
Abstract
New quinoline-pyridine hybrids were designed and synthesised as PIM-1/2 kinase inhibitors. Compounds 5b, 5c, 6e, 13a, 13c, and 14a showed in-vitro low cytotoxicity against normal human lung fibroblast Wi-38 cell line and potent in-vitro anticancer activity against myeloid leukaemia (NFS-60), liver (HepG-2), prostate (PC-3), and colon (Caco-2) cancer cell lines. In addition, 6e, 13a, and 13c significantly induced apoptosis with percentage more than 66%. Moreover, 6e, 13a, and 13c significantly induced caspase 3/7 activation in HepG-2 cell line. Furthermore, 5c, 6e, and 14a showed potent in-vitro PIM-1 kinase inhibitory activity. While, 5b showed potent in-vitro PIM-2 kinase inhibitory activity. Kinetic studies using Lineweaver–Burk double-reciprocal plot indicated that 5b, 5c, 6e, and 14a behaved as competitive inhibitors while 13a behaved as both competitive and non-competitive inhibitor of PIM-1 kinase enzyme. Molecular docking studies indicated that, in-silico affinity came in coherence with the observed in-vitro inhibitory activities against PIM-1/2 kinases.
Introduction
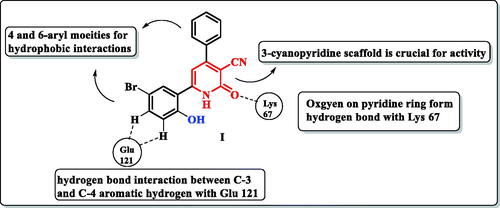
Human Proviral Integration Moloney (PIM) kinases are a family of serine/threonine kinases includes PIM-1, PIM-2, and PIM-3 isoforms. The PIM-1 kinase has direct role in tumorigenesis in various haematological malignanciesCitation1, such as leukaemia and lymphomaCitation2–5. Also, PIM-1 kinase is overexpressed in several solid tumours such as prostateCitation6, colonCitation7, hepaticCitation8, and breast cancersCitation9. In addition, it is involved in many biological processes such as cell cycle, cell proliferation, apoptosis, and drug resistanceCitation10. PIM-1 kinase has unusual adenosine triphosphate (ATP) binding pocket having proline 123 in the hinge region which is specific to PIM-1 kinase over other serine/threonine kinasesCitation11. Therefore, ATP could not bind to the hinge region by a second hydrogen bond as other protein kinasesCitation12,Citation13. In addition, most PIM-1 kinase inhibitors are ATP-competitive inhibitorsCitation14. PIM-1 kinase competitive inhibitors are classified into non-ATP mimetics and ATP-mimetics. ATP-mimetics bind directly to the hinge region via Glu121 while non-ATP mimetics bind to Lys67Citation15,Citation16. PIM-1 kinase inhibition is an attractive target to overcome PIM-1 kinase induced chemotherapy resistance caused by induction of hypoxia in tumour cellsCitation17. X-ray crystallographic study of the lead scaffold I showed on interaction to ATP-binding site of PIM-1 kinase through prominent Hydrogen bond interaction of the carbonyl group on the pyridone ring with the Lys67. Another weak H-bond was observed with the hinge region between the main chain carbonyl of Glu121 and an aromatic hydrogen (C-3 position and C-4-position) on 6-phenyl moiety. Besides, the hydrophobic interactions with aryl moieties at 4 and 6 positionsCitation18 ().
Figure 1. A schematic representation of pyridone derivative I complexed with PIM-1 kinase. Dashed lines indicated hydrogen bonds.
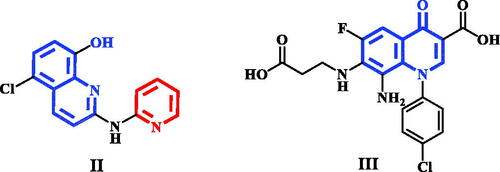
Pyridine-quinoline hybrid structure (compound II) showed potent inhibition of PIM-1 kinase, increase anticancer activity against human prostatic PC-3 cancer cell line as well as induction apoptosisCitation19. In addition, structure based drug design of PIM-1 kinase inhibitors revealed that electron deficient quinolone core, as in compound III, was able to form strong interactions with the hinge region of PIM-1 kinaseCitation20 ().
Figure 2. Quinoline and quinolone having PIM-1 kinase inhibitory activity.
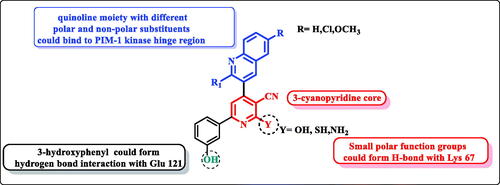
In continuation of our previously published workCitation21, the structure-based design strategy was rationalised via tethering the pyridine moiety with the quinoline moiety with keeping the common respective pharmacophoric features of the lead scaffold I () to improve the PIM kinase inhibitory activity. It must be pointed out that, the six-position of pyridine ring was selected to be substituted with the bioactive pharmacophore 3-hydroxyphenyl group which was reported to enhance the anticancer activity through hydrogen bond interactions. In addition, the designed scaffold was modified at numerous positions, where the pyridine ring was substituted with either oxygen or its isostere sulphur or its isostere amino group at position 2. Moreover, pyridine was also hybridised with electron deficient quinolone or quinoline substituted at position 2 with either piperidine or morpholine and substituted at six-position with either hydrophilic or hydrophobic groups to impart variety of enzyme interactions hoping to enhance binding affinity hence potency ().
Figure 3. Design of pyridine-quinoline hybrids.
Experimental
Chemistry
General
All chemicals and solvents were purchased from commercial suppliers. Melting points were determined in open-glass capillaries using a Griffin melting point apparatus and were uncorrected. The progress of the reactions was monitored by thin-layer chromatography (TLC) on commercially available precoated silica gel aluminium-backed plates and the spots were visualised by exposure to iodine vapours or UV-lamp at λ 254 nm for few seconds. Infra-red spectra (IR) were recorded, for KBr discs, on a PerkinElmer RXIFT-IR. Nuclear magnetic resonance spectra, 1H-NMR and 13C-NMR, were recorded on Bruker spectrometer (400 MHz) using DMSO-d6 as solvent using deuterated dimethyl sulfoxide as solvents. The data were reported as chemical shifts or δ values (ppm) relative to tetramethyl silane (TMS) as internal standard. Signals were indicated by the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartette and m = multiplet, dd = doublet of doublet and br. = broad. Electron impact mass spectra (EIMS) were run on a gas chromatograph/mass spectrophotometer Shimadzu GCMS/QP-2010 plus (70 eV). Relative intensity % corresponding to the most characteristic fragments is recorded. Elemental microanalyses were performed at the microanalytical unit, the Regional Centre of Microbiology and Biotechnology, Al-Azhar University, Egypt.
Compounds 2a–c, 3a–c, and 4a–c were prepared as reportedCitation22–24.
General experimental procedure for the synthesis of 4-(6-substituted-2-morpholino or (piperidin-1-yl)quinolin-3-yl)-6-(3-hydroxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile; (5a–f)
A mixture of 3-hydroxyacetophenone (1 mmol), substituted aldehyde 4a–f (1 mmol), ethyl cyanoacetate (1 mmol), and ammonium acetate (8 mmol) in 5 ml absolute ethanol was heated under reflux for 4 h during which the product was separated out. Then, the reaction mixture was cooled, and the yellow precipitate was filtered, washed with cold ethanol, air dried, and recrystallized from ethanol/DMF (9:1) mixture to give pure oxo-cyanopyridine derivatives.
6-(3-Hydroxyphenyl)-4-(morpholin-4-yl-quinolin-3-yl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5a)
Yield, 40%; m.p > 300 °C; IR (KBr, v cm−1): 3400 (OH), 3218 (NH), 2200 (C≡N), 1649 (C=O), 1596 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.20–3.30 (m, 4H, morpholine-C2,6-H); 3.63 (t, J = 4,4.8 Hz, 4H, morpholine-C3,5-H); 6.97–6.99 (m, 2H, phenyl-C4-H and pyridine-C5-H); 7.25 (s, 1H, phenyl-C2-H); 7.31–7.37 (m, 2H, phenyl-C5,6-H); 7.45 (t, J = 7.2,7.6 Hz, 1H, quinoline- C7-H); 7.73 (t, J = 7.2, 8 Hz, 1H, quinoloine-C6-H); 7.80 (d, J = 8.4 Hz, 1H, quinoline-C5-H); 7.91 (d, J = 8 Hz, 1H, quinoline-C8-H); 8.38 (s, 1H, quinoline-C4-H); 9.87 (s, 1H, OH, D2O-exchangeable); 12.70 (brs, 1H, NH, D2O-exchangeable). Elemental analysis Calcd for C25H20N4O3 (424.46): C 70.74; H 4.75; N 13.20. Found: C 71.02, H 4.89, N 13.43
4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-6-(3-hydroxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5b)
Yield, 40%; m.p > 300 °C; IR (KBr, v cm−1): 3400 (OH), 3233 (NH), 2220 (C≡N), 1649 (C=O), 1592 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.20–3.30 (m, 4H, morpholine-C2,6-H); 3.60–3.70 (m, 4H, morpholine-C3,5-H); 6.90 (brs, 1H, pyridine-C5-H); 6.98 (d, J = 6.8 Hz, 1H, phenyl-C4-H); 7.23 (s, 1H, phenyl-C2-H); 7.31–7.37 (m, 2H, phenyl-C5,6-H); 7.72 (d, J = 8.8 Hz, 1H, quinoline-C7-H); 7.80 (d, J = 8.8 Hz, 1H, quinoline-C8-H); 8.02 (s, 1H, quinoline-C5-H); 8.37 (s, 1H, quinoline-C4-H); 9.91 (s, 1H, OH, D2O-exchangeable); 12.83 (brs, 1H, NH, D2O-exchangeable). EI-MS m/z (% relative abundance): 460 [M+•+2] (66.47); 458 [M+•] (100); 400(59.39); 402 (53.11). Elemental analysis Calcd for C25H19ClN4O3 (458.90): C 65.43; H 4.17; N 12.21. Found: C 65.27, H 4.28, N 12.47.
6-(3-Hydroxyphenyl)-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5c)
Yield, 40%; m.p > 300 °C; IR (KBr, v cm−1): 3400 (OH), 3237 (NH), 2219 (C≡N), 1647 (C=O), 1596 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.15–3.22 (m, 4H, morpholine-C2,6-H); 3.57–3.62 (m, 4H, morpholine-C3,5-H); 3.87 (s, 3H, OCH3); 6.93 (brs, 1H, pyridine-C5-H); 6.98 (d, J = 6.8 Hz, 1H, phenyl-C4-H); 7.25 (s, 1H, phenyl-C2-H); 7.31 (d, J = 8 Hz, 1H, quinoline-C7-H); 7.34 (s, 1H, quinoline-C5-H); 7.37 (dist. t, J = 8 Hz, phenyl-C5-H); 7.40 (d, J = 4 Hz, 1H, phenyl-C6-H); 7.74 (d, J = 8.8 Hz, 1H, quinoline-C8-H); 8.29 (s, 1H, quinoline-C4-H); 9.86 (s, 1H, OH, D2O-exchangeable); 12.71 (brs, 1H, NH, D2O-exchangeable). 13C-NMR (100 MHz, DMSO-d6δ ppm): 49.95 (morpoline-C2,6); 55.97 (OCH3); 66.41 (morpholine-C3,5); 104.96 (pyridine-C5); 107.02 (quinoline-C5); 114.73 (phenyl-C2); 116.60 (CN); 118.75 (phenyl-C4); 123.13 (phenyl-C6); 123.30 (pyridine-C3); 124.00 (quinoline-C3); 125.26 (quinoline-C7); 128.87 (quinoline-C4a); 130.64 (quinoline-C8); 139.06 (phenyl-C5); 139.13 (quinoline-C4); 141.64 (phenyl-C1); 142.98 (quinoline-C8a); 152.64 (quinoline-C6); 155.92 (phenyl-C3); 156.53 (C=O); 158.22 (pyridine-C6); 162.92 (pyridine-C4); 163.22 (quinoline-C2);. Elemental analysis Calcd for C26H22N4O4 (454.49): C 68.71; H 4.88; N 12.33. Found: C 68.94, H 5.12, N 12.59.
6-(3-Hydroxyphenyl)-2-oxo-4-(2-(piperidin-1-yl)quinoline-3-yl)-1,2-dihydropyridine-3-carbonitrile (5d)
Yield, 40%; m.p 244–246 °C; IR (KBr, v cm−1): 3519 (OH), 3400 (NH), 2225 (C≡N), 1629 (C=O), 1595 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 1.53 (s, 6H, piperidine-C3,4,5-H); 3.23 (s, 4H, piperidine-C2,4-H); 6.94 (brs, 1H, pyridine-C5-H); 6.98 (d, J = 6.8 Hz, 1H, phenyl-C4-H); 7.25 (s, 1H, phenyl-C2-H); 7.31 (d, J = 7.6 Hz, 1H, phenyl-C6-H); 7.35 (dist. t, J = 7.6, 8 Hz, 1H, phenyl-C5-H); 7.41 (t, J = 7.2,7.6 Hz, 1H, quinoline-C6-H) 7.69 (t, J = 7.2, 8 Hz, 1H, quinoline-C7-H); 7.76 (d, J = 8.4 Hz, 1H, quinoline-C8-H); 7.87 (d, J = 8 Hz, 1H, quinoline-C5-H); 8.31 (s, 1H, quinoline-C4-H); 9.87 (s, 1H, OH, D2O-exchangeable); 12.76 (brs, 1H, NH, D2O-exchangeable). 13C-NMR (100 MHz, DMSO-d6δ ppm): 24.49 (piperdine-C4); 25.54 (piperdine-C3,5); 50.37 (piperdine-C2,6); 114.74 (pyridine-C5); 116.52 (phenyl-C2); 116.55 (CN); 116.57 (phenyl-C4); 118.75 (phenyl-C6); 123.86 (quinoline-C3); 124.09 (pyridine-C3); 124.10 (quinoline-C4a); 124.64 (quinoline-C6); 127.17 (quinoline-C8); 128.65 (quinoline-C5); 130.69 (phenyl-C5); 131.32 (quinoline-C7); 139.87 (quinoline-C4); 147.47 (phenyl-C1); 147.48 (quinoline-C8a); 156.97 (phenyl-C3); 158.20 (C=O); 158.23 (pyridine-C4); 158.24 (pyridine-C6); 158.27 (quinoline-C2). Elemental analysis Calcd for C26H22N4O2 (422.49): C 73.92; H 5.25; N 13.26. Found: C 74.19, H 5.39, N 13.53.
4-(6-Chloro-2-(piperidin-1-yl)quinoline-3-yl)-6-(3-hydroxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5e)
Yield, 40%; m.p > 300 °C; IR (KBr, v cm−1): 3454 (OH), 3210 (NH), 2224 (C≡N), 1634 (C=O), 1594 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 1.50 (s, 6H, piperidine-C3,4,5-H); 3.23 (s, 4H, piperidine-C2,4-H); 6.89 (s, 1H, pyridine-C5-H); 6.91 (d, J = 4.8 Hz, 1H, phenyl-C4-H); 7.26–7.33 (m, 3H, phenyl-C2,5,6-H); 7.65 (d, J = 8.4 Hz, 1H, quinoline- C8-H); 7.72 (d, J = 8.8 Hz, 1H, quinoline-C7-H); 7.95 (s, 1H, quinoline-C5-H); 8.19 (s, 1H, quinoline-C4-H); EI-MS m/z (% relative abundance): 458 [M+•+2] (24.70); 457 [M+•+1] (35.23); 456 [M+•] (100); 430 (70.40); 388 (54.53). Elemental analysis Calcd for C26H21ClN4O2 (456.93): C 68.34; H 4.63; N 12.26. Found: C 68.60, H 4.80, N 12.49.
