
Abstract
A new series of 2-aminobenzothiazole hybrids linked to thiazolidine-2,4-dione 4a–e, 1,3,4-thiadiazole aryl urea 6a–d, and cyanothiouracil moieties 8a–d was synthesised. The in vitro antitumor effect of the new hybrids was assessed against three cancer cell lines, namely, HCT-116, HEPG-2, and MCF-7 using Sorafenib (SOR) as a standard drug. Among the tested compounds, 4a was the most potent showing IC50 of 5.61, 7.92, and 3.84 µM, respectively. Furthermore, compounds 4e and 8a proved to have strong impact on breast cancer cell line with IC50 of 6.11 and 10.86 µM, respectively. The three compounds showed a good safety profile towards normal WI-38 cells. Flow cytometric analysis of the three compounds in MCF-7 cells revealed that compounds 4a and 4c inhibited cell population in the S phase, whereas 8a inhibited the population in the G1/S phase. The most promising compounds were subjected to a VEGFR-2 inhibitory assay where 4a emerged as the best active inhibitor of VEGFR-2 with IC50 91 nM, compared to 53 nM for SOR. In silico analysis showed that the three new hybrids succeeded to link to the active site like the co-crystallized inhibitor SOR.
Introduction
Cancer is a complicated world spread fatal illness that affects several organs in the human body and represents a serious challenge to the health and welfare of humanityCitation1–3. Rapid proliferation is the most common property shared among all types of cancerCitation4. This proliferation involves highly complex and interconnected molecular pathways, this multifarious nature forces using multi-target cancer treatment techniquesCitation5. So, the discovery and development of new therapeutic agents with this anti-proliferative activity with selective cytotoxicity are needed urgently. Unfortunately, chemotherapeutic anticancer treatment causes several adverse effects, including multiple drug resistance, unwanted side effects, adverse events, and bad selectivity. Due to the previously‐mentioned shortcomings, there is tremendous need to discover and develop new, potent, safe, and efficacious therapeutic agents with anti-proliferative activity and selective cytotoxicity. The rapid expansion in the number of new anti-cancer drugs with versatile mechanisms of action has successfully emphasised traditionally used chemotherapeutic agents; these agents are used in combination with traditional agents. Although they acquire various mechanisms, most new drugs are thought to induce apoptosis of cancer cells or their supportive vascularityCitation6.
Protein kinases (PKs) stand for cell function regulationCitation7. Dysfunction of PKs leads to several diseases such as inflammation, metabolic diseases and ultimately cancer. Thus the utilisation of PK-inhibitors is regarded as a promising strategy to inhibit cancer progressionCitation8, especially in cancers known with mutations and alterations of PKs which contribute to development of drug resistanceCitation9. VEGFR-2 is a PK receptor located on blood vessels and regulates angiogenesisCitation10. It is highly expressed in many types of cancer. Hence, it’s considered as a main target for many clinically approved anticancer agents ()Citation11.
Figure 1. The necessary pharmacophoric properties of some FDA and clinically approved VEGFR-2 inhibitors.
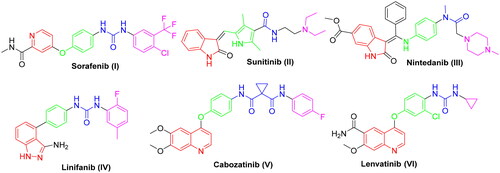
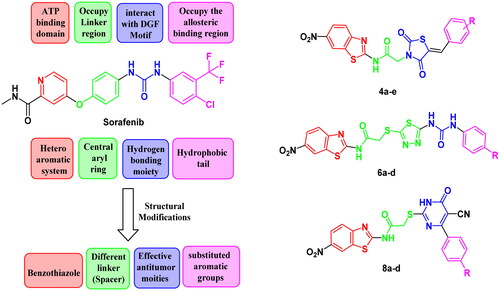
SAR analysis of various VEGFR-2 inhibitors has revealed four common pharmacophoric features as demonstrated in . These features include (i) hinge binder which is composed of a flat aromatic or heteroaromatic system containing one N at least and fills the catalytic ATP-binding pocketCitation12. Moreover, X-ray structure analysis in different inhibitors linked to VEGFR-2 enzyme demonstrated that there is a sufficient space around the terminal heteroaromatic core available for various substituentsCitation13. (ii) A spacer group that occupies the linker region between the hinge region and the DFG-domain of the enzymeCitation14. (iii) A pharmacophoric functional group consists of a moiety of hydrogen bond donor-acceptor pair (e.g. amide or urea) which binds with two essential amino acids (Glu885 and Asp1046) in the DFG (Asp-Phe-Gly) motifCitation15. (iv) A hydrophobic tail (present only in type II inhibitors) which occupies the back allosteric site created when phenylalanine residue of the DFG loop flips out of its lipophilic pocket making the DFG motif adopting the inactive conformation. So, various hydrophobic bindings mostly occur in this allosteric binding siteCitation16.
Generally, due to the nature of VEGFR-2 enzyme binding site, its inhibitors display wide diversity in structure such as picolinamide (e.g. Sorafenib) SOR (I), indole (e.g. Sunitinb II and Nintedanib III), indazole (e.g. Linifanib IV), and quinolone (e.g. Cabozatinib V and Lenvatinib VI) ()Citation17–23.
Benzothiazole is a privileged chemical scaffold possessing a wide spectrum of pharmacological effects including, anti-diabeticCitation24, fungicidalCitation25, anti-microbialCitation26, analgesicCitation27, CNS depressantCitation28, as well as anticancer activityCitation29–32. This moiety revealed outstanding and prevalent pharmacological and biological effect against various kinds of cancer cell lines as HeLa, SW480, HepG2, mammary and ovarian tumour cell lines, colon, non-small-cell lung and breast subpanels cell lines, and HCCCitation33–36.
The benzothiazole and its isosteres like the indole ring have proved to exhibit promising antitumor activity. The structure–activity relationship for the various derivatives revealed an excellent understanding of the behaviour of benzothiazole moiety in the field of cancer therapy against different cancer cell line. This moiety has also been explored for its therapeutic potential. Benzothiazole‐based derivatives have emerged as effective enzyme inhibitors against EGFR, VEGFR, PI3K, topoisomerases, and thymidylate kinases. Some of these inhibitors have entered different phases of clinical trials. In addition, aryl benzothiazole, aminobenzothiazole and other structural benzothiazole hybrids have attracted outstanding attention in the search for new chemotherapeutic agents as they exhibited effective cytotoxic activity in in vivo and in vitro modelsCitation37–42.
Furthermore, during the last years, there have been several attempts to develop different benzothiazole surrogates with unique and promising profile as antitumor agents through inhibition of protein kinasesCitation43–46. In this context, 2-aminobenzothiazole derivatives were found to be very effective scaffolds as will be shown in this workCitation47. Recently, V.G. Reddy et al. proposed a series of 2-aminobenzothiazole-pyrazoles as a new group of potent VEGFR-2 inhibitors with outstanding anticancer effect in the low micromolar range towards several cancer cell lines. Within this series, compound VII exerted the highest activity with a VEGFR-2 IC50 of 97 nM ()Citation48. Furthermore, K. El-Adl et al. disclosed the 2-aminobenzothiazole-thiazolidinedione hybrids VIII–XI as new VEGFR-2 inhibitors with IC50 150–210 nM, introducing them as a novel start to produce promising inhibitors against VEGFR-2 ()Citation45.
Figure 2. Representative examples of some reported anticancer VEGFR-2 inhibitors carrying 2-aminobenzothiazole scaffold, thiazolidine-2,4-dione, cyanothiouracil, and thiadiazole-urea pharmacophores.
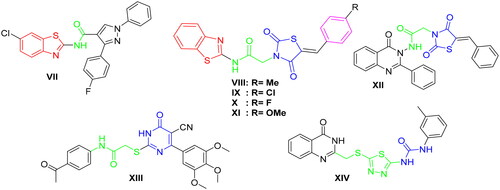
In addition, different scaffolds such as thiazolidine-2,4-dione (e.g. compound XII)Citation14, 6-aryl-5-cyanothiouracil (e.g. compound XIII)Citation49, and thiadiazole-urea (e.g. compound XIV)Citation50 were reported as anticancer activity enhancers with potent VEGFR-2 inhibitory activity (.
Counting on the ligand-based design of drugs, molecular hybridisation approach is concerned with the combination of two or more bioactive groups to produce hybrids showing enhanced bioactivity, particularly, when applied in the search for new effective antitumor candidatesCitation51. So that, the anticancer and VEGFR-2 inhibitory activity of benzothiazole are combined with anticancer activity enhancer in a new structure that is expected to exert more potent anticancer candidates that possess the same vital pharmacophoric characteristics of the old VEGFR-2 inhibitors (e.g. SOR) with bioisosteric alterations at four various sites.
