
Abstract
Herein, a new series of 2-chloro-N-(5-(2-oxoindolin-3-yl)-4H-pyrazol-3-yl) acetamide derivatives containing 1,3,4-thiadiazole (10a–i) and 4H-1,2,4-triazol-4-amine (11a–r) moiety was designed, synthesised as novel anticancer agents. The antiproliferative activity values indicated that compound 10 b stood as the most potent derivative with IC50 values of 12.0 nM and 10 nM against A549 and K562 cells, respectively. Mechanism investigation and docking studies of 10 b indicated that it possessed good apoptosis characteristic and dose-dependent growth arrest of A549 and K562 cells, blocked cell cycle into G2/M phase. Interestingly, 10 b suppressed the growth of A549 and K562 cells via modulation of EGFR and p53-MDM2 mediated pathway.
Introduction
Cancer, a kind of pathological proliferation of abnormal cells, fast, incontrollable, and unregulated, is the second leading cause of death worldwide after cardiovascular diseaseCitation1,Citation2. Worldwide, the cancer incidence, an estimated 19.3 million new cancer cases in 2020, compared to 2018, an estimated 18.1 million, increased by 6.2%. Lung cancer is the most commonly diagnosed cancer (11.6% of the total cases), closely followed by female breast cancer (11.6%), prostate cancer (7.1%), and colorectal cancer (6.1%) for incidence in 2018Citation3,Citation4. Lung cancer and leukaemia remain major public health concern among current all diseases due to the toxicity and side effects of the available commercially synthesised drugsCitation5,Citation6. Currently, chemotherapy, surgical resection, and radio therapy are the main means of cancer treatment, while chemotherapy plays an indispensable role in current clinical treatment. However, due to poor selectivity, drug resistance and serious side effects of chemotherapy, its clinical application is limited. Therefore, it is of great significance to find, design and synthesise novel and highly effective targeted small molecule antitumor drugs for cancer treatmentCitation7–9.
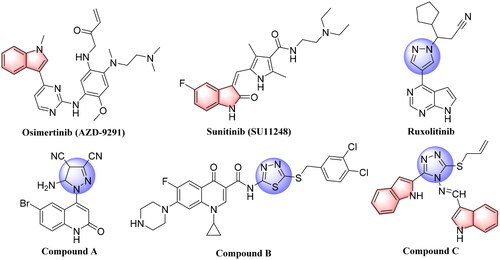
In recent studies, the development of novel scaffolds focussed on the mutant-selective EGFR-TKIs against T790M and C797S resistance, which was proven as an effective approachCitation10,Citation11. Indole core plays an important role in drug discovery, which has a wide range of biological activities, especially in the development of anticancer agents, such as Vinblastine (VLB), Vincristine (VCR), Osimertinib (AZD-9291), and Sunitinib (SU11248) ()Citation12–15. Penta-heterocycles framework is an exceptionally adaptable drug-like format that is being utilised broadly in the structure of cancer treatments and cellular apoptosis such as pyrazoles, which were reported in a variety of potent anti-cancer active agents as listed on RuxolitinibCitation16 and Compound ACitation17 (). Furthermore, 1,3,4-thiadiazoles such as Compound BCitation18 was reported as highly anti-cancer agent by inducing apoptosis significantly in MCF-7 cell (). Substituted 1,2,4-triazazole (Compound C)Citation19 also displayed remarkable epidermal growth factor receptor (EGFR) inhibition activity with IC50 value of 19.8 nM ().
Figure 1. Structures of Osimertinib (AZD-9291), Sunitinib (SU11248), approved anticancer drugs, and some previously reported penta-heterocycles scaffold as antiproliferative agents (Compound A, B and C).
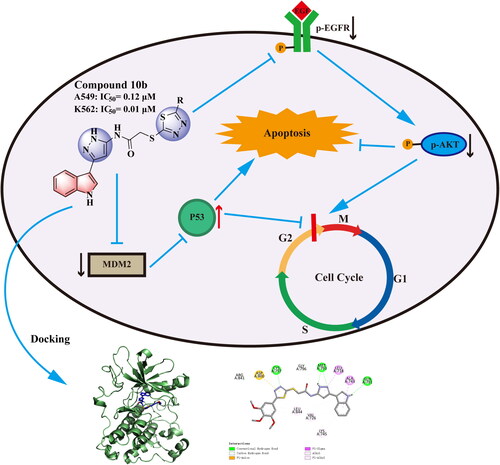
EGFR mutation and overexpression are closely related to tumorigenesis and development of non-small-cell lung cancer (NSCLC), which makes EGFR an important target for the design and development of antitumor agentsCitation20–22. MDM2 directly binds to and forms a complex with p53, inhibiting p53 transactivationCitation23. Several different therapeutic approaches have been attempted with the goal of restoring p53 functionCitation24–27. Among these, targeting the MDM2-p53 interaction by small molecules for the reactivation of p53 has emerged as a promising approach for the treatment of cancers that retain wild-type p53Citation28–30. Our previous work explored strategies for designing and synthesising a series of heterocycle antitumor inhibitorsCitation31–34. Based on structure guided molecular hybridisation, we designed and synthesised a series of novel indole derivatives in this research containing 1,3,4-thiadiazoles (10a–i) and 4H-1,2,4-triazol-4-amines (11a–r), expecting to discover compounds with more powerful activity. In particularly, we found the resulting most active antitumor candidates (10 b and 11 h) (), as well as the inhibition mechanisms of the said compounds by apoptosis, cell cycle, cell migration, EGFR and p53-MDM2 enzyme inhibition analysis, and docking studies, wherein it proved to be a highly potent antitumor agents in the future.
Figure 2. The design strategy of the target compounds in this study, and the resulting most active antitumor candidates (10 b and 11 h).
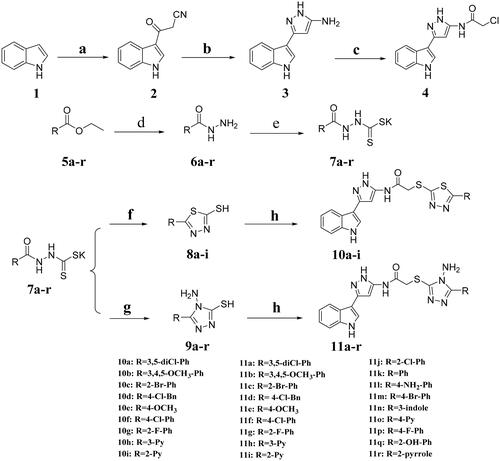
Reagents and conditions: a: Cyanoacetic acid, Ac2O, 40 °C, 4 h; b: Hydrazine hydrate (80%), EtOH, reflux; c: Chloroacetyl chloride, 1,4-dioxane, rt; d: Hydrazine hydrate (80%), EtOH, reflux; e: KOH/CS2, EtOH, reflux; f: (1) KOH/CS2, EtOH, reflux; (2) HCl, pH4-5; g: (1) Hydrazine hydrate (80%), reflux; (2) HCl, pH2-3; h: intermediate 4, KOH, EtOH, rt.
Results and discussion
Chemistry
The general route for the synthesis of title compounds 10a–i and 11a–r was illustrated in Scheme 1. Compound 2 was synthesised through indole as starting material added to cyanoacetic acid and acetic anhydride. Then, cyclisation reaction was carried out to obtain compound 3. The step of amino acylation was operated subsequently with chloroacetyl chloride to give intermediate 4. The 1,3,4-thiadiazoles (8a–i) and 1,2,4-triazoles (9a–r) were prepared according to a series of reactions of esterification, hydrazinolysis, salt formation and cyclisation. Target compounds were synthesised by binding intermediate 4 with 1,3,4-thiadiazoles (8a–i) and 1,2,4-triazoles (9a–r) using potassium hydroxide as a catalyst at room temperature. All the target derivatives were fully characterised by 1H NMR, 13C NMR and HR-MS.
Scheme 1 Synthetic route for the title compounds 10a–i and 11a–r.
