Med Chem. 2021;13(19):1619–1622; (c) Hewitt CS, Abutaleb NS, Elhassanny AEM, Nocentini A, Cao X, Amos DP, Youse MS, Holly KJ, Marapaka AK, An W, et al. Structure-activity relationship studies of acetazolamide-based carbonic anhydrase inhibitors with activity against Neisseria gonorrhoeae. ACS Infect Dis. 2021;7(7):1969–1984; (d) Abutaleb NS, Elhassanny AEM, Nocentini A, Hewitt CS, Elkashif A, Cooper BR, Supuran CT, Seleem MN, Flaherty DP. Repurposing FDA-approved sulphonamide carbonic anhydrase inhibitors for treatment of Neisseria gonorrhoeae. J Enzyme Inhib Med Chem. 2022;37(1):51–61; (e) An W, Holly KJ, Nocentini A, Imhoff RD, Hewitt CS, Abutaleb NS, Cao X, Seleem MN, Supuran CT, Flaherty DP, et al. Structure-activity relationship studies for inhibitors for vancomycin-resistant Enterococcus and human carbonic anhydrases. J Enzyme Inhib Med Chem. 2022;37(1):1838–1844.
- (a) Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs - antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzyme Inhib Med Chem. 2014;29(3):379–387; (b) Capasso C, Supuran CT. Anti-infective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin Ther Pat. 2013;23(6):693–704; (c) Nishimori I, Vullo D, Minakuchi T, Scozzafava A, Osman SM, AlOthman Z, Capasso C, Supuran CT. Anion inhibition studies of two new β-carbonic anhydrases from the bacterial pathogen Legionella pneumophila. Bioorg Med Chem Lett. 2014;24(4):1127–1132; (d) Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem. 2015;30(2):325–332; (e) Ferraroni M, Del Prete S, Vullo D, Capasso C, Supuran CT. Crystal structure and kinetic studies of a tetrameric type II β-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Acta Crystallogr D Biol Crystallogr. 2015;71(Pt 12):2449–2456.
- (a) Brogden RN, Heel RC, Speight TM, Avery GS. Cimetidine: a review of its pharmacological properties and therapeutic efficacy in peptic ulcer disease. Drugs. 1978;15(2):93–131; (b) Winship DH. Cimetidine in the treatment of duodenal ulcer: review and commentary. Gastroenterology. 1978;74(2 Pt 2):402–406.
- Yoo SE, Yi KY, Lee S, Suh J, Kim N, Lee BH, Seo HW, Kim SO, Lee DH, Lim H, et al. A novel anti-ischemic ATP-sensitive potassium channel (KATP) opener without vasorelaxation: N-(6-aminobenzopyranyl)-N‘-benzyl-N‘‘-cyanoguanidine analogue. J Med Chem. 2001;44(24):4207–4215.
- Hu Z, Ou L, Li S, Yang L. Synthesis and biological evaluation of 1-cyano-2-amino-benzimidazole derivatives as a novel class of antitumor agents. Med Chem Res. 2014;23(6):3029–3038.
- Li ZQ, Chen X, Wang Y. Small molecules targeting ubiquitination to control inflammatory diseases. Drug Discov Today. 2021;26(10):2414–2422.
- Kim KY, Kim BG, Kim S-O, Yoo S-E, Kwak Y-G, Chae S-W, Hong KW. Prevention of lipopolysaccharide-induced apoptosis by (2S,3S,4R)-N"-cyano-N-(6-amino-3,4-dihydro-3-hydroxy-2-methyl-2-dimethoxymethyl-2H-benzopyran-4-yl)-N′-benzylguanidine, a benzopyran analog. J Pharmacol Exp Ther. 2002;300(2):535–542.
- Deaton DN, Hassell AM, McFadyen RB, Miller AB, Miller LR, Shewchuk LM, Tavares FX, Willard DH, Wright LL. Novel and potent cyclic cyanamide-based cathepsin K inhibitors. Bioorg Med Chem Lett. 2005;15(7):1815–1819.
- (a) Lainé D, Palovich M, McCleland B, Petitjean E, Delhom I, Xie H, Deng J, Lin G, Davis R, Jolit A, et al. Discovery of novel cyanamide-based inhibitors of cathepsin C. ACS Med Chem Lett. 2011;2(2):142–147; (b) Falgueyret JP, Oballa RM, Okamoto O, Wesolowski G, Aubin Y, Rydzewski RM, Prasit P, Riendeau D, Rodan SB, Percival MD, et al. Novel, nonpeptidic cyanamides as potent and reversible inhibitors of human cathepsins K and L. J Med Chem. 2001;44(1):94–104.
