
Abstract
A library of novel alkyl/benzyl (4-sulphamoylphenyl)carbamimidothioates was synthesised by selective S-alkylation of the easily accessible 4-thioureidobenzenesulphonamide. The compounds were assayed as inhibitors of four human (h) carbonic anhydrase isoforms hCA I, II, VII, and XIII, as well as three bacterial enzymes belonging to the β-CA class, MscCA from Mammaliicoccus (Staphylococcus) sciuri and StCA1 and StCA2, from Salmonella enterica (serovar Typhimurium). Most compounds investigated here exhibited moderate to low nanomolar inhibition constants against hCA I, II, and VII. The cytosolic hCA XIII was also inhibited by these compounds, but not as effective as hCA I, II, and VII. Several compounds were very effective against MscCA and StCA1. StCA2 was less inhibited compared to MscCA and StCA1. Some compounds showed considerable selectivity for inhibiting some CA isoforms. They may thus be considered as interesting starting points for the discovery and development of novel therapeutic agents belonging to this class of enzyme inhibitors.
Introduction
Enzymes are drug targets for at least 50% of the clinically used drugs Citation1. At leasta third of all known enzymes contain one or more metallic ions (such as Ca, K, Mg, Mn, Fe, Co, Ni, Cu, Zn, and Mo), that are essential to their biological functionsCitation2,Citation3. Among the zinc-containing metalloenzymes, the carbonic anhydrases (or carbonate dehydratases) (CA, EC 4.2.1.1) are abundantly present in most life forms, e.g. mammalian tissues, plants, algae as well as all types of microorganisms, such as bacteria, protozoans, diatoms, etcCitation4. Human CAs occur in at least 15 isoforms (CA I, II, III, IV, VA, VB, VI, VII, VIII, IX, X, XI, XII, XIII, and XIV) depending upon the tissue, organ, or cell-type they are distributed inCitation5. This enzyme assists the chemical interconversion of carbon dioxide and water to bicarbonate and H+ ions, which are thereafter involved in mammals in both physiological and pathological processes, including respiration, pH homeostasis, signal transduction, electrolyte secretion, bone resorption, lipogenesis, calcification, etcCitation6–8. Due to broad roles of carbonic anhydrases in metabolism, selective inhibition of their activity is an excellent therapeutic target for several diseasesCitation5, including glaucoma (CA II, IV), edoema (CA II), obesity (CA VA), cancer (CA IX, XII), sterility (CA XIII), haemolytic anaemia (CA I), altitude sickness (CA II), and more recently in the management of neuropathic pain (CA VII) or as antiinfectives (when pathogenic organisms CAs are being targeted)Citation9.
Sulphonamide-containing compounds are the main class of CA inhibitorsCitation5. More than 70 currently marketed drugs contain this privileged moietyCitation10, and many researchers have been working to explore it, due to the potential of sulphonamides against diverse diseases. In Scheme 1, some of the effective CA inhibitors of the sulphonamide/sulphamate types (compounds 1–4) are shown.
Scheme 1. Chemical structures of some selected examples of effective CA inhibitors 1–4.
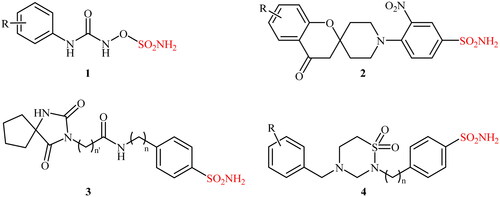
In continuation of our work on the developments of carbonic anhydrase inhibitorsCitation11, in this study we report the synthesis of a panel of 15 novel alkyl/benzyl (4-sulphamoylphenyl)carbamimidothioates, for which we explored their inhibitory effects against a panel of human and bacterial CA isoforms involved in various pathologies.
Results and discussion
Chemistry
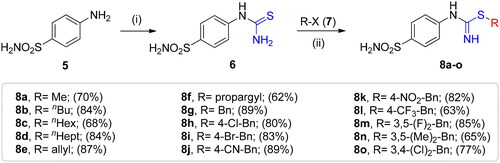
The synthetic route to obtain the target compounds reported here is outlined in Scheme 2. The 4-thioureidobenzenesulphonamide intermediate 6, prepared according to the literature procedureCitation12,Citation13 was upon treatment of sulphanilamide 5 with KSCN in a refluxing aqueous solution of HCl. The target alkyl/benzyl (4-sulphamoylphenyl)carbamimidothioates 8a–o were selectively synthesised by reaction of 4-thioureidobenzenesulphonamide 6 with the corresponding alkyl/benzyl halides 7 at elevated temperatures. Except for a few compounds which were purified by column chromatography, most of them were purified by simple extraction using ethyl acetate, which afforded the desired products with high purity and yields ranging from 62% to 89%. The analytical and spectroscopic data (1H and 13C NMR chemical shifts and mass spectra) of the purified compounds are in agreement with the purposed structures (see Experimental section for details).
Scheme 2. Reagents and conditions: (i) KSCN, aq. 3.5 M HCl, reflux, 3 h, 31%; (ii) DMF 20–100 °C, 2.5–24 h.
Carbonic anhydrase inhibition
Sulphonamides 8a–o incorporating carbamimidothioate moieties have been tested as inhibitors of four human (h) CA isoforms, the cytosolic hCA I, II, VII and XIII, as well as the bacterial β-CAs from Mammaliicoccus (Staphylococcus) sciuriCitation14 and Salmonella enterica (serovar Typhimurium), StCA1 and StCA2Citation15. It should be mentioned that the first bacterial enzyme, MscCA, was originally reported by us as Staphylococcus aureus β-CA, SauCA based on a genomic sequence annotated in the data bases in 2017Citation14. A recent reanalysis of that sequence revealed that the original annotation was erroneous, and that the sequence encodes a β-class CA from another Staphylococcaceae family member, i.e. Staphylococcus sciuri, which is a Gram-positive, oxidase-positive, coagulase-negative member of these infectious bacteria known to provoke disease in humans and animals (it was originally isolated from the squirrel)Citation16. In 2020, the species was renamed as belonging to a new genus, as Mammaliicoccus sciuriCitation16. The hCAs included in the study were the ubiquitous hCA I and II, as well as hCA VII and XIII which are found in fewer tissues compared to hCA I and II, and are involved in several pathologies in the CNS (hCA VII) or reproductive tract (hCA XIII)Citation17,Citation18.
The following structure-activity relationship (SAR) can be evidenced from the data of .