In vitro antiproliferative activities
To probe the in vitro cytotoxic impact of these synthesised indole derivatives (10a–i and 11a–r), four human cancer cell lines, human non-small cell lung cancer cells (A549), human prostate cancer cells (PC-3), human liver cancer cells (HepG2) and human chronic myeloid leukaemia cells (K562) were selected. The results expressed as IC50 (µM) were summarised in . The majority of the tested compounds exhibited moderate to potent antiproliferative activity, especially the series of compounds 10a–i. Remarkably, analogues 10a, 10 b, 10d, 10e, 10f, 11 h, 11 l exhibited potent antitumor efficacies against K562 (IC50 = 0.06–0.99 µM), while 10a, 10 b, 10e were found to be the most active compounds with the IC50 values of 0.12–0.83 µM against A549. Interestingly, 10 b was stood as the most potent derivative with IC50 values of 0.12 µM, 0.85 µM, 0.12 µM, 0.21 µM, 0.01 µM, respectively, against A549, PC-3, HepG2 and K562. Clearly, 10 b showed 82 folds and 1225 times superior potency to the reference drug Gefitinib against A549, and 5-Fluorouracil (5-Fu) against K562. On the other hand, except for compounds 11 h, 11i and 11 l in the series of compounds 11a–r, the rest of the compounds displayed weak to moderate anti-proliferative activities against the tested cell lines. Amazingly, compound 11 h (IC50 = 0.06 µM) had excellent anticancer activity against the K562 cell line. Next, we found that compound 1,3,4-thiadiazoles displayed dramatically potent anticancer activity than 1,2,4-triazoles against four cancer cell lines. However, after introducing an electron-withdrawing group, bromine and fluorine atoms to the ortho-position in the benzene group of 1,3,4-thiadiazole derivatives, compound 10 c and 10 g only kept a quite weak antiproliferative activity.
Table 1. Antiproliferative activity of the synthesised compounds 10a–i and 11a–r a.
The results of CC50 against normal cell line (HEK293) indicated these synthesised compounds were also moderately cytotoxic to normal cells, which were not currently the most promising candidate drugs. However, by calculating the corresponding relative selectivity indexes of compounds 10 b and 11 h in A549 and K562 cell lines, the results in showed that the possibility of drug development was still optimistic. The follow-up study for us is to optimise the structure of the compound to reduce the toxicity to normal cells
Table 2. The selectivity index for the compounds 10 b and 11 h.
Apoptosis analysis
Concerning the antitumor activity of derivative 10 b, we first examined whether the inhibitory effects of 10 b on A549 and K562 cell lines were due to apoptosis by DAPI staining and apoptosis morphological analysis. After 48 h incubation with 10 b at 1, 2, and 4 µM in DAPI staining, analogue 10 b induced apoptosis as shown by DAPI staining of micronuclei of proliferating A549 cells. As the nuclear membrane was ruptured, 10 b induced significant dense blue staining and apoptotic bodies formation, thereby indicating that 10 b had apoptotic activity against A549 cells.
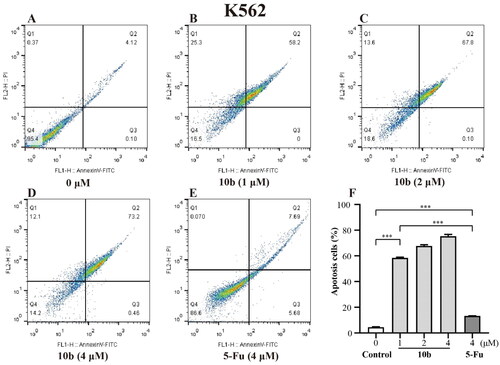
Meanwhile, the detailed cytotoxic effects of A549 and K562 cells were also performed through annexin V-FITC/PI double staining and quantified by flow cytometry for apoptotic analysis method. As shown in , we tested apoptosis caused by 10 b and compared it with 5-Fu in A549 and K562 cells. After 48 h incubation, the results showed that the percentage of the apoptotic cells of A549 and K562 treated with 10 b mainly lead to late apoptotic. Specifically, the numbers of late apoptotic cells of A549 were up to 8.36%, 16.0% and 35.7% at 1, 2 and 4 µM in 10 b treatment, respectively, whereas the total numbers of apoptotic cells were only 28.3% at 4 µM 5-Fu treatment (). There were special concerns associated with the percentage of late apoptotic cells of K562 were rapidly up to 58.2%, 67.8% and 73.2% in a dose dependent manner at 1, 2 and 4 µM 10 b treatment, respectively, whereas that the total numbers of apoptotic cells were only 13.4% at 4 µM 5-Fu treatment (). Indeed, cell apoptosis was increased for 10 b compared with control 5-Fu both in A549 and K562 cells.
Figure 3. The fluorescence microscopy images of A549 cells after DAPI staining. A549 cells treated with compound 10 b at 1, 2 and 4 μM for 48 h. Arrows indicated signs of nuclear shrinkage and chromatin condensation, the scar bar is 100 μm. And the apoptosis analysis through annexin V-FITC/PI double staining and following flow cytometry for the A549 cells treated with compound 10 b at 1, 2 and 4 μM for 48 h. DMSO was the negative control, as 5-Fu was the reference drug. (A) Control, (B) 10 b (1 μM), (C) 10 b (2 μM), (D) 10 b (4 μM) and (E) 5-Fu (4 μM). (F) The percentage of apoptosis cells was quantified in the segments of the bar chart. Three individual experiments were performed for each group. Data are expressed as the mean ± SD of three independent experiments. ***p < 0.005.
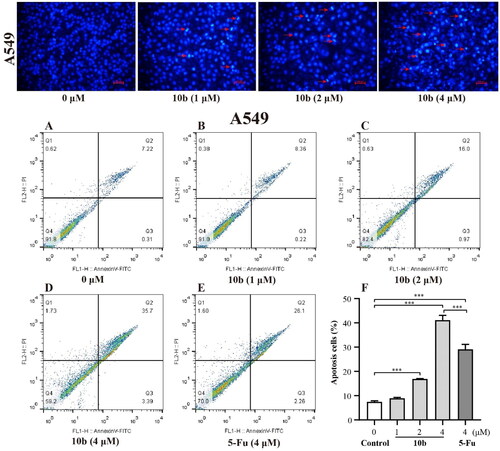
Figure 4. Apoptosis analysis through annexin V-FITC/PI double staining and following flow cytometry for the K562 cells treated with compound 10 b at 1, 2 and 4 μM for 48 h. DMSO was the negative control, as 5-Fu was the reference drug. (A) Control, (B) 10 b (1 μM), (C) 10 b (2 μM), (D) 10 b (4 μM) and (E) 5-Fu (4 μM). (F) The percentage of apoptosis cells was quantified in the segments of the bar chart. Three individual experiments were performed for each group. Data are expressed as the mean ± SD of three independent experiments. ***p < 0.005.
Cell cycle analysis
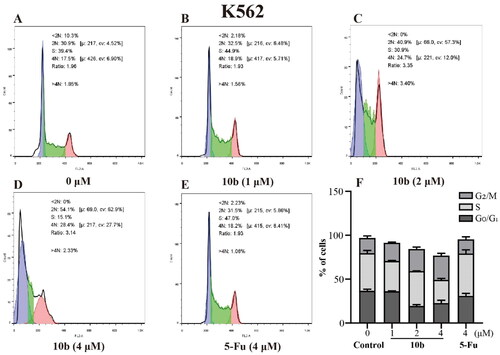
Cell cycle arrest was an important indication for inhibition of proliferationCitation35; therefore, we evaluated the effect of the most promising compound 10 b on the cell cycle of A549 and K562 cells using the flow cytometry assay. Thus, the influence of 10 b on the cell cycle progression was investigated, with 48 h following treatment at 1, 2, 4 µM, interpreted in and . When the A549 cell line was exposed to 10 b at 4 µM for 48 h, the DNA contents of the live population were 49.03%, 30.33% and 12.77% for untreated cells, 38.60%, 27.47% and 24.33% for 10 b-treated cells, and 46.87%, 34.63%, and 13.13% for 5-Fu-treated cells (4 µM), respectively, for G0/G1, S, and G2/M phase. Indeed, treatment with 10 b (4 µM) resulted in a 1.85-fold than 5-Fu treatment, respectively increased in the fraction of cells arrested at G2/M. Similar results were also observed for the K562 cell line. When the K562 cell line was exposed to 10 b at 4 µM for 48 h, the DNA contents of the live population were 36.70%, 42.80%, and 17.43% for untreated cells, 22.73%, 26.47%, and 27.60% for 10 b-treated cells, and 30.93%, 48.27%, and 16.07% for 5-Fu-treated cells (4 µM), respectively, for G0/G1, S, and G2/M phase. Meanwhile, treatment with 10 b (4 µM) resulted in a 1.72-fold than 5-Fu treatment, respectively increased in the fraction of cells arrested at G2/M. Taken together, these results suggested that 10 b delayed cell cycle progression by arresting cells in the G2/M phase of the cell cycle in a dose-dependent manner, regarding A549 and K562 cell growth.
