
Abstract
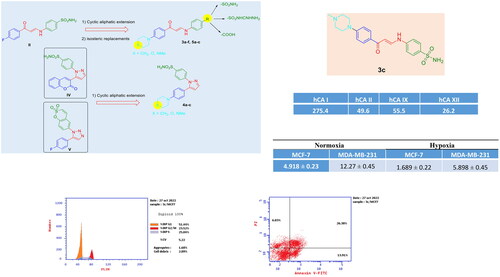
Design and synthesis of three novel series of aryl enaminones (3a–f and 5a–c) and pyrazole (4a-c) linked compounds with sulphonamides, sulfaguanidine, or carboxylic acid functionalities were reported as carbonic anhydrase inhibitors (CAIs) using the “tail approach” strategy in their design to achieve the most variable amino acids in the middle/outer rims of the hCAs active site. The synthesised compounds were assessed in vitro for their inhibitory activity against the following human (h) isoforms, hCA I, II, IX, and XII using stopped-flow CO2 hydrase assay. Enaminone sulphonamide derivatives (3a–c) potently inhibited the target tumour-associated isoforms hCA IX and hCA XII (KIs 26.2–63.7 nM) and hence compounds 3a and 3c were further screened for their in vitro cytotoxic activity against MCF-7 and MDA-MB-231 cancer cell lines under normoxic and hypoxic conditions. Derivative 3c showed comparable potency against both MCF-7 and MDA-MB-231 cancer cell lines under both normoxic ((IC50 = 4.918 and 12.27 µM, respectively) and hypoxic (IC50 = 1.689 and 5.898 µM, respectively) conditions compared to the reference drug doxorubicin under normoxic (IC50 = 3.386 and 4.269 µM, respectively) and hypoxic conditions (IC50 = 1.368 and 2.62 µM, respectively). Cell cycle analysis and Annexin V-FITC and propidium iodide double staining methods were performed to reinforce the assumption that 3c may act as a cytotoxic agent through the induction of apoptosis in MCF-7 cancer cells.
Graphical Abstract
Introduction
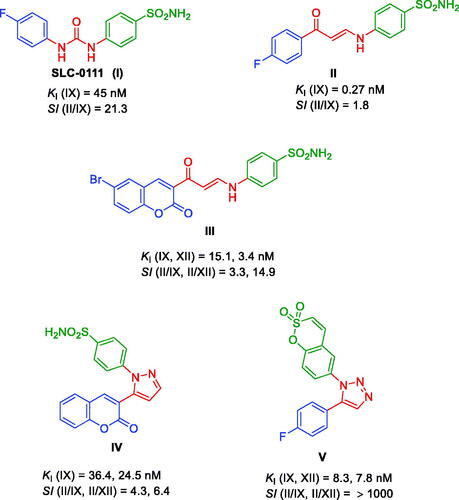
The reversible hydration of carbon dioxide into bicarbonate and a proton is an essential reaction for all living being and it is catalysed by the superfamily of metalloenzymes carbonic anhydrases (CAs, EC 4.2.1.1)Citation1. This pivotal reaction is involved in several physiological processes such as respiration, pH, and homeostasis regulation, CO2 and HCO3– transportation, electrolyte secretion in tissue and organs, biosynthetic reactions (e.g. gluconeogenesis, lipogenesis, and ureagenesis), photosynthesis (e.g. plants and cyanobacteria), bone resorption, calcification, and tumorigenesis. Nowadays, eight genetically unrelated CA families α, β, γ, δ, η, ζ, θ, and ιCitation1–8, were identified and only α-CAs are present in higher vertebrates and humans (h)Citation1. In particular, among the 15 hCAs only 12 are catalytically active, differing for catalytic activity, subcellular/tissue distribution, and physiological roleCitation9. Thus, several studies highlighted that abnormal levels or activity of hCAs are linked to many human diseases such as retinopathies (e.g. glaucoma and retinitis pigmentosa), retinal/cerebral oedema, stroke, altitude sickness, cariogenesis, epilepsy, osteoporosis, neurodegeneration, sterility, obesity, and cancerCitation10–12. Consequently, catalytically hCAs have been considered targets of great importance for the design of modulators, such as inhibitors (CAIs) and activators (CAAs), with biomedical applicationsCitation13. Whereas initially CAIs were used as diuretics, antiglaucoma agents, antiepileptics and for the management of altitude sicknessCitation1, new CAIs generations are being developed for the treatment of cancers, inflammation, obesity, neuropathic pain, infections, and neurodegenerative disordersCitation14–19. Sulphonamides are the most potent CAI chemotype acting as a zinc-binding inhibitor. The “tail approach” is one of the most used strategies in the design of CAIs, which consists in appending a tail of various nature on a CA modulator scaffold (e.g. benzenesulfonamide) to achieve the most variable amino acids in the middle/outer rims of the hCAs active siteCitation20–24. This strategy improves the CAIs selectivity towards specific hCAs isoforms, preventing the onset of side effects due to the inhibition of the other CA isozymes that are not involved in the pathophysiology of the treated disease. An important result of the “tail approach” application is SLC-0111: a ureido-benzenesulfonamide derivative selective against the tumour-associated hCA IX and XII, that displayed promising antiproliferative effects in cancer cells in vitro and in vivo decreasing the tumoural growth, progression and metastases. Currently, SLC-0111 is in phase Ib/II clinical development for the treatment of advanced solid hypoxic tumours in patients and no significant dose-limiting toxicities were encounteredCitation25–27. These extraordinary results led our several research efforts to optimise the SLC-0111 structure based on using different linkers or aryl analogues with the aim to identify more potent and selective inhibitors against the tumour-associated isoform CA IX and XIICitation28,Citation29. Within the region of trials to use flexible linker compounds analogous to SLC-0111, compound II was synthesised before and showed sub-nanomolar activity against hCA IX but with a low selectivity ratio compared to hCA IICitation30. Upon changing the phenyl group to coumarin to study the effect of the combination of both classical and non-classical inhibitors, compound (III) () illustrated a significant nanomolar activity against hCA IX and XII which is more potent than SLC-0111 but unfortunately, the selectivity indices were not promisingCitation31. On the other hand, the part of this flexible linker is rigidified by the use of pyrazole, as in compound IVCitation31 or even triazole, as in compound V ()Citation32,Citation33. Both of them with enhanced nanomolar activity against isoforms IX and XII but the selectivity ratio is in favour of compound V which is attributed to the presence of sulfocoumarin as ZBG.