So, in this work we depended on the following strategy to design novel VEGFR inhibitors. Firstly, we adopted a bioisosteric replacement of the pyridine ring of SOR with benzothiazole scaffold. In the second modification, various linkers with variable lengths were designed to occupy the spacer region. The linker may involve three or four atom bridges (acetamide and 2-sulfanylacetamide). In some instances, it is four atoms in addition to 1,3,4-thiadiazole moiety. These linkers were considered to take the place of the central aryl moiety of the lead structure to enhance the flexibility which consequently should improve the affinity of VEGFR-2 binding. The main modifications focussed on the region representing the HBA/HBD pharmacophore where moieties with effective anticancer potency such as thiazolidine-2,4-dione, cyanothiouracil, or urea were used instead of the pharmacophore of unsymmetrical diaryl urea scaffolds of SOR.
Then, the aromatic terminal of the lead structure was replaced by another different hydrophobic system attached to either electron withdrawing or electron donating group that may affect the activity of the new hybrids allowing studying SAR of such hybrids as potential anti-tumour candidates with potential inhibitory potencies against VEGFR-2. All alteration pathways and design rationale were illustrated in .
Figure 3. The fundamental structural requirements for SOR and rational of design of the novel postulated VEGFR-2 inhibitors.
Material and methods
Chemistry
The mp (°C) were recorded using Stuart melting point apparatus (SMP 30) and are uncorrected. IR (KBr) was measured on FT-IR 200 spectrophotometer (ύ cm−1), Faculty of Pharmacy, Mansoura University. 1H-NMR and 13C-NMR were performed in (DMSO-d6) at 1HNMR (400 MHz), 13CNMR (100 MHz) on an NMR spectrometer (δ ppm) using TMS as an internal standard, NMR Unit, Faculty of Pharmacy, Mansoura University. Abbreviations are as indicated: (s), singlet; (d), doublet; (t), triplet; (m), multiplet; (br), broad. Mass spectra were performed in Thermo Scientific GC/MS model ISQ at the Regional Centre for Mycology and Biotechnology (RCMB), Al-Azhar University, Egypt. Microanalyses were carried out at Cairo University on PerkinElmer 240 elemental analyser for elements C, H, and N, results were obtained within the accepted limits. Compounds were visualised with 254 nm UV lamp. Chemicals and reagents were obtained from Aldrich Chemicals Co, USA, and commercial sources. The key precursor compound 2 and intermediate compounds 3a–e, 5a–d, and 7a–d were synthesised following reported procedures in literatureCitation50,Citation52–54.
General method for the preparation of thiazolidine-2,4-dione derivatives (4a–e)
To a solution of the appropriate salt derivative 3a–e (0.3 mmol) in DMF, the chloride derivative 2 (0.3 mmol) was added. Then, the mixture was refluxed all night. After reaction was complete (TLC), the mixture was poured into crushed ice; the precipitate obtained was filtered off and recrystallized from ethanol.
2-(5-(4-Fluorobenzylidene)-2,4-dioxothiazolidin-3-yl)-N-(6-nitrobenzo[d]thiazol-2-yl)acetamide (4a)
Brown solid; (0.08 g, 58%). M.p. 199–201 °C. IR (νmax/cm−1): 3445 (NH), 1740, 1680 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.30 (s, 1H, NH), 9.10 (s, 1H, thiazole-H), 8.30 (d, J = 7.7 Hz, 1H, thiazole-H), 8.1 (s, 1H, benzylidene-H), 7.9 (d, J = 7.7 Hz, 1H, thiazole-H), 7.73 (d, J = 8.5 Hz, 2H, phenyl-2H), 7.44 (d, J = 8.5 Hz, 2H, phenyl-2H), 4.76 (s, 2H, CH2). 19F NMR (377 MHz, DMSO-d6) δ −108.91 (s). 13C NMR (101 MHz, DMSO-d6) δ 167.5, 166.9, 166.8, 165.6, 164.9, 164.0, 143.7, 133.4, 133.2, 132.9, 132.7, 122.4, 121.0, 119.7, 117.3, 117.0, 44.3. MS m/z (%):458.37 (M+, 29.33). Anal. Calcd for C19H11FN4O5S2 (458.44): C, 49.78; H, 2.42; N, 12.22. Found: C, 49.55; H, 2.48; N, 12.12%.
2-(5-(4-Methoxybenzylidene)-2,4-dioxothiazolidin-3-yl)-N-(6-nitrobenzo[d]thiazol-2-yl)acetamide (4b)
Orange solid; (0.092 g, 65%). M.p. 259–261 °C. IR (νmax/cm−1): 3451 (NH), 1739, 1683 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.24 (s, 1H, NH), 9.08 (s, 1H, thiazole-H), 8.30 (d, J = 7.7 Hz, 1H, thiazole-H), 8.0 (s, 1H, benzylidene-H), 7.91 (d, J = 7.7 Hz, 1H, thiazole-H), 7.65 (d, J = 7.1 Hz, 2H, phenyl-2H), 7.15 (d, J = 7.1 Hz, 2H, phenyl-2H), 4.40 (s, 2H, CH2), 3.85 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 167.7, 167.0, 165.7, 163.6, 162.8, 161.9, 143.6, 134.4, 132.9, 132.7, 132.5, 122.3, 121.2, 119.7, 117.9, 115.5, 56.0, 44.1. MS m/z (%):470.59 (M+, 24.65). Anal. Calcd for C20H14N4O6S2 (470.48): C, 51.06; H, 3.00; N, 11.91. Found: C, 51.15; H, 3.08; N, 11.88.
2-(5-(4-Bromobenzylidene)-2,4-dioxothiazolidin-3-yl)-N-(6-nitrobenzo[d]thiazol-2-yl)acetamide (4c)
Yellow solid; (0.099 g, 63%). M.p. 252–254 °C. IR (νmax/cm−1): 3450 (NH), 1747, 1691 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.30 (s, 1H, NH), 9.09 (s, 1H, thiazole-H), 8.31 (d, J = 7.7 Hz, 1H, thiazole-H), 8.0 (s, 1H, benzylidene-H), 7.90 (d, J = 7.7 Hz, 1H, thiazole-H), 7.77 (d, J = 8.5 Hz, 2H, phenyl-2H), 7.63 (d, J = 8.5 Hz, 2H, phenyl-2H), 4.40 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6) δ 172.6, 167.3, 166.9, 165.5, 163.5, 162.8, 143.7, 133.2, 132.9, 132.5, 132.2, 125.0, 122.4, 122.1, 121.3, 119.7, 44.3. MS m/z (%):519.53 (M+, 20.71). Anal. Calcd for C19H11BrN4O5S2 (519.35): C, 43.94; H, 2.13; N, 10.79. Found: C, 43.90; H, 2.14; N, 10.88%.
2-(5-(4-Methylbenzylidene)-2,4-dioxothiazolidin-3-yl)-N-(6-nitrobenzo[d]thiazol-2-yl)acetamide (4d)
Beige solid; (0.076 g, 56%). M.p. 201–203 °C. IR (νmax/cm−1): 3430 (NH), 1740, 1688 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.13 (s, 1H, NH), 9.09 (s, 1H, thiazole-H), 8.30 (d, J = 7.7 Hz, 1H, thiazole-H), 8.0 (s, 1H, benzylidene-H), 7.95 (d, J = 7.7 Hz, 1H, thiazole-H), 7.58 (d, J = 8.1 Hz, 2H, phenyl-2H), 7.40 (d, J = 8.1 Hz, 2H, phenyl-2H), 4.75 (s, 2H, CH2), 2.39 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 167.6, 167.0, 165.7, 163.6, 162.8, 143.6, 134.5, 132.7, 130.8, 130.6, 130.5, 130.4, 122.3, 121.3, 120.2, 119.7, 44.2, 21.6. MS m/z (%):454.14 (M+, 19.79). Anal. Calcd for C20H14N4O5S2 (454.48): C, 52.85; H, 3.10; N, 12.33. Found: C, 52.70; H, 3.15; N, 12.32%.
2-(2,4-Dioxo-5–(3,4,5-trimethoxybenzylidene)thiazolidin-3-yl)-N-(6-nitrobenzo[d]thiazol-2-yl)acetamide (4e)
Buff solid; (0.110 g, 69%). M.p. 149–151 °C. IR (νmax/cm−1): 3450 (NH), 1745, 1670 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.30 (s, 1H, NH), 9.10 (s, 1H, thiazole-H), 8.32 (d, J = 7.7 Hz, 1H, thiazole-H), 8.0 (s, 1H, benzylidene-H), 7.93 (d, J = 7.7 Hz, 1H, thiazole-H), 7.01 (s, 2H, phenyl-2H), 4.76 (s, 2H, CH2), 3.86 (s, 6H, 2OCH3), 3.76 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 167.6, 166.9, 165.6, 163.4, 162.8, 153.8, 143.7, 140.2, 134.6, 132.7, 128.8, 122.4, 121.4, 120.3, 119.8, 108.2, 60.7, 56.5, 44.1. MS m/z (%):530.70 (M+, 31.02). Anal. Calcd for C22H18N4O8S2 (530.53): C, 49.81; H, 3.42; N, 10.56. Found: C, 49.74; H, 3.42; N, 10.51%.