6-(3-Hydroxyphenyl)-4-(6-methoxy-2-(piperidin-1-yl)quinolin-3-yl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5f)
Yield, 40%; m.p 265–267 °C; IR (KBr, v cm−1): 3500 (OH), 3400 (NH), 2220 (C≡N), 1659 (C=O), 1598 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 1.51 (s, 6H, piperidine-C3,4,5-H); 3.15 (s, 4H, piperidine-C2,4-H); 3.86 (s, 3H, OCH3); 6.92 (brs, 1H, pyridine-C5-H); 6.98 (d, J = 7.2 Hz, 1H, phenyl-C4-H); 7.24 (s, 1H, phenyl-C2-H); 7.30–7.34 (m, 3H, quinoline-C5,7-H and phenyl-C5-H); 7.37 (d, J = 4 Hz, 1H, phenyl-C6-H); 7.70 (d, J = 9.2 Hz, 1H, quinoline-C8-H); 8.23 (s, 1H, quinoline-C4-H); 9.85 (s, 1H, OH, D2O-exchangeable); 12.73 (brs, 1H, NH, D2O-exchangeable). Elemental analysis Calcd for C27H24N4O3 (452.51): C 71.67; H 5.35; N 12.38. Found: C 71.90, H 5.26, N 12.56.
4-(6-Substituted-2-morpholino or (piperidin-1-yl) quinolin-3-yl)-6-(3-hydroxyphenyl)-2-mercaptonicotinonitrile; (6a–e)
A mixture of 3-hydroxyacetophenone (1 mmol), substituted aldehyde 4a–f (1 mmol), cyanothioacetamide (1 mmol) and ammonium acetate (8 mmol) was refluxed in ethanol (10 ml) with stirring for 3 h during which the product was obtained. The reaction mixture was cooled, and the yellow precipitate was filtered, washed with cold ethanol, air dried and recrystallized from ethanol/DMF (1:9) mixture.
6-(3-Hydroxyphenyl)-4-(2-morpholin-4-yl-quinolin-3-yl)-2-mercaptopyridine-3-carbonitrile (6a)
Yield, 40%; m.p 240–242 °C; IR (KBr, v cm−1): 3389 (OH), 2590 (SH), 2221 (C≡N), 1599 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.05–3.20 (m, 4H, morpholine-C2,6-H); 3.42–3.50 (m, 4H, morpholine-C3,5-H); 6.89 (dd, J = 2,8 Hz, 1H, phenyl-C4-H); 7.19 (t, J = 8,7.6 Hz, 1H, phenyl-C5-H); 7.47 (s, 1H, phenyl-C2-H); 7.51 (t, J = 7.6 Hz 1H, quinoline-C6-H); 7.59 (d, J = 8 Hz, 1H, quinoline-C5-H); 7.77 (t, J = 7.6,8 Hz, 1H, quinoline-C7-H); 7.86 (d, J = 8.4 Hz, 1H, phenyl-C6-H); 7.96 (d, J = 7.6 Hz, 1H, quinoline-C8-H); 8.28 (s, 1H, pyridine-C5-H); 8.47 (s, 1H, quinoline-C4-H); 9.68 (s, 1H, OH, D2O-exchangeable); 14.15 (s, 1H, SH, D2O-exchangeable). Elemental analysis Calcd for C25H20N4O2S (440.52): C 68.16; H, 4.58; N, 12.72. Found: C 68.42, H 4.75, N 12.94.
4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-6-(3-hydroxyphenyl)-2-mercaptopyridine-3-carbonitrile (6b)
Yield, 30%; m.p 260–261 °C; IR (KBr, v cm−1): 3451 (OH), 2595 (SH), 2202 (C≡N), 1633 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.60–3.65 (m, 4H, morpholine-C2,6-H, under DMSO); 3.75–3.78 (m, 4H, morpholine-C3,5-H, under DMSO); 6.96 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.35 (t, J = 4,8 Hz, 1H, phenyl-C5-H); 7.60–7.81 (m, 5H, quinoline-C5,7,8-H and phenyl-C2,6-H); 8.42 (s, 1H, pyridine-C5-H); 9.49 (s, 1H, quinoline-C4-H); 9.75 (s, 1H, OH, D2O-exchangeable); 14.56 (s, 1H, SH, D2O-exchangeable). MS m/z (% relative abundance): 476 [M+•+2] (6.71); 474 [M+•] (23.44); 425 (72.98); 334 (100); 190 (84.18); 161 (82.30). Elemental analysis Calcd for C25H19ClN4O2S (474.96): C 63.22, H 4.03, N 11.80. Found: C 63.45, H 4.11, N 11.94
6-(3-Hydroxyphenyl)-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)-2-mercaptopyridine-3-carbonitrile (6c)
Yield, 40%; m.p > 300 °C; IR (KBr, v cm−1): 3392 (OH), 2600 (SH), 2230 (C≡N), 1602 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 2.90–3.10 (m, 4H, morpholine-C2,6-H); 3.99–3.45 (m, 4H, morpholine-C3,5-H); 3.90 (s, 3H, OCH3); 6.89 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.19 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.37 (s, 1H, phenyl-C2-H); 7.42–7.47 (m, 2H, quinoline-C5,7-H); 7.59 (d, J = 8 Hz, 1H, quinoline-C8-H); 7.80 (d, J = 8 Hz, 1H, phenyl-C6-H); 8.27 (s, 1H, pyridine-C5-H); 8.37 (s, 1H, quinoline-C4-H); 9.67 (s, 1H, OH, D2O-exchangeable). 13C-NMR (100 MHz, DMSO-d6δ ppm): 49.99 (morpoline-C2,5); 56.01 (OCH3); 66.19 (morpholine-C3.5); 104.62 (pyridine-C5); 107.08 (quinoline-C5); 114.52 (pyridine-C3); 115.57 (CN); 118.53 (phenyl-C2); 118.59 (phenyl-C4); 119.00 (phenyl-C6); 123.40 (quinoline-C3); 123.66 (quinoline-C7); 125.73 (quinoline-C4a); 129.09 (quinoline-C8); 130.44 (phenyl-C5); 137.93 (quinoline-C4); 139.95 (phenyl-C1); 143.07 (quinoline-C8a); 153.49 (quinoline-C6); 156.49 (phenyl-C3); 156.76 (C=S); 158.41 (pyridine-C4); 158.73 (quinoline-C2); 159.12 (pyridine-C6). Elemental analysis Calcd for C26H22N4O3S (470.55): C 66.37, H 4.71, N 11.91. Found: C 66.59, H 4.83, N 11.75.
6-(3-Hydroxyphenyl)-4-(2-(piperidin-1-yl)quinolin-3-yl)-2-mercaptopyridine-3-carbonitrile (6d)
Yield, 40%; m.p > 300 °C; IR (KBr, v cm−1): 3400 (OH), 2600 (SH), 2220 (C≡N), 1610 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 1.10–1.50 (m, 6H, piperidine-C3,4,5-H); 3.07 (s, 4H, piperidine-C2,6-H); 6.88 (dd, J = 2, 8 Hz, 1H, phenyl-C4-H); 7.17 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.46–7.50 (m, 2H, quinoline-C6-H and phenyl-C2-H); 7.60 (d, J = 7.6 Hz, 1H, quinoline-C5-H); 7.74 (t, J = 7.6 Hz, 1H, quinoline-C7-H); 7.82 (d, J = 8.4 Hz, 1H, phenyl-C6-H); 7.92 (d, 1H, quinoline-C8-H); 8.24 (s, 1H, pyridine-C5-H); 8.40 (s, 1H, quinoline-C4-H); 9.65 (s, 1H, OH, D2O-exchangeable). Elemental analysis Calcd for C26H22N4OS (438.55): C 71.21, H 5.06, N 12.78. Found: C 71.47, H 5.82, N 13.02.
6-(3-Hydroxyphenyl)-4-(6-methoxy-2-(piperidin-1-yl)quinolin-3-yl)-2-mercaptopyridine-3-carbonitrile (6e)
Yield, 40%; m.p > 300 °C; IR (KBr, v cm−1): 3403 (OH), 2214 (C≡N), 1602 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 1.15–1.45 (m, 6H, piperidine-C3,4,5-H); 2.90–3.15 (m, 4H, piperidine-C2,6-H); 3.89 (s, 3H, OCH3); 6.88 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.17 (t, J = 8,8 Hz, 1H, phenyl-C5-H); 7.34 (s, 1H, quinoline-C5-H); 7.40 (d, J = 8 Hz, 1H, quinoline-C7-H); 7.51 (s, 1H, phenyl-C2-H); 7.61 (d, J = 8 Hz, 1H, quinoline-C8-H); 7.76 (d, J = 8 Hz, 1H, phenyl-C6-H); 8.24 (s, 1H, pyridine-C5-H); 8.31 (s, 1H, quinoline-C4-H); 9.68 (s, 1H, OH, D2O-exchangeable). 13C-NMR (100 MHz, DMSO-d6δ ppm): 24.36 (piperdine-C4); 25.46 (piperdine-C3,5); 50.69 (piperdine-C2,6); 55.98 (OCH3); 104.68 (pyridine-C5); 107.07 (quinoline-C5); 114.55 (pyridine-C3); 115.54 (CN); 116.66 (phenyl-C2); 118.55 (phenyl-C4); 118.93 (phenyl-C6); 123.14 (quinoline-C3); 124.05 (quinoline-C7); 125.47 (quinoline-C4a); 128.98 (quinoline-C8); 130.40 (phenyl-C5); 137.93 (quinoline-C4); 139.60 (phenyl-C1); 143.20 (quinoline-C8a); 153.92 (quinoline-C6); 156.49 (phenyl-C3); 157.49 (C=S); 158.44 (pyridine-C4); 158.55 (quinoline-C2); 159.14 (pyridine-C6). EI-MS m/z (% relative abundance): 468 [M+•] (22.75); 291 (73.83); 275 (100), 124 (79.94). Elemental analysis Calcd for C27H24N4O2S (468.58): C 69.21, H 5.16, N 11.96. Found: C 69.43, H 5.34, N 12.13.
General procedure for synthesis of 3-(5-Cyano-4-(2-morpholin-4-yl-6-subsitituedquinolin-3-yl)-6-oxo-1,6-dihydropyridin-2-yl)phenyl acetate; (7a–c)
To a suspension of pyridone derivatives 5a–c (1 mmol) in acetic acid (10 ml), acetic anhydride (0.141 ml, 1.5 mmol) was added. The reaction mixture was refluxed for 3–5 h, then was quenched with ice cold water. The precipitate formed was collected by filtration, air dried and crystallised from ethanol to afford the desired compounds.
3-(5-Cyano-4-(2-morpholin-4-yl-quinolin-3-yl)-6-oxo-1,6-dihydropyridin-2-yl)phenyl acetate (7a)
Yield, 93%; m.p > 300 °C; IR (KBr, v cm−1): 3400 (NH), 2220 (C≡N), 1762 (COCH3), 1643 (C=O), 1615 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 2.31 (s, 3H, CH3); 3.20–3.25 (m, 4H, morpholine-C2,6-H); 3.58–3.63 (m, 4H, morpholine-C3,5-H); 7.10 (brs, 1H, pyridine-C5-H); 7.35 (d, J = 8, 1H, phenyl-C4-H); 7.47 (t, J = 4,7.6 Hz, 1H, quinoline-C6-H); 7.60 (t, J = 4,7.6 Hz, 1H, quinoline-C7-H); 7.74 (t, J = 4,7.6 Hz,1H, phenyl-C5-H); 7.79–7.81 (m, 3H, phenyl-C2,6-H and quinoline-C8-H); 7.91 (d, J = 7.6 Hz, 1H, quinoline-C5-H); 8.39 (s, 1H, quinoline-C4-H); 12.87 (s, 1H, NH, D2O-exchangeable). Elemental analysis Calcd for C27H22N4O4 (466.50): C 69.52, H 4.75, N 12.01. Found: C 69.30, H 4.94, N 12.29.
3-(4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-5-cyano-6-oxo-1,6-dihydropyridin-2-yl)phenyl acetate (7b)
Yield, 92%; m.p 189–190 °C; IR (KBr, v cm−1): 3448 (NH), 2221(C≡N), 1763 (COCH3), 1644 (C=O), 1600 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 2.30 (s, 3H, CH3); 3.20–3.31 (m, 4H, morpholine-C2,6-H); 3.55–3.70 (m, 4H, morpholine-C3,5-H); 7.14 (brs, 1H, pyridine-C5-H); 7.36 (d, J = 7.6 Hz, 1H, phenyl-C4-H); 7.61 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.72 (dd, J = 2,8.8 Hz, 1H, quinoline-C7-H); 7.75–7.82 (m, 2H, phenyl-C2,6-H); 7.86 (d, J = 8 Hz, 1H, quinoline-C8-H); 8.02 (d, J = 2 Hz, 1H, quinoline-C5-H); 8.37 (s, 1H, quinoline-C4-H); 12.88 (s, 1H, NH, D2O-exchangeable). Elemental analysis Calcd for C27H21ClN4O4 (500.94): C 64.74, H 4.23, N 11.18. Found: C 64.58, H 4.59, N 11.34.
3-(5-Cyano-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)-6-oxo-1,6-dihydropyridin-2-yl)phenyl acetate (7c)
Yield, 91%; m.p > 300 °C; IR (KBr, v cm−1): 3450 (NH), 2218 (C≡N), 1760 (COCH3), 1646 (C=O), 1599 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 2.30 (s, 3H, CH3); 3.10–3.20 (m, 4H, morpholine-C2,6-H); 3.57–3.64 (m, 4H, morpholine-C3,5-H); 3.87 (s, 3H, OCH3); 7.04 (brs, 1H, pyridine-C5-H); 7.35–7.36 (m, 2H, quinoline-C5-H and phenyl-C4-H); 7.39 (dd, J = 2.8,9 Hz, 1H, quinoline-C7-H); 7.60 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.73–7.83 (m, 3H, phenyl-C2,6-H and quinoline-C8-H); 8.30 (s, 1H, quinoline-C4-H); 12.88 (s, 1H, NH, D2O-exchangeable). 13C-NMR (100 MHz, DMSO-d6δ ppm): 21.29, 49.93, 55.96, 66.38, 100.00, 106.98, 115.45, 115.93, 121.22, 122.43, 123.32, 125.21, 127.50, 128.35, 128.89, 130.67, 135.83, 139.57, 142.98, 145.00, 151.32, 152.51, 156.52, 163.97, 169.65, 174.50. Elemental analysis Calcd for C28H24N4O5 (496.52): C 67.73, H 4.87, N 11.28. Found: C 67.54, H 5.01, N 11.49.
General procedure for synthesis of ethyl 2-((6-(3-acetoxyphenyl)-4-(2-morpholin-4-yl-6-substituted quinolin-3-yl)-3-cyanopyridin-2-yl)oxy)acetate (8a–c)
A mixture of phenylacetate derivatives 7a–c (1 mmol), ethylbromoacetate (0.110 ml, 1 mmol) and anhydrous potassium carbonate (0.207 g, 1.5 mmol) in dry acetone (10 ml) was heated under reflux for 4 h then the reaction mixture was cooled and triturated with water till complete precipitation of the product. The green precipitate was filtered, washed with water, air dried and crystallised from ethanol.
Ethyl 2-((6-(3-acetoxyphenyl)-3-cyano-4-(2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)acetate (8a)
Yield, 92%; m.p 129–130 °C; IR (KBr, v cm−1): 2224 (C≡N), 1760, 1750 (C=O), 1592 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 1.21 (t, J = 7 Hz, 3H, CH3); 2.32 (s, 3H, COCH3); 3.10–3.19 (m, 4H, morpholine-C2,6-H); 3.55–3.62 (m, 4H, morpholine-C3,5-H); 4.20 (q, J = 7 Hz, 2H, OCH2); 5.22 (s, 2H, CH2); 7.34 (dd, J = 1.2,8 Hz, 1H, phenyl-C4-H); 7.49 (t, J = 7.1,7.8 Hz, 1H, quinoline-C6-H); 7.61 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.76 (t, J = 7.1,8.4 Hz, 1H, quinoline-C7-H); 7.85 (d, J = 8.4 Hz, 1H, quinoline-C8-H); 7.93–7.95 (m, 2H, quinoline-C5-H and pyridine-C5-H); 8.13 (d, J = 8 Hz, 1H, phenyl-C6-H); 8.20 (s, 1H, quinoline-C4-H); 8.45 (s, 1H, phenyl-C2-H) .
Ethyl 2-((6-(3-acetoxyphenyl)-4-(6-chloro-2-morpholin-4-yl-quinolin-3-yl)-3-cyanopyridin-2-yl)oxy)acetate (8b)
Yield, 89%; m.p 228–230 °C; IR (KBr, v cm−1): 2224 (C≡N), 1756 (C=O); 1593 (C=N).
