
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The copper-catalysed azide-alkyne cycloaddition was applied to prepare three enantiomeric pairs of heterodimers containing a tacrine residue and a 1,4-dideoxy-1,4-imino-D-arabinitol (DAB) or 1,4-dideoxy-1,4-imino-L-arabinitol (LAB) moiety held together via linkers of variable lengths containing a 1,2,3-triazole ring and 3, 4, or 7 CH2 groups. The heterodimers were tested as inhibitors of butyrylcholinesterase (BuChE) and acetylcholinesterase (AChE). The enantiomeric heterodimers with the longest linkers exhibited the highest inhibition potencies for AChE (IC50 = 9.7 nM and 11 nM) and BuChE (IC50 = 8.1 nM and 9.1 nM). AChE exhibited the highest enantioselectivity (ca. 4-fold). The enantiomeric pairs of the heterodimers were found to be inactive (GI50 > 100 µM), or to have weak antiproliferative properties (GI50 = 84–97 µM) against a panel of human cancer cells.
Introduction
Enzyme inhibition represents an attractive target for drug developmentCitation1. Because enzymes are built up by chiral building blocks, amino acids, it is not surprising if only one member of an enantiomeric pair causes inhibition upon binding. Another alternative is that both enantiomers display various degrees of inhibition, due to different interaction modesCitation2–4. One such example is the natural enantiomer 1a () of huperzine A, which is a 38- to 49-fold more potent AChE inhibitor than its unnatural enantiomer 1bCitation5,Citation6. In fact, inhibition of cholinesterases (ChEs) is an attractive target for treatment of Alzheimer’s disease (AD) and there are currently three ChE inhibitors on the list of FDA approved AD drugsCitation7. (–)-Huperzine (1a) is not on the list of FDA approved drugs, but it was approved in China as a symptomatic AD drugCitation8,Citation9.The much stronger AChE inhibition exhibited by 1a compared to 1b, was partially rationalised by comparison of the X-ray structures of Torpedo californica acetylcholinesterase (TcAChE) complexed with enantiomers 1a and 1b, which demonstrated the presence and absence of an interaction between the ethylidene methyl of 1a and 1b, respectively, with His440Citation10, which is a member of the catalytic triad almost on the bottom of a ca. 20 Å deep active gorge of TcAChECitation11.
Figure 1. Examples of enantiomeric pairs of ChE inhibitors of which the mirror images display different potencies.
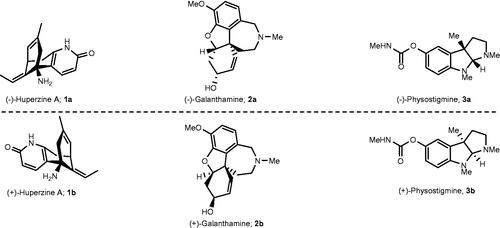
(-)-Galantamine (2a) () is an FDA approved ChE inhibitor drug for the treatment of mild-to-moderate ADCitation7. This alkaloid is a reversible AChE inhibitor and exhibits 53-times selectivity for AChE over BuChECitation12. X-ray studies of the TcAChE/(-)-galantamine complex revealed that the inhibitor binds in its acidic form at the base of the active gorge in the region between the acetyl hole and the catalytic anionic site (CAS)Citation13. The protonated amine group is quite remote from Trp84 in CAS and thereby is not involved in any cation–π interactions with the Trp84 residue, which is in stark contrast to acetylcholine (ACh), whose quaternary ammonium group establishes a cation–π interaction with Trp84. Instead, the high affinity of (-)-galantamine for AChE was attributed to multiple moderate and weak interactions with the enzymeCitation13. The inhibition of AChE by galantamine appears to be enantioselective, at 20 μM inhibitor concentration, as the natural enantiomer 2a (ca. 94% of inhibition) is a much stronger inhibitor than its unnatural antipode 2b (ca. 4% of inhibition)Citation14, which indicates that several interactions with the enzyme are eliminated or attenuated when the configuration in all stereogenic centres of 2a is reversed.
Significant enantioselectivity has also been observed for the inhibition of AChE by physostigmine; natural (-)-physostigmine (3a) () is a ca. 25- to 1000-fold stronger inhibitor, depending on the enzyme source, than (+)-physostigmine (3b)Citation15,Citation16.
Figure 2. Selected examples of iminosugars that have been investigated as ChE inhibitors.
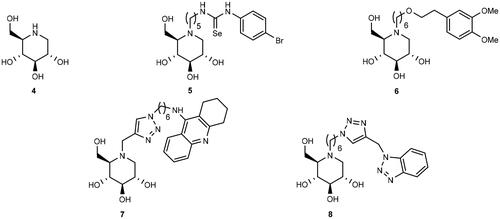
Iminosugars are glycomimetics in which the ring oxygen atom has been replaced by a nitrogen atomCitation17. Iminosugars are attractive as pharmaceutical candidates because they inhibit glycosidases without being metabolised by such enzymesCitation18. Such properties have made iminosugars attractive as synthetic targetsCitation19 and lead compounds for the treatment of various diseases such as viral infections, diabetes, type 2 diabetes, and lysosomal disordersCitation20. In addition, it has been found that iminosugars are able to inhibit the growth of cancer cellsCitation21,Citation22, without affecting the viability and mortality of normal cellsCitation21. To date, three iminosugars, namely, miglitolCitation23, miglustatCitation24, and migalastatCitation25 have been approved by FDA for the treatment of type 2 diabetes, Gaucher’s disease, and Fabry’s disease, respectively. Miglustat has also been found to reduce the production of amyloid β-peptide (Aβ)Citation26, which is a component of senile plaque in AD patients.
