
Abstract
Novel 5-deazaflavins were designed as potential anticancer candidates. Compounds 4j, 4k, 5b, 5i, and 9f demonstrated high cytotoxicity against MCF-7 cell line with IC50 of 0.5–190nM. Compounds 8c and 9g showed preferential activity against Hela cells (IC50: 1.69 and 1.52 μM respectively). However, compound 5d showed notable potency against MCF-7 and Hela cell lines of 0.1 nM and 1.26 μM respectively. Kinase profiling for 4e showed the highest inhibition against a 20 kinase panel. Additionally, ADME prediction studies exhibited that compounds 4j, 5d, 5f, and 9f have drug-likeness criteria to be considered promising antitumor agents deserving of further investigation. SAR study showed that substitutions with 2-benzylidene hydra zino have a better fitting into PTK with enhanced antiproliferative potency. Noteworthy, the incorporation of hydrazino or ethanolamine moieties at position 2 along with small alkyl or phenyl at N-10, respectively revealed an extraordinary potency against MCF-7 cells with IC50 values in the nanomolar range.
Graphical abstract
Introduction
Cancer is a major threat to public health, so our recent considerable interest is to design and develop new 5-deazaflavins as potent antitumor agents, with high selectivity towards different tumour cells. Our previous publicationsCitation1–7 reported the promising antitumor activities of 5-deazaflavins against different cell lines and their preferential binding affinities to PTK, with reasonably low binding free energies. The 10-Alkyl-2-deoxo-2-methylthio-5-deazaflavins and 10-alkyl-2-deoxo-2-dimethylamino-5-deazaflavins have been found to exhibit significant antitumor activitiesCitation3. The biological screening of a series of 5-deazaflavins revealed their ability to stabilise and activate p53, as it was found that they act as low molecular weight inhibitors of the E3 activity of HMD2 in tumours that retain wild-type p53Citation8,Citation9. 5-Deazaflavins (5-deaza isoalloxazines)Citation1,Citation2, a modified type of flavocoenzymeCitation10, are 10-alkylpyrimido[4,5-b]quinoline-2,4(3H,10H)-dioneCitation3, have been first synthesised as potential riboflavin antagonistsCitation11. Structurally, 5-deazaflavins can be considered analogs of flavin and nicotinamide nucleotide (), therefore it would be expected that 5-deazaflavins abstract hydrogen equivalents from hydrogen donors under certain conditionsCitation12.
Figure 1. Structure of 5-deazaflavin skeleton.
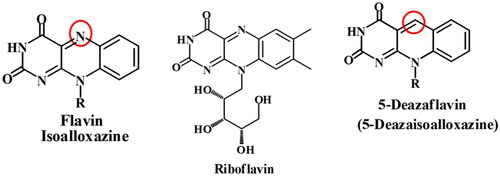
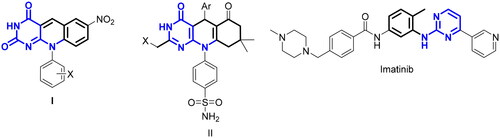
Naturally occurring coenzyme factor 420 (F420), isolated from the methane-producing bacteria, has the 5-deazaflavin skeleton as an 8-hydroxy-5-deazaflavin derivative and acts as coenzymes in the oxidoreductase system from CO2 to methaneCitation13–15. 5-Deazaflavins have long been known for their strong oxidising power which oxidises alcoholsCitation16, and amines to the corresponding carbonyl compoundsCitation17, and they behave as auto-recycling turnover catalysts in the redox reactionCitation12. The synthetic 5-deazaflavin derivatives have been reported to exhibit wide bioactivity as antimicrobial, antimalarial, anti-inflammatory, antioxidant, and antitumor activitiesCitation18,Citation19. A series of 5-deazaflavin derivatives have been reported as antitumor agents via a different mechanism of action. The 10-aryl-7-nitro-5-deazaflavins (I) were investigated as inducers of apoptosis and cell cycle arrest through activation of P538. In addition to the 5-deazaflavin analogs (II) incorporating sulphonamide moieties were reported to act as radiosensitizers and displayed a wide range of antiproliferative activityCitation20. Moreover, 5-deazaflavins have gained much attention as inhibitors of PTK due to their promising in vitro PTK inhibitory activity and potent antiproliferative activityCitation5. The PTK inhibitory activity of 5-deazaflavins may be attributed to the pyrimido[4,5-b]quinoline scaffold which could act as a hinge binder moiety and their structural similarity to imatinib as a rigid analog ().
Figure 2. Structures 5-deazaflavins anticancer agents and their structural similarity to the well-known kinase inhibitors imatinib.
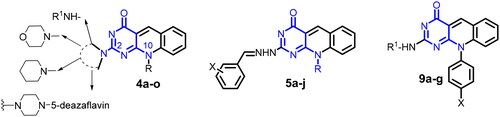
In the current study, we present the design and development of new series of 5-deazaflavins (4a–f, 5a–j, and 9a–g) (), through different modifications at C-2 and N-10 positions. The new derivatives were screened as potential antitumor agents against MCF-7 and Hela cell lines, hoping to introduce promising anticancer agents based on the 5-deazaflavin scaffold for further investigations. The proposed mechanism of action for the potential candidates was evaluated by docking into the active site of c-Kit kinase compared to its native co-crystalized ligand imatinib (PDB code: 1t46).
Figure 3. General structure of the targeted 5-deazaflavins as novel anticancer agents.
Results and discussion
Chemistry
Synthesis of 5-deazaflavins could be achieved from suitably functionalised pyrimidine derivatives such as barbituric acidCitation21, 6-(N-substituted)aminopyrimidinesCitation1,Citation2,Citation18,Citation19, and 6-chloro-5-formylpyrimidinesCitation22,Citation23. Alternatively, the appropriate quinoline derivatives provide another starting material for the synthesis of 5-deazaisoalloxazineCitation24,Citation25. The third pathway to prepare 5-deazaisoalloxazine derivatives involves the direct reaction of these compounds with the suitable reagent to get a certain target compound.Citation10,Citation24 As continuing our previous studies of 5-deazaflavinsCitation1–7, in this study, we explore the effect of different substituents at C-2 or N-10 positions on the antitumor activity of 5-deazaflavins. We illustrate the synthetic approaches utilised for the preparation of various novel 5-deazaflavin derivatives namely: 2-(N-substituted amino and heterocyclic amino)-7-(unsubstituted/substituted)-10-alkyl-2-deoxo-5-deazaflavins (4a–h) and (4k–m), 2-Hydrazino-10-alkyl-2-deoxo-5-deazaflavins (4i,j), 2,2′-(Piperazine-1,4-diyl)bis(10-alkyl-2-deoxo-5-deazaflavins), 2[(E)-2-(substituted)benzylidenehydrazino]-10-alkyl-2-deoxo-5-deazaflavins (5a–j), 10-aryl-2-deoxo-2-thioxo-5-deazaflavins (8a–c), and 2-(substituted amino)-10-aryl-2-deoxo-5-deazaflavins (9a–g) to afford new candidates as antitumor agents via PTK inhibition. Compounds 4a–o and 9a–g were prepared by the nucleophilic substitution of 10-alkyl-2-deoxo-2-methylthio-5-deazaflavins (3a–c) or 10-aryl-2-deoxo-2-thioxo-5-deazaflavins (8a–c), respectively, with various amines or hydrazine hydrate. While compounds 5a–j were synthesised by condensation of 4i or 4j with the appropriate aromatic aldehydes. The intermediates, 6-(N-alkyl substituted anilino)-2-methylthiopyrimidin-4(3H)-ones (2a-c) were synthesised by heating under reflux a mixture of 6-chloro-2-methylthiopyrimidin-4(3H)-one (1) with the appropriate N-alkyl aniline derivative in n-butanol for 24–48 h, according to the reported procedureCitation26,Citation27, as shown in Scheme 1. The commercially unavailable N-methyl-4-ethylaniline was synthesised from 4-ethylaniline in 2 steps, by reaction of 4-ethylaniline with excess ethyl formate for 15 h, followed by reduction of N-(4-ethyl phenyl)formamide with LiAlH4 in dry tetrahydrofuran according to the reported method.Citation28 The intermediates 10-alkyl-2-deoxo-2-methylthio-5-deazaflavin derivatives (3a–c) were prepared by the reaction of 6-(N-alkylamino)-2-methylthiopyrimidin-4(3H)-ones (2a–c) with Vilsmeier reagent (DMF-POCl3) at 90 °C for 1–1.5 h according to the reported methodCitation3. Literature survey indicated that nucleophilic substitution on 2-methylthiopyrimidine and 2-methylthiopteridine with an appropriate amine could be performed by heating with freshly distilled amine without solventCitation28,Citation29, or in presence of DMFCitation30. In the present investigation, the 2-methylthio group was replaced by appropriate amino or hydrazino moieties to afford the targeted compounds (4a–m), by heating under reflux a mixture of (3a–c) with an appropriate benzylamine, cyclohexylamine, morpholine or piperidine in absolute ethanol. The targeted compounds (4n,o) were synthesised by refluxing (3a,b) with piperazine (ratio 2:1) in absolute ethanol, as depicted in Scheme 1.
Scheme 1. General method for the preparation of 6-(N-Alkyl substituted anilino)-2-methylthiopyrimidin-4(3H)-ones (2a–c), 7-(unsubstituted/substituted)-10-alkyl-2-deoxo-2-methylthio-5-deazaflavins (3a–c), 2-(N-substituted amino-10-alkyl-2-deoxo-5-deazaflavins (4a–h), 2-hydrazino-10-alkyl-2-deoxo-5-deazaflavins (4i, j), 2-(heterocyclic amino)-7-(unsubstituted/substituted)-10-alkyl-2-deoxo-5-deazaflavins (4k-m), and 2,2'-(Piperazine-1,4-diyl)bis(10-alkyl-2-deoxo-5-deazaflavins) (4n,o). Reagent and conditions: (a) N-alkyl aniline, n-butanol, reflux, 24–48 h; (b) DMF-POCl3, 90 °C, 1–1.5 h; (c) primary aliphatic amine, benzylamine or hydrazine hydrate, EtOH, reflux, 2–4 h; (d) secondary heterocyclic amine, EtOH, reflux, 6–7 h; (e) piperazine, EtOH, reflux, 6.5–7 h.
The reaction of aromatic aldehydes with hydrazines could be conducted in glacial acetic acidCitation31, ethanolCitation32,Citation33, ethanol/H2SO4Citation34, ethanol/acetic acidCitation35, or ethanol/KOHCitation36. In the present work, the targeted Schiff bases (5a–j) were synthesised by refluxing the corresponding 2-hydrazino-10-alkyl-2-deoxy-5-deazaflavins (4i,j) with the appropriate aldehyde in absolute ethanol for 2–3 h, as shown in Scheme 2.
Scheme 2. General method for the preparation of 2[(E)-2-(substituted)benzylidenehydrazino]-10-alkyl-2-deoxo-5-deazaflavins (5a–j). Reagent and conditions: aromatic aldehyde, EtOH, reflux, 2–3 h.
![Scheme 2. General method for the preparation of 2[(E)-2-(substituted)benzylidenehydrazino]-10-alkyl-2-deoxo-5-deazaflavins (5a–j). Reagent and conditions: aromatic aldehyde, EtOH, reflux, 2–3 h.](/cms/asset/17f2db1d-3590-4110-a779-d64d62c6f716/ienz_a_2220570_sch0002_b.jpg)
The synthesis of different N-aryl substituted 5-deazaflavins (9a–h) is described in Scheme 3. The N-arylaminopyrimidine derivatives (7a–c) could be prepared via transamination reaction between the appropriate arylamine hydrochloride and the correspondi312ng aminopyrimidinesCitation37–39. In the present study, intermediates (7a–c) were prepared by heating a mixture of 6-amino-2-thiouracil (6) and the appropriate arylamine together with anilinium chloride at 170 °C, according to the reported method.Citation4 Then, intermediate 10-aryl-2-deoxo-2-thioxo-5-deazaflavins (8a–c) were prepared by heating the appropriate intermediate (7a–c) with o-bromobenzaldehyde in DMF activitiesCitation7,Citation18,Citation40. Finally, the targeted Compounds (9a–g) were synthesised by condensation of 10-aryl-2-deoxo-2-thioxo-5-deazaflavins (8a–c) with different amines or hydrazine hydrate via nucleophilic substitution reaction.
Scheme 3. General method for the preparation of 10-aryl-2-deoxo-2-thioxo-5-deazaflavins (8a–c) and 2-(substituted amino)-10-aryl-2-deoxo-5-deazaflavins (9a–h). Reagent and conditions: (a) arylamine, anilinium chloride, 170 °C, 9 h; (b) o-bromo-benzaldhyde, DMF, 110 °C, 1.5–2 h; (c) primary aliphatic amine, EtOH, reflux, 5–7 h.
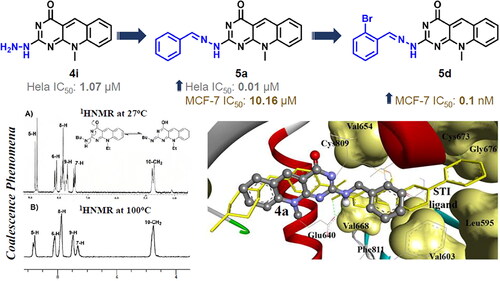
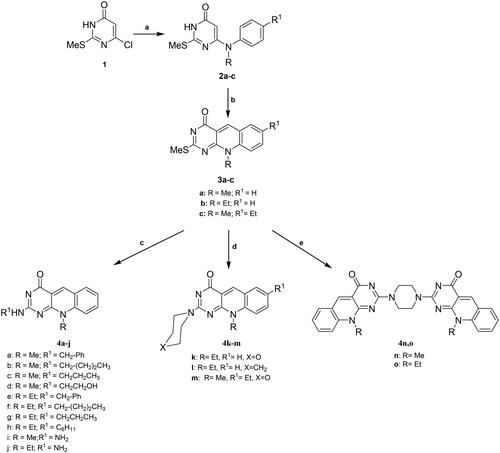
UV–vis, IR, NMR spectra, and elemental analyses were used for structural elucidation and identification of the newly assigned compounds. 1H NMR spectrum of the key intermediate 2c revealed a singlet signal at 4.76 ppm integrated for the characteristic proton at position 5 of the pyrimidine ring, in addition to the signals assigned for methyl, ethyl, aromatic, and NH protons. The cyclized 7- ethyl-10-methyl-2-deoxo-2-methylthio-5-deazaflavin (3c) was confirmed by the appearance of the characteristic singlet signal of the methine-proton at 9.19 ppm along with the disappearance of the singlet signal at 4.76 ppm. The structures of 2–(2-(N-substituted amino)-7-(unsubstituted/substituted)-10-alkyl-2-deoxy-5-deazaflavins (4a–m) were confirmed by the disappearance of the singlet signal of the 2-methyl thio group at δ2.51–2.63 ppm, also they showed a characteristic singlet signal of the C5-proton at the downfield of 8.45–8.98 ppm. Interestingly, the coalescence phenomenon, reversible inter-conversion of two isomers at 27 °C in the case of the 2-monoalkyl amino derivatives (4a–j), was observed as tautomerism in 1H-NMR spectra, as shown in the 1HNMR spectrum of compound 4f, . The twin overlapped spectra of approximately (1:2 or 2:1) ratio of the 2-monoalkyl amino and 2-monoalkyl imino tautomers in DMSO-d6 were obtained for the two tautomers of 4a–j. At a higher temperature of 100 °C, the coalescence of the duplicated signals was observed.
Figure 4. The 1H-NMR spectra for compound 4f at 27 °C exhibiting the coalescence phenomenon (A) and at 100 °C (B) in DMSO-d6, showing a single conformation spectrum.
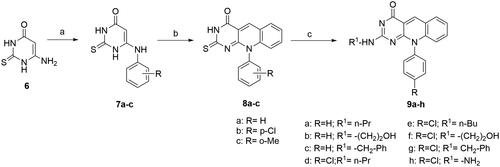
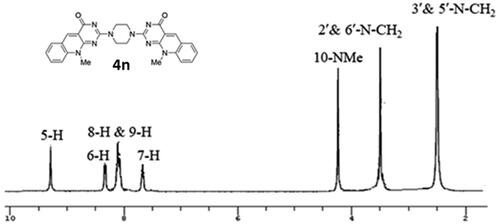
The structures of 2,2′-(piperazine-1,4-diyl) bis(10-alkyl-2-deoxo-5-deazaflavins) (4n,4o) were confirmed by the presence of signals attributed to the piperazine -CH2 protons at 2.46– 4.14 ppm in their 1H-NMR spectra, as depicted in . Moreover, the mass spectra showed the molecular ion peaks at m/z = 507 [M + 3] + (3% fragment due to protonated two nitrogen of the piperazine ring), and at m/z = 533[M + 1] +, respectively, which were consistent with their molecular weight.
Figure 5. 1HNMR spectrum of compound 4n.