Ethyl 2-((6-(3-acetoxyphenyl)-3-cyano-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)acetate (8c)
Yield, 90%; m.p 204–206 °C; IR (KBr, v cm−1): 2224 (C≡N), 1780, 1758 (C=O), 1595 (C=N).
General procedure for synthesis of 2-((3-Cyano-6-(3-hydroxyphenyl)-4-(2-morpholin-4-yl-6-substituted quinolin-3-yl)pyridin-2-yl)oxy)acetic acid (9a–c)
A mixture of compounds 8a–c (1 mmol), and sodium bicarbonate (0.756 g, 9 mmol) in methanol\water (1:1) was refluxed for 1 h. The reaction mixture was then cooled, triturated with water and neutralised with acetic acid (PH = 7). The precipitate of the product was collected, filtered, washed with water, air dried and crystallised from ethanol.
2-((3-Cyano-6-(3-hydroxyphenyl)-4-(2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)acetic acid (9a)
Yield, 90%; m.p 179–180 °C; IR (KBr, v cm−1): 3403 (OH), 3350–3050 (COOH), 2224 (C≡N), 1732 (C=O), 1643 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.09–3.19 (m, 4H, morpholine-C2,6-H); 3.55–3.62 (m, 4H, morpholine-C3,5-H); 5.15 (s, 2H, CH2); 6.95 (d, J = 7.6 Hz, 1H, phenyl-C4-H); 7.34 (t, J = 7.6,8 Hz, 1H, phenyl-C5-H); 7.48 (t, J = 7.2,7.6 Hz, 1H, quinoline-C6-H); 7.60 (s, 1H, phenyl-C2-H) ; 7.65 (d, J = 7.6 Hz, 1H, quinoline-C8-H); 7.75 (t, J = 7.2, 7.6 Hz, 1H, quinoline-C7-H); 7.84 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.93 (d, J = 7.6 Hz, 1H, quinoline-C5-H); 8.02 (s, 1H, pyridine-C5-H); 8.43 (s, 1H, quinoline-C4-H); 9.77, 13.22 (2brs, each for1H, 2OH, D2O-exchangeable). 13C-NMR (100 MHz, DMSO-d6δ ppm): 49.69, 63.77, 66.15, 93.33, 114.52, 114.78, 115.30, 118.44, 118.67, 123.70, 124.71, 125.21, 127.42, 128.71, 130.47, 131.43, 138.06, 140.78, 147.29, 155.38, 157.79, 158.08, 158.37, 163.38, 170.09. Elemental analysis Calcd for C27H22N4O5 (482.50): C 67.21, H 4.60, N 11.61. Found: C 67.49, H 4.83, N 11.88.
2-((4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-3-cyano-6-(3-hydroxyphenyl)pyridin-2-yl)oxy)acetic acid (9b)
Yield, 89%; m.p 279–280 °C; IR (KBr, v cm−1): 3448 (OH), 3600–3000 (COOH), 2225 (C≡N), 1626 (C=O), 1597 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.02–3.30 (m, 4H, morpholine-C2,6-H); 3.50–3.70 (4H, under DMSO, morpholine-C3,5-H); 4.96 (s, 2H, CH2); 6.89 (d, J = 7.6 Hz, 1H, phenyl-C4-H); 7.26 (t, J = 7.6,8 Hz, 1H, phenyl-C5-H); 7.58–7.60 (m, 2H, quinoline-C5-H and phenyl-C6-H); 7.73 (d, J = 9 Hz, 1H, quinoline-C7-H) ; 7.82 (d, J = 9 Hz, 1H, quinoline-C8-H); 7.88 (s, 1H, phenyl-C2-H); 8.03 (s, 1H, pyridine-C5-H); 8.32 (s, 1H, quinoline-C4-H); 10.09 (brs, 1H, OH, D2O-exchangeable). EI-MS m/z (% relative abundance): 518 [M+•+2] (21.67); 516 [M+•] (32.78); 413 (100); 391 (86.16). Elemental analysis Calcd for C27H21ClN4O5 (516.94): C 62.73, H 4.09, N 10.84. Found: C 62.96, H 4.13, N 11.06.
2-((3-Cyano-6-(3-hydroxyphenyl)-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)acetic acid (9c)
Yield, 92%; m.p 274–275 °C; IR (KBr, v cm−1): 3422 (OH), 3600–3050 (COOH), 2224 (C≡N), 1598 (C=O), 1549 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.02–3.12 (m, 4H, morpholine-C2,6-H); 3.55–3.60 (m, 4H, morpholine-C3,5-H); 3.87 (s, 3H, OCH3); 4.89 (s, 2H, -CH2); 6.92 (d, J = 8.4 Hz, 1H, phenyl-C4-H); 7.31 (t, J = 8,8.4 Hz, 1H, phenyl-C5-H); 7.38–7.41 (m, 2H, quinoline-C5,7-H); 7.60–7.67 (m, 2H, phenyl-C2-H and quinoline-C8-H); 7.77 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.90 (s, 1H, pyridine-C5-H); 8.30 (s, 1H, quinoline-C4-H); 9.84, 13.10 (2brs, each for 1H, 2OH, D2O-exchangeable). Elemental analysis Calcd for C28H24N4O6 (512.52): C 65.62, H 4.72, N 10.93. Found: C 65.90, H 4.89, N 11.24.
General procedure for synthesis of 3-(5-cyano-4-(2-morpholin-4-yl-6-substitiutedquinolin-3-yl)-6-(2-((4-substitutedphenyl)amino)-2-oxoethoxy)pyridin-2-yl)phenyl acetate; (10a–i)
To a mixture of compounds 7a–c (1 mmol), anhydrous potassium carbonate (0.207 g 1.5 mmol) in dry acetone (10 ml) the appropriate chloroacetanilide derivatives (1 mmol) were added and the reaction mixture was heated under reflux for 4 h. The reaction mixture was then cooled and treated with ice-cold water. The obtained product was collected by filtration and crystallised from ethanol.
3-(5-Cyano-4-(2-morpholin-4-yl-quinolin-3-yl)-6-(2-oxo-2-(phenylamino)ethoxy)pyridin-2-yl)phenyl acetate (10a)
Yield, 90%; m.p 178–180 °C; IR (KBr, v cm−1): 3447 (NH), 2220 (C≡N), 1761 (C=O), 1677 (CONH), 1651 (C=N).
3-(6-(2-((4-Chlorophenyl)amino)-2-oxoethoxy)-5-cyano-4-(2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)phenyl acetate (10b)
Yield, 89%; m.p 209–210 °C; IR (KBr, v cm−1): 3407 (NH), 2223 (C≡N), 1763 (C=O), 1679 (CONH), 1590 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 2.21 (s, 3H, COCH3); 3.10–3.20 (m, 4H, morpholine-C2,6-H); 3.50–3.65 (m, 4H, morpholine-C3,5-H); 5.21 (s, 2H, CH2); 7.26 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.39 (d, J = 8.4 Hz, 2H, p-chlorophenyl-C3,5-H); 7.47–7.51 (m, 2H, quinoline-C6-H and phenyl-C5-H); 7.65 (d, J = 8.4 Hz, 2H, p-chlorophenyl-C2,6-H); 7.76 (t, J = 6.8,8 Hz, 1H, quinoline-C7-H); 7.85 (d, J = 8 Hz, 1H, quinoline-C8-H); 7.93–7.95 (m, 2H, quinoline-C5-H and pyridine-C5-H); 8.12 (d, J = 7.6 Hz, 1H, phenyl-C6-H); 8.16 (s, 1H, quinoline-C4-H); 8.43 (s, 1H, phenyl-C2-H); 10.61 (s, 1H, NH, D2O-exchangeable).
3-(5-Cyano-6-(2-((4-methoxyphenyl)amino)-2-oxoethoxy)-4-(2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)phenyl acetate (10c)
Yield, 92%; m.p 268–270 °C; IR (KBr, v cm−1): 3414 (NH), 2225 (C≡N), 1764 (C=O), 1671 (CONH), 1591 (C=N).
3-(4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-5-cyano-6-(2-oxo-2-(phenylamino)ethoxy)pyridin-2-yl)phenyl acetate (10d)
Yield, 91%; m.p 197–198 °C; IR (KBr, v cm−1): 3416 (NH), 2224 (C≡N), 1744 (COCH3), 1680 (CONH), 1543 (C=N).
3-(4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-6-(2-((4-chlorophenyl)amino)-2-oxoethoxy)-5-cyanopyridin-2-yl)phenyl acetate (10e)
Yield, 89%; m.p 188–190 °C; IR (KBr, v cm−1): 3420 (NH), 2220 (C≡N), 1743 (OCOCH3), 1680 (CONH).
3-(4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-5-cyano-6-(2-((4-methoxyphenyl)amino)-2-oxoethoxy)pyridin-2-yl)phenyl acetate (10f)
Yield, 93%; m.p 188–190 °C; IR (KBr, v cm−1): 3414 (NH), 2223 (C≡N), 1761 (C=O), 1667 (CONH), 1592 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 2.23 (s, 3H, COCH3); 3.15–3.18 (m, 4H, morpholine-C2,6-H); 3.58–3.60 (m, 4H, morpholine-C3,5-H); 3.73 (s, 3H, OCH3); 5.20 (s, 2H, CH2); 6.91 (d, J = 8 Hz, 2H, p-methoxyphenyl-C3,5-H); 7.28 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.50–7.54 (m, 3H, p-methoxyphenyl-C2,6-H and phenyl-C5-H); 7.76 (d, J = 8 Hz, 1H, quinoline-C7-H); 7.86 (d, J = 8 Hz, 1H, quinoline-C8-H); 7.97 (s, 1H, quinoline-C5-H); 8.05 (s, 1H, pyridine-C5-H); 8.14 (d, J = 8 Hz, 1H, phenyl-C6-H); 8.17 (s, 1H, quinoline-C4-H); 8.41 (s, 1H, phenyl-C2-H); 10.33 (s, 1H, NH, D2O-exchangeable).
3-(5-Cyano-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)-6-(2-oxo-2-(phenylamino)ethoxy)pyridin-2-yl)phenyl acetate (10g)
Yield, 90%; m.p 284–285 °C; IR (KBr, v cm−1): 3420 (NH), 2223 (C≡N), 1759 (C=O), 1678 (CONH), 1597 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 2.20 (s, 3H, COCH3); 3.07–3.09 (m, 4H, morpholine-C2,6-H); 3.57–3.60 (m, 4H, morpholine-C3,5-H); 3.89 (s, 3H, OCH3); 5.23 (s, 2H, CH2); 7.09 (s, 1H, quinoline-C5-H); 7.25–7.49 (m, 5H, phenyl-C3,4,5-H, quinoline-C7-H and phenylacetate-C4-H); 7.61–7.64 (m, 2H, phenyl-C2,6-H); 7.77–7.81 (m, 2H, phenylacetate-C5-H and quinoline-C8-H); 7.98 (s, 1H, pyridine-C5-H); 8.12–8.16 (m, 2H, quinoline-C5-H and phenylacetate-C6-H); 8.36 (s, 1H, phenylacetae-C2-H); 10.48 (s, 1H, NH, D2O-exchangeable).
3-(6-(2-((4-Chlorophenyl)amino)-2-oxoethoxy)-5-cyano-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)phenyl acetate (10h)
Yield, 92%; m.p 264–265 °C; IR (KBr, v cm−1): 3421 (NH), 2225 (C≡N), 1748 (C=O), 1680 (CONH), 1651 (C=N).
3-(5-Cyano-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)-6-(2-((4-methoxyphenyl)amino)-2-oxoethoxy)pyridin-2-yl)phenyl acetate (10i)
Yield, 93%; m.p 270–271 °C; IR (KBr, v cm−1): 3419 (NH), 2222 (C≡N), 1762 (C=O), 1670 (CONH), 1599 (C=N).
General procedure for synthesis of 2-((3-Cyano-6-(3-hydroxyphenyl)4-(2-morpholin-4-yl-6-substituted quinolin-3-yl)pyridin-2-yl)oxy)-N-(4-substituted phenyl)acetamide (11a–i)
A mixture of compounds 10a–i (1 mmol), and sodium bicarbonate (0.756 g, 9 mmol) in equal amount of methanol\water (1:1) was refluxed for 1 h. The reaction mixture was then cooled, treated with water and neutralised with acetic acid (PH = 7). The obtained product was filtered, washed with water, air dried and crystallised from ethanol.
2-((3-Cyano-6-(3-hydroxyphenyl)-4-(2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)-N-phenylacetamide (11a)
Yield, 92%; m.p 243–245 °C; IR (KBr, v cm−1): 3550 (OH), 3392 (NH), 2220 (C≡N), 1670 (CONH), 1585 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.10–3.20 (m, 4H, morpholine-C2,6-H); 3.50–3.65 (m, 4H, morpholine-C3,5-H); 5.23 (s, 2H, CH2); 6.87 (d, J = 7.6 Hz, 1H, 3-hydroxyphenyl-C4-H); 7.06 (t, J = 7.2,7.6 Hz, 1H, 3-hydroxyphenyl-C5-H); 7.17 (t, J = 7.6,8 Hz, 1H, quinoline-C6-H); 7.31 (t, J = 7.2,7.6 Hz, 2H, phenyl-C3,5-H); 7.48 (t, J = 7.2 Hz, 1H, phenyl-C4-H); 7.58–7.63 (m, 4H, 3-hydroxyphenyl-C2,6-H and phenyl-C2,6-H); 7.74 (t, J = 7.6,7.2 Hz, 1H, quinoline-C7-H); 7.83 (d, J = 8 Hz, 1H, quinoline-C8-H); 7.93 (d, J = 8 Hz, 1H, quinoline-C5-H); 7.97 (s, 1H, pyridine-C5-H); 8.40 (s, 1H, quinoline-C4-H); 10.49 (s, 1H, NH, D2O-exchangeable). 13C-NMR (100 MHz, DMSO-d6δ ppm): 49.68, 65.82, 66.17, 93.23, 114.53, 114.77, 115.43, 118.19, 118.58, 120.01, 123.71, 123.97, 124.69, 125.18, 127.40, 128.71, 128.71, 130.26, 131.42, 137.92, 139.05, 140.77, 147.28, 155.31, 157.79, 158.18, 159.05, 163.46, 166.47. Elemental analysis Calcd for C33H27N5O4 (557.61): C 71.08, H 4.88, N 12.56. Found: C 70.89, H 4.96, N 12.31.
N-(4-Chlorophenyl)-2-((3-cyano-6-(3-hydroxyphenyl)-4-(2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)acetamide (11b)
Yield, 89%; m.p 279–280 °C; IR (KBr, v cm−1): 3500 (OH), 3396 (NH), 2224 (C≡N), 1659 (CONH), 1584 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.10–3.25 (m, 4H, morpholine-C2,6-H); 3.50–3.70 (m, 4H, morpholine-C3,5-H); 5.23 (s, 2H, CH2); 6.80–6.95 (m, 1H, phenyl-C4-H); 7.15–7.25 (m, 1H, phenyl-C5); 7.31–8.15 (m, 10H, p-chlorophenyl-C2,3,5,6-H, quinoline-C5,6,7,8-H and phenyl-C2,5-H); 8.00 (s, 1H, pyridine-C5-H); 8.42 (s, 1H, quinoline-C4-H); 9.66 (brs, 1H, OH, D2O-exchangeable); 10.59 (s, 1H, NH, D2O-exchangeable). Elemental analysis Calcd for C33H26ClN5O4 (592.05): C 66.95, H 4.43, N 11.83. Found: C 67.13, H 4.60, N 12.06.