The ester group of ACh is held in place for hydrolysis by the catalytic triad in the active gorge of AChE by the aid of cation − π interactions with a Trp residue in CASCitation27. Because many iminosugars are protonated at physiological pHCitation28, they were proposed to be capable of inhibiting ChEsCitation29. Thereby, a series of iminosugars of various stereochemistry and substitution patterns have been tested as ChE inhibitors, displaying particularly good BuChE inhibitionCitation29. Following this line, some of us have reported the synthesis and ChE inhibitory testing of bivalent inhibitors in which a 1-deoxynojirimycin (1-DNJ) (4) binding unit is connected to a second binding unit, namely, aryl-substituted selenourea (exemplified by 5)Citation30, catechol (exemplified by 6)Citation31, tacrine (exemplified by 7)Citation32, or benzotriazole (exemplified by 8)Citation33 binding unit (). Kinetic assays and modelling studies for the binding of 6, 7, and 8 to AChE indicated that they behave as dual binding site AChE inhibitors, which implies that they bind simultaneously to the peripheral anionic site (PAS) and CAS. A more surprising observation (given that the quaternary ammonium group of ACh participates in a cation–π interaction with a Trp residue in CAS) from the modelling studies was that when heterodimers 6 and 8 bind to AChE in their protonated states (on the 1-DNJ nitrogen atom), the positive charged nitrogen atom is not necessarily involved in cation − π interactions with the aromatic residues of the enzymeCitation31,Citation33.
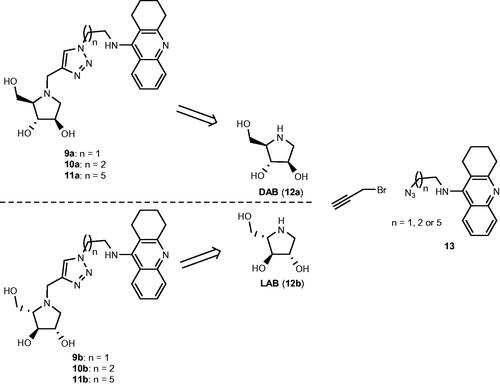
Thus far, five papers have been published, which demonstrate the potential of iminosugars as ChE inhibitorsCitation29–33. One entry of ChE inhibition by iminosugars that remains to be studied is whether iminosugars can achieve enantioselective ChE inhibition. Thus, in this paper, we present the synthesis of three pairs of optically pure iminosugar-tacrine heterodimer enantiomers, namely, 9a and 9b, 10a and 10b, and 11a and 11b (Scheme 1) and the evaluation of their performance as ChE inhibitors. The study also includes docking studies of the heterodimers to predict interaction with AChE and BuChE. Naturally occurring 1,4-dideoxy-1,4-imino-D-arabinitol (DAB) (12a) constitutes the iminosugar moiety in 9a, 10a, and 11a, whereas non-natural 1,4-dideoxy-1,4-imino-L-arabinitol (LAB) (12b) is the iminosugar moiety in 9b, 10b, and 11b. Because both iminosugarsCitation21,Citation22, and heterodimers containing a tacrine moietyCitation34 have been found to inhibit the growth of cancer cells, we also report the antiproliferative screening of 11a and 11b against a panel of six cancer cell lines.
Scheme 1. Retrosynthetic pathways to the optically pure pairs of enantiomers 9a and 9b, 10a and 10b, and 11a and 11b.
Materials and methods
General procedures
Dichloromethane (DCM), methanol (MeOH), acetone, dimethyl sulfoxide (DMSO) and dimethylformamide (DMF) were dried over 4 Å molecular sieves (oven dried). Petroleum ether (PE) from the 40–65 °C fraction was used for silica flash columns. All reactions were carried out under Ar atmosphere if not otherwise specified. Reactions performed at room temperature (rt) refer to the temperature range of 20 to 22 °C. TLC analyses were performed on Merck silica gel 60 F254 plates using UV light (λ = 254 nm) for detection. Silica gel NORMASIL 60® 40–63 µm was used for silica flash columns. A Bruker Avance NMR spectrometer was used to record 1H-NMR spectra (400.13 MHz) and 13C-NMR spectra (100.61 MHz) in CDCl3, CD3OD, or D2O. Chemical shifts (δ) are reported relative to residual DMSO (δ 2.50 ppm, 1H; δ 39.52 ppm, 13C), residual CHCl3 in CDCl3 (δ 7.26 ppm, 1H; δ 77.16 ppm, 13C), residual CD3OD (δ 3.31 ppm, 1H; δ 49.0 ppm, 13C), residual D2O (δ 4.79 ppm, 1H) and TMS as an internal standard in CDCl3. High-resolution mass spectra (HRMS) were recorded on a Qexactive spectrometer in positive electrospray ionisation (ESI) mode.
Synthetic protocols
General procedure for the preparation of compounds 20a–22a and 20b–22b
A mixture of 19b (1 equiv., 0.04 M for synthesis of 20b, 21b, and 22b) or 19a (1 equiv., 0.07 M for synthesis of 20a, 0.04 M for synthesis of 21a, and 0.05 M for synthesis of 22a), azide 13 (0.98 equiv.), and copper(II) sulphate pentahydrate (0.30 equiv.) in anhydrous DMF in an aluminium foil covered round bottom flask was degassed and introduced an argon atmosphere before the addition of sodium ascorbate (0.60 equiv.). After addition, the mixture was kept stirring for 48 h at rt. The solvent was then removed under reduced pressure and the residue obtained was purified by silica gel column chromatography (See Supplementary Material for details).
General procedure for the preparation of compounds 9a–11a and 9b–11b
To a mixture of 20a–22a (0.02 M, 1 equiv.) or 20b–22b (0.02 M, 1 equiv.) in anhydrous CH2Cl2 under an argon atmosphere at −78 °C was slowly added BCl3 (1 M in heptane, 15 equiv.). After addition, the mixture was kept stirring at −78 °C for 2 h and then at 0 °C overnight. The volatiles were then removed under reduced pressure and the concentrate underwent purification by gradient silica gel chromatography (MeCN/H2O/NH4OH 190:10:1 → 180:20:1) (column 1). The corresponding HCl salt was dissolved in MeOH (2 ml) and NH4OH (0.5 ml) and kept stirring for 48 h. The solvent was removed under reduced pressure and the resulting residue was purified by gradient silica gel chromatography (column 2) (the solvent gradient for column 2 for each single experiment is specified in the Supplementary Material).