As indicated in , the 1H-NMR spectrum for compound 4f at room temperature exhibited a complex duplicated pattern for all peaks, where C5-proton appear as duplicated signals at 8.77 and 8.84 ppm, N10–CH2 at 4.70 and 4.76 ppm, and for aromatic protons 6, 7, 8, and 9 at 7.46–8.10 ppm. Running the 1H-NMR experiment at 100 °C, the complex duplicated spectral pattern was resolved and the spectral signals for all duplicated protons coalesced as shown in . The 2-n-butyl imino tautomer is proposed predominantly at a higher temperature because it has a heat of formation (PM3-Mozyme) of 75.52 Kcal/mol, which is higher than that of 2-butylamine tautomer (65.62 Kcal/mol).
1H-NMR spectra of 2[(E)-2-(substituted)benzylidenehydrazino]-10-alkyl-2-deoxo-5-deazaflavins (5a–j) demonstrated a singlet signal at δ 8.31–8.67 ppm for the imine CH proton, for instance, compound 5c, the characteristic singlet signal of the C5 protons appeared more deshielded at δ: 8.72–8.97 ppm than those of their corresponding hydrazine derivatives 4i or 4j (δ: 8.45 ppm). Interestingly, the coalescence phenomenon was not observed in the 1HNMR spectra of 2-[2-(substituted) benzylidene hydrazino] derivatives (5a–j), due to the delocalisation of the proton on the nitrogen of the guanidine group with the conjugated system of the benzyl imino group by resonance.
The structure of 6-(p-Chloroanilino)-2-thiouracil (7b) was confirmed by the appearance of two doublet signals (AB spin system) of the aromatic protons at δ 7.24–7.43. The deazaflavins (8a–c) were confirmed by the appearance of the deshielded singlet signal of the C5-proton at δ 9.24–9.29, for instance, compound 8a, compared to the C-5 proton of (7a-c) which was appeared at δ 4.8–5.19. Mass spectra of compounds 8a and 8b revealed [M]+ and [M-1] at m/z = 305 and 338, respectively which were consistent with their molecular weight.
Moreover, The 1H NMR spectra of 2-(substituted amino)-10-aryl-2-deoxo-5-deazaflavins (9a–h) demonstrated the signals integrated for the N-alkyl protons in the aliphatic region of their 1H NMR spectra, also they showed the coalescence phenomenon for compound 9a, while the 10-(p-chlorophenyl)-2-hydrazinyl-2-deoxo-5-deazaflavin (9h), was confirmed by the appearance of two singlet signals in its 1H NMR spectrum corresponding to 2-NH and 2-NHNH2 which disappeared on the addition of D2O, by the appearance of a broad stretching band at 3420–3140 cm−1 of (NH–NH2) in the IR spectrum, and by the appearance of a characteristic peak for the 2-imino fragment at m/z = 322 in its mass spectrum.
The UV spectra of 2-(N-substituted amino)-10-alkyl-2-deoxo-5-deazaflavins (4a–m) showed four absorption maxima at 220–224, 270–276, 308–327, and 403–414 nm, together with an absorption shoulder at 418–437 nm. The absorption maxima for the 2-hydrazino derivatives (4i, j) showed at longer wavelengths than those for the 2-(N-substituted amino) derivatives (4a–m) due to the hydrazino moiety, which exhibited as an additional source for a lone pair of electrons. Generally, the UV spectra of 10-alkyl-2-deoxo-2-methylthio-5-deazaflavin (3a–c) exhibited the absorption band at a longer wavelength than those of 2-(N-substituted amino)-10-alkyl-2-deoxo-5-deazaflavins (4a–m) as shown in . This is attributed to the S-atom which causes a general redshift (bathochromic shift) in the spectrum due to its easier polarizability.
Figure 6. UV–VIS spectra of 3c, 4d, 4m, and 4o.
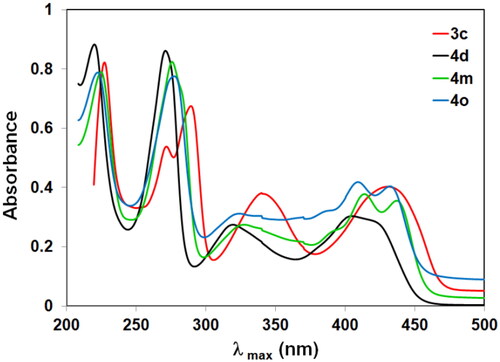
The UV spectra of compounds 5a–j exhibited longer wavelength than those of 2-Hydrazino-10-alkyl-2-deoxo-5-deazaflavins (4i, j). This is attributed to the benzylidine group at 2-position which increases the conjugated system, and so causes a generally bathochromic shift in the spectrum especially when the phenyl ring was substituted by an electron-withdrawing group (an auxochrome group). Additionally, UV spectra of compounds 8a–c showed that compound 8b exhibited longer wavelength than the other 10-aryl-2-deoxo-2-thioxo-5-deazaflavin analogs (8a, c), this attributed to the chlorine atom (an auxochrome group) at p-position which caused a bathochromic shift.
MTT antiproliferative activity assay
Out of the 36 synthesised compounds, as depicted in Schemes 1–3, 29 representative compounds (4a, 4e, 4i, 4j, 4k, 4m, 4n, 4o, 5a–j, 8a–c, and 9a–h) were validated against MCF-7 and HeLa cell lines for their in vitro growth inhibitory activities using MTT assay using 5-FU as a positive control. This study was conducted to enable a meaningful discussion of the structure-activity relationship (SAR).
Both MCF-7 and HeLa cells have been extensively characterised and widely used in cancer research, making them a convenient choice for evaluating the efficacy and toxicity of potential drug candidatesCitation41–43. Additionally, their response to drugs may provide insights into the mechanisms of action of the drugs, which can help to guide drug development and optimisation. MCF-7 and HeLa cells are two commonly used cancer cell lines that have been shown to express a variety of PTKs. MCF-7 cells are a widely used breast cancer cell line that has been extensively characterised over the years. These cells express several markers and genes that are characteristic of breast cancer, including the oestrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2)Citation44–46. HER2 is a growth factor receptor that is overexpressed in a subset of breast cancers and is associated with a more aggressive disease phenotype which was involved in our PTK profiling as a target of breast cancer therapy. Other genes and markers that are commonly expressed in MCF-7 cells include cytokeratin 18 (CK18), a marker of epithelial cells, and the cell cycle regulator p53, which is frequently mutated in breast cancerCitation47. These markers and gene expression profiles can be used to further characterise MCF-7 cells and to study the effects of compounds on breast cancer cell growth and proliferation.
On the other hand, HeLa cells are a cervical cancer cell line that is widely used in research due to their ability to grow rapidly and easily in culture. They have also been used in studies of the cell cycle, cancer genetics, and drug screeningCitation42. HeLa cells are known to express several kinases, including EGFR and FGFR1, as well as non-receptor tyrosine kinases such as Src and FAK. In addition, HeLa cells express several serine/threonine kinases such as AKT and MAPK that are involved in various signalling pathways regulating cell growth and survivalCitation48–51. It’s worth noting that the expression levels and activity of these kinases can vary depending on the specific HeLa cell line and experimental conditions.
As shown in ; substitution with N-Et at position 10 gives superior inhibitory activity against the MCF-7 cell line for compounds 4e, 4j, and 4k (IC50: 0.03, 0.0005 and 0.09 μM), respectively, compared to their N-Me analogs with 4a, 4i and 4m (IC50: 4.49, 0.58 and 0.20 μM), respectively. Additionally, substitution with a hydrazino moiety (-NHNH2) at position 2 revealed an outstanding inhibitory activity in a nanomolar range, as demonstrated by the IC50 value of the targeted compound 4j (IC50: 0.5 nM). To investigate the significance of the C-2 hydrazino moiety, it was allowed to react with different aromatic aldehydes to afford the corresponding imino compounds (5a–j). Generally, the imine substitution tends to decrease the potency of the N-Et-2-benzylidene derivatives (5f–j) (IC50: 0.001–5.73 μM) compared to the corresponding N-Et-2-hydrazino derivative 4j (IC50: 0.5 nM), this effect was more significant on the activity of the Para substituted benzylidene derivative 5h (IC50: 5.73 μM). This may be attributed to its bulky planar structure. In contrast, the imine substitution resulted in an exceptional increase in potency against the MCF-7 cell line, as demonstrated by the lowest IC50 value of the N-Me-2-benzylidene derivative 5d (IC50: <0.0001 μM) compared to the corresponding N-Me-2-hydrazino derivative 4i (IC50: 0.58 μM). A further modification was implemented to explore the effect of the substituent at C-2 and N-10 positions. Incorporation of C2-thioxo and N10-Aryl moieties resulted in diminished antiproliferative activities more significantly against the MCF-7 cell line than the Hela cell line, as indicated by the high IC50 values of compounds (8a–8c) (IC50: 23.15– >100 μM). Nevertheless, the nucleophilic substitution of the C2-thioxo group of 8a and 8b with different amines results in restoring the antiproliferative activity, as demonstrated by compounds 9a–9g. In general, our targeted compounds revealed a sort of preferential antiproliferative activities against the MCF-7 cell line than the Hela cell line. However, compounds 4i, 4o, 5d, 5g and 9d showed promising antiproliferative activities against both cell lines. The results considering the structure-activity relationship (SAR) revealed that the dimer compound incorporating ethyl group at N-10 position, 2,2′-(piperazine-1,4-diyl)bis(10-ethyl-2-deoxo-5-deazaflavin) (4o) showed significant growth inhibitory activity against MCF and HeLa tumour cell line of IC50: 0.98 and 1.13 μM, respectively, as shown in .
Table 1. Antiproliferative activity (IC50; µM) of compounds (4a, 4e, 4i, 4j, 4k, 4m, 4n, 4o, 5a–j, 8a–c, and 9a–h) against MCF-7 and Hela cell lines.
The differential selectivity of compounds 8c and 9c against MCF7 (>100 and 1.0 µM) and Hela cell lines (1.69 and 75.21 µM) can be postulated by the difference nature of these two compounds as illustrated below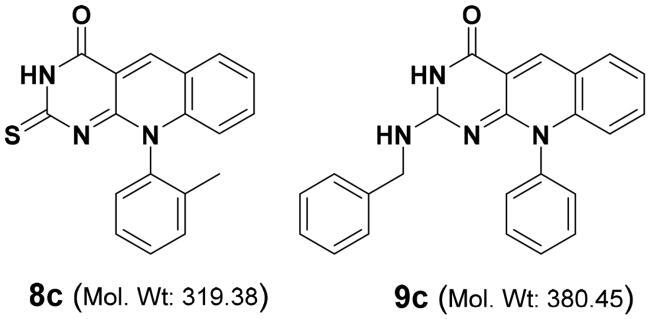
The differential activity of these two compounds may be due to various factors, such as differences in the expression of receptors, enzymes, or transporters in the two cell lines, as well as differences in the signalling pathways and gene expression profiles. Where the MCF-7 cell lines are invasive breast ductal carcinoma as Es+; Prog+; and HER2- cellsCitation52, while the Hela cell is an aggressive strain of cervical cancer cells that express different markers with a high degree of genomic instability, with multiple chromosomal rearrangements and aneuploidies, which contribute to their aggressive phenotype. Hela cell lines have been used to investigate the mechanisms underlying tumorigenesis, drug resistance, and metastasis. Due to their robust proliferation and transformation potentialCitation53.
Accordingly, compound 8c, with 2-thioxo moiety with potential antioxidant activity and a smaller molecular weight that enables it to avoid any potential resistance or efflux in the Hela cells revealing more potency (IC50 = 1.69 µM) than the MCF7 (IC50 > 100 µM). Identifying new scaffolds with differential selectivity against cancer cell lines may help in the development of more selective and effective antitumor agents
In vitro protein kinase assay
The protein kinase profiling for two compounds (4e and 4k) was conducted against 20 protein kinases at 10 µM concentration in single measurements including the reference compound imatinib, (also known as Gleevec) using the radiometric assay method. The protein kinases employed in the compound profiling process were cloned, expressed, and purified using proprietary methods. Quality control testing is routinely performed to ensure compliance with acceptable standards. 33P-ATP was purchased from PerkinElmer. All other materials were of standard laboratory grade. The compounds were supplied by the client in powder form. Kinexus Corp. evaluated the profile of 5 compounds against a panel of protein kinases by employing the standardised assay methodology outlined in the experimental part. The results observed as % activity change and % inhibition compared to the control are presented in . The intra-assay variability was determined to be less than 10%. Inhibition of target activity by the compound gives negative (−) values while activation of target activity gives positive (+) values. Kinexus considers only values of >25% change in activity compared to control to be significant. The profiling data for the two compounds tested against a panel of protein kinases using the radiometric assay method revealed inhibitions and activations at low to moderate levels, ranging from 1 to 22% for inhibitions and 1–24% for activations (). There was low to moderate inhibition (10–29%) with the compounds, as compared to the controls, against a select number of the kinases tested, including Compound 4e: ACVR2A, CAMK1, FAK, and SRC; Compound 4k: ACVR2A, and CAMK1; and Imatinib: BRAF, CAMK1, FAK, FLT1, and JNK. Overall, only 6 of the 20 kinases tested, including ACVR2A, CAMK1, EGFR, FAK, and SRC, showed varying levels of inhibition at 10 µM concentrations with the test compounds.
Table 2. % Activity or corresponding % inhibition of 20 kinases in the presence of 2 compounds and a reference compound (Imatinib) using the radiometric assay method.
Out of these 6 kinases, only CAMK1 was inhibited to varying extents (from 1% to 14% inhibition) by all three compounds. Compound 4e with a 14% inhibition. The compound Imatinib, which was included as a reference compound, gave high inhibitions with the kinases ABL1 (81%) and c-KIT (72%), and moderate inhibitions with BRAF (25%), CAMK1 (20%), FLT1 (29%) and JNK1 (21%). In addition, some of the kinases were shown to have moderate activations (8% to 24%), as compared to the controls, with the test compounds, namely Compound 4e: FLT3; Compound 4k: CDK1/Cyclin A1, and FLT3; and Imatinib: CK1 alpha and FLT3.
Only 3 of the 20 kinases tested, including CDK1/Cyclin A1, and FLT3 showed varying levels of activation at 10 µM concentration with some of the test compounds. Out of these 3 kinases, only CDK1/Cyclin was activated (from 9 to 19% inhibition) by all of the compounds. The compound Imatinib, which was included as a reference compound, also gave slight activations with the kinases CK1 alpha (11%) and FLT3 (17%). All the other measurements for activations or inhibitions, which were in the ± 10% range, were considered to be insignificant.
Apoptosis assay
Both kinase inhibition and apoptosis induction can be effective strategies for treating cancer, they have different modes of action. Therefore, the potential kinase selectivity that regulates cellular signalling pathways was initially conducted for compounds 4e and 4k from Scheme 1 (IC50 against MCF7 cells: 0.03 and 0.09 µM, respectively) at 10 µM concentration compared with imatinib as a positive control. This screening exhibited insignificant kinase inhibition that means kinase is not the potential target for our compounds. Therefore, Annexin V apoptosis assay using MCF7 cells was conducted for the most potent compounds 4j and 9f from Scheme 1 and 3 (IC50 against MCF7 cells: 0.5 and 4.57 nM, respectively) to screen their potential mechanism of action as apoptotic inducers.
MCF-7 is a commonly used cell line for breast cancer research, and it has been extensively characterised and utilised in a variety of studies due to (1) MCF-7 cells have been extensively characterised and their genetic characteristics are well understood. This may make them a valuable model for investigating the mechanisms of action of target compounds, (2) MCF-7 cells are derived from human breast cancer tissue, which makes them a clinically relevant model system for studying cancer biology and drug development, (3) MCF-7 cells are known to have a high proliferation rate, which makes them a suitable model system for studying cell cycle arrestCitation54, and (4) The well-established and extensively characterised model of MCF-7 cells is a widely used in cell cycle arrest and apoptosis assays, help ensure that the results are reliable and comparable with previous findingsCitation43,Citation52,Citation55.
Apoptosis induction is a process by which cancer cells undergo programmed cell death, which is a natural process that occurs in cells to maintain tissue homeostasis. Apoptosis is often dysregulated in cancer cells and inducing apoptosis can selectively eliminate cancer cells while sparing normal cells. In this study, compounds 4j and 9f induced late apoptosis to MCF-7 cells in a dose-dependent pattern. At the lowest concentration (1.0 µM), compounds 4j and 9f displayed a 24 and 44% increase in late-phase apoptosis compared to the control. However, compound 4j at the highest tested concentration (10 μM) demonstrated a 22 and 264% increase in early and late phase apoptosis compared to the control, respectively, as shown in . On the other hand, at the highest tested concentration (10 μM) of compound 9f, only a 22 and 44% increase in the early and late phase apoptosis was detected, respectively, . Therefore, the aforementioned results suggested that the compounds 4j and 9f revealed antitumor activity by inhibiting the proliferation and causing apoptosis of the cancer cells.
Figure 7. Annexin V FITC/PI apoptosis assay for MCF-7 cells treated separately with compounds (4d and 9f), at different concentrations (1, 5, and 10 μM) for 72 h. X-axis is annexin V and Y-axis is PI for each compound. C1 (upper left): necrotic cells (PI+/annexin V−); C2 (upper right): late apoptotic (PI+/annexin V+); C3 (lower left): live cells (PI−/annexin V−); C4 (lower right): early apoptotic cells (PI−/annexin V+). The data shown are the mean % cell number ± SD (n = 3).