Figure 5. Cell cycle analysis through PI staining and following flow cytometry for the A549 cells treated with compound 10 b at 1, 2 and 4 μM for 24 h. DMSO was the negative control, as 5-Fu was the reference drug. (A) Control, (B) 10 b (1 μM), (C) 10 b (2 μM), (D) 10 b (4 μM) and (E) 5-Fu (4 μM). (F) The proportion of the cell cycle was quantified in the segments of the bar chart. Three individual experiments were performed for each group. Data are expressed as the mean ± SD of three independent experiments. ***p < 0.005.
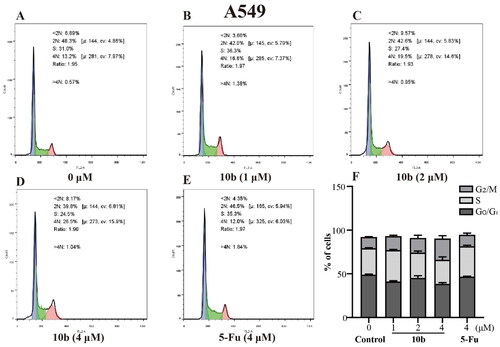
Figure 6. Cell cycle analysis through PI staining and following flow cytometry for the K562 cells treated with compound 10 b at 1, 2 and 4 μM for 24 h. DMSO was the negative control, as 5-Fu was the reference drug. (A) Control, (B) 10 b (1 μM), (C) 10 b (2 μM), (D) 10 b (4 μM) and (E) 5-Fu (4 μM). (F) The proportion of the cell cycle was quantified in the segments of the bar chart. Three individual experiments were performed for each group. Data are expressed as the mean ± SD of three independent experiments. ***p < 0.005.
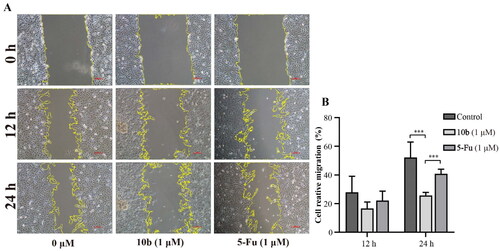
Cell migration assay
Analysis of cell migration in vitro is a useful assay to quantify alterations in cell migratory capacity in response to experimental manipulationsCitation36. Meanwhile, cell migration also is an important malignant behaviour and contributes to metastasis, a major cause of cancer-related deathCitation37. After incubation with compound 10 b of 1 µM for a few hours, the scratched areas of experimental groups were significantly larger than the control groups, indicating that compound 10 b could inhibit the migration of A549 cells ().
Figure 7. Cell migration and invasion assay carried out in A549 after 12 and 24 h of 1 μM 10 b or 5-Fu treatment. (A) Representative images of wound healing migration assay; (B) Semi-quantitative analysis of migration cells at non-covered area. The scar bar is 100 μm. Three individual experiments were performed for each group. Data are expressed as the mean ± SD of three independent experiments. ***p < 0.005.
Western blot assay
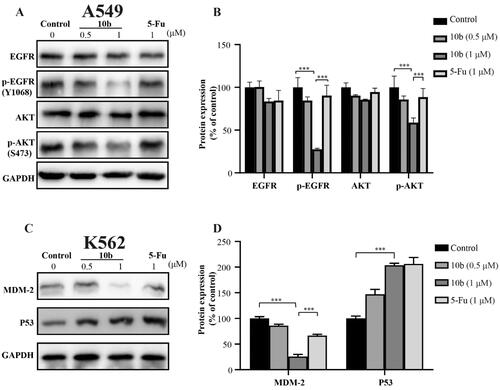
To better understand the mechanisms involved in compound 10 b-mediated antitumor activity, we further evaluated the effect of 10 b on expression level and phosphorylation of EGFR and AKT in A549 cells using western blot assayCitation38,Citation39. Because MDM2 is a primary cellular inhibitor of p53, it may be effective in the treatment of human cancer through retaining wild-type p53 by reactivating the p53 tumour suppressor function. Thus, the effect of 10 b on p53 in K562 cells was further investigatedCitation40. These results indicated that the p-EGFR and p-Akt proteins in A549 cells was downregulated in a dose dependently manner after 48 h treatment of 10 b. Moreover, after treated with compound 10 b at 0.5 µM in K562 cells, p53 protein was upregulated in a certain extent. Obviously, the expression of MDM2, as the negative regulator of p53 protein, was significantly downregulated. These data suggested that compound 10 b could inhibit the proliferation of A549 cells through EGFR-mediated pathway and regulate MDM2-p53 interaction to inhibit the K562 cells ().
Figure 8. Western blot analysis of proteins obtained from cells treated with compound 10 b at 0.5 and 1 μM for 48 h. DMSO was the negative control, as 5-Fu was the reference drug. (A) Western blotting analysis of EGFR, p-EGFR, Akt and p-Akt levels in A549. (B) Average fold change of EGFR, p-EGFR, Akt and p-Akt with respect to the housekeeping gene GAPDH in different treatment groups following densitometric analysis. (C) Western blotting analysis of MDM2 and p53 levels in K562. (D) Average fold change of MDM2 and p53 with respect to the housekeeping gene GAPDH in different treatment groups following densitometric analysis. Three individual experiments were performed for each group. Data are expressed as the mean ± SD of three independent experiments. ***p < 0.005.
Docking studies
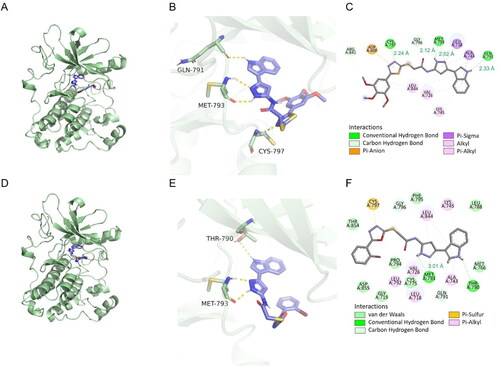
To corroborate the relation between in vitro cytotoxicity findings and binding affinities of analogues, docking analysis of the compound 10 b against EGFR (PDB code: 2ITY) was performed using docking studies, including the binding modes/orientations, affinities, and interactions analysis. The results of the docking study revealed nice fitting of the two compounds into EGFR with absence of any steric clashes or unfavourable interactions. Investigation of the binding interactions of compound 10 b showed two conventional hydrogen bonds with Met793 (2.12 Å and 2.52 Å), one conventional hydrogen bonds with Cys797 (2.24 Å) and Gln791 (2.33 Å), respectively. Compound 10 b also showed several hydrophobic interactions of the pi-sigma, pi-alkyl, pi-anion and alkyl types with Leu844, Val726, Lys745, Ala743, Leu718, Asp800 (). The docking score of delta G is minus 8.1, that negative binding affinity indicates the possibility of binding, while smaller value indicates stronger binding. We also examined whether this series of compounds such as 11 h have similar binding properties. Investigation of the binding interactions of compound 11 h showed one conventional hydrogen bonds with Gly796 (3.01 Å). Compound 11 h also showed several hydrophobic interactions of the pi-sigma, pi-alkyl, pi-anion and alkyl types with Met793, Leu718, Cys775, Leu844, Ala743, Val726, Lys745, Asp855 (). The docking score of delta G by 11 h is minus 8.9, which also has the fine combination ability. Compound 10 b and 11 h were superposed with gefitinib, found that although there were some differences in chemical structure, there was still certain superimposition, such as compound 10 b with an RMSD value of 4.87 Å, and compound 11 h with an RMSD value of 6.14 Å. Docking results showed that compounds 10 b and 11 h could successfully bind to the ATP pocket of EGFR protein, suggesting that it may be an ATP competitive inhibitor ().
Figure 9. The docking results were analysed visually and binding interactions of compounds 10 b and 11 h into EGFR. (A, D) Cartoon like representation showing the co-crystallized Gefitinib into EGFR of compound 10 b and 11 h. (B, E) 3 D binding mode of compound 10 b and 11 h into the active site of EGFR. (C, F) 2 D binding mode of compound 10 b and 11 h in the active site EGFR.