Figure 1. Different inhibitors of tumour-associated carbonic anhydrase XI including SLC-0111.
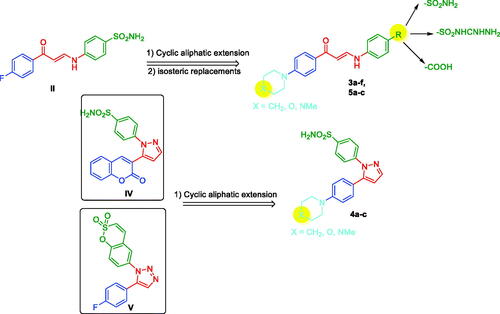
Cyclic secondary amines proved their substantial effect on different molecules considering the effect on both activity and pharmacokinetic characteristicsCitation34,Citation35. Cyclic secondary amines were employed to compound II to study the effect of these substitutions on the original inhibitory effect. These combinations were varied with different ZBGs as sulphonamide, sulfaguanidine, and carboxylic acid moieties as shown in compounds 3a–f, 5a–c in . The concept of using cyclic secondary amines was also applied to compounds with rigid linkers as pyrazole as in compounds 4a–c as in .
Figure 2. The rationale for the design of novel enaminones and pyrazoles as tumour-associated carbonic anhydrase inhibitors.
Materials and methods
Chemistry
General
The data of the devices used in the analytical experiments in the chemistry section were described in the supplementary data. Compounds 1a–cCitation36,Citation37 and 2a–cCitation38–40 were synthesised according to the reported procedures.
General procedure for the preparation of compounds 3a–f
The appropriate enaminone intermediate 2a–c (1 mmol) was added to a hot-stirred solution of either sulphanilamide or sulfaguanidine (1 mmol) in glacial acetic acid (15 ml). The reaction mixture was refluxed for 5 h. The obtained precipitate upon cooling was filtered off, washed with water, dried, and recrystallised from isopropanol to furnish compounds 3a–f.
(E/Z)-4-((3-oxo-3-(4-(piperidin-1-yl) phenyl) prop-1-en-1-yl)amino) benzenesulfonamide (3a)
Dark yellow crystals (yield 50%), m.p. 225–227 °C; IR (KBr, ν cm−1): 3356, 3267 (NH, NH2), 1635 (C═O), 1334 and 1153 (SO2); 1H NMR (DMSO-d6) δ ppm: 1.59 (s, 6H, piperidine H), 3.35 (s, 4H, piperidine H), 6.16, 6.55 (2d, 1H, J = 8.1, 12.6, COCH═CHNH), 6.96 (d, 2H, J = 8.6, Ar-H), 7.21–7.25 (m, 3H, Ar-H and NH2, D2O exchangeable), 7.43 (d, 1H, J = 8.6, Ar-H), 7.73–7.82 (m, 4H, Ar-H), 7.85, 8.07 (d, t, 1H, J = 8.5, 12.6, COCH═CHNH), 10.17, 12.04 (2d, 1H, J = 12.6, 12.2, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ ppm: 24.4, 25.3, 48.4, 95.3, 100.0, 113.7, 115.0, 115.8, 127.4, 127.9, 129.7, 136.9, 137.9, 141.8, 143.3, 143.7, 144.7, 153.8, 186.1, and 189.3; Anal. Calcd. for C20H23N3O3S (385.48): C, 62.32; H, 6.01; N, 10.90; found C, 62.51; H, 6.28; N, 11.14.
(E/Z)-4-((3-(4-morpholinophenyl)-3-oxoprop-1-en-1-yl) amino) benzenesulfonamide (3b)
Yellow crystals (yield 55%), m.p. 218–220 °C; IR (KBr, ν cm−1): 3379, 3251 (NH, NH2), 1635 (C═O), 1320 and 1149 (SO2); 1H NMR (DMSO-d6) δ ppm: 3.28 (t, 4H, J = 4.7, morpholine H), 3.75 (t, 4H, J = 4.5, morpholine H), 6.19, 6.55 (2d, 1H, J = 8.1, 12.6, COCH═CHNH), 7.01 (d, 2H, J = 8.7, Ar-H), 7.21–7.27 (m, 3H, Ar-H and NH2, D2O exchangeable), 7.44 (d, 1H, J = 8.6, Ar-H), 7.73–7.85 (m, 4H, Ar-H), 7.89, 8.09 (d, t, 1H, J = 8.8, 12.7, COCH═CHNH), 10.20, 12.04 (2d, 1H, J = 12.6, 11.9, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ ppm: 47.4, 66.3, 95.2, 99.9, 113.7, 115.1, 115.9, 127.9, 128.7, 129.5, 137.0, 138.1, 142.1, 143.6, 144.68, 153.9, 189.4, and 189.4; Anal. Calcd. for C19H21N3O4S (387.45): C, 58.90; H, 5.46; N, 10.85; found C, 59.21; H, 5.61; N, 11.08.
(E/Z)-4-((3-(4-(4-Methylpiperazin-1-yl)phenyl)-3-oxoprop-1-en-1-yl)amino)benzene sulphonamide (3c)
Yellow crystals (yield 46%), m.p. 248–250 °C; IR (KBr, ν cm−1): 3302, 3155 (NH, NH2), 1597 (C═O), 1319 and 1145 (SO2); 1H NMR (DMSO-d6) δ ppm: 2.23 (s, 3H, CH3), 2.45 (s, 4H, piperazine H), 3.32 (s, 4H, piperazine H), 6.17, 6.55 (2d, 1H, J = 7.7, 12.4, COCH═CHNH), 6.99 (d, 2H, J = 6.2, Ar-H), 7.21–7.25 (m, 3H, Ar-H and NH2, D2O exchangeable), 7.44 (d, 2H, J = 8.0, Ar-H), 7.74–7.88 (m, 3H, Ar-H), 7.87, 8.10 (d, t, 1H, J = 7.9, 12.0, COCH═CHNH), 10.19, 12.04 (2d, 1H, J = 12.0, 12.3, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ ppm: 13.7, 21.5, 23.5, 55.5, 112.6, 121.4, 127.2, 127.6, 134.3, 142.3, 148.8, 172.4; Anal. Calcd. for C20H24N4O3S (400.49): C, 59.98; H, 6.04; N, 13.99; found C, 60.24; H, 6.20; N, 14.17.