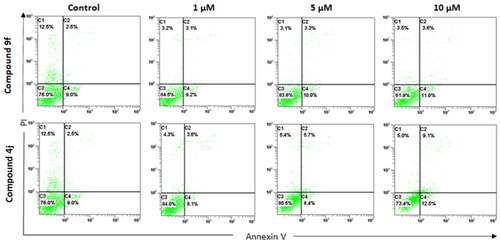
It is worth to note that performing an apoptosis assay at the IC50 value can be a useful starting point to determine the effect of the compound on apoptosis. However, it may not always be the optimal concentration for observing apoptotic effects, as different compounds can have different dose-response relationshipsCitation56–58. Therefore, for the apoptosis assay the best concentration depends on the specific experimental system being used, the type of cells being studied, and the particular assay being employed as reportedCitation58–60. Therefore, we screened the range of concentrations for compounds 4j and 9f that should be tested to determine the optimal concentration for inducing apoptosis. These optimal concentrations were determined by performing a dose-response curve to quantify the percentage of apoptotic cells in response to concentration increase which is 1–10 µM range in this study. The concentration that induces the highest percentage of apoptotic cells without causing excessive cell death can then be selected as the optimal concentration for the assay. These concentrations were selected to be high enough to cause a significant increase in the number of apoptotic cells, but not so high as to cause extensive cell death or necrosis. Accordingly, there was no detectable necrosis in : C1 (upper left): necrotic cells (PI+/annexin V−). It is also important to consider the time course of the assay, as some compounds may require longer exposure times to induce apoptosis, therefore we used 72 h as the time of exposure.
Molecular docking study
In the present investigation, we explored the binding affinities of the newly synthesised compounds using the molecular docking software Autodock 4.2Citation25 for future optimisation of promising compounds that demonstrate potent antiproliferative activities. The targeted 5-deazaflavin derivatives were docked into the 3D crystal structures of c-Kit receptor PTK (PDB: 1t46). The AutoDock binding affinities of the docked compounds were evaluated according to many parameters including, the binding free energies (ΔGb, kcal/mol), inhibition constants (Ki), hydrogen bonding, and RMSD values in comparison to the native co-crystallised ligand (STI-571(Imatinib).
Docking validation
Validation of the accuracy and reproducibility of the GOLD 5.2.2 docking program was conducted by redocking the co-crystallised ligands into their corresponding six kinases namely: Imatinib (STI571, Gleevec) (PDB: 2hyy)Citation61; Vemurafenib (PDB: 4rzv)Citation62; TI-571 c-kit kinase (PDB: 1t46)Citation63; 1(3,5-dihydro [1,2,4] triazino[3,4-c][1,4]benzoxazin-2(1H)-one) (PDB: 4q9s)Citation64; N-(4-Chlorophenyl)-2-((pyridin-4-ylmethyl)amino)benzamide (PDB: 3hng)Citation65; and Bosutinib (PDB: 4mxo)Citation66.
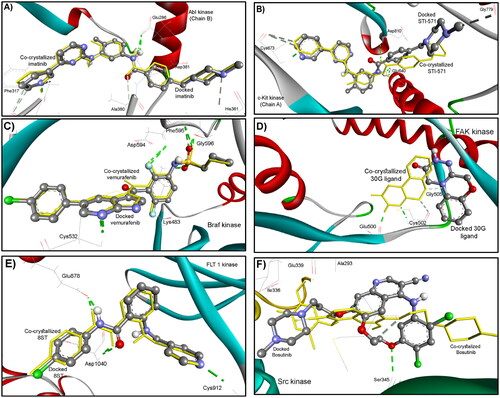
All these ligands were docked superimposed on the natively co-crystallised bound ligands within a root mean square deviation (RMSD) of 0.21, 1.30, 0.22, 3.97, 0.37, and 0.92 Å, respectively as shown in . This successful docking protocol with a similarity to the experimental findings confirms the accuracy of the Gold docking program, allowing for further docking of our test compounds in this study.
Figure 8. Docking validation of the reproducibility of GOLD 5.2.2. program for redocking of the co-crystalized ligands (ball and sticks coloured by elements) into their corresponding binding sites of (A) ABL (PDB: 2hyy); (B) c-Kit (PDB: 1t46), (C) B-Raf (PDB: 4rzv); (D) FAK (PDB: 4q9s), (E) FLT1 (PDB: 3hng); and (F) SRC (PDB: 4mxo) kinases compared to the native bound ligand (in sticks). The hydrogen bonds are shown in dashed lines.
The docking results of the test compounds into ABL, C-Kit, B-Raf, FAK, FLT1, and SRC kinases are tabulated in compared to the corresponding native bound ligand. As depicted in , the highest-scored and best-docked compounds are 4n, 5d,g, i,j, and 8c into Abl kinase; compounds 4e, 5b, 5d, i,j into B-Raf kinase, compounds 4e and 5d,i,j, and 9c,d,e,f,g into C-Kit kinase, compounds 4n,o, 5j, and 8b,c into FAK kinase, compounds 5i,j, and 9g into FLT1 kinase, and compounds 4m,n, 5a,b,c,d,e,f,g, and 8b into SRC kinase.
Table 3. Docking score results for the synthesised 5-deazaflavin analogs docked into six different kinases in comparison to the corresponding native bound inhibitor using the gold molecular docking program.
The most docked compounds in these kinases are 5d (), 5i, and 5j, followed by 4n, 9g, and 5b, whereas compound 9f is selectively bound into C-Kit and SRC kinases (). These results are correlated to some extent to the MTT antiproliferative assay, particularly for compounds 5d and 9f.
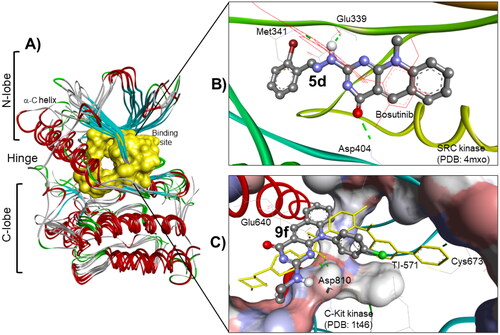
Figure 9. (A) The molecular overlay view of Abl (PDB: 2hyy), c-kit (PDB: 1t46), FAK (PDB: 4q9s), Src (PDB: 4mxo), B-raf (PDB: 4rzv), and FLT1 (PDB: 3hng) kinases showing the binding site in yellow surface view. (B) The binding mode of compound 5d into SCR kinase (PDB: 4mxo) reveals three hydrogen bonds with Met341, Glu339, and Asp404 amino acids. (C) The docking pose of compound 9f into C-Kit kinase (PDB: 1t46) within RMSD of 1.79 Å from the co-crystalized TI-571 ligand and exhibiting four hydrogen bonds with Glu640, Asp810, and Cys673 amino acids.
AutoDock binding affinities of the synthesised compounds into c-Kit receptor PTK
Further molecular docking protocol was conducted for the test compounds using AutoDock 4.2. program. This protocol was also validated by re-docking of the native bound ligand STI-571(Imatinib) in the c-Kit active site. The re-docking validation step reproduced the experimental binding mode of the co-crystallised ligand precisely demonstrating that the used docking protocol is suitable for the intended docking study. This was indicated by the small RMSD between the docked pose and the co-crystallised ligand 0.17 Å; and by the ability of the docking pose to reproduce all the key interactions accomplished by the co-crystallised ligands with the hot spots in the active sites; Thr670, Cys673, and Asp810. This validation step was followed by AutoDock binding affinities of the synthesised compounds considering the binding free energy (ΔGb, kcal/mol), inhibition constant (Ki), and hydrogen bonding, in comparison to the native co-crystallised ligand. Up to four hydrogen bonds were detected between the docked compounds and c-Kit receptor PTK. The main moieties of the docked 5-deazaflavin analogs that are involved in hydrogen binding are 4 C = O, 2NH, 3NH, and 6NH. The main amino acids of the PTK receptor involved are C = O, NH of Asp810, C = O of His790, NH of Cys673, OH of Thr670, C = O of Glu640, C = O of Ile789, SH of Cys788, and to a lower extent NH of Arg815. The formed hydrogen bonds are established within distances of 1.72–2.44 Å and angles of 104.6–171.5°. Where the binding free energies (ΔGb) revealed were in the range of (-8.12 to −14.51 kcal/mol), with corresponding inhibition constants (Ki) being 1.13 µM to 11.30 pM as represented in .
Table 4. Docking results for the synthesised 5-deazaflavin analogs docked into c-Kit receptor PTK (PDB: 1t46) in comparison to the native bound inhibitor STI-571(Imatinib), considering the binding free energies (ΔGb) and inhibition constants (Ki).
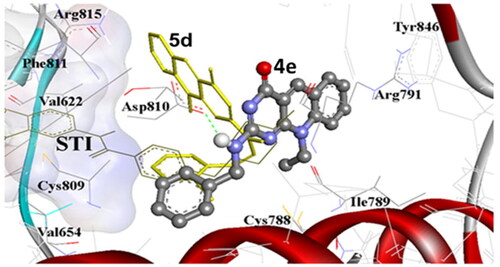
As represented in , many compounds in the present investigation exhibited good docking results into the binding site of c-Kit receptor PTK (PDB: 1t46) in comparison to the native co-crystallised STI-571 ligand, (Imatinib). Where the most promising compounds 4e, 4i, 4j, 5d, 5i, 9d, and 9f demonstrated binding free energies (ΔGb) range −12.08 to −14.27 kcal/mol, with up to four hydrogen bond interactions. These results are highly correlated to their antiproliferative potency against the MCF-7 cell line. Notably, compound 4i which exhibited potent activity against both MCF-7 and Hela cell lines with IC50 of 0.58 and 1.07 μM respectively, showed the maximum number of H-bonding interactions with binding free energy (ΔGb) of −12.63 kcal/mol. On the other hand, compound 4a which exhibited moderate antiproliferative activities displayed high binding affinity (ΔGb: −13.53 kcal/mol) with one hydrogen bond between its 2-amino group and C = O of Glu640 amino acid within RMSD of 2.78 Å. Thus, the docking result indicates that compound 4a is expected to be a reasonable candidate for c-Kit inhibition by its proper fitting into the binding site (). In addition, the docking of 2-morpholino-7-methyl-10-methyl-2-deoxo-5-deazaflavin (4m) exhibited binding free energy (ΔGb) of −11.65 with proper fitting into the binding site by one hydrogen bond between its 4-C = O and HS of Cys788. This result was also highly correlated to its antiproliferative potency against MCF-7 and HeLa cell lines, IC50: 0.20 and 4.31 µM, respectively (). Moreover, compounds 4e and 5d exhibited almost equal binding free energies (ΔGb) of −12.51 and −12.56 kcal/mol, respectively, and proper fitting into the binding site. These results were highly linked to their potency against MCF-7 and Hela tumour cell lines ().
Figure 10. The comparative AutoDock binding affinities of compound 4a (coloured by element, balls, and sticks) involving flexible docking into the binding pocket of the c-Kit PTK (1t46), shown as solid backbone ribbon with the STI ligand (dark sticks). The hydrogen bonds are shown as dashed lines.
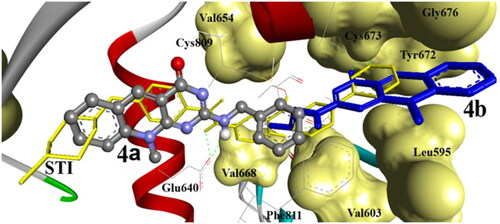
Figure 11. The binding affinity of compound 4m (balls and sticks) into c-Kit PTK (1t46). It exhibited one hydrogen bond with SH of Cys788. The binding site of the PTK is shown as a solid surface with labelled amino acids, STI cocrystallized ligand is shown as light sticks. The hydrogen bonds are shown as dashed lines.
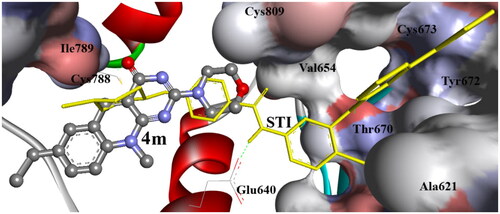
Figure 12. The comparative AutoDock binding affinities of compound 4e (coloured by element, balls, and sticks) and 5d (sticks) involving flexible docking into the binding pocket of the c-Kit PTK (1t46), shown as solid backbone ribbon with the STI ligand (lines) The hydrogen bonds are shown as dashed lines.
Compounds 4k, 4n, 4o, 5b, 5d, 5g, 5i, and 8b were docked within the binding pocket without any detectable hydrogen bonds. However, they were bound via hydrophobic electrostatic surface interaction of their planar pyrimidoquinoline ring, where it was sandwiched between the phenyl moieties of Phe 811, imidazole ring of His 790 and/or the terminal hydrocarbon chain of Leu 637, Val 643, Leu644, Val668, Lys 623 by hydrophobic attraction.
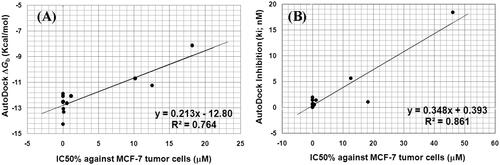
We tried to find a correlation between growth inhibitory activities (IC50, µM) of the synthesised compounds against MCF-7 and the binding affinities predicted by flexible docking. As shown in , The IC50 of compounds 4e, 4i, 4n, 5a, 5c, 5d, 5f, 5i, 5j, 9d, and 9f against (MCF-7), revealed a good correlation with their AutoDock binding free energies (ΔGb) being of correlation coefficient (R2) of 0.764 () and revealed a better correlation with their Autodock inhibition constant (Ki) of correlation coefficient (R2) of 0.861 ().
Figure 13. (A) Correlation between the IC50 (µM) against MCF-7 tumour cell lines and AutoDock binding free energy (ΔGb) for compounds 4e, 4i, 4n, 5a, 5c, 5d, 5f, 5i, 5j, 9d, and 9f. (B) correlation between the IC50 (µM) against MCF-7 tumour cell lines and AutoDock predicted inhibition (Ki) for compounds 4e, 4i, 4n, 5a, 5c, 5d, 5f, 5i, 5j, 9d, and 9f.
Physicochemical and predicted toxicity and pharmacokinetic properties of the synthesised derivatives
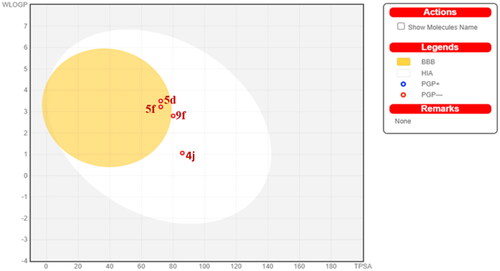
SwissADME online web tool provided by the Swiss Institute of Bioinformatics (SIB) was implemented for the calculation of the physicochemical properties in addition to the prediction of the pharmacokinetic properties and drug-likeness of the promising candidates 4j, 5d, 5f, and 9f. The submitted compounds are predicted to demonstrate promising physicochemical and pharmacokinetic properties. They exhibited a predicted wlogP in a range of 1.06–3.22, moderate water solubility, and high GIT absorption. Compounds 5d and 5f were expected to have BBB permeability. However, compounds 4j and 9f didn’t reveal a potential BBB permeability because of the high polarity attributed to the hydrazino or the terminal hydroxy ethanolamine moiety, respectively. The promising compounds fulfil the drug-likeness criteria set forth by the major pharmaceutical companies such as Ghose’s (Amgen), Lipinski’s (Pfizer), Muegge’s (Bayer), Veber’s (GSK) and Egan’s (Pharmacia). demonstrates the BOILED-Egg graph of the WLOGP vs TPSA (Topological Polar Surface Area) for the submitted compounds. All compounds are placed in the area of human intestinal absorption (HIA). The anticipated high GIT absorption of these compounds is due to their optimal physicochemical properties found in the suitable physicochemical properties range for oral bioavailability. Additionally, the graph shows that 4j, 5d, 5f, and 9f are not P-glycoprotein substrates (PGP-), hence they are not susceptible to the efflux process performed by this transporter, which is exploited as a drug-resistance mechanism by many tumour cell lines.
Figure 14. Predicted boiled-egg plot from SwissADME online web tool for compounds 4j, 5d, 5f, and 9f.
Regarding the potential toxicity of our most potent compounds against MCF-7 cell lines, namely 4e, 4j, 4k, 5d, and 9f with IC50 of 0.03, 0.0005, 0.09, <0.0001, and 0.00457 μM, respectively as listed in . This differential inhibitory effects of the most potent compounds without any toxic effects on Hela cell lines, particularly compounds 4j, 4k, and 9f, reflect the non-toxic actions of these compounds.
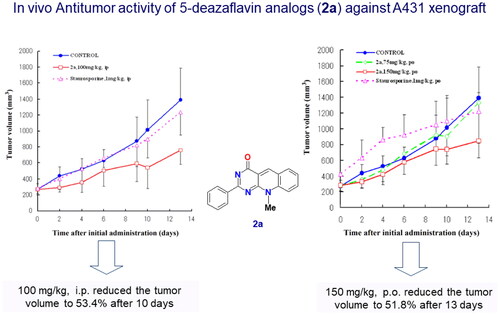
In addition, we have published 5-deazflavin scaffolds with different substitutionsCitation2,Citation3, in which we conducted in vivo study that revealed non-toxic effects on mice for 14 day I.P and P.O. administration as shown in the published figure belowCitation2().
