
Abstract
Inhibition of PI3K pathway has become a desirable strategy for cancer treatment. In this work, a series of 2, 6, 8-substituted Imidazo[1,2-a]pyridine derivatives were designed and screened for their activities against PI3Kα and a panel of PI3Kα-addicted cancer cells. Among them, compound 35 was identified as a PI3Kα inhibitor with nanomolar potency as well as acceptable antiproliferative activity. Flow cytometry analysis confirmed 35 induced cell cycle arrest and apoptosis in T47D cells. In addition, it also showed desirable in vitro ADME properties. The design, synthesis, and SAR exploration of 35 are described within.
Introduction
Malignant tumours have always been one of the major diseases threatening human health. At present, the commonly used treatment methods for cancer are inseparable from the support of chemotherapy drugsCitation1. Therefore, it is of great significance to find anti-tumour drugs with high efficiency and low side effectsCitation2.
Phosphatidylinositol-3-kinases (PI3Ks) are a family of lipid kinases that are responsible for catalysing the phosphorylation of inositol phospholipids to produce PIP3 (phosphatidylinositol triphosphate). This phosphorylation process results in recruiting cytosolic signalling enzymes such as Akt to the plasma membrane, triggering cell growth, proliferation, differentiation, and motilityCitation3–5. Aberrant activation of PI3K as well as its downstream effectors including Akt and mTOR has been linked to numerous forms of cancer including lymphatic tumours, breast, lung, brain, ovarian, melanoma, and prostate cancersCitation6–8. Moreover, the negative regulator PTENCitation9 which dephosphorylates PIP3 to PIP2, is often inactivated in many cancer types, leading to elevated levels of PIP3 and increased tumour survivalCitation3. With distinct sequence homology and substrate preferences, the eight known PI3K family members are divided into classes I, II, and III. The most extensively studied Class I PI3Ks are further subdivided into Class IA (α, β, and δ) and Class IB (PI3Kγ), based upon the types of regulatory subunits and the catalytic domains to which they bind in the active heterodimeric forms. The former Class IA PI3Ks are activated through tyrosine kinase signalling, whereas the sole PI3Kγ is mostly activated through GPCRsCitation10.
While there is growing evidence that small molecule inhibition of PI3Ks as an attractive strategy for oncology indicationCitation11,Citation12. Several small molecule inhibitors have been approved by FDA for use in patients or under active clinical development, including pan-PI3K inhibitors Copanlisib (1)Citation13,Citation14, Buparlisib (2)Citation15, and GDC-0941 (3)Citation16, α-specific PI3K inhibitor Alpelisib (4)Citation17,Citation18, PI3Kδ-selective inhibitor Idelalisib (5)Citation19, and so on (). The common pharmacophoric features of reported PI3Kα inhibitors are summarised as follows: First, the morpholine or heterocycle ring forms an important hydrogen bond with Val851 in the hinge region. Another hydrogen bond can be found between aromatic ring or lipophilic side chain with Tyr836, Asp810 and/or Lys802 at the affinity pocket. Furthermore, some derivatives extend to the solvent exposed region to form additional interaction with the surrounding amino acids as well as improve the druggability of designed compounds. Among them, PIK-75 (6) is an Imidazo[1,2-a]pyridine derivative that shows good selectivity for PI3Kα over the other related Class I PI3Ks as well as desirable activity in a human cancer xenograft modelCitation20. However, the SAR explorations on PIK-75 are focussed on either replacing the Imidazo[1,2-a]pyridine ring with other heterocycles or modifying the sulfonohydrazone side chainsCitation21, modifications on C-2 or C-8 position have barely been reported. Considering the sulfonohydrazide group as an alert structure, we were interested in removing this group, and introducing amide and substituted aryl group to the 2-, and 8-position of Imidazo[1,2-a]pyridine ring to improve the solubility as well as provide hydrogen binding group that was essential for PI3Kα activity ().
Figure 1. Representative structures of reported PI3K inhibitors.
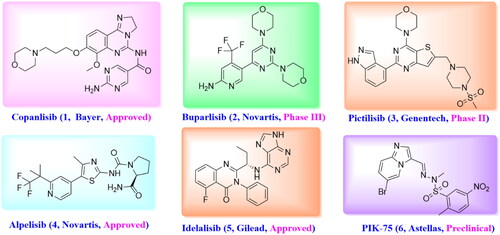
Figure 2. The design of novel Imidazo[1,2-a]pyridine derivatives based on PIK-75.
![Figure 2. The design of novel Imidazo[1,2-a]pyridine derivatives based on PIK-75.](/cms/asset/6078be0b-9943-4670-b6d3-b085e6c95ff1/ienz_a_2155638_f0002_c.jpg)
In this manuscript, we communicate the discovery and optimisation of a series of Imidazo[1,2-a]pyridine derivatives as PI3Kα selective inhibitors and their potential application in the treatment of cancers. The efforts leading to the discovery of the PI3Kα-specific inhibitor 35 are described herein.
Results and discussion
Chemistry
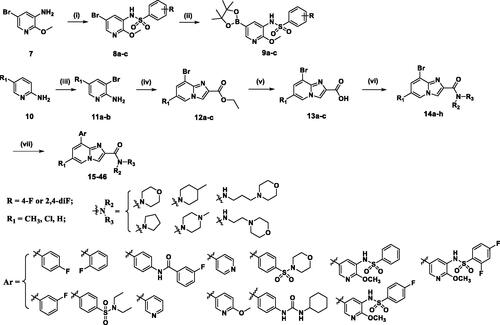
As depicted in Scheme 1, a simple five-step synthetic sequence was used for the preparation of compounds described herein. Firstly, 2-aminopyridine derivatives and N-Bromosuccinimide (NBS) undergo electrophilic aromatic substitution reaction in the presence of DMF to obtain 2-amino-3-bromo-pyridine derivatives (11a-b), 11a-b and commercially available 3-bromopyridin-2-amine were then cyclized with ethyl 3-bromopyruvate to give the ethyl 8-bromo-Imidazo[1,2-a]pyridine-2-carboxylate derivatives (12a-c). Intermediates 12a-c were further hydrolysed by NaOH to offer the corresponding carboxylic acids (13a-c). Subsequently, the key intermediates 14a-h were synthesised through amidation reaction of 13a-c with various amines and coupling reagent HBTU in DMF. Finally, compounds 14a-h undergo Suzuki-Miyaura reaction with the corresponding boronic acids or esters (9a-c) to obtain the corresponding target products 15–46. The chemical structures of the target compounds were confirmed with HRMS, 1H NMR, and 13C NMR.
Scheme 1. Reagents and conditions: (i) substituted benzene-1-sulfonyl chloride, pyridine, room temperature, overnight; (ii) (BPin)2, PdCl2(dppf), KOAc, dioxane, 100 °C, 8 h; (iii) NBS, CH3CN, 0 °C, 4 h; (iv) ethyl bromopyruvate, EtOH, 80 °C, 4 h; (v) NaOH, EtOH, 80 °C, 3 h; (vi) amines, HBTU, Et3N, DMF, rt, overnight; (vii) aryl boronic acid or 9a-c, Pd(PPh3)4, K2CO3, 1,4-Dioxane/H2O, 100 °C, overnight.
Biological evaluation
PI3Kα inhibition and SARs
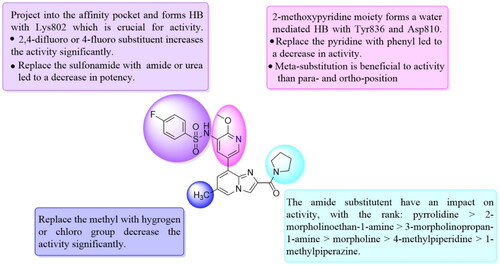
To screen the inhibitory activity of the target compounds against PI3Kα, the Kinase-Glo™ assay with ATP at 25 μM was utilised with PIK-75 as positive controlCitation22. We chose to explore the SARs at three positions on the Imidazo[1,2-a]pyridine core. To start with, a set of 8-position modifications were prepared while keeping the morpholinyl amide substituent on 2-position fixed, since it allowed quick structure − activity profiling via coupling reactions of bromide with aryl borates or boronic acids. The results are summarised in . Fluoro substituted phenyl was first incorporated, through scanning the 2, 3, 4-position of phenyl, it was clear that the ortho-position was not well tolerated (15, 16 vs 17), with only 14.1% inhibitory rate at 10 μM. Moreover, meta-fluoro substituted analog 16 showed a slightly enhanced PI3Kα activity compared to para-fluoro substituted analog 15. A similar trend can be observed for compound 23 vs 22. Replacement of fluorophenyl with 3-pyridinyl 20, 6-methoxy-3-pyridinyl 21 or 4-pyridinyl 30 did not increase the activity (20, 21 vs 16; 30 vs 29). We then turned our attention back to the phenyl substituents. Changing the fluoro group of compound 15 to amide 18, sulphonamide 19, or urea 33 led to an increase in potency, with the inhibitory rate range from 44.5 to 58.8, a similar trend can be found both at the pyrrolidinyl amide (26–28) and N-methylpiperidinyl series (29–33).