(E/Z)-N-Carbamimidoyl-4-((3-oxo-3-(4-(piperidin-1-yl)phenyl)prop-1-en-1-yl)amino) benzenesulfonamide (3d)
Yellow crystals (yield 52%), m.p. 256–258 °C; IR (KBr, ν cm−1): 3444, 3344, 3217 (NH, NH2), 1693 (C═O), 1369 and 1138 (SO2); 1H NMR (DMSO-d6) δ ppm: 1.59 (s, 6H, piperidine H), 3.35 (s, 4H, piperidine H), 6.13 (d, ½H, J = 8.2, COCH═CHNH), 6.51 (s, 1H, NH, D2O exchangeable), 6.53 (t, ½H, J = 12.6, COCH═CHNH), 6.68 (s, 2H, NH2, D2O exchangeable), 6.92–6.99 (m, 2H, Ar-H), 7.18 (d, 1H, J = 8.7, Ar-H), 7.35–7.40 (m, 2H, Ar-H and NH, D2O exchangeable), 7.66–7.70 (m, 2H, Ar-H), 7.76–7.85 (m, 2½ H, Ar-H and COCH═CHNH), 8.06 (t, ½ H, J = 12.6, COCH═CHNH), 10.12, 12.03 (2d, 1H, J = 12.7, 11.9, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ ppm: 21.5, 24.4, 25.3, 48.7, 95.0, 99.7, 113.7, 114.8, 127.8, 128.2, 129.6, 137.6, 138.6, 143.1, 143.5, 144.2, 153.8, 158.5, 172.4, 186.1, and 189.2; Anal. Calcd. for C21H25N5O3S (427.52): C, 59.00; H, 5.89; N, 16.38; found C, 58.79; H, 6.08; N, 16.62.
(E/Z)-N-Carbamimidoyl-4-((3-(4-morpholinophenyl)-3-oxoprop-1-en-1-yl)amino) benzenesulfonamide (3e)
Buff crystals (yield 48%), m.p. 168–170 °C; IR (KBr, ν cm−1): 3444, 3348, 3217 (NH, NH2), 1694 (C═O), 1369 and 1138 (SO2); 1H NMR (DMSO-d6) δ ppm: 3.28 (t, 4H, J = 4.7, morpholine H), 3.75 (t, 4H, J = 4.5, morpholine H), 6.16 (d, ½H, J = 8.1, COCH═CHNH), 6.51 (s, 1H, NH, D2O exchangeable), 6.53 (t, ½H, J = 12.6, COCH═CHNH), 6.67 (s, 2H, NH2, D2O exchangeable), 6.99–7.02 (m, 2H, Ar-H), 7.19 (d, 1H, J = 8.7, Ar-H), 7.36–7.38 (m, 2H, Ar-H and NH, D2O exchangeable), 7.66–7.70 (m, 2H, Ar-H), 7.79–7.90 (m, 2½ H, Ar-H and COCH═CHNH), 8.07 (t, ½ H, J = 12.5, COCH═CHNH), 10.15, 12.03 (2d, 1H, J = 12.8, 12, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ ppm: 47.3, 66.3, 112.7, 113.4, 118.7, 127.0, 127.7, 130.4, 131.2, 139.0, 142.1, 151.8, 158.2, 158.4, and 169.2; Anal. Calcd. for C20H23N5O4S (429.49): C, 55.93; H, 5.40; N, 16.31; found C, 56.15; H, 5.57; N, 16.58.
(E/Z)-N-Carbamimidoyl-4-((3-(4-(4-methylpiperazin-1-yl)phenyl)-3-oxoprop-1-en-1-yl)amino)benzenesulfonamide (3f)
Brown crystals (yield 50%), m.p. 265–267 °C; IR (KBr, ν cm−1): 3564, 3433, 3336 (NH, NH2), 1682 (C═O), 1315 and 1134 (SO2); 1H NMR (DMSO-d6) δ ppm: 2.28 (s, 3H, CH3), 2.51 (s, 4H, piperazine H), 3.34 (s, 4H, piperazine H), 6.15 (d, ½H, J = 7.2, COCH═CHNH), 6.52–6.68 (m, 3½H, COCH═CHNH, NH and NH2, D2O exchangeable), 6.99–7.00 (m, 2H, Ar-H), 7.19 (d, 1H, J = 7.0, Ar-H), 7.37 (m, 2H, Ar-H and NH, D2O exchangeable), 7.68–7.87 (m, 4½ H, Ar-H and COCH═CHNH), 8.07 (t, ½ H, J = 12.5, COCH═CHNH), 10.15, 12.03 (2d, 1H, J = 11.7, 11.6, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ ppm: 24.5, 46.1, 46.9, 54.7, 56.5, 113.5, 118.7, 118.9, 127.0, 139.0, 142.1, 142.9, 154.3, 154.4, 158.4, 169.2, and 169.4; Anal. Calcd. for C21H26N6O3S (442.53): C, 57.00; H, 5.92; N, 18.99; found C, 57.24; H, 6.04; N, 19.21.
General procedure for the preparation of compounds 4a–c
Sulphanilamide hydrazine hydrochloride (0.22 g, 1 mmol) was added to a solution of the corresponding enaminoketone 2a–c (1 mmol) dissolved in glacial acetic acid and the mixture was heated under reflux for 12–24 h. After cooling, the separated solid was filtered and crystallised from chloroform/methanol to afford the pyrazole derivatives 4a–c in 50–60% yield.
4-(5-(4-(Piperidin-1-yl)phenyl)-1H-pyrazol-1-yl)benzenesulfonamide (4a)
Light brown crystals (yield 48%), m.p. 103–105 °C; IR (KBr, ν cm−1): 3421 (NH2), 1315 and 1161 (SO2);1H NMR (DMSO-d6) δ ppm: 1.58–1.67 (m, 6H, piperidine H), 3.26 (s, 4H, piperidine H), 6.61 (s, 1H, pyrazole H), 7.12–7.20 (m, 3H, Ar-H and NH2, D2O exchangeable), 7.45–7.47 (m, 3H, Ar-H), 7.79–7.85 (m, 3H, pyrazole H and Ar-H), 8.58–8.70 (m, 2H, Ar-H); 13C NMR (DMSO-d6) δ ppm: 24.2, 25.2, 48.5, 107.1, 112.4, 122.3, 123.4, 127.1, 127.6, 130.6, 134.0, 140.2, 142.4, and 148.9; Anal. Calcd. for C20H22N4O2S (382.48): C, 62.80; H, 5.80; N, 14.65; found C, 63.06; H, 5.93; N, 14.86.