Table 1. Inhibition data of human CA isoforms CA I, II, VII, and XIII and bacterial β-CA isoforms MscCA, from Mammaliicoccus (Staphylococcus) sciuri, and StCA1 and StCA2, from Salmonella enterica (serovar Typhimurium), with compounds 8a–o using AAZ as standard drug. 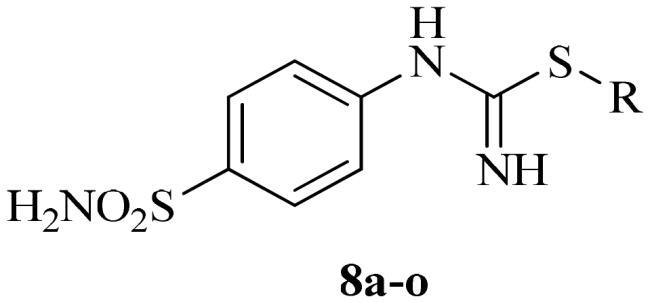
(i) The slow cytosolic human isoform hCA I was strongly inhibited by all the synthesised carbamimidothioate derivatives with KI ranging between 38.5 and 92.5 nM. These results indicated that all compounds reported here are more potent than the standard drug acetazolamide (AAZ) on this isoform. Overall the relative inhibitory rates of reported carbamimidothioates followed the order: allyl carbamimidothioate ≥ benzyl carbamimidothioates ≈alkyl carbamimidothioates ≥ propargyl carbamimidothioates. Although the SAR for the benzylic derivatives was not flat, the aliphatic derivatives showed a very flat SAR where the inhibition increased as the chains got longer.
(ii) The ubiquitous cytosolic human isoform hCA II was also effectively inhibited by all compounds in low to moderate nanomolar range with KI values ranging between 1.7 and 85.8 nM. Compound 8m having a 3,5-difluorobenzyl substitution was found to be the most effective amongst all reported derivatives, whereas the least effective inhibitor was the methyl-substituted compound 8a. The results showed that the presence of electron-withdrawing functionalities on the phenyl ring periphery of benzylic carbamimidothioates increased their inhibitory activities, whereas electron-donating groups reduced their potency. In the cases of aliphatic carbamimidothioates, it seems that the inhibitory activities increased with the increasing length of the carbon chain. For instance, the heptyl-substituted carbamimidothioate 8c was 34 times more potent as a CA II inhibitor compared to the methyl-substituted analogue 8a.
(iii) The brain-associated cytosolic isoform hCA VII showed a rather similar inhibition profile with hCA II, with most compounds investigated here. Thus, the 3,5-difluorobenzyl-substituted compound 8m was the strongest inhibitor with a KI of 1.2 nM whereas 3,5-dimethylbenzyl-, methyl-, and butyl-substituted carbamimidothioates 8o, 8a, and 8b, respectively, showed the weakest inhibitory capacities, with inhibition constants in the range of 13.8–25.6 nM (but they are still highly effective inhibitors). It is worth mentioning that more than half of the compounds investigated here (8c, 8e, 8g–8j, 8l, 8m), showed better inhibitory activities against hCA VII in comparison with the standard drug AAZ. Among them, half (8g–8j) showed more than 10 times a better selectivity against hCA VII vs hCA II.
(iv) The other cytosolic isoform hCA XIII was weakly inhibited by all investigated compounds (KI in the range of 69.3–925.9 nM) compared to AZA, which showed a KI of 16.0 nM. The best inhibitors were the 3,4-dichlorobenzyl- and 3,5-difluorobenzyl-substituted carbamimidothioates, 8n, 8m, which were however almost four times less potent inhibitor than acetazolamide.
(v) Against the bacterial β-CA isoform MscCA, except the benzyl- and 4-chlorobenzyl-substituted carbamimidothioates 8g, 8h, with KI values of 360.8 and 618.9 nM, respectively, the other compounds acted as weak inhibitors (KI in the range of 658.8–8761 nM) compared to the clinically used sulphonamide acetazolamide. It is noteworthy that while the allylic carbamimidothioate 8e showed the best result against hCA I, the worst result against MscCA were observed for this derivative. These results clearly indicated that the interaction of inhibitors with α- and β-CAs is rather different.
(vi) The bacterial β-CA isoform StCA1 was also inhibited by the synthesised compounds 8a–8n in a moderate nanomolar range with KI values of 52.2 to 750.9 nM. Similar to MscCA, the most potent inhibitor against this isoform was compound 8g with a potency similar to that of AAZ. Interestingly, the less effective inhibitor was again the benzyl-substituted derivative 8i with 4-bromo substitution. These results revealed that the inhibitory activity of benzyl-substituted carbamimidothioates 8g–8o against StCA1 significantly depended on the electronic effects of substituent on the phenyl ring. Generally, unsubstituted and strongly electron-withdrawing groups (e.g. CN, NO2) at the benzylcarbamimidothioate moiety induced higher inhibitory activities compared to electron-donating (e.g. Me) or less electron-withdrawing groups (e.g. Cl, Br).
(vii) StCA2 was the least inhibited bacterial CA among the three such enzymes investigated in this study. Indeed, the newly prepared compounds showed KI ranging between 270.2 and 9697 nM. Therefore, all of them were generally poor StCA2 inhibitors compared to AAZ. Interestingly, unlike StCA1, for which increasing the carbon chain-length of aliphatic carbamimidothioates led to an increased inhibition, for StCA2, the inhibition decreased by increasing the carbon chain-length in the synthesised derivatives.
Experimental protocols
Chemistry
Reagents, starting materials and solvents were obtained from commercial sources and used as received. Thin-layer chromatography was performed on silica gel, spots were visualised with UV light (254 and 365 nm). NMR spectra were recorded on Bruker 300 spectrometer with chemical shifts values (δ) in ppm relative to TMS using the residual DMSO-d6 signal (1H 2.50; 13C 39.52). High-resolution mass spectra (HRMS) were recorded on a mass spectrometer with a Q-TOF micro mass analyser using the ESI technique.
Synthesis
4-Thioureidobenzenesulphonamide (6)
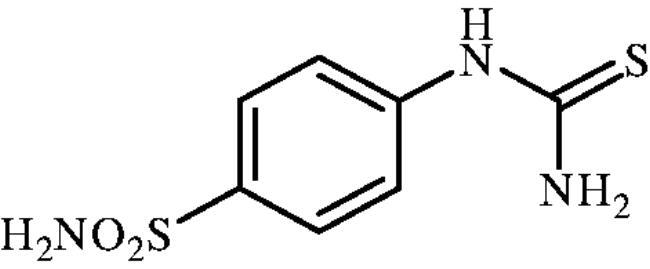
4-Aminobenzensulphonamide (5) (30 g, 174.3 mmol) was dissolved in 180 ml of 3.5 M HCl at 70 °C. After cooling to room temperature, KSCN (16.94 g, 174.3 mmol) was added and the mixture was refluxed for 3 h. After cooling to room temperature, the reaction mixture was diluted with ice-cooled water, filtered, washed with water. Solids formed were collected by filtration, washed with water, and air dried to afford 5 (12.1 g, 31%) as white powder.
1H NMR (300 MHz, DMSO-d6) δ = 7.32 (s, 2H), 7.69 (d, 2H, J = 8.6 Hz), 7.77 (d, 2H, J = 8.6 Hz), 10.02 (s, 1H) ppm 13C NMR (75 MHz, DMSO-d6) δ = 122.8, 127.3, 139.8, 143.9, 182.8 ppm MS (ESI) [M + H]+: m/z 232.0.