Inhibition assays
Measuring of the inhibition activity of compounds 9a–11a and 9b–11b against cholinesterases (AChE from Electrophorus electricus and BuChE from equine serum) was accomplished following minor modifications of the Ellman assayCitation35, as reported previouslyCitation36. A Thermo ScintificTM VarioskanTM LUX microplate reader and Greiner F-bottom 96-well plates were used. Cornish-Bowden plots (1/V vs. [I] and [S]/V vs. [I]) were used for the visualisation of the mode of inhibition. Calculation of the kinetic parameters (KM, Vmax) was accomplished using a nonlinear regression analysis (least squares fit) implemented in GraphPad Prism 8.01 software; such parameters were in turn used for calculating the inhibition constants of the mixed inhibitors using the following equations:
General method for docking simulations
Interactions of enzymes with compounds were analysed by computational docking using Molecular Operating Environment (MOE) software (Chemical Computing Group ULC, Montreal, Canada). Crystallographic structures of human AChE and human BuChE was obtained from Protein Data Bank (PDB code 4EY6Citation37 and 4AQDCitation38, respectively). Protein structures were prepared using Amber10 force field with EHT parameters, R-field solvation model, dielectric constant of 1 for the protein interior and 80 for exterior. Ligand structures were drawn in MOE software, and their energies were minimised using Amber10 force field with EHT parameters for small molecules, using as stop criterion an RMS gradient lower than 0.01 kcal/mol/Å. For the docking calculations: in the placement stage we used the Triangle Matcher algorithm with the London dG scoring scheme. In the refinement stage we kept the receptor rigid and used the GBVI/WSA dG scoring scheme. 2D diagrams were obtained from MOE software and 3D illustrations were obtained using Pymol software.
Antiproliferative activity assays
For the antiproliferative tests, we applied our implementation of the National Cancer Institute (NCI) screening protocolCitation39. As a model of human solid tumour cells, we selected the cell lines A549 (non-small cell lung), HBL-100 (breast), HeLa (cervix), SW1573 (non-small cell lung), T-47D (breast), and WiDr (colon). Cell seeding densities, based on the cell line doubling time, were 2500 (A549, HBL-100, HeLa and SW1573) or 5000 (T-47D and WiDr) cells/well. Compounds were initially dissolved in DMSO at 400 times the desired final maximum test concentration. Control cells were exposed to an equivalent concentration of DMSO (0.25% v/v, negative control). Each compound was tested in triplicate at different dilutions ranging from 1 to 100 μM. Drug treatment began on day 1 after sowing. The drug incubation times were 48 h, after which the cells were precipitated with ice-cold TCA (50% w/v) and fixed for 60 min at 4 °C. Then the SRB test was performed. The optical density (OD) of each well was measured at 530 nm using a microplate absorbance reader (PowerWave XS, BioTek Instruments Inc.). Values were corrected for background OD of wells containing medium onlyCitation39. The results were expressed as GI50, i.e. the dose that causes 50% growth inhibition after 48 h of exposure.
Synthesis
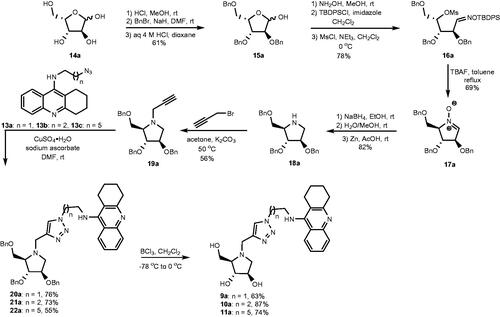
The synthesis of heterodimers 9a, 10a, and 11a commenced from L-xylose (14a), which was converted into 2,3,5-tri-O-benzyl-L-xylofuranose (15a) by following a reported three step procedure (Scheme 2)Citation40. The obtained furanose underwent three subsequent chemical modifications including: (1) aldoxime formation, (2) selective O-silylation of the oxime oxygen atom, and (3) mesylation to provide compound 16aCitation41 in 78% yield after purification by silica gel chromatography. When 16a was treated with F- ions it cyclized into nitrone 17aCitation42,Citation43 upon loss of the O-silyl group. Tetra-O-benzylated DAB 18a was obtained in 82% yield when nitrone 17a was reduced first by sodium borohydride and followed by zinc in acetic acidCitation44. In the following step, 18a underwent N-propargylation to form alkyne 19a when it was treated with propargyl bromide. This alkyne underwent copper-catalysed azide – alkyne cycloadditionCitation45 with azides 13aCitation46, 13bCitation32, and 13cCitation32 to form heterodimers 20a, 21a, and 22a, respectively. In the final step, heterodimers 20a, 21a, and 22a underwent BCl3 promoted de-O-benzylation to generate target compounds 9a, 10a, and 11a, respectively.
Scheme 2. Synthesis of heterodimers 9a–11a.
The synthesis of 9b, 10b, and 11b were performed in the same way as for 9a, 10a, and 11a by replacing L-xylose (14a) with D-xylose (14b) in the first step (Scheme 3).
Scheme 3. Synthesis of heterodimers 9b, 10b, and 11b.
ChE inhibitory testing
The minimum inhibitory concentrations of the enantiomeric pairs 9a and 9b, 10a and 10b, and 11a and 11b required to reach 50% inhibition (IC50) of eeAChE and eqBuChE are presented in . A minor modification of the Ellman assay was used in order to measure the IC50 valuesCitation35. The test series included (-)-galantamine (2a) and tacrine as positive references.
Table 1. IC50 values for the inhibition of eeAcHE and eqBuChE by 9a, 10a, and 11a and with their mirror images 9b, 10b, and 11b.
Both series of stereoisomers 9a–11a (incorporating a DAB moiety) and 9b–11b (incorporating a LAB moiety) displayed IC50 values from the submicromolar concentration range down to the nanomolar concentration range for the inhibition of eeAChE and eqBuChE. The only exception was 9b, which exhibits a IC50 value of 1480 nM for the inhibition of eeAChE. Thereby, 9b was the only compound in the testing series that is a less potent AChE inhibitor than (-)-galantamine, which is in in current use against ADCitation47.