Conclusion
Herein, a series of indole derivatives with penta-heterocycles substitutions were reported. The antiproliferative activity values indicated that compound 10 b stood as the most potent derivative with IC50 values of 120 nM and 10 nM, respectively, against A549 and K562 cells. To further investigate the mechanism underlying the antiproliferative effect of 10 b, the analysis of apoptosis, cell cycle arrest, cell migration, and the expression level and phosphorylation of EGFR and AKT, and p53 related proteins were investigated. Accordingly, it was evident that both DAPI staining and apoptosis morphological analysis observation strongly suggested 10 b induced apoptosis both in A549 and K562 cells. As illustrated in this study, the flow cytometric results showed that the exposure of A549 and K562 cells to compound 10 b gave rise to a significant rise in the cell populations at G2/M phase in a dose-dependent manner. Furthermore, 10 b displayed effects on suppression of EGFR-mediated proteins and promotion of p53 protein and indicating its anti-proliferation characteristics against A549 and K562 cells. In addition, the molecular docking assay of 10 b was performed to identify the protein-binding properties for the potential active sites of the EGFR. Interestingly enough, both 11 h and 10 b applied similar mode to bind with ATP pocket of EGFR protein. However, we still misunderstand the molecular pathway engaged by 11 h regular consumption and there are still a lot of studies in our future work. In conclusion, this series indole derivatives might serve as promising candidate for further development of potent A549 and K562 inhibitors.
Experimental section
Chemistry
The reagents were purchased from commercial sources, used directly. The melting points of the compounds were tested on an X-4D melting point apparatus. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX 400 MHz (Bruker, Germany), or on a JEOLECX-500 (JEOL, Japan) in DMSO-d6 solution. High-resolution mass spectra were obtained with Thermo Scientific Q Exactive (Thermo Scientific, USA).
General synthesis
Initially, indole as the starting material was reacted with cyanoacetic acid to obtain intermediate 2 as per the reported methodCitation41. The cyclic reaction of compound 2 with 80% hydrazine hydrate was carried out to obtain intermediate 3Citation42. The reaction of intermediate 4 with chloroacetyl chloride, in 1,4-dioxane was constructed according to the published procedures with some modificationsCitation43.
General procedure for the synthesis of compounds 10a–i
The different 1,3,4-thiadiazoles (8a–i) were prepared according to known methodsCitation44–46. Then, a mixture of the intermediate 1,3,4-thiadiazoles (10 mmol) and potassium hydroxide (20 mmol, 1.1 g) was stirred in anhydrous ethanol (50 ml). A solution of intermediate 4 (10 mmol, 2.7 g) was subsequently added into the mixture, which was stirred at room temperature for 3 h. Then, poured into 200 ml of ice water, when the reaction was completed. The solid was collected through filtration and then purified through crystallisation from absolute alcohol to give pure title compounds (10a–i).
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((5-(3,5-dichlorophenyl)-1,3,4-thiadiazol-2-yl)thio)acetamide (10a).
A green solid, yield 64.2%, m. p. 169–171 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.57 (s, 1H), 11.45 (s, 1H), 10.86 (d, J = 38.4 Hz, 1H), 8.08–7.69 (m, 4H), 7.46 (d, J = 7.8 Hz, 1H), 7.28–7.02 (m, 3H), 6.85 (d, J = 16.2 Hz, 1H), 4.27 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.2, 165.7, 164.7, 164.2, 136.8, 135.6, 132.8, 131.0, 126.4, 124.8, 123.9, 122.3, 121.7, 120.4, 119.5, 112.5, 105.7, 93.3, 43.5. HRMS (ESI)
[M + Na] + calcd for C21H14 Cl2N6OS2: 522.9924, found: 522.9940.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((5-(3,4,5-trimethoxyphenyl)-1,3,4-thiadiazol-2-yl)thio)acetamide (10 b).
A yellow solid, yield 27.2%, m. p 261–263 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.54 (s, 1H), 11.46 (s, 1H), 10.88 (s, 1H), 7.89–7.68 (m, 2H), 7.46 (d, J = 8.1 Hz, 1H), 7.32–6.99 (m, 4H), 6.83 (s, 1H), 4.35 (s, 2H), 3.91–3.84 (m, 6H), 3.76–3.63 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.5, 165.3, 164.8, 153.9, 148.1, 140.4, 138.2, 136.8, 125.1, 124.8, 123.8, 122.3, 120.4, 119.4, 112.5, 105.6, 105.3, 104.0, 93.3, 60.7, 56.6, 38.3. HRMS (ESI) [M-H] + calcd for C24H22N6O4S2 521.1060, found 521.1082.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((5–(2-bromophenyl)-1,3,4-thiadiazol-2-yl)thio)acetamide (10c).
A white solid, yield 21.2%, m. p. 159–161 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.51 (s, 1H), 11.42 (s, 1H), 10.89 (s, 1H), 7.93 (dd, J = 7.8, 1.8 Hz, 1H), 7.81 (dd, J = 8.0, 1.2 Hz, 1H), 7.73 (t, J = 5.1 Hz, 2H), 7.57–7.38 (m, 3H), 7.18–7.04 (m, 2H), 6.79 (s, 1H), 4.35 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 168.5, 164.9, 164.3, 164.3, 162.1, 148.1, 138.3, 136.8, 129.7, 124.8, 123.9, 123.9, 122.4, 122.3, 120.4, 119.5, 115.4, 112.6, 105.7, 93.4, 43.6. HRMS (ESI) [M-H] + calcd for C21H15BrN6OS2 508.9848, found 508.9865.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((5-(4-chlorobenzyl)-1,3,4-thiadiazol-2-yl)thio)acetamide (10d).
A yellow solid, yield 67.4%, m. p. 151–153 °C; 1H NMR (400 MHz, DMSO-d6) δ12.55 (s, 1H), 11.48 (s, 1H), 10.84 (s, 1H), 7.78 (d, J = 2.7 Hz, 1H), 7.41 (ddd, J = 22.3, 21.4, 8.2 Hz, 6H), 7.20–7.11 (m, 2H), 6.82 (d, J = 15.7 Hz, 1H), 4.44 (s, 2H), 4.30–4.24 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 169.9, 165.9, 164.8, 136.8, 136.8, 132.4, 131.2, 129.2, 124.8, 123.9, 123.8, 122.3, 120.3, 119.5, 112.5, 105.7, 93.3, 49.1, 43.5. HRMS (ESI) [M-H] + calcd for C22H17ClN6OS2: 479.0510, found: 479.0519.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((5-(4-methoxyphenyl)-1,3,4-thiadiazol-2-yl)thio)acetamide (10e).
A brown solid, yield 38.6%, m. p. 220–222 °C; 1HNMR (500 MHz, DMSO-d6) δ 12.51 (d, J = 12.5 Hz, 1H), 11.42 (s, 1H), 10.81 (d, J = 31.5 Hz, 1H), 7.84–7.69 (m, 4H), 7.42 (dd, J = 7.9, 4.2 Hz, 1H), 7.17–7.02 (m, 4H), 6.79 (d, J = 6.3 Hz, 1H), 4.26 (d, J = 31.2 Hz, 3H), 3.78 (s, 2H). 13CNMR (126 MHz, DMSO-d6) δ 168.5, 164.9, 164.3, 164.3, 162.1, 148.1, 138.3, 136.8, 129.7, 124.8, 123.9, 123.9, 122.4, 122.3, 120.4, 119.5, 115.4, 112.6, 105.7, 93.4, 43.6. HRMS (ESI) [M-H] + calcd for C22H18N6O2S2: 461.0849, found: 461.0863.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((5-(4-chlorophenyl)-1,3,4-thiadiazol-2-yl)thio)acetamide (10f).
A reddish-brown solid, yield 40.4%, m. p. 175–177 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 11.47 (s, 1H), 10.92 (s, 1H), 7.93 (d, J = 7.6 Hz, 2H), 7.77 (s, 2H), 7.61 (d, J = 7.5 Hz, 2H), 7.46 (d, J = 6.7 Hz, 1H), 7.15 (dd, J = 14.8, 7.0 Hz, 2H), 6.84 (s, 1H), 4.37 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.4, 166.0, 164.8, 148.1, 138.2, 136.8, 136.4, 130.0, 129.6, 128.6, 124.8, 123.8, 122.3, 120.4, 119.4, 112.5, 105.6, 93.2, 56.5. HRMS (ESI) [M-H] + calcd for C21H15ClN6OS2: 465.0354, found: 465.0363.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((5-(2-fluorophenyl)-1,3,4-thiadiazol-2-yl)thio)acetamide (10 g).