Table 1. In vitro enzyme inhibitory activity of target compounds on PI3Kα at 10 μM.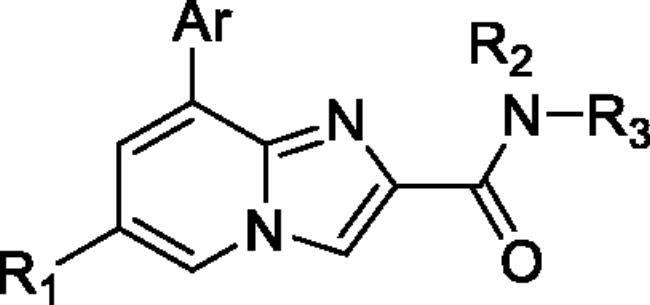
Unable to significantly improve the potency on the phenyl group at the 8-position of the Imidazo[1,2-a]pyridine template, we examined the binding mode of PIK-75 to PI3Kα, the removing sulfonohydrazide group formed an important H-bond with PI3Kα. To compensate for this loss, we speculated that the pyridinephenylsulfonamide on the 8-position would project into the affinity binding pocket of PI3Kα, making H-bond with Lys802 which is beneficial for potency. Thus, compounds 34–36 were prepared. However, compound 34 offered no advantage over the amide or sulphonamide analogs (26–28). Gratifying, a substantial increase in potency was observed when 4-fluoro or 2, 4-difluoro group were further incorporated (35, 36 vs 34). Having established the pyridinesulfonamide on the 8-position in 35 as the optimal substituent, different amines on the 2-position of the Imidazo[1,2-a]pyridine core were also designed and explored (40–44), since they may have influence on potency as well as physicochemical properties. The results revealed that all the compounds exhibit moderate to excellent inhibitory rate, ranging from 53.4 to 90.1, with the rank: pyrrolidine > 2-morpholinoethan-1-amine > 3-morpholinopropan-1-amine > morpholine > 4-methylpiperidine > 1-methylpiperazine, which was further demonstrated by compounds 36–39. Finally, the impact of 6-position on activity was also explored, neither the chloro nor the hydrogen substituent showed positive effect on the potency compared to methyl substituent (45 vs 39; 46 vs 43). Overall, The SAR studies depicted above (see also ) led to the identification 35, which was the most potent compound that we had obtained.
Figure 3. SAR of compound 35 represented in diagram.
In vitro antiproliferative activity
Following the direction, Compounds 35, 36, and 40 with inhibitory rate higher than 85% at 10 μM were progressed into full IC50 value determination with Alpelisib as positive control. As shown in , we detected these three compounds exhibit submicromolar activity for PI3Kα, with IC50 of 0.15, 1.12, 0.50 μM, respectively. Given the desirable enzyme activity profile, these compounds were further assayed for their antiproliferative activities against five independent human tumour cell line collections harbouring PIK3CA mutationCitation23, including human ovarian cancer cell line SKOV-3, human breast cancer T47D and MCF-7, human lung cancer H1975 and H460. As shown in , all compounds exhibited antiproliferative effects at micromolar concentrations. In general, the cellular activity of tested derivatives corrected with their effects on kinase activity. Interestingly, these PI3K inhibitors were more sensitive with human breast cancer than non-small cell lung cancer and ovarian cancer. Notably, compound 35 potently inhibited the proliferation of human breast cancer lines T47D and MCF-7 with IC50 of 7.9 and 9.4 μM, respectively. Hence, compound 35 was selected for further evaluation.
Table 2. Enzymatic and cellular effects of compounds against PI3Kα and five Human Cancer Cells.
Flow cytometry
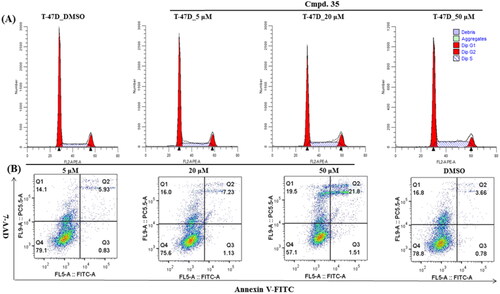
Considering compound 35 was most sensitive in T47D cells, we decided to get some mechanistic insight into the mode of action in T47D cell line. As such, the annexin V/7-AAD staining and cell cycle phase distribution have been carried out. In line with its activity, compounds 35 induced a S cell cycle arrest in T47D cells. Furthermore, T47D cells were also treated with different concentrations of 35, and the percentage of apoptotic cells was measured. Exposure to 50 μM of 35 resulted in an induction of early (1.51%) and late (21.8%) apoptosis of T47D cells ().
Figure 4. Compound 35 induces S cell cycle arrest and apoptosis in T47D cells. Exponentially growing cells were exposed to DMSO or indicated concentration of compound 35 for 24 h in T47D cells.
In vitro ADME properties of compound 35
To assess its druggability, In vitro ADME properties of compound 35 were also determined. As depicted in , compound 35 showed acceptable stability both in human liver microsomes (HLM) and mouse liver microsomes (RLM), with T1/2 of 45.1 and 31.9 min, respectively. To screen its membrane permeability, Parallel artificial membrane permeability assay (PAMPA) was conducted and the result revealed that compound 35 was highly permeable, with permeability rates (Pe) higher than 10 nm/s. Besides, to avoid any drug-drug interaction, compound 35 was further screened for its cytochrome P450 inhibition activity, and 35 showed little or minimal inhibitions for 5 major cytochrome P450 (CYP1A2, 2D6, 2C9, 2C19, and 3A4) enzymes.
Table 3. In Vitro ADME properties for compound 35.
Molecular modelling and dynamic simulation
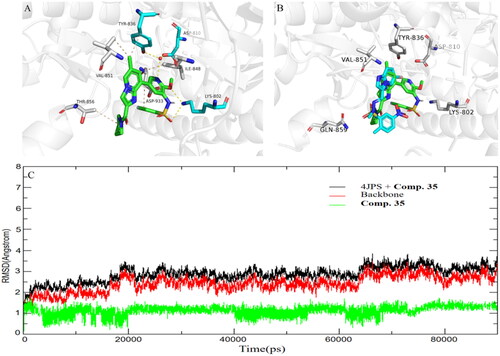
To better understand the activity of 35 against PI3Kα, the binding mode between the 35 and PI3Kα was then studied, and the structure of PI3Kα (PDB ID code: 4JPS)Citation18 was selected as the docking model. Compound 35 adopted a similar binding mode as previously reported PI3Kα inhibitorsCitation24,Citation25. As shown in , the methoxy in 2-position of pyridine and the oxygen of sulphonamide in compound 35 formed two H-bond donor–acceptor interactions with Lys802, and the pyridine nitrogen atom of 35 is part of a hydrogen bond network involving the conserved water molecule with the side chains of residues Tyr836 and Asp810. In addition, compound 35 also made hydrophobic interactions within Ile848, Val851, Thr856, and Asp933, respectively. The overlay of 35 with PIK-75 revealed the pyrrolidine amide moiety formed weaker interactions with PI3Kα than that of PIK-75 (). However, the binding affinity of compound 35 with PI3Kα was partly compensated by forming strong hydrogen bonds with the backbone of Tyr836 and Asp810 than that of a bromine atom in the Imidazopyridine ring of PIK-75. To further access the binding stability of compound 35/PI3Kα adducts, a 90 ns long molecular dynamics simulation was carried out. The RMSD values of protein backbone atoms relative to the initial structure were calculated to examine the protein stability over the course of the simulation period. As shown in , the RMSD did not fluctuate significantly, and the value of RMSD converged to 3.0 Å at 90 ns, which revealed that compound 35 could stably bind to PI3Kα. In general, the docking result confirm the rationality of our design strategy.
Figure 5. (A) Proposed binding mode of compound 35 to PI3Kα (4JPS). Hydrogen bonds are indicated by yellow dashed lines, and hydrophobic interactions are indicated in wheat lines. Images are generated using PyMol. (B) The overlay of compound 35 with PIK-75 in PI3Kα (4JPS). (C) Dynamics of compound 35 bound to FLT3 PI3Kα (4JPS) during 90 ns simulation time.
Conclusion
In summary, 32 novel compounds bearing Imidazo[1,2-a]pyridine scaffold were designed and synthesised. The design strategy of PI3Kα inhibitor involved modification of the 2,6,8-position of Imidazo[1,2-a]pyridine ring. Systematic SAR studies led us identified 35 as the most promising compound, with IC50 of 150 nM against PI3Kα in enzymatic assay. By screening a panel of cancer cell lines that harboured PIK3CA mutation, compound 35 was found to be more potent against breast cancer than others. Further in vitro ADME evaluation demonstrated 35 was metabolic stable in RLM and HLM, highly permeable in PAMPA, and not a CYP450 inhibitor. More work regarding the optimisation of 35 to achieve a more potent PI3Kα inhibitor will be disclosed in due course.
Experimental
Chemistry
Unless otherwise specified, all the reagents and solvents were obtained from commercial sources and used without further purification. Reactions’ time and purity of the products were monitored by TLC on FLUKA silica gel aluminium cards (0.2 mm thickness) with fluorescent indicator 254 nm. All melting points were obtained on a Büchi Melting Point B-540 apparatus (Büchi Labortechnik, Flawil, Switzerland) and were uncorrected. Flash chromatography was performed using silica gel (200–300 mesh) from Qingdao ocean Chemicals (Qingdao, Shandong, China). The positive control PIK-75 and Alpelisib were purchased from TargetMol (Shanghai, China). Mass spectra (MS) were taken in ESI mode on Waters e2695. 1H NMR and 13C NMR spectra were recorded on Bruker ARX-400 or ARX-600, 400 or 600 MHz spectrometers (Bruker Bioscience, Billerica, MA, USA) with TMS as an internal standard.