General method for the preparation of 1,3,4-thiadiazole derivatives (6a–d)
The appropriate thiol derivative 5a–d (0.3 mmole) with K2CO3 (0.45 mmole) in acetone were left for stirring at rt for 30 min. Then, an equivalent amount of the chloride derivative 2 was added. The resulting mixture was refluxed overnight. Then it was poured into crushed ice; the formed solid was obtained and recrystallized from ethanol to afford the required hybrids.
2-((5-(3-(4-Chlorophenyl)ureido)-1,3,4-thiadiazol-2-yl)thio)-N-(6-nitrobenzo[d]thiazol-2-yl)acetamide (6a)
Brown solid; (0.098 g, 62%). M.p. 241–243 °C. IR (νmax/cm−1): 3270 (NH), 1685 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 12.91 (s, 1H, NH), 11.8 (br s, 1H, NH), 9.51 (s, 1H, NH), 9.08 (s, 1H, thiazole-H), 8.30 (d, J = 8.8 Hz, 1H, thiazole-H), 7.94 (d, J = 8.8 Hz, 1H, thiazole-H), 7.54 (d, J = 8.5 Hz, 2H, phenyl-2H), 7.35 (d, J = 8.5 Hz, 2H, phenyl-2H), 4.38 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6) δ 168.4, 163.9, 157.9, 153.9, 143.5, 138.0, 133.1, 132.7, 129.7, 129.2, 127.0, 122.3, 121.2, 120.9, 119.6, 37.4. MS m/z (%): 521.08 (M+, 60.75). Anal. Calcd for C18H12ClN7O4S3 (521.98): C, 41.42; H, 2.32; N, 18.78. Found: C, 41.46; H, 2.12; N, 18.74%.
2-((5-(3-(4-Methoxyphenyl)ureido)-1,3,4-thiadiazol-2-yl)thio)-N-(6-nitrobenzo[d]thiazol-2-yl)acetamide (6 b)
Yellow solid; (0.093 g, 60%). M.p. 269–271 °C. IR (νmax/cm−1): 3290 (NH), 1682 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.11 (s, 1H, NH), 11.4 (br s, 1H, NH), 9.07 (br s, 2H, NH + thiazole-H), 8.30 (d, J = 8.0 Hz, 1H, thiazole-H), 7.93 (d, J = 8.0 Hz, 1H, thiazole-H), 7.38 (d, J = 7.1 Hz, 2H, phenyl-2H), 6.89 (d, J = 7.1 Hz, 2H, phenyl-2H), 4.37 (s, 2H, CH2), 3.73 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 167.4, 166.9, 163.2, 161.5, 158.9, 153.5, 143.8, 142.1, 132.5, 122.3, 119.9, 119.7, 119.6, 119.4, 114.3, 37.6, 56.1. MS m/z (%):517.52 (M+, 25.81). Anal. Calcd for C19H15N7O5S3 (517.56): C, 44.09; H, 2.92; N, 18.94. Found: C, 44.11; H, 2.91; N, 18.94%.
N-(6-Nitrobenzo[d]thiazol-2-yl)-2-((5–(3-(p-tolyl)ureido)-1,3,4-thiadiazol-2-yl)thio)acetamide (6c)
Brown solid; (0.085 g, 56%). M.p. 238–240 °C. IR (νmax/cm−1): 3283, 3173 (NH), 1683 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.13 (s, 1H, NH), 11.02 (s, 1H, NH), 9.09 (s, 1H, NH), 8.97 (s, 1H, thiazole-H), 8.30 (d, J = 8.9 Hz, 1H, thiazole-H), 7.94 (d, J = 8.9 Hz, 1H, thiazole-H), 7.35 (d, J = 8.1 Hz, 2H, phenyl-2H), 7.13 (d, J = 8.1 Hz, 2H, phenyl-2H), 4.38 (s, 2H, CH2), 2.26 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 168.4, 166.4, 163.8, 161.5, 153.9, 143.6, 142.4, 136.1, 132.7, 129.8, 122.3, 121.3, 119.7, 119.6, 119.4, 37.4, 20.9. MS m/z (%):501.66 (M+, 23.56). Anal. Calcd for C19H15N7O4S3 (501.56): C, 45.50; H, 3.01; N, 19.55. Found: C, 45.55; H, 2.99; N, 19.59%.
N-(6-Nitrobenzo[d]thiazol-2-yl)-2-((5–(3-phenylureido)-1,3,4-thiadiazol-2-yl)thio)acetamide (6d)
Beige solid; (0.094 g, 64%). M.p. 282–284 °C. IR (νmax/cm−1): 3290 (NH), 1683 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.13 (s, 1H, NH), 11.03 (s, 1H, NH), 9.08 (br s, 2H, NH + thiazole-H), 8.30 (d, J = 8.0 Hz, 1H, thiazole-H), 7.94 (d, J = 8.0 Hz, 1H, thiazole-H), 7.47 (d, J = 6.6 Hz, 2H, phenyl-2H), 7.33 (d, J = 6.6 Hz, 2H, phenyl-2H), 7.11– 7.05 (m, 1H, phenyl-1H), 4.39 (s, 2H, CH2). 13C NMR (101 MHz, DMSO-d6) δ 168.4, 164.3, 163.8, 161.2, 153.9, 143.6, 138.9, 132.7, 129.4, 124.6, 123.7, 122.3, 121.3, 119.6, 119.4, 37.4. MS m/z (%):487.43 (M+, 28.83). Anal. Calcd for C18H13N7O4S3 (487.54): C, 44.34; H, 2.69; N, 20.11. Found: C, 44.24; H, 2.60; N, 20.31%.
General method for the preparation of cyanothiouracil derivatives (8a–d)
The appropriate thiol derivative 7a–d (0.3 mmole) with K2CO3 (0.45 mmole) in acetone were stirred at rt for half an hour. An equivalent amount of the chloride 2 was then added. The obtained mixture was refluxed overnight. The precipitate was filtered, washed with acetone to furnish the target compounds.
2-((4-(4-Chlorophenyl)-5-cyano-6-oxo-1,6-dihydropyrimidin-2-yl)thio)-N-(6-nitrobenzo[d] thiazol-2-yl)acetamide (8a)
Beige solid; (0.082 g, 55%). M.p. 252–254 °C. IR (νmax/cm−1): 3496, 3155 (NH), 2219 (CN), 1667 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.11 (s, 1H, NH), 9.05 (s, 1H, thiazole-H), 8.33 (d, J = 8.9 Hz, 1H, thiazole-H), 7.95 (d, J = 8.9 Hz, 1H, thiazole-H), 7.76 (d, J = 8.3 Hz, 2H, phenyl-2H), 7.26 (d, J = 8.3 Hz, 2H, phenyl-2H), 4.34 (s, 2H, CH2), 1.2 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 168.2, 166.5, 166.4, 163.8, 162.1, 153.9, 143.6, 136.6, 134.4, 132.6, 130.7, 128.8, 122.4, 121.2, 119.6, 116.3, 93.8, 35.1. MS m/z (%):498.99 (M+, 23.03). Anal. Calcd for C20H11ClN6O4S2 (498.92): C, 48.15; H, 2.22; N, 16.84. Found: C, 48.28; H, 2.20; N, 16.77%.
2-((4-(4-Bromophenyl)-5-cyano-6-oxo-1,6-dihydropyrimidin-2-yl)thio)-N-(6-nitrobenzo[d] thiazol-2-yl)acetamide (8b)
White solid; (0.114 g, 70%). M.p. 229–231 °C. IR (νmax/cm−1): 3450 (NH), 2217 (CN), 1668 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.11 (s, 1H, NH), 9.04 (s, 1H, thiazole-H), 8.32 (d, J = 8.9 Hz, 1H, thiazole-H), 7.96 (d, J = 8.9 Hz, 1H, thiazole-H), 7.68 (d, J = 8.1 Hz, 2H, phenyl-2H), 7.38 (d, J = 8.1 Hz, 2H, phenyl-2H), 4.34 (s, 2H, CH2), 1.2 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 168.2, 166.6, 166.3, 163.7, 161.9, 153.9, 143.6, 134.7, 132.6, 131.7, 130.9, 125.6, 122.4, 121.2, 119.6, 116.2, 93.9, 35.1. MS m/z (%):543.57 (M+, 19.70). Anal. Calcd for C20H11BrN6O4S2 (543.37): C, 44.21; H, 2.04; N, 15.47. Found: C, 44.02; H, 2.08; N, 15.55%.