4-(5-(4-Morpholinophenyl)-1H-pyrazol-1-yl)benzenesulfonamide (4b)
Brown crystals (yield 45%), m.p. 281–283 °C; IR (KBr, ν cm−1): 3394 (NH2), 1327 and 1161 (SO2); 1H NMR (DMSO-d6) δ ppm: 3.14 (t, 4H, J = 4.4, morpholine H), 3.72 (t, 4H, J = 4.4, morpholine H), 6.59 (d, 1H, J = 1.48, pyrazole H), 6.95 (d, 2H, J = 8.6, Ar-H), 7.10 (d, 2H, J = 8.6, Ar-H), 7.45 (d, 4H, J = 8.4, Ar-H and NH2, D2O exchangeable), 7.78 (d, 1H, J = 1.48, pyrazole H), 7.84 (d, 2H, J = 8.5, Ar-H); 13C NMR (DMSO-d6) δ ppm: 48.1, 66.4, 108.4, 115.0, 120.3, 125.4, 127.0, 129.7, 141.4, 142.8, 142.9, 143.5, and 151.1; Anal. Calcd. for C19H20N4O3S (384.45): C, 59.36; H, 5.24; N, 14.57; found C, 59.58; H, 5.31; N, 14.68.
4-(5-(4-(4-Methylpiperazin-1-yl)phenyl)-1H-pyrazol-1-yl)benzenesulfonamide (4c)
Dark brown crystals (yield 51%), m.p. 285–287 °C; IR (KBr, ν cm−1): 3379 (NH2), 1319 and 1157 (SO2); 1H NMR (DMSO-d6) δ ppm: 2.21 (s, 3H, CH3), 2.42 (t, 4H, J = 6.8, piperazine H), 3.31 (t, 4H, J = 4.7, piperazine H), 5.83 (br s, 1H, pyrazole-H), 6.59 (d, 1H, J = 8.5, Ar H), 6.90–6.98 (m, 3H, pyrazole-H and Ar H), 7.25 (s, 2H, NH2, D2O exchangeable), 7.45 (d, 2H, J = 8.5, Ar-H), 7.73–7.80 (m, 3H, Ar-H), 13C NMR (DMSO-d6) δ ppm: 24.5, 26.5, 46.1, 46.9, 54.7, 112.8, 113.5, 118.8, 127.0, 127.8, 130.5, 138.4, 142.7, 152.4, 154.3, and 169.4; Anal. Calcd. for C20H23N5O2S (397.49): C, 60.43; H, 5.83; N, 17.62; found C, 60.55; H, 5.97; N, 17.89.
General procedure for the preparation of compounds 5a–c
The para amino benzoic acid (1 mmol) was added to a solution of the enaminone derivatives 2a–c (1 mmol) dissolved in glacial acetic acid (10 ml) then the reaction mixture was heated under reflux for 6 h. After cooling, the precipitate was filtered off, washed with water, dried and recrystallised from isopropanol.
(E/Z)-4-((3-Oxo-3-(4-(piperidin-1-yl)phenyl)prop-1-en-1-yl)amino)benzoic acid (5a)
Yellow crystals (yield 44%), m.p. 270–272 °C; IR (KBr, ν cm−1): 3465 (NH), 2665–2542 (OH), 1678, 1635 (2C═O); 1H NMR (DMSO-d6) δ ppm: 1.59 (s, 6H, piperidine H), 3.33 (s, 4H, piperidine H), 6.16, 6.55 (2d, 1H, J = 8.2, 12.6, COCH═CHNH), 6.94–6.98 (m, 2H, Ar-H), 7.18 (d, 1H, J = 8.6, Ar-H), 7.35 (d, 1H, J = 8.7, Ar-H), 7.76 (d, 1H, J = 8, Ar-H), 7.81–7.90 (m, 3½ H, Ar-H and COCH═CHNH), 8.07 (t, ½H, J = 12.6, COCH═CHNH), 10.17, 12.06 (2d, 1H, J = 12.6, 11.9, NH, D2O exchangeable), 12.61 (s, 1H, OH, D2O exchangeable); 13C NMR (DMSO-d6) δ ppm: 24.4, 25.3, 48.3, 95.3, 100.0, 113.7, 114.8, 115.6, 123.7, 124.8, 127.4, 128.1, 129.7, 131.6, 141.7, 143.2, 144.7, 145.8, 153.8, 167.3, and 189.3; Anal. Calcd. for C21H22N2O3 (350.41): C, 71.98; H, 6.33; N, 7.99; found C, 71.79; H, 6.45; N, 8.21.
(E/Z)-4-((3-(4-Morpholinophenyl)-3-oxoprop-1-en-1-yl)amino)benzoic acid (5b)
Yellow powder (yield 48%), m.p. >300 °C; IR (KBr, ν cm−1): 3441 (NH), 2646–2549 (OH), 1697, 1643 (2C═O), 1369 and 1138 (SO2); 1H NMR (DMSO-d6) δ ppm: 3.27–3.29 (m, 4H, morpholine H), 3.75 (t, 4H, J = 4.6, morpholine H), 6.18, 6.57 (2d, 1H, J = 8.2, 12.6, COCH═CHNH), 6.99 (dd, 2H, J = 9.0, 3.2, Ar-H), 7.19 (d, 1H, J = 8.7, Ar-H), 7.37 (d, 1H, J = 8.7, Ar-H), 7.79 (d, J = 8.9, 1H, Ar-H), 7.85–7.91 (m, 3½H, Ar-H and COCH═CHNH), 8.09 (t, ½H, J = 12.6, COCH═CHNH), 10.12, 12.05 (2d, 1H, J = 12.7, 11.9, NH, D2O exchangeable), 12.33 (s, 1H, OH, D2O exchangeable); 13C NMR (DMSO-d6) δ ppm: 53.5, 66.3, 113.7, 114.9, 115.7, 129.5, 131.6, 131.6, 136.4, 143.7, 144.7, 153.8, 172.6, and 189.6; Anal. Calcd. for C20H20N2O4 (352.38): C, 68.17; H, 5.72; N, 7.95; found C, 67.98; H, 5.85; N, 8.12.