Methyl (4-sulphamoylphenyl)carbamimidothioate (8a)
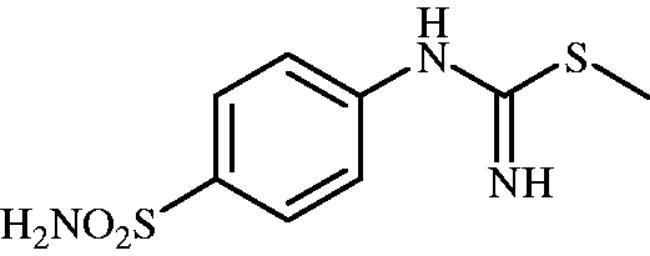
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) MeI (0.08 ml, 1.3 mmol) at room temperature was added and the mixture was heated at 40 °C for 2.5 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4. Solvent evaporation under vacuum afforded 8a (223 mg, 70%) as white powder.
1H NMR (300 MHz, DMSO-d6) δ = 2.37 (s, 3H), 6.63 (s, 2H), 6.94 (s, 2H), 7.22 (s, 2H), 7.71 (d, 2H, J = 8.4 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 14.2, 122.8, 127.7, 138.0, 153.9, 157.0 ppm HRMS (ESI) [M + H]+: m/z calcd for (C8H12N3O2S2) 246.0371. Found 246.0372.
Butyl (4-sulphamoylphenyl)carbamimidothioate (8b)
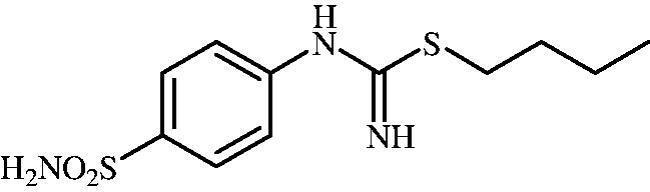
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) 1-iodobutane (0.176 ml, 1.6 mmol) at room temperature was added and the mixture was stirred at 30 °C for 24 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4. Solvent evaporation under vacuum afforded 8b (314 mg, 84%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 0.91 (t, 3H, J = 7.2 Hz), 1.33–1.45 (m, 2H), 1.54–1.64 (m, 2H), 2.98 (t, 2H, J = 7.2 Hz), 6.59 (s, 2H), 6.92 (d, 2H, J = 7.2 Hz), 7.19 (s, 2H), 7.68–7.72 (m, 2H) ppm 13C NMR (75 MHz, DMSO-d6) δ = 13.9, 21.7, 30.0, 31.7, 122.2, 127.2, 137.4, 153.4, 155.5 ppm HRMS (ESI) [M + H]+: m/z calcd for (C11H18N3O2S2) 288.0840. Found 288.0853.
Hexyl (4-sulphamoylphenyl)carbamimidothioate (8c)
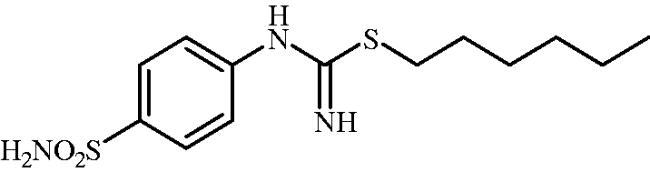
1-Bromohexane (0.181 ml, 1.3 mmol) and potassium iodide (215 mg, 1.3 mmol) were added to DMF (4 ml) and stirred at 100 °C for 1 h to in situ generate 1-iodohexane. After cooling to room temperature, 4-thioureidobenzenesulphonamide (6) (250 mg, 1.08 mmol) was added to the reaction mixture and the mixture was stirred at 100 °C for 3 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4 and solvent was evaporated under vacuum. The residue was purified by column chromatography on silica gel (DCM:MeOH, 95:5) to afford 8c (232 mg, 68%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 0.89 (t, 3H, J = 6.7 Hz), 1.30–1.42 (m, 6H), 1.56–1.65 (m, 2H), 2.95 (t, 2H, J = 7.3 Hz), 6.58 (s, 2H), 6.91 (d, 2H, J = 8.3 Hz), 7.19 (s, 2H), 7.71 (d, 2H, J = 8.3 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 14.8, 22.9, 28.7, 30.0, 30.8, 31.6, 122.9, 127.6, 137.9, 154.1, 156.0 ppm HRMS (ESI) [M + H]+: m/z calcd for (C13H22N3O2S2) 316.1153. Found 316.1161.
Heptyl (4-sulphamoylphenyl)carbamimidothioate (8d)
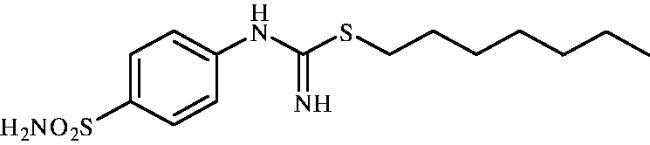
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) 1-iodoheptane (0.213 ml, 1.3 mmol) was added at room temperature. The mixture was stirred at 30 °C for 24 h. After cooling to room temperature aq. sat. NaHCO3 (20 ml) was added to the reaction mixture. A white precipitate formed was collected by filtration, followed by washing with water and drying on air to afford 8d (358 mg, 84%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 0.88 (d, 3H, J = 6.3 Hz), 1.28 (br. s, 8H), 1.56–1.65 (m, 2H), 2.94 (t, 2H, J = 6.9 Hz), 6.59 (s, 2H), 6.92 (d, 2H, J = 7.9 Hz), 7.21 (s, 2H), 7.71 (d, 2H, J = 7.9 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 14.4, 22.5, 28.5, 28.6, 29.6, 30.3, 31.6, 122.4, 127.2, 137.4, 153.6, 155.5 ppm HRMS (ESI) [M + H]+: m/z calcd for (C14H24N3O2S2) 330.1310. Found 330.1314.
Allyl (4-sulphamoylphenyl)carbamimidothioate (8e)
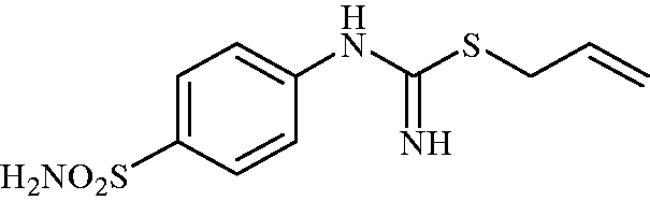
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) allyl bromide (0.112 ml, 1.3 mmol) was added and the mixture was stirred at 30 °C for 24 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4. Solvent evaporation under vacuum afforded 8e (304 mg, 87%) a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 3.66 (d, 2H, J = 6.7 Hz), 5.13 (d, 1H, J = 9.9 Hz), 5.28 (d, 1H, J = 16.9 Hz), 5.85–5.99 (m, 1H), 6.64 (s, 2H), 6.95 (s, 2H), 7.20 (s, 2H), 7.72 (d, 2H, J = 8.5 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 33.7, 118.5, 122.8, 127.7, 134.9, 138.0, 153.7, 155.3 ppm HRMS (ESI) [M + H]+: m/z calcd for (C10H14N3O2S2) 272.0527. Found 272.0538.