2-((3-Cyano-(3-hydroxyphenyl)-4-(2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)-N-(4-methoxyphenyl)acetamide (11c)
Yield, 90%; m.p 268–270 °C; IR (KBr, v cm−1): 3550 (OH), 3401 (NH), 2223 (C≡N), 1649 (CONH), 1583 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.05–3.20 (m, 4H, morpholine-C2,6-H); 3.50–3.60 (m, 4H, morpholine-C3,5-H); 3.72 (s, 3H, OCH3); 5.14 (s, 2H, CH2); 6.87 (d, J = 8 Hz, 2H, p-methoxyphenyl-C3,5-H); 6.90 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.24 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.44–7.49 (m, 3H, p-methoxyphenyl-C2,6-H and quinoline-C6); 7.55 (s, 1H, phenyl-C2-H); 7.64 (d, J = 8 Hz,1H, quinoline-C8-H); 7.74 (t, J = 8 Hz, 1H, quinoline-C7-H); 7.83 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.92 (d, J = 8 Hz, 1H, quinoline-C5-H); 7.95 (s, 1H, pyridine-C5-H); 8.33 (s, 1H, quinoline-C4-H); 9.71 (s, 1H, OH, D2O-exchangeable); 10.26 (s, 1H, NH, D2O-exchangeable). 13C-NMR (100 MHz, DMSO-d6δ ppm): 49.69; 55.62, 65.82, 66.17, 93.39, 100.00, 114.37, 114.58, 118.77, 119.02, 121.64, 122.48, 123.71, 124.69, 127.41, 128.71, 130.37, 131.15, 131.45, 132.04, 139.05, 140.79, 147.29, 155.35, 155.90, 157.80, 158.03, 158.32, 163.52, 165.99. Elemental analysis Calcd for C34H29N5O5 (587.64): C 69.49, H 4.97, N 11.92. Found: C 69.32, H 5.13, N 12.18.
2-((4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-3-cyano-6-(3-hydroxyphenyl)pyridin-2-yl)oxy)-N-phenylacetamide (11d)
Yield, 88%; m.p 277–280 °C; IR (KBr, v cm−1): 3550 (OH), 3437 (NH), 2225 (C≡N), 1657 (CONH), 1580 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.10–3.20 (m, 4H, morpholine-C2,6-H); 3.50–3.65 (m, 4H, morpholine-C3,5-H); 5.18 (s, 2H, -CH2); 6.89 (d, J = 8 Hz, 1H, 3-hydroxyphenyl-C4-H); 7.07 (t, J = 8 Hz, 1H, phenyl-C4-H); 7.20 (t, J = 8 Hz, 1H, 3-hydroxyphenyl-C5-H); 7.30 (t, J = 8 Hz, 2H, phenyl-C3,5-H); 7.56 (d, J = 8 Hz, 2H, phenyl-C2,6-H); 7.61 (d, J = 8 Hz, 1H, 3-hydroxyphenyl-C6-H); 7.72 (d, J = 8 Hz, 1H, quinoline-C5-H); 7.78–7.84 (m, 2H, 3-hydroxyphenyl-C2-H and quinoline-C7-H); 7.95 (s, 1H, pyridine-C5-H); 7.98 (d, J = 8 Hz, 1H, quinoline-C8-H); 8.31 (s, 1H, quinoline-C4-H); 9.88 (s, 1H, OH, D2O-exchangeable); 10.62 (s, 1H, NH, D2O-exchangeable). Elemental analysis Calcd for C33H26ClN5O4 (592.05): C 66.95, H 4.43, N 11.83. Found: C 66.81, H 4.62, N 11.94.
2-((4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-3-cyano-6-(3-hydroxyphenyl)pyridin-2-yl)oxy)-N-(4-chlorophenyl)acetamide (11e)
Yield, 87%; m.p > 300 °C; IR (KBr, v cm−1): 3500 (OH), 3413 (NH), 2218 (C≡N), 1654 (CONH), 1595 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.10–3.20 (m, 4H, morpholine-C2,6-H); 3.50–3.65 (m, 4H, morpholine-C3,5-H); 5.23 (s, 2H, CH2); 6.91 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.22 (t, J = 7.6,8 Hz, 1H, phenyl-C5-H); 7.38 (d, J = 8.8 Hz, 2H, p-chlorophenyl-C3,5-H); 7.58–7.66 (m, 1H, phenyl-C6-H); 7.65 (d, J = 8.8 Hz, 2H, p-chlorophenyl-C2,6-H); 7.75 (d, J = 8.8 Hz, 1H, quinoline-C7-H); 7.84 (d, J = 8.8 Hz, 1H, quinoline-C8-H); 8.00 (s, 1H, phenyl-C2-H); 8.04 (s, 1H, quinoline-C5-H); 8.32 (s, 1H, pyridine-C5-H); 8.39 (s, 1H, quinoline-C4-H); 9.75 (brs, 1H, OH, D2O-exchangeable); 10.59 (s, 1H, NH, D2O-exchangeable). EI-MS m/z (% relative abundance): 630 [M+•+4] (35.76); 626 [M+•] (40.95); 267 (92.07); 127 (100). Elemental analysis Calcd for C33H25Cl2N5O4 (626.49): C 63.27, H 4.02, N 11.18. Found: C 63.44, H 4.21, N 11.39.
2-((4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-3-cyano-6-(3-hydroxyphenyl)pyridin-2-yl)oxy)-N-(4-methoxyphenyl)acetamide (11f)
Yield, 89%; m.p 228–230 °C; IR (KBr, v cm−1): 3450 (OH), 3403 (NH), 2224 (C≡N), 1658 (CONH), 1593 (C=N). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.11–3.21 (m, 4H, morpholine-C2,6-H); 3.53–3.58 (m, 4H, morpholine-C3,5-H); 3.68 (s, 3H, OCH3); 5.14 (s, 2H, -CH2); 6.85–6.91 (m, 3H, phenyl-C4-H and p-methoxyphenyl-C3,5-H); 7.23 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.46 (d, J = 8 Hz, 2H, p-methoxyphenyl-C2,6-H); 7.54 (s, 1H, quinoline-C5-H); 7.62 (d, J = 4 Hz, 1H, phenyl-C2-H); 7.71 (d, J = 8 Hz, 1H, quinoline-C7-H); 7.81 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.93–7.98 (m, 2H, pyridine-C5-H and quinoline-C8-H); 8.28 (s, 1H, quinoline-C4-H); 9.73 (s, 1H, OH, D2O-exchangeable); 10.27 (s, 1H, NH, D2O-exchangeable). EI-MS m/z (% relative abundance): 624 [M+•+2] (29.04); 622 [M+•] (36.76); 366 (84.92); 182 (100). Elemental analysis Calcd for C34H28ClN5O5 (622.08): C 65.65, H 4.54, N 11.26. Found: C 65.47, H 4.63, N 11.45.
2-((3-Cyano-6-(3-hydroxyphenyl)-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)-N-phenylacetamide (11g)
Yield, 91%; m.p 269–270 °C; IR (KBr, v cm−1): 3550 (OH), 3409 (NH), 2220 (C≡N), 1650 (CONH). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 2.95–3.15 (m, 4H, morpholine-C2,6-H); 3.50–3.60 (m, 4H, morpholine-C3,5-H); 3.88 (s, 3H, OCH3); 5.23 (s, 2H, CH2); 6.91 (d, J = 8 Hz, 1H, 3-hydroxyphenyl-C4-H); 7.02–7.13 (m, 1H, quinoline-C7-H); 7.21 (s, 1H, quinoline-C5-H); 7.30–7.45 (m, 4H, phenyl-C3,4,5-H and 3-hydroxyphenyl-C5-H); 7.51–7.70 (m, 3H, phenyl-C2,6-H and 3-hydroxyphenyl-C2-H); 7.75–7.82 (m, 2H, 3-hydroxyphenyl-C6-H and quinoline-C8-H); 7.99 (s, 1H, pyridine-C5-H); 8.32 (s, 1H, quinoline-C4-H); 9.69 (brs, 1H, OH, D2O-exchangeable); 10.45 (s, 1H, NH, D2O-exchangeable). Elemental analysis Calcd for C34H29N5O5 (587.64): C 69.49, H 4.97, N 11.92. Found: C 69.31, H 5.14, N 12.13.
N-(4-Chlorophenyl)-2-((3-cyano-6-(3-hydroxyphenyl)-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)acetamide (11h)
Yield, 90%; m.p 230–232 °C; IR (KBr, v cm−1): 3550 (OH), 3428 (NH), 2220 (C≡N), 1650 (CONH). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.09–3.20 (m, 4H, morpholine-C2,6-H); 3.53 (s, 4H, morpholine-C3,5-H); 3.87 (s, 3H, OCH3); 4.68, 5.54 (2 s, each for 1H, CH2); 6.90–7.00 (m, 1H, phenyl-C4-H); 7.25–7.50 (m, 4H, p-chlorophenyl-C3,5-H and quinoline-C5,7-H); 7.60–7.70 (m, 2H, phenyl-C2,5-H); 7.75–7.90 (m, 2H, p-chlrophenyl-C2,6-H); 7.90–7.95 (m, 1H, quinoline-C8-H); 8.00–8.10 (m, 1H, phenyl-C6-H); 8.20–8.35 (m, 2H, quinoline-C4-H and pyridine-C5-H); 9.87 (brs, 1H, OH, D2O-exchangeable); 10.37 (s, 1H, NH, D2O-exchangeable). Elemental analysis Calcd for C34H28ClN5O5 (622.08): C 65.65, H 4.54, N 11.26. Found: C 65.92, H 4.67, N 11.52.
2-((3-Cyano-6-(3-hydroxyphenyl)-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)-N-(4-methoxyphenyl)acetamide (11i)
Yield, 93%; m.p > 300 °C; IR (KBr, v cm−1): 3500 (OH), 3435 (NH), 2220 (C≡N), 1654 (CONH). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.00–3.15 (m, 4H, morpholine-C2,6-H); 3.56 (s, 4H, morpholine-C3,5-H); 3.72 (s, 3H, OCH3); 3.88 (s, 3H, OCH3); 5.21 (s, 2H, -CH2); 6.80–6.95 (m, 3H, phenyl-C4-H and p-methoxyphenyl-C3,5-H); 7.22 (s, 1H, quinoline-C5-H); 7.35–7.43 (m, 2H, p-methoxyphenyl-C2,6-H); 7.50–7.60 (m, 1H, phenyl-C5-H); 7.67–7.79 (m, 3H, phenyl-C2,6-H and quinoline-C7-H); 7.98–8.00 (m, 2H, pyridine-C5-H and quinoline-C8-H); 8.33 (s, 1H, quinoline-C4-H); 9.77 (brs, 1H, OH, D2O-exchangeable); 10.41 (s, 1H, NH, D2O-exchangeable). Elemental analysis Calcd for C35H31N5O6 (617.66): C 68.06, H 5.06, N 11.34. Found: C 68.34, H 5.24, N 11.49.
Compounds 12a–c were prepared as reported 21,Citation25.
General procedure for synthesis of 6-(3-Hydroxyphenyl)-4-(2-oxo-6-substituted-1,2-dihydroquinolin-3-yl)-2-oxo-1,2-dihydropyridine-3-carbonitriles; (13a–c)
A mixture of 3-hydroxyacetophenone (0.136 g, 1 mmol), substituted aldehyde 12a–c (1 mmol), ethyl cyanoacetate (0.106 ml, 1 mmol) and ammonium acetate (0.616 g, 8 mmol) in 10 ml absolute ethanol was heated under reflux for 4 h. It was then allowed to cool and the obtained solid was filtered, washed with cold ethanol, air dried, and crystallised from ethanol/DMF (9:1) mixture.
6-(3-Hydroxyphenyl)-2-oxo-4-(2-oxo-1,2-dihydroquinolin-3-yl)-1,2-dihydropyridine-3-carbonitrile (13a)
Yield, 45%; m.p > 300 °C; IR (KBr, v cm−1): 3411 (OH), 3400 (NH), 2221 (C≡N), 1647 (C=O). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 6.82 (brs, 1H, pyridine-C5-H); 6.97 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.20 (s, 1H, phenyl-C2-H); 7.25–7.28 (m, 2H, phenyl-C5,6 -H); 7.34 (t, J = 7.6, 8 Hz, 1H, quinoline-C6-H); 7.39 (d, J = 8.4 Hz,1H, quinoline- C5-H); 7.62 (t, J = 7.6, 8 Hz, 1H, quinoline-C7-H); 7.78 (d, J = 8 Hz, 1H, quinoline-C8-H); 8.30 (s, 1H, quinoline-C4-H); 9.84 (s, 1H, OH, D2O-exchangeable); 12.22, 12.74 (2 s, each for 1H, 2NH, D2O-exchangeable). 13C-NMR (100 MHz, DMSO-d6δ ppm): 107.50, 114.73, 115.66, 116.50, 118.63, 118.75, 118.97, 122.89, 128.84, 129.27, 130.56. 130.63, 132.31, 134.16, 139.73, 141.11, 156.74, 158.21, 158.27, 159.74, 162.09. Elemental analysis Calcd for C21H13N3O3 (355.35): C 70.98, H 3.69, N 11.83. Found: C 70.69, H 3.84, N 11.97.
4-(6-Chloro-2-oxo-1,2-dihydroquinolin-3-yl)-6-(3-hydroxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (13b)
Yield, 30%; m.p > 300 °C; IR (KBr, v cm−1): 3415 (OH), 3304 (NH), 2219 (C≡N), 1647 (C=O). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 6.85 (s, 1H, pyridine-C5-H); 6.98 (t, J = 4,8 Hz, 1H, phenyl-C4-H); 7.18–7.28 (m, 2H, phenyl-C2,6-H); 7.35 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.40 (d, J = 8 Hz,1H, quinoline-C7-H); 7.67 (d, J = 8 Hz, 1H, quinoline-C8-H); 7.91 (s, 1H, quinoline-C5-H); 8.28 (s, 1H, quinoline-C4-H); 9.87 (s, 1H, OH, D2O-exchangeable); 11.52, 12.37 (2 s, each for 1H, 2NH, D2O-exchangeable). Elemental analysis Calcd for C21H12ClN3O3 (389.80): C 64.71, H 3.10, N 10.78. Found: C 64.89, H 3.32, N 10.85.
6-(3-Hydroxyphenyl)-4-(6-methoxy-2-oxo-1,2-dihydroquinolin-3-yl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (13c)
Yield, 42%; m.p > 300 °C; IR (KBr, v cm−1): 3427 (OH), 3320 (NH), 2217 (C≡N), 1645 (C=O). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.81(s,3H, OCH3); 6.76 (brs, 1H, pyridine-C5-H); 6.96 (d, J = 8.4 Hz, 1H, phenyl-C4-H); 7.15–7.16 (m, 1H, phenyl-C6-H); 7.20–7.24 (m, 1H, phenyl-C5-H); 7.28 (dd, J = 2.8,8.8 Hz,1H, quinoline- C7-H); 7.32 (s, 1H, phenyl-C2-H); 7.35 (d, J = 8 Hz, 1H, quinoline-C8-H); 7.95 (s, 1H, quinoline-C5-H); 8.22 (s, 1H, quinoline-C4-H); 9.87 (s, 1H, OH, D2O-exchangeable); 11.30, 12.15 (2 s, each for 1H, 2NH, D2O-exchangeable). EI-MS m/z (% relative abundance): 386 [M+•+1] (27.16); 385 [M+•] (100); 188 (76.41). Elemental analysis Calcd for C22H15N3O4 (385.38): C 68.57, H 3.92, N 10.90. Found: C 68.80, H 3.85, N 10.79.
General procedure for synthesis of 6-(3-Hydroxyphenyl)-2-mercapto-4-(2-oxo-6-substituted-1,2-dihydroquinolin-3-yl)pyridine-3-carbonitriles; (14a–c)
A mixture of 3-hydroxyacetophenone (0.136 g, 1 mmol), substituted aldehyde 12a–c (1 mmol), cyanothioacetamide (0.100 g, 1 mmol) and anhydrous ammonium acetate (0.616 g, 8 mmol) in ethanol was stirred and heated under reflux for 4 h. The reaction mixture allowed to cool, and the obtained precipitate was filtered, washed with cold ethanol, air dried and crystallised from ethanol/DMF (9:1) mixture.
6-(3-Hydroxyphenyl)-2-mercapto-4-(2-oxo-1,2-dihydroquinolin-3-yl)pyridine-3-carbonitrile (14a)
Yield, 49%; m.p 278–280 °C; IR (KBr, v cm−1): 3500 (OH), 3396 (NH), 2600 (SH), 2219 (C≡N), 1659 (C=O). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 6.92 (d, J = 6.8 Hz, 1H, phenyl-C4-H); 7.10 (s, 1H, phenyl-C2-H); 7.23–7.28 (m, 2H, phenyl-C5,6-H); 7.29–7.33 (m, 2H, pyridine-C5-H and quinoline-C6-H); 7.37 (d, J = 8.4 Hz,1H, quinoline- C5-H); 7.60 (t, J = 7.2, 8 Hz, 1H, quinoline-C7-H); 7.76 (d, J = 7.6 Hz, 1H, quinoline-C8-H); 8.22 (s, 1H, quinoline-C4-H); 9.77 (s, 1H, OH, D2O-exchangeable); 12.18 (s, 1H, NH, D2O-exchangeable); 14.25 (brs, 1H, SH, D2O-exchangeable). EI-MS m/z (% relative abundance): 371 [M+•] (33.99); 165 (100), 143 (97.90); 97 (93.44). Elemental analysis Calcd for C21H13N3O2S (371.41): C 67.91, H 3.53, N 11.31. Found: C 67.69, H 3.75, N 11.48.