A white solid, yield 53.7%, m. p. 163–165 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.57 (s, 1H), 11.47 (s, 1H), 10.93 (s, 1H), 8.22 (s, 1H), 7.77 (d, J = 2.4 Hz, 2H), 7.63 (s, 1H), 7.54–7.37 (m, 3H), 7.15 (dd, J = 14.4, 7.6 Hz, 2H), 6.84 (s, 1H), 4.39 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 167.1, 164.9, 160.7, 160.2, 158.2, 148.1, 138.3, 136.8, 133.9, 128.9, 126.1, 124.9, 123.9, 122.3, 120.4, 119.5, 117.7, 117.2, 112.5, 93.4, 56.6. HRMS (ESI) [M-H] + calcd for C21H15FN6OS2: 449.0649, found: 449.0657.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((5-(pyridine-3-yl)-1,3,4-thiadiazol-2-yl)thio)acetamide (10 h).
A chartreuse solid, yield 77.8%, m. p. 233–235 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.52 (s, 1H), 11.43 (s, 1H), 10.89 (s, 1H), 9.06 (s, 1H), 8.70 (d, J = 4.2 Hz, 1H), 8.27 (dd, J = 7.9, 1.6 Hz, 1H), 7.73 (s, 2H), 7.62–7.35 (m, 2H), 7.22 −7.00 (m, 2H), 6.80 (s, 1H), 4.36 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.5, 165.8, 164.7, 152.4, 148.4, 136.8, 135.4, 126.1, 124.9, 123.8, 122.3, 120.4, 119.4, 112.5, 105.6, 93.3, 38.2. HRMS (ESI) [M-H] + calcd for C20H15N7OS2: 432.0696, found: 432.0709.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((5-(pyridine-2-yl)-1,3,4-thiadiazol-2-yl)thio)acetamide (10i).
A chartreuse solid, yield 87.6%, m. p 226–228 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 11.47 (s, 1H), 10.92 (s, 1H), 9.10 (s, 1H), 8.73 (d, J = 3.8 Hz, 1H), 8.39–8.22 (m, 1H), 7.85–7.35 (m, 4H), 7.26–7.03 (m, 2H), 6.83 (s, 1H), 4.39 (s, 2H). 13 C NMR (101 MHz, DMSO-d6) δ 166.5, 165.7, 164.8, 152.3, 148.4, 148.0, 138.2, 136.7, 135.5, 126.1, 124.9, 124.8, 123.8, 122.3, 120.4, 119.4, 112.5, 105.6, 93.3, 56.5. HRMS (ESI) [M–H] + calcd for C20H15N7OS2: 432.0696, found: 432.0707.
General procedure for the synthesis of compounds 9a–r
A mixture of the potassium dithiocarbazinate as the respective aromatic esters (7a–r, 0.1 mol) and 80% hydrazine hydrate (0.3 mol, 18.9 ml) in water (5 ml) was stirred was refluxed for 5–8 h. After completion, the reaction was quenched with ice water. Then, the mixture was acidified with dilute HCl to pH 2 to 3. Finally, the crude products were filtered and recrystallized from ethanol to give intermediate 9a–rCitation47,Citation48. Similarly, compounds 11a–r was obtained according to the procedures of compounds 8a–i.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(3,5-dichlorophenyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11a)
A white solid, yield 45.6%, m. p. 258–260 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.47 (s, 1H), 11.41 (s, 1H), 10.75 (s, 1H), 8.03 (s, 2H), 7.75 (s, 2H), 7.42 (d, J = 7.8 Hz, 1H), 7.11 (dt, J = 14.3, 6.9 Hz, 2H), 6.78 (s, 1H), 6.28 (s, 2H), 4.14 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 164.4, 163.7, 148.0, 136.8, 133.7, 132.2, 131.6, 131.6, 124.8, 123.8, 122.6, 122.3, 120.3, 119.4, 112.5, 105.6, 93.2, 36.6. HRMS (ESI)
[M-H] + calcd for C21H16Cl2N8OS: 497.0461 found: 497.0473.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11 b)
A white solid, yield 69.3%, m. p. 259–261 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.53 (s, 1H), 11.48 (s, 1H), 10.78 (s, 1H), 7.76 (d, J = 6.6 Hz, 2H), 7.46 (d, J = 7.9 Hz, 1H), 7.27 (s, 2H), 7.15 (dd, J = 13.8, 7.5 Hz, 2H), 6.83 (d, J = 1.4 Hz, 1H), 6.27 (s, 2H), 4.16 (s, 2H), 3.83 (s, 7H), 3.72 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.7, 154.5, 153.7, 153.4, 148.1, 139.2, 138.1, 136.8, 124.8, 123.8, 122.5, 122.3, 120.3, 119.4, 112.5, 106.0, 105.6, 93.3, 49.0. HRMS (ESI) [M-H] + calcd for C24H24N8O4S: 519.1557 found: 519.1565.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(2-bromophenyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11c)
A white solid, yield 27.7%, m. p. 194–195 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.51 (s, 1H), 11.45 (s, 1H), 10.81 (s, 1H), 7.93–7.67 (m, 3H), 7.65–7.39 (m, 4H), 7.16 (dd, J = 12.9, 7.4 Hz, 2H), 6.84 (s, 1H), 5.93 (s, 2H), 4.18 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.6, 155.1, 152.6, 148.1, 138.1, 136.8, 133.2, 133.1, 132.4, 128.9, 128.0, 124.8, 123.9, 123.8, 122.3, 120.3, 119.4, 112.5, 105.7, 93.3, 67.5, 36.3. HRMS (ESI) [M-H] + calcd for C21H17BrN8OS: 507.0346 found: 507.0361.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(4-chlorobenzyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11d)
A white solid, yield 16.9%, m. p. 240–241 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.49 (s, 1H), 11.44 (s, 1H), 10.72 (s, 1H), 7.76 (d, J = 6.4 Hz, 2H), 7.46 (d, J = 7.9 Hz, 1H), 7.33 (dt, J = 25.0, 6.7 Hz, 4H), 7.15 (dt, J = 14.6, 7.0 Hz, 2H), 6.96 (d, J = 120.6 Hz, 1H), 5.97 (s, 2H), 4.08 (d, J = 4.8 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 165.7, 155.5, 151.5, 148.1, 138.1, 136.8, 136.0, 131.7, 131.1, 128.8, 124.8, 123.8, 122.3, 120.3, 119.4, 112.5, 105.6, 93.3, 36.3. HRMS (ESI) [M-H] + calcd for C22H19ClN8OS: 477.1007 found: 477.1018.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(4-methoxyphenyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11e)
A white solid, yield 86.8%, m. p. 238–240 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.51 (s, 1H), 11.45 (s, 1H), 10.78 (s, 1H), 8.10–7.89 (m, 2H), 7.77 (t, J = 4.5 Hz, 2H), 7.46 (d, J = 7.9 Hz, 1H), 7.21–7.05 (m, 4H), 6.82 (s, 1H), 6.20 (s, 2H), 4.14 (s, 2H), 3.82 (s, 3H).13C NMR (101 MHz, DMSO-d6) δ 165.8, 160.8, 154.4, 153.1, 148.1, 138.1, 136.8, 129.8, 124.8, 123.8, 122.3, 120.3, 119.7, 119.5, 114.4, 112.5, 105.7, 93.3, 55.8, 36.2. HRMS (ESI) [M-H] + calcd for C22H20N8O2S: 459.1346 found: 459.1365.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(4-chlorophenyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11f)
A white solid, yield 74.1%, m. p. 214–215 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.48 (s, 1H), 11.42 (s, 1H), 10.76 (s, 1H), 8.13–7.90 (m, 2H), 7.72 (t, J = 4.3 Hz, 2H), 7.59–7.53 (m, 2H), 7.42 (d, J = 8.0 Hz, 1H), 7.20–7.02 (m, 2H), 6.78 (s, 1H), 6.22 (s, 2H), 4.13 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.7, 154.0, 153.6, 136.8, 135.0, 129.9, 129.1, 126.2, 124.8, 123.8, 122.3, 120.3, 119.5, 112.5, 93.3, 36.2. HRMS (ESI) [M-H] + calcd for C21H17ClN8OS: 463.0851 found: 463.0877.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(2-fluorophenyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11 g)