General procedures
General procedure for preparation of intermediate 8a-c
To a solution of 5-bromo-2-methoxypyridin-3-amine (2.0 g, 10 mmol) in pyridine (30 mL) was added substituted benzene-1-sulfonyl chloride (15 mmol), the mixture was stirred at room temperature overnight. Upon completion, the mixture was then concentrated and redissolved in 30 mL CH2Cl2, washed with aqueous citric acid and then brine. After drying over Na2SO4, the solvent was evaporated and used without further purification.
N-(5-bromo-2-methoxypyridin-3-yl)benzenesulfonamide (8a)
White solid; Yield: 67.8%; HRMS(ESI) calculated for C12H12BrN2O3S [M + H]+ m/z 342.9674, found: 342.9679.
N-(5-bromo-2-methoxypyridin-3-yl)-4-fluorobenzenesulfonamide (8b)
White solid; Yield: 63.5%; HRMS(ESI) calculated for C12H11BrFN2O3S [M + H]+ m/z 360.9580, found: 360.9589.
N-(5-bromo-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (8c)
Yellow solid; Yield: 75.4%; HRMS(ESI) calculated for C12H10BrF2N2O3S [M + H]+ m/z 378.9485, found: 378.9480.
General procedure for preparation of intermediate 9a-c
A solution of 8a-c (3.2 mmol), bis(pinacolato)diboron (1.6 g, 6.4 mmol), PdCl2(dppf)-DCM (258 mg, 0.32 mmol), potassium acetate (932 mg, 9.4 mmol) in 1,4-dioxane (20 mL) was heated at 100 °C under N2 for 8 h. Upon cooling, the mixture was diluted with CH2Cl2 (100 mL) and then filtered, the organic layers were washed with brine, dried over Na2SO4, and concentrated. The residue was then subjected to flash chromatography to give pure 9a-c.
N-(2-methoxy-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)ben zenesulfonamide (9a)
White solid; Yield: 81.2%; HRMS(ESI) calculated for C18H23BN2O5S [M + H]+ m/z 391.1499, found: 391.1508.
4-Fluoro-N-(2-methoxy-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine-3-yl)benzenesulfonamide (9 b)
White solid; Yield: 78.5%; HRMS(ESI) calculated for C18H22BFN2O5S [M + H]+ m/z 409.1405, found: 409.1411.
2,4-Difluoro-N-(2-methoxy-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide (9c)
White solid; Yield: 76.7%; HRMS(ESI) calculated for C18H21BF2N2O5S [M + H]+ m/z 427.1311, found: 427.1323.
General procedure for preparation of intermediate 11a-b
NBS (32.9 g, 184.9 mmol) was slowly added into the solution of 2-aminopyridine derivatives (184.9 mmol) in N, N-dimethylformamide (200 mL) under ice bath, the reaction was then stirred at 0 °C for 4 h. The mixture was poured into ice water (400 mL) when the reaction was completed by TLC detection, the aqueous phase was then extracted with methyl tert-butyl ether (80 mL × 3), washed with brine, and concentrated under reduced pressure to give 11a-b.
3-Bromo-5-methyl-1,2-dihydropyridin-2-amine (11a)
Brown solid; Yield: 89.5%; HRMS(ESI) calculated for C6H10BrN2 [M + H]+ m/z 189.0027, found: 189.0035.
3-Bromo-5-chloro-1,2-dihydropyridin-2-amine (11b)
White solid; Yield: 86.9%; HRMS(ESI) calculated for C5H7BrClN2 [M + H]+ m/z 208.9481, found: 208.9496.
General procedure for preparation of intermediate 12a-c
Commercially available 2-aminopyridine or 11a-b (181.8 mmol), ethyl bromopyruvate (44.3 g, 227.2 mmol) were successively added to ethanol (300 mL), the mixture was heated to 80 °C for 4 h. Upon completion, the reaction solution was cooled to room temperature, concentrated under reduced pressure to obtain a brownish red solid, which was then triturated with acetone to give 12a-c.
Ethyl 8-bromo-6-methylImidazo[1,2-a]pyridine-2-carboxylate (12a)
Light yellow solid; Yield: 83.3%; HRMS(ESI) calculated for C11H12BrO2N2 [M + H]+ m/z 283.0082, found: 283.0091.
Ethyl 8-bromo-6-chloroImidazo[1,2-a]pyridine-2-carboxylate (12b)
White solid, Yield: 76.6%; HRMS(ESI) calculated for C10H9ClBrO2N2 [M + H]+ m/z 302.9536, found: 302.9548.
Ethyl 8-bromoImidazo[1,2-a]pyridine-2-carboxylate (12c)
Light yellow solid; Yield: 71.1%; HRMS(ESI) calculated for C10H10BrO2N2 [M + H]+ 268.9926, found: 268.9937.
General procedure for preparation of intermediate 13a-c
To a solution of corresponding carboxylate 12a-c (37.2 mmol) in ethanol (50 mL) was added NaOH solution (15 mL, 1 N), the reaction was heated to 80 °C for 3 h. After cooling, the reaction solution was concentrated under reduced pressure to remove the solvent and the pH was adjusted to 5 with diluted hydrochloric acid (15 mL, 1 N), a large amount of solid was precipitated. The precipitant was filtered, washed with water, and dried under vacuum to afford the products 13a-c.
8-Bromo-6-methylImidazo[1,2-a]pyridine-2-carboxylic acid (13a)
Light yellow solid; Yield: 74.4%; HRMS(ESI) calculated for C9H6ClBrO2N2 [M + H]+ m/z 252.9613, found: 252.9625.
8-Bromo-6-chloroImidazo[1,2-a]pyridine-2-carboxylic acid (13b)
Light yellow solid; Yield: 85.0%; HRMS(ESI) calculated for C8H3ClBrO2N2 [M + H]+ m/z 272.9066, found: 272.9079.
8-Bromoimidazo[1,2-a]pyridine-2-carboxylic acid (13c)
Light yellow solid; Yield: 77.3%; HRMS(ESI) calculated for C8H4ClBrO2N2 [M + H]+ m/z 238.9456, found: 238.9473.
General procedure for preparation of intermediate 14a-h
To a solution of corresponding carboxyl acid 13a-c (1.2 mmol) in DMF was subsequently added morpholine (0.31 g, 3.5 mmol), triethylamine (0.17 g, 1.8 mmol) and HBTU (0.67 g, 1.8 mmol). The reaction was stirred at room temperature overnight. Upon completion, the mixture was then poured into ice-water, extracted with EtOAc (15 mL × 3). The combined extracts were then washed with brine, dried over Na2SO4, and concentrated. The crude products were then subjected to flash chromatography to give pure products 14a-h.
(8-Bromo-6-methylImidazo[1,2-a]pyridin-2-yl)(morpholino)methanone (14a)
White solid; Yield: 45.3%; HRMS(ESI) calculated for C13H15BrO2N3 [M + H]+ m/z 324.0348, found: 324.0376.
(8-Bromo-6-methylImidazo[1,2-a]pyridin-2-yl)(4-methylpiperazin-1-yl)methanone (14 b)
Gray white solid; Yield: 50.3%; HRMS(ESI) calculated for C14H18BrON4 [M + H]+ m/z 337.0664, found: 337.0711.
(8-Bromo-6-methylImidazo[1,2-a]pyridin-2-yl)(pyrrolidin-1-yl)methanone (14c)
Light yellow solid; Yield: 48.9%; HRMS(ESI) calculated for C13H15BrON3 [M + H]+ m/z 308.0398, found: 308.0408.
(8-Bromo-6-methylImidazo[1,2-a]pyridin-2-yl)(4-methylpiperidin-1-yl)methanone (14d)
White solid; Yield: 63.1%; HRMS(ESI) calculated for C15H19BrON3 [M + H]+ m/z 336.0712, found: 336.0765.
8-Bromo-6-methyl-N-(2-morpholinoethyl)imidazo[1,2-a]pyridine-2-carboxamide (14e)
White solid; Yield: 69.4%; HRMS(ESI) calculated for C15H20BrO2N4 [M + H]+ m/z 367.0770, found: 367.0780.
8-Bromo-6-methyl-N-(3-morpholinopropyl)imidazo[1,2-a]pyridine-2-carboxamide (14f)
Light yellow solid; Yield: 57.6%; HRMS(ESI) calculated for C16H22BrO2N4 [M + H]+ m/z 381.0926, found: 381.0911.
8-Bromo-6-chloro-N-(3-morpholinopropyl)imidazo[1,2-a]pyridine-2-carboxamide (14 g)
White solid; Yield: 80.3%; HRMS(ESI) calculated for C15H19BrClO2N4 [M + H]+ m/z 401.0380, found: 401.0392.
(8-Bromoimidazo[1,2-a]pyridin-2-yl)(4-methylpiperidin-1-yl)methanone (14 h)
White solid; Yield: 62.3%; HRMS(ESI) calculated for C14H17BrF2ON3 [M + H]+ m/z 322.0555, found: 322.0578.