(E/Z)-4-((3-(4-(4-Methylpiperazin-1-yl)phenyl)-3-oxoprop-1-en-1-yl)amino)benzoic acid (5c)
Buff crystals (yield 50%), m.p. 260–262 °C; IR (KBr, ν cm−1): 3425 (NH), 2669–2453 (OH), 1678, 1643 (2C═O); 1H NMR (DMSO-d6) δ ppm: 2.33 (s, 3H, CH3), 2.34 (s, 4H, piperazine H), 3.33 (s, 4H, piperazine H), 6.18, 6.55 (2d, 1H, J = 8.1, 12.6, COCH═CHNH), 7.02 (dd, 1H, J = 8.8, 2.4, Ar-H), 7.18–7.25 (m, 2H, Ar-H), 7.37 (d, 1H, J = 8.6, Ar-H), 7.79–7.98 (m, 4½ H, Ar-H and COCH═CHNH), 8.19 (t, ½H, J = 12.6, COCH═CHNH), 10.21, 12.05 (2d, 1H, J = 12.68, 12.0, NH, D2O exchangeable), 12.5 (s, 1H, OH, D2O exchangeable); 13C NMR (DMSO-d6) δ ppm: 46.1, 46.9, 54.7, 96.4, 113.0, 118.6, 119.6, 128.4, 130.8, 131.6, 137.4, 143.9, 148.5, 170.8, and 188.4; Anal. Calcd. for C21H23N3O3 (365.43): C, 69.02; H, 6.34; N, 11.50; found C, 68.94; H, 6.51; N, 11.74.
Biological evaluation
CA inhibitory assay
For CA inhibitory assay, refer to Supplementary data.
Anti-proliferative activity towards hormonal breast MCF-7 and non-hormonal breast MDA-MB-231 cancer cell lines
The MTT assay was used to determine the IC50 of the substances that were examined. All other details were provided in the supplementary data.
Cell cycle analysis
To predict the effect of the enaminone-sulphonamide 3c on cell cycle progression in MCF-7, cell cycle analysis assay was accomplished, and the experimental assay was discussed in the supplementary data.
Apoptotic assay
Enaminone-sulphonamide 3c was assayed using Annexin V-FITC/PI apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ) with propidium iodide double staining method, more details in the supplementary data.
Results and discussion
Chemistry
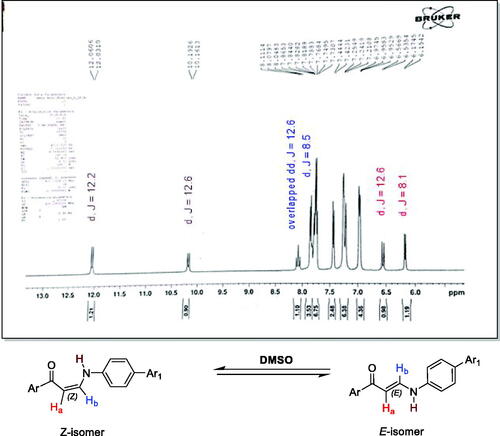
The synthetic pathways used to synthesise the new target derivatives are represented in Scheme 1. The starting point refluxed the substituted acetophenones 1a–c with the modified Vilsmeier–Haack reagent; dimethylformamide dimethyl acetal (DMF-DMA) in xylene to obtain the reported enaminoketones 2a–cCitation38–40. Reaction of compounds 2a–c with p-aminobenzenesulfonamides namely sulphanilamide and sulfaguanidine in glacial acetic acid furnished the desired sulphonamide compounds 3a–fCitation30,Citation41. Their IR spectra revealed the appearance of the NH absorption bands of the amino benzene sulphonamide moiety in the region 3564–3155 cm−1 besides prominent two SO2 bands at 1369–1315 and 1153–1134 cm−1. 1H NMR spectra of the 4-substituted enaminones 3a–f confirmed their presence as Z/E geometric isomers around their olefinic bond CHa═CHbCitation30,Citation41. The up-field doublet signal of olefinic hydrogen Z-Ha appeared at δ 6.16–6.19 ppm with JZ-Ha=Hb = 7.2–8.6 Hz whereas the down-field E-Ha appeared at δ 6.55 ppm with JE-Ha=Hb = 12.4 and 12.6 Hz. Another characteristic feature was the presence of two sets of doublet signals assigned to the NH proton at δ 10.12–10.20 and 12.03–12.04 ppm with J values 11.7–12.8 and 11.6–12.3 Hz, respectively ().
Scheme 1. Reagents and reaction conditions: (i) DMF-DMA, xylene reflux 8h; (ii) sulphanilamide or sulfaguanidine/gl. acetic acid, stirring overnight; (iii) sulphanilamide hydrazine HCl/gl. acetic, reflux; (iv) 4-aminobenzoic acid, gl. acetic acid, stirring overnight.
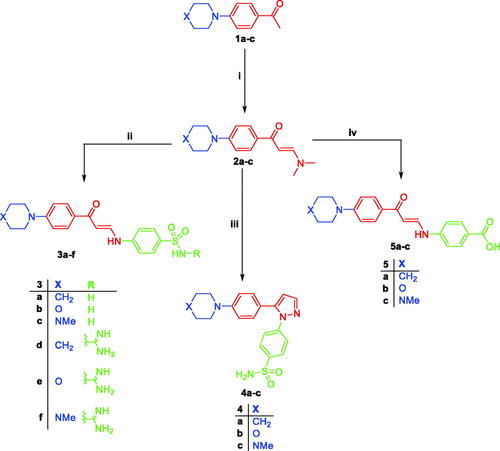
Figure 3. 1H NMR of compound 3a which showed the existing of Z/E geometric isomers in DMSO as represented example for 3a–f and 5a–c.
On the other hand, the obtained enaminoketones 2a–c were reacted with 4-sulphanilamide hydrazine hydrochloride via an amine exchange heterocyclisation reactionCitation42,Citation43 to afford exclusively the diarylpyrazoles 4a–c. These compounds revealed new absorption bands corresponding to the NH and SO2 groups at 3424–3379 and 1327–1315, 1161–1157 cm−1, respectively, along with the disappearance of the C═O group characterising the parent enaminones 2a–c. Interestingly, 1H NMR of these pyrazole derivatives demonstrated two signals at δ 5.83–6.61 and 6.90–7.79 ppm, respectively, corresponding to pyrazole protons.
