
Abstract
Parkinson’s disease (PD) is characterised by progressive death of dopamine (DA) neurons in the substantia nigra pars compacta (SNpc) and pathological accumulation of α-synuclein fibrils, as well as central nervous system inflammation. Elevated levels of central inflammatory factors in PD disrupt the kynurenine pathway (KP) and favour the activation of excitotoxic branches, leading to a significant reduction in the neuroprotective metabolite kynurenic acid (KYNA) and a significant increase in the neurotoxic metabolite quinolinic acid (QUIN), which exacerbates excitotoxicity and amplifies the inflammatory response, closely related to the occurrence and development of PD. KYNA analogs, precursor drugs, and KP enzyme modulators may represent a new therapeutic strategy for PD. This article reviews the role of KP in the neurodegenerative pathology of PD and its prevention and treatment, aiming to provide necessary theoretical basis and new ideas for the study of the neurobiological mechanisms underlying PD-related behavioural dysfunction and targeted interventions.
Parkinson’s disease (PD) is a complex multifactorial neurodegenerative disease with a prevalence second only to Alzheimer’s disease. The main pathological feature of the disease is the degeneration and loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the accumulation of misfolded α-synuclein (α-Syn) to form Lewy bodies (LB) in the brain, accompanied by the occurrence of neuroinflammationCitation1. Up to 10 million people worldwide are affected, with an estimated lifetime risk of about 2%Citation2. China is entering an ageing society, and it is estimated that by 2030, the number of PD patients in China will increase to 4.94 million, accounting for half of the global PD patientsCitation3. PD patients typically have four motor symptoms, including resting tremor, bradykinesia, postural instability, and rigidity of the neck, trunk, and limbsCitation4. Although PD was first described by James Parkinson in 1817, its aetiology has not been fully elucidated to this day. Most scholars believe that the degeneration and necrosis of SNpc dopaminergic neurons lead to the dysfunction of the basal ganglia circuit, resulting in motor disordersCitation5. Currently, there are two main treatment methods for PD, surgical treatment (such as deep brain stimulation) and drug treatment (such as levodopa and dopamine agonists)Citation6, which can only temporarily relieve PD symptoms but cannot stop the progression of the disease. Therefore, the development of new treatments for PD has always been an urgent task for clinical doctors and scientists worldwide.
Although the molecular pathological mechanisms underlying PD are not yet clear, in recent years, researchers have found abnormal activation of microglia and enhanced inflammatory responses in brain tissue, both in post-mortem results of PD patients and in PD animal modelsCitation7–9. In the 6-OHDA animal model, it was found that the activation of microglia occurred earlier than the loss of DA neurons, and the release of inflammatory factors by activated microglia could further promote abnormal aggregation of α-synuclein, suggesting that the activation of microglia may be the primary factor in the onset of PDCitation10. The kynurenine pathway (KP) of tryptophan metabolism is one of the main regulatory factors of immune response and may be related to the inflammation and neurotoxicity events of PDCitation11. Several neuroactive compounds produced by KP can be neurotoxic, neuroprotective or immunomodulatory. Among these metabolites, kynurenic acid (KYNA) produced by astrocytes is considered to have neuroprotective effects, while quinolinic acid (QUIN) released by activated microglia can activate the N-methyl-D-aspartate (NMDA) receptor signalling pathway, leading to excitotoxicity and amplifying the inflammatory responseCitation12. Previous studies have shown that NMDA receptor antagonists can alleviate PD symptoms in vivo and in vitro and exert neuroprotective effectsCitation13. Multiple lines of evidence suggest that some KP intermediates are related to the neuro-pathogenesis of PDCitation14–16. In addition, some KP metabolites may serve as prognostic biomarkers, and pharmacological modulators of KP enzymes may represent a new therapeutic strategy for PDCitation17. This article reviews the role of KP in PD neurodegeneration and PD motor prevention and treatment, in order to provide necessary theoretical basis and new ideas for the study of the neurobiological mechanisms of exercise intervention in alleviating PD-related behavioural dysfunction and targeted intervention.