4-(6-Chloro-2-oxo-1,2-dihydroquinolin-3-yl)-6-(3-hydroxyphenyl)-2-mercaptopyridine-3-carbonitrile (14b)
Yield, 45%; m.p 227–230 °C; IR (KBr, v cm−1): 3550 (OH), 3421 (NH), 2540 (SH), 2211 (C≡N), 1658 (C=O). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 6.87–6.97 (m, 1H, phenyl-C4-H); 7.14–7.50 (m, 4H, phenyl-C2,5-H, pyridine-C5-H and quinoline-C5-H); 7.65–7.76 (m, 1H, quinoline-C7-H); 7.90 (d, J = 9 Hz, 1H, phenyl-C6-H); 8.11–8.36 (m, 2H, quinoline-C4-H and quinoline-C8-H); 9.69 (s, 1H, OH, D2O-exchangeable); 12.39 (s, 1H, NH, D2O-exchangeable); 14.23 (s, 1H, SH, D2O-exchangeable). Elemental analysis Calcd for C21H12ClN3O2S (405.86): C 62.15, H 2.98, N 10.35. Found: C 62.41, H 3.16, N 10.44.
6-(3-Hydroxyphenyl)-2-mercapto-4-(6-methoxy-2-oxo-1,2-dihydroquinolin-3-yl)pyridine-3-carbonitrile (14c)
Yield, 50%; m.p 278–280 °C; IR (KBr, v cm−1): 3550 (OH), 3421 (NH), 2590 (SH), 2219 (C≡N), 1663 (C=O). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.81 (s, 3H, OCH3); 6.99 (d, J = 7.6 Hz, 1H, phenyl-C4-H); 7.16–7.18 (m, 2H, phenyl-C2,6-H); 7.23 (d, J = 8.4 Hz, 1H, quinoline-C7-H); 7.27–7.37 (m, 4H, pyridine-C5-H, phenyl-C5-H and quinoline-C5,8-H); 8.25 (s, 1H, quinoline-C4-H); 9.91 (s, 1H, OH, D2O-exchangeable); 12.18 (s, 1H, NH, D2O-exchangeable); 14.17 (brs, 1H, SH, D2O-exchangeable). 13C-NMR (100 MHz, DMSO-d6δ ppm): 56.00, 110.11, 114.16, 115.09, 115.45, 116.93, 117.08, 118.94, 119.44, 119.52, 121.92, 128.97, 130.52, 133.20, 134.39, 140.80, 152.70, 153.21, 154.93, 158.02, 159.24, 179.45. Elemental analysis Calcd for C22H15N3O3S (401.44): C 65.82, H 3.77, N 10.47. Found: C 65.70, H 3.98, N 10.60.
General procedure for synthesis of 2-Amino-6-(3-hydroxyphenyl)-4-(2-oxo-6-substitiuted-1,2-dihydroquinolin-3-yl)pyridine-3-carbonitriles; (15a–c)
An equimolar amount of 3-hydroxyacetophenone (0.136 g, 1 mmol), substituted aldehyde 12a–c (1 mmol), malononitrile (0.066 g, 1 mmol) and ammonium acetate (0.616 g, 8 mmol) in 10 ml absolute ethanol was heated under reflux for 6 h during which the product was separated out. Then the reaction mixture was cooled, and the yellow precipitate was filtered, washed with cold ethanol, air dried and crystallised from ethanol/DMF (9:1).
2-Amino-6-(3-hydroxyphenyl)-4-(2-oxo-1,2-dihydroquinolin-3-yl)pyridine-3-carbonitrile (15a)
Yield, 50%; m.p > 300 °C; IR (KBr, v cm−1): 3459 (OH), 3400 (νasN-H), 3356 (νsN-H), 3244 (NH), 2215 (C≡N), 1651 (C=O). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 6.88 (d, J = 7.6 Hz, 1H, phenyl-C4-H); 6.94 (s, 2H, NH2, D2O-exchangeable); 7.24 (s, 1H, pyridine-C5-H); 7.25–7.30 (m, 2H, phenyl-C5-H and quinoline-C6-H); 7.38 (d, J = 8.4 Hz,1H, quinoline- C5-H); 7.48–7.52 (m, 2H, phenyl-C2-H and quinoline-C8-H); 7.59 (t, J = 7.6, 8 Hz, 1H, quinoline-C7-H); 7.76 (d, J = 8 Hz, 1H, phenyl-C6-H); 8.20 (s, 1H, quinoline-C4-H); 9.61 (s, 1H, OH, D2O-exchangeable); 12.16 (s, 1H, NH, D2O-exchangeable). 13C-NMR (100 MHz, DMSO-d6δ ppm): 89.02, 110.64, 114.36, 115.57, 117.13, 117.59, 118.43, 119.21, 122.74, 129.05, 129.99, 130.16, 131.85, 134.49, 139.55, 140.59, 151.32, 158.16, 158.99, 160.21, 160.56. Elemental analysis Calcd for C21H14N4O2 (354.37): C 71.18; H 3.98; N 15.81. Found: C 71.49, H 4.06, N 15.95.
2-Amino-4-(6-chloro-2-oxo-1,2-dihydroquinolin-3-yl)-6-(3-hydroxyphenyl)pyridine-3-carbonitrile (15b)
Yield, 45%; m.p > 300 °C; IR (KBr, v cm−1): 3530(OH), 3418 (νasN-H), 3355 (νsN-H), 3250 (NH), 2215 (C≡N), 1662 (C=O). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 6.81 (s, 2H, NH2, D2O-exchangeable); 6.84 (s, 1H, phenyl-C2-H); 6.92 (d, J = 9 Hz, 1H, phenyl-C4-H); 6.98–7.01 (m, 1H, phenyl-C5-H); 7.24–7.50 (m, 3H, pyridine-C5-H and quinoline-C4,5-H); 7.63 (d, J = 8 Hz, 1H, quinoline-C7-H); 7.87 (d, J = 9 Hz, 1H, phenyl-C6-H); 8.18 (d, J = 8 Hz, 1H, quinoline-C8-H); 9.79 (s, 1H, OH, D2O-exchangeable); 12.30 (s, 1H, NH, D2O-exchangeable). MS m/z (% relative abundance): 390 [M+•+2] (24.76); 388 [M+•] (42.93); 315 (100); 63 (76.44). Elemental analysis Calcd for C21H13ClN4O2 (388.81): C 64.87, H 3.37, N 14.41. Found: C 64.71, H 3.19, N 14.62.
2-Amino-6-(3-hydroxyphenyl)-4-(6-methoxy-2-oxo-1,2-dihydroquinolin-3-yl)pyridine-3-carbonitrile (15c)
Yield, 53%; m.p 259–260 °C; IR (KBr, v cm−1): 3450 (OH), 3380 (νasN-H), 3290 (νsN-H), 3176 (NH), 2189 (C≡N), 1651 (C=O). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 3.81 (s, 3H, OCH3); 6.88 (d, J = 7.6 Hz, 1H, phenyl-C4-H); 6.93 (s, 2H, NH2, D2O-exchangeable); 7.22 (s, 1H, pyridine-C5-H); 7.24–7.38 (m, 4H, quinoline-C5,7-H and phenyl-C5,6-H); 7.48–7.51 (m, 2H, phenyl-C2-H and quinoline-C8-H); 8.13 (s, 1H, quinoline-C4-H); 9.61 (s, 1H, OH, D2O-exchangeable); 12.05 (s, 1H, NH, D2O-exchangeable). Elemental analysis Calcd for C22H16N4O3 (384.40): C 68.74, H 4.20, N 14.58. Found: C 68.95, H 4.32, N 14.67.
Biological evaluation
All chemicals, solvents, media and kits were purchased from commercial suppliers. Biological evaluation procedures were performed in Medical Biotechnology Department, Genetic Engineering and Biotechnology Research Institute, City of Scientific Research and Technological Applications (SRTA-City), Egypt.
Cytotoxicity screening
Cytotoxicity of the tested compounds was evaluated using normal human lung fibroblast Wi-38 cell line compared to currently used anticancer drug (doxorubicin) using MTT method was used for assaying cell viabilityCitation26 (Supplementary Page S55).
Determination of the anticancer activity
Anticancer effect of the above-mentioned compounds was evaluated using four human cancer cell lines. Colon cancer cells (Caco-2), myeloid leukaemia cell line (NFS-60), liver cancer cell line (HepG-2) and prostate cancer cell line (PC-3) using MTT method was used for assaying cell viabilityCitation26 (Supplementary Page S55).
Flow cytometric analysis of apoptosis
The apoptosis-dependent c active compounds was determined by quantification of annexin-stained apoptotic cells using the FITC signal detector (FL1) against the phycoerythrin emission signal detector (FL2) (Supplementary Page S55).
Caspase 3/7 activation assay
The percentage of caspase 3/7 activation was quantified using the Apo-ONE ® caspase 3/7 kit following the manufacturer’s instructionsCitation27 (Supplementary Page S56).
Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM). Statistical significance was estimated by the multiple comparisons Tukey post-hoc analysis of variance (ANOVA) using the SPSS16 programCitation28 (Supplementary Page S56).
PIM-1 and PIM-2 kinase inhibitory activity
The most active anticancer compounds were tested for their ability to in-vitro inhibit PIM-1 and PIM-2 kinase utilising PIM-1 and PIM-2 Kinase Assay Kit – Promega Corporation catalogue #V4032, following the manufacturer’s instructionsCitation29.
Molecular modelling
Molecular docking study
The X-ray structures of PIM-1 and PIM-2 enzymes were retrieved from the Protein Data Bank (PDB) with PDB IDs: 2OBJCitation18 and 4X7QCitation30, respectively. The PDB structures were prepared using OpenEye’s MakeReceptor GUI of OEDOCKING 3.5.0.4 of OpenEye Scientific SoftwareCitation31,Citation32 at default levels. Redundant chains, non-essential ions, water molecules and ligands were discarded. The structural water molecule of PIM-1 kinase was kept. The search box around the X-ray co-crystal ligand was set 15.33 Å x 17.00 Å x 19.33 Å (5039 Å3) as box dimensions (box volume) for PIM-1 kinase, and 19.00 Å x 16.67 Å x 16.00 Å (5066 Å3) for PIM-2 kinase. Site shape potentials were calculated via setting the binding site contours in a balance between the protein and the solvent. The protein files were saved as OEDU format for FRED docking.
The studied structures were built via Marvin Sketch 17.2.6.0 ChemAxon (http://www.chemaxon.com), the lowest energy conformer was generated using MMFF94 Force Field, with normal optimisation limit at default settings. Then different conformations were generated for FRED docking via OpenEye OMEGA conformer generation softwareCitation33,Citation34. The docking was carried out using OpenEye’s FRED docking software at default levelsCitation31,Citation32.
The 3 D and 2 D depictions of the docking poses in PIM-1 and PIM-2 binding sites were generated via Molecular Operating Environment (MOE)Citation35.
Results and discussion
Chemistry
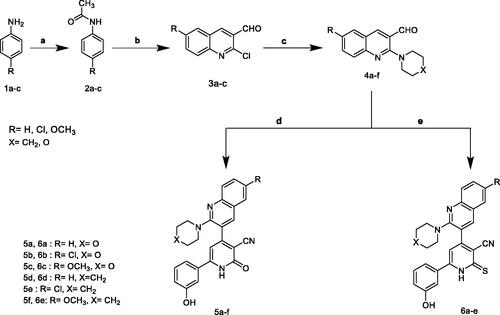
In our concernment for the synthesis of biologically active compounds, the synthetic strategies used for the preparation of the target compounds were outlined in three schemes. We illustrated herein the study for the synthesis of two new series of 4,6-diaryl −2-oxo or thioxo-1,2-dihydropyridine-3-carbonitriles 5a–f and 6a–f outlined in Scheme 1. The first step in this work was acetylation of aromatic amines 1a–c with acetic anhydride gave the corresponding substituted N-phenylacetamides 2a–c, which after treatment with Vilsmeier’s reagent afforded the substituted 2-chloro-3-formylquinolines 3a–c. After that, 2-chloro-3-formylquinolines 3a–c were reacted with morpholine or piperdine to give 2-(morpholine/piperdine-4-yl)quinoline-3-carbaldehyde 4a–f according to reported reaction conditionsCitation22–24. 2-(morpholine/piperdine-4-yl)quinoline-3-carbaldehyde 4a–f was then subjected to one pot reaction with the 3-hydroxyacetophenone and ethyl cyanoacetate or cyanothioacetamide in the presence of excess ammonium acetate to give the corresponding 3-cyanopyridones 5a–f or 3-cyanothiopyridines 6a–e, respectively. Structures of the synthesised compounds 5a–f and 6a–e were confirmed by elemental microanalysis, IR, 1H NMR for all compounds, beside EI/MS for compounds 5b,e and 6b,e and 13C NMR spectra for compound 5b,d and 6c,e. The IR spectra of all synthesised compounds showed bands at a stretching frequency (ν) around 3400 cm−1 corresponding to the N–H group and around 2200 cm−1 corresponding to CN group. The 1H-NMR spectra of the pyridone derivatives showed a methine proton as a deshielded singlet peak at the downfield region δ (6.9 − 7.3) ppm while for thiopyridine derivatives appeared around 8.25 ppm. 13C-NMR spectra revealed signals for C≡N and a highly deshielded signal corresponding to C=O or C=S. EI-MS of 5b, 5e, and 6e showed [M+•] peak at 458, 456, and 468, respectively in addition to [M+•+2] peak for 5b at 460 and for 5e at 458.
Scheme 1, the obtained results declared that, compounds 5a, 5b, 5c, 5d, and 6e had the most potent activity against Caco-2 cell line () even superior to doxorubicin. While compounds 5e, 5f, and 6b illustrated nearly equipotent activity. Also, compounds 5b and 5c exhibited high sensitivity against PC-3 cell line compared to doxorubicin (). Moreover, compounds 5b and 5c exerted higher anticancer activity than doxorubicin against HepG-2 cell line. While compound 6e displayed promising anticancer activity nearly equal to doxorubicin (). Furthermore, compound 5b represented high potency against NFS-60 cell line rather than doxorubicin. Whereas compound 5c showed nearly equipotent activity (). Consequently, compounds 5b and 5c exerted the highest anticancer activity against all tested human cancer cell lines, correlated to other compounds and doxorubicin. Indeed, compound 6e demonstrated higher anticancer activity against Caco-2 and HepG-2 compared to doxorubicin and nearly equipotent activity against NFS-60 and PC-3 cell lines. The selectivity index (SI) was calculated for the tested compounds as shown (Table 1 in Supplementary Material). The results indicated that, compounds 5b, 5c, and 6e showed the highest SI values. Compound 5c was found to have the highest SI values against all cancer lines. Consequently, it was the most selective compound towards all cancer cell lines as well as the most non-toxic towards Wi-38. Additionally, morphological changes on the four cancer cell lines (HepG-2, Caco-2, NFS-60, and PC-3) were investigated before and after treatment with the most active and safe compounds 5b, 5c, and 6e in comparison with cells treated with the reference doxorubicin. As shown, these compounds caused severe shrinking and collapsing in the normal shape of four studied cancer cell lines as shown in ().
Scheme 1. Synthesis of 2-pyridone/2-thioxopyridine – quinoline hybrids. (a) Ac2O; (b) POCI3/DMF; (c) morpholine or piperdiine/ K2CO3/DMF; (d) 3-hydroxyacetophenone / ammonium acetate/ CNCH2COOC2H5/ ethanol, reflux; (e) 3-hydroxyacetophenone /ammonium acetate / CNCH2CSNH2/ ethanol, reflux.