General procedure for preparation of target compounds 15–46
Method A: a mixture of corresponding amide (3.1 mmol), xylene: H2O = 5:1 (30 mL), arylboric acids (4.8 mmol), potassium carbonate (0.85 g, 6.2 mmol) and Pd(PPh3)4 (0.25 g, 0.31 mmol) was heated to 80 °C for 8 h under the protection of N2. Upon completion, the solvent was removed under reduced pressure, and then poured into water to precipitate solids. The precipitant was filtered, washed with tert-Butyl methyl ether, dried under vacuum to afford the target compounds.
Method B: a mixture of corresponding amide (0.93 mmol), corresponding boric acid ester (1.39 mmol), potassium carbonate (0.26 g, 1.86 mmol), 1,4-dioxane: H2O = 3:1 and Pd(PPh3)4 (0.076 g, 0.093 mmol) was stirred at 90 °C for 12 h under nitrogen atmosphere. After cooling to room temperature, water (30 mL) was added followed by extraction with EtOAc (10 mL × 3). The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The crude products were purified using silica gel column chromatography to give pure compounds.
(8–(4-Fluorophenyl)-6-methylImidazo[1,2-a]pyridine-2-yl)(morpholino)methanone (15)
White solid; Yield: 75.0%; m.p.: 1 8 0 ∼ 182 °C; 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 8.12 − 7.98 (m, 2H), 7.96 (s, 1H), 7.23 (s, 1H), 7.22 − 7.18 (m, 2H), 4.52 − 4.42 (m, 2H), 3.89 − 3.79 (m, 6H), 2.43 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 163.83, 162.77, 161.39, 141.25, 140.01, 132.34, 131.09, 127.41, 124.32, 123.11, 117.44, 115.74, 66.92, 40.99, 18.1. HRMS (ESI) calculated for C19H19FN3O2 [M + H]+ m/z 340.1461, found: 340.1447.
(8–(3-Fluorophenyl)-6-methylImidazo[1,2-a]pyridin-2-yl)(morpholino)methanone (16)
White solid; Yield: 79.6%; m.p.: 1 7 7 ∼ 180 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.45 (s, 1H), 8.38 (s, 1H), 8.11 (d, J = 11.3 Hz, 1H), 8.02 (d, J = 7.6 Hz, 1H), 7.62 (s, 1H), 7.60 − 7.54 (m, 1H), 7.30 (t, J = 7.6 Hz, 1H), 4.27 − 4.21 (m, 2H), 3.73 − 3.65 (m, 6H), 2.39 (s, 3H). HRMS (ESI) calculated for C19H19FN3O2 [M + H]+ m/z 340.1461, found: 340.1530.
(8–(2-Fluorophenyl)-6-methylImidazo[1,2-a]pyridin-2-yl)(morpholino)methanone (17)
White solid; Yield: 76.4%; m.p.: 1 8 6 ∼ 189 °C; 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 8.00 (s, 1H), 7.85 (t, J = 7.4 Hz, 1H), 7.45 (dd, J = 13.6, 7.3 Hz, 1H), 7.29 − 7.21 (m, 3H), 4.44 − 4.36 (m, 2H), 3.85 − 3.73 (m, 6H), 2.43 (s, 3H). HRMS (ESI) calculated for C19H19FN3O2 [M + H]+ m/z 340.1461, found: 340.1451.
3-Fluoro-N-(4–(6-methyl-2-(morpholine-4-carbonyl)Imidazo[1,2-a]pyridin-8-yl)phenyl)benzamide (18)
White solid; Yield: 70.9%; m.p.: 2 6 8 ∼ 270 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 8.40 (s, 1H), 8.37 (s, 1H), 8.20 (d, J = 7.7 Hz, 2H), 7.94 (d, J = 7.9 Hz, 2H), 7.90 − 7.82 (m, 2H), 7.64 (d, J = 5.4 Hz, 1H), 7.54 − 7.48 (m, 2H), 4.34 − 4.24 (m, 2H), 3.74 − 3.65 (m, 6H), 2.39 (s, 3H). HRMS (ESI) calculated for C26H24FN4O3 [M + H]+ m/z 459.1832, found: 459.1937.
N,N-Diethyl-3–(6-methyl-2-(morpholine-4-carbonyl)Imidazo[1,2-a]pyridin-8-yl)benzenesulfonamide (19)
White solid; Yield: 72.2%; m.p.: 1 7 0 ∼ 173 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 8.48 (s, 1H), 8.42 (s, 1H), 8.34 (d, J = 7.6 Hz, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.77 (t, J = 7.7 Hz, 1H), 7.68 (s, 1H), 4.41 − 4.31 (m, 2H), 3.75 − 3.63 (m, 6H), 3.23 (q, J = 6.7 Hz, 4H), 2.40 (s, 3H), 1.09 (t, J = 6.9 Hz, 6H). HRMS (ESI) calculated for C23H29N4O4S [M + H]+ m/z 457.1910, found: 457.1897.
(6-Methyl-8-(pyridin-3-yl)Imidazo[1,2-a]pyridin-2-yl)(morpholino)methanone (20)
White solid; Yield: 77.5%; m.p.: 1 9 5 ∼ 198 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.30 (s, 1H), 8.64 (d, J = 3.4 Hz, 1H), 8.53 (d, J = 7.9 Hz, 1H), 8.46 (s, 1H), 8.39 (s, 1H), 7.61 (s, 1H), 7.57 (dd, J = 7.6, 4.9 Hz, 1H), 4.26 − 4.18 (s, 2H), 3.72 − 3.62 (s, 6H), 2.39 (s, 3H). HRMS (ESI) calculated for C18H19N4O2 [M + H]+ m/z 323.1508, found: 323.1541.
(8–(6-Methoxypyridin-3-yl)-6-methylImidazo[1,2-a]pyridin-2-yl)(morpholino)methanone (21)
White solid; Yield: 50.2%; 1H NMR (600 MHz, DMSO-d6) δ 8.92 (d, J = 2.2 Hz, 1H), 8.44 (dd, J = 8.7, 2.5 Hz, 1H), 8.37 (t, J = 1.1 Hz, 1H), 8.34 (s, 1H), 7.48 (d, J = 1.4 Hz, 1H), 6.96 (d, J = 8.7 Hz, 1H), 4.25 − 4.15 (m, 2H), 3.93 (s, 3H), 3.71 − 3.59 (m, 6H), 2.34 (s, 3H). 13C NMR (151 MHz, DMSO) δ 163.9, 162.8, 147.0, 141.1, 139.9, 139.5, 126.7, 125.5, 125.3, 124.2, 123.1, 117.5, 110.5, 67.0, 66.8, 53.8, 47.6, 43.0, 18.1. HRMS (ESI) calculated. for C19H21O3N4 [M + H]+ m/z 353.1614, found: 353.1619.
(8–(4-Fluorophenyl)-6-methylImidazo[1,2-a]pyridin-2-yl)(pyrrolidin-1-yl)methanone (22)
White solid; Yield: 78.9%; m.p.: 1 8 8 ∼ 191 °C; 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 8.12 (t, J = 7.0 Hz, 2H), 7.97 (s, 1H), 7.29 − 7.17 (m, 3H), 4.20 (t, J = 6.7 Hz, 2H), 3.74 (t, J = 6.8 Hz, 2H), 2.43 (s, 3H), 2.06 − 2.00 (m, 2H), 1.99 − 1.93 (m, J = 4.5 Hz, 2H). HRMS (ESI) calculated for C19H19FN3O [M + H]+ m/z 324.1512, found: 324.1500.
(8–(3-Fluorophenyl)-6-methylImidazo[1,2-a]pyridin-2-yl)(pyrrolidin-1-yl)methanone (23)
White solid; Yield: 81.4%; m.p.: 1 5 3 ∼ 155 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 1H), 8.64 (s, 1H), 8.55 (d, J = 7.4 Hz, 1H), 8.45 (s, 1H), 8.32 (s, 1H), 7.62 (s, 1H), 7.60 − 7.52 (m, 1H), 4.03 (t, J = 6.7 Hz, 2H), 3.63 (t, J = 6.8 Hz, 2H), 2.39 (s, 3H), 1.70 − 1.64 (m, 2H), 1.60 − 1.54 (m, 2H). HRMS (ESI) calculated for C19H19FN3O [M + H]+ m/z 324.1512, found: 321.1741.
(6-Methyl-8-(pyridin-3-yl)Imidazo[1,2-a]pyridin-2-yl)(pyrrolidin-1-yl)methanone (24)
White solid; Yield: 79.7%; m.p.: 1 5 9 ∼ 162 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.39 (s, 1H), 8.66 (d, J = 3.4 Hz, 1H), 8.59 (d, J = 7.9 Hz, 1H), 8.48 (s, 1H), 8.42 (s, 1H), 7.63 (s, 1H), 7.61 − 7.56 (m, 1H), 4.05 (t, J = 6.4 Hz, 2H), 3.55 (t, J = 6.6 Hz, 2H), 2.40 (s, 3H), 1.99 − 1.93 (m, 2H), 1.90 − 1.85 (m, 2H). HRMS (ESI) calculated for C18H19N4O [M + H]+ m/z 307.1559, found: 307.1658.