2-((5-Cyano-4-(4-methoxyphenyl)-6-oxo-1,6-dihydropyrimidin-2-yl)thio)-N-(6-nitrobenzo[d] thiazol-2-yl)acetamide (8c)
Off white solid; (0.105 g, 71%). M.p. 273–275 °C. IR (νmax/cm−1): 3451(NH), 2221 (CN), 1700, 1659 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.17 (s, 1H, NH), 9.07 (s, 1H, thiazole-H), 8.32 (d, J = 8.2 Hz, 1H, thiazole-H), 7.97 (d, J = 8.2 Hz, 1H, thiazole-H), 7.81 (d, J = 8.4 Hz, 2H, phenyl-2H), 6.71 (d, J = 8.4 Hz, 2H, phenyl-2H), 4.37 (s, 2H, CH2), 3.61 (s, 3H, OCH3), 1.2 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 168.5, 167.1, 165.2, 163.9, 160.5, 159.8, 153.9, 143.5, 132.9, 132.5, 130.1, 122.3, 121.1, 119.6, 116.9, 114.6, 93.1, 35.1, 55.8. MS m/z (%):494.61 (M+, 25.45). Anal. Calcd for C21H14N6O5S2 (494.50): C, 51.01; H, 2.85; N, 16.99. Found: C, 51.12; H, 2.80; N, 16.93%.
2-((5-Cyano-6-oxo-4-(p-tolyl)-1,6-dihydropyrimidin-2-yl)thio)-N-(6-nitrobenzo[d]thiazol-2-yl)acetamide (8d)
Beige solid; (0.090 g, 63%). M.p. 210–212 °C. IR (νmax/cm−1): 3486, 3182 (NH), 2227 (CN), 1684 (C = O). 1H NMR (400 MHz, DMSO-d6) δ 13.13 (s, 1H, NH), 9.06 (s, 1H, thiazole-H), 8.33 (d, J = 8.9 Hz, 1H, thiazole-H), 7.97 (d, J = 8.9 Hz, 1H, thiazole-H), 7.66 (d, J = 8.0 Hz, 2H, phenyl-2H), 6.98 (d, J = 8.0 Hz, 2H, phenyl-2H), 4.36 (s, 2H, CH2), 2.12 (s, 3H, CH3), 1.2 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 167.6, 165.7, 163.8, 161.9, 153.97, 143.5, 142.1, 132.8, 132.7, 129.3, 128.9, 122.3, 121.1, 119.6, 116.4, 93.1, 35.1, 21.3. MS m/z (%):478.79 (M+, 19.92). Anal. Calcd for C21H14N6O4S2 (478.50): C, 52.71; H, 2.95; N, 17.56; S, 13.40. Found: C, 52.71; H, 2.92; N, 17.65; S, 13.47%.
Biological evaluation
Antitumor screening
The in vitro anticancer activity of the new benzothiazole hybrids was evaluated by performing MTT assay as in the literatureCitation51,Citation55,Citation56.
In vitro VEGFR-2 kinase inhibitory assay
VEGFR-2 enzyme assay was performed as reportedCitation57.
Flow cytometry analysis of the cell cycle distribution
It was carried out using MCF-7 cell lines stained with PI and analysed by FACS Calibur flow cytometer as reportedCitation51,Citation58.
Analysis of cellular apoptosis
The extent of apoptosis was evaluated on MCF-7 cells and Annexin V-FITC/PI apoptosis detection kit following the reported methodCitation51,Citation59.
In silico evaluation and ADME prediction
Molecular docking
Since compounds 4a, 4e, and 8a showed significant inhibition to VEGFR-2, Molecular docking was utilised for shedding the light on the mode of binding of these compounds. Briefly, chemical structure of hybrids of interest was sketched by Chemoffice softwareCitation60, converted to SDF file, then their 3 D structure was generated by MOE software and the file was saved as mol2 fileCitation61. The 3 D structure of (VEGFR-2) was obtained from PDB using the code (4ASD) and subjected to protein preparation protocol in MOE using default options.
The prepared PDB was exported to Leadit softwareCitation62,Citation63 and the active site was set as sphere with radius 6.5 Å around the co-crystallized ligand. The software was validated as previously reportedCitation64 and the RMSD was found to be 0.8. The 3 D structure of the compounds was selected as a library and was docked to the active site. Finally, the docked poses were inspected using Discovery studio visualiser to study their interaction with the binding siteCitation65.
In silico prediction of physicochemical properties and pharmacokinetic profile
For ADMET profiling, the free access of website (https://preadmet.bmdrc.kr/)Citation66 was used for estimation.
Results and discussion
Chemistry
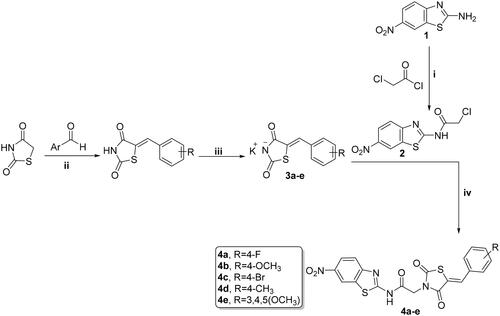
Synthesis of new analogs, thiazolidine-2,4-diones 4a–e, 1,3,4-thiadiazoles 6a–d, and cyanothiouracils 8a–d, is illustrated in Schemes 1, 2, and 3. Starting with 6-nitrobenzo[d]thiazol-2-amine (1) which was subjected to acetylation using chloroacetyl chloride and triethylamine as a base, furnished the chloride derivative 2, which is considered as the key precursor in the next nucleophilic substitution reactionsCitation52. 5-Arylidene-thiazolidine-2,4-diones were formed through a Knoevenagel condensation reaction, and then refluxed with ethanolic KOH to obtain the potassium salts 3a–f, that were further substituted with the chloride derivative 2 to obtain the target benzothiazole/thiazolidine-2,4-dione hybrids 4a–e (Scheme 1)Citation53.
Scheme 1. Preparation of benzothiazole/thiazolidine-2,4-dione hybrids 4a–e; reagents and conditions: (i) CH2Cl2/Et3N 0 °C, rt; (ii) AcOH, NaOAc, reflux 14 h; (iii) EtOH, KOH, reflux 2 h; and (iv) DMF, K2CO3, reflux overnight.
The key intermediates 5a–d were obtained via refluxing the 2-thiol derivative with various 4-substituted phenyl isocyanate derivatives in acetonitrileCitation50. The formed urea derivatives 5a–d underwent reaction with the chloride derivative 2 yielding the target benzothiazole/thiadiazole-aryl urea hybrids 6a–d (Scheme 2).
Scheme 2. Synthesis of benzothiazole/1,3,4-thiadiazole-aryl urea hybrids 6a–d; reagents and conditions: (i) CS2, EtOH, reflux overnight; (ii) CH3CN, reflux overnight; and (iii) acetone, K2CO3, reflux overnight.
The 6-phenyl-2-thiouracil-5-carbonitrile derivatives 7a–d were obtained by prolonged heating of different aldehydes, thiourea, and ethyl cyanoacetate with K2CO3 (Scheme 3)Citation54. Finally, compounds 7a–d were refluxed with the chloride derivative 2 in acetone using potassium carbonate to furnish the corresponding cyanothiouracils 8a–d (Schemes 3).
Scheme 3. Synthesis of benzothiazole/cyanothiouracil hybrids 8a–d; reagents and conditions: (i) EtOH, K2CO3, reflux 12 h and (ii) acetone, K2CO3, reflux overnight.
The newly synthesised benzothiazole-based derivatives (4a–e, 6a–d, and 8a–d) were characterised using different means of IR, melting point, elemental analysis and spectroscopic data (1H-NMR, 19F-NMR, 13C-NMR, and MS). The IR charts of the new targets exhibited characteristic bands ranging from 1670 to 1730 cm−1 verifying the two amidic − C=O groups. 1H-NMR spectra for all the newly synthesised target compounds showed a characteristic singlet signal ranging from 4.3 to 4.8 ppm corresponding to methylene protons –N/SCH2CO– guaranteeing the synthesis of our new targets. In addition, 13C-NMR revealed the appearance of three peaks (4a–e) or two peaks (6a–d and 8a–d) at ∼160–169 ppm equivalent to the carbonyl groups. Moreover, all final targets exhibited characteristic peak in the aliphatic region at approximately 37–44 ppm for the methyl carbon of acetamide moiety. All other data were consistent with the proposed structures. The mass spectra of target compounds showed the right molecular ion peaks (M+). The synthetic methods and the yields are presented in experimental part.