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci USA. 2007;104(25):10655–10660.
- Montague-Cardoso K, Malcangio M. Cathepsin S as a potential therapeutic target for chronic pain. Med Drug Discov. 2020;7:100047.
- (a) Tars K, Vullo D, Kazaks A, Leitans J, Lends A, Grandane A, Zalubovskis R, Scozzafava A, Supuran CT. Sulfocoumarins (1, 2-benzoxathiine-2, 2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem. 2013;56(1):293–300; (b) Leitans J, Kazaks A, Balode A, Ivanova J, Zalubovskis R, Supuran CT, Tars K. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J Med Chem. 2015;58(22):9004–9009; (c) Grandane A, Tanc M, Di Cesare Mannelli L, Carta F, Ghelardini C, Žalubovskis R, Supuran CT. 6-Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J Med Chem. 2015;58(9):3975–3983; (d) Grandāne A, Nocentini A, Domračeva I, Žalubovskis R, Supuran CT. Development of oxathiino [6, 5-b] pyridine 2, 2-dioxide derivatives as selective inhibitors of tumor-related carbonic anhydrases IX and XII. Eur J Med Chem. 2020;200:112300; (e) Abdoli M, Angeli A, Bozdag M, Carta F, Kakanejadifard A, Saeidian H, Supuran CT. Synthesis and carbonic anhydrase I, II, VII, and IX inhibition studies with a series of benzo [d] thiazole-5-and 6-sulfonamides. J Enzyme Inhib Med Chem. 2017;32(1):1071–1078; (f) Abdoli M, Bozdag M, Angeli A, Supuran C. Benzamide-4-sulfonamides are effective human carbonic anhydrase I, II, VII, and IX inhibitors. Metabolites. 2018;8(2):37; (g) Abdoli M, Giovannuzzi S, Supuran CT, Žalubovskis R. 4-(3-Alkyl/benzyl-guanidino) benzenesulfonamides as selective carbonic anhydrase VII inhibitors. J Enzyme Inhib Med Chem. 2022;37(1):1568–1576.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.
- (a) Pastorekova S, Casini A, Scozzafava A, Vullo D, Pastorek J, Supuran CT. Carbonic anhydrase inhibitors: the first selective, membrane-impermeant inhibitors targeting the tumor-associated isozyme IX. Bioorg Med Chem Lett. 2004;14(4):869–873; (b) Vullo D, Voipio J, Innocenti A, Rivera C, Ranki H, Scozzafava A, Kaila K, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the human cytosolic isozyme VII with aromatic and heterocyclic sulfonamides. Bioorg Med Chem Lett. 2005;15(4):971–976; (c) Gieling RG, Babur M, Mamnani L, Burrows N, Telfer BA, Carta F, Winum J-Y, Scozzafava A, Supuran CT, Williams KJ, et al. Antimetastatic effect of sulfamate carbonic anhydrase IX inhibitors in breast carcinoma xenografts. J Med Chem. 2012;55(11):5591–5600; (d) Grandane A, Nocentini A, Werner T, Zalubovskis R, Supuran CT. Benzoxepinones: a new isoform-selective class of tumor associated carbonic anhydrase inhibitors. Bioorg Med Chem. 2020;28(11):115496.
- (a) Krasavin M, Sharonova T, Sharoyko V, Zhukovsky D, Kalinin S, Žalubovskis R, Tennikova T, Supuran CT. Combining carbonic anhydrase and thioredoxin reductase inhibitory motifs within a single molecule dramatically increases its cytotoxicity. J Enzyme Inhib Med Chem. 2020;35(1):665–671; (b) Ivanova J, Balode A, Žalubovskis R, Leitans J, Kazaks A, Vullo D, Tars K, Supuran CT. 5-Substituted-benzylsulfanyl-thiophene-2-sulfonamides with effective carbonic anhydrase inhibitory activity: Solution and crystallographic investigations. Bioorg Med Chem. 2017;25(3):857–863; (c) Alterio V, Tanc M, Ivanova J, Zalubovskis R, Vozny I, Monti SM, Di Fiore A, De Simone G, Supuran CT. X-ray crystallographic and kinetic investigations of 6-sulfamoyl-saccharin as a carbonic anhydrase inhibitor. Org Biomol Chem. 2015; 13(13):4064–4069.