Finally, the reaction of enaminoketones 2a–c with 4-aminobenzoic acid yielded the final compounds 5a–c. IR spectra of 5a–c exhibited the NH band at 3465–3425 cm−1 in addition to the OH broadband at 2669–2453 cm−1 and C═O band at 1635–1678 cm−1. 1H NMR of the derivatives 5a–c exhibited the Z/E geometric isomers around the olefinic bond CHa═CHb with two sets of up-field doublets of Z-Ha proton at δ 6.16–6.18 ppm and E-Hb δ 6.55–6.75 ppm with J = 8.1–8.2 and 12.6 Hz, respectively, along with two exchangeable doublets for the NH proton at δ 10.12–10.21 and 12.05–12.06 ppm with J = 12.6–12.7 and 11.9–12.05 Hz.
The additional signal for the OH group in all these series was remarked at 12.16–13.80 ppm. All the target compounds 3a–f, 4a–c, and 5a–c demonstrated the expected aliphatic signals assigned for the piperidine, morpholine, and methyl piperazine rings. 13C NMR of all the synthesised compounds was in full agreement with their predicted carbon skeleton.
Biological evaluation
Carbonic anhydrase inhibition
Acetazolamide (AAZ) was used as a standard inhibitor for the measurement of the CA inhibitory activities of sulphonamide derivatives 3a–3f, 4a–4c, and carboxylic acid derivatives 5a–5c against the isoforms hCA I, and II (cytosolic) and hCA IX and XII (transmembrane, tumour-associated isoform) using a stopped-flow CO2 hydrase assayCitation44 ().
Table 1. Inhibition data of human CA isoforms I, II, IX, and XII for the target compounds (3a–f, 4a–c, and 5a–c), using (AAZ) as a standard drug.
The ubiquitous off-target hCA I was moderately inhibited by the enaminone-sulphonamide analogues 3a–3c with inhibition constants between 211.2 and 289.1 nM while the enaminone-sulfaguanidine analogues 3d–3f possessed slightly inferior inhibitory activities with inhibition constants between 643.0 and 822.3. The enaminone-benzene carboxylic acid derivatives 5a–5c failed to produce detectable inhibition properties. It is noteworthy, the presence of sulphonamide functionality is optimal for activity while substitution with COOH completely abolished activity. On the other hand, the substitution of the heterocycloalkyl moiety was detrimental to the activity of pyrazole-sulphonamide analogues 4a–4c probably causing unfavourable steric clashes with the active site.
The ubiquitous and physiologically predominant isoform hCA II was significantly inhibited by the enaminone-sulphonamide analogues 3a–3c with inhibition constants between 49.6 and 65.3 nM. The replacement of sulphonamide by sulfaguanidine group as in analogues 3d–3f, significantly reduced the activity with inhibition constants between 607.9 and 822.3 nM while replacement with COOH as in analogues 5a–5c completely abolished the activity. The heterocycloalkyl substituents in compounds 4a–4c exhibited moderate inhibitory activity with inhibition constants between 586.7 and 689.2 nM probably due to unfavourable steric clashes.
The target tumour-associated isoforms hCA IX and hCA XII are potently inhibited by the enamine-sulphonamide derivatives 3a–3c (KIs 26.2–63.7 nM). On the other hand, the guanidino group of 3d–3f decreased the activity (102.6–149.2 nM) probably due to less efficient zinc binding in the active sites of hCA IX and hCA XII. The COOH substituted derivatives 5a–5c were the least potent inhibitors (12 090–27 460 nM) probably because of the lack of zinc binding to the active site. Heterocycloalkyl substituted 4a–4c gave moderate inhibitory effects.
The selectivity index (SI) () shows that, while actually all compounds 3a–f are selective CAIs for the tumour-associated isoforms over hCA I (SI in the range 3.32–6.87 for hCA IX and 4.24–10.51 for hCA XII), significant selectivity for the target CAs over hCA II exists within this series 3a–f ranging from 0.86 to 1.55 for hCA IX and 1.21 to 1.89 for hCA XII, which indicates that these targets could represent interesting candidates for further anti-tumour assessments.
Table 2. Selectivity ratios for the inhibition of hCA IX and XII over hCA I and II for compounds (3a–f, 4a–c, and 5a–c) and acetazolamide.
Anti-proliferative activity towards hormonal breast MCF-7 and non-hormonal breast MDA-MB-231 cancer cell lines
The in vitro cytotoxic IC50 assessment against two hormonal and non-hormonal breast cancer cell lines (MCF-7 and MDA-MB-231) was performed for the best two derivatives 3a and 3c, in terms of the CA inhibition assay under both normoxic and hypoxic conditions. Derivative 3c showed comparable potency against both MCF-7 and MDA-MB-231 cancer cell lines under both normoxic (IC50 = 4.918 and 12.27 µM, respectively) and hypoxic (IC50 = 1.689 and 5.898 µM, respectively) conditions compared to the reference drug doxorubicin under normoxic (IC50 = 3.386 and 4.269 µM, respectively) and hypoxic conditions (IC50 = 1.368 and 2.62 µM, respectively). Compound 3a, on the other hand, exhibited weaker inhibitory activity against both MCF-7 and MDA-MB-231 cancer cell lines under both normoxic (IC50 = 21.09 and 35.04 µM, respectively), and hypoxic conditions (IC50 = 16.73 and 26.68 µM, respectively) (). Therefore, it was found that the MCF-7 cancer cell line was the most affected cell to compound 3c under both normoxic and hypoxic conditions.
Table 3. In vitro anti-proliferative activity of 3a and 3c towards breast MCF-7 and MDA-MB-231 cancer cell lines.
Cell cycle analysis
For more investigation, cell cycle analysis was achieved for MCF-7 cells treated with 3c derivative at IC50 concentration using untreated MCF-7 cells as a negative control. It shows slight decrease in G0–G1 and S phases (51.44 and 25.04%, respectively) compared to the control cells (56.49 and 28.61%, respectively) whereas it increase G2/M phase, 23.52% compared to the control cells, 14.9% ( and ).
Figure 4. The cell cycle of treated MCF-7 cells with 3c.
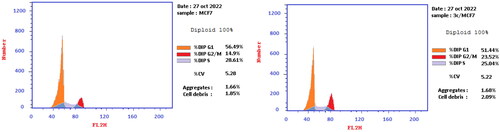
Table 4. Cell cycle analysis of the treated MCF-7 cells with 3c.