The length of the linker between the tacrine ring and iminosugar moiety had a significant impact on the inhibition potency of both eeAChE and eqBuChE in which a longer linker provided higher inhibition potencies. This was demonstrated by the result that 11a (n = 5, IC50 = 9.7 nM against eeAChE) is a ca. 43-fold more potent eeAChE inhibitor than 9a (n = 1, IC50 = 420 nM against eeAChE) and a 55-fold more potent eeAChE inhibitor than 10a (n = 2, IC50 = 530 nM against eeAChE). A similar trend was observed when the enantiomer of 11a, namely, 11b (n = 5, 10.7 nM against eeAChE) was compared with 9b (n = 1, 1480 nM against eeAChE) and 10b (n = 2, 150 nM against eeAChE) for the inhibition of the same enzyme as 11b is a ca. 138- and 14-fold stronger inhibitor than 9b and 10b, respectively. Six CH2-groups between the 1,2,3-triazole and tacrine moiety was also most favourable for the inhibition of eqBuChE as 11a (n = 5, IC50 = 9.1 nM against eqBuChE) is a roughly 11-fold stronger inhibitor than 9a (n = 1, IC50 = 96 nM against eqBuChE) and a 20-fold stronger inhibitor than 10a (n = 2, IC50 = 184 nM against eqBuChE). Likewise, 11b (n = 5, IC50 = 8.1 nM against eqBuChE) is a 22- and 29-fold stronger eqBuChE inhibitor than 9b (n = 1, IC50 = 179 nM against eqBuChE) and 10b (n = 2, IC50 = 232 μM against eqBuChE), respectively.
No obvious enantioselectivity of eeAChE and eqBuChE was observed for the three pairs of enantiomeric inhibitors included in this study. In addition, no preferential inhibitory activity trend was found for the enantiomers incorporating a DAB or LAB moiety. For instance, 9a is a ca. 4-fold more potent eeAChE inhibitor than its enantiomer 9b, whereas 10b is a ca. 4-fold more potent eeAChE inhibitor than its enantiomer 10a. For the enantiomeric pair 11a and 11b, we observed essentially equal eeAChE inhibitory activities. These observations indicate that the impact on the eeAChE inhibitory potency of our heterodimers by switching between a DAB and LAB moiety is small compared to the contribution from the tacrine ring.
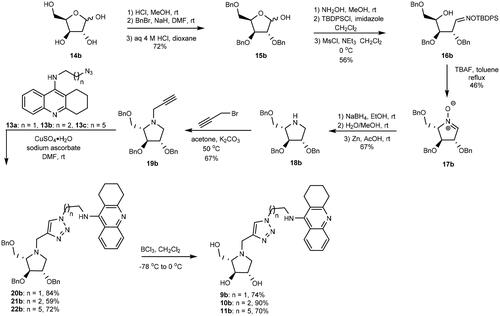
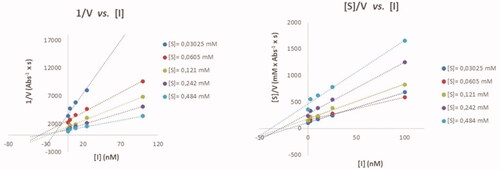
The inhibition modes of eeAChE and eqBuChE by heterodimer 11b were investigated by using the Cornish-Bowden method, that is, by creating two plots (1/V vs. [I] and [S]/V vs. [I]) for the inhibition of both enzymes (). The two plots for the inhibition of each enzyme included a point of intersection at different [I]-coordinates, which implies that 11b is a mixed inhibitor of both enzymesCitation47. The competitive inhibition constant, Ki, and uncompetitive inhibition constant, αKi, for eeAChE by 11b is 19.0 ± 1.8 nM and 21.9 ± 7.2 nM, respectively. The inhibition constants of eqBuChE are Ki =10.0 ± 2.7 nM and αKi = 14.3 ± 3.3 nM. The mixed inhibition modes of eeAChE and eqBuChE by 11b were interpreted to indicate that 11b behaves as a dual binding site inhibitor of both enzymes; it is tempting to think that 11b binds simultaneously to the active site and PAS of both eeAChE and eqBuChE. However, in this context it is worth mentioning that the architecture of PAS in the two enzymes is different as it is richer on aromatic amino acid residues in eeAChECitation48,Citation49, which allow formation of π–π interactions and cation–π interactions with ligandsCitation50.
Figure 3. Cornish-Bowden plots for analysing the inhibition mode of eeAChE by 11b.