- (a) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal beta-class (Cab) and gamma-class (Cam) carbonic anhydrases. Curr Top Med Chem. 2007;7(9):901–908; (b) Supuran CT, Clare BW. Carbonic anhydrase inhibitors. Part 57. Quantum chemical QSAR of a group of 1,3,4-thiadiazole and 1,3,4-thiadiazoline disulfonamides with carbonic anhydrase inhibitory properties. Eur J Med Chem. 1999;34(1):41–50; (c) Supuran CT, Barboiu M, Luca C, Pop E, Brewster ME, Dinculescu A. Carbonic anhydrase activators. Part 14. Synthesis of mono- and bis- pyridinium salt derivatives of 2-amino-5-(2-aminoethyl)- and 2-amino-5-(3-aminopropyl)-1,3,4-thiadiazole, and their interaction with isozyme II. Eur J Med Chem. 1996;31(7-8):597–606; (d) Aspatwar A, Barker H, Aisala H, Zueva K, Kuuslahti M, Tolvanen M, Primmer CR, Lumme J, Bonardi A, Tripathi A, et al. Cloning, purification, kinetic and anion inhibition studies of a recombinant β-carbonic anhydrase from the Atlantic salmon parasite platyhelminth Gyrodactylus salaris. J Enzyme Inhib Med Chem. 2022;37(1):1577–1586.
- (a) Urbanski LJ, Vullo D, Parkkila S, Supuran CT. An anion and small molecule inhibition study of the β-carbonic anhydrase from Staphylococcus aureus. J Enzyme Inhib Med Chem. 2021;36(1):1088–1092; (b) Urbanski LJ, Bua S, Angeli A, Kuuslahti M, Hytönen VP, Supuran CT, Parkkila S. Sulphonamide inhibition profile of Staphylococcus aureus β-carbonic anhydrase. J Enzyme Inhib Med Chem. 2020;35(1):1834–1839.
- (a) Nishimori I, Minakuchi T, Vullo D, Scozzafava A, Supuran CT. Inhibition studies of the β-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium with sulfonamides and sulfamates. Bioorg Med Chem. 2011;19(16):5023–5030; (b) Vullo D, Nishimori I, Minakuchi T, Scozzafava A, Supuran CT. Inhibition studies with anions and small molecules of two novel β-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium. Bioorg Med Chem Lett. 2011;21(12):3591–3595; (c) Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol. 2011;2:34.
- (a) De Simone G, Di Fiore A, Truppo E, Langella E, Vullo D, Supuran CT, Monti SM. Exploration of the residues modulating the catalytic features of human carbonic anhydrase XIII by a site-specific mutagenesis approach. J Enzyme Inhib Med Chem. 2019;34(1):1506–1510; (b) Monti DM, De Simone G, Langella E, Supuran CT, Di Fiore A, Monti SM. Insights into the role of reactive sulfhydryl groups of carbonic anhydrase III and VII during oxidative damage. J Enzyme Inhib Med Chem. 2017;32(1):5–12.
- (a) Gramoli JL, Wilkinson BJ. Characterization and identification of coagulase-negative, heat-stable deoxyribonuclease-positive Staphylococci. J Gen Microbiol. 1978;105(2):275–285; (b) Fungwithaya P, Boonchuay K, Narinthorn R, Sontigun N, Sansamur C, Petcharat Y, Thomrongsuwannakij T, Wongtawan T. First study on diversity and antimicrobial-resistant profile of Staphylococci in sports animals of Southern Thailand. Vet World. 2022;15(3):765–774; (c) Cai Y, Zheng L, Lu Y, Zhao X, Sun Y, Tang X, Xiao J, Wang C, Tong C, Zhao L, et al. Inducible resistance to β-lactams in oxacillin-susceptible mecA1-positive Staphylococcus sciuri isolated from retail pork. Front Microbiol. 2021; 12:721426; (d) Chen S, Wang Y, Chen F, Yang H, Gan M, Zheng SJ. A highly pathogenic strain of Staphylococcus sciuri caused fatal exudative epidermitis in piglets. PLoS One. 2007;2(1):e147.
- Madhaiyan M, Wirth JS, Saravanan VS. Phylogenomic analyses of the Staphylococcaceae family suggest the reclassification of five species within the genus Staphylococcus as heterotypic synonyms, the promotion of five subspecies to novel species, the taxonomic reassignment of five Staphylococcus species to Mammaliicoccus gen. nov., and the formal assignment of Nosocomiicoccus to the family Staphylococcaceae. Int J Syst Evol Microbiol. 2020;70(11):5926–5936.
- Angeli A, Urbański LJ, Capasso C, Parkkila S, Supuran CT. Activation studies with amino acids and amines of a β-carbonic anhydrase from Mammaliicoccus (Staphylococcus) sciuri previously annotated as Staphylococcus aureus (SauBCA) carbonic anhydrase. J Enzyme Inhib Med Chem. 2022;37(1):2786–2792.