Kynurenine pathway
KP is the main metabolic pathway for the essential amino acid tryptophan (TRP) in the body. This pathway is responsible for 95%-99% of TRP metabolism in the body. The remaining TRP is used for protein synthesis or for the formation of serotonin and melatonin. The generation of kynurenine from TRP is the first rate-limiting step of KP, which is mediated by indoleamine 2,3-dioxygenase (IDO) 1, IDO2, and tryptophan 2,3-dioxygenase (TDO). These three enzymes catalyse the production of kynurenine from TRP but are activated by different stimuli and have different tissue distributionsCitation18 (). IDO1 is widely expressed in immune and non-immune tissues and is activated by certain immune and inflammatory stimuli. IDO2 is mainly located in the liver and kidneys, and has lower catalytic activity than IDO1Citation21. TDO is mainly located in the liver, but also exists in nanomolar concentrations in brain astrocytesCitation22. Under normal physiological conditions, IDO activity is low, and KP basic metabolism is mainly mediated by TDO in the liver, which is the rate-limiting enzyme for the core metabolic product of KP, kynurenine. When the body is in an immune response or stress state, the activity of TDO and its isozyme IDO is overactivated by pro-inflammatory cytokines such as interferon (IFN)-γ, tumour necrosis factor (TNF)-α, lipopolysaccharides (LPS), interleukin (IL)-1,2,6, IL-1β, and prostaglandins. IDO then becomes the rate-limiting enzyme for the core metabolic product of KP, kynurenine, leading to an increase in kynurenine levels in peripheral and central nervous tissuesCitation23,Citation24. Peripheral kynurenine can cross the blood-brain barrier and enter the central nervous system (60% of kynurenine in the central nervous system comes from the periphery)Citation25. Subsequently, depending on the cell type, kynurenine is further metabolised into neurotoxic products mainly composed of QUIN and neuroprotective products mainly composed of KYNA. In microglia and macrophages, kynurenine is mainly converted to 3-hydroxykynurenine (3-HK), 3-hydroxyanthranilic acid (3-HAA), and QUIN by kynurenine-3-monooxygenase (KMO) and 3-hydroxyanthranilate-3,4-dioxygenase (HAAO)Citation26. Like TRP and kynurenine, 3-HK can cross the blood-brain barrier and, in addition to increasing the availability of substrates for QUIN production, is a neurotoxic intermediate that plays a neurotoxic role at nanomolar concentrations by generating free radicals and causing neuronal degeneration and apoptosisCitation15. In addition, 3-HK can generate superoxide and hydrogen peroxide, promoting copper-dependent oxidative protein damageCitation27. 3-HK can also significantly synergize with QUIN neurotoxicityCitation28. QUIN promotes neurodegeneration by inducing excitotoxicity of ionotropic glutamate receptors (iGluRs) (N-methyl-D-aspartate receptors (NMDAR), α-amino − 3 - hydroxy − 5 - methy l − 4 - isoxazole propionic acid receptors (AMPAR), and kainate receptors (KAR)) and α7-nicotinic acetylcholine receptors (α7-nAChR). In addition, QUIN can promote the release of glutamate (Glu) in neurons, inhibit the reuptake of Glu by astrocytes, and block the expression of glutamine synthetase, thereby further activating iGluRs and disrupting the dynamic balance of the Glu-glutamine-γ-aminobutyric acid cycle, exacerbating the excitotoxicity of Glu and causing neuronal damageCitation29. QUIN can also induce immune imbalance and inflammation, leading to the production of various pro-inflammatory cytokines such as IFN-α, IFN-γ, TNF-α, IL-1,2,6, and IL-1β, activating IDO, while KMO is also upregulated under inflammatory conditions, leading to a vicious cycle of KP formation and further promoting the neurotoxic metabolite pathway of KP. In astrocytes and peripheral skeletal muscle cells, kynurenine is catalysed to KYNA by kynurenine aminotransferase (KAT), KAT II is responsible for synthesising the majority of KYNA in the central nervous system (CNS). In contrast to the neurotoxic activity of QUIN, KYNA is a neuroprotective metabolite that acts as a broad-spectrum antagonist of iGluRs (AMPAR, NMDAR, and KAR) and a potent negative allosteric modulator of α7-nAChR, thereby blocking the excitotoxic effects of QUIN on iGluRs and α7-nAChR. KYNA also has the ability to clear reactive oxygen and nitrogen species and extracellular glutamate, exerting a neuroprotective effectCitation30. Under inflammatory conditions, although IDO-1 is activated and overexpressed in astrocytes, QUIN cannot be synthesised because astrocytes do not express KMO. However, KAT II, which is specifically expressed in astrocytes, can synthesise neuroprotective KYNA. Although the neuroprotective effect of KYNA can counteract the toxic effects of QUIN, a three-fold concentration of KYNA is required to counteract the same amount of QUINCitation31. Under normal physiological conditions, the KP is in a state of good metabolic balance, producing all intermediate metabolites of the KP and ultimately generating the coenzyme nicotinamide adenine dinucleotide (NAD+). However, under inflammatory conditions, the balance of KP metabolites or enzyme expression levels is disrupted in the pathological process driven by inflammation, mainly manifested as an imbalance in the QUIN/KYNA balance towards overexpression of neurotoxic and/or pro-inflammatory molecules of KP.