Biological evaluation
In vitro cytotoxicity and antitumor activity:
The in vitro cytotoxic activity of the new benzothiazole hybrids 4a–e, 6a–d, and 8a–d was evaluated by standard MTT assay against three human cancer cell lines: HCT-116 (colorectal carcinoma), HEPG-2 (hepatocellular carcinoma), and MCF-7 (breast cancer) cell linesCitation55, using SOR as a positive drug control. IC50 values were calculated and summarised in .
Table 1. Cytotoxicity (IC50) of the target hybrids 4a–e, 6a–d, and 8a–d towards HCT-116, HepG-2, MCF-7, and WI-38 cell lines.
The results showed that the investigated derivatives displayed different degrees of cytotoxic effect against the tested cancer cell lines.
Structure–activity correlation
The IC50 results of benzothiazole/thiazolidine-2,4-dione hybrids 4a–e revealed that compounds 4a and 4e showed wide spectrum cytotoxic potencies and are considered the most potent members in this series with IC50 values range of 3.84–9.04 μM towards the three cell lines. 4a is considered the best member of all compounds with IC50 of 5.61, 7.92, and 3.84 μM against HCT-116, HEPG-2, and MCF-7, respectively, compared to SOR (IC50 of 5.23, 4.50, and 4.17 μM, respectively), while, compounds 4d and 4b exhibited moderate cytotoxicity against the three cell lines with IC50 of 26.78, 21.13, and 15.48 for compound 4d and IC50 of 29.92, 34.68, and 22.27 for compound 4b against HCT-116, HEPG-2, and MCF-7 cell lines. In addition, compound 4c displayed a diminished activity with IC50 range of 47.94 to 65.47 compared to the other analogs.
It is observed that the chemistry of hybrids 4a–e influenced their anticancer activity. Compound 4a with small electron withdrawing substituent (F) on para position of benzylidene group showed higher antitumor activity (IC50 range of 3.84–5.61 μM) than compound 4c which contains a bulky substituent (Br) (IC50 range of 47.94–65.47 μM). However, the introduction of electron donating substituent on para position (OCH3, CH3) as in compounds 4b and 4d resulted in a drop of the antitumor activity (IC50 range of 22.27–34.68 and 15.48–26.78 μM, respectively). Moreover, the 3,4,5-trimethoxy substitution resulted in a sharp increase in the antitumor activity as in compound 4e with IC50 of 6.11– 9.04 μM.
Regarding IC50 values of benzothiazole/1,3,4-thiadiazole-aryl urea hybrids 6a–d, they generally revealed a much weaker cytotoxic activity towards the examined cell lines (IC50 range: 39.79–92.45 μM) compared to thiazolidine-2,4-dione 4 and cyanothiouracil 8 hybrids.
The benzothiazole/cyanothiouracil hybrids 8a–d showed a variable degree of cytotoxic potencies. Compound 8a is considered the most active member in this series with IC50 of 13.89, 18.10, and 10.86 μM against HCT-116, HEPG-2, and MCF-7, respectively.
8a (IC50 of 10.86–18.10 μM) with small π-rich substituent (CH3) on para position of phenyl group had the highest activity against the three cell lines than those containing bulky substituent (OCH3) as in compound 8d (IC50 of 78.22 - >100 μM). On the other hand, the introduction of π-deficient para substituted phenyl moiety (Cl and Br) decreased the activity as in compounds 8b and 8c with IC50 range of 27.94–54.06 μM).
In vitro cytotoxicity against human normal cell
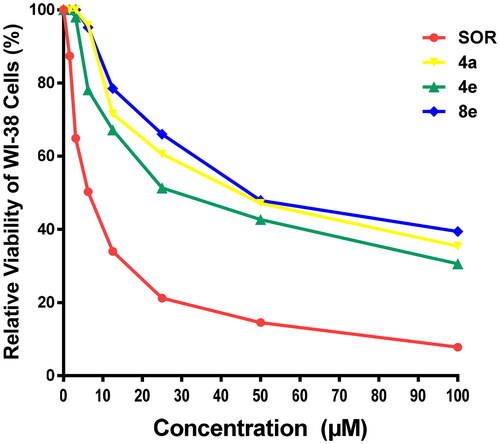
All tested compounds were studied for their cytotoxic activity on the WI-38 normal fibroblast WI-38 cells to assess their therapeutic safety. As depicted in , all the investigated hybrids showed lower toxicity against normal fibroblast cells WI-38. Accordingly, compounds 4a, 4e, and 8a exhibited low cytotoxicity on WI-38 with IC50 values of 45.23, 32.81, and 53.10 μM, respectively in comparison to SOR (IC50 = 6.72 μM) (.
Figure 4. Cytotoxic effect of the most active compounds 4a, 4e, 8e, and SOR on human normal WI-38 cell line.
VEGFR-2 enzyme inhibition assay
The promising hybrids, displaying the highest anticancer potencies were selected to further assess their dose-dependent VEGFR-2 enzyme inhibition at four various concentrations (10 nM–100 nM–1 μM–10 μM) to determine their IC50 values. As depicted in , all tested compounds had high % inhibition values against VEGFR-2 ranging from 70.11 to 77.94% at concentration of 1 μM, and range of 83.47–91.96% at concentration of 10 μM which is very close to the standard drug SOR. At concentration of 10 μM, 4a showed VEGFR-2% inhibition of 91.96% which is very close to the standard drug used (92.17%).
Table 2. Inhibitory effects of compounds 4a, 4e, and 8a against VEGFR-2.
In addition, consistent with the cytotoxic activity, compound 4a showed strong VEGFR-2 inhibitory activity with IC50 of 91 nM, in comparison with 53 nM for SOR. The benzothiazole derivative 4e exhibited lower inhibitory effect with IC50 of 161 nM. Also, the IC50 of compound 8a was 266 nM which could explain the moderate cytotoxic activity observed in MTT assay. It is worth mentioning that compound 4a (IC50 of 91 nM), with 6-nitrobenzothiazole and F atom on para position of benzylidene group, was more potent than the corresponding non-nitrated benzothiazole derivative X with IC50 of 190 nMCitation45.
Cell cycle analysis
For further investigation of the mode of antitumor activity of the most potent candidates 4a, 4e, and 8a, cell cycle analysis and apoptosis induction were done using propidium iodide (PI) stainingCitation67. MCF-7 breast cancer cells were incubated with IC50 concentration of compounds 4a, 4e, and 8a for 24 h, stained with PI and DNA contents were measured by flow cytometry (FCM). The cells were treated with DMSO as a control.
Quantification of the results (, ) showed that compounds 4a and 4e arrested MCF-7 cells at S phase. The cell population in S phase increased from 31.89 in the untreated cell to 49.85 and 42.66% in the cell treated with compounds 4a and 4e, respectively. On the other hand, compound 8a led to an outstanding growth in ratio of MCF-7 cells in G0/G1 phase from 57.92% in the untreated cells to 61.36% and in S phase from 31.89% in the untreated cells to 36.11% with a decrease in the ratio of cells in G2/M phase by 2.53% in comparison with untreated control (10.19%). These findings obviously show that 8a arrested the cell cycle in MCF-7 at a G1/S phase.
Figure 5. Effect of compounds 4a, 4e, and 8a on DNA-ploidy flow cytometric analysis of MCF-7 cells. The cells were treated with DMSO as control and 4a, 4e, and 8a for 24 h.
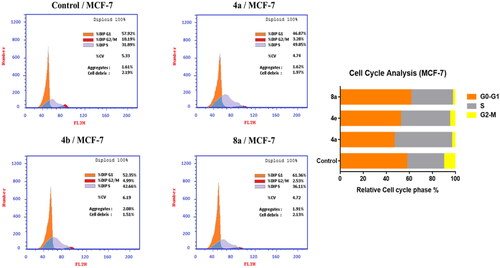
Table 3. Effect of compounds 4a, 4e, and 8a on the cell cycle progression in MCF-7 cells compared to SOR.
Detection of apoptosis
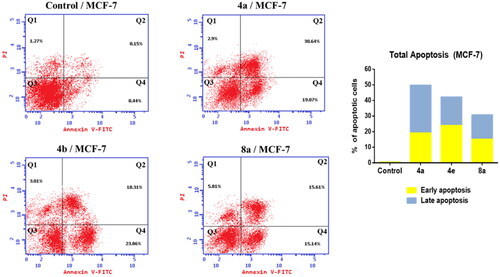
The development of apoptosis inducers is highly attractive therapeutic strategy for the identification of powerful anticancer drugsCitation67,Citation68. So that, the ability of the most active compounds 4a, 4e, and 8a to induce apoptosis in MCF-7 cells was evaluated using AnnexinV and PI double staining flow cytometry. As shown in , MCF-7 cells treated with compounds 4a, 4e, and 8a exhibited the accumulation of early and late apoptotic cells, as compared to the control. For example, the ratio of total apoptotic cells (early and late apoptotic cells) after treatment of MCF-7 cells with compounds 4a, 4e, and 8a for 24 was sharply increased to 49.71, 42.17, and 30.75%, respectively as compared to 0.59% of apoptotic cells in the untreated control. These results indicated that these compounds could inhibit cell growth through induction of cell apoptosis.