Figure 15. In vivo study of compound 2a with a 5-deazaflavin scaffold indication non-toxic effect after 14 days administration in nude mice. Adopted from our research group publication (PubMed PMID: 17049252)Citation2.
Furthermore, we have conducted Toxicity prediction using ProTox-II softwareCitation67 (https://tox-new.charite.de/protox_II/index.php?site=compound_input) and Swiss ADMECitation68 (http://www.swissadme.ch/). The results cited below for compounds (4e, 4j, 4k, 5d and 9f) compared with Methotrexate (MTX) indicated that our compounds have the safest toxicity class of 6 in comparison with MTX of class 1 (highly toxic) as listed in . In addition, our compounds (4e, 4j, 4k, 5d and 9f) did not reveal any hepatotoxicity except compound 5d which has imine moiety, however its predicted class of 6 and predicted high LD50 of 10,000 mg/kg provide an enough safety profile that might need further study in future.
Table 5. Predicted toxicity study of the most potent compounds (4e, 4j, 4k, 5d, and 9f) using toxicity model computation and SWISS ADME calculation.
Conclusion
In this study, various 10-alkyl-2-deoxo-2-methylthio-5-deazaflavins (3a–c) were synthesised from 6-(N-alkylanilino)-2-methylthiopyrimidin-4(3H)-ones (2a–c) by the reaction with Vilsmeier reagent. The 2-(alkylamino, benzyl amino, hydrazino, piperidino, and morpholino)-10-alkyl-2-deoxo-5-deazaflavins (4a–m) were synthesised by the facile replacement of C2-methylthio by different amines. Additionally, 2,2′-(Piperazine-1,4-diyl)bis(10-alkyl-2-deoxo-5-deazaflavins) (4n,o) were prepared by refluxing compound 3a,b, independently, with piperazine (ratio 2:1) in absolute ethanol. The Schiff’s bases, 2[(E)-2-(substituted) benzylidene-hydrazino]-10-alkyl-2-deoxo-5-deazaflavins (5a–j) were synthesised by refluxing the appropriate hydrazino derivative 4i,j with certain aldehydes in absolute ethanol. Various novel 10-aryl-2-deoxo-2-thioxo-5-deazaflavins (8a–c) were prepared by condensation of certain 6-(substituted)anilino-2,3-dihydro-2-thioxopyrimidin-4(1H)-ones (7a–c) with o-bromobenzaldehyde in DMF. Similarly, the 2-(substituted amino)-10-aryl-2-deoxo-5-deazaflavins (9a–h) were prepared by condensation of 10-aryl-2-deoxo-2-thioxo-5-deazaflavins (8a–c) with different amines or hydrazine hydrate. In Vitro growth, inhibitory activities of the synthesised compounds were evaluated against MCF-7, HeLa, CCRF-HSB-2, and KB cell lines. SAR study revealed that the C-2 and N-10 positions are critical for the antiproliferative activity of 5-deazflavins. Hence ethyl substitution at N-10 position (4e) results in superior activity over the methyl (4a) and phenyl (9c and 9g) analogs. The amino moiety at the C-2 position was confirmed to be essential for activity by observing the diminished antitumor activities of compounds (8a–c) lacking the amino functionality at position 2. Moreover, the hydrazino substitution gave rise to the potent compound 4j of IC50 0.0005 μM against the MCF-7 cell line. Incorporation of a 2-bromobenzylidene moiety at the terminal amino group of the hydrazine derivative (4i) resulted in dramatic enhancement of the antiproliferative activity against both MCF-7 and Hela cell lines as noted in the most potent compound 5d of IC50 <0.0001 μM against MCF-7 cell line and 1.26 μM against Hela cell line. Prediction of the pharmacokinetic and drug-likeness properties of the most potent candidates 4j, 5d, 5f, and 9f was proved to be satisfactory. Finally, the present investigation identified compounds 4j, 5d, 5f, and 9f as promising antitumor agents that deserve further investigations to shed line on their potential molecular targets.
Compound kinase inhibition profiling was undertaken to measure the effect of 20 protein kinases against two test compounds and the reference compound Imatinib (or Gleevec). The aim was to identify to what extent the compounds were able to inhibit the protein kinase tested. The profiling data for these two compounds showed low to moderate inhibition (>10%) with 6 of the kinases including ACVR2A, CAMK1, EGFR, FAK, FLT3, and SRC. Compound 4e showed the highest inhibition of all the compounds tested, with a 22% inhibition of FAK. The reference compound, Imatinib gave the highest inhibitions (>72%) with ABL1 and c-KIT, and moderate inhibitions (>20%) with BRAF, CAMK1, FLT1, and JNK1. All the other inhibitions observed were less than 20%. In addition to inhibitions, some of the compounds gave low to moderate activations with a few of the kinases tested, including CDK1/Cyclin A1, FLT3, and SRC. Compound 4k was observed to give the greatest activation with FLT3 at 24%. All the other activations noted were less than 20%.
Experimental
Chemistry
Melting points were determined on the electrothermal capillary melting point apparatus. IR spectra were recorded on Perkin Elmer model 137 infra-red spectrophotometer, using a KBr disc. The 1H-NMR spectra were obtained using Varian mercury VXR-300 MHz and VXR-500MHz spectrophotometers at both room temperature or at 100 °C, and chemical shift values were expressed in δ values (ppm) relative to tetramethylsilane (TMS) as internal standard. Coupling constants are given in Hz. All NH and OH protons were exchangeable with D2O. The mass spectra were recorded on GCMS-QP 1000 EX Shimadzu Gas Chromatography MS spectrometer, E.I0.70 ev. Microanalyses were measured by Automatic CHN analyser, Vario E1Ш, Elementary-Germany at the Microanalytical unit, faculty of Science, Cairo University, Egypt.
All reagents were of commercial quality and were used without further purification. Organic solvents were dried in the presence of an appropriate drying agent and were stored over suitable molecular sieves. Reaction progress was monitored by analytical thin layer 18 chromatography (TLC) on pre-coated glass plates silica gel (60F254-plate-Merck) and the products were visualised by UV light.
General procedure for the preparation of 6-(N-alkyl anilino)-2-methylthiopyrimidin-4(3H)-ones (2a–c)
A mixture 6-chloro-4-hydroxy-2-methylthiopyrimidineCitation43 (1, 3.0 g, 0.02 mol) and an appropriate N-alkylaniline (0.05–0.09 mol) in n-butanol (10 ml) was refluxed with stirring for 24–48 h according to the reported methodCitation3. After cooling, the precipitated solid was filtered off, washed with isopropyl alcohol, and then washed with water to get the first crop. The filtrate was concentrated in vacuo and the residue was crushed with diethyl ether to precipitate powdery crystals, which were filtered off, and treated as done for the first crop to afford the second crop. The combined crops were recrystallized from ethanol to afford the product as colourless crystals in 65–77% yields.
6-(N-Methylanilino)-2-methylthiopyrimidin-4(3H)-one (2a)
Yield, 3.24 g (77%); mp 268–270 °C as reportedCitation3.
6-(N-Ethylanilino)-2-methylthiopyrimidin-4(3H)-one (2b)
Yield, 2.89 g (65%); mp 235–236 °C as reportedCitation3.
6-(N-Methyl-4-ethylanilino)-2-methylthiopyrimidin-4(3H)-one (2c)
Yield, 2.89 g (65%); mp 235–236 °C; UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 242 (4.67), 256sh (4.58), 370sh (2.27); IR (νmax/cm−1): 3207 (NH), 1624 (C = O); 1HNMR (DMSO-d6): δ 1.1 (3H, t, J = 7.5 Hz, p-CH2CH3), 2.49 (3H, s, 2-SCH3), 2.60 (2H, q, J = 7.5 Hz, p-CH2CH3), 3.34 (3H, s, 6-NMe), 4.76 (1H, s, 5-H), 7.17 (2H, d, J2',3′ = J5',6′ = 8.4 Hz, Ar-oH), 7.26 (2H, d, J2',3′= J5',6′ = 8.4 Hz, Ar-mH), 11.7 (1H, br s, 3-NH; exchangeable with D2O); Anal. calcd for C14H17N3OS: C, 61.06; H, 6.22; N, 15.26. Found: C, 60.86; H, 5.89; N, 15.15.
General procedure for the preparation of 7-(unsubstituted/substituted)-10-alkyl-2-deoxo-2-methylthio-5-deazaflavins (3a–c)
A mixture of 6-(N-alkylanilino)-2-methylthiopyrimidin-4(3H)-one (2, 0.01 mol) and phosphoryl chloride (7.7 g, 0.05 mol) in anhydrous dimethyl formamide (10 ml) was heated at 90 °C with stirring for 1–1.5 h. The reaction mixture was poured onto ice and neutralised with aqueous ammonia (pH 7) according to the reported procedureCitation3. The yellow crystals separated were filtered off, washed with water, dried, and recrystallized from ethanol to afford the product as yellow needles in 84–99% yields.
10-Methyl-2-deoxo-2-methylthio-5-deazaflavin (3a)
Yield, 2.44 g (95%); mp 281–282 °C, as reportedCitation3.
10-Ethyl-2-deoxo-2-methylthio-5-deazaflavin (3b)
Yield, 2.69 g (99%); mp 262–265 °C, as reportedCitation3.
7-Ethyl-10-methyl-2-deoxo-2-methylthio-5-deazaflavin (3c)
Yield, 2.40 g (84%); mp 227–228 °C; UV (EtOH):): λmax nm (log ε/dmCitation3 mol−1 cm−1): 227 (4.97), 271sh (4.79), 289 (4.89), 340 (4.64), 430 (4.66); IR (νmax/cm−1): 1647 (C = O); 1HNMR (DMSO-d6): δ 1.28 (3H, t, J = 7.5 Hz, 7-CH2CH3), 2.51 (3H, s, 2-SCH3), 2.8 (2H, q, J = 7.5 Hz, 7-CH2CH3), 4.23 (3H, s, 10-NMe), 7.94 (1H, d, J8,9 = 9 Hz, 9-H), 8.02 (1H, d, J8,9 = 9 Hz, 8-H), 8.12 (1H, s, 6-H), 9.19 (1H, s, 5-H). Anal. calcd for C15H15N3OS: C, 63.13; H, 5.30; N, 14.73. Found: C, 63.23; H, 5.00; N, 14.39.
General procedure for the preparation of 2-(N-substituted amino and heterocyclic amino)-7-(unsubstituted/substituted)-10-alkyl-pyrimido[4,5-b]quinolin-4(10H)-one (4a-h) and (4k–m)
A mixture of the appropriate 10-alkyl-2-deoxo-2-methylthio-5-deazaflavin (3a–c, 5.0 mmol) and the appropriate primary aliphatic amine (25–35 mmol) or secondary heterocyclic amine (35–50 mmol) in ethanol (50 ml) was refluxed with stirring for 2–7 h according to the reported methodCitation3. After the clear yellow solution was kept overnight in the refrigerator, the precipitated solid crystals were collected by filtration and washed with little water to get the first crop. The filtrate was concentrated in vacuo and the residue was treated with ether then water to remove the excess amine to get the second crop, dried and recrystallized from ethanol to give the corresponding products as bright yellow needles in 66–91% yields.
2-Benzylamino-10-methylpyrimido[4,5-b]quinolin-4(10H)-one (4a)
Yield, 1.11 g (70%); mp 292–294 °C (dec); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 220 (4.57), 271 (4.56), 319 (4.06), 405 (4.11), 418sh (4.09); IR (νmax/cm-1): 3433 (NH), 1610 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 4.08 (3H, s, 10-NMe), 4.62 (2H, s, 2-NHCH2Ph), 7.22–7.40 (5H, m, o,m,p Ph-H), 7.47–7.52 (1H, m, 9-H), 7.88–7.91 (2H, m, 7 and 8-H), 8.16 (1H, d, J 6,7 = 9 Hz, 6-H), 8.25 (1H, br s, 2-NH, exchangeable with D2O), 8.85 (1H, s, 5-H); Anal. calcd for C19H16N4O: C, 72.13; H, 5.10; N, 17.71. Found: C, 71.94; H, 4.86; N, 17.56.
2-(Butylamino)-10-methylpyrimido[4,5-b]quinolin-4(10H)-one (4b)
Yield, 1.20 g (85%); mp 257–260 °C (decomp. From DMF); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 222 (4.64), 270 (4.75), 321 (4.17), 404 (4.29), 418sh (4.27); IR (νmax/cm−1): 3185 (NH), 1612 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 0.87 (3H, t, J = 7.4 Hz, 2–N(CH2)3CH3), 1.30 (2H, sextet, J = 7.4 Hz, 2-N(CH2)2CH2–), 1.48 (2H, quintet, J = 7.4 Hz, 2–NCH2CH2–), 3.30–3.55 (2H, m, 2-NCH2-), 4.10 (3H, s, 10-NMe), 7.45 (1H, t, J6,7 = J7,8 = 8 Hz, 7-H), 7.70–7.73 (1H, m, 9-H), 7.72 (1H, t, J7,8 = J8,9 = 8 Hz, 8-H), 7.90 (1H, br s, 2-NH, exchangeable with D2O), 8.05 (1H, d, J6,7 = 8 Hz, 6-H), 8.80 (1H, s, 5-H); Anal. calcd for C16H18N4O.0.3DMF: C, 66.71; H, 6.66; N, 19.79. Found: C, 66.38; H, 6.80; N, 20.05.
10-methyl-2-(propylamino)pyrimido[4,5-b]quinolin-4(10H)-one (4c)
Yield, 0.97 g (72%); mp 252–254 °C (decomp. From EtOH); UV (EtOH): λmax nm (log ε/dm3 mol−1 cm−1): 221 (4.81), 270 (4.83), 321 (4.28), 404 (4.40), 419sh (4.37); IR (νmax/cm−1): 3181 (NH), 1649 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 0.95 (3H, t, J = 5.6 Hz, 2-N(CH2)2CH3), 1.60 (2H, sextet, J = 5.6 Hz, 2-NCH2CH2–), 3.86–4.03 (2H, m, 2-NCH2–), 4.18 (3H, s, 10-NMe), 6.43 (1H, br s, 2-NH, exchangeable with D2O), 7.50–7.74 (1H, t, J6,7 = J7,8 =9 Hz, 7-H), 7.86 (1H, t, J7,8 = J8,9 = 9 Hz, 8-H), 7.93 (1H, m, 9-H), 8.15 (1H, d, J6,7 = 9 Hz, 6-H), 8.85 (1H, s, 5-H); Anal. calcd for C15H16N4O: C, 67.15; H, 6.01; N, 20.88. Found: C, 67.38; H, 6.36; N, 21.17.
2-((2-Hydroxyethyl)amino)-10-methylpyrimido[4,5-b]quinolin-4(10H)-one (4d)
Yield, 1.23 g (91%); mp 253–256 °C (decomp. From EtOH); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 220 (4.49), 270 (4.51), 322 (3.99), 405 (4.03), 425sh (3.96); IR (νmax/cm−1): 3302 (OH), 3190 (NH), 1649 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 3.37–3.54 (2H, m, 2-NCH2CH2OH), 3.61 (2H, m, 2-NCH2CH2OH), 4.01 (3H, s, 10-NMe), 4.76 (1H, br s, 2-NCH2-CH2OH, exchangeable with D2O), 7.46 (1H, t, J6,7 = J7,8 = 7.4 Hz, 7-H), 7.60–7.66 (1H, m, 9-H), 7.84 (1H, t, J7,8 = J8,9 = 7.4 Hz, 8-H), 7.90 (1H, br s, 2-NH, exchangeable with D2O), 8.09 (1H, d, J6,7 = 7.4 Hz, 6-H), 8.79 (1H, s, 5-H); Anal. calcd for C14H14N4O2: C, 62.21; H, 5.22; N, 20.73. Found: C, 61.86; H, 5.05; N, 20.47.
2-Benzylamino-10-ethylpyrimido[4,5-b]quinolin-4(10H)-one (4e)
Yield, 1.15 g (70%); mp 299–302 °C (dec); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 221 (4.62), 271 (4.61), 319 (4.14), 404 (4.16), 418sh (4.14); IR (νmax/cm-1): 3174 (NH), 1650 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 1.28 (3H, t, J = 6.6 Hz, 10-NCH2CH3), 4.56 (2H, s, 2-NCH2Ph), 4.75 (2H, q, J = 6.6 Hz,10-NCH2CH3), 7.20–7.38 (5H, m, o,m,pPh-H), 7.46–7.50 (1H, m, 9-H), 7.90–7.96 (2H, m, 7 and 8-H), 8.15 (1H, d, J6,7 = 8.7 Hz, 6-H), 8.40 (1H, br s, 2-NH, exchangeable with D2O), 8.83 (1H, s, 5-H); Anal. calcd for C20H18N4O: C, 72.71; H, 5.49; N, 16.96. Found: C, 72.46; H, 5.31; N, 16.70.