(8–(6-Methoxypyridin-3-yl)-6-methylImidazo[1,2-a]pyridin-2-yl)(pyrrolidin-1-yl)methanone (25)
Gray white solid; Yield: 46.5%; 1H NMR (600 MHz, DMSO-d6) δ 9.01 (d, J = 2.4 Hz, 1H), 8.48 (dd, J = 8.7, 2.5 Hz, 1H), 8.37 (t, J = 1.5 Hz, 1H), 8.35 (s, 1H), 7.49 (d, J = 1.3 Hz, 1H), 6.96 (d, J = 8.7 Hz, 1H), 4.01 (t, J = 6.8 Hz, 2H), 3.93 (s, 3H), 3.52 (t, J = 6.9 Hz, 2H), 2.34 (d, J = 1.3 Hz, 3H), 1.96 − 1.88 (m, 2H), 1.87 − 1.80 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 163.9, 162.1, 147.1, 141.3, 141.1, 139.4, 126.4, 125.5, 125.4, 124.2, 123.1, 117.0, 110.5, 53.9, 48.7, 47.0, 26.8, 23.8, 18.1. HRMS (ESI) calculated. for C19H21O2N4 [M + H]+ m/z 337.1665, found: 337.1674.
3-Fluoro-N-(4–(6-methyl-2-(pyrrolidine-1-carbonyl)Imidazo[1,2-a]pyridin-8-yl)phenyl)benzamide (26)
White solid; Yield: 68.6%; m.p.: 2 6 3 ∼ 265 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H), 8.43 − 8.25 (m, 4H), 8.00 − 7.90 (m, 2H), 7.65 − 7.46 (m, 5H), 4.10 (t, J = 6.8 Hz, 2H), 3.55 (t, J = 6.8 Hz, 2H), 2.42 (s, 3H), 1.96 − 1.88 (m, 4H); HRMS (ESI) calculated for C26H24FN4O2 [M + H]+ m/z 443.1883, found: 443.1995.
(6-Methyl-8–(3-(morpholinosulfonyl)phenyl)Imidazo[1,2-a]pyridin-2-yl)(pyrrolidin-1-yl)methanone (27)
White solid; Yield: 71.1%; m.p.: 1 7 0 ∼ 172 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.97 (s, 1H), 8.51 − 8.45 (m, 2H), 8.43 (s, 1H), 7.87 − 7.80 (m, 2H), 7.72 (s, 1H), 4.13 (t, J = 6.4 Hz, 2H), 3.70 − 3.62 (m, 4H), 3.55 (t, J = 6.6 Hz, 2H), 2.98 − 2.90 (m, 4H), 2.41 (s, 2H), 2.00 − 1.88 (m, 3H), 1.88 − 1.83 (m, 2H). HRMS (ESI) calculated for C23H27N4O4S [M + H]+ m/z 455.1753, found: 455.1736.
N,N-Diethyl-3–(6-methyl-2-(pyrrolidine-1-carbonyl)Imidazo[1,2-a]pyridin-8-yl)benzenesulfonamide (28)
White solid; Yield: 73.7%; m.p.: 1 6 4 ∼ 167 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.83 (s, 1H), 8.48 (s, 1H), 8.40 − 8.35 (m, 2H), 7.88 (d, J = 7.2 Hz, 1H), 7.77 (t, J = 7.6 Hz, 1H), 7.66 (s, 1H), 5.13 (s, 1H), 4.53 (s, 1H), 3.30 − 3.22 (m, 5H), 2.46 − 2.38 (m, 2H), 1.76 − 1.68 (m, 2H), 1.14 − 1.02 (m, 7H), 0.96 (d, J = 5.3 Hz, 3H). HRMS (ESI) calculated for C23H29N4O3S [M + H]+ m/z 441.1960, found: 441.1976.
(8–(3-Fluorophenyl)-6-methylImidazo[1,2-a]pyridin-2-yl)(4-methylpiperidin-1-yl)methanone (29)
White solid; Yield: 85.9%; 1H NMR (400 MHz, DMSO-d6) δ 8.44 (s, 1H), 8.31 (s, 1H), 8.18 (d, J = 11.3 Hz, 1H), 8.03 (d, J = 7.7 Hz, 1H), 7.62 (s, 1H), 7.60 − 7.54 (m, 1H), 7.30 (t, J = 8.2 Hz, 1H), 5.03 (s, 1H), 4.51 (s, 1H), 3.14 (s, 1H), 2.78 (s, 1H), 2.39 (s, 3H), 1.76 − 1.68 (m, 3H), 1.24 − 1.16 (m, 2H), 0.96 (d, J = 5.5 Hz, 3H). HRMS (ESI) calculated for C21H23FN3O [M + H]+ m/z 352.1825, found: 352.1854.
(6-Methyl-8-(pyridin-4-yl)Imidazo[1,2-a]pyridin-2-yl)(4-methylpiperidin-1-yl)methanone (30)
White solid; Yield: 76.5%; m.p.: 1 7 4 ∼ 177 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.72 (d, J = 5.4 Hz, 2H), 8.50 (s, 1H), 8.33 (s, 1H), 8.23 (d, J = 5.4 Hz, 2H), 7.74 (s, 1H), 4.97 (s, 1H), 4.52 (s, 1H), 3.15 (s, 1H), 2.78 (s, 1H), 2.40 (s, 3H), 1.76 − 1.66 (m, 3H), 1.18 − 1.10 (m, 2H), 0.96 (d, J = 5.8 Hz, 3H). HRMS (ESI) calculated for C20H23N4O [M + H]+ m/z 335.1872, found: 335.1955.
(6-Methyl-8-(pyridin-3-yl)Imidazo[1,2-a]pyridin-2-yl)(4-methylpiperidin-1-yl)methanone (31)
White solid; Yield: 79.5%; m.p.: 1 4 4 ∼ 147 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.72 (d, J = 5.4 Hz, 2H), 8.50 (s, 1H), 8.33 (s, 1H), 8.23 (d, J = 5.4 Hz, 2H), 7.74 (s, 1H), 4.97 (s, 1H), 4.52 (s, 1H), 3.15 (s, 1H), 2.78 (s, 1H), 2.40 (s, 3H), 1.77 − 1.67 (m, 3H), 1.17 − 1.11 (m, 2H), 0.96 (d, J = 5.8 Hz, 3H). HRMS (ESI) calculated for C20H23N4O [M + H]+ m/z 335.1872, found: 335.1959.
N,N-Diethyl-3–(6-methyl-2–(4-methylpiperidine-1-carbonyl)Imidazo[1,2-a]pyridin-8-yl)benzenesulfonamide (32)
White solid; Yield: 73.9%; m.p.: 1 3 1 ∼ 133 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.81 (s, 1H), 8.47 (s, 1H), 8.39 − 8.33 (m, 2H), 7.87 (d, J = 6.4 Hz, 1H), 7.76 (t, J = 7.6 Hz, 1H), 7.65 (s, 1H), 4.13 (t, J = 6.7 Hz, 2H), 3.64 (t, J = 6.8 Hz, 2H), 3.29 − 3.21 (m, 5H), 2.41 (s, 3H), 1.69 − 1.63 (m, 2H), 1.61 − 1.55 (m, 4H), 1.14 − 1.06 (m, 7H); 13C NMR (100 MHz, DMSO-d6) δ 162.6, 140.9, 140.8, 140.5, 136.9, 132.4, 129.9, 128.0, 127.2, 126.8, 126.4, 125.1, 122.9, 117.2, 42.5, 40.9, 39.1, 31.0, 22.1, 18.1, 14.7. HRMS (ESI) calculated for C25H33N4O3S [M + H]+ m/z 469.2273, found: 469.2343.
1-Cyclohexyl-3–(3-(6-methyl-2–(4-methylpiperidine-1-carbonyl)Imidazo[1,2-a]pyridin-8-yl)phenyl)urea (33)
White solid; Yield: 75.0%; m.p.: 1 6 8 ∼ 170 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.50 (s, 1H), 8.39 (s, 1H), 8.29 (s, 1H), 8.25 (s, 1H), 7.59 (s, 1H), 7.44 − 7.32 (m, 3H), 6.21 (s, 1H), 5.08 (s, 1H), 4.51 (s, 1H), 3.11 (s, 1H), 2.76 (s, 1H), 2.37 (s, 3H), 1.86 − 1.78 (m, 3H), 1.73 − 1.65 (m, 4H), 1.55 (s, 1H), 1.36 − 1.28 (m, 2H), 1.27 − 1.21 (m, 7H), 0.98 − 0.90 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 162.8, 154.9, 141.3, 141.0, 140.7, 137.2, 136.5, 128.9, 127.0, 124.1, 122.8, 121.7, 118.6, 118.1, 116.7, 48.2, 40.9, 38.8, 33.5, 31.1, 25.7, 24.8, 22.1, 18.1. HRMS (ESI) calculated for C28H36N5O2 [M + H]+ m/z 474.2869, found: 474.2998.