A white solid, yield 43.3%, m. p. 226–227 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.49 (s), 11.43 (s), 10.79 (s), 7.73 (d, J = 7.2 Hz), 7.64 (dd, J = 7.3, 6.0 Hz), 7.56 (d, J = 7.9 Hz), 7.36 (ddd, J = 20.1, 19.5, 7.7 Hz), 7.11 (dd, J = 16.9, 7.6 Hz), 6.80 (d, J = 1.1 Hz), 6.02 (s), 4.15 (s). 13C NMR (126 MHz, DMSO-d6) δ 165.7, 161.2, 159.2, 153.5, 151.6, 148.2, 138.1, 136.8, 133.0, 132.9, 132.2, 125.1, 124.8, 123.9, 122.4, 120.4, 119.5, 116.7, 116.6, 115.5, 115.4, 112. 6, 105.7, 93.4, 49.1. HRMS (ESI) [M-H] + calcd for C21H17FN8OS: 447.1146 found: 447.1152.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5-(pyridine-3-yl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11 h)
A yellow solid, yield 62.1%, m. p. 195–197 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.51 (d, J = 24.6 Hz), 11.42 (s), 10.76 (d, J = 9.0 Hz), 9.11 (d, J = 1.7 Hz), 8.65 (dd, J = 4.8, 1.6 Hz), 8.38–8.28 (m), 7.77–7.67 (m), 7.53 (ddd, J = 8.0, 4.9, 0.8 Hz), 7.42 (d, J = 8.0 Hz), 7.14–7.06 (m), 6.86 − 6.71 (m), 6.27 (s), 4.19 (d, J = 44.2 Hz). 13C NMR (126 MHz, DMSO-d6) δ 165.7, 164.3, 154.4, 152.7, 151.0, 148.7, 148.1, 138.1, 136.8, 135.6, 124.8, 124.2, 123.9, 123.9, 123.6, 122.3, 120.4, 119.5, 112.5, 105.7, 93.4, 43.6. HRMS (ESI) [M-H] + calcd for C20H17N9OS: 430.1193 found: 430.1213.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5-(pyridine-2-yl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11i)
A white solid, yield 43.8%, m. p. 208–210 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.48 (s, 1H), 11.42 (s, 1H), 10.77 (s, 1H), 8.67 (d, J = 4.3 Hz, 1H), 8.15–7.84 (m, 2H), 7.72 (s, 2H), 7.45 (dd, J = 35.6, 6.7 Hz, 2H), 7.11 (dd, J = 18.0, 7.5 Hz, 2H), 6.80 (s, 1H), 6.59 (s, 2H), 4.17 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.6, 153.5, 151.5, 149.5, 148.2, 147.4, 146.8, 138.3, 138.1, 136.8, 125.0, 124.8, 123.8, 122.7, 122.3, 120.4, 119.5, 112.5, 105.7, 93.3, 56.6. HRMS (ESI) [M-H] + calcd for C20H17N9OS: 430.1193 found: 430.1120.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(2-chlorophenyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11j)
A white solid, yield 59.9%, m. p. 136–138 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.55 (s, 1H), 11.48 (s, 1H), 10.84 (s, 1H), 7.78 (d, J = 6.7 Hz, 2H), 7.68–7.41 (m, 5H), 7.16 (dt, J = 14.6, 6.9 Hz, 2H), 6.85 (s, 1H), 5.98 (s, 2H), 4.20 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.6, 154.0, 152.9, 148.1, 138.1, 136.8, 133.9, 132.9, 132.28, 130.1, 127.6, 126.7, 124.8, 123.8, 122.3, 120.4, 119.4, 112.5, 105.7, 93.3, 36.2. HRMS (ESI) [M + Na] + calcd for C21H17ClN8OS: 487.0826 found: 487.0818.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5-phenyl-4H-1,2,4-triazol-3-yl)thio)acetamide (11k)
A white solid, yield 32.6%, m. p. >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.62 (s, 1H), 11.64 (s, 1H), 10.81 (s, 1H), 8.14–7.93 (m, 2H), 7.79 (dd, J = 23.4, 4.4 Hz, 2H), 7.58–7.44 (m, 4H), 7.23–7.04 (m, 2H), 6.80 (s, 1H), 6.28 (s, 2H), 4.16 (s, 2H).13C NMR (101 MHz, DMSO-d6) δ 165.7, 154.6, 153.7, 136.8, 130.1, 129.0, 128.3, 127.3, 124.8, 123.9, 122.2, 120.3, 119.4, 112.5, 93.2, 36.2. HRMS (ESI) [M-H] + calcd for C21H18N8OS: 429.1241 found: 429.1262.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(4-aminophenyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11 l)
A white solid, yield 21.1%, m. p. 213–215 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.50 (s, 1H), 11.44 (s, 1H), 10.76 (s, 1H), 7.76 (d, J = 2.6 Hz, 1H), 7.68 (d, J = 8.6 Hz, 2H), 7.46 (d, J = 7.9 Hz, 1H), 7.21–7.09 (m, 2H), 6.63 (d, J = 8.7 Hz, 2H), 6.09 (s, 2H), 5.53 (s, 2H), 4.10 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.9, 155.2, 152.3, 150.7, 148.1, 136.8, 129.3, 124.9, 123.9, 122.3, 120.4, 119.5, 114.3, 113.7, 112.5, 105.7, 93.4, 43.6. HRMS (ESI) [M-H] + calcd for C21H19N9OS: 444.1350 found: 444.1371.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(4-bromophenyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11 m)
A yellow solid, yield 53.4%, m. p. 270–272 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.47 (s, 1H), 11.41 (s, 1H), 10.74 (s, 1H), 7.94–7.91 (m, 2H), 7.73–7.68 (m, 4H), 7.42 (d, J = 8.0 Hz, 1H), 7.16 −7.06 (m, 2H), 6.78 (d, J = 1.9 Hz, 1H), 6.21 (s, 2H), 4.12 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.7, 154.1, 153.7, 136.8, 132.1, 130.1, 126.5, 124.8, 123.9, 123.8, 122.3, 120.4, 119.5, 112.5, 105.7, 93.4, 36.2. HRMS (ESI)
[M-H] + calcd for C21H17BrN8OS: 507.0346 found 507.0367.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5-(1H-indol-3-yl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11n)
A white solid, yield 42.5%, m. p. 230–232 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.72 (s, 1H), 12.54 (s, 1H), 11.46 (s, 1H), 10.84 (s, 1H), 8.26 (d, J = 8.1 Hz, 1H), 7.88–7.59 (m, 4H), 7.48 (t, J = 8.2 Hz, 2H), 7.29 (t, J = 7.5 Hz, 1H), 7.21–7.09 (m, 2H), 6.85 (s, 1H), 6.40 (s, 2H), 4.23 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.7, 152.8, 149.7, 140.8, 136.8, 133.4, 127.6, 124.8, 123.8, 122.3, 122.3, 122.1, 121.4, 120.3, 119.5, 112.5, 111.0, 93.3, 35.4. HRMS (ESI) [M + H] + calcd for C23H19N9OS: 470.1506 found: 470.1477.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11o)
A reddish-brown solid, yield 41.1%, m. p. 170–172 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.54 (s, 1H), 11.47 (s, 1H), 10.80 (d, J = 18.0 Hz, 1H), 8.91–8.41 (m, 2H), 7.89 (dd, J = 101.8, 4.1 Hz, 4H), 7.46 (d, J = 7.9 Hz, 1H), 7.31– 6.98 (m, 2H), 6.82 (s, 1H), 6.27 (d, J = 83.6 Hz, 2H), 4.20 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.7, 165.6, 155.2, 152.5, 150.6, 148.1, 146.8, 138.1, 136.7, 134.4, 124.8, 123.8, 122.3, 121.8, 120.4, 119.4, 112.5, 105.6, 93.3, 36.0. HRMS (ESI) [M-H] + calcd for C20H17N9OS: 430.1193 found: 430.1200.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(4-fluorophenyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11p)
A brown solid, yield 33.2%, m. p. 209–212 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.46 (s, 1H), 11.40 (s, 1H), 10.75 (s, 1H), 8.01 (s, 2H), 7.71 (s, 2H), 7.48–7.27 (m, 3H), 7.22–6.97 (m, 3H), 6.77 (s, 1H), 6.20 (s, 2H), 4.11 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.75, 164.4, 162.4, 153.8, 148.2, 138.1, 136.8, 130.7, 130.6, 124.8, 123.9, 122.3, 120.4, 119.5, 116.2, 116.1, 112.5, 105.71, 93.3, 36.2. HRMS (ESI) [M-H] + calcd for C21H17FN8OS: 447.1146 found: 447.1156.