2-Butylamino-10-ethylpyrimido[4,5-b]quinolin-4(10H)-one (4f)
Yield, 1.15 g (78%); mp 196–198 °C (decomp. From EtOH); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 223 (4.83), 270 (4.94), 322 (4.42), 403 (4.51), 418sh (4.48); IR (νmax/cm−1): 3188 (NH), 1653 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 0.88 (3H, t, J = 6.8 Hz, 2-N(CH2)3CH3), 1.32 (2H, sextet, J = 6.8 Hz, 2-N(CH2)2CH2–), 1.49 (2H, quintet, J = 6.8 Hz, 2-NCH2CH2–), 1.56 (3H, t, J = 6.8 Hz, 10-NCH2CH3), 3.33–3.41 (2H, m, 2-NCH2–), 4.71 (2H, q, J = 6.8 Hz,10-NCH2–), 7.46 (1H, t, J6,7 = J7,8 = 7.6 Hz, 7-H), 7.16–7.73 (1H, m, 9-H), 7.76 (1H, br s, 2-NH, exchangeable with D2O), 7.90 (1H, dt, J7,8 = J8,9 = 7.6 Hz, J6,8 =1.2 Hz, 8-H), 8.10 (1H, d, J6,7 = 7.6 Hz, 6-H), 8.77 (1H, s, 5-H); Anal. calcd for C17H20N4O: C, 68.89; H, 6.80; N, 18.90. Found: C, 69.16; H, 7.12; N, 19.01.
10-Ethyl-2-(propylamino)pyrimido[4,5-b]quinolin-4(10H)-one (4 g)
Yield, 1.20 g (85%); mp 217–220 °C (decomp. From EtOH); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 222 (4.87), 270 (4.88), 322 (4.34), 403 (4.44), 418sh (4.41); IR (νmax/cm−1): 3186 (NH), 1610 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 0.91 (3H, t, J = 6.9 Hz, 2-N(CH2)2CH3), 1.34 (3H, t, J = 6.9 Hz, 10-NCH2CH3), 1.57 (2H, sextet, J = 6.9 Hz, 2-NCH2CH2–), 3.27 (2H, t, J = 6.9 Hz, 2-NCH2–), 4.73–4.81 (2H, m,10-NCH2CH3), 7.48–7.52 (1H, m, 9-H), 7.72 (1H, br s, 2-NH, exchangeable with D2O), 7.89–7.96 (2H, m, 7 and 8-H), 8.14 (1H, d, J6,7 = 8.1 Hz, 6-H), 8.8 (1H, s, 5-H); Anal. calcd for C16H18N4O.0.2 H2O: C,67.21; H, 6.49; N, 19.59. Found: C, 67.42; H, 6.65; N, 19.78.
2-(Cyclohexylamino)-10-ethylpyrimido[4,5-b]quinolin-4(10H)-one (4h)
Yield, 1.10 g (68%); mp 233–237 °C (decomp. From EtOH); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 220 (4.64), 270 (4.65), 326 (4.14), 404 (4.06), 418sh (4.04); IR (νmax/cm−1): 3229 (NH), 1667 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 1.16–1.21 (6H, m, 3′, 4′, and 5′-CH2), 1.58–1.78 (3H, m, 10-NCH2CH3), 1.82–1.98 (4H, m, 2′and 6′-CH2), 3.6–4.0 (1H, m,1′-CH), 4.69–4.81 (2H, m,10-NCH2CH3), 6.39 (1H, br s, 2-NH, exchangeable with D2O), 6.86–7.09 (1H, m, 9-H), 7.48–7.59 (2H, m, 7 and 8-H), 7.88–8.09 (1H, br s, 6-H), 8.78 (1H, s, 5-H),; Anal. calcd for C19H22N4O.0.5 H2O: C, 68.86; H, 7.00; N, 16.91. Found: C, 69.07; H, 7.12; N, 17.19.
10-Ethyl-2-morpholinopyrimido[4,5-b]quinolin-4(10H)-one (4k)
Yield, 1.13 g (73%); mp 198–200 °C (decomp. From EtOH); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 223 (4.69), 273 (4.69), 322 (4.22), 407 (4.35), 434sh (4.29); IR (νmax/cm-1): 1648 (C = O); 1HNMR (DMSO-d6): δ 1.49 (3H, t, J = 6.9 Hz, 10-NCH2CH3), 3.78 (4H, br s, 2′ and 6′-NCH2), 4.08 (4H, br d, 3′ and 5′-OCH2), 4.77 (2H, q, J = 6.9 Hz, 10-NCH2CH3), 7.26 (1H, br s, 9-H), 7.46 (1H, t, J6,7 = J7,8 = 7.2 Hz, 7-H), 7.67 (1H, d, J6,7 = 7.2 Hz, 6-H), 7.87 (1H, dt, J7,8 = J8,9 = 7.2 Hz, J6,8 = 1.2 Hz, 8-H), 8.98 (1 H, s, 5-H); Anal. calcd for C17H18N4O2. 0.2 H2O: C, 65.04; H, 5.91; N, 17.85. Found: C, 64.92; H, 5.73; N, 17.72.
10-Ethyl-2-(piperidin-1-yl)pyrimido[4,5-b]quinolin-4(10H)-one (4l)
Yield, 1.02 g (66%); mp 197–198 °C (decomp. From EtOH); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 222 (4.71), 274 (4.71), 320 (4.27), 410 (4.30), 433sh (4.24); IR (νmax/cm−1): 1644 (C = O); 1HNMR (DMSO-d6): δ 1.46 (3H, t, J = 6.9 Hz, 10-NCH2CH3), 1.60–1.85 (6H, m, 3′, 4′ and 5′-NCH2), 4.0–4.05 (4H, m, 2′ and 6′-NCH2), 4.74 (2H, q, J = 6.9 Hz, 10-NCH2CH3), 7.27 (1H, br s, 9-H), 7.42 (1H, t, J6,7 = J7,8 = 7.8 Hz, 7-H), 7.63 (1H, d, J6,7 = 7.8 Hz, 6-H), 7.83 (1H, dt, J6,7 = J7,8 = 7.8 Hz, J6,8 = 1.2 Hz, 8-H), 8.92 (1 H, s, 5-H); Anal. calcd for C18H20N4O: C, 70.11; H, 6.54; N, 18.17. Found: C, 70.35; H, 6.67; N, 18.53.
7-Ethyl-10-methyl-2-morpholinopyrimido[4,5-b]quinolin-4(10H)-one (4m)
Yield, 1.19 g (73%); mp 275–278 °C (decomp. From EtOH); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 224 (4.68), 276 (4.70), 327 (4.22), 414 (4.36), 437sh (4.33); IR (νmax/cm-1): 1651 (C = O); 1HNMR (DMSO-d6): δ 1.22 (3H, t, J = 7.6 Hz, 7-CH2CH3), 2.72 (2H, q, J = 7.6 Hz, 7-CH2CH3), 3.81 (4H, br s, 2′ and 6′-NCH2), 3.92 (4H, br s, 3′ and 5′-OCH2), 4.05 (3H, s, 10-NMe), 7.76 (1H, d, J8,9 = 9 Hz, 9-H), 7.82 (1H, d, J9,8 = 9 Hz, 8-H), 7.92 (1H, s, 6-H), 8.79 (1H, s, 5-H); Anal. calcd for C18H20N4O2: C, 66.65; H, 6.21; N, 17.27. Found: C, 66.41; H, 6.05; N, 16.98.
General procedure for the preparation of 2-Hydrazino-10-alkyl-pyrimido[4,5-b]quinolin-4(10H)-one (4i,j)
A mixture of the appropriate 10-alkyl-2-deoxo-2-methylthio-5-deazaflavin (3a, b, 10.0 mmol) and hydrazine hydrate (3.85 g, 0.12 mol) in ethanol (50 ml) was refluxed with stirring for 2.5–3 h. After cooling, the precipitated solid crystals were collected by filtration and washed with a little amount of water to get the first crop, dried, and recrystallized from ethanol to afford 4i,j.
2-Hydrazineyl-10-methylpyrimido[4,5-b]quinolin-4(10H)-one (4i)
Yield, 2.16 g (90%); mp 283–286 °C (dec); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 221 (4.03), 271(3.94), 309 (3.72), 414 (3.64), 430sh (3.61); IR (νmax/cm-1): 3357, 3255 (NH), 1670 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 3.90 (3H, s, 10-NMe), 4.33 (2H, br s, 2-NHNH2, exchangeable with D2O), 7.66 (1H, br s, 2-NH, exchangeable with D2O), 7.35 (1H, br t, J6,7 = J7,8 = 7.5 Hz, 7-H), 7.67 (1H, d, J8,9 = 7.5 Hz, 9-H), 7.76–7.88 (1H, m, 8-H), 7.92 (1H, d, J6,7 = 7.5 Hz, 6-H), 8.45 (1H, s, 5-H); Anal. calcd for C12H11N5O: C, 59.74; H, 4.60; N, 29.03, Found: C, 60.0; H, 4.72; N, 29.24.
10-Ethyl-2-hydrazino-pyrimido[4,5-b]quinolin-4(10H)-one (4j)
Yield, 2.14 g (84%); mp 270–274 °C (dec); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 222 (4.18), 272 (4.09), 308 (3.89), 413 (3.77), 430sh (3.73); IR (νmax/cm-1): 3357, 3255 1670 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 1.03 (3H, t, J = 7.2 Hz, 10-NCH2CH3), 4.59 (2H, q, J = 7.2 Hz, 10-NCH2CH3), 4.69 (2H, br s, 2-NHNH2, exchangeable with D2O), 7.35 (1H, br s, 2-NH, exchangeable with D2O), 7.72–7.76 (1H, m, 9-H), 7.87–7.95 (2H, m, 7 and 8-H), 8.1 (1H, d, J6,7 = 8 Hz, 6-H), 8.45 (1H, s, 5-H); Anal. calcd for C13H13N5O: C, 61.17; H, 5.13; N, 27.43, Found: C, 61.49; H, 5.33; N, 27.70.
General procedure for the preparation of 2,2'-(piperazine-1,4-diyl)bis(10-alkyl-2-deoxo-5-deazaflavins) (4n,o)
A mixture of the appropriate 10-alkyl-2-deoxo-2- methylthio-5-deazaflavin (3a,b, 5.0 mmol) and piperazine (0.22 g, 2.5 mmol) in ethanol (25 ml) was refluxed with stirring for 6.5–7 h. After cooling, the precipitated solid was filtered off, washed with a little amount of ethanol then washed with water, dried, and recrystallized from DMF to give 4n,o.
2,2'-(Piperazine-1,4-diyl)bis(10-methylpyrimido[4,5-b]quinolin-4(10H)-one) (4n)
Yield, 1.83 g (73%); mp 271–274 °C (dec); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 223 (5.09), 287 (4.94), 340 (4.68), 423 (4.69); IR (νmax/cm−1): 1654 (C = O); 1HNMR (DMSO-d6): δ 2.46 (4H, br s, 3′ and 5′-NCH2), 3.09 (4H, br s, 2′ and 6′-NCH2), 4.22 (6H, s, 10-NMe), 7.62 (2H, t, J6,7 = J7,8 = 9 Hz, 7-H), 8.00–8.07 (4H, m, 8 and 9-H), 8.26 (2H, d, J6,7 = 9 Hz, 6-H), 9.19 (2H, s, 5-H); MS., EI m/z.: 507 (M + 3, 3% due to protonated two nitrogen of the piperazine ring), 257 (100%); Anal. calcd for C28H24N8O2. 0.5 DMF: C, 65.48; H, 5.12; N, 22.00. Found: C, 65.24; H, 4.97; N, 22.40.
2,2'-(Piperazine-1,4-diyl)bis(10-ethylpyrimido[4,5-b]quinolin-4(10H)-one) (4o)
Yield, 1.70 g (64%); mp 290–292 °C (dec); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 222 (4.32), 277 (4.32), 409 (4.05), 432 (4.03); IR (νmax/cm−1): 1655 (C = O); 1HNMR (DMSO-d6): δ 1.3 (6H, t, J = 6.9 Hz, 10-NCH2CH3), 3.15 (4H, br s, 3 and 5-NCH2), 4.04– 4.14 (4H, m, 2 and 6-NCH2), 4.74–4.79 (4H, m, 10-NCH2CH3), 7.5 (2H, t, J6,7 = J7,8 = 7.6 Hz, 7-H), 7.88 (2H, d, J8,9 = 7.6 Hz, 9-H), 7.94 (2H, t, J7,8 = J8,9 = 7.6 Hz, 8-H), 8.14 (2H, d, J6,7 = 7.6 Hz, 6-H), 8.86 (2H, s, 5-H); MS., EI m/z.: 533.20 (M + 1, 4%), 268 (33%), 202 (100%); Anal. calcd for C30H28N8O2. 0.1DMF: C, 67.41; H, 5.36; N, 21.01. Found: C, 67.68; H, 5.49; N, 21.37.
General procedure for the preparation of 2[(E)-2-(substituted) benzylidenehydrazino]-10-alkyl-2-deoxo-5-deazaflavins (5a–j)
A mixture of 10-alkyl-2-hydrazino-2-deoxo-5-deazaflavin (4i,j, 2 mmol) and the appropriate aromatic aldehyde (1.29 mmol) in ethanol (30 ml) was refluxed for 2–3 h. After cooling the precipitated solid was collected by filtration, washed with hot ethanol, dried, and recrystallized from the appropriate solvent to afford the corresponding product in 71–84% yields.
2-[(E)-2-Benzylidenehydrazino]-10-methylpyrimido[4,5-b]quinolin-4(10H)-one (5a)
Yield, 0.51 g (77%); mp 308–310 °C (dec); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 220 (4.76), 263 (4.74), 321 (4.25), 398 (4.23), 421sh (4.15); IR (νcm−1): 3441 (NH), 1697 (C = O), 1614 (C = N); 1HNMR (DMSO-d6): δ 4.02 (3H, s, 10-NMe), 7.52 (1H, br s, 9-H), 7.58–7.64 (2H, m, 7 and 8-H),7.86–7.98 (3H, m, m,p Ph-H), 8.10 (1H, br s, 6-H), 8.15 (2H, d, J2',3′ = J5',6′ = 6.9 Hz, oPh-H), 8.38 (1H, s, N=CH-Ph), 8.97 (1H, s, 5-H), 11.04 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C19H15N5O: C, 69.29; H, 4.59; N, 21.26. Found: C, 69.60; H, 4.84; N, 21.55.
2-[(E)-2–(2-Chlorobenzylidene(hydrazino]-10-methylpyrimido[4,5-b]quinolin-4(10H)-one (5b)
Yield, 0.6 g (83%); mp 280–283 °C (dec); UV (EtOH): λmax nm (log ε/dm3 mol−1 cm−1): 226 (4.54), 267sh (4.03), 343 (4.55), 373sh (4.29), 446 (4.35); IR (νcm−1): 3435(NH), 1690 (C = O), 1617 (C = N); 1HNMR (DMSO-d6): δ 3.97 (3H, s, 10-NMe), 7.38 (1H, br s, 9-H), 7.42–7.51 (2H, m, 7 and 8-H), 7.78–7.86 (3H, m, 3′,4′,5′ Ph-H), 8.05 (1H, d, J6,7 = 8.1 Hz, 6-H), 8.57 (1H, br s, 6′ Ph-H), 8.64 (1H, s, N=CH-Ph), 8.78 (1H, s, 5-H), 10.89 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C19H14ClN5O: C, 62.73; H, 3.88; N, 19.25. Found: C, 63.12; H, 4.00; N, 19.59.
2-[(E)-2–(4-Chlorobenzylidene(hydrazino]-10-methylpyrimido[4,5-b]quinolin-4(10H)-one (5c)
Yield, 0.53 g (73%); mp 267–270 °C (dec); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 224 (4.33), 267sh (4.02), 340 (4.27), 371sh (4.03), 441 (4.04); IR (νcm−1): 3433 (NH), 1677 (C = O), 1611 (C = N); 1HNMR (DMSO-d6): δ 3.92 (3H, s, 10-NMe), 7.4 (1H, br s, 9-H), 7.43 (1H, d, J6,7 = 7.6 Hz, 6-H), 7.48 (2H, d, J2',3′ = J5',6′ = 7.6 Hz, oPh-H), 7.75 (1H, t, J6,7 = J7,8 = 7.6 Hz, 7-H), 7.81–7.85(1H, m, 8-H), 8.02 (2H, br s, mPh-H), 8.31 (1H, s, N=CH-Ph), 8.72 (1H, s, 5-H), 10.94 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C19H14ClN5O: C, 62.73; H, 3.88; N, 19.25. Found: C, 62.47; H, 3.72; N, 18.94.
