
Abstract
Phenethyl-based edelfosine-analogs with saturated, monounsaturated, or polyunsaturated alkoxy substituents on phenyl ring were designed as novel antitumor lipids modulating p38 MAPK. Evaluation of the synthesised compounds against nine panels of diverse cancer cells presented saturated and monounsaturated alkoxy-substituted derivatives as the most active than other derivatives. In addition, ortho-substituted compounds were more active than meta- or ortho-substituted compounds. They were potential anticancer agents against blood, lung, colon, CNS, ovary, renal, and prostate cancers but not against skin nor breast cancers. Compounds, 1b and 1a emerged as the most potential anticancer agents. Assessment of compound 1b impact on p38 MAPK and AKT confirmed it as an inhibitor of p38 MAPK but not AKT. In silico study suggested compounds 1b and 1a as possible binders to the lipid binding pocket of p38 MAPK. Overall, compounds 1b and 1a as novel broad spectrum antitumor lipids modulating activity of p38 MAPK for further development.
Graphical abstract
Introduction
Despite the recent advancement in diagnosis and treatment, cancer diseases remain imposing health problems. Significant high fatality rates because of cancer diseases are still occurring and/or anticipated amongst populations of both developed and developing countries. Being a multifactorial, heterogeneous group of diseases and, in addition, the evolvement of resistance render cancer treatment more difficult and complex which necessitate searching for novel therapeutic agents.
Although lipids play crucial roles within cells, lipid-based therapeutics received relatively little attention in comparison with classical small molecules or peptide-based therapeutics which represent the principal focus of most of conducted drug discovery and development programs. In fact, lipids are not only major structural components of biological systems (e.g., glycerophospholipids in the cytoplasmic and cellular organelles’ membranes), but several lipids; such as endocannabinoids, eicosanoids, prostanoids, sphingomyelin, ceramides, are known as endogenous bioactive molecules triggering and/or modulating diverse biological functions and signalling pathways within cellsCitation1–4. Development of lipid analogs might offer new opportunities to modulate such lipid-mediated functions and signalsCitation3–6.
Targeted chemotherapeutics modulating oncogenic signalling pathways such as receptor-tyrosine kinase/rat sarcoma (RAS)/mitogen-activated protein kinase (MAPK) signalling pathway has been found to be safer and more promising in comparison with conventional anticancer therapeutics. Considering MAPK, it refers to multiple kinases amongst is p38 MAPK. Accumulated evidences indicate that p38 MAPK plays key roles at different levels in cancer diseases such as blood, lung, colon, breast, prostate, bladder and renal cancersCitation7. It can directly phosphorylate and activate multiple transcription factors involved in gene expression and, thus, promoting survival and proliferation of cancer cellsCitation8. Besides, p38 MAPK was found to contribute to epithelial-to-mesenchymal transition, migration and invasion of tumour resulting in tumour metastasisCitation7,Citation8. Moreover, roles of p38 MAPK in chemotherapy resistance were reported and co-administration of p38 MAPK inhibitors and chemotherapeutic agents was found to overcome chemotherapy resistanceCitation8. In addition to the roles of p38 MAPK in primary and metastatic cancer cells, its roles were found to extend to cancer stem cells and p38 MAPK inhibitors repress stemness factors expressionCitation9. Considering the low side effects for systemic p38 MAPK inhibition coupled with the multiple roles of p38 MAPK in cancer, it might be desirable to develop anticancer p38 MAPK inhibitors.
As the ATP-binding pocket exist in all kinases, selectivity is a major issue in the development of kinase inhibitors that bind to this pocket. Consequently, developing type IV kinase inhibitors that target non-ATP allosteric pockets, which are unique structural features outside the ATP-binding site, would offer better selectivityCitation10–13. Recently, a hydrophobic non-ATP allosteric pocket in MAPKs sandwiched between the C-lobe and the MAPK insert domain was discovered to bind and accommodate the glycolipid 1-O-n-octyl-β-D-glucopyranosideCitation14–18. This pocket might offer an opportunity to develop novel inhibitors of MAPKs. Interestingly, small molecule inhibitors of p38 MAPK targeting this pocket have been reportedCitation18. Moreover, p38 MAPK inhibitors targeting this allosteric lipid binding pocket were recently reported to show antiproliferative activity in cutaneous T-cell lymphomaCitation19. Accordingly, we tentatively anticipated that developing lipid-based allosteric inhibitors of P38 MAPK might be a new promising approach for access novel antitumor lipids (ATL) for cancer treatment. Herein, we present our interesting results.
Results and discussion
Design rational
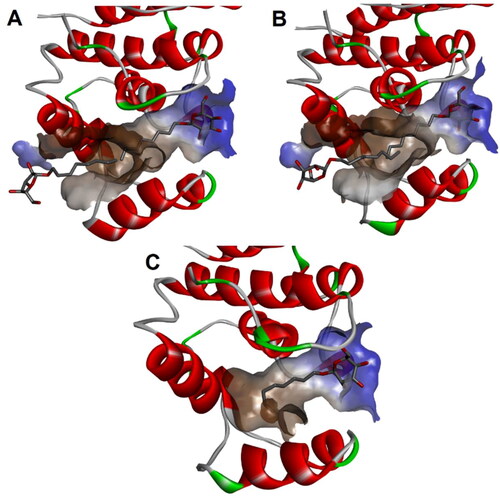
Several crystal structures of p38 MAPK have been co-crystallized with 1-O-n-octyl-β-D-glucopyranoside bound within lipid-binding pocket and have been deposited in protein databank. Exploration of these structures reveals an interesting plasticity of this pocket. Thus, this pocket might accommodate a channel-like open configuration harbouring two 1-O-n-octyl-β-D-glucopyranoside molecules (p38 MAPK crystal structures with PDB codes 2npg and 2fst; ). It might also show a closed-end pocket conformation harbouring only one 1-O-n-octyl-β-D-glucopyranoside molecule (p38 MAPK crystal structure with PDB code 1zyj; ).
Figure 1. Lipid binding pocket at the MAPK insertion domain from different crystal structures of p38 MAPK: (A) Crystal structure of p38 MAPK (PDB: 2npg); (B) Crystal structure of p38 MAPK (PDB: 2fst); (C) Crystal structure of p38 MAPK (PDB: 1zyj).
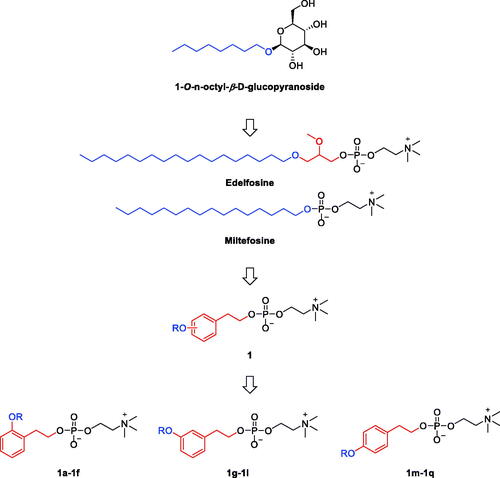
The structure of the lipid pocket binder; glycolipid 1-O-n-octyl-β-D-glucopyranoside, involves a hydrophobic C8-alkyl tail connected to a polar glucose sugar head (. Interestingly, edelfosine is an alkyllysophospholipid ATL derived from the metabolically unstable 1-stearoyl-lysophosphatidylcholine that possesses a C18-long stearyl alkyl tail connected to 2-O-methyl-sn-glycerophosphorylcholine polar head (. Edelfosine and miltefosine are alkylphospholipids well known as anticancer agents through inhibition of AKT. They possess similar structural features to the glycolipid 1-O-n-octyl-β-D-glucopyranoside but with longer tails. As the ligand binding pocket in some crystal structures (e.g., 2npg and 2fst) was found to accommodate up to two molecules of 1-O-n-octyl-β-D-glucopyranoside in linear position, molecules having longer tails might also be capable of binding within this pocket. Consequently, edelfosine was selected as a starting point for structural modifications to design new lipids-based molecules anticipated as antitumor agents binding the allosteric lipid binding pocket and, thus, might inhibit MAPKs. Considering that biological activity of flexible lipid molecules is affected by controlling their conformational preferences via substituents introduction or conformational restrictionCitation20, the proposed design addressed introduction of conformational restrictions via incorporation of the cyclic phenyl ring moiety coupled or not with double bonds. As shown in , the rotatability and the high flexibility near the polar phosphocholine head was reduced in the designed molecules through replacing the glycerol moiety of edelfosine by the ring-containing phenethyl moieties that bears alkoxy tails attached at three different configurations (ortho, meta or para) and thus would represent conformational restrictions that imitate different conformational configurations. In addition, the saturated stearyl alkyl moiety of edelfosine was maintained or replaced with unsaturated chains that incorporate one or more double bonds at different positions. These double bonds would restrict the free rotation at their introduced positions and, thus, fixing the stereochemistry in configurations equivalent to different conformations. Such variations might enable exploration of pocket’s plasticity, stereochemical limitations and their impact on activity.
Figure 2. Design rational of phenethyl-based edelfosine analogs as antitumor lipids.
Chemistry
As ideal or efficient synthesis should be practical, concise and economicCitation21–23, synthesis of the designed compounds was planned and achieved in 4 linear steps from commercially available starting materials. First, commercially available 2-, 3-, or 4-hydroxyphenylacetic acids (2) were converted to the corresponding O-alkylated phenylacetate esters (3) through esterification with methanol followed by O-alkylation with 1-bromo derivative of the appropriate hydrocarbon chain (Scheme 1). Metal hydride reduction using lithium aluminium hydride in THF medium efficiently converted the ester derivatives (3) into alcohol derivatives (4). Similar to the reported methodCitation3–6, alcohol derivatives (4) were converted into the targeted compounds in two sequential steps involving reaction with 2-chloro-2-oxo-1,3,2-dioxaphospholane followed by opening of the 1,3,2-dioxaphospholane ring with trimethylamine to obtain the desired phosphocholine derivatives (1). Spectroscopic and analytical data confirmed the structures of the prepared final compounds ().
Scheme 1. Reagents and conditions: (a) (i) MeOH, AcCl, 65 °C; (ii) RBr, K2CO3, DMF, 60 °C; (b) LiAlH4, THF, 0 °C; (c) 2-Chloro-2-oxo-1,3,2-dioxaphospholane, TEA, benzene; (d) TMA, CH3CN, 65 °C.
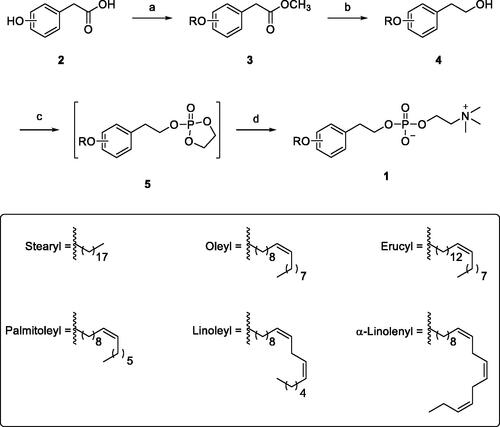
Table 1. Structures of the prepared final compounds submitted for biological evaluations.
Biological evaluations
Evaluation of spectrum of activity and structure-activity relationship
To investigate the structure activity relationship of this class of compounds as well as their activity spectrum, the antiproliferative activities of the prepared compounds derivatives bearing diverse chains attached to the ring of the phenethyl moiety at different positions were evaluated against cancer cells using panels of cancer cell lines belonging to nine cancer diseases including blood, lung, colon, CNS, skin, ovary, renal, prostate and breast cancers. To unveil potentially active compounds, evaluations were conducted at 10 µM concentration.