Modelling studies
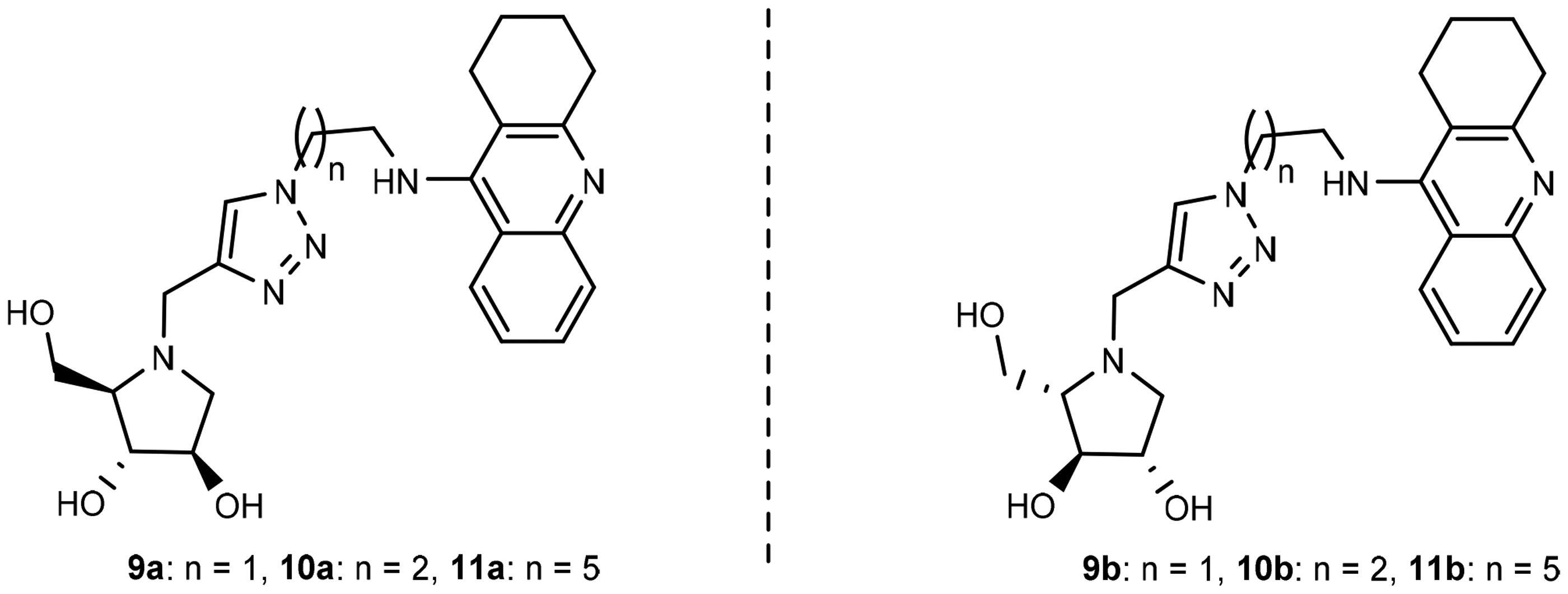
The preferred binding poses for enantiomers 11a and 11b in recombinant human acetylcholinesterase (rhAChE) are presented in and , respectively, whereas the preferred binding poses for the enantiomeric pairs 9a and 9b, and 10a and 10b are presented in Figure SI2 and Figure SI3, respectively. A common trend for all energetically preferred binding poses is that the tacrine moiety and the iminosugar moiety bind to the active site and PAS, respectively. Such preferred binding pose is not very surprising given that X-ray analysis has shown that tacrine is bound to the active site of AChECitation51.
Figure 4. (a) Docking simulations for the interactions in the 11a-rhAChE complex. (b) Three-dimensional structure of rhAChE showing the binding mode of compound 11a. The residues, Ser203, His447, and Glu334 corresponding to the catalytic triad are depicted in sticks.
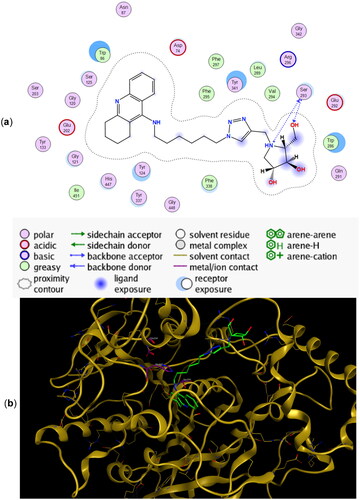
Figure 5. (a) Docking simulations for the interactions in the 11b-rhAChE complex. (b) Three-dimensional structure of rhAChE showing the binding mode of compound 11b. The residues, Ser203, His447, and Glu334 corresponding to the catalytic triad are depicted in sticks.
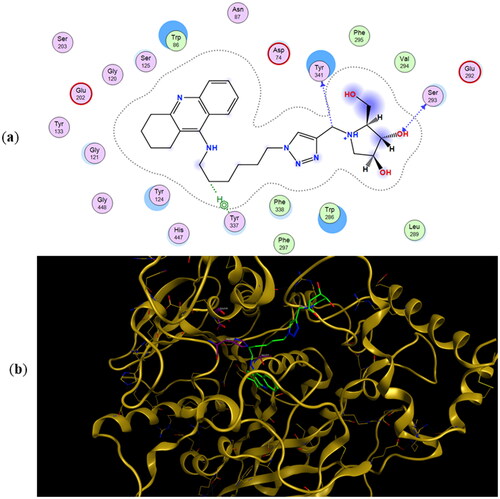
Hydrogen bonding interactions between one of the hydroxyl groups of the iminosugar moiety and Ser293 in rhAChE are observed in both 11a () and 11b (). Interestingly, the protonated imino group of 11a showed another hydrogen bond with the same Ser293, a feature not observed in 11b. This helps explain the lower binding energy for 11a (−10.45 kcal/mol) compared to its antipode 11b (−9.92 kcal/mol) (). Slight differences in binding energies were also observed between enantiomers 9a and 9b (−9.25 kcal/mol for 9a vs. −9.05 kcal/mol for 9b) and between 10a and 10b (−9.52 kcal/mol for 10a vs. −8.91 kcal/mol for 10b) when they are bound to rhAChE. The hydroxyl groups of 9a showed interactions with Tyr341 and Arg296 meanwhile there is an arene cation interaction between one of the hydroxyl groups in 9b and Trp286 (Figure SI2). Hydrogen bond interaction between Ser293 and the iminosugar moiety is observed for 10a but is lacking in its antipode 10b (Figure SI3).
Table 2. Binding energies for 9a, 9b, 10a, 10b, 11a, and 11b to rhAChE and hBuChE.
We found that our measured IC50 values () for the inhibition of eeAChE by 11a and 11b are in agreement with the calculated binding energies (), which predict 11a and 11b to possess the highest affinity for the enzyme. However, IC50 is not a true measure of binding affinity of a ligand to an enzymeCitation52, which explains why the calculated binding energies in fail in predicting the relative IC50 values for the inhibition of eeAChE by the heterodimers (9a–11a and 9b–11b) in our series.