2-[(E)-2–(2-Bromobenzylidene(hydrazino]-10-methylpyrimido[4,5-b]quinolin-4(10H)-one (5d)
Yield, 0.58 g (71%); mp 272–274 °C (dec), crystalise from DMF-H2O); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 224 (3.96), 267sh (3.53), 344 (3.94), 372sh (3.72), 446 (3.77); IR (νcm−1): 3432 (NH), 1689 (C = O), 1621 (C = N); 1H-NMR [DMSO-d6]: δ 3.98 (3H, s, 10-NMe), 7.32 (1H, t, J6,7 = J7,8 = 7.2 Hz, 7-H), 7.39–7.45 (2H, m, 8 and 9-H), 7.65 (1H, d, J6,7 = 7.2 Hz, 6-H), 7.78–7.88 (2H, m, 4′,5′ Ph-H), 8.08 (1H, br s, 3′ Ph-H), 8.54 (1H, br s, 6′ Ph-H), 8.60 (1H, s, N=CH-Ph), 8.79 (1H, s, 5-H), 10.98 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C19H14BrN5O: C, 55.90; H, 3.46; N, 17.15. Found: C, 56.24; H, 3.66; N, 17.39.
2-[(E)-2–(4-Nitrobenzylidene(hydrazino]-10-methylpyrimido[4,5-b]quinolin-4(10H)-one (5e)
Yield, 0.54 g (72%); mp 298–301 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 223 (3.81), 267sh (3.59), 318 (3.65), 360sh (3.67), 444 (3.63); IR (νcm−1): 3437 (NH), 1692 (C = O), 1619 (C = N); 1HNMR (DMSO-d6): δ 4.0 (3H, s, 10-NMe), 7.49 (1H, br s, 9-H), 7.80–7.85 (1H, m, 7-H), 8.0–8.09 (1H, m, 8-H), 8.18 (1H, br d, 6-H), 8.23 (2H, br s, oPh-H), 8.38 (2H, br d, mPh-H), 8.43 (1H, s, N=CH-Ph), 8.82 (1H, s, 5-H), 11.05 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C19H14N6O3.0.5 DMF: C, 59.92; H, 4.29; N, 22.16. Found: C, 60.30; H, 4.52; N, 22.43.
2-[(E)-2-Benzylidenehydrazino]-10-ethylpyrimido[4,5-b]quinolin-4(10H)-one (5f)
Yield, 0.58 g (85%); mp 213–216 °C (decomp. From DMF); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1):227 (4.60), 252sh (4.22), 338 (4.63), 366sh (4.33), 448 (4.37); IR (νcm−1): 3423 (NH), 1693 (C = O), 1614 (C = N); 1H-NMR [DMSO-d6]: δ 1.35 (3H, t, J = 6.3 Hz, 10-NCH2CH3), 4.64 (2H, q, J = 6.3 Hz, 10-NCH2CH3), 7.37 (1H, d, J8,9 = 7.5 Hz, 9-H), 7.4–7.51(2H, m, 7 and 8-H),7.80–7.89(3H, m, m,pPh-H), 7.96 (1H, d, J6,7 = 7.5 Hz, 6-H), 8.05 (2H, d, J2',3′ = J5',6′ = 7.5 Hz, oPh-H), 8.36 (1H, s, N=CH-Ph), 8.74 (1H, s, 5-H), 10.65 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C20H17N5O.0.1 DMF: C, 69.52; H, 5.09; N, 20.37. Found: C, 69.25; H, 4.98; N, 20.13.
2-[(E)-2–(2-Chlorobenzylidene(hydrazino]-10-ethylpyrimido[4,5-b]quinolin-4(10H)-one (5 g)
Yield, 0.61 g (81%); mp 232–234 °C (decomp. From DMF); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 227 (4.72), 264sh (4.24), 344 (4.74), 374sh (4.48), 446 (4.55); IR (νcm−1): 3431 (NH), 1691 (C = O), 1606 (C = N)); 1HNMR (DMSO-d6): δ 1.36 (3H, t, J = 6.6 Hz, 10-NCH2CH3), 4.66 (2H, q, J = 6.6 Hz, 10-NCH2CH3), 7.41 (1H, d, J8,9 = 8.1 Hz, 9-H), 7.47–7.54 (2H, m, 7 and 8-H), 7.81–7.89 (3H, m, 3′,4′,5′ Ph-H), 8.07 (1H, d, J6,7 = 8.1 Hz, 6-H), 8.54 (1H, br s, 6′ Ph-H), 8.67 (1H, s, N=CH-Ph), 8.79 (1H, s, 5-H), 10.9 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C20H16ClN5O: C, 63.58; H, 4.27; N, 18.54. Found: C, 63.82; H, 4.43; N, 18.85.
2-[(E)-2–(4-Chlorobenzylidene(hydrazino]-10-ethylpyrimido[4,5-b]quinolin-4(10H)-one (5h)
Yield, 0.57 g (75%); mp 246–250 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 227 (4.74), 262sh (4.33), 340 (4.80), 369sh (4.55), 448 (4.56); IR (νcm−1): 3434 (NH), 1691 (C = O), 1613 (C = N)); 1H-NMR [DMSO-d6]: δ 1.32 (3H, t, J = 7.5 Hz, 10-NCH2CH3), 4.66 (2H, q, J = 7.5 Hz, 10-NCH2CH3), 7.42(1H, br s, 9-H), 7.44 (1H, d, J6,7 = 7.6 Hz, 6-H), 7.47 (2H, d, J2',3′ = J5',6′ = 7.6 Hz, oPh-H), 7.81–7.9 (2H, m, 7 and 8-H), 8.05 (2H, br s, mPh-H), 8.34 (1H, s, N=CH-Ph), 8.75 (1H, s, 5-H), 10.95 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C20H16ClN5O.0.25 DMF: C, 62.92; H, 4.52; N, 18.56. Found: C, 63.13; H, 4.68; N, 18.80.
2-[(E)-2–(2-Bromobenzylidene(hydrazino]-10-ethylpyrimido[4,5-b]quinolin-4(10H)-one (5i)
Yield, 0.60 g (71%); mp 240–242 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 226 (4.73), 263sh (4.33), 344 (4.75), 374sh (4.52), 447 (4.58); IR (νcm−1): 3432 (NH), 1689 (C = O), 1604 (C = N); 1HNMR (DMSO-d6): δ 1.4 (3H, m, 10-NCH2CH3), 4.62–4.78 (2H, m, 10-NCH2CH3), 7.31 (1H, t, J6,7 = J7,8 = 7.8 Hz, 7-H), 7.40–7.45 (2H, m, 8 and 9-H), 7.64 (1H, d, J6,7 = 7.8 Hz, 6-H), 7.83–7.96 (2H, m, 4′,5′ Ph-H), 8.06 (1H, d, J3',4′ = 7.8 Hz, 3′ Ph-H), 8.46 (1H, br s, 6′ Ph-H), 8.63 (1H, s, N=CH-Ph), 8.76 (1H, s, 5-H), 10.78 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C20H16BrN5O: C, 56.89; H, 3.82; N, 16.58. Found: C, 57.26; H, 3.96; N, 16.84.
2-[(E)-2–(2-Nitrobenzylidene(hydrazino]-10-ethylpyrimido[4,5-b]quinolin-4(10H)-one (5j)
Yield, 0.59 g (76%); mp 251–254 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 225 (4.65), 260sh (4.26), 319 (4.51), 364sh (4.35), 444 (4.48); IR (νcm−1): 3434 (NH), 1689 (C = O), 1624 (C = N); 1HNMR (DMSO-d6): δ 1.38 (3H, t, J = 6.6 Hz, 10-NCH2CH3), 4.69 (2H, q, J = 6.6 Hz, 10-NCH2CH3), 7.44 (1H, br s, 9-H), 7.61 (1H, t, J6,7 = J7,8 = 7.5 Hz, 7-H), 7.73 (1H, t, J6,7 = J7,8 = 7.5 Hz, 8-H),7.83–7.91 (2H, m, 4′,5′ Ph-H), 7.99 (1H, d, J6,7 = 7.5 Hz, 6-H), 8.10 (1H, d, J3',4′ = 7.5 Hz, 3′ Ph-H), 8.61 (1H, s, N=CH-Ph), 8.71 (1H, d, J5',6′ = 7.5 Hz, 6′ Ph-H), 8.81 (1H, s, 5-H), 10.91 (1H, s, 2-NH, exchangeable with D2O); Anal. calcd for C20H16N6O3: C, 61.85; H, 4.15; N, 21.64. Found: C, 62.19; H, 4.28; N, 21.93.
General procedure for the preparation of 6-(substituted)anilino-2-thiouracils (7a–c)
A mixture of 6-amino-2-thiouracil (6, 7.10 g, 0.05 mol), the appropriate arylamine (0.1 mol) together with anilinium chloride (0.05 mol) was heated at 170 °C for 9 h. The warm mixture was diluted with 65% ethanol (200 ml). The formed precipitate was collected by filtration, washed with ether then water, dissolved in 5% NaOH solution by heating on a steam bath, and reprecipitated by neutralisation with 10% HCl. The solid formed was collected by filtration, washed with water, dried, and crystallised from DMF-H2O to afford 7a–c in 55–71% yields.
6-Anilino-2-thiouracil (7a)
Yield, 6.0 g (55%); mp 296–298 °C as reportedCitation4.
6-(p-Chloroanilino)-2-thiouracil (7b)
Yield, 8.98 g (71%); mp 248–250 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 265 (4.40), 298sh (4.29); IR (νcm−1): 3420, 3300, 3250 (NH), 1620 (C = O); 1HNMR (DMSO-d6): δ 4.96 (1H, s, 5-H), 7.24 (2H, d, J2',3′ = J5',6′ = 8.7 Hz, m-Ph-H), 7.43 (2H, d, J2',3′ = J5',6′ = 8.7 Hz, o-Ph-H), 8.28 (1H, s, 6-NH, exchangeable with D2O), 11.60 (1H, br s, 1-NH, exchangeable with D2O), 11.93(1H, br s, 3-NH, exchangeable with D2O); Anal. calcd for C10H8ClN3OS: C, 47.34; H, 3.18; N, 16.56. Found: C, 47.72; H, 3.49; N, 16.33.
6-(o-Tolylamino)-2-thiouracil (7c)
Yield, 8.0 g (69%); mp 178–180 °C as reportedCitation44.
General procedure for the preparation of 10-aryl-2-deoxo-2-thioxo-5-deazaflavins (8a-c)
A mixture of 6-anilino-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (7, 0.01 mol), o-bromobenzaldhyde (1.3 g, 0.01 mol), and DMF(6 ml) was heated at 110 °C for 1.5–2 h according to the reported procedureCitation8,Citation69. The reddish brown solution was cooled to afford a precipitate which was collected by filtration, washed with water, and dried to get the first crop. The filtrate was poured onto water, and the resulting precipitate was filtered off, washed with water, and dried. The combined product was recrystallized from DMF to afford 8a-c in 89–94% yields.
10-Phenyl-2-thioxo-2,10-dihydropyrimido[4,5-b]quinolin-4(3H)-one (8a)
Yield, 1.64 g (89%); mp 237–238 °C (decomp. From DMF); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 231 (4.25), 268sh (3.90), 317 (4.30), 446 (4.08); IR (νcm−1): 3384 (NH), 1689 (C = O); 1H-NMR [DMSO-d6]: δ 6.73 (1H, d, J8,9 = 7.6 Hz, 9-H), 7.44 (2H, d, J2',3′ = 7.6 Hz, oPh-H), 7.55 (1H, t, J3',4′ = 7.6 Hz, pPh-H), 7.6 (1H, t, J7,8 = J8,9 = 7.6 Hz, 8-H), 7.67 (2H, t, J2',3′ = J3',4′ = 7.6 Hz, mPh-H), 7.77 (1H, t, J6,7 = J7,8 = 7.6 Hz, 7-H), 8.26 (1H, d, J6,7 = 7.6 Hz, 6-H), 9.24 (1H, s, 5-H), 12.2 (1H, s, 3-NH, exchangeable with D2O); MS., EI m/z: 305 (M+, 100%); Anal. calcd for C17H11N3OS: C, 66.87; H, 3.63; N, 13.76. Found: C, 67.26; H, 3.92; N, 13.52.
10-(p-Chlorophenyl)-2-thioxo-2,10-dihydropyrimido[4,5-b]quinolin-4(3H)-one (8b)
Yield, 1.92 g (94%); mp 262–264 °C (decomp. From DMF); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 230 (4.41), 262sh (4.05), 292 (4.43), 328sh (4.06), 396 (3.85), 452sh (3.48); IR (νcm−1): 3441 (NH), 1693 (C = O); 1HNMR (DMSO-d6): δ 6.84 (2H, d, J2',3′ = J5',6′ = 8.4 Hz, oPh-H), 7.30 (1H, d, J8,9 = 8.4 Hz, 9-H), 7.52 (1H, dt, J7,8 = J8,9 = 8.4 Hz, J6,8 = 1.5 Hz, 8-H), 7.78 (1H, t, J6,7 = J7,8 = 8.4 Hz, 7-H), 7.89 (2H, d, J2',3′ = J5',6′ = 8.4 Hz, mPh-H), 8.28 (1H, d, J6,7 = 8.4 Hz, 6-H), 9.25 (1H, s, 5-H), 12.22 (1H, s, 3-NH, exchangeable with D2O); MS., EI m/z: 338 (M-1, 6%), 73 (100%); Anal. calcd for C17H10ClN3OS.1.0 H2O: C, 57.06; H, 3.38; N, 11.74. Found: C, 56.62; H, 3.81; N, 12.01.
2-Thioxo-10-(o-tolyl)-2,10-dihydropyrimido[4,5-b]quinolin-4(3H)-one (8c)
Yield, 1.69 g (90%); mp 324–326 °C (decomp. From DMF); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 231 (3.73), 260sh (3.17), 317 (3.81), 450 (3.58); IR (νcm−1): 3434 (NH), 1690 (C = O); 1HNMR (DMSO-d6): δ 1.88 (3H, s, o-CH3), 6.68 (1H, d, J8,9 = 9 Hz, 9-H), 7.43 (1H, dt, J7,8 = J8,9 = 9 Hz, J6,8 = 1.5 Hz, 8-H), 7.53–7.59 (4H, m, o,m,p Ph-H), 7.82 (1H, t, J6,7 = J7,8 = 9 Hz, 7-H), 8.32 (1H, d, J6,7 = 9 Hz, 6-H), 9.29 (1H, s, 5-H), 12.28 (1H, s, 3-NH, exchangeable with D2O); Anal. calcd for C18H13N3OS: C,67.69; H, 4.10; N, 13.16. Found: C, 67.36; H, 4.40; N, 13.32.
General procedure for the preparation of 2-(substituted amino)-10-aryl-2-deoxo-5-deazaflavins (9a–g)
A mixture of the appropriate 10-aryl-2-deoxo-2-thioxo-5-deazaflavins (8a,b, 2.0 mmol) and an appropriate primary amine or hydrazine (0.02 mol) in ethanol (10 ml) was refluxed for 5–7 h. After cooling, the resulting solid was collected by filtration and washed with EtOH-H2O (1:2) mixture to get the product as yellowish powder, dried and crystallised from DMF-H2O to afford 9a–h in 77–89% yields.
10-Phenyl-2-(propylamino)-2,10-dihydropyrimido[4,5-b]quinolin-4(3H)-one (9a)
Yield, 0.54 g (82%); mp 278–280 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 221 (4.12), 271 (4.00), 322 (3.61), 407 (3.48), 424sh (3.44); IR (νcm−1): 3397 (NH), 1653 (C = O); 1H-NMR [DMSO-d6]: at 100 °C: δ 0.54 (3H, t, J = 7.6 Hz, 2-N(CH2)2CH3), 1.13–1.18 (2H, m, 2-NCH2CH2-), 2.64–2.70 (2H, m, 2-NCH2-), 6.02 (1H, d, J8,9 = 8.4 Hz, 9-H), 6.84 (1H, t, J3',4′ = 8.4 Hz, pPh-H), 7.12 (2H, d, J2',3′ = 8.4 Hz, oPh-H), 7.28–7.33 (1H, m, 8-H), 7.45 (2H, t, J2',3′ = J5',6′ =8.4 Hz, mPh-H), 7.6 (1H, t, J6,7 = J7,8 = 8.4 Hz, 7-H), 8.13 (1H, d, J6,7 = 8.4 Hz, 6-H), 8.90 (1H, s, 5-H), 10.45 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C20H18N4O: C, 72.71; H, 5.49; N, 16.96. Found: C, 73.03; H, 5.66; N, 17.25.
2-((2-Hydroxyethyl)amino)-10-phenyl-2,10-dihydropyrimido[4,5-b]quinolin-4(3H)-one (9b)
Yield, 0.51 g (77%); mp 241–245 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 223 (4.62), 275 (4.62), 409 (4.11); IR (νcm−1): 3402 (OH), 3249 (NH) 1687 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 3.18–3.20 (2H, m, 2-NCH2–), 3.31–3.57 (2H, m, -CH2OH), 4.84 (1H, br s, -CH2OH, exchangeable with D2O), 6.65 (1H, d, J8,9 = 8.7 Hz, 9-H), 6.7–7.0 (1H, m, 8-H), 7.20–7.28 (1H, m, pPh-H), 7.43 (2H, d, J2',3′ = 8.7 Hz, oPh-H), 7.64 (2H, t, J2',3′ = J5',6′ =8.7 Hz, mPh-H), 7.80 (1H, dt, J6,7 = J7,8 = 8.7 Hz, J7,9 =1.4 Hz, 7-H), 8.17 (1H, d, J6,7 = 8.7 Hz, 6-H), 8.95 (1H, s, 5-H), 10.50 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C19H16N4O2: C, 68.66; H, 4.85; N, 16.86. Found: C, 69.05; H, 5.01; N, 17.09.