Annexin V-FITC apoptosis assay
Annexin V-FITC and propidium iodide double staining methods were performed to reinforce the assumption that 3c may act as a cytotoxic agent through apoptotic induction in MCF-7 cancer cells. There was a significant increase in both early and late population percentages of 3c treated MCF-7 cancer cells (13.91% and 26.38%, respectively) compared to the negative control MCF-7 cells (0.34% and 0.19%, respectively). This result supports the proposed 3c antiproliferative mechanism of action as an apoptotic inducer ( and ).
Figure 5. Effect of compound 3c on the percentage of annexin V-FITC-positive staining in MCF-7 cells.
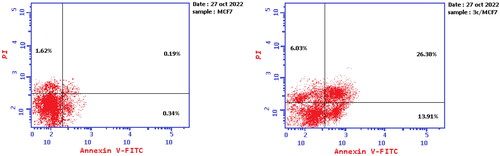
Table 5. Apoptotic cells sub-population percentage in treated MCF-7 cells with 3c.
Conclusions
Novel series of aryl enaminones (3a–f and 5a–c) and pyrazole (4a–c) linked compounds with sulphonamides, sulfaguanidine, or carboxylic acid functionalities as human carbonic anhydrases inhibitors (hCAIs) were designed, synthesised, and screened for their inhibitory activity against the isoforms hCA I, and II (cytosolic) and hCA IX and XII (transmembrane, tumour-associated isoform) using a stopped-flow CO2 hydrase assay. The target tumour-associated isoforms hCA IX and hCA XII are potently inhibited by the enamine-sulphonamide derivatives 3a–c (KIs 26.2–63.7 nM). Moreover, the most selective hCA IX and XII inhibitors 3a and 3c were assessed for their potential anticancer against (MCF-7 and MDA-MB-231) cancer cell lines. Derivative 3c showed comparable potency against both MCF-7 and MDA-MB-231 cancer cell lines under both normoxic (IC50 = 4.918 and 12.27 µM, respectively) and hypoxic (IC50 = 1.689 and 5.898 µM, respectively) conditions. Therefore, compound 3c was additionally subjected to cell cycle analysis and apoptotic assay at IC50 concentration using untreated MCF-7 cells as a negative control. It shows slight decrease in G0–G1 and S phases (51.44 and 25.04%, respectively) compared to the control cells (56.49 and 28.61%, respectively), while, it increased G2/M phase, 23.52% compared to the control cells, 14.9%. Whereas, there were significant increases in both early and late apoptotic percentages of 3c treated MCF-7 cancer cells (13.91% and 26.38%, respectively) compared to the negative control MCF-7 cells (0.34% and 0.19%, respectively), which supports the proposed 3c antiproliferative mechanism of action as an apoptotic inducer.
Supplemental Material
Download PDF (1.2 MB)Disclosure statement
CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7(2):168–181.
- Ferry JF. The gamma class of carbonic anhydrases. Biochim Biophys Acta. 2010;1804(2):374–381.
- Kikutani S, Nakajima K, Nagasato C, Tsuji Y, Miyatake A, Matsuda Y. Thylakoid luminal θ-carbonic anhydrase critical for growth and photosynthesis in the marine diatom Phaeodactylum tricornutum. Proc Natl Acad Sci U S A. 2016;113(35):9828–9833.
- Del Prete S, Vullo D, Fisher GM, Andrews KT, Poulsen SA, Capasso C, Supuran CT. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum—the η-carbonic anhydrases. Bioorg Med Chem Lett. 2014;24(18):4389–4396.
- Alterio V, Langella E, Viparelli F, Vullo D, Ascione G, Dathan NA, Morel FM, Supuran CT, De Simone G, Monti SM. Structural and inhibition insights into carbonic anhydrase CDCA1 from the marine diatom Thalassiosira weissflogii. Biochimie. 2012;94(5):1232–1241.
- Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin Ther Pat. 2018;28(10):745–754.
- Jensen EL, Clement R, Kosta A, Maberly SC, Gontero B. A new widespread subclass of carbonic anhydrase in marine phytoplankton. ISME J. 2019;13(8):2094–2106.
- Del Prete S, Nocentini A, Supuran CT, Capasso C. Bacterial ι-carbonic anhydrase: a new active class of carbonic anhydrase identified in the genome of the Gram-negative bacterium Burkholderia territorii. J Enzyme Inhib Med Chem. 2020;35(1):1060–1068.
- Alterio V, Di Fiore A, D'Ambrosio K, Supuran CT, De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev. 2012;112(8):4421–4468.
- De Simone G, Scozzafava A, Supuran CT. Which carbonic anhydrases are targeted by the antiepileptic sulfonamides and sulfamates? Chem Biol Drug Des. 2009;74(3):317–321.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J. 2016;473(14):2023–2032.
- Nocentini A, Donald WA, Supuran CT. Chapter 8 - Human carbonic anhydrases: tissue distribution, physiological role, and druggability. In: Supuran CT, Nocentini A, editors. Carbonic anhydrases. Academic Press; 2019. p. 151–185.
- Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov. 2019;14(11):1175–1197.
- Supuran CT, Alterio V, Di Fiore A, D' Ambrosio K, Carta F, Monti SM, De Simone G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev. 2018;38(6):1799–1836.
- Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008–2018). Expert Opin Ther Pat. 2018;28(10):729–740.
- Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother. 2016;16(8):961–968.
- De Simone G, Di Fiore A, Supuran CT. Are carbonic anhydrase inhibitors suitable for obtaining antiobesity drugs? Curr Pharm Des. 2008;14(7):655–660.
- Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets. 2015;19(12):1689–1704.
- Scozzafava A, Menabuoni L, Mincione F, Briganti F, Mincione G, Supuran CT. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring? J Med Chem. 1999;42(14):2641–2650.
- Nocentini A, Ferraroni M, Carta F, Ceruso M, Gratteri P, Lanzi C, Masini E, Supuran CT. Benzenesulfonamides incorporating flexible triazole moieties are highly effective carbonic anhydrase inhibitors: synthesis and kinetic, crystallographic, computational, and intraocular pressure lowering investigations. J Med Chem. 2016;59(23):10692–10704.