Evaluation of antiproliferative effects on blood cancers
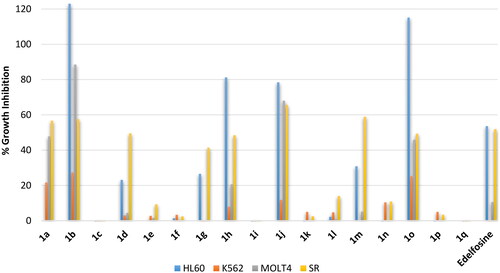
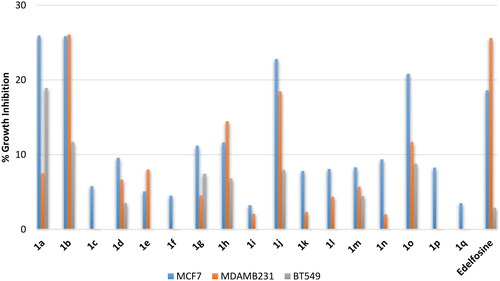
Blood cancer is not a single disease but involves several heterogenous types that could be of lymphoblastic or myeloid progenitor cells and, furthermore, could be acute or chronic. Accordingly, evaluation of activity against blood cancer involved four different cell lines namely: (1) HL60, an acute myeloid leukaemia (AML); (2) K562, a chronic myelogenous leukaemia (CML); (3) MOLT4, an acute lymphoblastic leukaemia (ALL); (4) SR, an anaplastic large cell lymphoma (ALCL) which is a type of non-Hodgkin lymphoma (NHL). At the tested 10 µM concentration, the reference drug elicited percent growth inhibition near 50% against two cells; the AML cell line HL60 and the ALCL cell line SR, while it showed a very weak activity or no growth inhibition of the other two cell lines; the CML cell line K562 and the ALCL cell line SR (. Analysis of the results revealed significantly high influence for the alkyl chain type on the activity. Compound 1a possessing in ortho-position the same saturated C18 stearyl hydrocarbon chain found in edelfosine showed approximately similar activity to edelfosine against the ALCL cell line SR but it showed higher activity against the ALL cell line MOLT4. Meanwhile, introduction of cis-double bond at 9-position of the hydrocarbon chain converting it into the monounsaturated C18 oleyl hydrocarbon chain afforded the more active compound 1b that showing high activity against AML, ALL and ALCL cancers (123%, 88.5% and 57.6 growth inhibitions against AML cell line HL60, ALL cell line MOLT4, and ALCL cell line SR, respectively, . Unfortunately, the analog 1c having the longer monounsaturated C22 erucyl hydrocarbon chain possessing a cis-double bond at 13-position showed no activity at the tested concentration. Meanwhile, shorting the hydrocarbon chain length through replacement with the monounsaturated C16 palmitoleyl chain possessing a cis-double bond at 9-position resulted in activity decrease (compound 1d, . Maintaining the C18 length of the hydrocarbon chain but introducing additional cis-double bonds resulted in very low active compounds (compounds 1e and 1f having linoleyl and α-linolenyl chains with two or three cis-double bonds, respectively, . It can be deduced that introduction of a cis-double at 9-position of C18 chain at ortho-position is favourable for activity while introduction of additional double bonds or alerting chain length are detrimental for activity. The results showed that shifting the chain to meta-position resulted in relatively less active compounds. Amongst, meta-oleyl and meta-palmitoleyl derivatives were the most active (compounds 1h and 1j, respectively, . Similar to observed activity trend for ortho-derivatives, meta-derivative bearing erucyl chain was inactive while linoleyl and α-linolenyl meta-derivatives were of very low activity. Interestingly, shifting the hydrocarbon chain resulted in further activity increase for para-palmitoleyl derivative 1o relative to corresponding ortho- and meta-derivative but opposite trend was noticed for oleyl derivatives. However, para-derivative bearing erucyl, linoleyl or α-linolenyl chains were inactive or of very low activity. Collectively, the revealed structure-activity relationship indicates that the saturated stearyl C18 chain and the monounsaturated C18 oleyl chain afforded the most potential ortho-derivatives while the monounsaturated C16 palmitoleyl chain afforded the most potential para-derivative with comparable activity to ortho-oleyl derivative. In addition, the results showed that CML cell line K562 was resistant to this class of compounds including the used standard drug, while other cell lines were sensitive with varying degrees.
Figure 3. % Growth inhibition of growth of diverse blood cancer cell lines triggered by 10 µM dose of the prepared compounds 1a–1q and the reference standard drug edelfosine. All data are the average of duplicates.
Evaluation of antiproliferative effects on non-small cell lung cancers
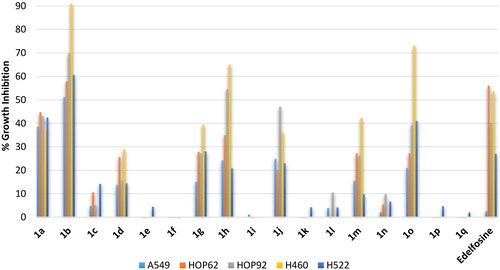
Non small lung cancer is the most threating lung cancer. It involves multiple subtypes including adenocarcinoma, bronchoalveolar carcinoma and large-cell lung carcinoma. Accordingly, activity profiling involved different types of lung cancers involving in addition to three lung adenocarcinoma cell lines (A549, HOP62 and H522) and two human large-cell lung carcinoma cell lines (HOP92 and H460). As shown in , the standard edelfosine drug showed average inhibition of three cell lines (adenocarcinoma cell line HOP62 large-cell lung carcinoma cell lines HOP92 and H460), while it showed low activity against the other two cell lines. Investigation of compounds activities showed that activity trend against lung cancers similar to the trend that has been observed against blood cancers. Thus, compound 1a having the ortho-C18 saturated stearyl chain possessed potential activity but compound 1b having the ortho-oleyl chain showed higher potential activity that reached more than 90% inhibition against large-cell lung carcinoma cell line H460. In addition, activity of derivatives bearing the stearyl C18 chain and the monounsaturated C18 oleyl chain was found to decrease upon shifting from ortho- to meta- then to para-positions. Meanwhile, there was a relative increase in activity for the monounsaturated C16 palmitoleyl chain upon shifting from ortho- to meta- then to para-positions. Furthermore, all derivative bearing erucyl, linoleyl or α-linolenyl chains were inactive or of very low activity.
Figure 4. % Growth inhibition of diverse non-small cell lung cancer cell lines triggered by 10 µM dose of the prepared compounds 1a–1q and the reference standard drug edelfosine. All data are the average of duplicates.
Evaluation of antiproliferative effects on colorectal cancers
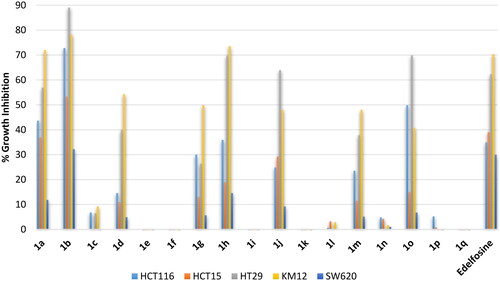
Colon cancers are the third most cancer diagnosed in both men and women. Despite that several types of colon cancer were identified such as gastrointestinal Stromal Tumours (GIST) and carcinoids, the most common colon cancers are of adenocarcinoma type. Consequently, evaluation was conducted employing five colorectal adenocarcinoma cell lines (HCT116, HCT15, H29, KM12 and SW620). The standard reference edelfosine drug showed potential inhibition against two cell lines and less than 40% inhibition of the growth of the other three cell lines (. Similar activity trend was observed here also. Thus, all derivative bearing erucyl, linoleyl or α-linolenyl chains were inactive or of very low activity. Meanwhile, compound 1a having the ortho-C18 saturated stearyl chain possessed potential activity comparable to edelfosine but compound 1b having the ortho-oleyl chain showed relatively higher potential activity. Also, the activity of the C18 saturated stearyl chain or the monounsaturated oleyl chain derivatives decreased upon shifting from ortho- to meta- then to para-positions. The results also showed that amongst the employed colon cancer cells, HT29 and KM12 were the most sensitive cell lines to this class of compounds while SW620 cell line was the least responsive.
Figure 5. % Growth inhibition of diverse colorectal cancer cell lines triggered by 10 µM dose of the prepared compounds 1a–1q and the reference standard drug edelfosine. All data are the average of duplicates.
Evaluation of antiproliferative effects on brain cancers
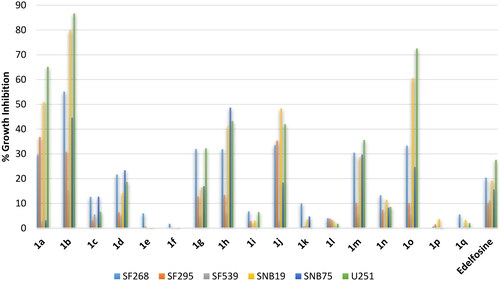
Gliomas are common brain cancers that originate from glial cells. As different subtypes of gliomas are known coupled with heterogeneity, profiling was conducted against six cell lines including: (1) SF268, an anaplastic astrocytoma cell line; (2) SNB75, an astrocytoma cell line; (3) SF295; a glioblastoma multiforme cell line; (4) SF539, a glioblastoma cell line; (5) SNB19, a glioblastoma cell line; (6) U251, a glioblastoma cell line. The refence standard edelfosine drug showed low activity below 30% inhibition against all tested cell lines (). Meanwhile, compounds 1a, 1b and 1o bearing C18 ortho-stearyl, C18 ortho-oleyl or C16 para-palmitoleyl chains elicited potential activities. SAR noted herein was similar to that observed against the above-discussed cancers. Thus, all derivative bearing erucyl, linoleyl or α-linolenyl chains were inactive or of very low activity; and activity decreases for stearyl and oleyl but increases for palmitoleyl derivatives upon shifting the chain from ortho- to meta- then to para-positions. The results showed that glioblastoma cell lines SNB19 and U251 were the most sensitive cells to this class of compounds while the glioblastoma multiforme SF295 and glioblastoma SF539 cell lines were the least responsive.
Figure 6. % Growth inhibition of diverse brain cancer cell lines triggered by 10 µM dose of the prepared compounds 1a–1q and the reference standard drug edelfosine. All data are the average of duplicates.
Evaluation of antiproliferative effects on melanoma skin cancers
Amongst skin cancers, melanomas are the most aggressive form that often metastasise to different sites. Accordingly, the activity was investigated using six diverse melanoma cell lines including: (1) LOXIMVI, a metastatic amelanotic melanoma derived from axillary lymph node; (2) MDAMB435, a metastatic amelanotic melanoma derived from subcutaneous site; (3) MALME3M, a metastatic melanoma derived from lung; (4) SKMEL2, a metastatic melanoma derived from the skin of the thigh; (5) SKMEL5, a metastatic melanoma derived from axillary lymph node; (6) UACC62, a malignant melanoma. Despite more or less activity trend similar to above-noted trend, the results demonstrated that all of the standard reference edelfosine drug and the tested compounds elicited weak % growth inhibition below 46% against tested melanoma skin cancer cell lines (. It might be deduced that this class of compounds might be ineffective or of low efficacy against melanoma skin cancers.
Figure 7. % Growth inhibition of diverse melanoma skin cancer cell lines triggered by 10 µM dose of the prepared compounds 1a–1q and the reference standard drug edelfosine. All data are the average of duplicates.
Evaluation of antiproliferative effects on ovarian cancers
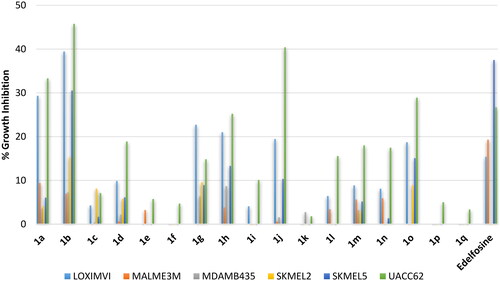
Amongst ovarian cancers, high-grade serous ovarian carcinoma (HGSOC) is the most common and is characterised by poor prognosis. Meanwhile endometrioid carcinoma, a subtype of epithelial ovarian cancers, is known for its malignant and invasive characteristics. Accordingly, evaluation was conducted employing the endometrioid carcinoma cell line; IGROV1, and three high grade ovarian serous adenocarcinoma (HGSOC) cell lines; OVCAR3, OVCAR4, and ADRRES. As revealed from results, the standard reference drug was effective only against the endometrioid carcinoma IGROV1 cell line (. Similarly, the active compounds amongst tested compounds showed potential high activity against the endometrioid carcinoma IGROV1 cell line but they elicited better inhibition relative to edelfosine against the high grade ovarian serous adenocarcinoma cell lines, despite still very low. As found in above-shown activity trends, stearyl and oleyl as well as palmitoleyl derivatives were the most active while erucyl, linoleyl or α-linolenyl derivatives were inactive or of very low activity (.