2-(Benzylamino)-10-phenyl-2,10-dihydropyrimido[4,5-b]quinolin-4(3H)-one (9c)
Yield, 0.64 g (85%); mp 271–274 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 223 (4.70), 277 (4.69), 406 (4.10); IR (νcm−1): 3409 (NH), 1694 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 4.67 (2H, s, 2-NCH2Ph), 6.67 (2H, d, J2',3′ = J5’,6′= 8.7 Hz, oPh-H), 6.86 (1H, d, J8,9 = 8.1 Hz, 9-H), 7.13–7.19 (2H, m, mPh-H), 7.28 (1H, dt, J6,7 = J7,8 = 8.1 Hz, J7,9 = 1.4 Hz, 7-H), 7.38–7.42 (3H, m, m,pPhCH2-H), 7.45–7.53 (1H, m, pPh-H), 7.64–7.69 (1H, m, 8-H), 7.86 (2H, m, oPhCH2-H), 8.2 (1H, d, J6,7 = 8.1 Hz, 6-H), 8.97 (1H, s, 5-H), 10.97 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C24H18N4O: C, 76.17; H, 4.79; N, 14.81. Found: C, 75.83; H, 4.63; N, 14.54.
10–(4-Chlorophenyl)-2-(propylamino)-2,10-dihydropyrimido[4,5-b]quinolin-4(3H)-one (9d)
Yield, 0.65 g (89%); mp 259–262 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm (log ε/dmCitation3 mol−1 cm−1): 220 (4.56), 271 (4.48), 407 (3.99); IR (νcm−1): 3425 (NH), 1693 (C = O); 1H-NMR [DMSO-d6]: at 100 °C: δ 0.59 (3H, t, J = 6.9 Hz, 2-N(CH2)2CH3), 2.77 (2H, t, J = 6.9 Hz, 2-NCH2CH2-), 3.15–3.22 (2H, m, 2-NCH2-), 6.22 (1H, d, J8,9 = 7.8 Hz, 9-H), 6.78 (2H, d, J2',3′ = J5’,6′ = 7.8 Hz, oPh-H), 7.39–7.59 (1H, m, 8-H), 7.76 (1H, dt, J6,7 = J7,8 = 7.8 Hz, J7,9 = 1.6 Hz, 7-H), 7.86 (2H, d, J2',3′ = J5’,6′ = 7.8 Hz, mPh-H), 8.23 (1H, d, J6,7 = 7.8 Hz, 6-H), 8.99 (1H, s, 5-H), 9.13 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C20H17ClN4O.0.4DMF: C, 64.62; H, 5.06; N, 15.64. Found: C, 64.94; H, 5.29; N, 15.79.
2-(Butylamino)-10–(4-chlorophenyl)-2,10-dihydropyrimido[4,5-b]quinolin-4(3H)-one (9e)
Yield, 0.61 g (81%); mp 264–267 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm nm (log ε/dmCitation3 mol−1 cm−1): 221 (4.71), 272 (4.65), 406 (4.21); IR (νcm−1): 3426 (NH), 1693 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 0.74 (3H, t, J = 8 Hz, 2-N(CH2)3CH3), 0.87–0.98 (2H, m, 2-N(CH2)2CH2-), 1.17–1.23 (2H, m, 2-NCH2CH2–), 3.17–3.25 (2H, m, 2-NCH2–), 6.71 (1H, d, J8,9 = 9 Hz, 9-H), 7.25–7.37 (2H, m, oPh-H), 7.46–7.55 (1H, m, 8-H), 7.66 (1H, dt, J6,7 = J7,8 = 9 Hz, J7,9 = 1.4 Hz, 7-H), 7.89 (2H, d, J2',3′ = J5’,6′= 9 Hz, mPh-H), 8.16 (1H, d, J6,7 = 9 Hz, 6-H), 8.92 (1H, s, 5-H), 9.80 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C21H19ClN4O.0.2H2O: C, 65.95; H, 5.11; N, 14.65. Found: C, 66.19; H, 5.42; N, 14.91.
10–(4-Chlorophenyl)-2-((2-hydroxyethyl)amino)-2,10-dihydropyrimido[4,5-b]quinolin-4(3H)-one (9f)
Yield, 0.64 g (87%); mp 270–272 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm nm (log ε/dmCitation3 mol−1 cm−1): 221 (4.62), 271 (4.59), 409 (4.07); IR (νcm−1): 3410 (OH), 3261 (NH), 1608 (C = O); 1H-NMR [DMSO-d6]: at 100 °C: δ 2.86–2.98 (2H, m, 2-NCH2–), 3.45–3.59 (2H, m, –CH2OH), 4.81 (1H, br s, –CH2OH, exchangeable with D2O), 6.29 (1H, br s, 2-NH, exchangeable with D2O), 6.79 (1H, d, J8,9 = 8.5 Hz, 9-H), 7.51 (1H, dt, J6,7 = J7,8 = 8.5 Hz, J7,9 = 1.5 Hz, 7-H), 7.59 (2H, d, J2',3′ = J5’,6′= 8.5 Hz, oPh-H), 7.65 (1H, t, J7,8 = J8,9 = 8.5 Hz, 8-H), 7.79 (2H, d, J2',3′ = J5’,6′= 8.5 Hz, mPh-H), 8.18 (1H, d, J6,7 = 8.5 Hz, 6-H), 8.94 (1H, s, 5-H),; Anal. calcd for C19H15ClN4O2.0.25 H2O: C, 61.46; H, 4.21; N, 15.09. Found: C, 61.80; H, 4.46; N, 15.38.
2-(Benzylamino)-10–(4-chlorophenyl)-2,10-dihydropyrimido[4,5-b]quinolin-4(3H)-one (9 g)
Yield, 0.68 g (82%); mp 288–291 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm nm (log ε/dmCitation3 mol−1 cm−1): 220 (4.79), 252 (4.56), 273 (4.70), 301sh (4.32), 407 (4.11); IR (νcm−1): 3412 (NH), 1693 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 4.65 (2H, br s, 2-NCH2–), 6.82 (1H, d J8,9 = 8.7 Hz, 9-H), 6.83–6.88 (2H, m, 7 and 8-H), 7.19–7.30 (5H, m, o,m,pPhCH2-H), 7.63–7.79 (2H, m, oPh-H), 7.93 (1H, d, J6,7 = 8.7 Hz, 6-H), 8.20 (2H, d, J2',3′ = J5’,6′= 8.7 Hz, mPh-H), 8.97 (1H, s, 5-H), 10.42 (1H, br s, 2-NH, exchangeable with D2O); Anal. calcd for C24H17ClN4O.0.4H2O: C, 68.62; H, 4.27; N, 13.34. Found: C, 68.91; H, 4.41; N, 13.45.
10–(4-Chlorophenyl)-2-hydrazineyl-2,10-dihydropyrimido[4,5-b]quinolin-4(3H)-one (9h)
Yield, 0.54 g (80%); mp 277–280 °C (decomp. From DMF-H2O); UV (EtOH): λmax nm nm (log ε/dmCitation3 mol−1 cm−1): 219 (4.04), 259 (3.92), 265 (3.91), 312sh (3.79); IR (νcm−1): 3420–3140 (NH, NH2), 1604 (C = O); 1HNMR (DMSO-d6): at 100 °C: δ 4.57 (1H, br s, 2-NH, exchangeable with D2O), 6.93 (1H, d, J8,9 = 8.4 Hz, 9-H), 6.98–7.02 (2H, m, 7 and 8-H), 7.06 (2H, d, J2',3′ = J5’,6′= 8.4 Hz, oPh-H), 7.17 (1H, d, J6,7 = 8.4 Hz, 6-H), 7.48 (2H, d, J2',3′ = J5’,6′= 8.4 Hz, mPh-H), 8.25 (2H, br s, 2NHNH2, exchangeable with D2O), 9.46 (1H, s, 5-H); MS., EI m/z: 338 (M++H, 1%), 322 (M-16, 25%), 64 (M-275, 100%); Anal. calcd for C17H12ClN5O. 0.2H2O: C, 59.46; H, 4.23; N, 20.40. Found: C, 59.83; H, 4.43; N, 20.57.
Biological screenings
Growth inhibitory activities of the test compounds against CCRF-HSB and KB tumour cell lines
The modified MTT assayCitation70 developed by T. MosmannCitation71, was used to determine the inhibitory effects of test compounds on cell growth in vitro. Medium containing 2 cells (CCRF-HSB-2 or KB cells) was seeded into each well of a 96-well microplate, with the compound solution added simultaneously to triplicate wells, before the final volume was made up to 100µl. The plate was incubated at 37 °C for 72 h in a humidified atmosphere of 5% CO2. After the incubation, 10 µl of MTT solution (5 mg/ml in phosphate-buffered saline lacking calcium and magnesium) was added to each well. After a further incubation for 4 h at 37 °C, 100 µl of 0.02 N HCl, 50% N,N-dimethylformamide, and 20% SDS was added to solubilise any MTT-formazan that had formed. The optical density of each well was measured at 570 nm (OD570) with a microplate reader and the inhibition of cell growth (%) was calculated as (1 – T/C) × 100, where C is the mean OD570 of the control group and T is that of the treated group. The IC50 was determined from the dose–response curve.
Growth inhibitory activities of the test compounds against MCF-7 and HeLa tumour cell lines
Cells were seeded onto sterile 96-well cell culture plates at densities of 5x103/well for HeLa cells and 2 × 104/well for MCF-7 cells. Cells were incubated for 24 h at 37 °C and 5% CO2, after which media was aspirated from cells and 100 µl fresh media with/without treatments were added. Control wells were treated by media with DMSO concentrations equivalent to those of treatment peptides. Cells were further incubated at 37 °C and 5% CO2 for 72 h, followed by the addition of MTT (10 µl of a 10 mg/ml solution) to each well. Following 2 h of incubation of the plate at 37 °C and 5% CO2 and shielded from light with tin foil, the media was removed, and the produced formazan crystals were solubilised by the addition of 200 µl of DMSO. The optical density of each well was measured quantitatively at 550 nm (OD550) by EL808 HiTeck™ Spectra Thermo plate reader (HiTeck Instruments, USA) and the inhibition of cell growth (%) was calculated as (1 – T/C) x 100, where C is the mean OD550 of the control group and T is that of the treated group. The IC50 was determined from the dose–response curve.
[γ-32P] ATP radiometric protein kinase assay method
The protein kinase profiling for compounds (4e and 4k) was conducted using the following protocol of KINEXUS Bioinformatics Corporation, Vancouver, British Columbia, Canada as we previously reportedCitation72,Citation73. Briefly, this assay was applied for a single measurement at 10 µM concentration utilising [γ-32P] ATP radioisotope against 20 protein kinases panel. Imatinib was used as a positive reference. All experiments have been carried out in a designated radioactive environment and at room temperature for 30 min in a total volume of 25.0 µL in the following sequence: component 1:5 µL of the active kinase (ca 10–50 nM final concentration), component 2:5 µL of the substrate solution. component 3: 5 µL of a buffer, component 4: 5 µL of test compounds and reference at different concentrations or 10% DMSO as a blank control, component 5: 5 µL of [γ-32P] ATP (from 50 µM stock solution, 0.8µCi). This assay was started with the addition of [γ-32P] ATP to a mixture of other components and then incubation for 30 min. The reaction was terminated by adding 10 µL of the mixture onto a MultiScreen phosphocellulose P81 plate, washing 3 times with 1% solution of phosphoric acid solution. Using a Trilux scintillation counter, the radioactivity was measured for the test samples in comparison to the blank control which contains all components except that the kinase substrate was replaced by the buffer solution.
Annexin V PI/FITC apoptosis assay
Apoptosis was quantified by detecting cell surface exposure of phosphatidylserine (PS) in apoptotic cells using Annexin V Fluorescein Isothiocyanate/Propidium Iodide (FITC/PI)Citation74. The seeded MCF-7 cells in 6 well plates at 1 × 105 cells/well in 2 ml medium. They were left to attach overnight at 37 °C, before treatment with compounds 4j and 9f for 72 h incubation periods at final concentrations of 0, 1, 5, and 10 µM. Cells were inspected microscopically before and after treatment to observe morphological changes. Floating cells were collected in tubes and kept on ice while the remaining cells were detached with trypsin, incubated at 37 °C for 3 min, and pooled. Cells were centrifuged at 1200 rpm for 5 min at 4 °C. Supernatants were discarded and 2 ml fresh medium was added to each tube and kept on ice. Each sample was syringed with a 23 G needle, and 1 × 105 cells were transferred to new 12 × 75 mm tubes. Cell solutions were washed (1 ml PBS) and centrifuged again. Supernatants were discarded and pellets re-suspended in 100 ml binding buffer (10 mM 4–(2-hydroxyethyl)-1- piperazine ethane sulphonic acid (HEPES), 0.14 M NaCl, 2.5 mM CaCl2, pH 7.4, diluted in dH2O 1:10, stored at 4 °C), and shaken gently. Annexin V FITC (10 ml, stored at 4 °C) was added to each sample and allowed to incubate for 15 min in the dark at room temperature. A further 400 ml of binding buffer and 10 ml PI (0.05 mg/mL in PBS) were added. Tubes were shaken gently and left to set for 10 min in the dark on ice. Annexin-PI positive cells were assessed within 1.0 h using a Becton Dickinson FACScan flow cytometer (BD Biosciences, San Jose, CA, USA). Data were acquired and analysed using CellQuest Software. Viable cells were differentiated from early and late apoptotic/necrotic cells by Annexin V (x-axis) and PI staining (y-axis). The percentage of apoptosis was calculated according to the reported methodsCitation75 by the formula: Sample – Control/Control × 100. The results were expressed as the percentages of apoptosis and necrosis, calculated from the ratio of absorbance of treated (apoptotic) samples to that of untreated (control) ones.
Molecular docking study
This study was performed using the advanced and widely distributed molecular grid-based docking program; Autodock 4.2Citation76 for docking of a flexible ligand within a flexible protein, where the flexibility of the target protein is taken into account in the later type and ignored in the former one. Autodock scans the active site for low energy binding models and orientations of the probe molecule, using a modified genetic algorism that employs a local search (GALS) and pre-computed grids for the evaluation of the interaction energy. The target PTK enzyme (PDB code, 1t46) protein was separated alone by using Accelyrs Discovery Studio visualise v3.1 client software [Accelrys inc., San Diego, CA,USA (2005)], and representative amino acids of the ligand-binding site were selected within 5 Å neighbourhood surrounding the embedded native ligand, STI-571 (Imatinib or Gleevec). The results of 10 randomly seeded runs were analysed for each of the docked inhibitors. The docked inhibitors were assigned to a cluster if the atomic coordinates of the docked inhibitors exhibited a root mean square deviation (RMSD) of less than 0.5 Å difference from each other (RMSD-tolerance of 0.5 Å). The clusters were ranked from the average lowest energy obtained for members of the cluster to the highest. The analysis was carried out for the top 10 docking clusters. Each of the clusters that exhibited significant negative interaction energies (binding free energies) was examined by the Accelyrs Discovery Studio visualiser.
Preparation of ligands and target protein tyrosine kinase
The three-dimensional structures of the aforementioned compounds were constructed using Chem3D Ultra 8.0 software [Chemical Structure Drawing Standard; Cambridge Soft Corporation, USA (2003)] to obtain standard 3D structures (pdb format), then they were energetically minimised by using MOPAC with 100 iterations and minimum RMS gradient of 0.10. It is recommended to confirm whether all hydrogen atoms are in the file before working with the AutoDock tool (ADT). After opening the ligand, it can be visualised, and ADT now automatically computes Gasteiger charges (empirical atomic partial charges) and distinguishes between the hybridisation state and type of each atom. As a part of the preparation, the program determines rotatable bonds of the ligand to be able to generate different conformers for the docking. The crystal structure of c-Kit receptor protein-tyrosine kinase in complex with STI-571 (Imatinib or Gleevec) was obtained from the Protein Data Bank (PDB) website: http://www.rcsb.org/ in pdb format. All bound waters and ligands were removed from the protein. For the target, the amino acids of the ligand binding site were defined according to the literatureCitation3. The flexible residues are Glu640, Thr670, and Asp810 in our system. The polar hydrogen model was chosen for the crystal structure. The proteins were assigned with Kollman-united charge. The polar hydrogens and charges were added.
Calculation of affinity maps by using a 3D grid embracing the protein and ligand
AutoGrid calculates the energy of the non-covalent interactions between the protein and probe atoms that are located in the different grid points of a lattice that defines the area of interest (i.e., the area of the macromolecule where the possibility of ligand binding is studied). AutoGrid builds as many files as the number of probe atoms used. There are about 30 different types of grid maps. The created three-dimensional grids of 60 × 60 × 60 Å size (x,y,z) with a spacing of 0.375 Å centred at 27.696, 26.657, and 39.342 Å that encompassed the active site where the co-crystallised ligand; STI-571 was embedded, was used to guide the docked inhibitors within PTK receptor. In this part of modelling, we need to determine the area where the ligand interacts with the enzyme, the size of that area, and the particular types of atoms participating in the interaction of the PTK enzyme. The first two parameters are determined by the size and position of the grid box; the third parameter is given by map type. Once those parameters were set in one file, AutoGrid calculates grid parameter files for each type of atom within a given area, then AutoDock is carried out the docking automatically using the newest docking algorithm (Lamarckian Genetic Algorithm). As a result of AutoDock calculations, we obtain the output file with ten conformers of the protein-ligand complex with flexible residues and the ligand located within the binding pocket. Each structure is scored and ranked by the program by the calculated interaction energy.