Prop-2-yn-1-yl (4-sulphamoylphenyl)carbamimidothioate (8f)
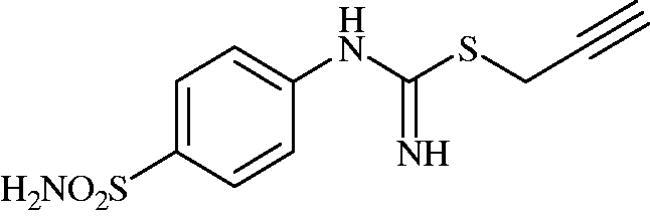
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) 3-bromoprop-1-yne (80% in toluene) (0.144 ml, 1.3 mmol) was added at 20 °C and the mixture was stirred at 20 °C for 7 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4. Solvent evaporation under vacuum afforded 8f (216 mg, 62%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 3.21 (s, 1H), 3.85 (s, 2H), 6.76 (s, 2H), 6.94 (d, 2H, J = 6.6 Hz), 7.23 (s, 2H), 7.72 (dd, 2H, J = 6.6, 1.5 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 19.2, 74.5, 81.2, 123.0, 127.7, 138.2, 153.5, 154.5 ppm HRMS (ESI) [M + H]+: m/z calcd for (C10H12N3O2S2) 270.0371. Found 270.0374.
Benzyl (4-sulphamoylphenyl)carbamimidothioate (8g)
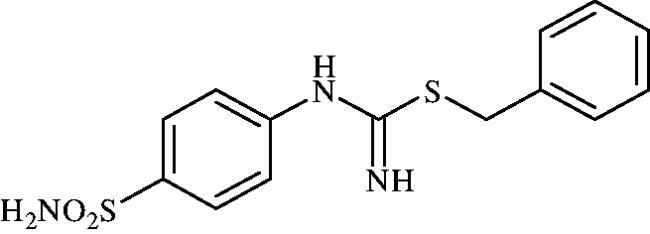
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) benzyl bromide (0.153 ml, 1.3 mmol) was added at room temperature and the mixture was stirred at 40 °C for 3 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4. Solvent evaporation under vacuum afforded 8g (370 mg, 89%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 4.25 (s, 2H), 6.71 (s, 2H), 6.91 (d, 2H, J = 8.3 Hz), 7.21 (s, 2H), 7.28–7.41 (m, 5H), 7.71 (d, 2H, J = 8.3 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 35.0, 122.8, 127.7, 127.9, 129.3, 129.8, 138.1, 138.9, 153.2, 155.8 ppm HRMS (ESI) [M + H]+: m/z calcd for (C14H16N3O2S2) 322.0684. Found 322.0693.
4-Chlorobenzyl (4-sulphamoylphenyl)carbamimidothioate(8h)
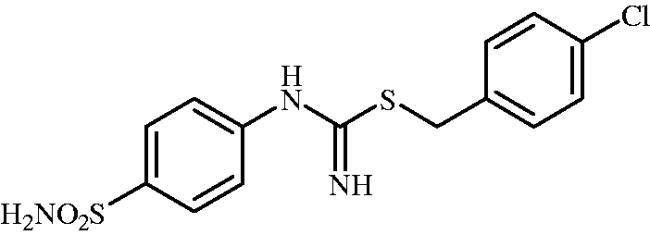
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) 4-chlorobenzyl bromide (266 mg, 1.3 mmol) was added at room temperature and the mixture was stirred at 30 °C for 6 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4. Solvent evaporation under vacuum afforded 8h (271 mg, 80%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 4.25 (s, 2H), 6.80 (s, 2H), 6.96–6.98 (m, 2H), 7.23 (s, 2H), 7.39–7.45 (m, 4H), 7.72 (d, 2H, J = 8.5 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 34.0, 122.8, 127.7, 129.2, 131.6, 132.5, 138.1, 138.4, 153.5, 155.5 ppm HRMS (ESI) [M + H]+: m/z calcd for (C14H15N3O2S2Cl) 356.0294. Found 356.0301.
4-Bromobenzyl (4-sulphamoylphenyl)carbamimidothioate (8i)
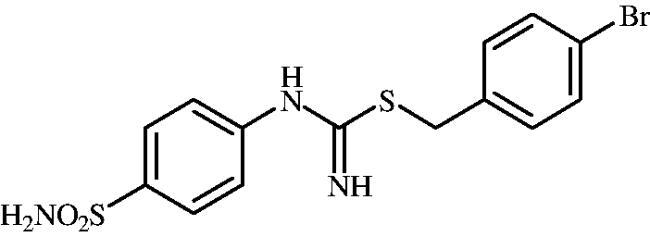
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) 4-bromobenzyl bromide (324 mg, 1.3 mmol) was added at room temperature and the mixture was stirred at 30 °C for 6 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4. Solvent evaporation under vacuum afforded 8i (430 mg, 83%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 4.24 (s, 2H), 6.81 (s, 2H), 6.95–6.98 (m, 2H), 7.23 (s, 2H), 7.37 (d, 2H, J = 8.3 Hz), 7.55 (d, 2H, J = 8.3 Hz), 7.71–7.74 (m, 2H, m) ppm 13C NMR (75 MHz, DMSO-d6) δ = 34.0, 121.0, 122.8, 127.7, 132.0, 132.1, 138.1, 138.9, 153.4, 155.5 ppm HRMS (ESI) [M + H]+: m/z calcd for (C14H15N3O2S2Br) 399.9789. Found 399.9798.
4-Cyanobenzyl (4-sulphamoylphenyl)carbamimidothioate(8j)
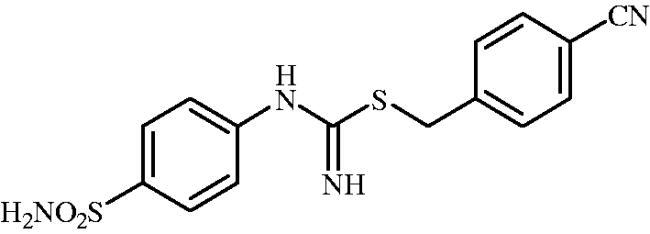
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) 4-cyanobenzyl bromide (255 mg, 1.3 mmol) was added at room temperature and the mixture was stirred at 30 °C for 6 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4 and solvent was evaporated under vacuum. The residue was purified by column chromatography on silica gel (DCM:MeOH, 95:5) to afford 8j (401 mg, 89%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 4.33 (s, 2H), 6.75 (s, 2H), 6.91 (d, 2H, J = 8.2 Hz), 7.24 (s, 2H), 7.61 (d, 2H, J = 8.2 Hz), 7.72 (d, 2H, J = 8.1 Hz), 7.82 (d, 2H, J = 8.2 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 34.3, 110.5, 119.8, 122.9, 127.8, 130.8, 133.2, 138.2, 145.7, 153.7, 155.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C15H15N4O2S2) 347.0636. Found 347.0645.