Figure 8. % Growth inhibition of diverse ovarian cancer cell lines triggered by 10 µM dose of the prepared compounds 1a–1q and the reference standard drug edelfosine. All data are the average of duplicates.
Evaluation of antiproliferative effects on renal cancers
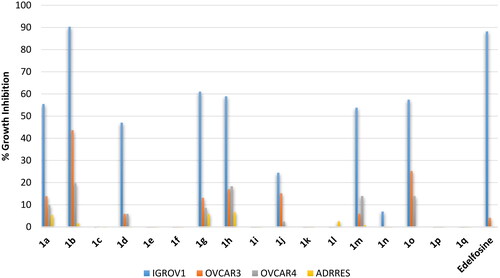
Renal cell carcinomas (RCC) are classified into two major subtypes; clear cell renal cell carcinomas (ccRCC), which represents the ma jority of renal cell cancers, and non-clear cell renal cell carcinomas (nccRCC) which difficult to treat and have poor long-term survival. nccRCC are heterogenous cancers and involve several types including papillary, clear cell papillary, medullary, chromophobe and sarcomatoid renal cell carcinomasCitation24. Consequently, a panel of six cell lines were employed for activity evaluation including: (1) 786 O, a primary ccRCC cell line; (2) ACHN, a metastatic papillary renal cell carcinoma (a subtype of nccRCC); (3) CAKI1, a metastatic ccRCC cell line; (4) SN12C, an unclassified renal cell carcinoma; (5) TK10, a clear cell renal cell carcinoma; (6) UO31, an unclassified renal cell carcinoma. As illustrated in , the standard edelfosine triggered modest inhibition below 37% against all the tested cell lines. On the opposite, tested compounds showed potential activity against the metastatic ccRCC CAKI1 cell line and the clear cell renal cell carcinoma TK10 cell line while the activity was modest against other tested cell lines. The activity pattern of these compounds followed the activity trend found against other cancers. Thus, compounds 1a, 1b, 1d, 1 g, 1h, 1j, 1 m and 1o possessing stearyl and oleyl as well as palmitoleyl derivatives were the most active while erucyl, linoleyl or α-linolenyl derivatives were inactive or of very low activity. This outcome supports the revealed importance number and position of double length as well as the length of the chain.
Figure 9. % Growth inhibition of diverse renal cancer cell lines triggered by 10 µM dose of the prepared compounds 1a–1q and the reference standard drug edelfosine. All data are the average of duplicates.
Evaluation of antiproliferative effects on prostate cancers
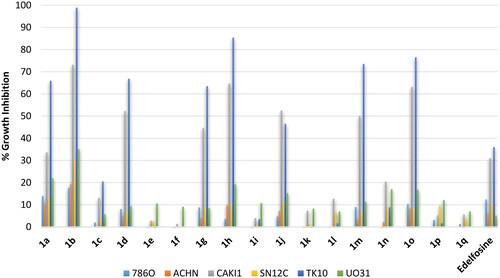
Amongst prostate carcinomas, the hormone-resistant prostate cancer is the most difficult to treat and show poor prognosis. Accordingly, evaluation was conducted employing two metastatic prostate adenocarcinoma cell lines: (1) PC3, a grade IV metastatic prostate adenocarcinoma derived from bone; (2) DU145, a metastatic prostate adenocarcinoma derived from brain. As shown in , edelfosine showed potential inhibition of the metastatic PC3 derived from bone (around 70% inhibition) by not the metastatic DU145 derived from brain prostate cancer cells. Similarly, the active compounds amongst the tested compounds showed potential inhibition of the bone-derived metastatic PC3 by not the brain-derived metastatic DU145 prostate cancer cells. Such differential response of DU145 and PC3 prostate cancer cells is interesting PC3 and DU145 were claimed similar in biological behaviour and gene expressionCitation25. Thus, stearyl and oleyl as well as palmitoleyl derivatives 1a, 1b, 1 g, 1h, 1 m and 1o showed better growth inhibition of PC3 cells than the standard edelfosine drug (99.3–85.2% growth inhibition values versus 70.5% for edelfosine). Meanwhile, erucyl, linoleyl or α-linolenyl derivatives were of very low activity. Based on the high activity of derivatives 1a, 1b, 1 g, 1h, 1 m and 1o against PC3 suggests these compounds as potential compounds for further development antiprostate cancer agents.
Figure 10. % Growth inhibition of diverse prostate cancer cell lines triggered by 10 µM dose of the prepared compounds 1a–1q and the reference standard drug edelfosine. All data are the average of duplicates.
Evaluation of antiproliferative effects on breast cancers
Breast cancer is the most common cancer amongst females. It can be hormone sensitive or resistant and, in addition, a primary or metastatic. Accordingly, profiling of the antiproliferative activities of involved three breast cancer cell lines: (1) MCF7, a metastatic invasive hormone-responsive breast adenocarcinoma; (2) MDAMB231, a metastatic triple negative breast adenocarcinoma; (3) BT549, a primary triple negative invasive breast adenocarcinoma. As shown in , the standard reference drug edelfosine possessed low activity against the tested breast cancer cells. Unfortunately, none of the tested compounds showed potential activity against any or the tested breast cancer cell lines. Despite their low activity, the tested compounds still showed the found above-described activity trend in which erucyl, linoleyl or α-linolenyl derivatives were of very low activity relative to stearyl, oleyl and palmitoleyl derivatives. However, the low activity of this class of compounds indicate that its activity spectrum does not involve breast cancer.
Figure 11. % Growth inhibition of diverse breast cancer cell lines triggered by 10 µM dose of the prepared compounds 1a–1q and the reference standard drug edelfosine. All data are the average of duplicates.
Potency assessment for compound 1b against diverse cancer cells
To assess the potency of the promising compound 1b, determination of IC50 values was addressed using diverse cancer cells in comparison with miltefosine as a reference standard. It might be conferred from results in that, in general, compound 1b has stronger potencies than the standard miltefosine drug. Its potencies varied from a tissue cancer to another and from a cell line to another cell line of the same tissue cancer. Amongst employed tissue cancers, the strongest potency for compound 1b was against colorectal cancer while the weakest potency was against cervical cancer. Accordingly, compound 1b might be a promising lead compound for further development of more promising anticancer compounds.
Table 2. Assessment of potency of compound 1b against diverse cancers.
Assessment of impact on p38 MAPK and AKT activities
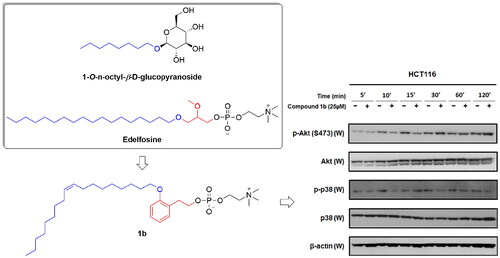
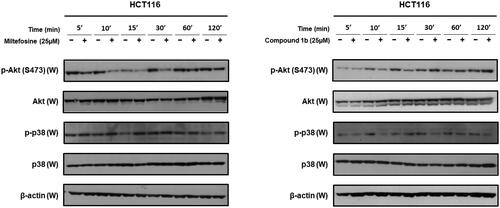
While the synthesised compounds were anticipated to modulate p38 MAPK, they were developed through structural modification of alkyl phospholipids which are known inhibitors of AKT. Consequently, the most promising compound 1b was selected for further assessment of its impact on AKT and p38 MAPK activity in HCT116 colon cancer cells in comparison with miltefosine as a reference standard well-known for AKT inhibitory activity. As in , miltefosine time-dependently inhibited phosphorylation of AKT and conversion into the active form (p-AKT) but did not inhibit phosphorylation of p38 MAPK and conversion into the active form (p-p38). On the contrary, compound 1b triggered time-dependent decline of phosphorylative activation of p38 MAPK in HCT116 cells but did not inhibit phosphorylation and activation of AKT. Consequently, it might be deduced that inhibition of AKT is not involved in the anticancer activity of compound 1b but inhibition of p38 MAPK is involved in its antiproliferative activity in HCT116 colon cancer cells.
Figure 12. Time-dependent effects of compound 1b and miltefosine on phosphorylation of AKT and p38 MAPK in HCT116 colon cancer cells.
In silico investigation of binding mode to p38 MAPK lipid binding pocket
According to deposited crystals structures of p38 MAPK in protein data bank, the lipid binding pocket might accommodate two distinct conformations: either a closed-end or channel-like open conformation. To anticipate which conformation of the lipid binding pocket might be the co-partner for the unveiled potential compounds 1a and 1b, in silico simulation study was conducted employing the closed-end conformation (PDB code: 1zyj) and the channel-like open conformation (PDB code: 2fst) crystal structures.
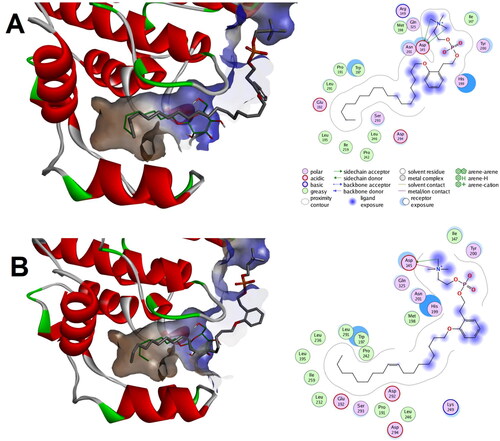
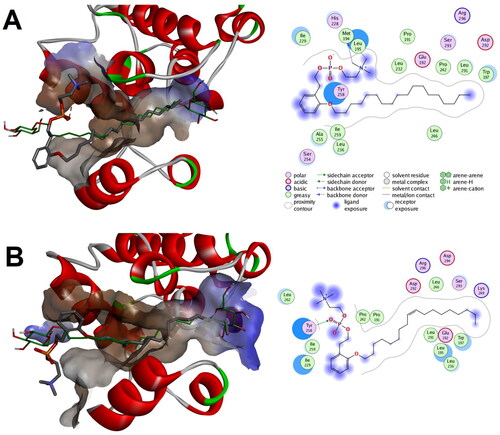
In silico prediction showed that both compound 1a and 1b were successfully docked to the closed-end conformation (PDB code: 1zyj) with calculated binding scores of −8.82 and −9.75 Kcal/mol, respectively. As illustrates, both alkyl chains of compound 1a and 1b were situated in the lipid binding pocket showing good alignment with the co-crystallized ligand and establishing a network of favourable hydrophobic interactions. Meanwhile the phenethyl and phosphocholine moieties of both compounds 1a and 1b were establishing favourable polar charged as well as hydrophobic interactions in the outside vicinity of the lipid binding pocket. Noticeably, the number of favourable interactions in the outside vicinity of the lipid binding pocket was higher and more intricated for compound 1b which might be a contributor to its calculated better interaction score. These results suggest that both compounds can bind to the closed end conformation of lipid binding pocket of p38 MAPK as well as to the outside vicinity of the pocket.
Figure 13. Predicted binding modes of compounds 1a and 1b with the closed-end conformation of ligand binding pocket of p38 MAPK (PDB code: 1zyj): (a) Calculated binding mode for compound 1a relative to the native binding mode of the co-crystallized ligand (green color); (b) Calculated binding mode for compound 1b relative to the native binding mode of the co-crystallized ligand (green color).