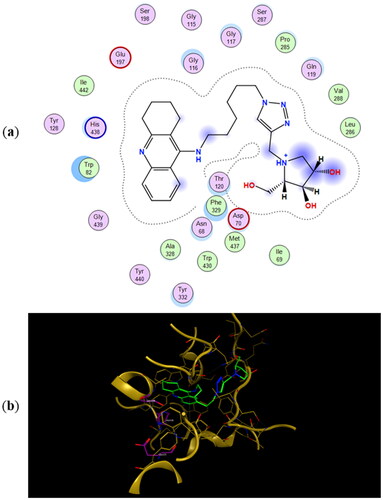
The most energetically favourable binding poses of enantiomers 11a and 11b to human butyrylcholinesterase (hBuChE) are presented in and , respectively. The preferred binding poses of the enantiomeric pairs 9a and 9b, and 10a and 10b to the same enzyme are presented in Figures SI5 and SI6, respectively. The number of CH2-groups between the tacrine ring and 1,2,3-triazole ring appears to control whether the iminosugar moiety is bound to the active site or PAS. In fact, the tacrine ring of 9a, 9b, 11a, and 11b is accommodated in the active site whereas their iminosugar moiety is bound to PAS. For heterodimers 10a and 10b the binding scenarios are different, as the tacrine ring is accommodated in PAS and the iminosugar moiety in the active site. As for the inhibition of eeAChE, even though the calculated binding energies in predict 11a and 11b to be the most potent BuChE inhibitors, they fail in predicting the relative IC50 values for the whole testing series.
Figure 6. (a) Docking simulations for the interactions in the 11a-hBuChE complex. (b) Three-dimensional structure of hBuChE showing the binding mode of compound 11a. The residues, Ser198, His438 and Glu325, corresponding to the catalytic triad are depicted in sticks.
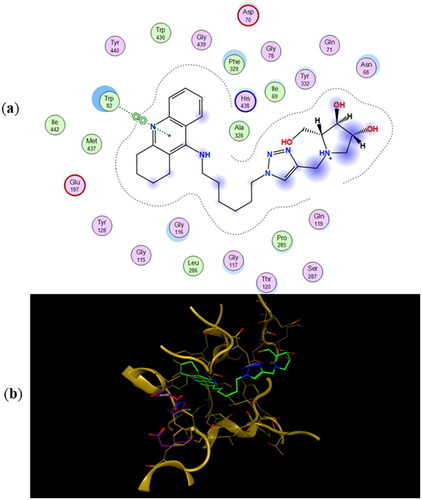
Figure 7. (a) Docking simulations for the interactions in the 11b-hBuChE complex. (b) Three-dimensional structure of hBuChE showing the binding mode of compound 11b. The residues, Ser198, His438, and Glu325, corresponding to the catalytic triad are depicted in sticks.
Antiproliferative activity
Antiproliferative activity of our heterodimers was investigated for six cancer cell lines including A549, HBL-100, HeLa, SW1573, T-47D and WiDr. The inhibition of cancer cell growth by each heterodimer is expressed in concentration of heterodimer required to lower the cell growth by 50% (GI50). The antiproliferative activity of a compound is only significant when GI50 ˂ 100 μM. The measured GI50 values demonstrated that those heterodimers with two CH2-groups (9a and 9b) and three CH2-groups (10a and 10b) between the tacrine and 1,2,3-triazole rings display no significant antiproliferative activity (GI50 > 100 μM). 11a and 11b on the other hand that contain six CH2-groups between the tacrine and 1,2,3-triazole rings display GI50 values below 100 μM for the inhibition of A549 cancer cell growth (). In addition, 11a and 11b display weak but significant inhibition of cell growth of HeLa and SW1573 cancer cells, respectively.
Table 3. Antiproliferative activity (GI50) of 9a, 9b, 10a, 10b, 11a, and 11b against human cancer cells.
Conclusions
In contrast to the enantiomeric pairs 1a and 1b of huperzine, 2a and 2b of galantamine, and 3a and 3b of physostigmine, our enantiomeric pairs 9a and 9b, 10a and 10b, and 11a and 11b of iminosugar-tacrine heterodimers displayed low enantioselectivity (˂4) for the inhibition of eeAChE and eqBuChE. The following three observations: (1) 9a is a ca. 3.5-fold stronger eeAChE inhibitor than 9b, (2) 10a is a ca. 3.5-fold less potent eeAChE inhibitor than 10b, and (3) 11a and 11b are essentially equipotent eeAChE inhibitors, show that eeAChE exhibits no consequent preference for any of the enantiomeric heterodimers, which include a DAB or LAB moiety. These observations can either be interpreted as the tacrine moiety contributes much more to the eeAChE inhibitory potencies than the DAB or LAB moieties or that the LAB and DAB moieties have similar contribution to the inhibition potencies when they are connected to a tacrine ring. However, the latter interpretation is to some extent contradicted by the modelling studies, which show that the DAB and LAB moieties display different interaction modes with the enzymes.
Like in our earlier studies when we connected an iminosugar to a tacrine ring to obtain ChE inhibitors of type 7 in Citation32, heterodimers 11a and 11b with the longest linkers exhibited the highest inhibition potencies. From modelling studies for the binding to BuChE, it appeared that the length of the linker between the tacrine ring and DAB or LAB moiety controls whether the tacrine ring binds to the active site of PAS. On the other hand, because the trend of the measured IC50 values do not parallel the calculated binding energies, it is possible that the title compounds are not bound in their most energetically favourable poses when they inhibit the enzymes in our testing series.
Author contributions
Conceptualisation, E.L.; methodology, I.C.V. and Ó.L.; funding acquisition, E.L, M.O.S., Ó.L., J.M.P., and J.G.F.B.; investigation, E.L, I.C.V, A.P., M.X.F., and Ó.L.; project administration, E.L.; resources, E.L., Ó.L., M.O.S., J.M.P., and J.G.F.B.; supervision, E.L. and M.O.S.; writing – original draft, E.L., I.C.V.; writing – review & editing, E.L., Ó.L., I.C.V., M.O.S., J.M.P., and J.G.F.B.
Supplemental Material
Download PDF (3 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Ramsay RR, Tipton KF. Assessment of enzyme inhibition: a review with examples from the development of monoamine oxidase and cholinesterase inhibitory drugs. Molecules. 2017;22(7):1192.