N-(2-methoxy-5–(6-methyl-2-(pyrrolidine-1-carbonyl)Imidazo[1,2-a]pyridin-8-yl)pyridin-3-yl)benzenesulfonamide (34)
Light yellow solid; Yield: 49.5%; 1H NMR (600 MHz, DMSO-d6) δ 9.93 (s, 1H), 8.73 − 8.70 (m, 1H), 8.55 (d, J = 2.3 Hz, 1H), 8.39 − 8.35 (m, 1H), 8.35 − 8.33 (m, 1H), 7.75 (t, t, J = 1.8 Hz, 1H), 7.73 (t, J = 1.8 Hz, 1H), 7.64 − 7.58 (m, 1H), 7.55 − 7.50 (m, 2H), 7.43 − 7.41 (m, 1H), 4.03 (t, J = 7.3 Hz, 2H), 3.63 (s, 3H), 3.51 (t, J = 6.9 Hz, 2H), 2.33 (s, 3H), 1.93 − 1.86 (m, 2H), 1.82 (q, J = 6.8 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 162.1, 157.4, 143.4, 141.2, 141.1, 140.9, 134.2, 133.3, 129.4, 127.1, 126.6, 125.6, 124.8, 124.5, 123.0, 120.3, 117.1, 53.9, 48.8, 47.1, 26.8, 23.8, 18.1. HRMS (ESI) calculated. for C25H26N5O4S [M + H]+ m/z 492.1706, found: 492.1709.
4-Fluoro-N-(2-methoxy-5–(6-methyl-2-(pyrrolidine-1-carbonyl)Imidazo[1,2-a]pyridin-8-yl)pyridin-3-yl)benzenesulfonamide (35)
White solid; Yield: 52.3%; 1H NMR (600 MHz, DMSO-d6) δ 10.01 (s, 1H), 8.76 (d, J = 2.2 Hz, 1H), 8.60 (d, J = 2.2 Hz, 1H), 8.40 (t, J = 1.2 Hz, 1H), 8.36 (s, 1H), 7.85 − 7.79 (m, 2H), 7.47 (d, J = 0.9 Hz,1H), 7.43 − 7.36 (m, 2H), 4.05 (t, J = 6.8 Hz, 2H), 3.67 (s, 3H), 3.53 (t, J = 6.9 Hz, 2H), 2.35 (s, 3H), 1.95 − 1.88 (m, 2H), 1.87 − 1.76 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 162.1, 157.5, 143. 7, 141.2, 141.1, 137.3, 134.7, 130.3, 126.6, 125.7, 124.7, 124.5, 123.0, 120.0, 117.1, 116.6, 53.9, 48.8, 47.1, 26.8, 23.8, 18.1. HRMS (ESI) calculated. for C25H25FN5O4S [M + H]+ m/z 510.1611, found: 510.1631.
2,4-Difluoro-N-(2-methoxy-5–(6-methyl-2-(pyrrolidine-1-carbonyl)Imidazo[1,2-a]pyridin-8-yl)pyridin-3-yl)benzenesulfonamide (36)
White solid; Yield: 50.6%; 1H NMR (600 MHz, DMSO-d6) δ 10.27 (s, 1H), 8.83 − 8.81 (m, 1H), 8.63 (d, J = 2.2 Hz, 1H), 8.41 − 8.38 (m, 1H), 8.37 − 8.35 (m, 1H), 7.74 (q, J = 8.3 Hz, 1H), 7.62 − 7.54 (m, 1H), 7.53 − 7.50 (m, 1H), 7.19 (td, J = 8.5, 2.3 Hz, 1H), 4.07 (t, J = 6.7 Hz, 2H), 3.66 (s, 3H), 3.53 (t, J = 6.9 Hz, 2H), 2.35 (s, 3H), 1.96 − 1.90 (m, 2H), 1.87 − 1.81 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 165.8, 161.9, 159.9, 158.4, 144.5, 141.1, 141.0, 136.9, 132.3, 126.5, 125.8, 125.7, 124.5, 124.4, 123.1, 119.3, 117.2, 112.2, 106.2, 53.9, 48.9, 47.1, 26.8, 23.8, 18.1. HRMS (ESI) calculated. for C25H24F2N5O4S [M + H]+ m/z 528.1517, found: 528.1516.
2,4-Difluoro-N-(2-methoxy-5–(6-methyl-2-(morpholine-4-carbonyl)Imidazo[1,2-a]pyridin-8-yl)pyridin-3-yl)benzenesulfonamide (37)
White solid; Yield: 55.2%; 1H NMR (600 MHz, DMSO-d6) δ 10.30 (s, 1H), 8.72 (d, J = 2.3 Hz, 1H), 8.62 (d, J = 2.1 Hz, 1H), 8.40 (s, 1H), 8.37 (s, 1H), 7.75 (q, J = 8.4 Hz, 1H), 7.63 − 7.55 (m, 1H), 7.53 (s, 1H), 7.19 (td, J = 8.5, 2.3 Hz, 1H), 4.36 − 4.24 (m, 2H), 3.71 − 3.66 (m, 6H), 3.66 (s, 3H), 2.35 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 165.5, 162.5, 159.6, 158.4, 144.4, 140.9, 140.1, 136.9, 132.3, 126.9, 125.7, 125.7, 124.6, 124.4, 123.2, 119.3, 117.9, 112.2, 106.2, 67.1, 66.8, 53.9, 47.5, 43.0, 18.1. HRMS (ESI) calculated. for C25H24F2N5O5S [M + H]+ m/z 544.1466, found: 544.1511.
8–(5-((2,4-Difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-6-methyl-N-(2-morpholinoethyl)Imidazo[1,2-a]pyridine-2-carboxamide (38)
White solid; Yield: 51.9%; 1H NMR (600 MHz, DMSO-d6) δ 10.32 (s, 1H), 9.00 (s, 1H), 8.46 (d, J = 2.4 Hz, 1H), 8.39 (s, 1H), 8.35 (s, 1H), 8.20 (t, J = 5.4 Hz, 1H), 7.77 (q, J = 8.1 Hz, 1H), 7.52 − 7.46 (s, 2H), 7.20 (t, J = 7.7 Hz, 1H), 3.70 (s, 3H), 3.58 (t, J = 4.7 Hz, 4H), 3.44 (q, J = 6.4 Hz, 2H), 3.39 − 3.30 (m, 2H), 2.47 − 2.39 (m, 4H), 2.35 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 165.5, 162.3, 159.9, 158.0, 144.6, 141.5, 139.7, 135.6, 132.4, 127.0, 125.6, 125.4, 124.8, 124.4, 123.1, 119.9, 115.1, 112.2, 106.3, 66.6, 57.6, 53.8, 53.6, 35.8, 18.1. HRMS (ESI) calculated. for C27H29F2N6O5S [M + H]+ m/z 587.1888, found: 587.1896.
8–(5-((2,4-Difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-6-methyl-N-(3-morpholinopropyl)Imidazo[1,2-a]pyridine-2-carboxamide (39)
White solid; Yield: 49.7%; 1H NMR (600 MHz, DMSO-d6) δ 8.88 (d, J = 2.2 Hz, 1H), 8.51 (d, J = 2.2 Hz, 1H), 8.39 (t, J = 1.4 Hz, 1H), 8.34 (s, 1H), 8.24 (t, J = 6.0 Hz, 1H), 7.79 − 7.74 (m, 1H), 7.60 − 7.53 (m, 1H), 7.48 (d, J = 1.2 Hz, 1H), 7.19 (td, J = 8.5, 2.3 Hz, 1H), 3.71 (s, 3H), 3.52 (t, J = 4.6 Hz, 4H), 3.36 (q, J = 6.7 Hz, 2H), 2.40 − 2.32 (m, 9H), 1.75 − 1.66 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 165.5, 162.5, 159.9, 157.9, 144.1, 141.5, 139.9, 135.4, 132.3, 127.0, 125.7, 125.5, 124.8, 124.6, 123.1, 120.1, 115.1, 112.3, 106.3, 66.4, 56.6, 53.9, 53.7, 37.6, 26.4, 18.1. HRMS (ESI) calculated. for C28H31F2N5O5S [M + H]+ m/z 601.2045, found: 601.2063.
8–(5-((4-Fluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-6-methyl-N-(2-morpholinoethyl)Imidazo[1,2-a]pyridine-2-carboxamide (40)
White solid; Yield: 50.9%; 1H NMR (600 MHz, Chloroform-d) δ 9.11 (d, J = 2.1 Hz, 1H), 8.55 (d, J = 2.1 Hz, 1H), 8.12 (s, 1H), 8.09 (t, J = 6.1 Hz, 1H), 7.97 − 7.95 (m, 1H), 7.94 − 7.87 (m, 2H), 7.29 (s, 1H), 7.11 − 7.05 (m, 2H), 3.87 (s, 3H), 3.70 (t, J = 4.6 Hz, 4H), 3.62 (q, J = 6.3 Hz, 2H), 2.64 (t, J = 6.6 Hz, 2H), 2.57 − 2.47 (m, 4H), 2.41 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 162.4, 157.0, 143.6, 141.5, 139.7, 137.0, 133.5, 130.3, 126.8, 125.3, 124.8, 124.5, 123.1, 120.4, 116.7, 115.0, 66.6, 57.6, 53.9, 53.6, 35.8, 18.1. HRMS (ESI) calculated. for C27H30FN6O5S [M + H]+ m/z 569.1982, found: 569.1980.