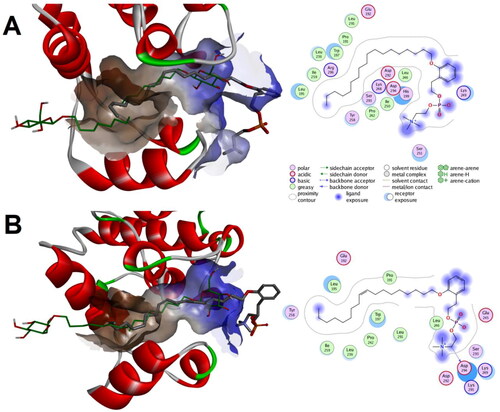
Investigation of whether a possible binding mode to the channel-like open conformation (PDB code: 2fst) could be produced in which the phenethyl phosphocholine moiety maintain direction towards the always-open end of the lipid binding pocket of p38 MAPK showed that both compounds 1a and 1b might elicit such binding mode with calculated favourable interaction scores of −9.30 and −8.93 Kcal/mol, respectively (. In these predicted bind modes, the phenethyl phosphocholine moiety of both compounds 1a and 1b was bound to the outside vicinity of the lipid binding pocket while their alkyl chains were placed within the lipid binding pocket good alignment with almost one of the co-crystallized ligands. It was noticeable that the phenethyl phosphocholine moiety of compounds 1b established better interactions network relative to that of compound 1a.
Figure 14. Predicted binding modes for compounds 1a and 1b with p38 MAPK that maintain orientation of the phenethyl phosphocholine moiety towards the always-open end of the lipid binding channel in the channel-like open conformation (PDB code: 2fst): (a) Calculated binding mode for compound 1a relative to the native binding mode of the co-crystallized ligand (green color); (b) Calculated binding mode for compound 1b relative to the native binding mode of the co-crystallized ligand (green color).
Investigation of whether a possible binding mode to the channel-like open conformation (PDB code: 2fst) could be produced where the phenethyl phosphocholine moiety is directed towards the lipid binding channel end that tends to close in the one-ended pocket conformation showed that both compounds 1a and 1b might elicit such binding mode with calculated favourable interaction scores of −8.98 and −9.64 Kcal/mol, respectively (. In these predicted binding modes, the alkyl chains of compounds 1a and 1b showed overlaying with not just one, but with both co-crystallized ligands establishing a network of favourable hydrophobic interactions. Meanwhile the phenethyl phosphocholine moiety for compounds 1a and 1b were protruding out of the pocket and interacting favourably with the outside vicinity of the lipid binding channel.
Figure 15. Predicted binding modes for compounds 1a and 1b within the channel-like open conformation of lipid binding pocket of p38 MAPK (PDB code: 2fst) in which the phenethyl phosphocholine moiety is directed towards the end that might close in the other conformation: (a) Calculated binding mode for compound 1a relative to the native binding mode of the co-crystallized ligand (green color); (b) Calculated binding mode for compound 1b relative to the native binding mode of the co-crystallized ligand (green color).
Collectively, the overall outcome of the three predicted binding modes for compounds 1a and 1b shows that their scores of the mode in which the phenethyl phosphocholine moiety maintain direction towards the always-open end of the lipid binding pocket in the channel-like open conformation of p38 MAPK is higher for compound 1a relative to 1b which do not correlates with the found higher activity of the latter. In addition, the alkyl chains in this binding mode overlayed only one of the two co-crystallized ligands (. Meanwhile, the calculated scores for predicted binding mode in which the phenethyl phosphocholine moiety is directed towards the lipid binding channel end that tends to close in the one-ended pocket conformation () was correlated with the found higher activity of compound 1a relative to 1b and, furthermore, the molecules overlayed with both co-crystallized ligands suggesting that this latter binding mode might contribute to mediating the activity. However, the calculated scores for binding to the closed-end conformation was close enough to suggest that compounds 1a and 1b might be able to bind this conformation also.
Conclusion
A non-ATP allosteric lipid binding pocket was discovered in p38 MAPK which might offer an opportunity to develop novel anticancer non-ATP inhibitors of p38 MAPK. To the best of our knowledge, only a recent single literature report has addressed development of such inhibitors. To develop novel ATL modulating p38 MAPK, novel phenethyl-based edelfosine analogs bearing saturated, monounsaturated, and polyunsaturated alkyl chains substituted at variable positions of the phenyl ring were designed and synthesised. The synthetic pathways to access target compounds were conducted efficiently in 4 linear steps employing commercially available starting materials. Evaluation of the anticancer activity of the prepared compounds revealed potential anticancer activity of saturated and monounsaturated ortho-substituted compounds against seven out of the nine cancer diseases’ panels. In general, the activity pattern was found to decrease upon number of saturation or translocating the alkyl chain substituent to meta- or para-positions. Investigation of the impact of potential compound 1b on p38 MAPK and AKT in colorectal cancer cells confirmed its inhibitory activity on the phosphorylative activation of p38 MAPK but not on AKT. In silico predictions of binding of compounds 1a and 1b with different conformations of the lipid binding site of p38 MAPK suggested their binding to the channel-like open conformation of lipid binding pocket of p38 MAPK in a mode that the phenethyl phosphocholine moiety is directed towards the lipid binding channel end that tends to close in the one-ended pocket conformation of p38 MAPK. However, binding to the closed-end conformation of p38 MAPK might still contribute to binding of compounds 1a and 1b. Collectively, the current effort suggests compounds 1a and 1b as promising broad spectrum ATL binding the lipid pocket of p38 MAPK and sowing inhibitory modulation of its activity.
Experimental
Chemistry
General
All solvents were purified and used on scrupulously dry condition. NMR spectra of all new compounds were recorded on Bruker AC 400 spectrometer (400 MHz for 1H NMR and 100 MHz for 13C NMR). Chemical shifts (δ) were reported in ppm, downfield from internal TMS standard. Chromatographic purifications were performed using Merck Kiesegel 60 Art 9385 (230–400 mesh).
General method for preparation of methyl 2-alkoxyphenylacetates 3a–3q
To a solution of the appropriate hydroxyphenylacetic acid (2.00 g, 13.14 mmol) in methanol (20 ml), acetyl chloride (1.8 ml, 25.62 mmol) was added at 0 °C, stirred for 1 h then heated to 65 °C for 24 h. After cooling to room temperature, the solvent was evaporated in vacuo then water was added to the residue and extracted thrice with methylene chloride, washed with brine, dried over anhydrous MgSO4, and evaporated in vacuo to afford the corresponding crude methyl hydroxyphenylacetate ester (98–95% yield). To the appropriate methyl hydroxyphenylacetate ester (0.50 g, 3.01 mmol) in DMF (5 ml), K2CO3 (1.25 g, 9.03 mmol) was added and stirred for 30 min at room temperature then the appropriate bromohydrocarbon (3.97 mmol) was added. The reaction mixture was heated at 60 °C for 7 h, cooled to room temperature, diluted with water and extracted thrice with EtOAc, washed with brine and dried over anhydrous MgSO4. The residue obtained after solvent removal in vacuo was purified using column chromatography (EtOAc/Hexane = 1:20) to afford methyl 2-alkoxyphenylacetates 3a–3q.
Methyl 2-stearoxyphenylacetate (3a)
Compound 3a was obtained from using methyl 2-hydroxyphenylacetate ester and stearyl bromide (yield 45%).
1H NMR (400 MHz, CD3OD) δ 7.25–7.20 (m, 1H), 7.19–7.15 (m, 1H), 6.92–6.87 (m, 1H), 6.86–6.82 (m, 1H), 3.95 (t, J = 6.4 Hz, 2H), 3.68 (s, 3H), 3.63 (s, 2H), 1.80–1.70 (m, 2H), 1.49–1.38 (m, 2H), 1.38–1.01 (m, 32H), 0.88 (t, J = 6.8 Hz, 3H).
Methyl 2-oleoxyphenylacetate (3b)
Compound 3b was obtained from methyl 2-hydroxyphenylacetate ester and oleyl bromide (yield 61%).
1H NMR (400 MHz, CD3OD) δ 7.25–7.20 (m, 1H), 7.20–7.12 (m, 1H), 6.94–6.87 (m, 1H), 6.87–6.81 (m, 1H), 5.42–5.28 (m, 2H), 3.95 (t, J = 6.3 Hz, 2H), 3.68 (s, 3H), 3.63 (s, 2H), 2.06–1.96 (m, 4H), 1.80–1.70 (m, 2H), 1.50–1.39 (m, 2H), 1.39–1.11 (m, 20H), 0.88 (t, J = 6.8 Hz, 3H).
Methyl 2-erucoxyphenylacetate (3c)
Compound 3c was obtained from methyl 2-hydroxyphenylacetate ester and erucyl bromide (yield 38%).
1H NMR (400 MHz, CD3OD) δ 7.25–7.19 (m, 1H), 7.19–7.15 (m, 1H), 6.92–6.86 (m, 1H), 6.86–6.82 (m, 1H), 5.39–5.30 (m, 2H), 3.95 (t, J = 6.4 Hz, 2H), 3.68 (s, 3H), 3.63 (s, 2H), 2.08–1.93 (m, 4H), 1.80–1.70 (m, 2H), 1.49–1.38 (m, 2H), 1.38–1.16 (m, 28H), 0.88 (t, J = 6.8 Hz, 3H).
Methyl 2-palmitoleoxyphenylacetate (3d)
Compound 3d was obtained from methyl 2-hydroxyphenylacetate ester and palmitoleyl bromide (yield 48%).
1H NMR (400 MHz, CD3OD) δ 7.26–7.20 (m, 1H), 7.19–7.15 (m, 1H), 6.93–6.86 (m, 1H), 6.86–6.82 (m, 1H), 5.41–5.27 (m, 2H), 3.95 (t, J = 6.4 Hz, 2H), 3.68 (s, 3H), 3.63 (s, 2H), 2.07–1.93 (m, 4H), 1.80–1.70 (m, 2H), 1.50–1.39 (m, 2H), 1.39–1.15 (m, 16H), 0.88 (t, J = 6.8 Hz, 3H).
Methyl 2-linoleoxyphenylacetate (3e)
Compound 3e was obtained from methyl 2-hydroxyphenylacetate ester and linoleyl bromide (yield 30%).
1H NMR (400 MHz, CD3OD) δ 7.25–7.20 (m, 1H), 7.20–7.15 (m, 1H), 6.93–6.87 (m, 1H), 6.87–6.82 (m, 1H), 5.43–5.28 (m, 4H), 3.95 (t, J = 6.4 Hz, 2H), 3.68 (s, 3H), 3.63 (s, 2H), 2.78 (t, J = 6.4 Hz, 2H), 2.10–1.97 (m, 4H), 1.80–1.70 (m, 2H), 1.49–1.40 (m, 2H), 1.40–1.13 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H).
Methyl 2-α-linolenoxyphenylacetate (3f)
Compound 3f was obtained from methyl 2-hydroxyphenylacetate ester and α-linolenyl bromide (yield 30%).
1H NMR (400 MHz, CD3OD) δ 7.25–7.20 (m, 1H), 7.20–7.15 (m, 1H), 6.93–6.87 (m, 1H), 6.87–6.82 (m, 1H), 5.48–5.27 (m, 6H), 3.95 (t, J = 6.4 Hz, 2H), 3.68 (s, 3H), 3.63 (s, 2H), 2.88–2.64 (m, 4H), 2.13–1.94 (m, 4H), 1.81–1.69 (m, 2H), 1.49–1.39 (m, 2H), 1.39–1.11 (m, 8H), 1.01–0.93 (m, 3H).
Methyl 3-stearoxyphenylacetate (3 g)
Compound 3 g was obtained from methyl 3-hydroxyphenylacetate ester and stearyl bromide (yield 89%).
1H NMR (400 MHz, CD3OD) δ 7.22 (t, J = 7.8 Hz, 1H), 6.86–6.77 (m, 3H), 3.94 (t, J = 6.4 Hz, 2H), 3.69 (s, 3H), 3.59 (s, 2H), 1.82–1.71 (m, 2H), 1.49–1.39 (m, 2H), 1.39–1.11 (m, 32H), 0.88 (t, J = 6.8 Hz, 3H).
Methyl 3-oleoxyphenylacetate (3h)
Compound 3h was obtained using methyl 3-hydroxyphenylacetate ester (0.11 g, 0.69 mmol), K2CO3 (0.23 g, 1.65 mmol) and oleyl bromide (0.18 g, 0.55 mmol). Yield (0.19 g, 66%).
1H NMR (400 MHz, CD3OD) δ 7.22 (t, J = 7.8 Hz, 1H), 6.86–6.77 (m, 3H), 5.41–5.29 (m, 2H), 3.94 (t, J = 6.6 Hz, 2H), 3.69 (s, 3H), 3.59 (s, 2H), 2.07–1.94 (m, 4H), 1.82–1.71 (m, 2H), 1.51–1.40 (m, 2H), 1.40–1.21 (m, 20H), 0.88 (t, J = 6.8 Hz, 3H).