4-Nitrobenzyl (4-sulphamoylphenyl)carbamimidothioate(8k)
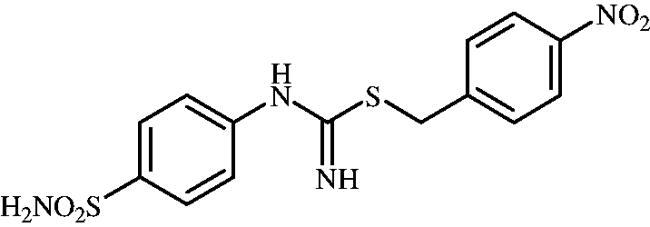
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) 4-nitrobenzyl bromide (280 mg, 1.3 mmol) was added at room temperature and the mixture was stirred at 30 °C for 6 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4. Solvent evaporation under vacuum afforded 8k (390 mg, 82%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 4.39 (s, 2H), 6.79 (s, 2H), 6.95 (s, 2H), 7.24 (s, 2H), 7.67–7.74 (m, 4H), 8.23 (d, 2H, J = 8.7 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 34.0, 122.8, 124.4, 127.8, 131.0, 138.2, 147.3, 148.0, 153.4, 155.0 ppm HRMS (ESI) [M + H]+: m/z calcd for (C19H15N2O2S2) 367.0575. Found 367.0558.
4-(Trifluoromethyl)benzyl (4-sulphamoylphenyl)carbamimidothioate(8l)
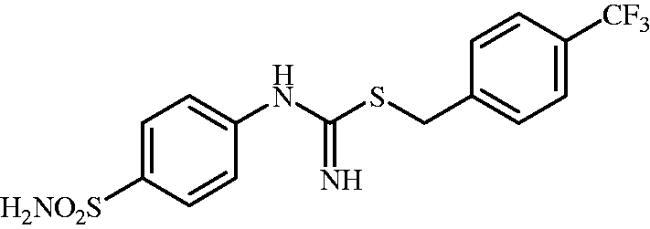
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) 1-(bromomethyl)-4-(trifluoromethyl)benzene (310 mg, 1.3 mmol) was added at room temperature and the mixture was stirred at 30 °C for 7 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO and solvent was evaporated under vacuum. The residue was purified by column chromatography on silica gel (DCM:MeOH, 95:5) to afford 8l (317 mg, 63%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 4.35 (s, 2H), 6.75 (s, 2H), 6.93 (d, 2H, J = 7.9 Hz), 7.27 (s, 2H), 7.64 (d, 2H, J = 7.9 Hz), 7.71–7.74 (m, 4H) ppm 13C NMR (75 MHz, DMSO-d6) δ = 34.1, 123.0, 126.1 (d, J = 3.8 Hz), 127.8, 128.5 (q, J = 31.1 Hz), 130.6, 138.2, 144.6, 153.8, 155.2 ppm 19F NMR (470 MHz, DMSO-d6) δ = −60.86 ppm HRMS (ESI) [M + H]+: m/z calcd for (C15H15N3O2S2F3) 390.0558. Found 390.0564.
3,5-Difluorobenzyl (4-sulphamoylphenyl)carbamimidothioate(8m)
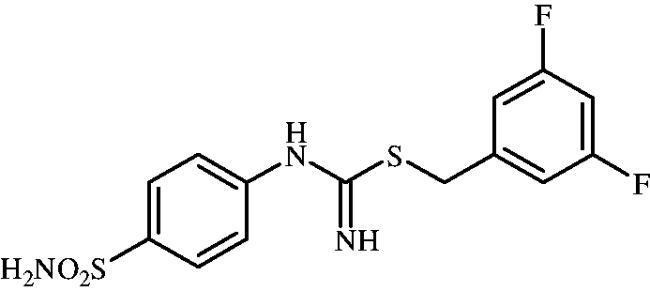
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) 1-(bromomethyl)-3,5-difluorobenzene (168 ml, 1.3 mmol) was added at room temperature and the mixture was stirred at 30 °C for 8 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4. Solvent evaporation under vacuum afforded 8m (395 mg,85%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 4.28 (s, 2H), 6.79 (s, 2H), 6.95 (d, 2H, J = 8.5 Hz), 7.13–7.17 (m, 3H), 7.24 (s, 2H), 7.76 (d, 2H, J = 8.5 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 33.9, 103.4 (app t, J = 26.8 Hz), 112.9 (dd, J = 9.5 Hz, 7.7 Hz), 122.8, 127.8, 138.2, 144.4, 153.4, 155.2, 161.1 (dd, J = 232.6 Hz, 13.2 Hz) ppm 19F NMR(470 MHz, DMSO-d6) δ = −110.06 ppm HRMS (ESI) [M + H]+: m/z calcd for (C14H14N3O2S2F2) 358.0495. Found 358.0497.
3,5-Dimethylbenzyl (4-sulphamoylphenyl)carbamimidothioate (8n)
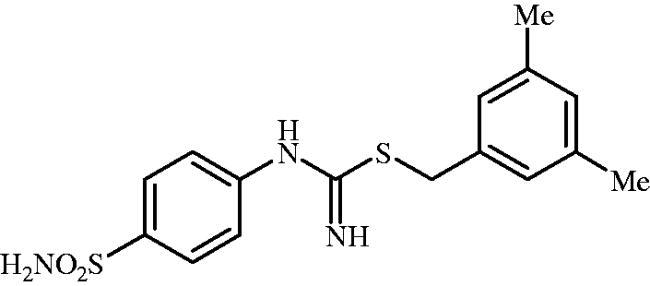
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) 1-(bromomethyl)-3,5-dimethylbenzene (261 mg, 1.3 mmol) was added at room temperature and the mixture was stirred at 30 °C for 8 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4 and solvent was evaporated under vacuum. The residue was purified by column chromatography on silica gel (DCM:MeOH, 95:5) to afford 8n (294 mg, 65%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 2.27 (s, 6H), 4.17 (s, 2H), 6.69 (s, 2H), 6.90–6.93 (m, 3H), 6.98 (s, 2H), 7.21 (s, 2H), 7.70–7.73 (m, 2H) ppm 13C NMR (75 MHz, DMSO-d6) δ = 21.3, 34.4, 122.4, 127.1, 127.2, 128.9, 137.5, 137.8, 138.0, 153.5, 155.2 ppm HRMS (ESI) [M + H]+: m/z calcd for (C16H20N3O2S2) 350.0997. Found 350.1007.