8–(5-((4-Fluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-6-methyl-N-(3-morpholinopropyl)Imidazo[1,2-a]pyridine-2-carboxamide (41)
Light yellow solid; Yield: 55.2%. 1H NMR (600 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.78 (d, J = 2.2 Hz, 1H), 8.69 (d, J = 2.2 Hz, 1H), 8.40 (t, J = 1.5 Hz, 1H), 8.36 (s, 1H), 8.24 (t, J = 6.0 Hz, 1H), 7.88 − 7.83 (m, 2H), 7.49 (d, J = 1.3 Hz, 1H), 7.44 − 7.38 (m, 2H), 3.71 (s, 3H), 3.49 (t, J = 4.5 Hz, 4H), 3.36 (q, J = 6.7 Hz, 2H), 2.35 (d, J = 6.1 Hz, 3H), 2.34 − 2.23 (m, 6H), 1.71 − 1.64 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 164.81, 162.4, 156.8, 143.2, 141.6, 139.8, 137.1, 133.4, 130.2, 126.8, 125.4, 124.8, 124.6, 123.1, 120.5, 116.7, 115.1, 66.5, 56.6, 53.9, 53.7, 37.6, 26.5, 18.1. HRMS (ESI) calculated. for C28H32FN6O5S [M + H]+ m/z 583.2139, found: 583.2141.
4-Fluoro-N-(2-methoxy-5–(6-methyl-2-(morpholine-4-carbonyl)Imidazo[1,2-a]pyridin-8-yl)pyridin-3-yl)benzenesulfonamide (42)
White solid; Yield: 60.8%; 1H NMR (600 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.66 (d, J = 2.2 Hz, 1H), 8.63 (d, J = 2.2 Hz, 1H), 8.40 (d, J = 1.7 Hz, 1H), 8.37 (s, 1H), 7.84 (dd, J = 8.7, 5.2 Hz, 2H), 7.50 − 7.48 (m, 1H), 7.44 − 7.37 (m, 2H), 4.35 − 4.25 (m, 2H), 3.74 − 3.62 (m, 9H), 2.35 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 162.7, 157.3, 143.4, 140.9, 140.0, 137.2, 134.6, 130.3, 126.9, 125.6, 124.6, 124.5, 123.1, 120.1, 117.8, 116.6, 67.1, 66.8, 53.9, 47.6, 43.0, 18.1. HRMS (ESI) calculated. for C25H25FN5O5S [M + H]+ m/z 526.1560, found: 526.1552.
4-Fluoro-N-(2-methoxy-5–(6-methyl-2–(4-methylpiperidine-1-carbonyl)Imidazo[1,2-a]pyridin-8-yl)pyridin-3-yl)benzenesulfonamide (43)
White solid; Yield: 50.4%; 1H NMR (600 MHz, DMSO-d6) δ 10.02 (s, 1H), 8.70 (t, J = 2.1 Hz, 1H), 8.62 − 8.57 (m, 1H), 8.41 − 8.35 (m, 1H), 8.28 (d, J = 1.3 Hz, 1H), 7.88 − 7.81 (m, 2H), 7.50 − 7.44 (m, 1H), 7.39 (t, J = 8.9 Hz, 2H), 4.95 (d, J = 12.7 Hz, 1H), 4.50 (d, J = 12.5 Hz, 1H), 3.67 (s, 3H), 3.12 (t, J = 12.2 Hz, 1H), 2.75 (t, J = 12.2 Hz, 1H), 2.38 − 2.30 (m, 3H), 1.68 (s, 3H), 1.22 − 1.10 (m, 2H), 0.92 (d, J = 6.3 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 162.8, 157.3, 143.5, 140.9, 140.6, 137.2, 134.3, 132.0, 131.9, 130.3, 125.7, 124.5, 122.9, 120.1, 116.7, 116.5, 53.9, 47.0, 42.8, 35.1, 34.2, 31.1, 22.2, 18.1. HRMS (ESI) calculated. for C27H29FN5O4S [M + H]+ m/z 538.1924, found: 538.1952.
4-Fluoro-N-(2-methoxy-5–(6-methyl-2–(4-methylpiperazine-1-carbonyl)Imidazo[1,2-a]pyridin-8-yl)pyridin-3-yl)benzenesulfonamide (44)
White solid; Yield: 59.9%; 1H NMR (600 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.66 (d, J = 2.2 Hz, 1H), 8.60 (d, J = 2.2 Hz, 1H), 8.40 (t, J = 1.4 Hz, 1H), 8.34 (s, 1H), 7.87 − 7.81 (m, 2H), 7.47 (s, 1H), 7.43 − 7.36 (m, 2H), 4.25 − 4.15 (m, 2H), 3.73 − 3.63 (m, 5H), 2.40 (t, J = 5.1 Hz, 4H), 2.35 (s, 3H), 2.21 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.7, 162.7, 157.3, 143.1, 140.9, 140.2, 137.5, 134.1, 130.2, 126.8, 125.6, 124.7, 124.5, 123.0, 120.6, 117.4, 116.5, 55.6, 54.9, 53.9, 46.7, 46.0, 42.3, 18.1. HRMS (ESI) calculated. for C26H28FN6O4S [M + H]+ m/z 539.1875, found: 539.1877.
6-Chloro-8–(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-N-(3-morpholinopropyl)Imidazo[1,2-a]pyridine-2-carboxamide (45)
White solid; Yield: 47.9%; 1H NMR (600 MHz, Chloroform-d) δ 8.92 (d, J = 2.1 Hz, 1H), 8.46 (d, J = 2.1 Hz, 1H), 8.20 (d, J = 1.7 Hz, 1H), 8.17 (s, 1H), 8.03 (t, J = 5.9 Hz, 1H), 7.88 − 7.81 (m, 1H), 7.33 (d, J = 1.7 Hz, 1H), 6.97 − 6.90 (m, 1H), 6.89 − 6.85 (m, 1H), 3.96 (s, 3H), 3.64 (t, J = 4.4 Hz, 4H), 3.56 (q, J = 6.7 Hz, 2H), 2.48 (t, J = 7.0 Hz, 3H), 2.44 (s, 3H), 1.89 − 1.81 (m, 2H). 13C NMR (151 MHz, Chloroform-d) δ 166.2, 162.4, 159.9, 154.9, 141.8, 141.3, 140.9, 132.4, 129.3, 126.9, 124.4, 123.8, 123.4, 123.3, 121.7, 119.9, 114.8, 112.0, 105.8, 66.7, 56.8, 54.2, 53.6, 37.9, 26.2. HRMS (ESI) calculated. for C27H28ClF2N6O5S [M + H]+ m/z 621.1498, found: 621.1503.
4-Fluoro-N-(2-methoxy-5–(2-(4-methylpiperidine-1-carbonyl)Imidazo[1,2-a]pyridin-8-yl)pyridin-3-yl)benzenesulfonamide (46)
White solid; Yield: 58.1%; 1H NMR (600 MHz, DMSO-d6) δ 10.03 (s, 1H), 8.68 (d, J = 2.2 Hz, 1H), 8.63 − 8.61 (m, 1H), 8.59 (d, J = 6.7 Hz, 1H), 8.38 (s, 1H), 7.88 − 7.83 (m, 2H), 7.62 − 7.58 (m, 1H), 7.41 − 7.37 (m, 2H), 7.10 − 7.05 (m, 1H), 4.89 (d, J = 11.0 Hz, 1H), 4.51 (d, J = 10.2 Hz, 1H), 3.68 (s, 3H), 3.13 (s, 1H), 2.76 (s, 1H), 1.68 (d, J = 10.7 Hz, 3H), 1.19 − 1.05 (m, 2H), 0.95 − 0.85 (m, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.8, 162.9, 157.2, 143.4, 141.8, 140.7, 137.2, 134.0, 130.3, 127.2, 125.7, 125.3, 123.7, 120.2, 117.0, 116.6, 113.8, 53.9, 47.1, 42.7, 35.0, 34.2, 31.0, 22.2. HRMS (ESI) calculated. for C26H27FN5O4S [M + H]+ m/z 524.1768, found: 524.1852.
Pharmacology
In vitro enzymatic assay
In vitro enzymatic assay of compounds 15–46 against PI3Kα was evaluated by ADP-Glo™ Kinase Assay. Briefly, the compound, PI3K enzyme (PI3Kα from Invitrogen), the PIP2 (Life Technologies) substrate, and ATP (25 μM, Sigma) were diluted in kinase buffer to the indicated concentrations. The assay plate was covered and incubated at room temperature for 1 h. Then, the Kinase-Glo reagent (Promega) was added to the PI3Kα plate to stop the reaction, mixed briefly with centrifuge, shaken slowly on the shaker and then equilibrate for 120 min. Finally, add Kinase detection reagent to each well, shake 1 min, equilibrate for 30 min before reading on a plate reader for luminescence. The data were collected on Envision and presented in Excel. IC50 values were calculated from the inhibition curves.
Cell proliferation assays
All target compounds were evaluated for antiproliferative potency against SKOV-3, T47D,NCI-H1975, NCI-H460,and MCF-7 tumour cell lines using a CellTiter-Glo® Luminescent Cell Viability Assay. The human tumour cell lines used were obtained from the ATCC or Shanghai ZhongQiaoXinZhou Biotech. All the mediums and FBS were Gibco. T47D, H1975 and NCI-H460 tumour cells were cultured in RPMI1640 medium supplemented with 10% FBS. SKOV-3 was cultured in McCoy’s 5 A medium supplemented with 10%FBS. The day before treatment with compounds, cells were seeded at a density of 2000 or 5000 in each well of a 96-well plate. The tumour cells were then treated with 3-fold serial diluted compound or DMSO control in the incubator at 37 °C and 5% CO2 for 3 days, prior to the addition of CellTiter-Glo reagents (Promega) and reading of luminescence using a PerkinElmer Envision plate reader. Data were analysed using GraphPad Prism 7.0.