Methyl 3-erucoxyphenylacetate (3i)
Compound 3i was obtained from methyl 3-hydroxyphenylacetate ester and erucyl bromide (yield 49%).
1H NMR (400 MHz, CD3OD) δ 7.22 (t, J = 7.8 Hz, 1H), 6.90–6.76 (m, 3H), 5.40–5.30 (m, 2H), 3.94 (t, J = 6.6 Hz, 2H), 3.69 (s, 3H), 3.59 (s, 2H), 2.08–1.94 (m, 4H), 1.82–1.71 (m, 2H), 1.50–1.39 (m, 2H), 1.39–1.17 (m, 28H), 0.88 (t, J = 6.8 Hz, 3H).
Methyl 3-palmitoleoxyphenylacetate (3j)
Compound 3j was obtained from methyl 3-hydroxyphenylacetate ester and palmitoleyl bromide (yield 71%).
1H NMR (400 MHz, CD3OD) δ 7.22 (t, J = 7.8 Hz, 1H), 6.88–6.77 (m, 3H), 5.40–5.31 (m, 2H), 3.94 (t, J = 6.6 Hz, 2H), 3.69 (s, 3H), 3.59 (s, 2H), 2.08–1.95 (m, 4H), 1.82–1.72 (m, 2H), 1.50–1.40 (m, 2H), 1.40–1.20 (m, 16H), 0.88 (t, J = 6.8 Hz, 3H).
Methyl 3-linoleoxyphenylacetate (3k)
Compound 3k was obtained from methyl 3-hydroxyphenylacetate ester and linoleyl bromide (yield 40%).
1H NMR (400 MHz, CD3OD) δ 7.22 (t, J = 7.8 1H), 6.87–6.77 (m, 3H), 5.44–5.28 (m, 4H), 3.94 (t, J = 6.4 Hz, 2H), 3.69 (s, 3H), 3.59 (s, 2H), 2.78 (t, J = 6.4 Hz, 2H), 2.11–1.99 (m, 4H), 1.81–1.72 (m, 2H), 1.51–1.40 (m, 2H), 1.40–1.20 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H).
Methyl 3-α-linolenoxyphenylacetate (3 l)
Compound 3 l was obtained from methyl 3-hydroxyphenylacetate ester and α-linolenyl bromide (yield 37%).
1H NMR (400 MHz, CD3OD) δ 7.22 (t, J = 7.8 Hz, 1H), 6.88–6.78 (m, 3H), 5.54–5.27 (m, 6H), 3.94 (t, J = 6.3 Hz, 2H), 3.69 (s, 3H), 3.59 (s, 2H), 2.86–2.66 (m, 4H), 2.13–1.95 (m, 4H), 1.85–1.72 (m, 2H), 1.51–1.40 (m, 2H), 1.40–1.21 (m, 8H), 1.03–0.93 (m, 3H).
Methyl 4-oleoxyphenylacetate (3 m)
Compound 3 m was obtained from methyl 4-hydroxyphenylacetate ester and oleyl bromide (yield 50%).
1H NMR (400 MHz, CD3OD) δ 7.18 (d, J = 8.7 Hz, 2H), 7.84 (d, J = 8.7 Hz, 2H), 5.40–5.30 (m, 2H), 3.93 (t, J = 6.3 Hz, 2H), 3.68 (s, 3H), 3.56 (s, 2H), 2.08–1.94 (m, 4H), 1.81–1.71 (m, 2H), 1.49–1.39 (m, 2H), 1.39–1.18 (m, 20H), 0.88 (t, J = 6.8 Hz, 3H).
Methyl 4-erucoxyphenylacetate (3n)
Compound 3n was obtained from methyl 4-hydroxyphenylacetate ester and erucyl bromide (yield 47%).
1H NMR (400 MHz, CD3OD) δ 7.18 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.40–5.30 (m, 2H), 3.93 (t, J = 6.6 Hz, 2H), 3.68 (s, 3H), 3.56 (s, 2H), 2.08–1.94 (m, 4H), 1.82–1.71 (m, 2H), 1.50–1.39 (m, 2H), 1.39–1.16 (m, 28H), 0.88 (t, J = 6.8 Hz, 3H).
Methyl 4-palmitoleoxyphenylacetate (3o)
Compound 3o was obtained from methyl 4-hydroxyphenylacetate ester and palmitoleyl bromide (yield 82%).
1H NMR (400 MHz, CD3OD) δ 7.18 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 5.38–5.32 (m, 2H), 3.93 (t, J = 6.8 Hz, 2H), 3.68 (s, 3H), 3.56 (s, 2H), 2.06–1.96 (m, 4H), 1.80–1.71 (m, 2H), 1.49–1.39 (m, 2H), 1.39–1.21 (m, 16H), 0.88 (t, J = 6.8 Hz, 3H).
Methyl 4-linoleoxyphenylacetate (3p)
Compound 3p was obtained from methyl 4-hydroxyphenylacetate ester and linoleyl bromide (yield 29%).
1H NMR (400 MHz, CD3OD) δ 8.02–7.96 (m, 1H), 7.20–7.14 (m, 1H), 6.98–6.92 (m, 1H), 6.87–6.81 (m, 1H), 5.43–5.28 (m, 4H), 4.04 (t, J = 6.6 Hz, 2H), 3.98–3.90 (m, 3H), 3.70–3.53 (m, 2H), 2.78 (t, J = 6.4 Hz, 2H), 2.10–1.99 (m, 4H), 1.89–1.70 (m, 2H), 1.52–1.41 (m, 2H), 1.41–1.19 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H).
Methyl 4-α-linolenoxyphenylacetate (3q)
Compound 3q was obtained from methyl 4-hydroxyphenylacetate ester and α-linolenyl bromide (yield 43%).
1H NMR (400 MHz, CD3OD) δ 7.18 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 5.54–5.27 (m, 6H), 3.93 (t, J = 6.4 Hz, 2H), 3.68 (s, 3H), 3.56 (s, 2H), 2.86–2.66 (m, 4H), 2.13–1.95 (m, 4H), 1.82–1.71 (m, 2H), 1.50–1.40 (m, 2H), 1.40–1.23 (m, 8H), 1.01–0.93 (m, 3H).
General method for preparation of 2-alkoxyphenethanols 4a–4q
Lithium aluminium hydride (0.11 g, 2.99 mmol) was added to a cooled solution of the appropriate methyl 2-alkoxyphenylacetate derivative 3a–3q (1.19 mmol) in THF (10 ml) at 0 °C. The reaction mixture was stirred for 30 min at room temperature, quenched with ethyl acetate, stirred for 10 min, minimal amount of water was added, filtered using celite then washed with ethyl acetate. The obtained residue after evaporation in vacuo was purified using column chromatography (EtOAc:Hexane = 1:9) to afford pure 2-alkoxyphenethanols 4a–4q.
2-Stearoxyphenethanol (4a)
Compound 4a was obtained from compound 3a (yield 73%).
1H NMR (400 MHz, CD3OD) δ 7.23–7.13 (m, 2H), 6.93–6.82 (m, 2H), 3.97 (t, J = 6.4 Hz, 2H), 3.84 (q, J = 12.2 Hz, 2H), 2.92 (t, J = 6.4 Hz, 2H), 1.85–1.74 (m, 2H), 1.71 (t, J = 5.8 Hz, 1H), 1.51–1.40 (m, 2H), 1.40–1.16 (m, 32H), 0.88 (t, J = 6.8 Hz, 3H).
2-Oleoxyphenethanol (4b)
Compound 4b was obtained from compound 3b (yield (0.14 g, 0.86 mmol, 80%).
1H NMR (400 MHz, CD3OD) δ 7.22–7.08 (m, 2H), 6.92–6.78 (m, 2H), 5.40–5.28 (m, 2H), 3.97 (t, J = 6.4 Hz, 2H), 3.87–3.79 (m, 2H), 2.92 (t, J = 6.3 Hz, 2H), 2.05–1.93 (m, 4H), 1.84–1.74 (m, 2H), 1.51–1.40 (m, 2H), 1.40–1.16 (m, 20H), 0.88 (t, J = 6.8 Hz, 3H).
2-Erucoxyphenethanol (4c)
Compound 4c was obtained from compound 3c (yield 67%).
1H NMR (400 MHz, CD3OD) δ 7.24–7.10 (m, 2H), 6.94–6.81 (m, 2H), 5.43–5.29 (m, 2H), 3.97 (t, J = 6.6 Hz, 2H), 3.84 (q, J = 12.2, 2H), 2.92 (t, J = 6.4 Hz, 2H), 2.08–1.96 (m, 4H), 1.85–1.75 (m, 2H), 1.52–1.41 (m, 2H), 1.41–1.16 (m, 28H), 0.88 (t, J = 6.8 Hz, 3H).
2-Palmitoleoxyphenethanol (4d)
Compound 4d was obtained from compound 3d (yield 82%).
1H NMR (400 MHz, CD3OD) δ 7.23–7.13 (m, 2H), 6.92–6.82 (m, 2H), 5.40–5.30 (m, 2H), 3.97 (t, J = 6.4 Hz, 2H), 3.84 (q, J = 12.2, 2H), 2.92 (t, J = 6.4 Hz, 2H), 2.06–1.97 (m, 4H), 1.84–1.72 (m, 2H), 1.51–1.41 (m, 2H), 1.41–1.19 (m, 16H), 0.88 (t, J = 6.8 Hz, 3H).
2-Linoleoxyphenethanol (3e)
Compound 3e was obtained from compound 3e (yield 78%).
1H NMR (400 MHz, CD3OD) δ 7.23–7.13 (m, 2H), 6.92–6.82 (m, 2H), 5.43–5.28 (m, 4H), 3.97 (t, J = 6.4 Hz, 2H), 3.84 (q, J = 12.2, 2H), 2.92 (t, J = 6.4 Hz, 2H), 2.78 (t, J = 6.4 Hz, 2H), 2.09–2.00 (m, 4H), 1.84–1.76 (m, 2H), 1.51–1.41 (m, 2H), 1.41–1.22 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H).
2-α-Linolenoxyphenylacetate (4f)
Compound 4f was obtained from compound 3f (yield 54%).
1H NMR (400 MHz, CD3OD) δ 7.23–7.14 (m, 2H), 6.92–6.83 (m, 2H), 5.53–5.27 (m, 6H), 3.97 (t, J = 6.4 Hz, 2H), 3.84 (t, J = 6.4, 2H), 2.92 (t, J = 6.4 Hz, 2H), 2.84–2.71 (m, 4H), 2.13–1.94 (m, 4H), 1.84–1.74 (m, 2H), 1.51–1.41 (m, 2H), 1.41–1.23 (m, 8H), 1.00–0.93 (m, 3H).
3-Stearoxyphenethanol (4 g)
Compound 4 g was obtained from compound 3 g (yield 76.4%).
1H NMR (400 MHz, CD3OD) δ 7.25–7.18 (m, 1H), 6.85–6.74 (m, 3H), 3.94 (t, J = 6.4 Hz, 2H), 3.91–3.82 (m, 2H), 2.84 (t, J = 6.4 Hz, 2H), 1.84–1.72 (m, 2H), 1.52–1.40 (m, 2H), 1.40–1.19 (m, 32H), 0.88 (t, J = 6.8 Hz, 3H).
3-Oleoxyphenethanol (4h)
Compound 4h was obtained from compound 3h (yield 85%).
1H NMR (400 MHz, CD3OD) δ 7.24–7.19 (m, 1H), 6.82–6.74 (m, 3H), 5.40–5.30 (m, 2H), 3.94 (t, J = 6.6 Hz, 2H), 3.86 (q, J = 12.2, 2H), 2.84 (t, J = 6.4 Hz, 2H), 2.06–1.94 (m, 4H), 1.82–1.73 (m, 2H), 1.50–1.42 (m, 2H), 1.42–1.12 (m, 20H), 0.88 (t, J = 6.8 Hz, 3H).
3-Erucoxyphenethanol (4i)
Compound 4i was obtained from compound 3i (yield 80%).