- Fokkens J, Klebe G. A simple protocol to estimate differences in protein binding affinity for enantiomers without prior resolution of racemates. Angew Chem Int Ed Engl. 2006;45(6):985–989.
- Mentel M, Blankenfeldt W, Breinbauer R. The active site of an enzyme can host both enantiomers of a racemic ligand simultaneously. Angew Chem Int Ed Engl. 2009;48(48):9084–9087.
- Brooks WH, Guida WC, Daniel KG. The significance of chirality in drug design and development. Curr Top Med Chem. 2011;1:760–770.
- McKinney M, Miller JH, Yamada F, Tuckmantel W, Kozikowski AP. Potencies and stereoselectivities of enantiomers of huperzine A for inhibition of rat cortical acetylcholinesterase. Eur J Pharmacol. 1991;203(2):303–305.
- Zhang HY, Liang YQ, Tang XC, He XC, Bai DL. Stereoselectivities of enantiomers of huperzine A in protection against beta-amyloid(25-35)-induced injury in PC12 and NG108-15 cells and cholinesterase inhibition in mice. Neurosci Lett. 2002;317(3):143–146.
- Anand P, Singh B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res. 2013;36(4):375–399.
- Zangara A. The psychopharmacology of huperzine A: an alkaloid with cognitive enhancing and neuroprotective properties of interest in the treatment of Alzheimer’s disease. Pharmacol Biochem Behav. 2003;75(3):675–686.
- Orhan IE, Orhan G, Gurkas E. An overview on natural cholinesterase inhibitors–a multi-targeted drug class–and their mass production. Mini Rev Med Chem. 2011;11(10):836–842.
- Dvir H, Jiang HL, Wong DM, Harel M, Chetrit M, He XC, Jin GY, Yu GL, Tang XC, Silman I, et al. X-ray structures of Torpedo californica acetylcholinesterase complexed with (+)-huperzine A and (-)-huperzine B: structural evidence for an active site rearrangement. Biochemistry. 2002;4:10810–10818.
- Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science. 1991;253(5022):872–879.
- Sramek JJ, Frackiewicz EJ, Cutler NR. Review of the acetylcholinesterase inhibitor galanthamine. Expert Opin Investig Drugs. 2000;9(10):2393–2402.
- Greenblatt HM, Kryger G, Lewis T, Silman I, Sussman JL. Structure of acetylcholinesterase complexed with (-)-galanthamine at 2.3 A resolution. FEBS Lett. 1999;463(3):321–326.
- Kimura H, Kawai T, Hamashima Y, Kawashima H, Miura K, Nakaya Y, Hirasawa M, Arimitsu K, Kajimoto T, Ohmomo Y, et al. Synthesis and evaluation of (–)- and (+)-[11C]galanthamine as PET tracers for cerebral acetylcholinesterase imaging. Bioorg Med Chem. 2014;22(1):285–291.
- Brossi A, Schönenberger B, Clark OE, Ray R. Inhibition of acetylcholinesterase from electric eel by (-)-and (+)-physostigmine and related compounds. FEBS Lett. 1986;201(2):190–192.
- Triggle DJ, Mitchell JM, Filler R. The pharmacology of physostigmine. CNS Drug Rev. 1998;4(2):87–136.
- McNaught AD. Nomenclature of carbohydrates (IUPAC recommendations 1996). Pure Appl Chem. 1996;68(10):1919–2008.
- Horne G, Wilson FX, Tinsley J, Williams DH, Storer R. Iminosugars past, present and future: medicines for tomorrow. Drug Discov Today. 2011;16(3-4):107–118.
- Lillelund VH, Jensen HH, Liang X, Bols M. Recent developments of transition-state analogue glycosidase inhibitors of non-natural product origin. Chem Rev. 2002;102(2):515–553.
- Asano N, Nash RJ, Molyneux RJ, Fleet GWJ. Sugar-mimic glycosidase inhibitors: natural occurrence, biological activity and prospects for therapeutic application. Tetrahedron Asymmetry. 2000;11:49–58.
- Sánchez-Fernández EM, Gonçalves-Pereira R, Rísquez-Cuadro R, Plata GB, Padrón JM, García Fernández JM, Ortiz Mellet C. Influence of the configurational pattern of sp(2)-iminosugar pseudo N-, S-, O- and C-glycosides on their glycoside inhibitory and antitumor properties. Carbohydr Res. 2016;429:113–122.
- Sánchez-Fernández EM, García-Hernández R, Gamarro F, Arroba AI, Aguilar-Diosdado M, Padrón JM, García Fernández JM, Ortiz Mellet C. Synthesis of sp2-iminosugar selenoglycolipids as multitarget drug candidates with antiproliferative, leishmanicidal and anti-inflammatory properties. Molecules. 2021;26(24):7501.
- Sugimoto S, Nakajima H, Kosaka K, Hosoi H. Review: miglitol has potential as a therapeutic drug against obesity. Nutr Metab. 2015;12:51.
- Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, Levade T, Astudillo L, Serratrice J, Brassier A, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. IJMS. 2017;18(2):441.
- Sunder-Plassmann G, Schiffmann R, Nicholls K. Migalastat for the treatment of Fabry disease. Expert Opin Orphan Drugs. 2018;6:303–309.
- Noel A, Ingrand S, Barrier L. Anti-amyloidogenic effects of glycosphingolipid synthesis inhibitors occur independently of ganglioside alterations. Mol Cell Neurosci. 2016;75:63–70.
- Macdonald IR, Martin E, Rosenberry TL, Darvesh S. Probing the peripheral site of human butyrylcholinesterase. Biochemistry. 2012;51(36):7046–7053.
- Gloster TM, Meloncelli P, Stick RV, Zechel D, Vasella A, Davies GJ. Glycosidase inhibition: an assessment of the binding of 18 putative transition-state mimics. J Am Chem Soc. 2007;129(8):2345–2354.