Cell cycle and apoptosis analysis
For cell cycle analysis, T47D cell (1 × 106) were seeded in 6-well plates overnight and treated with different concentrations of compound 35 and DMSO control on the next day. After 24 h, the cells were collected by EDTA-free trypsinization, centrifuged at 1500 rpm for 5 min, and fixed in 75% ethanol at −20 °C for 1 h. The cells were then centrifuged at 2000 rpm for 5 min, resuspended in 500 µL PI/Rnase Staining Buffer, incubated for 30 min at room temperature, and analysed using flow cytometry. The data were analysed using Modfit software. For cell apoptosis analysis, T47D cell were seeded in 6 well-plates and treated with DMSO or compound 35 for 24 h. The treated cells were collected and washed with cold PBS for three times, resuspended in 185 µL binding buffer containing 5 μL Annexin V-FITC Apoptosis Detection Kit and 10 μL 7-AAD Viability Staining Solution for 15 min at room temperature in the dark. Apoptotic cells were analysed by flow cytometer.
Liver microsomal stability assay
The metabolic stability of compound 35 was determined in human or SD rat liver microsomes, with and without the NADPH regenerating system. Briefly, compound 35 was incubated with microsomes (human microsome, CORNING, lot No. 38296; rat microsome, Xenotech, Lot No. 2110178) (0.5 mg protein/mL) at 1 μM at 37 °C in potassium phosphate buffer (100 mM at pH 7.4 with 10 mM MgCl2). The reactions were initiated by adding prewarmed cofactors (1 mmol NADPH). After incubation for different times (0, 5, 10, 20, 30, and 60 min) at 37 °C, cold acetonitrile containing 200 ng/mL tolbutamide and 200 ng/mL labetalol as internal standards was added to precipitate the protein. Then, the samples were centrifuged, and the supernatants were transferred into HPLC water, mixed by plate shaker for 10 min prior to LC − MS/MS analysis.
Parallel artificial membrane permeability assay (PAMPA)
10.0 μM donor solution (5% DMSO) was prepared by diluting of working solution with PBS. The donor solution was added to each well of the donor plate, whose PVDF membrane was precoated with 5 µL of 1% lecithin/dodecane mixture. Duplicates were prepared.Then, PBS was added to each well of the PTFE acceptor plate. The donor plate and acceptor plate were combined and incubated for 4 h at room temperature with shaking at 300 rpm. Acceptor samples and donor samples were prepared and analysed by LC-MS/MS.
Cyp 450 inhibition assay
Cytochrome P450 inhibition was evaluated in human liver microsomes (0.253 mg/mL, Lot No. 38292) using five specific probe substrates (CYP1A2, 10 μM phenacetin; CYP2D6, 5 μM dextromethorphan; and CYP3A4, 2 μM midazolam; 2C9, 5 μM Diclofenac; 2C19, 30 μM S-mephenytoin) and positive control in the presence of multiple concentrations of the test compound. After pre-warm at 37 °C for 10 min, the reaction was initiated by the addition of of NADPH. The mixture was incubated at 37 °C for 10 min, terminated by the addition of cold stop solution (200 ng/mL tolbutamide and 200 ng/mL labetalol in acetonitrile). The samples were centrifuged, and the supernatants were analysed by LC − MS/MS.
Molecular docking and dynamics simulation
The protein coordinate (PDB: 4JPS), downloaded from the Protein Data Bank (http://www.rcsb.org/pdb/), was chosen as templates to compare the docking mode among compound 35 bound to PI3Kα. Molecular docking calculations were conducted using the Dock6 protocol in Yinfo Cloud Platform (http://cloud.yinfotek.com/). Briefly, the structure of compound was built with energy minimisation in MMFF94 force field, and PI3Kα was assigned hydrogen atoms and partial charges in Amber ff14SB force field and partial charges in Chimaera. The binding pocket of the crystal ligand was assumed to be analogous to that of 35 in PI3Ks. The box centre and the dimensions were thus set. The DOCK 6.7 program was utilised to conduct semiflexible docking with 10 000 different orientations generated. Then, the Grid-based score was calculated for each pose. The image files were generated by Pymol. To investigate the combined stability of inhibitors to PI3K kinase, the MD simulations were conducted on the PI3K kinase in complex with compound 35 by Amber 16 software. The restrained electrostatic potential (RESP) calculated by the Gaussian16 package was used to fit the charges for the inhibitors. The ff14SB force field and the general AMBER force field (gaff2) were used for PI3K kinase and the inhibitors, respectively. Build a solvent octahedral box with a boundary 15 Å away from the protein and use the TIP3P water model to fill the entire octahedral box. 7 Na+ ions were added to neutralise the system. The MD simulation protocol comprised the following stepsCitation1: The limiting potentials of proteins, ligands, and counterions were all restricted by the force constant of 200 kcal/(mol Å), and the energy of the solvent water molecules was minimised to make the water molecules reach a relaxed state. (2) The energy of the system is further minimised. Protein, ligand and ions were subjected to the limiting potential with a force constant of 300 kcal/(molÅ). (3) The restriction potential of the protein backbone was restricted by the force constant of 20 kcal/(mol Å). (4) Then the system was minimised without any restriction. The cpptraj module in AMBER16 was employed for the RMSD calculations.
Supplemental Material
Download PDF (831.2 KB)Acknowledgements
We would also like to thank Guangzhou Yinfo Information Technology Co., Ltd. for providing a friendly and versatile web server (https://cloud.yinfotek.com) to aid the docking studies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Corrie PG. Cytotoxic chemotherapy: clinical aspects. Medicine. 2008;36(1):24–28.
- Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther. 2016;38(7):1551–1566.
- Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675.
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619.
- Carrera AC, Anderson R. The cell biology behind the oncogenic PIP3 lipids. J Cell Sci. 2019;132:jcs228395.
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004.
- Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170(4):605–635.
- Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20(1):87–90.
- Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13(5):283–296.
- Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15(1):7–24.
- Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13(2):140–156.
- Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36(7):422–439.
- Markham A. Copanlisib: first global approval. Drugs. 2017;77(18):2057–2062.
- Scott WJ, Hentemann MF, Rowley RB, Bull CO, Jenkins S, Bullion AM, Johnson J, Redman A, Robbins AH, Esler W, et al. Discovery and SAR of Novel 2,3-DihydroImidazo[1,2-c]quinazoline PI3K Inhibitors: identification of Copanlisib (BAY 80-6946). ChemMedChem. 2016;11(14):1517–1530.
- Burger MT, Pecchi S, Wagman A, Ni Z-J, Knapp M, Hendrickson T, Atallah G, Pfister K, Zhang Y, Bartulis S, et al. Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer. ACS Med Chem Lett. 2011;2(10):774–779.
- Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51(18):5522–5532.
- André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380(20):1929–1940.
- Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J, Aichholz R, et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013;23(13):3741–3748.
- Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, Flinn IW, Flowers CR, Martin P, Viardot A, et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370(11):1008–1018.
- Kendall JD, O’Connor PD, Marshall AJ, Frédérick R, Marshall ES, Lill CL, Lee W-J, Kolekar S, Chao M, Malik A, et al. Discovery of pyrazolo[1,5-a]pyridines as p110a-selective PI3 kinase inhibitors. Bioorg Med Chem. 2012;20(1):69–85.
- Gao GR, Liu JL, Mei DS, et al. Design, synthesis and biological evaluation of acylhydrazone derivatives as PI3K inhibitors. Chin Chem Lett. 2015; 26:562–570.
- Liao W, Wang Z, Han Y, Qi Y, Liu J, Xie J, Tian Y, Lei Q, Chen R, Sun M, et al. Design, synthesis and biological activity of novel 2,3,4,5-tetra-substituted thiophene derivatives as PI3Kα inhibitors with potent antitumor activity. Eur J Med Chem. 2020; 197:112309.
- Tanaka H, Yoshida M, Tanimura H, Fujii T, Sakata K, Tachibana Y, Ohwada J, Ebiike H, Kuramoto S, Morita K, et al. The selective Class I PI3K inhibitor CH5132799 targets human cancers harboring oncogenic PIK3CA mutations. Clin Cancer Res. 2011;17(10):3272–3281.
- Knight SD, Adams ND, Burgess JL, Chaudhari AM, Darcy MG, Donatelli CA, Luengo JI, Newlander KA, Parrish CA, Ridgers LH, et al. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the Mammalian target of Rapamycin. ACS Med Chem Lett. 2010;1(1):39–43.
- Lin S, Wang C, Ji M, Wu D, Lv Y, Zhang K, Dong Y, Jin J, Chen J, Zhang J, et al. Discovery and optimization of 2‑Amino-4-methylquinazoline derivatives as highly potent phosphatidylinositol 3‑kinase inhibitors for cancer treatment. J Med Chem. 2018;61(14):6087–6109.