3,4-Dichlorobenzyl (4-sulphamoylphenyl)carbamimidothioate (8o)
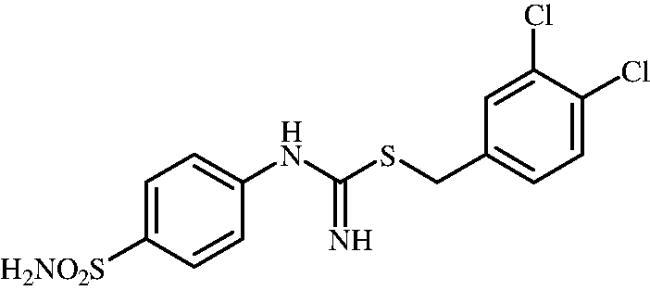
To a solution of 4-thioureidobenzenesulphonamide (6) (300 mg, 1.3 mmol) in DMF (4 ml) 4-(bromomethyl)-1,2-dichlorobenzene (189 ml, 1.3 mmol) was added at room temperature and the mixture was stirred at 30 °C for 6 h. After cooling to room temperature water (30 ml) was added and the mixture was extracted with EtOAc (3 × 20 ml). Combined organic layer was washed with aq. sat. NaHCO3 (2 × 20 ml) and aq. sat. NH4Cl (20 ml), and dried over Na2SO4 and solvent was evaporated under vacuum. The residue was purified by column chromatography on silica gel (DCM:MeOH, 95:5) to afford 8o (391 mg, 77%) as a white powder.
1H NMR (300 MHz, DMSO-d6) δ = 4.25 (s, 2H), 6.75 (s, 2H), 6.92 (d, 2H, J = 8.0 Hz), 7.24 (s, 2H), 7.41 (d, 1H, J = 8.2 Hz), 7.62 (d, 1H,J = 8.2 Hz), 7.69 (s, 1H), 7.73 (d, 2H, J = 8.0 Hz) ppm 13C NMR (75 MHz, DMSO-d6) δ = 33.4, 122.9, 127.8, 130.2, 130.4, 131.4, 131.6, 131.7, 138.2, 141.1, 153.7, 155.1 ppm HRMS (ESI) [M + H]+: m/z calcd for (C14H14N3O2S2Cl2) 389.9904. Found 389.9911.
Ca inhibitory assay
An applied photophysics stopped-flow instrument has been used for assaying the CA-catalysed CO2 hydration activityCitation19. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer for α-CAs or 20 mM TRIS (pH 8.4) as buffer for β-CAs, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10 – 100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5 – 10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled – deionised water, and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 6 h at room temperature prior to assay in order to allow for the formation of the E – I complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation12,Citation13,Citation20,Citation21, and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation22,Citation23 and their concentrations in the assay system ranged between 7.6–12.5 nM.
Conclusions
A new series of novel alkyl/benzyl (4-sulphamoylphenyl)carbamimidothioates (8a–o) was designed, synthesised and evaluated for their ability to inhibit four pharmacologically significant cytosolic hCA isozymes, hCA I, hCA II, hCA VII and hCA XIII as well as three β-CAs from the bacterial pathogens Mammaliicoccus (Staphylococcus) sciuri, MscCA, and Salmonella enterica (serovar Typhimurium), StCA1 and StCA2. Listed below are some important information of the bioassays: (i) all investigated compounds 8a–o showed strong inhibitory activities against hCA I (KIs, 38.5–92.5 nM) compared to that of AAZ (KI, 250.0 nM); (ii) up to half of the compounds investigated here (8c–f, 8k–n) showed better inhibitory activities against hCA II (KIs, 1.7–9.4 nM) in comparison with AAZ (KI, 12.5 nM); (iii) Among the 15 compounds examined, 8 of them (8c, 8e, 8g–8j, 8l, and 8m) showed better inhibitory activities against hCA VII (KIs, 1.2–2.4 nM) in comparison with AAZ (KI, 2.5 nM). Importantly, among these 8 compounds, half (8g–8j) showed more than 10 times much better selectivity against hCA VII vs hCA II; (iv) All investigated compounds (8a-o) exhibited weaker inhibitory activity (KIs, 69.3–925.9 nM) than AAZ (KI, 16.0 nM); (v) Against the bacterial β-CA isoform MscCA, except compounds 8g and 8h with KI values of 360.8 and 618.9 nM, respectively, other compounds acted as weak inhibitors (KIs, 658.8–8761 nM) compared to AAZ (KI, 625 nM); (vi) Against the bacterial β-CA isoform StCA1, only benzyl- and 4-cyanobenzyl-substituted carbamimidothioates 8g and 8j with KI values of 52.2 and 55.7 nM, respectively, showed better inhibitory activities compared to AAZ (KI, 59 nM); and (vii) These compounds were generally poor inhibitors of StCA2 with KI values ranging between 270.2 and 9697 nM.
Disclosure statement
No potential conflict of interest was reported by all author(s) except CTS. CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- a) Vermelho AB, Mori M, Donald WA, Supuran CT. Challenges and promises for obtaining new antiprotozoal drugs: What’s going wrong? In: Vermelho AB, Supuran CT, editors. Antiprotozoal drug development and delivery. Cham (Switzerland): Springer Nature; 2022. p. 321–330. b) Supuran CT, Capasso C. Carbonic anhydrases from pathogens: bacterial carbonic anhydrases and their inhibitors as potential antiinfectives. In: Supuran CT, Nocentini A, editors. Carbonic anhydrases – biochemistry and pharmacology of an evergreen pharmaceutical target. London (UK): Elsevier – Academic Press; 2019. p. 387–417. c) Capasso C, Supuran CT. Dihydropteroatesynthase (sulfonamides) and dihydrofolate reductase inhibitors. In: Bonev BB, Brown NM, editors. Bacterial resistance to antibiotics - from molecules to man. West Sussex (UK): John Wiley & Sons Ltd; 2020, p. 163–172.
- Wang S, Blahut M, Wu Y, Philipkosky KE, Outten FW. Communication between binding sites is required for YqjI regulation of target promoters within the yqjH-yqjI intergenic region. J Bacteriol. 2014;196(17):3199–3207.
- a) Dupont CL, Butcher A, Valas RE, Bourne PE, Caetano-Anollés G. History of biological metal utilization inferred through phylogenomic analysis of protein structures. Proc Natl Acad Sci USA. 2010; 107 (23):10567–10572. b) Supuran CT, Winum JY. Introduction to zinc enzymes as drug targets. In: Supuran CT, Winum JY, editors. Drug design of zinc-enzyme inhibitors: functional, structural, and disease applications. Hoboken (NJ): Wiley; 2009. p. 3–12.
- a) Supuran CT. Emerging role of carbonic anhydrase inhibitors. Clin Sci. 2021;135(10):1233–1249. b) Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem. 2007;15(13):4336–4350. c) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat. 2018;28(10):709–712. d) Supuran CT. Novel carbonic anhydrase inhibitors. Future Med Chem. 2021;13(22):1935–1937.
- a) Alterio V, Di Fiore A, D'Ambrosio K, Supuran CT, De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev. 2012;112(8):4421–4468. b) Supuran CT. Anti-obesity carbonic anhydrase inhibitors: challenges and opportunities. J Enzyme Inhib Med Chem. 2022;37(1):2478–2488. c) McDonald PC, Chafe SC, Supuran CT, Dedhar S. Cancer therapeutic targeting of hypoxia induced carbonic anhydrase IX: from bench to bedside. Cancers. 2022;14(14):3297. d) Supuran CT. Multitargeting approaches involving carbonic anhydrase inhibitors: hybrid drugs against a variety of disorders. J Enzyme Inhib Med Chem. 2021;36(1):1702–1714.