1H NMR (400 MHz, CD3OD) δ 7.25–7.18 (m, 1H), 6.85–6.74 (m, 3H), 5.40–5.30 (m, 2H), 3.94 (t, J = 6.8 Hz, 2H), 3.90–3.82 (m, 2H), 2.84 (t, J = 6.4 Hz, 2H), 2.06–1.96 (m, 4H), 1.82–1.72 (m, 2H), 1.51–1.40 (m, 2H), 1.40–1.21 (m, 28H), 0.88 (t, J = 6.8 Hz, 3H).
3-Palmitoleoxyphenethanol (4j)
Compound 4j was obtained from compound 3j (yield 84%).
1H NMR (400 MHz, CD3OD) δ 7.25–7.18 (m, 1H), 6.82–6.74 (m, 3H), 5.40–5.30 (m, 2H), 3.94 (t, J = 6.4 Hz, 2H), 3.86 (q, J = 12.2, 2H), 2.84 (t, J = 6.4 Hz, 2H), 2.06–1.96 (m, 4H), 1.82–1.72 (m, 2H), 1.50–1.42 (m, 2H), 1.42–1.21 (m, 16H), 0.88 (t, J = 6.8 Hz, 3H).
3-Linoleoxyphenethanol (4k)
Compound 4k was obtained from compound 3k (yield 58%).
1H NMR (400 MHz, CD3OD) δ 7.25–7.18 (m, 1H), 6.83–6.74 (m, 2H), 5.43–5.29 (m, 4H), 3.94 (t, J = 6.6 Hz, 2H), 3.90–3.82 (m, 2H), 2.94 (t, J = 6.4 Hz, 2H), 2.78 (t, J = 6.4 Hz, 2H), 2.10–2.00 (m, 4H), 1.82–1.73 (m, 2H), 1.51–1.42 (m, 2H), 1.42–1.18 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H).
3-α-Linolenoxyphenethanol (4 l)
Compound 4 l was obtained from compound 3 l (yield 58%).
1H NMR (400 MHz, CD3OD) δ 7.25–7.19 (m, 1H), 6.83–6.74 (m, 2H), 5.50–5.27 (m, 6H), 3.94 (t, J = 6.4 Hz, 2H), 3.86 (t, J = 6.4, 2H), 2.87–2.71 (m, 6H), 2.13–1.95 (m, 4H), 1.82–1.72 (m, 2H), 1.51–1.41 (m, 2H), 1.41–1.25 (m, 8H), 1.01–0.93 (m, 3H).
4-Oleoxyphenethanol (4 m)
Compound 4 m was obtained from compound 3 m (yield 89%).
1H NMR (400 MHz, CD3OD) δ 7.13 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.40–5.30 (m, 2H), 3.93 (t, J = 6.6 Hz, 2H), 3.82 (q, J = 12.2, 2H), 2.81 (t, J = 6.4 Hz, 2H), 2.06–1.95 (m, 4H), 1.81–1.72 (m, 2H), 1.49–1.39 (m, 2H), 1.39–1.19 (m, 20H), 0.88 (t, J = 6.8 Hz, 3H).
4-Erucoxyphenethanol (4n)
Compound 4n was obtained from compound 3n (yield 78%).
1H NMR (400 MHz, CD3OD) δ 7.13 (d, J = 8.3 Hz, 2H), 6.85 (d, J = 8.3 Hz, 2H), 5.40–5.30 (m, 2H), 3.93 (t, J = 6.8 Hz, 2H), 3.82 (q, J = 12.2, 2H), 2.81 (t, J = 6.4 Hz, 2H), 2.10–1.95 (m, 4H), 1.84–1.70 (m, 2H), 1.50–1.39 (m, 2H), 1.39–1.18 (m, 28H), 0.88 (t, J = 6.8 Hz, 3H).
4-Palmitoleoxyphenethanol (4o)
Compound 4o was obtained from compound 3o (yield 88%).
1H NMR (400 MHz, CD3OD) δ 7.13 (d, J = 8.3 Hz, 2H), 6.85 (d, J = 8.3 Hz, 2H), 5.40–5.30 (m, 2H), 3.93 (t, J = 6.4 Hz, 2H), 3.82 (q, J = 12.2, 2H), 2.81 (t, J = 6.4 Hz, 2H), 2.06–1.96 (m, 4H), 1.81–1.72 (m, 2H), 1.49–1.40 (m, 2H), 1.40–1.21 (m, 16H), 0.88 (t, J = 6.8 Hz, 3H).
4-Linoleoxyphenethanol (4p)
Compound 4p was obtained from compound 3p (yield 42%).
1H NMR (400 MHz, CD3OD) δ 7.13 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.3 Hz, 2H), 5.44–5.28 (m, 4H), 3.93 (t, J = 6.4 Hz, 2H), 3.82 (q, J = 12.2, 2H), 2.84–2.70 (m, 4H), 2.11–1.96 (m, 4H), 1.82–1.72 (m, 2H), 1.50–1.40 (m, 2H), 1.40–1.22 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H).
4-α-Linolenoxyphenethanol (4q)
Compound 4q was obtained from compound 3q (yield 75%).
1H NMR (400 MHz, CD3OD) δ 7.13 (d, J = 8.3 Hz, 2H), 6.85 (d, J = 8.3 Hz, 2H), 5.53–5.27 (m, 6H), 3.93 (t, J = 6.4 Hz, 2H), 3.82 (t, J = 6.4, 2H), 2.85–2.71 (m, 6H), 2.13–1.94 (m, 4H), 1.81–1.71 (m, 2H), 1.50–1.40 (m, 2H), 1.40–1.22 (m, 8H), 1.01–0.93 (m, 3H).
General method for preparation of 2-alkoxyphenethyl phosphocholines 1a–1q
2-Chloro-2-oxo-1,3,2-dioxaphospholane (0.24 ml, 2.38 mmol) was added to a cooled stirred solution of the appropriate 2-alkoxyphenethanol derivative 4a–4q (0.79 mmol) and triethylamine (0.34 ml, 2.38 mmol) in benzene (5 ml) at 0 °C, stirred at room temperature for 5 h, filtered, washed with benzene, and concentrated. In a pressure tube, trimethylamine (2 ml) was added to a cooled solution of the obtained crude intermediate 2–(2-alkoxyphenethoxy)-2-oxo-1,3,2-dioxaphospholane in acetonitrile (10 ml) at −78 °C. The mixture was reacted at 65 °C for 15 h, cooled to room temperature, vented then evaporated in vacuo. The residue was purified by column chromatography first eluting the less polar impurities (CHCl3/MeOH = 9:1), then the more polar product (CHCl3/MeOH/H2O = 65:25:4) to obtain the desired titled compounds.
2-Stearoxyphenethyl phosphocholine (1a)
Compound 1a was obtained from compound 4a (yield 55%).
1H NMR (400 MHz, CD3OD) δ 7.25–7.14 (m, 2H), 6.95–6.82 (m, 2H), 4.14–4.03 (m, 4H), 4.00 (t, J = 6.4 Hz, 2H), 3.54–3.48 (m, 2H), 3.19–3.13 (s, 9H), 2.98 (t, J = 6.8 Hz, 2H), 1.86–1.78 (m, 2H), 1.56–1.46 (m, 2H), 1.46–1.21 (m, 32H), 0.92 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 158.5, 132.1, 128.9, 127.9, 121.3, 112.4, 69.1, 67.4, 66.4, 60.2, 54.7(3 C), 33.1, 30.8(13 C), 30.5(2 C), 27.3, 23.8, 14.5; HRMS: m/z calculated for C31H59NO5P: 556.41308 [M + H]+, Found 556.41092.
2-Oleoxyphenethyl phosphocholine (1b)
Compound 1b was obtained from compound 4b (yield 45%).
1H NMR (400 MHz, CD3OD) δ 7.16–7.00 (m, 2H), 6.85–6.64 (m, 2H), 5.31–5.17 (m, 2H), 4.03–3.90 (m, 4H), 3.86 (t, J = 6.4 Hz, 2H), 3.43–3.35 (m, 2H), 3.08–2.99 (s, 9H), 2.86 (t, J = 6.4 Hz, 2H), 1.98–1.86 (m, 4H), 1.74–1.64 (m, 2H), 1.45–1.34 (m, 2H), 1.34–1.09 (m, 20H), 0.79 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 157.1, 130.7, 129.5, 129.4, 127.4, 126.5, 119.9, 111.0, 67.7, 66.0, 64.9, 58.7, 53.2(3 C), 31.7, 29.2(10 C), 26.8(2 C), 25.9, 22.4, 13.1; HRMS: m/z calculated for C31H57NO5P: 554.39743 [M + H]+, Found 554.39727.
2-Erucoxyphenethyl phosphocholine (1c)
Compound 1c was obtained from compound 4c (yield (0.10 g, 49%).
1H NMR (400 MHz, CD3OD) δ 7.13–7.09 (m, 1H), 7.09–7.02 (m, 1H), 6.81–6.70 (m, 2H), 5.29–5.18 (m, 2H), 4.04–3.90 (m, 4H), 3.86 (t, J = 6.4 Hz, 2H), 3.42–3.35 (m, 2H), 3.06–3.00 (s, 9H), 2.86 (t, J = 6.4 Hz, 2H), 1.97–1.87 (m, 4H), 1.74–1.64 (m, 2H), 1.44–1.33 (m, 2H), 1.33–1.10 (m, 28H), 0.79 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 157.1, 130.7, 129.5(2 C), 127.5, 126.5, 120.0, 111.0, 67.7, 66.0, 64.9, 58.7, 53.2(3 C), 31.7, 29.5(14 C), 28.2(2 C), 27.3, 23.8, 14.8; HRMS: m/z calculated for C35H65NO5P: 610.46003 [M + H]+, Found 610.45857.
2-Palmitoleoxyphenethyl phosphocholine (1d)
Compound 1d was obtained from compound 4d (yield 79%).
1H NMR (400 MHz, CD3OD) δ 7.14–7.01 (m, 2H), 6.83–6.68 (m, 2H), 5.30–5.16 (m, 2H), 4.02–3.90 (m, 4H), 3.88 (t, J = 6.4 Hz, 2H), 3.42–3.35 (m, 2H), 3.07–3.00 (s, 9H), 2.86 (t, J = 6.4 Hz, 2H), 1.98–1.86 (m, 4H), 1.74–1.64 (m, 2H), 1.45–1.34 (m, 2H), 1.34–1.12 (m, 16H), 0.80 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 158.5, 132.2, 130.9(2 C), 128.9, 121.3, 112.4, 69.1, 67.3, 66.3, 60.2, 54.7(3 C), 33.0, 30.9(8 C), 28.2(2 C), 27.4, 23.8, 14.6; HRMS: m/z calculated for C29H53NO5P: 526.36613 [M + H]+, Found 526.36672.
2-Linoleoxyphenethyl phosphocholine (1e)
Compound 1e was obtained from compound 4e (yield 94%).
1H NMR (400 MHz, CD3OD) δ 7.28–7.13 (m, 2H), 6.98–6.82 (m, 2H), 5.50–5.25 (m, 4H), 4.22–4.02 (m, 4H), 4.00 (t, J = 6.4 Hz, 2H), 3.56–3.47 (m, 2H), 3.21–3.12 (s, 9H), 2.98 (t, J = 6.4 Hz, 2H), 2.80 (t, J = 6.0 Hz, 2H), 2.16–1.96 (m, 4H), 1.90–1.74 (m, 2H), 1.59–1.47 (m, 2H), 1.47–1.20 (m, 14H), 0.92 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CD3OD) δ 157.3, 130.9(2 C), 129.8, 127.9(2 C), 127.7, 126.7, 120.1, 111.2, 67.9, 66.2, 65.1, 59.0, 53.5(3 C), 31.8, 31.5, 29.2(6 C), 27.0, 26.2, 26.1, 25.4, 22.5, 13.3; HRMS: m/z calculated for C31H55NO5P: 552.38124 [M + H]+, Found 552.37984.
2-α-Linolenoxyphenethyl phosphocholine (1f)
Compound 1f was obtained from compound 4f (yield 84%).