- Wilkinson BL, Bornaghi LF, Houston TA, Innocenti A, Supuran CT, Poulsen S-A. A novel class of carbonic anhydrase inhibitors: glycoconjugate benzene sulfonamides prepared by “click-tailing”. J Med Chem. 2006;49(22):6539–6548.
- Pacchiano F, Carta F, McDonald PC, Lou Y, Vullo D, Scozzafava A, Dedhar S, Supuran CT. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem. 2011;54(6):1896–1902.
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov. 2013;8(7):793–810.
- Supuran CT. Drug interaction considerations in the therapeutic use of carbonic anhydrase inhibitors. Expert Opin Drug Metab Toxicol. 2016;12(4):423–431.
- Eloranta K, Pihlajoki M, Liljeström E, Nousiainen R, Soini T, Lohi J, Cairo S, Wilson DB, Parkkila S, Heikinheimo M. SLC-0111, an inhibitor of carbonic anhydrase IX, attenuates hepatoblastoma cell viability and migration. Front Oncol. 2023;13:1118268.
- McDonald PC, Chia S, Bedard PL, Chu Q, Lyle M, Tang L, Singh M, Zhang Z, Supuran CT, Renouf DJ, et al. A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am J Clin Oncol. 2020;43(7):484–490.
- Peppicelli S, Andreucci E, Ruzzolini J, Bianchini F, Nediani C, Supuran CT, Calorini L. The carbonic anhydrase IX inhibitor SLC-0111 as emerging agent against the mesenchymal stem cell-derived pro-survival effects on melanoma cells. J Enzyme Inhib Med Chem. 2020;35(1):1185–1193.
- Allam HA, Fahim SH, Abo-Ashour MF, Nocentini A, Elbakry ME, Abdelrahman MA, Eldehna WM, Ibrahim HS, Supuran CT. Application of hydrazino and hydrazido linkers to connect benzenesulfonamides with hydrophilic/phobic tails for targeting the middle region of human carbonic anhydrases active site: selective inhibitors of hCA IX. Eur J Med Chem. 2019;179:547–556.
- Abo-Ashour MF, Eldehna WM, Nocentini A, Ibrahim HS, Bua S, Abdel-Aziz HA, Abou-Seri SM, Supuran CT. Novel synthesized SLC-0111 thiazole and thiadiazole analogues: determination of their carbonic anhydrase inhibitory activity and molecular modeling studies. Bioorg Chem. 2019;87:794–802.
- Eldehna WM, Abo-Ashour MF, Berrino E, Vullo D, Ghabbour HA, Al-Rashood ST, Hassan GS, Alkahtani HM, Almehizia AA, Alharbi A, et al. SLC-0111 enaminone analogs, 3/4-(3-aryl-3-oxopropenyl) aminobenzenesulfonamides, as novel selective subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform IX. Bioorg Chem. 2019;83:549–558.
- Ibrahim HS, Abdelrahman MA, Nocentini A, Bua S, Abdel-Aziz HA, Supuran CT, Abou-Seri SM, Eldehna WM. Insights into the effect of elaborating coumarin-based aryl enaminones with sulfonamide or carboxylic acid functionality on carbonic anhydrase inhibitory potency and selectivity. Bioorg Chem. 2022;126:105888.
- Grandane A, Tanc M, Zalubovskis R, Supuran CT. 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem Lett. 2014;24(5):1256–1260.
- Krasavin M, Žalubovskis R, Grandāne A, Domračeva I, Zhmurov P, Supuran CT. Sulfocoumarins as dual inhibitors of human carbonic anhydrase isoforms IX/XII and of human thioredoxin reductase. J Enzyme Inhib Med Chem. 2020;35(1):506–510.
- Bolleddula J, DeMent K, Driscoll JP, Worboys P, Brassil PJ, Bourdet DL. Biotransformation and bioactivation reactions of alicyclic amines in drug molecules. Drug Metab Rev. 2014;46(3):379–419.
- Stepan AF, Mascitti V, Beaumont K, Kalgutkar AS. Metabolism-guided drug design. Med Chem Commun. 2013;4(4):631–652.
- Magdolen P, Mečiarová M, Toma Š. Ultrasound effect on the synthesis of 4-alkyl-(aryl)aminobenzaldehydes. Tetrahedron. 2001;57(22):4781–4785.
- Ibrahim HS, Albakri ME, Mahmoud WR, Allam HA, Reda AM, Abdel-Aziz HA. Synthesis and biological evaluation of some novel thiobenzimidazole derivatives as anti-renal cancer agents through inhibition of c-MET kinase. Bioorg Chem. 2019;85:337–348.
- Ahmad Bhat M, Al-Omar MA, Naglah AM. Synthesis and in vivo anti-ulcer evaluation of some novel piperidine linked dihydropyrimidinone derivatives. J Enzyme Inhib Med Chem. 2018;33(1):978–988.
- Bhat MA, Al-Omar MA, Ghabbour HA, Naglah AM. A one-pot biginelli synthesis and characterization of novel dihydropyrimidinone derivatives containing piperazine/morpholine moiety. Molecules. 2018;23(7):1559–1572.
- Bingham AH, Davenport RJ, Fosbeary R, Gowers L, Knight RL, Lowe C, Owen DA, Parry DM, Pitt WR. Synthesis and structure–activity relationship of aminopyrimidine IKK2 inhibitors. Bioorg Med Chem Lett. 2008;18(12):3622–3627.
- Alkhaldi AAM, Al-Sanea MM, Nocentini A, Eldehna WM, Elsayed ZM, Bonardi A, Abo-Ashour MF, El-Damasy AK, Abdel-Maksoud MS, Al-Warhi T, et al. 3-Methylthiazolo[3,2-a]benzimidazole-benzenesulfonamide conjugates as novel carbonic anhydrase inhibitors endowed with anticancer activity: design, synthesis, biological and molecular modeling studies. Eur J Med Chem. 2020;207:112745–112757.
- Hernández S, Moreno I, SanMartin R, Gómez G, Herrero MT, Domínguez E. Toward safer processes for C–C biaryl bond construction: catalytic direct C–H arylation and tin-free radical coupling in the synthesis of pyrazolophenanthridines. J Org Chem. 2010;75(2):434–441.
- Olivera R, SanMartin R, Domínguez E. A combination of tandem amine-exchange/heterocyclization and biaryl coupling reactions for the straightforward preparation of phenanthro[9,10-d]pyrazoles. J Org Chem. 2000;65(21):7010–7019.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase: I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.