Figure 6. Effect of compounds 4a, 4e, and 8a on the percentage of annexin V-FITC-positive staining in MCF-7 cells. The cells were treated with DMSO as control and 4a, 4e, and 8a for 24 h. Q1 quadrant represents dead (necrotic) cells; Q2 quadrant represents late apoptosis; Q3 quadrant represent live cells; Q4 quadrant represents early apoptosis.
In silico evaluation and ADME prediction
Molecular docking results
Molecular docking proved to be a versatile tool in the field of drug discovery due to its ability to explain the interaction of small molecules with various biological targets accelerating the discovery and development of new potential therapeuticsCitation69–74. Since VEGFR-2 is one of the most investigated targets using in silico tools, Molecular docking was preformed to identify the interaction of compounds 4a, 4e, and 8a with the active site. All the three compounds were able to occupy the same active site of SOR, the co-crystallized ligand as depicted in but showed different interaction profile with the binding site. Compound 4a showed the lowest binding energy followed by compound 4e and 8a, respectively as shown in .
Figure 7. Compound 4a compound 4e compound 8a docked in the active site and aligned to the co-crystallized ligand SOR. Please refer to the online version for colored figure.
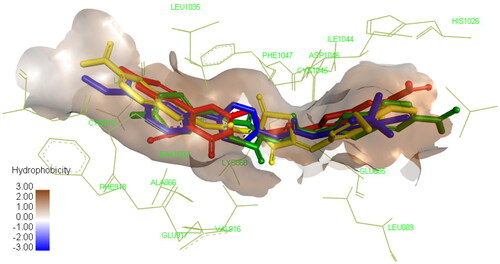
Table 4. Binding energy of the top three compounds docked in VEGFR-2 (PDB: 4ASD) in comparison to the co-crystallized ligand SOR.
Concerning compound 4a, interaction with important residues in the active site was achieved through hydrogen bonding such as Asp1046 through its carbonyl group and Glu885 through the NH in the amide group which is a known requirement to achieve good inhibitory activity against this enzyme. In addition, certain hydrophobic interactions have been achieved with IL888, ILE892, Leu1019, ILE1025, and His1026 residues. Furthermore, the nitro group of benzothiazole was able to form hydrogen bond with Ile1025, and ionic bond with ASP814 and Arg1027 which could explain the superior activity of our compound 4a over the previously reported non-nitrated derivative XCitation45. The thiazolidine-2,4-dione moiety was found to occupy the linker area in the active site interacting with Lys868, Val 899, Val916, and Phe1047 allowing the fluoro-phenyl ring to interact with several residues in the hydrophobic groove such as Leu840, Ala866, and Leu1035 through hydrophobic interactions and with Cys919 through halogen hydrogen bond as demonstrated in Figure S1. These extensive interactions with the active site may explain its ability to inhibit the enzyme experimentally at low nanomolar concentration.
Despite the structural similarity between compounds 4a and 4e, the later showed different binding pattern, as it was only able to form one hydrogen bond with Asp1046 but not with Glu885 which explains the significant increase of IC50 shown in the experimental enzyme inhibition assay as this interaction is important for achieving potent inhibitory activity. Still, this was compensated by the interaction of the compound with several amino acid residues in the active site through hydrophobic interaction of benzothiazole ring with Ala866, Leu840, Phe918, Cys919, and Leu1035, while the thiazolidine-2,4-dione formed hydrophobic interaction with Val899, Val916, Cys1045, and Lys868. Finally, the trimethoxy substituted phenyl occupied the hydrophobic allosteric site so it was able to interact with Ile892, Val898, Ile1044, and Leu1019 as demonstrated in Figure S2.
Regarding compound 8a, it showed similar binding pattern to that of compound 4a since it was able to form two hydrogen bonds between the amide moiety and Asp1046 and Glu885. Still, it protrudes towards the ATP active site allowing the interaction of cyanothiouracil with Cys919 through the cyanide group and with Leu840, Val 899, Ala866, Cys919, Phe1047, Leu1035, and Cys1045 through hydrophobic interactions. Consequently, the benzothiazole ring was not able to fully occupy the hydrophobic pocket in the allosteric site, so it interacted only with His1026 (Figure S3) which explains the moderate inhibitory activity demonstrated in the experimental enzyme inhibition assay.
ADMET analysis
Predicting the pharmacokinetic characteristics (ADMET) is important in early stage of drug development to enhance the efficacy and safety profile and to avoid the failure of therapeutic agents to be effective clinical candidatesCitation75. Thus, the ADMET profile of the most active hybrids 4a, 4e, and 8a was calculated theoretically using online Pre-ADMET serverCitation76.
As shown in , the obtained results revealed that all the tested hybrids predicted to have negative carcinogenic activities and exerted excellent intestinal absorption with HIA values ranging from 83.30 to 91.96%, demonstrating their potency as oral anticancer candidates. They also showed low CNS absorption, low cell permeability in the CaCo2 cell model (CaCo2 values < 4 nm/s) and low cell permeability in the MDCK cell model (MDCK < 25 nm/s). Moreover, all hybrids are expected to be not incorporated in drug-drug interactions as they are non-inhibitors of CYP3A4 enzyme, in contrast to reference drug which has CYP3A4 inhibitory activity. Similarly, as reference drug, all the hybrids are inhibitors to P-glycoprotein (PgP), increasing bioavailability of co-administered drugs and reversing multi-drug resistance in cancer cells.
Table 5. ADMET profile for active compounds 4a, 4e, 8a, and SOR.
Conclusion
In summary, three series of novel benzothiazole-based hybrids bearing thiazolidine-2,4-dione 4a–e, thiadiazole-aryl urea 6a–d and cyanothiouracil 8a–d were synthesised, their structure was confirmed and evaluated for their cytotoxic activity against three human cancer cell lines: HCT-116, HEPG-2, and MCF-7. Thereafter, the most effective anti-proliferative members were explored for their VEGFR-2 inhibitory activity. Compounds 4a, 4e, and 8a were the most active hybrids. Hybrid 4a was found to be the most active one with wide spectrum of cytotoxic activity against the three cell lines with IC50 of 5.61, 7.92, and 3.84 µM, respectively. The most effective Hybrids 4a, 4e, and 8a also displayed an evident apoptosis in MCF-7 cell lines and induced cell cycle arrest at the S phase (compounds 4a and 4e) and at G1/S phase (compound 8a). Consistent with the cytotoxic activity, compound 4a showed strong VEGFR-2 inhibition with IC50 91 nM, in comparison with 53 nM for SOR. The cytotoxic and VEGFR-2 inhibition activities were illustrated using molecular docking study which revealed that 4a involves the key fragment important for the interaction with the DFG-binding domain that is essential for VEGFR-2 hindrance. In silico study involving ADMET analysis were performed where compound 4a showed acceptable physicochemical and pharmacokinetic properties. Still, further chemical optimisation and pharmacokinetic studies are needed before considering this class of compounds as potential candidate for the development of future anticancer agents.
Supplemental Material
Download PDF (5.2 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Varmus H. The new era in cancer research. Science. 2006;312(5777):1162–1165.
- Wang Z, Shi X-H, Wang J, Zhou T, Xu Y-Z, Huang T-T, Li Y-F, Zhao Y-L, Yang L, Yang S-Y. Synthesis, structure–activity relationships and preliminary antitumor evaluation of benzothiazole-2-thiol derivatives as novel apoptosis inducers. Bioorganic Med Chem Lett. 2011;21:1097–1101.
- Hamdi A, Said E, Farahat AA, Aa El-Bialy S, Am Massoud M. Synthesis and in vivo antifibrotic activity of novel leflunomide analogues. Lett Drug Des Discov. 2016;13:912–920.
- Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–348.
- Branca MA. Multi-kinase inhibitors create buzz at ASCO. Nat Biotechnol. 2005;23(6):639–640.
- Blankenberg FG. Apoptosis imaging: anti-cancer agents in medicinal chemistry. Anticancer Agents Med Chem. 2009;9(9):944–951.
- Roskoski R Jr, Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res Commun. 2016;103:26–48.