N-(3-(1H-indol-3-yl)-1H-pyrazol-5-yl)-2-((4-amino-5–(2-hydroxyphenyl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11q)
A brown solid, yield 43.9%, m. p. 263–266 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.53 (s, 1H), 11.47 (s, 1H), 11.08 (s, 1H), 10.83 (s, 1H), 7.88–7.73 (m, 3H), 7.47 (d, J = 7.9 Hz, 1H), 7.42–7.34 (m, 1H), 7.21–7.10 (m, 2H), 7.00 (ddd, J = 15.1, 8.2, 4.3 Hz, 2H), 6.85 (s, 1H), 6.11 (s, 2H), 4.19 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.6, 156.1, 154.3, 153.3, 148.1, 138.1, 136.8, 132.0, 130.0, 124.8, 123.8, 122.29, 120.3, 119.7, 119.5, 116.9, 113.3, 112.5, 105.7, 93.3, 35.6. HRMS (ESI) [M-H] + calcd for C21H18N8O2S: 445.1190 found: 445.1194.
N-(3-(1H-indol-3–yl)-1H-pyrazol-5-yl)-2-((4-amino-5-(1H-pyrrol-2-yl)-4H-1,2,4-triazol-3-yl)thio)acetamide (11r)
A grey solid, yield 45.4%, m. p. 241–243 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.48 (s), 11.60 (s), 11.42 (s), 10.74 (s), 7.73 (d, J = 7.6 Hz), 7.42 (d, J = 7.9 Hz), 7.11 (dd, J = 16.6, 7.5 Hz), 6.95–6.72 (m), 6.21–6.08 (m), 4.08 (s). 13C NMR (126 MHz, DMSO-d6) δ 165.9, 152.0, 150.1, 148.1, 138.2, 136.8, 124.8, 123.9, 122.4, 121.3, 120.4, 119.5, 118.3, 112.6, 110.1, 109.3, 105.7, 93.4, 36.3. HRMS (ESI) [M-H] + calcd for C19H17N9OS: 418.1193 found: 418.1203.
Evaluation of biological activity
MTT assay
A549, PC-3, HepG2 and K562 cells were purchased from the Kunming cell bank of the Chinese academy of sciences. All cell lines were cultured in DMEM or RPMI 1640 complete medium. Logarithmic growth cells were cultured in 96-well plates at a concentration of 104 cells per well and pre-cultured for 24 h before adding test compounds. The MTT test with 5-Fluorouracil and Gefitinib as reference compounds were used to evaluate antiproliferative activity of target compounds in four human cancer line cells. For the estimation of the inhibition concentration (IC50), the Graph Pad Prism (GraphPad Software, San Diego, CA) was used.
Apoptosis analysis
DAPI staining assay, A549 or K562 cells were separately seeded at 4.0 × 105 cells per well in six–well plates and incubated for 24 h until adherent. Next, the cells were treated with 10 b at 0, 1, 2 and 4 µM for 48 h. Then the cells were fixed with 4% paraformaldehyde for 15 min, and 1 ml/well of DAPI staining dye was added for 20 min in the dark. After adding the anti-fluorescence quencher, the cells can be imaged by an inverted fluorescence microscope.
Annexin-V/FITC assay, after the adherent cells grew stably in the 6-well plate, each group was reacted with different concentrations of compound solutions for 48 h. Then, cells were collected by trypsinization without EDTA, rinsed twice with PBS, followed by the Annexin V-FITC/PI apoptosis detection kit (Solarbio, Bei Jing, China) according to the kit instructions, flow cytometry by BD FACSCalibur instrument (American BD Company Shanghai Co., Ltd.).
Cell cycle analysis
A549 or K562 cells were separately seeded at 4.0 × 105cells/well in six-well plates and incubated for 24 h until adherent. Compound 10 b were added at 0, 1, 2 and 4 µM for 24 h. After treatment, the cells were collected, and washed twice with pre-cooled PBS and then fixed with ice-cold ethanol (70%), subsequently kept 4 °C for overnight. After washing twice with PBS, cells were incubated at 37 °C with 100 µ L RNase A for 30 min. After this, cells were incubated with 400 µL PI for 30 min at 4 °C. Finally, the cell cycle distribution of compound 10 b was analysed via the BD FACSCalibur flow cytometry (American BD Corporation Shanghai Co. Ltd.)
Western blot assay
Cells were treated with different concentrations of compound for 48 h. After incubation, the cells were washed and collected. Total cell extracts were prepared with RIPA buffer, containing protease inhibitor and phosphatase inhibitor. The lysate was incubated at 0 °C for 60 min, and centrifuged at 12,000 rpm for 15 min at 4 °C to remove cellular debris. Next, the protein concentration was detected using BCA protein concentration assay kit (Beyotime, Shanghai). 20 µg of total protein was subjected to gel electrophoresis and transferred to PVDF membranes (Millipore, Burlington, MA, USA). After blocking in 5% BSA blocking buffer for 60 min, then incubated with primary antibody overnight at 4 °C, washed three times with TBST for 5 min each, followed by incubation with secondary antibody and washed. The blot was developed and exposed by chemiluminescence developer solution, which was placed on a chemiluminescence developer (Pierce, Rockford, Illinois, USA).
Wound healing assay
After 24 h of growth, cells were placed in a 6-well plate, scratched using a 10 µL pipet tip on the cell surface. Then, gently wash twice with medium to remove the detached cells. The cells were treated with compound 10 b with 0.5 and 1.0 µM followed by 48 h incubation and photographed using an inverted microscope.
Molecular docking
The computational docking study into the active site of EGFR (PDB code: 2ITY) was performed by PyMol 2.3.2 and AutoDock Vina 1.1.2Citation49,Citation50. The crystal structure was retrieved from the RCSB protein data bank (www.rcsb.org). Prior to docking, the receptor protein was treated with PyMol 2.5Citation51, including removal of water molecules, salt ions, and small molecule Gefitinib, with the centre of mass of crystal ligand as the box centre, the box size was 22.5 * 22.5 * 22.5 cubic angstroms, subsequently the polar hydrogen atoms were added to the crystal structure. In addition, all processed small molecules and receptor proteins were converted to the required PDB QT format using ADFR suite 1.0 softwareCitation52. Then, the global search detail was set to 32, other parameters ware default. The docking conformation with the highest output score was considered as the binding conformation, and the discovery Studio Viewer docking result was used for visual analysisCitation53.
Supplemental Material
Download PDF (1.2 MB)Acknowledgements
The authors acknowledge the financial supports of National Natural Science Foundation of China [21867004, 22007022], Science and Technology Foundation of Guizhou Province [ZK[2021]034], Top Science and Technology Talent Program of Guizhou Education Department (2022), Guizhou Provincial Department of Education [Qjh KYZi[2021]041].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary data
1H NMR, 13C NMR and HR-MS spectra for the target compounds (Figures S1−S81), 1H NMR and 13C NMR spectra for t key intermediate 4(Figures S82, S83), the results of superimposition on molecular docking (Figures S84, S85) associated with this article can be found in the online version.
References
- Yahya EB, Alqadhi AM. Recent trends in cancer therapy: a review on the current state of gene delivery. Life Sci. 2021;269:119087.
- You WP, Henneberg M. Cancer incidence increasing globally: the role of relaxed natural selection. Evol Appl. 2018;11(2):140–152.
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249.
- Bansal A, Saleh-E-In MM, Kar P, Roy A, Sharma NR. Synthesis of carvacrol derivatives as potential new anticancer agent against lung cancer. Molecules. 2022;27(14):4597.
- Meggendorfer M, Haferlach T, Alpermann T, Jeromin S, Haferlach C, Kern W, Schnittger S. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia. Haematologica. 2014;99(12):e244–e246.
- Ali R, Mirza Z, Ashraf GMD, Kamal MA, Ansari SA, Damanhouri GA, Abuzenadah AM, Chaudhary AG, Sheikh IA. New anticancer agents: recent developments in tumor therapy. Anticancer Res. 2012;32(7):2999–3005.
- Zhou X, Liu J, Meng J, Fu Y, Wu Z, Ouyang G, Wang Z. Discovery of facile amides-functionalized rhodanine-3-acetic acid derivatives as potential anticancer agents by disrupting microtubule dynamics. J Enzyme Inhib Med Chem. 2021;36(1):1996–2009.
- Kumar V, Kaur K, Gupta GK, Sharma AK. Pyrazole containing natural products: synthetic preview and biological significance. Eur J Med Chem. 2013;69:735–753.
- Minnelli C, Laudadio E, Sorci L, Sabbatini G, Galeazzi R, Amici A, Semrau MS, Storici P, Rinaldi S, Stipa P, et al. Identification of a novel nitroflavone-based scaffold for designing mutant-selective EGFR tyrosine kinase inhibitors targeting T790M and C797S resistance in advanced NSCLC. Bioorg Chem. 2022;129:106219.
- Park H, Jung H-Y, Mah S, Hong S. Discovery of EGFR(d746-750/T790M/C797S) mutant-selective inhibitors via structure-based de novo design. Angew Chem Int Ed Engl. 2017;56(26):7634–7638.