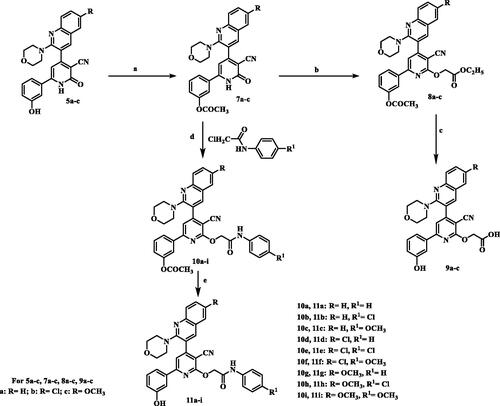
The synthesis of 2-O-substitutedpyridine derivatives is very straightforward and demonstrated in Scheme 2. The key intermediates, phenyl acetate derivatives 7a–c, were synthesised via heating compounds 5a–c with excess acetic anhydride. IR spectra of newly synthesised compounds 7a–c displayed absence of the band characteristic to OH function and presence of the absorption band for carbonyl moiety of the acetyl group (COOCH3) at 1760–1763 cm−1. 1H-NMR spectra for compounds 13a–c revealed presence of shielded singlet signal attributed to CH3 protons at a range of δ 2.30–2.31 ppm and in addition, absence of D2O exchangeable signal corresponding to OH. 13C-NMR spectrum for compound 13c showed signals assigned for a highly shielded signal corresponding to acetyl CH3 at δ 21.29 ppm and highly deshielded signal corresponding to carbonyl group of the ester at δ 174.50 ppm. First alkylation takes place by treatment of intermediated 7a–c with ethyl bromoacetate in presence of potassium carbonate to give compounds 8a–c. IR spectra of compounds 8a–c displayed a characteristic two absorbing bands for two esters C=O at 1750–1780 cm−1. 1H-NMR spectrum for compound 8a revealed disappearance of D2O exchangeable signal corresponding to NH. In addition, it showed shielded triplet signal integrated for three protons assigned for CH3 at δ 1.21 ppm, a singlet signal for two protons attributed to CH2 at δ 2.32 ppm and a quartette band integrated for two protons corresponding to CH2 at δ 4.20 ppm. Compounds 8a–c were hydrolysed using sodium bicarbonate in methanol\water (1:1) to produce the acid derivatives 9a–c. IR spectra of compounds 9a–c lacked the absorption bands of the carbonyl groups of the starting compounds. Moreover, a broad band characteristic to carboxylic (OH) appeared at 3000–3600 cm−1 in addition to an absorption band assigned for acidic carbonyl function observed at 1598–1732 cm−1 and an absorbing band for OH function was obtained at 3403–3448 cm−1. 1H-NMR spectra for compounds 9a–c revealed absence of triplet and quartette signals corresponding to C2H5 of ester and singlet attributed to CH3 of acetyl group. Instead, spectra showed two D2O exchangeable signal resonated at downfield corresponding to 2OH protons at range of δ 9.77–9.84 and 13.10–13.22 ppm. 13C-NMR spectrum for compound 9a showed appearance of highly deshielded signal corresponding to C=O for acid at δ 170.09 ppm. EI/MS for 9b showed [M+•+2] at m/z 518, molecular ion peak [M+•] at m/z 516 and base peak at m/z 413. Furthermore, the intermediates 7a–c were alkylated with N-aryl-2-chloroacetamides in the presence of anhydrous potassium carbonate in dry acetone to yield the intermediates 10a–i which were further deprotected by sodium bicarbonate in aqueous methanol (1:1) to prepare the final compounds 11a–i. IR spectra of the target compounds 10a–i displayed stretching absorption bands for amide NH and C=O functions at 3407–3447 and 1667–1680 cm−1, respectively. 1H-NMR spectra for compounds 10b, 10f, and 10g demonstrated upfield singlet signal corresponding to CH2 at a range of δ 5.20–5.23 ppm, and D2O exchangeable signal corresponding to NH for amide linkage at a range of δ 10.33–10.61 ppm. While, IR spectra of compounds 11a–i showed disappearance of a characteristic absorption band due to acetyl C=O function and appearance of a characteristic absorption band due to OH function at 3450–3550 cm−1. 1H-NMR spectra for compounds 11a–i demonstrated presence of upfield singlet signal corresponding to CH3 of acetyl group. Furthermore, the spectra displayed two D2O exchangeable signals corresponding to OH at a range of δ 9.66–9.88 ppm and amide NH at a range of δ 10.26–10.62 ppm. 13C-NMR spectra for compounds 11a,c displayed shielded signal corresponding to OCH2 at δ 65.82 ppm and a highly deshielded signal corresponding to C=O at δ 166.47 and 165.99 ppm, respectively. EI/MS for 11e showed [M+•+4] at m/z 630, the molecular ion peak [M+•] at m/z 626 and the base peak at m/z 127. EI/MS for 11f showed [M+•+2] at m/z 624, the molecular ion peak [M+•] at m/z 622 and the base peak at m/z 182.
Scheme 2. Synthesis of 2-O-substitutedpyridine – quinoline hybrids. (a) Ac2O, reflux; (b) BrCH2COOC2H5/K2CO3, reflux; (c) NaHCO3 / reflux; (d) K2CO3, reflux; (e) NaHCO3/ reflux.
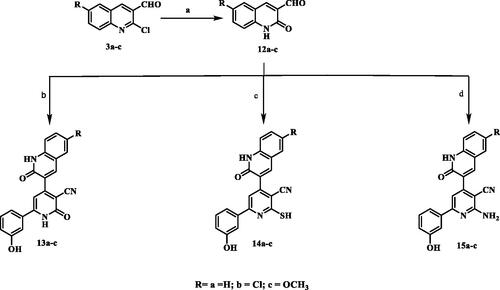
The synthesis of the target pyridine-quinolone hybrids was outlined in Scheme 3. This scheme discussed the chemical pathway used to prepare quinolone aldehydes 12a–c via hydrolysis of 2-chloro-6-substitutedquinoline-3-carbaldehydes 3a–c which were prepared by the action of Vilsmeier’s reagent on acetanilide derivatives 2a–cCitation25. Those target quinolone aldehydes 12a–c were reacted with 3-hydroxyacetophenone, ammonium acetate and ethyl cyanoacetate or cyanothioacetamide or malononitrile to give the corresponding 3-cyanopyridones 13a–c or 3-cyanothiopyridines 14a–c, or 3-cyano-2-aminopyridines 15a–c, respectively. The IR spectra of compounds 13a–c showed absorption bands at 3304–3400 cm−1 attributed to NH, in addition to absorption bands at 2217–2221 cm−1 and 1645–1647 cm−1 assigned to C≡N and C=O functions, respectively. Moreover, IR of compounds 14a–c showed characteristic SH band at 2540–2600 cm−1. Compounds 15a–c displayed the presence of two strong bands of asymmetrical and symmetrical stretching frequency of NH2 function at 3380–3418 and 3290– 3356 cm-1, respectively. The 1H-NMR spectra of the all-synthesised compounds showed a methine proton as a deshielded singlet peak at the downfield region δ 6.76–7.50 ppm. In addition, 1H-NMR spectra of compounds 14a–c showed highly deshielded D2O exchangeable SH proton appeared at a range δ 14.17–14.25 ppm which confirm the presence of all derivatives in thiol tautomer, while compounds 15a–c showed D2O singlet for two protons corresponding to NH2 protons at δ 6.81–6.94 ppm. 13C-NMR spectra showed signals for C≡N and highly deshielded signal corresponding to C=O, C=S or C-NH2 determined to be in accordance with the recommended molecule structures. EI-MS of 13c and 14a showed [M+•] peak at 385 and 371, respectively.
Scheme 3. Synthesis of 2-pyridone/2-thiopyridine/2-aminopyridine – quinolone hybrids. (a) CH3COOH, reflux; (b) 3-hydroxyacetophenone / ammonium acetate/ CNCH2COOC2H5/ ethanol, reflux; (c) 3-hydroxyacetophenone /ammonium acetate / CNCH2CSNH2/ ethanol, reflux; (d) 3-hydroxyacetophenone /ammonium acetate / CNCH2CN/ ethanol, reflux.
Biological evaluation
Cytotoxicity screening
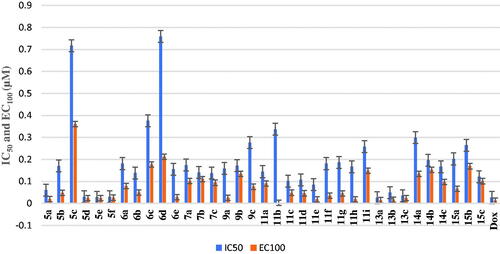
Cytotoxicity test is one of the biological evaluations and screening tests that use human normal and cancer cells in-vitro to observe the effect of the studied compounds on their growth using MTT-based cytotoxicity. Microculture MTT methodCitation26 was used to determine cytotoxic activity of the newly synthesised compounds against normal human lung fibroblast (Wi-38) using doxorubicin (Dox) as standard anticancer drug. The highest IC50 and EC100 values illustrate that the compounds were safe on the proliferation of normal human cells. All the tested compounds showed significant noncytotoxic effect on human lung fibroblasts (Wi-38) with IC50 ranging from 0.0255 to 0.759 µM compared to doxorubicin (IC50 = 0.0266 µM). Compounds 5b, 5c, 6a–e, 7a–c, 9a–c, 11a–d, 11f–i, 14a–c, and 15a–c showed the highest IC50 values > 0.099 µM which revealed their safety on normal human cells (Table 1 in Supplementary Material, ).
Figure 4. IC50 and EC100 (μM) of the tested compounds against normal human cells (wi-38).
In-vitro anticancer screening
All final compounds were evaluated against four human cancer cell lines namely; liver cancer (HepG2) (ATCC® HB-8065™), colon cancer (Caco-2) (ATCC® HTB-37™), leukaemia cancer (NFS-60) (ATCC® CRL-1838™), and prostate cancer (PC-3) (ATCC® CRL-1435™) using doxorubicin (Dox) as the reference drug being widely used in tumour management utilising microculture MTT assay, declared by MosmannCitation26 (Table 1 in Supplementary Material, ). The provided data revealed that most of the tested compounds exerted remarkable broad antitumor activity against all tested cancer cell lines.
As a result, it can be concluded that pyridone moiety is important for high anticancer activity Moreover, compounds bearing 4-morpholinyl moiety are more crucial than that having 1-piperdinyl moiety. Therefore, new compounds were designed to keep morpholinyl − 2-oxopyridine scaffold basic structure with various substitutions (Scheme 3) aiming to potentiate the anticancer activity and hence their PIM kinase inhibitory activity. Compounds 7a–c were designed to acetylate 3-hydroxyphenyl group to study the effect of free OH on the activity. Such structural modification was found to abolish the anticancer activity compared with the hydroxyl derivatives 5a–c which confirmed the importance of the free hydroxyl group for maintaining high anticancer activity. Our study was excreted to include carboxylic acid moiety on 2-oxopyridine aiming to increase its solubility and hence its bioavailability. Such structural modification doesn’t possess superior anticancer activity. On the other hand, compound 9b showed low anticancer activity but with equipotent with the reference drug against HepG-2 cell line with IC50= 0.0454 µM. The O-(N-aryl acetamide) derivatives 11d, 11e, and 11f showed potent anticancer activity even more potent than doxorubicin against HepG-2 cell line with IC50= 0.0458, 0.0416 and 0.0363 µM, respectively (). Those compounds showed reasonable anticancer activity against NFS-60, PC-3 and Caco-2 cell lines with IC50 < 0.1 µM (). This activity may be due to an increased lipophilicity (6-chloroquinoline) that might enhance the binding pattern to protein hydrophobic moieties. Moreover, compound 11i showed nearly equipotent activity to the reference drug against HepG-2 cell line with IC50= 0.0486 µM. Finally, compounds 11b, 11g, and 11h displayed moderate anticancer activity against the four cancer cell lines.
Figure 5. In vitro anticancer activity, IC50 (μM), of the tested compounds against Caco-2.
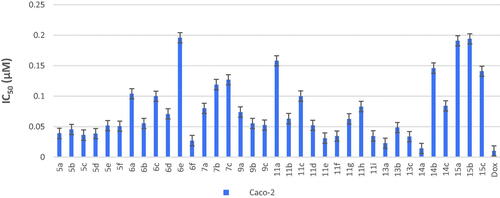
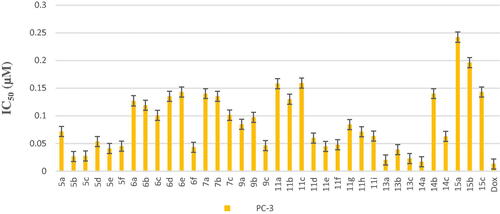
Figure 6. In vitro anticancer activity, IC50 (μM), of the tested compounds against HepG-2.
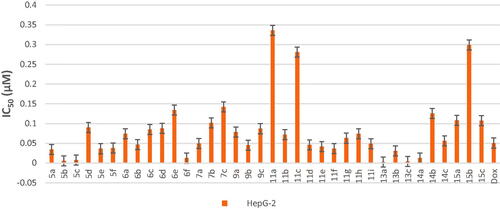
Figure 7. In vitro anticancer activity, IC50 (μM), of the tested compounds against NFS-60.
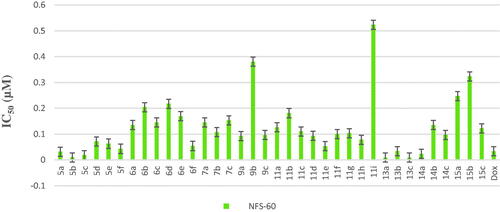
Figure 8. In vitro anticancer activity, IC50 (μM), of the tested compounds against PC-3.
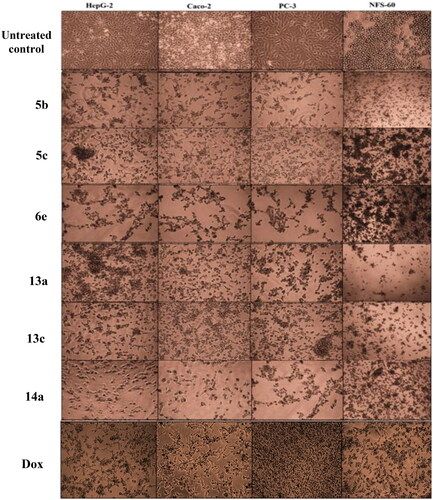
Another modification was performed through tethering the pyridine moiety with quinolone to study their effect on anticancer activity (Scheme 3). The results indicated that most of the tested compounds 13a–c, 14a–c, and 15a–c exerted potent broad antitumor activity against all tested cancer cell lines. Especially, the unsubstituted pyridone derivative 13a that displayed extraordinary increase in anticancer activity. It was twenty-one-fold more active against HepG-2 cell line (IC50=0.00238 μM) and with three-fold against NFS-60 cell line (IC50 = 0.009 μM) compared to the reference (IC50=0.0504 and 0.0332 μM, respectively) (). In addition, 6-methoxy derivative 13c exhibited thirteen-fold high in the activity against HepG-2 cell line and with three-fold high against NFS-60 cell line. Furthermore, the 6- chloro derivative 13b was equipotent activity with the reference drug against HepG-2 and NFS-60 cell lines with IC50 = 0.0309, 0.03373 μM, respectively, and with pronounced anticancer activity against PC-3 and Caco-2 cell lines. In addition, replacing of 2-oxo group on pyridine ring with 2-mercapto function 14a–c resulted in slightly decreasing in the anticancer activity. It was found that, the unsubstituted derivative 14a was the most active one against HepG-2 and NFS-60 cell lines with IC50= 0.0127and 0.0232 μM, respectively, and showed moderate activity against PC-3 and Caco-2 cell lines. Moreover, the 6-methoxy derivative 14c possessed anticancer activity against HepG-2 with IC50=0.0557 μM, in a manner that is equipotent to doxorubicin. On the other hand, a dramatic decline in the anticancer activity was recorded with replacement of 2-mercapto group with 2-amino group against the four cancer cell lines. Furthermore, the SI results (Table 1 in Supplementary Material) revealed that, compounds 13a, 13c, and 14a showed the highest SI values than the reference drug doxorubicin. They were the least cytotoxic and highest anticancer activity with high SI value compared to the Dox. These results indicated a successful discovery of new anticancer drug candidates and confirmed that they could be safe drugs against tumours. Additionally, morphological changes on the four cancer cell lines (HepG-2, Caco-2, NFS-60 and PC-3) were demonstrated before and after treatment with the most active and safe compounds 13a, 13b, and 14a in comparison with cells treated with the reference doxorubicin. those compounds caused severe shrinking and collapsing in the normal shape of four studied cancer cell lines as shown in .