- a) Saad AE, Ashour DS, Rayia DMA, Bedeer AE. Carbonic anhydrase enzyme as a potential therapeutic target for experimental trichinellosis. Parasitol Res. 2016;115(6):2331–2339. b) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7(2):168–181. c) Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother. 2016;16(8):961–968. d) Carta F, Scozzafava A, Supuran CT. Sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat. 2012;22(7):747–758. e) Mishra CB, Tiwari M, Supuran CT. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev. 2020;40(6):2485–2565.
- a) Nocentini A, Angeli A, Carta F, Winum JY, Žalubovskis R, Carradori S, Capasso C, Donald WA, Supuran CT. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J Enzyme Inhib Med Chem. 2021;36(1):561–580. b) Abdoli M, Bonardi A, Supuran CT, Žalubovskis R. 4-Cyanamidobenzenesulfonamide derivatives: a novel class of human and bacterial carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2023;38(1):156–165. c) Supuran CT. Carbonic anhydrase inhibitors: an update on experimental agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs. 2021;30(12):1197–1208. d) Supuran CT. Experimental carbonic anhydrase inhibitors for the treatment of hypoxic tumors. J Exp Pharmacol. 2020;12:603–617.
- a) Angeli A, Velluzzi A, Selleri S, Capasso C, Spadini C, Iannarelli M, Cabassi CS, Carta F, Supuran CT. Seleno containing compounds as potent and selective antifungal agents. ACS Infect Dis. 2022;8(9):1905–1919. b) D'Agostino I, Mathew GE, Angelini P, Venanzoni R, Angeles Flores G, Angeli A, Carradori S, Marinacci B, Menghini L, Abdelgawad MA, et al. Biological investigation of N-methyl thiosemicarbazones as antimicrobial agents and bacterial carbonic anhydrases inhibitors. J Enzyme Inhib Med Chem. 2022;37(1):986–993. c) De Luca V, Angeli A, Mazzone V, Adelfio C, Carginale V, Scaloni A, Carta F, Selleri S, Supuran CT, Capasso C, et al. Heterologous expression and biochemical characterisation of the recombinant β-carbonic anhydrase (MpaCA) from the warm-blooded vertebrate pathogen malassezia pachydermatis. J Enzyme Inhib Med Chem. 2022;37(1):62–68. d) De Luca V, Angeli A, Mazzone V, Adelfio C, Carta F, Selleri S, Carginale V, Scaloni A, Supuran CT, Capasso C, et al. Inhibitory effects of sulfonamide derivatives on the β-carbonic anhydrase (MpaCA) from Malassezia pachydermatis, a commensal, pathogenic fungus present in domestic animals. IJMS. 2021;22(22):12601. e) Supuran CT, Capasso C. A highlight on the inhibition of fungal carbonic anhydrases as drug targets for the antifungal armamentarium. IJMS. 2021;22(9):4324.
- a) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal beta-class (Cab) and gamma-class (Cam) carbonic anhydrases. Curr Top Med Chem. 2007;7(9):901–908. b) Supuran CT, Clare BW. Carbonic anhydrase inhibitors. Part 57. Quantum chemical QSAR of a group of 1,3,4-thiadiazole and 1,3,4-thiadiazoline disulfonamides with carbonic anhydrase inhibitory properties. Eur J Med Chem. 1999;34(1):41–50. c) Supuran CT, Barboiu M, Luca C, Pop E, Brewster ME, Dinculescu A. Carbonic anhydrase activators. Part 14. Synthesis of mono- and bis- pyridinium salt derivatives of 2-amino-5-(2-aminoethyl)- and 2-amino-5-(3-aminopropyl)-1,3,4-thiadiazole, and their interaction with isozyme II. Eur J Med Chem. 1996;31(7-8):597–606. d) Aspatwar A, Barker H, Aisala H, Zueva K, Kuuslahti M, Tolvanen M, Primmer CR, Lumme J, Bonardi A, Tripathi A, et al. Cloning, purification, kinetic and anion inhibition studies of a recombinant β-carbonic anhydrase from the Atlantic salmon parasite platyhelminth Gyrodactylussalaris. J Enzyme Inhib Med Chem. 2022;37(1):1577–1586.
- Scott KA, Njardarson JT. Analysis of US FDA-approved drugs containing sulfur atoms. Top Curr Chem. 2018;376(1):5.
- a) Abdoli M, Giovannuzzi S, Supuran CT, Žalubovskis R. 4-(3-Alkyl/benzyl-guanidino)benzenesulfonamides as selective carbonic anhydrase VII inhibitors. J Enzyme Inhib Med Chem. 2022;37(1):1568–1576. b) Abdoli M, Angeli A, Bozdag M, Carta F, Kakanejadifard A, Saeidian H, Supuran CT. Synthesis and carbonic anhydrase I, II, VII, and IX inhibition studies with a series of benzo [d] thiazole-5-and 6-sulfonamides. J Enzyme Inhib Med Chem. 2017;32(1):1071–1078. c) Abdoli M, Bozdag M, Angeli A, Supuran CT. Benzamide-4-sulfonamides are effective human carbonic anhydrase i, ii, vii, and ix inhibitors. Metabolites. 2018;8(2):37.
- a) Petreni A, Iacobescu A, Simionescu N, Petrovici AR, Angeli A, Fifere A, Pinteala M, Supuran CT. Carbonic anhydrase inhibitors bearing organotelluride moieties as novel agents for antitumor therapy. Eur J Med Chem. 2022;244:114811. b) Artasensi A, Angeli A, Lammi C, Bollati C, Gervasoni S, Baron G, Matucci R, Supuran CT, Vistoli G, Fumagalli L. Discovery of a potent and highly selective dipeptidyl peptidase IV and carbonic anhydrase inhibitor as "antidiabesity" agents based on repurposing and morphing of WB-4101. J Med Chem. 2022;65(20):13946–13966. c) Najm MAA, Mahmoud WR, Taher AT, Abbas SE, Awadallah FM, Allam HA, Vullo D, Supuran CT. Design and synthesis of some new benzoylthioureido phenyl derivatives targeting carbonic anhydrase enzymes. J Enzyme Inhib Med Chem. 2022;37(1):2702–2709.