- Han Y, Dong W, Guo Q, Li X, Huang L. The importance of indole and azaindole scaffold in the development of antitumor agents. Eur J Med Chem. 2020;203:112506.
- Dhuguru J, Skouta R. Role of indole scaffolds as pharmacophores in the development of Anti-lung cancer agents. Molecules. 2020;25(7):1615.
- More KN, Hong VS, Lee A, Park J, Kim S, Lee J. Discovery and evaluation of 3,5-disubstituted indole derivatives as pim kinase inhibitors. Bioorg Med Chem Lett. 2018;28(14):2513–2517.
- Dadashpour S, Emami S. Indole in the target-based design of anticancer agents: a versatile scaffold with diverse mechanism. Eur J Med Chem. 2018;150:9–29.
- Koca İ, Özgür A, Coşkun KA, Tutar Y. Synthesis and anticancer activity of acyl thioureas bearing pyrazole moiety. Bioorg Med Chem. 2013;21(13):3859–3865.
- Elbastawesy MAI, Aly AA, Ramadan M, Elshaier YAMM, Youssif BGM, Brown AB, El-Din A Abuo-Rahma G. Novel pyrazoloquinolin-2-ones: design, synthesis, docking studies, and biological evaluation as antiproliferative EGFR-TK inhibitors. Bioorg Chem. 2019;90:103045.
- Ahadi H, Shokrzadeh M, Hosseini-Khah Z, Ghassemi Barghi N, Ghasemian M, Emadi E, Zargari M, Razzaghi-Asl N, Emami S. Synthesis and biological assessment of ciprofloxacin-derived 1,3,4-thiadia-zoles as anticancer agents. Bioorg Chem. 2020;105:104383.
- Nafie MS, Boraei ATA. Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole schiff bases for targeting breast cancer. Bioorg Chem. 2022;122:105708.
- Pallis AG, Serfass L, Dziadziusko R, van Meerbeeck JP, Fennell D, Lacombe D, Welch J, Gridelli C. Targeted therapies in the treatment of advanced/metastatic NSCLC. Eur J Cancer. 2009;45(14):2473–2487. −
- Ramalingam SS, Owonikoko TK, Khuri FR. Lung cancer: new biological insights and recent therapeutic advances. CA Cancer J Clin. 2011;61(2):91–112. −
- Zheng D, Wang R, Ye T, Yu S, Hu H, Shen X, Li Y, Ji H, Sun Y, Chen H, et al. MET exon 14 skipping defines a unique molecular class of non-small cell lung cancer. Oncotarget. 2016;7(27):41691–41702. −
- Shangary S, Wang SM. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223–241.
- Bykov VJN, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8(3):282–288.
- Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LGGC, Masucci M, Pramanik A, Selivanova G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10(12):1321–1328.
- Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13(1):23–31.
- Wiman KG. Strategies for therapeutic targeting of the p53 pathway in cancer. Cell Death Differ. 2006;13(6):921–926.
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848.
- Ding K, Lu Y, Nikolovska-Coleska Z, Qiu S, Ding Y, Gao W, Stuckey J, Krajewski K, Roller PP, Tomita Y, et al. Structure-based design of potent non-peptide MDM2 inhibitors. J Am Chem Soc. 2005;127(29):10130–10131.
- Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A. 2008;105(10):3933–3938.
- Yan L, Li Y, Deng M, Chen A, Du Z, Dong C, Chen H. Design, synthesis and biological activities of compounds containing 1,3,4-oxadiazole or 1,3,4-thiadiazole. Chin J Org Chem. 2020;40(3):731–739.
- Zhang Y, Wang Q, Li L, Le Y, Liu L, Yang J, Li Y, Bao G, Yan L. Synthesis and preliminary structure-activity relationship study of 3-methylquinazolinone derivatives as EGFR inhibitors with enhanced antiproliferative activities against tumor cells. J Enzyme Inhib Med Chem. 2021;36(1):1205–1216.
- Le Y, Gan Y, Fu Y, Liu J, Li W, Zou X, Zhou Z, Wang Z, Ouyang G, Yan L, et al. Design, synthesis and in vitro biological evaluation of quinazolinone derivatives as EGFR inhibitors for antitumor treatment. J Enzyme Inhib Med Chem. 2020;35(1):555–564.
- Shao L-H, Fan S-L, Meng Y-F, Gan Y-Y, Shao W-B, Wang Z-C, Chen D-P, Ouyang G-P. Design, synthesis, biological activities and 3D-QSAR studies of quinazolinone derivatives containing hydrazone structural units. New J Chem. 2021;45(10):4626–4631.
- Zhao N, Tian K-T, Cheng K-G, Han T, Hu X, Li D-H, Li Z-L, Hua H-M. Antiproliferative activity and apoptosis inducing effects of nitric oxide donating derivatives of evodiamine. Bioorg Med Chem. 2016;24(13):2971–2978.
- Grada A, Otero-Vinas M, Prieto-Castrillo F, Obagi Z, Falanga V. Research techniques made simple: analysis of collective cell migration using the wound healing assay. J Invest Dermatol. 2017;137(2):e11–e16.
- Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3(5):362–374.
- Abou-Zied HA, Youssif BGM, Mohamed MFA, Hayallah AM, Abdel-Aziz M. EGFR inhibitors and apoptotic inducers: design, synthesis, anticancer activity, and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg Chem. 2019;89:102997.
- Hisham M, Youssif BGM, Osman EEA, Hayallah AM, Abdel-Aziz M. Synthesis, and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents. Eur J Med Chem. 2019;176:117–128.
- Wang S, Zhao Y, Aguilar A, Bernard D, Yang C-Y. Targeting the MDM2-p53 protein-protein interaction for new cancer therapy: progress and challenges. Cold Spring Harb Perspect Med. 2017;7(5):a026245.
- Slaett J, Romero I, Bergman J. Cyanoacetylation of indoles, pyrroles and aromatic amines with the combination cyanoacetic acid and acetic anhydride. Synthesis. 2004;16:2760–2765.
- El-Mekabaty A, Etman HA, Mosbah A. Synthesis of some new fused pyrazole derivatives bearing indole moiety as antioxidant agents. J Heterocyclic Chem. 2016;53(3):894–900.
- Kaftory M, Botoshansky M, Sheinin Y. Dimormorphism and cocrystal formation of 3-(chloroacetamido)pyrazole. Crystal Growth & Design. 2005;5(6):2242–2247.
- Chen X, Gan X, Chen J, Chen Y, Wang Y, Hu D, Song B. Synthesis and Nematicidal activity of novel 1,3,4-oxadiazole (thiadiazole)thioether derivatives containing trifluorobuten moiety. Chin J Org Chem. 2017;37(9):2343–2351.
- Altıntop M, Ciftci H, Radwan M, Sever B, Kaplancıklı Z, Ali T, Koga R, Fujita M, Otsuka M, Özdemir A, et al. Design, synthesis, and biological evaluation of novel 1,3,4-thiadiazole derivatives as potential antitumor agents against chronic myelogenous leukemia: striking effect of nitrothiazole moiety. Molecules. 2017;23(1):59–75.
- Yang C, Yang S, Song BA, et al. Synthesis and antifungal activity of 2-substituted sulfinyl (sulfonyl)-5-(4-nitro or 4-methoxyphenyl)-1,3,4-thiadiazole derivatives. Chin J Org Chem. 2010;30:1327–1334.
- Sami AA, Thoraya AF, Magdi EAZ, et al. Discovery of novel indolyl-1,2,4-triazole hybrids as potent vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors with potential anti-renal cancer activity. Bioorg Chem. 2020;105:104330.
- Mathew V, Giles D, Keshavayya J, Vaidya VP. Studies on synthesis and pharmacological activities of 1,2,4-triazolo[3,4-b]1,3,4-thiadiazoles and their dihydro analogues. Arch Pharm (Weinheim)). 2009;342(4):210–222.
- Yun C-H, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11(3):217–227.
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461.
- Seeliger D, Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24(5):417–422.
- Ravindranath PA, Forli S, Goodsell DS, Olson AJ, Sanner MF. AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput Biol. 2015;11(12):e1004586.
- Mohamed FAM, Gomaa HAM, Hendawy OM, Ali AT, Farghaly HS, Gouda AM, Abdelazeem AH, Abdelrahman MH, Trembleau L, Youssif BGM, et al. Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-scarboxamide scaffold with apoptotic antiproliferative activity. Bioorg Chem. 2021;112:104960.