- Decroocq C, Stauffert F, Pamlard O, Oulaïdi F, Gallienne E, Martin OR, Guillou C, Compain P. Iminosugars as a new class of cholinesterase inhibitors. Bioorg Med Chem Lett. 2015;25(4):830–833.
- Olsen JI, Plata GB, Padrón JM, López Ó, Bols M, Fernández-Bolaños JG. Selenoureido-iminosugars: a new family of multitarget drugs. Eur J Med Chem. 2016;123:155–160.
- Ahuja-Casarín AI, Merino-Montiel P, Vega-Baez JL, Montiel-Smith S, Fernandes MX, Lagunes I, Maya I, Padrón JM, López Ó, Fernández-Bolaños JG. Tuning the activity of iminosugars: novel N-alkylated deoxynojirimycin derivatives as strong BuChE inhibitors. J Enzyme Inhib Med Chem. 2021;36(1):138–146.
- de Santana QLO, Santos Evangelista TC, Imhof P, Baptista Ferreira S, Fernández-Bolaños JG, Sydnes MO, Lopéz Ó, Lindbäck E. Tacrine-sugar mimetic conjugates as enhanced cholinesterase inhibitors. Org Biomol Chem. 2021;19(10):2322–2337.
- Santos Evangelista TC, López Ó, Puerta A, Fernandes MX, Baptista Ferreira S, Padrón JM, Fernández-Bolaños JG, Sydnes MO, Lindbäck E. A hybrid of 1-deoxynojirimycin and benzotriazole induces preferential inhibition of butyrylcholinesterase (BuChE) over acetylcholinesterase (AChE). J Enzyme Inhib Med Chem. 2022;37(1):2395–2402.
- Janockova J, Korabecny J, Plsikova J, Babkova K, Konkolova E, Kucerova D, Vargova J, Koval J, Jendzelovsky R, Fedorocko P, et al. In vitro investigating of anticancer activity of new 7-MEOTA-tacrine heterodimers. J Enzyme Inhib Med Chem. 2019;34(1):877–897.
- Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95.
- Moutayakine A, Marques C, López Ó, Bagetta D, Leitzbach L, Hagenow S, Carreiro EP, Stark H, Alcaro S, Fernández-Bolaños JG, et al. Evaluation of chromane derivatives: promising privileged scaffolds for lead discovery within Alzheimer’s disease. Bioorg Med Chem. 2022;68:116807.
- Lagunes I, Martín-Batista E, Silveira-Dorta G, Fernandes MX, Padrón JM. Differential mechanism of action of the CK1ε inhibitor GSD0054. J Mol Clin Med. 2018;1:77–84.
- Cheung J, Rudolph MJ, Burshteyn F, Cassidy MS, Gary EN, Love J, Franklin MC, Height JJ. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J Med Chem. 2012;55(22):10282–10286.
- Brazzolotto X, Wandhammer M, Ronco C, Trovaslet M, Jean L, Lockridge O, Renard PY, Nachon F. Human butyrylcholinesterase produced in insect cells: huprine-based affinity purification and crystal structure. FEBS J. 2012;279(16):2905–2916.
- Santos Evangelista TC, Lopéz Ó, Sydnes MO, Fernández-Bolaños JG, Baptista Ferreira S, Emil Lindbäck E. Bicyclic 1-azafagomine derivatives: synthesis and glycosidase inhibitory testing. Synthesis. 2019;51(21):4066–4077.
- Desvergnes S, Py S, Vallée Y. Total synthesis of (+)-hyacinthacine A2 based on SmI2-induced nitrone umpolung. J Org Chem. 2005;70(4):1459–1462.
- Carmona AT, Whigtman RH, Robina I, Vogel P. Synthesis and glycosidase inhibitory activity of 7-deoxycasuarine. HCA. 2003;86(9):3066–3073.
- Cardona F, Faggi E, Liguori F, Cacciarini M, Goti A. Total syntheses of hyacinthacine A2 and 7-deoxycasuarine by cycloaddition to a carbohydrate derived nitrone. Tetrahedron Lett. 2003;44(11):2315–2318.
- D'Adamio G, Matassini C, Parmeggiani C, Catarzi S, Morrone A, Goti A, Paoli P, Cardona F. Evidence for a multivalent effect in inhibition of sulfatases involved in lysosomal storage disorders (LSDs). RSC Adv. 2016;6(69):64847–64851.
- Breugst M, Reissig HU. The Huisgen reaction: milestones of the 1,3-dipolar cycloaddition. Angew Chem Int Ed Engl. 2020;59(30):12293–12307.
- Oukoloff K, Coquelle N, Bartolini M, Naldi M, Le Guevel R, Bach S, Josselin B, Ruchaud S, Catto M, Pisani L, et al. Design, biological evaluation and X-ray crystallography of nanomolar multifunctional ligands targeting simultaneously acetylcholinesterase and glycogen synthase kinase-3. Eur J Med Chem. 2019;168:58–77.
- Cornish-Bowden A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem J. 1974;137(1):143–144.
- Rosenberry TL, Brazzolotto X, Macdonald IR, Wandhammer M, Trovaslet-Leroy M, Darvesh S, Nachon F. Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: a crystallographic, kinetic and calorimetric study. Molecules. 2017;22(12):2098.
- Radić Z, Pickering NA, Vellom DC, Camp S, Taylor P. Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry. 1993;32(45):12074–12084.
- Barak D, Kronman C, Ordentlich A, Ariel N, Bromberg A, Marcus D, Lazar A, Velan B, Shafferman A. Acetylcholinesterase peripheral anionic site degeneracy conferred by amino acid arrays sharing a common core. J Biol Chem. 1994;269(9):6296–6305.
- Harel M, Schalk I, Ehret-Sabatier L, Bouet F, Goeldner M, Hirth C, Axelsen PH, Silman I, Sussman JL. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc Natl Acad Sci U S A. 1993;90(19):9031–9035.
- Holdgate G, Meek T, Grimley R. Mechanistic enzymology in drug discovery: a fresh perspective. Nat Rev Drug Discov. 2018;17(2):115–132.