Author contributions
Participated in research design: Hamed I. Ali and Mosaad S. Mohamed. Conducted experiments: Walaa A. Bedewy, Ahmed M. Abdelhameed, Noriyuki Ashida, Mohamed A. Elsawy, Mohammed Al-Muhur. Performed data analysis: Hamed I. Ali, Tamer A. Elwaie, Ashraf N Abdalla, Noriyuki Ashida, Tomohisa Nagamatsu. Wrote or contributed to the writing of the manuscript: Hamed I.Ali, Walaa A. Bedewy, Tamer A. Elwaie, Ahmed M. Abdelhameed, Ashraf N Abdalla
Supplemental Material
Download PDF (3 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Shrestha AR, Ali HI, Ashida N, Nagamatsu T. Antitumor studies. Part 5: synthesis, antitumor activity, and molecular docking study of 5-(monosubstituted amino)-2-deoxo-2-phenyl-5-deazaflavins. Bioorg Med Chem. 2008;16(20):9161–9170.
- Ali HI, Tomita K, Akaho E, Kambara H, Miura S, Hayakawa H, Ashida N, Kawashima Y, Yamagishi T, Ikeya H, et al. Antitumor studies. Part 1: design, synthesis, antitumor activity, and AutoDock study of 2-deoxo-2-phenyl-5-deazaflavins and 2-deoxo-2-phenylflavin-5-oxides as a new class of antitumor agents. Bioorg Med Chem. 2007;15(1):242–256.
- Ali HI, Ashida N, Nagamatsu T. Antitumor studies. part 3: design, synthesis, antitumor activity, and molecular docking study of novel 2-methylthio-, 2-amino-, and 2-(N-substituted amino)-10-alkyl-2-deoxo-5-deazaflavins. Bioorg Med Chem. 2007;15(19):6336–6352.
- Ali HI, Ashida N, Nagamatsu T. Antitumor studies. Part 4: Design, synthesis, antitumor activity, and molecular docking study of novel 2-substituted 2-deoxoflavin-5-oxides, 2-deoxoalloxazine-5-oxides, and their 5-deaza analogs. Bioorg Med Chem. 2008;16(2):922–940.
- Ali HI, Tomita K, Akaho E, Kunishima M, Kawashima Y, Yamagishi T, Ikeya H, Nagamatsu T. Antitumor studies – part 2: structure-activity relationship study for flavin analogs including investigations on their in vitro antitumor assay and docking simulation into protein tyrosine kinase. Eur J Med Chem. 2008;43(7):1376–1389.
- Ali HI, Nagamatsu T, Akaho E. Structure-based drug design and AutoDock study of potential protein tyrosine kinase inhibitors. Bioinformation. 2011;5(9):368–374.
- Malki WH, Gouda AM, Ali HEA, Al-Rousan R, Samaha D, Abdalla AN, Bustamante J, Abd Elmageed ZY, Ali HI. Structural-based design, synthesis, and antitumor activity of novel alloxazine analogues with potential selective kinase inhibition. Eur J Med Chem. 2018;152:31–52.
- Wilson JM, Henderson G, Black F, Sutherland A, Ludwig RL, Vousden KH, Robins DJ. Synthesis of 5-deazaflavin derivatives and their activation of p53 in cells. Bioorg Med Chem. 2007;15(1):77–86.
- Dickens MP, Roxburgh P, Hock A, Mezna M, Kellam B, Vousden KH, Fischer PM. 5-Deazaflavin derivatives as inhibitors of p53 ubiquitination by HDM2. Bioorg Med Chem. 2013;21(22):6868–6877.
- Fenner H, Grauert R, Hemmerich P, Michel H, Massey V. 5-Thia-5-deazaflavin, a 1e–transferring flavin analog. Eur J Biochem. 1979;95(1):183–191.
- Kimachi T, Yoneda F, Sasaki T. New synthesis of 5-amino-5-deazaflavin derivatives by direct coupling of 5-deazaflavins and amines. J Heterocycl Chem. 1992;29(4):763–765.
- Yoneda F, Sakuma Y, Hemmerich P. Dehydrogenation of alcohols by pyrimido[4,5-b]quinoline-2(3H),4(10H)-dione (5-deazaflavin). J Chem Soc, Chem Commun. 1977;22(22):825–826.
- Eirich LD, Vogels GD, Wolfe RS. Proposed structure for coenzyme F420 from Methanobacterium. Biochemistry. 1978;17(22):4583–4593.
- Hossain MS, Le CQ, Joseph E, Nguyen TQ, Johnson-Winters K, Foss FW. Jr. Convenient synthesis of deazaflavin cofactor FO and its activity in F(420)-dependent NADP reductase. Org Biomol Chem. 2015;13(18):5082–5085.
- Philmus B, Decamps L, Berteau O, Begley TP. Biosynthetic versatility and coordinated action of 5'-deoxyadenosyl radicals in deazaflavin biosynthesis. J Am Chem Soc. 2015;137(16):5406–5413.
- Nagamatsu T,H, Yoneda F. Synthesis of pyridodipyrimidines as a redox catalyst and their autorecycling oxidation of alcohols. J Pharmacobio-Dyn. 1992;15:s-42.
- Yoneda F. Organic catalysts and autorecycling reactions. Yakugaku Zasshi. 1984;104(2):97–126.
- el-Gazzar AB, Hafez HN, Nawwar GA. New acyclic nucleosides analogues as potential analgesic, anti-inflammatory, anti-oxidant and anti-microbial derived from pyrimido[4,5-b]quinolines. Eur J Med Chem. 2009;44(4):1427–1436.
- El-Gohary NS. Synthesis and in vitro antitumor activity of new quinoline, pyrimido[4,5-b]quinoline, [1,2,3]triazino[4,5-b]quinoline, and [1,2,4]triazolo[2′,3′:3,4]pyrimido[6,5-b]quinoline analogs. Med Chem Res. 2013;22(11):5236–5247.
- Ghorab MM, Ragab FA, Heiba HI, Arafa RK, El-Hossary EM. In vitro anticancer screening and radiosensitizing evaluation of some new quinolines and pyrimido[4,5-b]quinolines bearing a sulfonamide moiety. Eur J Med Chem. 2010;45(9):3677–3684.
- O'Brien DE, Weinslock LT, Cheng CC. Synthesis of 10-deazariboflavin and related 2,4-dioxopyrimido[4,5-b]quinolones. J Heterocycl Chem. 1970;7(1):99–105.
- Yoneda F, Sakuma Y, Mizumoto S, Ito R. Syntheses of 5-deazaflavines. J Chem Soc Perkin Trans 1. 1976;(16):1805–1805.
- Chen X, Tanaka K, Yoneda F. A new synthetic approach to 5-deazaflavin and 5-deaza-10-thiaflavin. Chem Pharm Bull. 1990;38(3):612–615.
- Lacroix A, Fleury J-P. A new synthesis of 5-deazaflavins. Tetrahedron Lett. 1978;19(37):3469–3470.
- Marjani AP, Khalafy J, Ebrahimlo ARM, Prager RH. Synthesis of some pyrimido[4,5-b]quinoline derivatives. Bull Korean Chem Soc. 2011;32(7):2183–2186.
- Knabe J, Heckmann R. Dihydroisochinolinumlagerung, 30. Mitt. Synthese einiger Furo[3,2-c]pyridine. Arch Pharm Pharm Med Chem. 1980;313(9):809–811.
- Moriyama K, Nagamatsu T, Yoneda F. Synthesis of 8-methylpyrido[2,3-d:6,5-d’X]dipyrimidine-2,4,6(3H,10H,7H)-triones and their use in the oxidation of alcohols. J Heterocycl Chem. 1986;23(1):241–243.
- Taylor EC, Cain CK. Pteridines. VII. The synthesis of 2-alkylaminopteridines. J Am Chem Soc. 1952;74(7):1644–1647.
- Taylor EC, Cain CK. Pteridines. VI. Replacement reactions of amino, hydroxyl, and mercapto groups in the pteridine series. J Am Chem Soc. 1951;73(9):4384–4387.
- Coppola GM, Schuster HF. The chemistry of 2H-3,1-benzoxazine-2,4-(1H)-dione (isatoic anhydride). XXI: [1]. A mild process for the preparation of 10-alkyl-9-acridanones and it’s application to the synthesis of acridone alkaloids. J Heterocycl Chem. 1989;26(4):957–964.
- Kadry AM, Aal EHA, Abdel-Fattah HA, Al-Mahmoudy AM. Synthesis an antimicrobial activity of new triazolopyrimidinecarbonitrile derivatives. Arkivoc. 2008;2008(10):127–134.
- Farghaly AH., Abdel-Rahman Synthesis, reactions and antimicrobial activity of some new indolyl-1,3,4-oxadiazole, triazole and pyrazolederivatives. Jnl Chinese Chemical Soc. 2004;51(1):147–156.
- Irfan MS. S. Synthesis, characterization and antimicrobial activity of some substituted Ǹ-arylidene-2-(quinolin-8-yloxy)acetohydrazide. Acta Pharm Sci. 2009;51:163–168.
- Bhat I, Chaithanya S, Satyanarayana PD, Kalluraya B. The synthesis and antimicrobial study of some azetidinone derivatives with the para-anisidine moiety. J Serb Chem Soc. 2007;72(5):437–442.
- Cacic M, Trkovnik M, Cacic F, Has-Schon E. Synthesis and antimicrobial activity of some derivatives of (7-hydroxy-2-oxo-2H-chromen-4-yl)-acetic acid hydrazide. Molecules. 2006;11(2):134–147.
- Dabholkar V,SASA. Synthesis of novel oxazoles and their hydrazones. Rasayan J Chem. 2010;3:761–765.
- Nogimori T, Emerson CH, Braverman LE, Wu CF, Gambino J, Wright GE. Synthesis of 6-anilino-2-thiouracils and their inhibition of human placenta iodothyronine deiodinase. J Med Chem. 1985;28(11):1692–1694.
- Hübsch W, Pfleiderer W. Part LXXXVII. Synthesis and properties of 8-substituted 2-Thiolumazines. HCA. 1988;71(6):1379–1391.
- Fathalla W, Čajan M, Pazdera P. Regioselectivity of electrophilic attack on 4-methyl-1-thioxo-1,2,4,5-tetrahydro[1,2,4]triazolo[4,3-a]quinazolin-5-one. Part 1: reactions at the sulfur atom. Molecules. 2001;6(6):557–573.
- Israel M, Protopapa HK, Schlein HN, Modest EJ. Pyrimidine derivatives. v. synthesis of substituted pyrimidines from 4-amino-6-chloro-2-methylthiopyrimidine. J Med Chem. 1964;7:5–10.
- Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr., Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50(18):6075–6086.
- Masters JR. HeLa cells 50 years on: the good, the bad and the ugly. Nat Rev Cancer. 2002;2(4):315–319.
- Holliday DL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13(4):215–20110812.
- Singh JK, Simões BM, Howell SJ, Farnie G, Clarke RB. Recent advances reveal IL-8 signaling as a potential key to targeting breast cancer stem cells. Breast Cancer Res. 2013;15(4):210.
- Abdel-Fatah TM, Middleton FK, Arora A, Agarwal D, Chen T, Moseley PM, Perry C, Doherty R, Chan S, Green AR, et al. Untangling the ATR-CHEK1 network for prognostication, prediction and therapeutic target validation in breast cancer. Mol Oncol. 2015;9(3):569–585.
- Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. Embo J. 2000;19(13):3159–3167.
- Gillet JP, Calcagno AM, Varma S, Marino M, Green LJ, Vora MI, Patel C, Orina JN, Eliseeva TA, Singal V, et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc Natl Acad Sci USA. 2011;108(46):18708–18713.
- Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villén J, Li J, Cohn MA, Cantley LC, Gygi SP. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci USA. 2004;101(33):12130–12135.
- Watson NA, Cartwright TN, Lawless C, Cámara-Donoso M, Sen O, Sako K, Hirota T, Kimura H, Higgins JMG. Kinase inhibition profiles as a tool to identify kinases for specific phosphorylation sites. Nat Commun. 2020;11(1):1684–20200403.
- Wu C, Macleod I, Su AI. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 2013;41(Database issue):D561–5.
- Grueneberg DA, Degot S, Pearlberg J, Li W, Davies JE, Baldwin A, Endege W, Doench J, Sawyer J, Hu Y, et al. Kinase requirements in human cells: I. Comparing kinase requirements across various cell types. Proc Natl Acad Sci USA. 2008;105(43):16472–16477.
- Soule HD, Vazguez J, Long A, Albert S, Brennan M. A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst. 1973;51(5):1409–1416.
- Landry JJ, Pyl PT, Rausch T, Zichner T, Tekkedil MM, Stütz AM, Jauch A, Aiyar RS, Pau G, Delhomme N, et al. The genomic and transcriptomic landscape of a HeLa cell line. G3. 2013;3(8):1213–1224.
- Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9(9):631–643.
- Shajahan-Haq AN, Cook KL, Schwartz-Roberts JL, Eltayeb AE, Demas DM, Warri AM, Facey CO, Hilakivi-Clarke LA, Clarke R. MYC regulates the unfolded protein response and glucose and glutamine uptake in endocrine resistant breast cancer. Mol Cancer. 2014;13:239–20141023.
- Hafner M, Niepel M, Chung M, Sorger PK. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat Methods. 2016;13(6):521–527.
- Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol. 2012;920:613–626.
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516.
- Green DR. The death receptor pathway of apoptosis. Cold Spring Harb Perspect Biol. 2022;14(2):a041053.
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752.
- Cowan-Jacob SW, Fendrich G, Floersheimer A, Furet P, Liebetanz J, Rummel G, Rheinberger P, Centeleghe M, Fabbro D, Manley PW. Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukaemia. Acta Crystallogr D Biol Crystallogr. 2007;63(Pt 1):80–93.
- Karoulia Z, Wu Y, Ahmed TA, Xin Q, Bollard J, Krepler C, Wu X, Zhang C, Bollag G, Herlyn M, et al. An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell. 2016;30(3):485–498.
- Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC, Wilson KP. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279(30):31655–31663.
- George DM, Breinlinger EC, Friedman M, Zhang Y, Wang J, Argiriadi M, Bansal-Pakala P, Barth M, Duignan DB, Honore P, et al. Discovery of selective and orally bioavailable protein kinase Cθ (PKCθ) inhibitors from a fragment hit. J Med Chem. 2015;58(1):222–236.
- Tresaugues L, Roos A, Arrowsmith CH, Berglund H, Bountra C, Collins R, Edwards AM, Flodin S, Flores A, Graslund S, et al. Crystal structure of VEGFR1 in complex with N-(4-chlorophenyl)-2-((pyridin-4-ylmethyl)amino)benzamide. (To Be Published). https://www.rcsb.org/structure/3HNG.
- Levinson NM, Boxer SG. A conserved water-mediated hydrogen bond network defines bosutinib’s kinase selectivity. Nat Chem Biol. 2014;10(2):127–132.
- Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–W263.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717–20170303.
- Nagamatsu T, Hashiguchi Y, Yoneda F. A new, general, and convenient synthesis of 5-deazaflavins (5-deazaisoalloxazines) and bis-(5-deazaflavin-10-yl) alkanes. J Chem Soc Perkin Trans 1. 1984;1:561–565.
- Miura S, Yoshimura Y, Endo M, Machida H, Matsuda A, Tanaka M, Sasaki T. Antitumor activity of a novel orally effective nucleoside, 1-(2-deoxy-2-fluoro-4-thio-beta-D-arabinofuranosyl)cytosine. Cancer Lett. 1998;129(1):103–110.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63.
- Mahmoud S, Samaha D, Mohamed MS, Abou Taleb NA, Elsawy MA, Nagamatsu T, Ali HI. Design, synthesis, antitumor activity and molecular docking study of novel 5-deazaalloxazine analogs. Molecules. 2020;25(11):2518.
- Elwaie TA, Abbas SE, Aly EI, George RF, Ali H, Kraiouchkine N, Abdelwahed KS, Fandy TE, El Sayed KA, Abd Elmageed ZY, et al. HER2 kinase-targeted breast cancer therapy: design, synthesis, and in vitro and in vivo evaluation of novel lapatinib congeners as selective and potent her2 inhibitors with favorable metabolic stability. J Med Chem. 2020;63(24):15906–15945.
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184(1):39–51.
- Sineh Sepehr K, Baradaran B, Mazandarani M, Yousefi B, Abdollahpour Alitappeh M, Khori V. Growth-inhibitory and apoptosis-inducing effects of Punica granatum L. var. spinosa (Apple Punice) on fibrosarcoma cell lines. Adv Pharm Bull. 2014;4(Suppl 2):583–590.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791.