Figure 9. Morphological alterations of the most active compounds-treated cancer cells lines in comparison with the untreated cancer cells.
Flow cytometric analysis of apoptosis
Flow cytometric annexin V/propidium iodide analysisCitation37 was used for detecting the apoptotic effects of the most active and non-toxic anticancer compounds 5b, 5c, 6e, 13a, 13c, and 14a. Results ( and and ) showed that all tested compounds except 5c possessed apoptosis-dependent death by over 42% in the treated HepG-2, Caco-2, NFS-60, and PC-3 cancer cell lines higher than doxorubicin which showed less than 39% apoptotic cell. It is worth mentioning that, compounds 6e, 13a, and 13c exerted the highest percentage of annexin-stained population cells (>66%) in comparison with the other most effective studied compounds.
Figure 10. Flow charts of Annexin-PI analysis of 5b, 5c, and 6e – treated cancer cell lines in comparison with the untreated cancer cells.
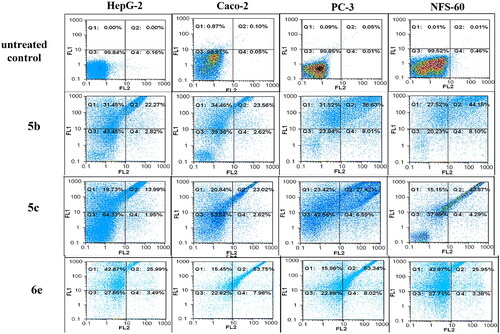
Figure 11. Flow charts of Annexin-PI analysis of 13a, 13c, and 14a – treated cancer cell lines in comparison with the untreated cancer cells.
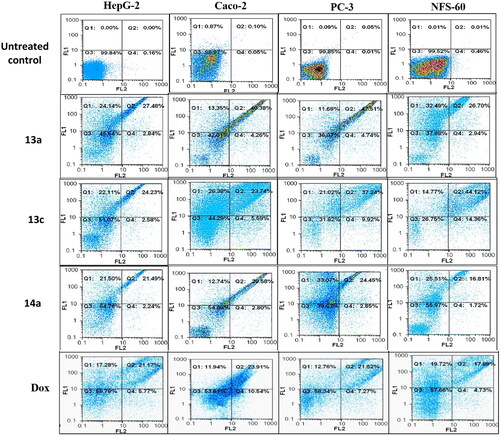
Figure 12. Lineweaver–Burk double-reciprocal plot for PIM kinase inhibition by 5c, 5b, and 6e in comparison with quercetin as reference inhibitor.
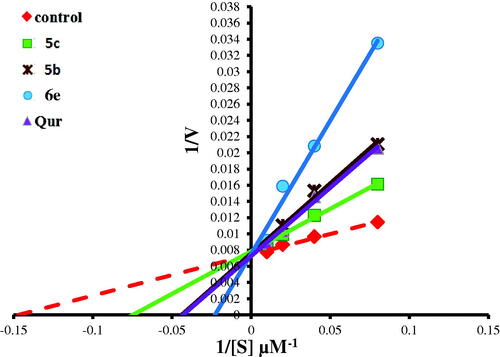
Table 1. The total percentage of the apoptotic cell population in the most effective compounds-treated cancer cells lines.
Caspase 3/7 activation assay
It’s a fluorescence assay that measures caspase-3 and −7 activities. Caspase 3/7 activators are well known as apoptotic inducers. In view of this, the apoptotic induction mechanism displayed by the most active compounds 5b, 5c, 6e, 13a, 13c, and 14a was explained by estimating the fold increase in caspase 3/7 activation in the treated cancer cell line relative to the untreated HepG-2 cells. Results () revealed that compounds 6e and 13a exhibited the highest caspase 3/7 up regulation by more than 3 folds. In addition, compounds 5b, 5c, and 13c significantly induced caspase 3/7 activation by more than two folds in HepG-2 cancer cell lines.
Table 2. Illustrates relative fold increase in caspase activity by the most effective compounds relative to untreated HepG2 cancer cells.
In-vitro PIM-1 and PIM-2 kinase inhibitory activity
The active synthesised compounds 5b, 5c, and 6e, were evaluated for their PIM-1 and PIM-2 kinase inhibitory activities using Quercetin, as reference drug (). All compounds showed potent PIM-1 kinase inhibitory activity comparable to the reference quercetin. The PIM-2 kinase inhibitory activity results showed that, compound 5b showed the highest inhibitory activity among other compounds compared to quercetin. While compound 5c and 6e excreted slightly less activity than the reference. Therefore, PIM-1 kinase inhibitory activities were tested for the other active synthesised compounds 13a, 13c, and 14a using Quercetin, as reference drug (). Compounds 13a and 14a showed potent PIM-1 kinase inhibitory activity.
Table 3. In-vitro PIM-1 and PIM-2 kinase inhibition data of the most active compounds.
Mechanism of PIM-1 kinase inhibition
To investigate the mechanism of inhibition of the most active compounds 5b, 5c, 6e, 13a, and 14a on PIM-1 kinase activity, enzyme kinetic parameters (maximum activity “Vmax” and substrate concentration at half Vmax “Km”) were assessed by Lineweaver–Burk double-reciprocal plot. As it was shown in this and . Lineweaver–Burk double-reciprocal plot for compounds 5b, 5c, 6e, and 14a showed the same Y-intercept with the control and quercetin (the same Vmax in comparison with control while the X-intercept has the negative sign (right to the control) which indicated an increasing in Km value from 7.24 µM (in case of control) to 14.16, 24.69, 27.84, 8.92, and 19.59 µM, for 5b, 5c, 6e, 14a, and quercetin. respectively. These results indicated that, those compounds are competitive inhibitors for PIM-1 kinase enzyme. In addition, the highest K was recorded in 6e and 5b comparing to 5c, 14a and reference PIM kinase inhibitor (quercetin) indicates the strongest inhibition effect of 6e and 5b via lowering the binding affinity of PIM kinase to its substrate by more than three folds. On the other hand, compound 13a revealed Vmax at a higher position on the axis (reduced in Vmax value) and also has the negative sign on the X-intercept (right to control) which indicated increase in Km value therefore, this compound is both competitive and non-competitive inhibitor for PIM-1 kinase enzyme.
Figure 13. Lineweaver–Burk double-reciprocal plot for PIM kinase inhibition by 13a and 14a in comparison with quercetin as reference inhibitor.
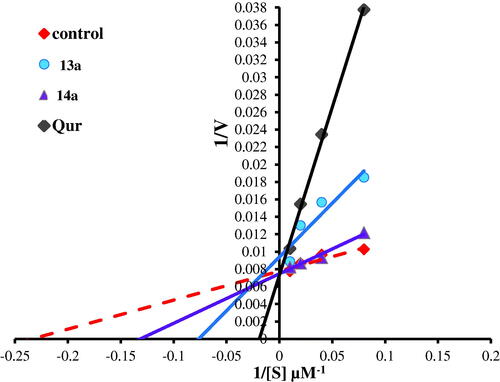
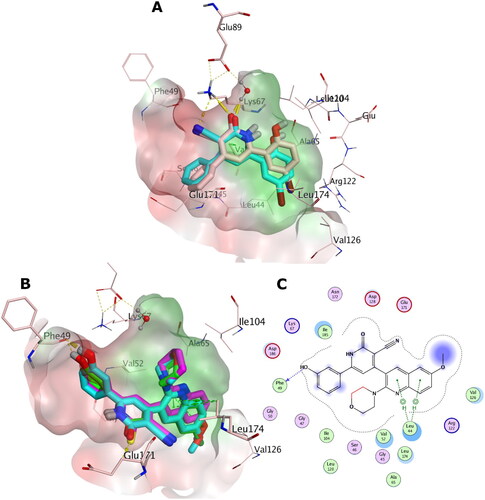
Figure 14. (A) Overlay of the X-ray co-crystal (simon sticks) on its docked pose (cyan sticks) in the binding site of PIM-1 kinase (PDB ID: 2OBJ). (B) Overlay of the docking poses of 5b, 5c, and 6e as green, cyan, and magenta sticks, respectively, in the binding site of PIM-1 kinase. (C) Interaction pattern of 5c with PIM-1 residues in 2D depictions. Polar and non-polar regions of the binding site were presented by red and green coloured molecular surface, respectively. Dashed lines indicate favourable interactions. Non-polar hydrogen atoms were omitted for clarity.
Structure activity relationship
The obtained results explained that most of the tested compounds exerted remarkable broad antitumor activity against all tested cancer cell lines. Compounds 5a, 5b, 5c, and 5d that contain pyridone moiety hybridised with quinoline derivatives and compound 6e which comprises thiopyridine moiety showed the most potent activity against Caco-2 cell line superior to doxorubicin which confirm the importance of the presence of 2-oxo or 2-thiopyridine core in the structure. Also, compounds 5b and 5c exerted the highest anticancer activity against all tested human cancer cell lines, correlated to other compounds and doxorubicin. Therefore, compounds containing morpholino moiety showed higher activity compared to piperdine-1-yl moiety. Except for, compound 6e which substituted with 2-piperidine and 6-methoxy group on quinoline ring demonstrated also higher anticancer activity against Caco-2 and HepG-2 than doxorubicin and nearly equipotent activity against NFS-60 and PC-3 cell lines. Finally, substitution at 6-position of quinoline with either lipophilic chloro group or hydrophilic methoxy group showed higher activity rather than unsubstituted one. On the other hand, acetylation of 3-hydroxyphenyl group was found to abolish the anticancer activity compared with the hydroxyl derivatives 5a–c which confirmed the importance of the free hydroxyl group for maintaining high anticancer activity. Furthermore, SAR study on different alkyl group on 2-oxopyridine revealed that, insertion of N-aryl acetamide showed potent anticancer activity rather than presence of carboxylic acid group which may be due to an increased lipophilicity that might enhance the binding pattern to the enzyme active site by hydrophobic interaction. In addition, linking pyridine to the electron deficient quinolone moiety achieved the goal and exhibited broad in-vitro antitumor activity against all tested cancer cell lines. Moreover, pyridone derivatives (13a and 13c) and thiopyridine derivative 14a showed the highest in-vitro anticancer activity against HepG-2 and NFS-60 cell lines superior to reference doxorubicin. Also, compounds 13a and 14a revealed potent PIM-1 kinase competitive inhibitory activity. These findings confirmed the importance of substitution at position 2 of pyridine with either oxo or thioxo rather than amino group in anticancer and PIM-1 kinase inhibitory activity. Besides, substitution at position 6 of quinolone with hydrophilic methoxy group or unsubstituted derivatives showed the highest activity rather than the hydrophobic chloro derivatives which could be due to increased polar interaction between the quinolone and PIM-1 kinase hinge region.
Molecular modelling
Optimisation methods
Ligand efficiency (LE)
A correlation between the potency of a compound (IC50) and the number of its heavy atoms (non-hydrogen atoms) is called Ligand Efficiency. It is a measurement of the binding energy which related to the potency of the compound (pIC50) with consider its molecular sizeCitation38. The suggested LE value for lead-likeness should be in the range of 0.3 while the acceptable LE value for drug-likeness should be higher than 0.3 ().
Table 4. LE and LLE values for the most active anticancer compounds against the four cancer cell lines (HepG-2, Caco-2, PC-3, NFS-60).
Ligand lipophilic efficiency (LLE)
LLE is a criterion that evaluate the potency in terms of lipophilicity. In other words, it measures the binding of a ligand to a given target in respect to its lipophilicityCitation38. LLE value should be ≥ 3 for lead compound to be acceptable and recommended to be LLE value ≥ 5 for drug like candidate ().
According to the results, compounds 5b and 5c obeyed acceptable ranges of LE (>0.3) and LLE values (> 3). Therefore, these compounds fulfilled the criteria of lipophilicity and ligand efficacy to be lead-like compounds. On the other hand, compounds 13a, 13c, and 14a showed drug like criteria (LE >0.3, LLE >5). Therefore, these compounds could impart improved potency along with convenient lipophilicity which make these compounds promising drug-like candidates in anticancer drug discovery.
Docking with PIM-1 and PIM-2 kinases
The aim of this section in our study is to provide in-silico insights to rationalise the observed in-vitro PIM-1 and PIM-2 inhibition activity based on our design approach.
For this we performed molecular docking experiments utilising the high-resolution X-ray structures for PIM-1 and PIM-2 kinases, namely, PDB IDs: 2OBJ and 4X7Q, respectively. To validate the docking setup, a pose-retrieval docking experiment was conducted for the co-crystal ligand against its respective X-ray coordinates. All pose-retrieval experiments were successful ( and with RMSD value < 2 Å indicating high docking accuracy for the docking setup for pose prediction purposes.
Figure 15. (A) Overlay of the docking poses of 13a, 13c, and 14a as magenta, green, and cyan sticks, respectively, in the binding site of PIM-1 kinase. (B) Interaction pattern of 13a, 13c, and 14a with PIM-1 residues in 2D depictions. Polar and non-polar regions of the binding site were presented by red and green coloured molecular surface, respectively. Dashed lines indicate favourable interactions. Non-polar hydrogen atoms were omitted for clarity.
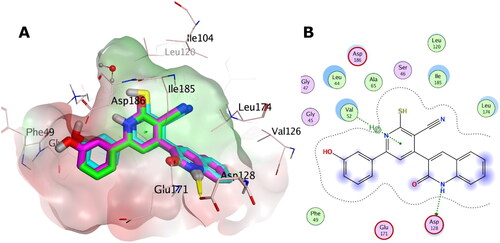
Concerning PIM-1 kinase, the docking score distribution () exhibits narrow range of scores for 5b, 5c, and 6e. This indicates comparable in-silico affinity which came in coherence with the observed in-vitro inhibitory activities (IC50 range: 1 − 1.31 µM) against PIM-1 kinase. However, the score distribution of the series of 13a, 13c, and 14a displayed superior scores compared to the 5 and 6 candidates, as well as the reference quercetin. One explanation for this is the topological differences between both series that affects pose orientations (as discussed later). It is obvious that 14a exhibited the best score among all compounds which agrees with the observed in-vitro inhibitory activities (14a is the best active one).
Table 5. The docking score distribution of 5b, 5c, 6e, 13a, 13c, 14a and Quercetin against PIM-1 and PIM-2 kinases.
Regarding PIM-2 kinase, the docking score of 5b and Quercetin indicate superior in-silico binding compared to 5c and 6e, as shown in . Again, this observation is in consistency with the measured in-vitro data where both 5b and Quercetin are the most active against PIM-2 kinase with a sub-micromolar range of activity compared to 5c and 6e (low micromolar range). It is worth mentioning that 5c, 6e, and Quercetin (except 5b) display superior docking score for PIM-1 compared to PIM-2 rationalising their observed in-vitro activities. The docking experiments for 13a, 13c, and 14a against PIM-2 were omitted due to the lack of PIM-2 inhibitory activity data.
Based on our analysis on the crystal PIM-1 protein structure, a structural water molecule appears to mediate a H-bonding bonding interaction of the pyridone scaffold of the co-crystal ligand with the side chain of Glu89. Therefore, we kept the structural water molecule in our docking setup. The pose retrieval of the co-crystal ligand was successful implying an acceptable accuracy of the docking setup.
Although both the co-crystal ligand and the selected candidates (5b, 5c, and 6e) share pyridone/thiopyridine moiety, the docking pose of the core pyridone scaffold of the selected candidates showed a flip towards Glu171. This is attributable to the topology difference between the co-crystal ligand which is 4,6-diaryl pyridone derivative and our quinolinyl – pyridone/thiopyridine hybrids.
Elucidating the docking poses of 5b, 5c, and 6e, they all produce comparable interactions in the binding site of PIM-1 kinase, as seen in . For instance, the core pyridone/thiopyridine demonstrate a favourable hydrophobic interaction with Glu171. Besides, the hydroxyphenyl group shows H-bonding interaction with the backbone of Phe49, e.g. as seen in for 5c. The methoxy/chloro quinoline moiety is well packed in the hydrophobic area (green area in ) indicating hydrophobic interactions with Leu174 and Leu44. Interestingly, the morpholine/piperidine moiety appeared to be partially included in a hydrophobic interaction with Ile104 and Ala45, and partially solvent-exposed. Overall, these findings exhibited comparable docking score for 5b, 5c, and 6e, as shown in . These results are consistent with observed in-vitro inhibitory activity against PIM-1 kinase since these compounds exhibited comparable IC50 values.