- a) Al-Warhi T, Elbadawi MM, Bonardi A, Nocentini A, Al-Karmalawy AA, Aljaeed N, Alotaibi OJ, Abdel-Aziz HA, Supuran CT, Eldehna WM. Design and synthesis of benzothiazole-based SLC-0111 analogues as new inhibitors for the cancer-associated carbonic anhydrase isoforms IX and XII. J Enzyme Inhib Med Chem. 2022;37(1):2635–2643. b) Biagiotti G, Angeli A, Giacomini A, Toniolo G, Landini L, Salerno G, Di Cesare Mannelli L, Ghelardini C, Mello T, Mussi S, et al. Glyco-coated CdSe/ZnS quantum dots as nanoprobes for carbonic anhydrase IX imaging in cancer cells. ACS Appl Nano Mater. 2021;4(12):14153–14160. c) Vannozzi G, Vullo D, Angeli A, Ferraroni M, Combs J, Lomelino C, Andring J, Mckenna R, Bartolucci G, Pallecchi M, et al. One-pot procedure for the synthesis of asymmetric substituted ureido benzene sulfonamides as effective inhibitors of carbonic anhydrase enzymes. J Med Chem. 2022;65(1):824–837.
- a) Urbanski LJ, Vullo D, Parkkila S, Supuran CT. An anion and small molecule inhibition study of the β-carbonic anhydrase from Staphylococcus aureus. J Enzyme Inhib Med Chem. 2021;36(1):1088–1092. b) Urbanski LJ, Bua S, Angeli A, Kuuslahti M, Hytönen VP, Supuran CT, Parkkila S. Sulphonamide inhibition profile of Staphylococcus aureus β-carbonic anhydrase. J Enzyme Inhib Med Chem. 2020;35(1):1834–1839.
- a) Nishimori I, Minakuchi T, Vullo D, Scozzafava A, Supuran CT. Inhibition studies of the β-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium with sulfonamides and sulfamates. Bioorg Med Chem. 2011;19(16):5023–5030. b) Vullo D, Nishimori I, Minakuchi T, Scozzafava A, Supuran CT. Inhibition studies with anions and small molecules of two novel β-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium. Bioorg Med Chem Lett. 2011;21(12):3591–3595. c) Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol. 2011;2:34.
- Angeli A, Urbański LJ, Capasso C, Parkkila S, Supuran CT. Activation studies with amino acids and amines of a β-carbonic anhydrase from Mammaliicoccus (Staphylococcus) sciuri previously annotated as Staphylococcus aureus (SauBCA) carbonic anhydrase. J Enzyme Inhib Med Chem. 2022;37(1):2786–2792.
- a) De Simone G, Di Fiore A, Truppo E, Langella E, Vullo D, Supuran CT, Monti SM. Exploration of the residues modulating the catalytic features of human carbonic anhydrase XIII by a site-specific mutagenesis approach. J Enzyme Inhib Med Chem. 2019;34(1):1506–1510. b) Monti DM, De Simone G, Langella E, Supuran CT, Di Fiore A, Monti SM. Insights into the role of reactive sulfhydryl groups of carbonic anhydrase III and VII during oxidative damage. J Enzyme Inhib Med Chem. 2017;32(1):5–12.
- a) Payaz DÜ, Küçükbay FZ, Küçükbay H, Angeli A, Supuran CT. Synthesis carbonic anhydrase enzyme inhibition and antioxidant activity of novel benzothiazole derivatives incorporating glycine, methionine, alanine, and phenylalanine moieties. J Enzyme Inhib Med Chem. 2019;34(1):343–349. b) José O, Torres-Rodríguez P, Forero-Quintero LS, Chávez JC, De la Vega-Beltrán JL, Carta F, Supuran CT, Deitmer JW, Treviño CL. Carbonic anhydrases and their functional differences in human and mouse sperm physiology. Biochem Biophys Res Commun. 2015;468(4):713–718. c) Bootorabi F, Jänis J, Hytönen VP, Valjakka J, Kuuslahti M, Vullo D, Niemelä O, Supuran CT, Parkkila S. Acetaldehyde-derived modifications on cytosolic human carbonic anhydrases. J Enzyme Inhib Med Chem. 2011;26(6):862–870.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.
- a)Pastorekova S, Casini A, Scozzafava A, Vullo D, Pastorek J, Supuran CT. Carbonic anhydrase inhibitors: the first selective, membrane-impermeant inhibitors targeting the tumor-associated isozyme IX. Bioorg Med Chem Lett. 2004;14(4):869–873. b) Vullo D, Voipio J, Innocenti A, Rivera C, Ranki H, Scozzafava A, Kaila K, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the human cytosolic isozyme VII with aromatic and heterocyclic sulfonamides. Bioorg Med Chem Lett. 2005;15(4):971–976. c) Gieling RG, Babur M, Mamnani L, Burrows N, Telfer BA, Carta F, Winum J-Y, Scozzafava A, Supuran CT, Williams KJ, et al. Antimetastatic effect of sulfamate carbonic anhydrase IX inhibitors in breast carcinoma xenografts. J Med Chem. 2012;55(11):5591–5600. d) Grandane A, Nocentini A, Werner T, Zalubovskis R, Supuran CT. Benzoxepinones: a new isoform-selective class of tumor associated carbonic anhydrase inhibitors. Bioorg Med Chem. 2020;28(11):115496.
- a) Krasavin M, Sharonova T, Sharoyko V, Zhukovsky D, Kalinin S, Žalubovskis R, Tennikova T, Supuran CT. Combining carbonic anhydrase and thioredoxin reductase inhibitory motifs within a single molecule dramatically increases its cytotoxicity. J Enzyme Inhib Med Chem. 2020;35(1):665–671. b) Ivanova J, Balode A, Žalubovskis R, Leitans J, Kazaks A, Vullo D, Tars K, Supuran CT. 5-Substituted-benzylsulfanyl-thiophene-2-sulfonamides with effective carbonic anhydrase inhibitory activity: Solution and crystallographic investigations. Bioorg Med Chem. 2017;25(3):857–863. c) Alterio V, Tanc M, Ivanova J, Zalubovskis R, Vozny I, Monti SM, Di Fiore A, De Simone G, Supuran CT. X-ray crystallographic and kinetic investigations of 6-sulfamoyl-saccharin as a carbonic anhydrase inhibitor. Org Biomol Chem. 2015; 13(13):4064–4069.
- a) Luchinat E, Barbieri L, Cremonini M, Pennestri M, Nocentini A, Supuran CT, Banci L. Determination of intracellular protein-ligand binding affinity by competition binding in-cell NMR. Acta Crystallogr D Struct Biol. 2021;77(Pt 10):1270–1281. b) Berrino E, Michelet B, Martin-Mingot A, Carta F, Supuran CT, Thibaudeau S. Modulating the efficacy of carbonic anhydrase inhibitors through fluorine substitution. Angew Chem Int Ed Engl. 2021;60(43):23068–23082.
- a) Bouzina A, Berredjem M, Nocentini A, Bua S, Bouaziz Z, Jose J, Le Borgne M, Marminon C, Gratteri P, Supuran CT. Ninhydrins inhibit carbonic anhydrases directly binding to the metal ion. Eur J Med Chem. 2021;209:112875. b) Angeli A, Carta F, Nocentini A, Winum JY, Zalubovskis R, Akdemir A, Onnis V, Eldehna WM, Capasso C, Simone G, et al. Carbonic Anhydrase Inhibitors Targeting Metabolism and Tumor Microenvironment. Metabolites. 2020;10(10):412.