1H NMR (400 MHz, CD3OD) δ 7.26–7.15 (m, 2H), 6.96–6.82 (m, 2H), 5.51–5.27 (m, 6H), 4.13–4.03 (m, 4H), 4.00 (t, J = 6.4 Hz, 2H), 3.54–3.48 (m, 2H), 3.20–3.11 (s, 9H), 2.98 (t, J = 6.4 Hz, 2H), 2.87–2.70 (m, 4H), 2.15–1.96 (m, 4H), 1.87–1.76 (m, 2H), 1.58–1.46 (m, 2H), 1.46–1.25 (m, 8H), 1.02–0.94 (m, 3H); 13C NMR (125 MHz, CD3OD) δ 157.3, 131.6, 131.0(2 C), 130.0, 128.1, 127.7(2 C), 127.1, 126.7, 120.1, 111.2, 67.9, 66.2, 65.1, 59.0, 53.4(3 C), 31.8, 29.6(5 C), 27.0, 26.1, 25.4, 25.3, 20.3, 13.5; HRMS: m/z calculated for C31H53NO5P: 550.36559 [M + H]+, Found 550.36389.
3-Stearoxyphenethyl phosphocholine (1 g)
Compound 1 g was obtained from compound 4 g (yield 79%).
1H NMR (400 MHz, CD3OD) δ 7.19 (t, J = 7.8 Hz, 1H), 6.88–6.83 (m, 2H), 6.79–6.73 (m, 1H), 4.13–4.05 (m, 4H), 3.96 (t, J = 6.4 Hz, 2H), 3.54–3.49 (m, 2H), 3.19–3.12 (s, 9H), 2.93 (t, J = 6.8 Hz, 2H), 1.82–1.72 (m, 2H), 1.55–1.44 (m, 2H), 1.44–1.22 (m, 32H), 0.92 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 159.3, 140.1, 129.0, 121.0, 115.1, 112.1, 77.9, 72.5, 63.0, 58.8, 53.2(3 C), 36.8, 31.7, 29.4(13 C), 25.9, 22.4, 13.2; HRMS: m/z calculated for C31H59NO5P: 556.41308 [M + H]+, Found 556.40882.
3-Oleoxyphenethyl phosphocholine (1h)
Compound 1h was obtained from compound 4h (yield 84%).
1H NMR (400 MHz, CD3OD) δ 7.12–7.01 (m, 1H), 6.78–6.58 (m, 3H), 5.32–5.16 (m, 2H), 4.10–3.90 (m, 4H), 3.84 (t, J = 6.4 Hz, 2H), 3.43–3.36 (m, 2H), 3.07–3.01 (s, 9H), 2.80 (t, J = 6.8 Hz, 2H), 1.98–1.86 (m, 4H), 1.70–1.60 (m, 2H), 1.42–1.32 (m, 2H), 1.32–1.12 (m, 20H), 0.79 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 159.3, 140.1, 129.5, 129.4, 129.0, 121.0, 115.1, 112.1, 67.5, 66.2, 66.0, 58.8, 53.2(3 C), 36.8, 32.2, 29.3(9 C), 28.8(2 C), 25.9, 22.4, 13.1; HRMS: m/z calculated for C31H57NO5P: 554.39743 [M + H]+, Found 554.39918.
3-Erucoxyphenethyl phosphocholine (1i)
Compound 1i was obtained from compound 4i (yield 52%).
1H NMR (400 MHz, CD3OD) δ 7.11–7.02 (m, 1H), 6.78–6.59 (m, 3H), 5.30–5.16 (m, 2H), 4.04–3.90 (m, 4H), 3.84 (t, J = 6.4 Hz, 2H), 3.43–3.35 (m, 2H), 3.09–2.98 (s, 9H), 2.80 (t, J = 6.8 Hz, 2H), 1.98–1.86 (m, 4H), 1.70–1.59 (m, 2H), 1.42–1.32 (m, 2H), 1.32–1.10 (m, 28H), 0.80 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 160.7, 141.5 130.9(2 C), 130.4, 122.4, 116.5, 113.5, 68.9, 67.6, 67.4, 60.2, 54.7(3 C), 38.3, 33.2, 30.9(14 C), 28.2, 27.3, 23.8, 14.6; HRMS: m/z calculated for C35H65N4O4: 610.46003 [M + H]+, Found 610.45857.
3-Palmitoleoxyphenethyl phosphocholine (1j)
Compound 1j was obtained from compound 4j (yield 86%).
1H NMR (400 MHz, CD3OD) δ 7.10–7.04 (m, 1H), 6.76–6.70 (m, 2H), 6.67–6.61 (m, 1H), 5.29–5.19 (m, 2H), 4.02–3.91 (m, 4H), 3.83 (t, J = 6.4 Hz, 2H), 3.42–3.36 (m, 2H), 3.06–3.01 (s, 9H), 2.80 (t, J = 6.8 Hz, 2H), 1.97–1.88 (m, 4H), 1.69–1.60 (m, 2H), 1.42–1.32 (m, 2H), 1.32–1.14 (m, 28H), 0.79 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CD3OD) δ 140.4, 129.7, 129.2, 121.2, 115.3(2 C), 112.3(2 C), 67.7, 66.4, 66.2, 59.0, 53.4(3 C), 37.0, 31.8, 29.7(8 C), 27.0, 26.1, 22.6, 13.4; HRMS: m/z calculated for C29H53NO5P: 526.36613 [M + H]+, Found: 526.36495.
3-Linoleoxyphenethyl phosphocholine (1k)
Compound 1k was obtained from compound 4k (yield 71%).
1H NMR (400 MHz, CD3OD) δ 7.23–7.16 (m, 1H), 7.89–7.82 (m, 2H), 6.80–6.73 (m, 2H), 5.44–5.29 (m, 4H), 4.16–4.02 (m, 4H), 3.97 (t, J = 6.4 Hz, 2H), 3.54–3.48 (m, 2H), 3.20–3.12 (s, 9H), 2.93 (t, J = 6.8 Hz, 2H), 2.80 (t, J = 6.0 Hz, 2H), 2.14–2.02 (m, 4H), 1.82–1.73 (m, 2H), 1.54–1.45 (m, 2H), 1.45–1.25 (m, 14H), 0.92 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CD3OD) δ 159.5, 140.4, 129.8(2 C), 129.2, 127.9(2 C), 121.3, 115.3, 112.3, 67.7, 66.4, 66.2, 59.0, 53.4(3 C), 37.0, 31.5, 29.3(6 C), 27.0(2 C), 26.1, 25.4, 22.5, 13.3; HRMS: m/z calculated for C31H55NO5P: 552.38124 [M + H]+, Found 552.37936.
3-α-Linolenoxyphenethyl phosphocholine (1 l)
Compound 1 l was obtained from compound 4 l (yield 91%).
1H NMR (400 MHz, CD3OD) δ 7.19 (t, J = 7.8 Hz, 1H), 6.88–6.82 (m, 2H), 6.79–6.73 (m, 1H), 5.48–5.27 (m, 4H), 4.13–4.01 (m, 4H), 3.97 (t, J = 6.4 Hz, 2H), 3.53–3.46 (m, 2H), 3.20–3.09 (s, 9H), 2.92 (t, J = 6.8 Hz, 2H), 2.87–2.71 (m, 4H), 2.18–1.96 (m, 4H), 1.82–1.70 (m, 2H), 1.55–1.44 (m, 2H), 1.44–1.27 (m, 8H), 1.03–0.89 (m, 3H); 13C NMR (125 MHz, CD3OD) δ 159.5, 140.4, 131.6, 130.0, 129.2(2 C), 128.0, 127.7, 127.1, 121.3, 115.4, 112.3, 67.7, 66.4, 66.2, 59.0, 53.4(3 C), 37.0, 29.4(5 C), 27.0, 26.1, 25.4, 25.3, 20.4, 13.6; HRMS: m/z calculated for C31H53NO5P: 550.36559 [M + H]+, Found 550.36408.
4-Oleoxyphenethyl phosphocholine (1 m)
Compound 1 m was obtained from compound 4 m (yield 61%).
1H NMR (400 MHz, CD3OD) δ 7.06 (d, J = 8.8 Hz, 2H), 6.72 (d, J = 8.8 Hz, 2H), 5.30–5.18 (m, 2H), 4.04–3.88 (m, 4H), 3.81 (t, J = 6.4 Hz, 2H), 3.45–3.36 (m, 2H), 3.09–2.99 (s, 9H), 2.76 (t, J = 6.8 Hz, 2H), 1.98–1.85 (m, 4H), 1.68–1.58 (m, 2H), 1.41–1.31 (m, 2H), 1.31–1.12 (m, 20H), 0.79 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 159.2, 131.8, 131.6, 131.2, 130.9(2 C), 115.5(2 C), 69.1, 67.8, 67.4, 60.2, 54.7(3 C), 37.4, 33.7, 30.7(9 C), 28.2(2 C), 27.3, 23.8, 14.6; HRMS: m/z calculated for C31H57NO5P: 554.39743 [M + H]+, Found 554.39896.
4-Erucoxyphenethyl phosphocholine (1n)
Compound 1n was obtained from compound 4n (yield 79%).
1H NMR (400 MHz, CD3OD) δ 7.18 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 5.41–5.31 (m, 2H), 4.15–4.00 (m, 4H), 3.93 (t, J = 6.4 Hz, 2H), 3.56–3.50 (m, 2H), 3.20–3.12 (s, 9H), 2.89 (t, J = 6.8 Hz, 2H), 2.09–2.00 (m, 4H), 1.80–1.72 (m, 2H), 1.52–1.43 (m, 2H), 1.43–1.26 (m, 28H), 0.92 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 157.8, 130.3, 129.8(2 C), 129.5(2 C), 114.1(2 C), 67.6, 66.4, 66.0, 58.8, 53.3(3 C), 36.0, 31.8, 29.5(14 C), 28.2, 27.3, 23.8, 14.6; HRMS: m/z calculated for C35H65N4O4: 610.46003 [M + H]+, Found 610.45869.
4-Palmitoleoxyphenethyl phosphocholine (1o)
Compound 1o was obtained from compound 4o (yield 79%).
1H NMR (400 MHz, CD3OD) δ 7.06 (d, J = 8.8 Hz, 2H), 6.72 (d, J = 8.8 Hz, 2H), 5.30–5.18 (m, 2H), 4.05–3.88 (m, 4H), 3.80 (t, J = 6.4 Hz, 2H), 3.46–3.38 (m, 2H), 3.10–3.00 (s, 9H), 2.76 (t, J = 6.8 Hz, 2H), 1.98–1.87 (m, 4H), 1.68–1.58 (m, 2H), 1.41–1.31 (m, 2H), 1.31–1.11 (m, 16H), 0.79 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 159.2, 131.8, 131.2(2 C), 130.9(2 C), 115.5(2 C), 69.0, 67.8, 67.4, 60.2, 54.7(3 C), 37.4, 33.0, 30.9(8 C), 28.2, 27.3, 23.8, 14.6; HRMS: m/z calculated for C29H53NO5P: 526.36613 [M + H]+, Found: 526.36432.
4-Linoleoxyphenethyl phosphocholine (1p)
Compound 1p was obtained from compound 4p (yield 90%).
1H NMR (400 MHz, CD3OD) δ 7.19 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 5.43–5.30 (m, 4H), 4.13–4.01 (m, 4H), 3.95 (t, J = 6.4 Hz, 2H), 3.55–3.49 (m, 2H), 3.20–3.13 (s, 9H), 2.89 (t, J = 6.8 Hz, 2H), 2.80 (t, J = 6.0 Hz, 2H), 2.13–2.02 (m, 4H), 1.81–1.72 (m, 2H), 1.54–1.45 (m, 2H), 1.45–1.13 (m, 14H), 0.92 (t, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CD3OD) δ 158.0, 130.6, 130.0(2 C), 129.8(2 C), 128.0(2 C), 114.3(2 C), 67.8, 66.6, 64.7, 59.0, 53.4(3 C), 36.1, 31.5, 29.3(6 C), 27.0(2 C), 26.1, 25.4, 22.5, 13.3; HRMS: m/z calculated for C31H55NO5P: 552.38124 [M + H]+, Found 552.37982.
4-α-Linolenoxyphenethyl phosphocholine (1q)
Compound 1q was obtained from compound 4q (yield 90%).