On the other hand, the postulated binding poses of the hybrids of pyridone/thiopyridine with 2-quinolone derivatives, namely 13a, 13c, and 14a, displayed flipped poses compared to 5 and 6 candidates, as exhibited in . This is attributable to the differences of topology and substitution pattern between both series. For instance, unlike 5b, 5c, and 6e poses, the pyridone/thiopyridine poses of 13a, 13c, and 14a point towards the structural water molecule (). This orientation enables the 2-quinolone moiety to have the advantage of making H-bonding interactions with the side chain of Asp128, and to be packed between the hydrophobic side chains of Val126 and Leu174 implying favourable hydrophobic interactions (). Likewise, the pyridone/thiopyridine moiety of 13a, 13c, and 14a is packed between the hydrophobic side chains of Val52 and Ile185. Overall, this postulated orientation produced superior in-silico binding particularly for 13a and 14a, as observed in .
Figure 16. (A) Overlay of the X-ray co-crystal (grey sticks) on its docked pose (cyan sticks) in the binding site of PIM-2 kinase (PDB ID: 4X7Q). (B) Overlay of the docking pose of 5b, 5c, and 6e as green, cyan, and magenta sticks, respectively, in the binding site of PIM-2 kinase. (C) Interaction pattern of 5b with PIM-2 residues in 2D depictions. Polar and non-polar regions of the binding site were presented by red and green coloured molecular surface, respectively. Dashed lines indicate favourable interactions. Non-polar hydrogen atoms were omitted for clarity.
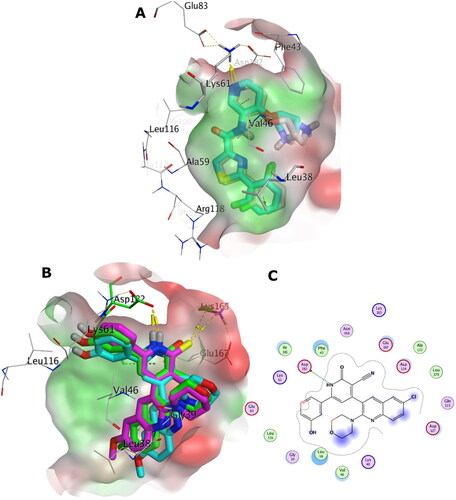
The postulated binding pose of 5b illustrates favourable interactions in the binding site of PIM-2 kinase as shown in ). The pyridone scaffold demonstrates H-bonding interaction with the side chain of Asp182 and hydrophobic interaction with the side chain of Ile181. The chloroquinoline moiety is packed in the hydrophobic area (green area in ) formed by Leu38, Ala59, Val46 and Asp124, while the morpholine moiety is directed towards the polar area (red area in ) formed by the side chains of Glu167 and Lys40. The hydroxyphenol group appeared to show hydrophobic interactions with the side chains of Val46. Like 5b, the docking poses of 5c and 6e showed similar interactions in the binding site of PIM-2 kinase. Interestingly, the chloroquinoline moiety of 5b appeared to be well packed in the hydrophobic area avoiding the possible steric bumps of the bigger methoxyquinoline moiety of the congeneric 5c and 6e. Globally, these observations contribute to the superior score of 5b towards the binding site of PIM-2 kinase compared to 5c and 6e.
In-silico physicochemical predictions
The purpose of this section is to deliver hints about the drug-likeness and pharmacokinetic properties of the active compounds 5b, 5c, and 6e. Explicating these properties can be supportive in the context of medicinal chemistry. Typically, a molecule that fails to fulfil the drug-likeness standards also fail as a clinical candidate due to poor bioavailability, adverse effects or other concerns. We employed the automated SwissADMECitation36 for the pharmacokinetics and drug-likeness predictions. The evaluations against passive human gastrointestinal absorption (GIA) and blood-brain barrier (BBB) permeation were extracted from the BOILED-Egg modelCitation36,Citation39, which represent some parameters for the pharmacokinetics predictions. The compounds presented high gastrointestinal absorption except 6e and 14a, as seen in . Also, these molecules were suggested to fail to permeate through BBB, and hence, they are expected to show low incidence for central nervous system (CNS) adverse effects. Permeability glycoprotein (P-gp) is proposed to be the most important member among ATP-binding cassette transporters or ABC-transportersCitation36 since it is part of the resistance mechanisms against drugsCitation36,Citation40. 5b, 5c, and 6e molecules were foreseen to be substrate for the P-gp which introduce a chance of being resistant against the human biological membranes. However, 13a, 13c, and 14a were predicted to escape the P-gp indicating low incidence of developing resistance against human biological membranes.
Table 6. In-silico predictions of the pharmacokinetics and drug-likeness properties for 5b, 5c, 6e, 13a, 13c and 14a.
Commonly, 50–90% of therapeutic molecules are judged to be a substrate of at least one of the five major isoforms of Cytochrome P (CYP) enzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4)Citation36,Citation42,Citation43. Inhibition of these isoenzymes is unquestionably one cause of pharmacokinetics-related drug-drug interactions leading to toxic or adverse effectsCitation44,Citation45. As shown in , all molecules were predicted to display inhibition of at least one of the CYP isoforms, with exception of 13a. This recommends administering these candidates in a sole regime and not in combination with other therapeutic agents. Fortunately, all molecules showed no alert to be a possible PAINS (pan-assay interference compounds)Citation46, as shown in . This stresses that their chemical structures would not interfere in protein assays denoting the in-vitro results to be robust with minimum artefacts. Lastly, drug-likeness properties underlined that all molecules do not display violations for the Lipinski and Veber rulesCitation47 highlighting that these candidates would demonstrate good bioavailability profiles.
Conclusion
New pyridine-quinoline hybrids were designed and synthesised as PIM-1 kinase inhibitors. They were evaluated against four human cancer cell lines namely; HepG2, Caco-2, NFS-60, and PC-3 using Dox as the reference. Compounds 5b, 5c, 13a, 13c, and 14a showed the highest anticancer activity against all tested human cancer cell lines, correlated to other compounds and doxorubicin. Also compound 6e demonstrated higher anticancer activity against Caco-2 and HepG-2 compared to Dox and nearly equipotent activity against NFS-60 and PC-3 cell lines. On the other hand, flow cytometric annexin V/propidium iodide analysis observed that, compound 6e revealed the highest percentage of annexin-stained population cells (>67%) in the four tested cancer cell lines in comparison with the other most effective studied compounds. In addition, caspase 3/7 activation assay elicited that, all tested compounds significantly induced caspase 3/7 activation in HepG-2 cancer cell lines. Furthermore, compounds 5b, 5c, 6e, 13a, and 14a showed potent PIM-1 kinase inhibitory activity comparable to the reference quercetin. Besides, kinetic studies using Lineweaver–Burk double-reciprocal plot for the most active compounds on PIM-1 kinase proved that, compounds 5b, 5c, 6e, and 14a and quercetin behaved as competitive inhibitors for PIM-1 kinase. While, compound 13a was both competitive and non-competitive inhibitor of PIM-1 kinase enzyme. In addition, molecular modelling study indicated that, compounds 5b and 5c fulfilled the criteria of lipophilicity and ligand efficacy to be lead-like compounds. Moreover, drug-likeness properties underlined that all molecules do not display violations for the Lipinski and Veber rules highlighting that these candidates would demonstrate good bioavailability profiles. Molecular docking studies indicated that, in-silico affinity came in coherence with the observed in-vitro inhibitory activities against PIM-1/2 kinases.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supplemental Material
Download PDF (2.6 MB)Acknowledgement
TMI acknowledges OpenEye Scientific Software Inc. for providing a free academic license. Also, he thanks the lab of Prof. Frank Boeckler (Eberhard Karls University Tuebingen) for offering a free access to perform some computational calculations.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Puente-Moncada N, Costales P, Antolin I, Nunez LE, Oro P, Hermosilla MA, Perez-Escuredo J, Rios-Lombardia N, Sanchez-Sanchez AM, Luno E, et al. Inhibition of FLT3 and PIM kinases by EC-70124 exerts potent activity in preclinical models of acute myeloid leukemia. Mol Cancer Ther. 2018;17(3):614–624.
- Ogawa N, Yuki H, Tanaka A. Insights from PIM1 structure for anti-cancer drug design. Expert Opin Drug Discov. 2012;7(12):1177–1192.
- Schenone S, Tintori C, Botta M. Using insights into PIM1 structure to design new anticancer drugs. Curr Pharm Des. 2010;16(35):3964–3978.
- Blanco-Aparicio C, Carnero A. PIM kinases in cancer: diagnostic, prognostic and treatment opportunities. Biochem Pharmacol. 2013;85(5):629–643.
- Mohamed EA, Ismail NSM, Hagras M, Refaat H. Medicinal attributes of pyridine scaffold as anticancer targeting agents. Futur J Pharm Sci. 2021;7(1):24.
- Ye C, Zhang C, Huang H, Yang B, Xiao G, Kong D, Tian Q, Song Q, Song Y, Tan H, et al. The natural compound myricetin effectively represses the malignant progression of prostate cancer by inhibiting PIM1 and disrupting the PIM1/CXCR4 interaction. Cell Physiol Biochem. 2018;48(3):1230–1244.
- Liu K, Gao H, Wang Q, Wang L, Zhang B, Han Z, Chen X, Han M, Gao M. Hispidulin suppresses cell growth and metastasis by targeting PIM1 through JAK2/STAT3 signaling in colorectal cancer. Cancer Sci. 2018;109(5):1369–1381.
- Stafman LL, Mruthyunjayappa S, Waters AM, Garner EF, Aye JM, Stewart JE, Yoon KJ, Whelan K, Mroczek-Musulman E, Beierle EA. Targeting PIM kinase as a therapeutic strategy in human hepatoblastoma. Oncotarget. 2018;9(32):22665–22679.
- Brasó-Maristany F, Filosto S, Catchpole S, Marlow R, Quist J, Francesch-Domenech E, Plumb DA, Zakka L, Gazinska P, Liccardi G, et al. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat Med. 2016;22(11):1303–1313.
- Abnous K, Manavi H, Mehri S, Alibolandi M, Kamali H, Ghandadi M, Hadizadeh F. In vitro evaluation of dihydropyridine-3-carbonitriles as potential cytotoxic agents through PIM-1 protein kinase inhibition. Res Pharm Sci. 2017;12(3):196–203.
- Walhekar V, Bagul C, Kumar D, Muthal A, Achaiah G, Kulkarni R. Topical advances in PIM kinases and their inhibitors: medicinal chemistry perspectives. Biochim Biophys Acta Rev Cancer. 2022;1877(3):188725.
- Mori M, Tintori C, Christopher RS, Radi M, Schenone S, Musumeci F, Brullo C, Sanita P, Delle Monache S, Angelucci A, et al. A combination strategy to inhibit PIM-1: synergism between noncompetitive and ATP-competitive inhibitors. ChemMedChem. 2013;8(3):484–496.
- Morwick T. PIM kinase inhibitors: a survey of the patent literature. Expert Opin Ther Pat. 2010;20(2):193–212.
- Nair JR, Caserta J, Belko K, Howell T, Fetterly G, Baldino C, Lee KP. Novel inhibition of PIM2 kinase has significant anti-tumor efficacy in multiple myeloma. Leukemia. 2017;31(8):1715–1726.
- Wähler K, Kräling K, Steuber H, Meggers E. Non-ATP-mimetic organometallic protein kinase inhibitor. ChemistryOpen. 2013;2(5-6):180–185.
- Lee SJ, Han B-G, Cho J-W, Choi J-S, Lee J, Song H-J, Koh JS, Lee BI. Crystal structure of PIM1 kinase in complex with a pyrido[4,3-d]pyrimidine derivative suggests a unique binding mode. PLos One. 2013;8(7):1–7.
- Chen J, Kobayashi M, Darmanin S, Qiao Y, Gully C, Zhao R, Kondo S, Wang H, Wang H, Yeung S-CJ, et al. Hypoxia-mediated up-regulation of PIM-1 contributes to solid tumor formation. Am J Pathol. 2009;175(1):400–411.
- Cheney IW, Yan S, Appleby T, Walker H, Vo T, Yao N, Hamatake R, Hong Z, Wu JZ. Identification and structure-activity relationships of substituted pyridones as inhibitors of PIM-1 kinase. Bioorg Med Chem Lett. 2007;17(6):1679–1683.
- Li K, Li Y, Zhou D, Fan Y, Guo H, Ma T, Wen J, Liu D, Zhao L. Synthesis and biological evaluation of quinoline derivatives as potential anti-prostate cancer agents and PIM-1 kinase inhibitors. Bioorg Med Chem. 2016;24(8):1889–1897.
- Swellmeen L, Shahin R, Al-Hiari Y, Alamiri A, Hasan A, Shaheen O. Structure based drug design of PIM-1 kinase followed by pharmacophore guided synthesis of quinolone-based inhibitors. Bioorg Med Chem. 2017;25(17):4855–4875.
- Abdelaziz ME, El-Miligy MMM, Fahmy SM, Mahran MA, Hazzaa AA. Design, synthesis and docking study of pyridine and thieno[2,3-b] pyridine derivatives as anticancer PIM-1 kinase inhibitors. Bioorg Chem. 2018;(80):674–692.
- Howard JC. 2-amino-3-nitrotoluene. Org Synth. 1955:42–44.
- JJaS B. Synthesis of novel substituted quinoline derivatives with 2-amino-5-methylthiophene-3-carbonitrile. Indo Am J Pharm Res. 2014; 45(5):2496–2502.
- Hamama WS, Ibrahim ME, Gooda AA, Zoorob HH. Recent advances in the chemistry of 2-chloroquinoline-3-carbaldehyde and related analogs. RSC Adv. 2018;8(16):8484–8515.
- Abdel-Wahab BF, Khidre RE. 2-chloroquinoline-3-carbaldehyde II: synthesis, reactions, and applications. J Chem. 2013;2013:1–13.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983; 65(1-2):55–63.
- Xu T, Niu C, Zhang X, Dong M. β-Ecdysterone protects SH-SY5Y cells against β-amyloid-induced apoptosis via c-Jun N-terminal kinase- and Akt-associated complementary pathways. Lab Invest. 2018;98(4):489–499.
- Brown AM. A new software for carrying out one-way ANOVA post hoc tests. Comput Methods Programs Biomed. 2005;79(1):89–95.
- Available from: www.promega.com
- Ishchenko A, Zhang L, Le Brazidec J-Y, Fan J, Chong JH, Hingway A, Raditsis A, Singh L, Elenbaas B, Hong VS, et al. Structure-based design of low-nanomolar PIM kinase inhibitors. Bioorg Med Chem Lett. 2015;25(3):474–480.
- McGann M. Fred pose prediction and virtual screening accuracy. J Chem Inf Model. 2011;51(3):578–596.
- McGann M. Fred and hybrid docking performance on standardized datasets. J Comput Aided Mol Des. 2012;26(8):897–906.
- Andre CM, Hausman J-F, Guerriero G. Cannabis sativa: the plant of the thousand and one molecules. Front Plant Sci. 2016;7:19.
- Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer generation with omega: algorithm and validation using high quality structures from the protein databank and Cambridge structural database. J Chem Inf Model. 2010;50(4):572–584.
- Pazouki L, Niinemets Ü. Multi-substrate terpene synthases: their occurrence and physiological significance. Front Plant Sci. 2016;7:1019.
- Daina A, Michielin O, Zoete V. Swissadme: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717.
- Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1(3):1458–1461.
- Hopkins AL, Keseru GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13(2):105–121.
- Daina A, Zoete V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11(11):1117–1121.
- Szakacs G, Varadi A, Ozvegy-Laczka C, Sarkadi B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discov Today. 2008;13(9-10):379–393.
- Cheng T, Zhao Y, Li X, Lin F, Xu Y, Zhang X, Li Y, Wang R, Lai L. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J Chem Inf Model. 2007;47(6):2140–2148.
- Farghaly A-R. Synthesis, reactions and antimicrobial activity of some new indolyl-1,3,4-oxadiazole, triazole and pyrazole derivatives. J Chin Chem Soc. 2004;51(1):147–156.
- Di L. The role of drug metabolizing enzymes in clearance. Expert Opin Drug Metab Toxicol. 2014;10(3):379–393.
- Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34(1-2):17–35.
- Huang SM, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, Abraham S, Habet SA, Baweja RK, Burckart GJ, et al. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48(6):662–670.
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53(7):2719–2740.
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623.