- Fabbro D, Ruetz S, Buchdunger E, Cowan-Jacob SW, Fendrich G, Liebetanz J, Mestan J, O'Reilly T, Traxler P, Chaudhuri B, et al. Protein kinases as targets for anticancer agents: from inhibitors to useful drugs. Pharmacol Ther. 2002;93(2-3):79–98.
- Mohamed AR, El Kerdawy AM, George RF, Georgey HH, Gawad NMA. Design, synthesis and in silico insights of new 7, 8-disubstituted-1, 3-dimethyl-1H-purine-2, 6 (3H, 7H)-dione derivatives with potent anticancer and multi-kinase inhibitory activities. Bioorg Chem. 2021;107:104569.
- Drąg-Zalesińska M, Drąg M, Poręba M, Borska S, Kulbacka J, Saczko J. Anticancer properties of ester derivatives of betulin in human metastatic melanoma cells (Me-45). Cancer Cell Int. 2017;17:1–7.
- El-Sherief HA, Youssif BG, Bukhari SNA, Abdelazeem AH, Abdel-Aziz M, Abdel-Rahman HM. Synthesis, anticancer activity and molecular modeling studies of 1, 2, 4-triazole derivatives as EGFR inhibitors. Eur J Med Chem. 2018;156:774–789.
- McTigue M, Murray BW, Chen JH, Deng Y-L, Solowiej J, Kania RS. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc Natl Acad Sci U S A. 2012;109(45):18281–18289.
- Aziz MA, Serya RAT, Lasheen DS, Abdel-Aziz AK, Esmat A, Mansour AM, Singab ANB, Abouzid KAM. Discovery of Potent VEGFR-2 Inhibitors based on Furopyrimidine and Thienopyrimidne Scaffolds as Cancer Targeting Agents. Sci Rep. 2016;6:24460.
- Mahdy HA, Ibrahim MK, Metwaly AM, Belal A, Mehany ABM, El-Gamal KMA, El-Sharkawy A, Elhendawy MA, Radwan MM, Elsohly MA, et al. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4(3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorg Chem. 2020;94:103422.
- Lee K, Jeong K-W, Lee Y, Song JY, Kim MS, Lee GS, Kim Y. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur J Med Chem. 2010;45(11):5420–5427.
- Wang Y, Peng C, Wang G, Xu Z, Luo Y, Wang J, Zhu W. Exploring binding mechanisms of VEGFR2 with three drugs lenvatinib, sorafenib, and sunitinib by molecular dynamics simulation and free energy calculation. Chem Biol Drug Des. 2019;93(5):934–948.
- Modi SJ, Kulkarni VM. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med Drug Discov. 2019;2:100009.
- Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5(10):835–844.
- Roskoski R. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. 2007;356(2):323–328.
- Zhu C, Ma X, Hu Y, Guo L, Chen B, Shen K, Xiao Y. Safety and efficacy profile of lenvatinib in cancer therapy: a systematic review and meta-analysis. Oncotarget. 2016;7(28):44545–44557.
- Abdelaziz A, Vaishampayan U. Cabozantinib for the treatment of kidney cancer. Expert Rev Anticancer Ther. 2017;17(7):577–584.
- Richeldi L, Cottin V, Flaherty KR, Kolb M, Inoue Y, Raghu G, Taniguchi H, Hansell DM, Nicholson AG, Le Maulf F, et al. Design of the INPULSIS™ trials: two phase 3 trials of nintedanib in patients with idiopathic pulmonary fibrosis. Respir Med. 2014;108(7):1023–1030.
- Tan E-H, Goss GD, Salgia R, Besse B, Gandara DR, Hanna NH, Yang JCH, Thertulien R, Wertheim M, Mazieres J, et al. Phase 2 trial of linifanib (ABT-869) in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2011;6(8):1418–1425.
- Meltzer-Mats E, Babai-Shani G, Pasternak L, Uritsky N, Getter T, Viskind O, Eckel J, Cerasi E, Senderowitz H, Sasson S, et al. Synthesis and mechanism of hypoglycemic activity of benzothiazole derivatives. J Med Chem. 2013;56(13):5335–5350.
- Liu Y, Wang Y, Dong G, Zhang Y, Wu S, Miao Z, Yao J, Zhang W, Sheng C. Novel benzothiazole derivatives with a broad antifungal spectrum: design, synthesis and structure–activity relationships. Med Chem Comm. 2013;4:1551–1561.
- Singh M, Singh SK, Gangwar M, Nath G, Singh SK. Design, synthesis and mode of action of some benzothiazole derivatives bearing an amide moiety as antibacterial agents. RSC Adv. 2014;4:19013–19023.
- Mahesh G, Kumar KA, Reddanna P. Overview on the discovery and development of anti-inflammatory drugs: should the focus be on synthesis or degradation of PGE2? J Inflamm Res. 2021;14:253–263.
- Siddiqui N, Pandeya SN, Khan SA, Stables J, Rana A, Alam M, Arshad MF, Bhat MA. Synthesis and anticonvulsant activity of sulfonamide derivatives-hydrophobic domain. Bioorg Med Chem Lett. 2007;17(1):255–259.
- Rana A, Siddiqui N, Khan S. Benzothiazoles: a new profile of biological activities. Indian J Pharm Sci. 2007;69:10.
- Shi D-F, Bradshaw TD, Wrigley S, McCall CJ, Lelieveld P, Fichtner I, Stevens MF. Antitumor benzothiazoles. 3. Synthesis of 2-(4-aminophenyl) benzothiazoles and evaluation of their activities against breast cancer cell lines in vitro and in vivo. J Med Chem. 1996;39(17):3375–3384.
- Hutchinson I, Chua M-S, Browne HL, Trapani V, Bradshaw TD, Westwell AD, Stevens MF. Antitumor benzothiazoles. 14. Synthesis and in vitro biological properties of fluorinated 2-(4-aminophenyl) benzothiazoles. J Med Chem. 2001;44(9):1446–1455.
- Kok SHL, Gambari R, Chui CH, Yuen MCW, Lin E, Wong RSM, Lau FY, Cheng GYM, Lam WS, Chan SH, et al. Synthesis and anti-cancer activity of benzothiazole containing phthalimide on human carcinoma cell lines. Bioorg Med Chem. 2008;16(7):3626–3631.
- Shi X-H, Wang Z, Xia Y, Ye T-H, Deng M, Xu Y-Z, Wei Y-Q, Yu L-T. Synthesis and biological evaluation of novel benzothiazole-2-thiol derivatives as potential anticancer agents. Molecules. 2012;17(4):3933–3944.
- Leong CO, Suggitt M, Swaine DJ, Bibby MC, Stevens MF, Bradshaw TD. In vitro, in vivo, and in silico analyses of the antitumor activity of 2-(4-amino-3-methylphenyl)-5-fluorobenzothiazoles. Mol Cancer Ther. 2004;3(12):1565–1575.
- Mortimer CG, Wells G, Crochard J-P, Stone EL, Bradshaw TD, Stevens MF, Westwell AD. Antitumor benzothiazoles. 26. 2-(3, 4-Dimethoxyphenyl)-5-fluorobenzothiazole (GW 610, NSC 721648), a simple fluorinated 2-arylbenzothiazole, shows potent and selective inhibitory activity against lung, colon, and breast cancer cell lines. J Med Chem. 2006;49(1):179–185.
- Baffy G. Editorial: hepatocellular carcinoma in type 2 diabetes: more than meets the eye. Off J Am Coll Gastroenterol ACG. 2012;107:53–55.
- Turan-Zitouni G, Özkay Y, Özdemir A, Asim Kaplancıklı Z, Dilek Altıntop M. Synthesis of some benzothiazole based piperazine-dithiocarbamate derivatives and evaluation of their anticancer activities. Lett Drug Des Discov. 2011;8:830–837.
- Altintop MD, Sever B, Özdemir A, Ilgın S, Atlı Ö, Turan-Zitouni G, Kaplancıklı ZA. Synthesis and evaluation of a series of 1, 3, 4-thiadiazole derivatives as potential anticancer agents. Anticancer Agents Med Chem. 2018;18(11):1606–1616.
- Tariq S, Kamboj P, Amir M. Therapeutic advancement of benzothiazole derivatives in the last decennial period. Arch Pharm. 2019;352:1800170.
- Irfan A, Batool F, Zahra Naqvi SA, Islam A, Osman SM, Nocentini A, Alissa SA, Supuran CT. Benzothiazole derivatives as anticancer agents. J Enzyme Inhib Med Chem. 2020;35(1):265–279.
- Pathak N, Rathi E, Kumar N, Kini SG, Rao CM. A review on anticancer potentials of benzothiazole derivatives. Mini Rev Med Chem. 2020;20(1):12–23.