1H NMR (400 MHz, CD3OD) δ 7.19 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 5.46–5.27 (m, 4H), 4.13–4.01 (m, 4H), 3.95 (t, J = 6.4 Hz, 2H), 3.55–3.48 (m, 2H), 3.20–3.11 (s, 9H), 2.89 (t, J = 6.8 Hz, 2H), 2.86–2.70 (m, 4H), 2.15–1.97 (m, 4H), 1.82–1.70 (m, 2H), 1.55–1.44 (m, 2H), 1.44–1.28 (m, 8H), 1.02–0.92 (m, 3H); 13C NMR (125 MHz, CD3OD) δ 158.1, 131.6, 130.6, 130.0, 129.9(2 C), 128.1, 128.0, 127.7, 127.1, 114.3(2 C), 67.9, 66.6, 66.2, 59.0, 53.4(3 C), 36.1, 29.3(5 C), 27.0, 26.1, 25.4, 25.3, 20.4, 13.6; HRMS: m/z calculated for C31H53NO5P: 550.36559 [M + H]+, Found 550.36413.
Biological evaluations
Biological evaluations were conducted following literature protocolsCitation26–31 as in supplementary information.
Statistical analysis
Whenever applied, statistical significance was assessed using one-way ANOVA and Dunnett’s post hoc test. P values less than 0.05 were considered statistically significant.
In silico studies
In silico studies were conducted following literature protocolsCitation32–36 as in supplementary information.
Supplemental Material
Download PDF (1.6 MB)Acknowledgement
National Cancer Institute, Bethesda, Maryland, USA is acknowledged for performing NCI-60 human tumor cell lines screen.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hassan AHE, Cho MC, Kim HI, Yang JS, Park KT, Hwang JY, Jang C-G, Park KD, Lee YS. Synthesis of oxidative metabolites of CRA13 and their analogs: Identification of CRA13 active metabolites and analogs thereof with selective CB2R affinity. Bioorg Med Chem. 2018;26(18):5069–5078.
- Hassan AHE, Park KT, Kim HJ, Lee HJ, Kwon YH, Hwang JY, Jang C-G, Chung JH, Park KD, Lee SJ, et al. Fluorinated CRA13 analogues: Synthesis, in vitro evaluation, radiosynthesis, in silico and in vivo PET study. Bioorg Chem. 2020;99:103834.
- Hassan AHE, Park HR, Yoon YM, Kim HI, Yoo SY, Lee KW, Lee YS. Antiproliferative 3-deoxysphingomyelin analogs: Design, synthesis, biological evaluation and molecular docking of pyrrolidine-based 3-deoxysphingomyelin analogs as anticancer agents. Bioorg Chem. 2019;84:444–455.
- Hassan AHE, Phan T-N, Yoon S, Lee CJ, Jeon HR, Kim S-H, No JH, Lee YS. Pyrrolidine-based 3-deoxysphingosylphosphorylcholine analogs as possible candidates against neglected tropical diseases (NTDs): identification of hit compounds towards development of potential treatment of Leishmania donovani. J Enzyme Inhib Med Chem. 2021;36(1):1922–1930.
- Alam MM, Hassan AHE, Kwon YH, Lee HJ, Kim NY, Min KH, Lee S-Y, Kim D-H, Lee YS. Design, synthesis and evaluation of alkylphosphocholine-gefitinib conjugates as multitarget anticancer agents. Arch Pharm Res. 2018;41(1):35–45.
- Alam MM, Hassan AHE, Lee KW, Cho MC, Yang JS, Song J, Min KH, Hong J, Kim D-H, Lee YS. Design, synthesis and cytotoxicity of chimeric erlotinib-alkylphospholipid hybrids. Bioorg Chem. 2019;84:51–62.
- Koul HK, Pal M, Koul S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer. 2013;4(9-10):342–359.
- Igea A, Nebreda AR. The Stress Kinase p38α as a Target for Cancer Therapy. Cancer Res. 2015;75(19):3997–4002.
- Kudaravalli S, den Hollander P, Mani SA. Role of p38 MAP kinase in cancer stem cells and metastasis. Oncogene. 2022;41(23):3177–3185.
- Fang Z, Grütter C, Rauh D. Strategies for the Selective Regulation of Kinases with Allosteric Modulators: exploiting exclusive structural features. ACS Chem Biol. 2013;8(1):58–70.
- Elkamhawy A, Kim Ny, Hassan AHE, Park J-e, Yang J-E, Oh K-S, Lee BH, Lee MY, Shin KJ, Lee K-T, et al. Design, synthesis and biological evaluation of novel thiazolidinedione derivatives as irreversible allosteric IKK-β modulators. Eur J Med Chem. 2018;157:691–704.
- Elkamhawy A, Kim Ny, Hassan AHE, Park J-e, Paik S, Yang J-E, Oh K-S, Lee BH, Lee MY, Shin KJ, et al. Thiazolidine-2,4-dione-based irreversible allosteric IKK-β kinase inhibitors: optimization into in vivo active anti-inflammatory agents. Eur J Med Chem. 2020;188:111955.
- Elkamhawy A, Youn Kim N, Hassan AHE, Park J-e, Yang J-E, Elsherbeny MH, Paik S, Oh K-S, Lee BH, Lee MY, et al. Optimization study towards more potent thiazolidine-2,4-dione IKK-β modulator: synthesis, biological evaluation and in silico docking simulation. Bioorg Chem. 2019;92:103261.
- Diskin R, Engelberg D, Livnah O. A Novel Lipid Binding Site Formed by the MAP Kinase Insert in p38α. J Mol Biol. 2008;375(1):70–79.
- Kumar GS, Clarkson MW, Kunze MBA, Granata D, Wand AJ, Lindorff-Larsen K, Page R, Peti W. Dynamic activation and regulation of the mitogen-activated protein kinase p38. Proc Natl Acad Sci U S A. 2018;115(18):4655–4660.
- Perry JJP, Harris RM, Moiani D, Olson AJ, Tainer JA. p38α MAP Kinase C-Terminal Domain Binding Pocket Characterized by Crystallographic and Computational Analyses. J Mol Biol. 2009;391(1):1–11.
- Comess KM, Sun C, Abad-Zapatero C, Goedken ER, Gum RJ, Borhani DW, Argiriadi M, Groebe DR, Jia Y, Clampit JE, et al. Discovery and Characterization of Non-ATP Site Inhibitors of the Mitogen Activated Protein (MAP) Kinases. ACS Chem Biol. 2011;6(3):234–244.
- Bührmann M, Wiedemann BM, Müller MP, Hardick J, Ecke M, Rauh D. Structure-based design, synthesis and crystallization of 2-arylquinazolines as lipid pocket ligands of p38α MAPK. PLOS One. 2017;12(9):e0184627.
- Zhang XH, Chen C-H, Li H, Hsiang J, Wu X, Hu W, Horne D, Nam S, Shively J, Rosen ST. Targeting the non-ATP-binding pocket of the MAP kinase p38γ mediates a novel mechanism of cytotoxicity in cutaneous T-cell lymphoma (CTCL). FEBS Lett. 2021;595(20):2570–2592.
- Boshkow J, Fischer S, Bailey AM, Wolfrum S, Carreira EM. Stereochemistry and biological activity of chlorinated lipids: a study of danicalipin A and selected diastereomers. Chem Sci. 2017;8(10):6904–6910.
- Gaich T, Baran PS. Aiming for the Ideal Synthesis. J Org Chem. 2010;75(14):4657–4673.
- Jo H, Hassan AHE, Jung SY, Lee JK, Cho YS, Min S-J. Construction of 8-Azabicyclo[3.2.1]octanes via Sequential DDQ-Mediated Oxidative Mannich Reactions of N-Aryl Pyrrolidines. Org Lett. 2018;20(4):1175–1178.
- Seo JM, Hassan AHE, Lee YS. An expeditious entry to rare tetrahydroimidazo[1,5-c]pyrrolo[1,2-a]pyrimidin-7(8H)-ones: a single-step gateway synthesis of glochidine congeners. Tetrahedron. 2019;75(51):130760.
- Lue H-w, Derrick DS, Rao S, Van Gaest A, Cheng L, Podolak J, Lawson S, Xue C, Garg D, White R, et al. Cabozantinib and dasatinib synergize to induce tumor regression in non-clear cell renal cell carcinoma. Cell Rep Med. 2021;2(5):100267.
- Liu AY. Differential Expression of Cell Surface Molecules in Prostate Cancer Cells. Cancer Res. 2000;60:3429–3434.
- Farag AK, Hassan AHE, Chung K-S, Lee J-H, Gil H-S, Lee K-T, Roh EJ. Diarylurea derivatives comprising 2,4-diarylpyrimidines: discovery of novel potential anticancer agents via combined failed-ligands repurposing and molecular hybridization approaches. Bioorg Chem. 2020;103:104121.
- Hassan AHE, Lee K-T, Lee YS. Flavone-based arylamides as potential anticancers: design, synthesis and in vitro cell-based/cell-free evaluations. Eur J Med Chem. 2020;187:111965.
- Farag AK, Hassan AHE, Ahn BS, Park KD, Roh EJ. Reprofiling of pyrimidine-based DAPK1/CSF1R dual inhibitors: identification of 2,5-diamino-4-pyrimidinol derivatives as novel potential anticancer lead compounds. J Enzyme Inhib Med Chem. 2020;35(1):311–324.
- Hassan AHE, Choi E, Yoon YM, Lee KW, Yoo SY, Cho MC, Yang JS, Kim HI, Hong JY, Shin J-S, et al. Natural products hybrids: 3,5,4′-Trimethoxystilbene-5,6,7-trimethoxyflavone chimeric analogs as potential cytotoxic agents against diverse human cancer cells. Eur J Med Chem. 2019;161:559–580.
- Elkamhawy A, Paik S, Hassan AHE, Lee YS, Roh EJ. Hit discovery of 4-amino-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide: a novel EGFR inhibitor from a designed small library. Bioorg Chem. 2017;75:393–405.
- Hassan AHE, Yoo SY, Lee KW, Yoon YM, Ryu HW, Jeong Y, Shin J-S, Kang S-Y, Kim S-Y, Lee H-H, et al. Repurposing mosloflavone/5,6,7-trimethoxyflavone-resveratrol hybrids: discovery of novel p38-α MAPK inhibitors as potent interceptors of macrophage-dependent production of proinflammatory mediators. Eur J Med Chem. 2019;180:253–267.
- Hassan AHE, Phan T-N, Choi Y, Moon S, No JH, Lee YS. Design, Rational Repurposing, Synthesis, In Vitro Evaluation, Homology Modeling and In Silico Study of Sulfuretin Analogs as Potential Antileishmanial Hit Compounds. Pharmaceuticals. 2022;15(9):1058.
- Lee J-H, Lee H-H, Ryu KD, Kim M, Ko D, Chung K-S, Hassan AHE, Lee SH, Lee JY, Lee K-T. KCP10043F Represses the Proliferation of Human Non-Small Cell Lung Cancer Cells by Caspase-Mediated Apoptosis via STAT3 Inactivation. JCM. 2020;9(3):704.
- Hong JY, Chung K-S, Shin J-S, Lee J-H, Gil H-S, Lee H-H, Choi E, Choi J-H, Hassan AHE, Lee YS, et al. The Anti-Proliferative Activity of the Hybrid TMS-TMF-4f Compound Against Human Cervical Cancer Involves Apoptosis Mediated by STAT3 Inactivation. Cancers. 2019;11(12):1927.
- Hassan AHE, Mahmoud K, Phan T-N, Shaldam MA, Lee CH, Kim YJ, Cho SB, Bayoumi WA, El-Sayed SM, Choi Y, et al. Bestatin analogs-4-quinolinone hybrids as antileishmanial hits: design, repurposing rational, synthesis, in vitro and in silico studies. Eur J Med Chem. 2023;250:115211.
- Hassan AHE, Phan T-N, Moon S, Lee CH, Kim YJ, Cho SB, El-Sayed SM, Choi Y, No JH, Lee YS. Design, synthesis, and repurposing of O6-aminoalkyl-sulfuretin analogs towards discovery of potential lead compounds as antileishmanial agents. Eur J Med Chem. 2023;251:115256.