- Sever B, Altıntop MD, Özdemir A, Akalın Çiftçi G, Ellakwa DE, Tateishi H, Radwan MO, Ibrahim MAA, Otsuka M, Fujita M, et al. In vitro and in silico evaluation of anticancer activity of new indole-based 1,3,4-oxadiazoles as EGFR and COX-2 inhibitors. Molecules. 2020;25:5190.
- Haider K, Shrivastava N, Pathak A, Prasad Dewangan R, Yahya S. Shahar Yar, M. Recent advances and SAR study of 2-substituted benzothiazole scaffold based potent chemotherapeutic agents. Results Chem. 2022;4:100258.
- Dhadda S, Raigar AK, Saini K, Manju, Guleria A. Benzothiazoles: from recent advances in green synthesis to anti-cancer potential. Sustain Chem Pharm. 2021;24:100521.
- El-Helby A-GA, Sakr H, Eissa IH, Al-Karmalawy AA, El-Adl K. Benzoxazole/benzothiazole-derived VEGFR-2 inhibitors: design, synthesis, molecular docking, and anticancer evaluations. Arch Pharm. 2019;352:1900178.
- Racané L, Ptiček L, Fajdetić G, Tralić-Kulenović V, Klobučar M, Kraljević Pavelić S, Perić M, Paljetak HČ, Verbanac D, Starčević K. Green synthesis and biological evaluation of 6-substituted-2-(2-hydroxy/methoxy phenyl)benzothiazole derivatives as potential antioxidant, antibacterial and antitumor agents. Bioorg Chem. 2020;95:103537.
- Ammazzalorso A, Carradori S, Amoroso R, Fernández IF. 2-substituted benzothiazoles as antiproliferative agents: novel insights on structure-activity relationships. Eur J Med Chem. 2020;207:112762.
- Reddy VG, Reddy TS, Jadala C, Reddy MS, Sultana F, Akunuri R, Bhargava SK, Wlodkowic D, Srihari P, Kamal A. Pyrazolo-benzothiazole hybrids: synthesis, anticancer properties and evaluation of antiangiogenic activity using in vitro VEGFR-2 kinase and in vivo transgenic zebrafish model. Eur J Med Chem. 2019;182:111609.
- Marzouk AA, Abdel-Aziz SA, Abdelrahman KS, Wanas AS, Gouda AM, Youssif BGM, Abdel-Aziz M. Design and synthesis of new 1,6-dihydropyrimidin-2-thio derivatives targeting VEGFR-2: Molecular docking and antiproliferative evaluation. Bioorg Chem. 2020;102:104090.
- Faraji A, Motahari R, Hasanvand Z, Oghabi Bakhshaiesh T, Toolabi M, Moghimi S, Firoozpour L, Boshagh MA, Rahmani R, Ketabforoosh SHME, et al. Quinazolin-4(3H)-one based agents bearing thiadiazole-urea: synthesis and evaluation of anti-proliferative and antiangiogenic activity. Bioorg Chem. 2021;108:104553.
- El-Shafey HW, Gomaa RM, El-Messery SM, Goda FE. Synthetic approaches, anticancer potential, HSP90 inhibition, multitarget evaluation, molecular modeling and apoptosis mechanistic study of thioquinazolinone skeleton: promising antibreast cancer agent. Bioorg Chem. 2020;101:103987.
- Yang S-K, Kang JS, Oelschlaeger P, Yang K-W. Azolylthioacetamide: a highly promising scaffold for the development of metallo-β-lactamase inhibitors. ACS Med Chem Lett. 2015;6(4):455–460.
- Tilekar K, Upadhyay N, Schweipert M, Hess JD, Macias LH, Mrowka P, Meyer-Almes F-J, Aguilera RJ, Iancu CV, Choe J-Y, et al. Permuted 2, 4-thiazolidinedione (TZD) analogs as GLUT inhibitors and their in-vitro evaluation in leukemic cells. Eur J Pharm Sci. 2020;154:105512.
- Al-Wahaibi LH, Chakraborty K, Al-Shaalan NH, Majeed MYAS, Blacque O, Al-Mutairi AA, El-Emam AA, Percino MJ, Thamotharan S. Quantitative analysis of hydrogen and chalcogen bonds in two pyrimidine-5-carbonitrile derivatives, potential DHFR inhibitors: an integrated crystallographic and theoretical study. RSC Adv. 2020;10(60):36806–36817.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1-2):55–63.
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival: modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89:271–277.
- Abou-Seri SM, Eldehna WM, Ali MM, Abou El Ella DA. 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: synthesis and in vitro biological evaluation. Eur J Med Chem. 2016;107:165–179.
- Thornton TM, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci. 2009;5(1):44–51.
- Kumar CP, Reddy TS, Mainkar PS, Bansal V, Shukla R, Chandrasekhar S, Hügel HM. Synthesis and biological evaluation of 5, 10-dihydro-11H-dibenzo [b, e][1, 4] diazepin-11-one structural derivatives as anti-cancer and apoptosis inducing agents. Eur J Med Chem. 2016;108:674–686.
- ChemOffice. Version 12.0. Cambridge (MA): C.J.C. Cambridge Scientific Computing Inc.
- C.C.G.C. Inc. Molecular operating environment (MOE). Montreal: C.C.G.C. Inc; 2016.
- Böhm H-J. Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J Comput Aided Mol Des. 1998;12(4):309–323.
- Kramer B, Rarey M, Lengauer T. Evaluation of the FLEXX incremental construction algorithm for protein–ligand docking. Proteins Struct Funct Genet. 1999;37:228–241.
- Elimam DM, Elgazar AA, El-Senduny FF, El-Domany RA, Badria FA, Eldehna WM. Natural inspired piperine-based ureas and amides as novel antitumor agents towards breast cancer. J Enzyme Inhib Med Chem. 2022;37(1):39–50.
- Studio, D.J.A.I.S.D., CA, USA. Version 2.5. 2009.
- PreADMET. Prediction of ADM/Tox; 2022; [accessed 2022 June 21]. https://preadmet.bmdrc.kr/.
- Hamdi A, El-Shafey HW, Othman DIA, El-Azab AS, AlSaif NA, Abdel-Aziz AAM. Design, synthesis, antitumor, and VEGFR-2 inhibition activities of novel 4-anilino-2-vinyl-quinazolines: molecular modeling studies. Bioorg Chem. 2022;122:105710.
- Hamdi A, Elhusseiny WM, Othman DI, Haikal A, Bakheit AH, El-Azab AS, Al-Agamy MH, Alaa A-M. Synthesis, antitumor, and apoptosis-inducing activities of novel 5-arylidenethiazolidine-2, 4-dione derivatives: histone deacetylases inhibitory activity and molecular docking study. Eur J Med Chem. 2022;244:114827.
- Selim NM, Elgazar AA, Abdel-Hamid NM, Abu El-Magd MR, Yasri A, El Hefnawy HM, Sobeh MJA. Chrysophanol, physcion, hesperidin and curcumin modulate the gene expression of pro-inflammatory mediators induced by LPS in HepG2: in silico and molecular studies. Antioxidants 2019;8:371.
- Elsbaey M, Ibrahim MAA, Bar FA, Elgazar AA. Chemical constituents from coconut waste and their in silico evaluation as potential antiviral agents against SARS-CoV-2. S Afr J Bot. 2021;141:278–289.
- Elgazar AA, Selim NM, Abdel-Hamid NM, El-Magd MA, El Hefnawy HM. Isolates from Alpinia officinarum Hance attenuate LPS-induced inflammation in HepG2: evidence from in silico and in vitro studies. Phytother Res. 2018;32(7):1273–1288.
- Elgazar AA, Knany HR, Ali MS. Insights on the molecular mechanism of anti-inflammatory effect of formula from Islamic traditional medicine: an in-silico study. J Tradit Complement Med. 2019;9(4):353–363.
- Badria FA, Elgazar AA. Chapter 37 - revealing the molecular mechanism of Olea europaea L. in treatment of cataract. In: Preedy VR, Watson RR, editors. Olives and olive oil in health and disease prevention. 2nd ed. San Diego (CA): Academic Press; 2021. p. 445–456.
- Othman DI, Hamdi A, Abdel-Aziz MM, Elfeky SM. Novel 2-arylthiazolidin-4-one-thiazole hybrids with potent activity against Mycobacterium tuberculosis. Bioorg Chem. 2022;124:105809.
- El-Shafey HW, Gomaa RM, El-Messery SM, Goda FE. Quinazoline based HSP90 inhibitors: synthesis, modeling study and ADME calculations towards breast cancer targeting. Bioorg Med Chem Lett. 2020;30(15):127281.
- Wadapurkar RM, Shilpa MD, Katti AKS, Sulochana MB. In silico drug design for Staphylococcus aureus and development of host-pathogen interaction network. Inform Med Unlocked. 2018;10:58–70.