Figure 1. (A B). Pathway of canine uric acid. Schematic diagram of the canine uric acid pathway in human peripheral and brain. Modified fromCitation19,Citation20. (A) In the periphery, tryptophan is metabolised into uric acid mainly by liver tryptophan 2,3-dioxygenase (TDO) (activated by glucocorticoids) and indoleamine 2,3-dioxygenase 1 (IDO1) (activated by inflammatory cytokines). After the first rate-limiting step, kynurenine is metabolised into kynurenic acid by kynurenine aminotransferase (KAT) through the neuroprotective branch (A: 1-2-3), or into quinone by kynurenine 3-monooxygenase (KMO) through the potential neurotoxic branch (A: 1-2-4-6-8). (B) Cytokines, tryptophan, kynurenin, and 3-hydroxyuric acid (3-HK) can cross the blood-brain barrier. Once in the brain, cytokines and glucocorticoids stimulate the uric acid pathway, and inflammation increases the favourable neurotoxic quinone branch due to its activation of KMO. The quinone branch of the uric acid pathway mainly occurs in activated and resting microglia [branch] and infiltrating macrophages (B:1-2-3-5-7), while the kynurenic acid branch mainly occurs in astrocytes (B:1-2-3). HAA: 3-hydroxyanthranilic acid; 3-HAO: 3,4-dioxygenase of 3-hydroxyanthranilic acid; 3-HK: 3-hydroxyuric acid; IDO1: indoleamine 2,3-dioxygenase 1; IFN: interferon; IL: interleukin; KA: uric acid; KAT: kynurenine aminotransferase; KMO: kynurenine 3-monooxygenase; KYN: kynurenine; KYNU: kynureninase; NAD +: nicotinamide adenine dinucleotide; PicA: picolinic acid; TDO: tryptophan 2,3-dioxygenase; TNF: tumour necrosis factor; TRP: tryptophan; QUIN: quinone; XA: xanthine acid.
![Figure 1. (A B). Pathway of canine uric acid. Schematic diagram of the canine uric acid pathway in human peripheral and brain. Modified fromCitation19,Citation20. (A) In the periphery, tryptophan is metabolised into uric acid mainly by liver tryptophan 2,3-dioxygenase (TDO) (activated by glucocorticoids) and indoleamine 2,3-dioxygenase 1 (IDO1) (activated by inflammatory cytokines). After the first rate-limiting step, kynurenine is metabolised into kynurenic acid by kynurenine aminotransferase (KAT) through the neuroprotective branch (A: 1-2-3), or into quinone by kynurenine 3-monooxygenase (KMO) through the potential neurotoxic branch (A: 1-2-4-6-8). (B) Cytokines, tryptophan, kynurenin, and 3-hydroxyuric acid (3-HK) can cross the blood-brain barrier. Once in the brain, cytokines and glucocorticoids stimulate the uric acid pathway, and inflammation increases the favourable neurotoxic quinone branch due to its activation of KMO. The quinone branch of the uric acid pathway mainly occurs in activated and resting microglia [branch] and infiltrating macrophages (B:1-2-3-5-7), while the kynurenic acid branch mainly occurs in astrocytes (B:1-2-3). HAA: 3-hydroxyanthranilic acid; 3-HAO: 3,4-dioxygenase of 3-hydroxyanthranilic acid; 3-HK: 3-hydroxyuric acid; IDO1: indoleamine 2,3-dioxygenase 1; IFN: interferon; IL: interleukin; KA: uric acid; KAT: kynurenine aminotransferase; KMO: kynurenine 3-monooxygenase; KYN: kynurenine; KYNU: kynureninase; NAD +: nicotinamide adenine dinucleotide; PicA: picolinic acid; TDO: tryptophan 2,3-dioxygenase; TNF: tumour necrosis factor; TRP: tryptophan; QUIN: quinone; XA: xanthine acid.](/cms/asset/edab6447-9969-4aed-8b83-c1255d0a9d76/ienz_a_2225800_f0001_c.jpg)
KP and PD
Research has shown that immune dysfunctionCitation32, excitotoxicity of GluCitation33, and depletion of DACitation34 are all related to the increased risk and development of PD. The potential intersection of the immune system, Glu system, and monoaminergic system involved in these factors is KPCitation35. Under the induction of pro-inflammatory cytokines, KP activation disrupts the balance between neuroprotection and neurotoxicity branches, tending to produce neurotoxic products 3-HK (a strong inducer of neurotoxic free radicals) and QUIN, which play an important role in the pathogenesis of PD through excitotoxicity, oxidative stress, and/or inflammatory reactions. The first description of KP abnormalities in PD patients was in the early 1990s. Ogawa et al.Citation36 reported that the concentration of KYN and KYNA in the substantia nigra pars compacta (SNpc), striatum, and prefrontal cortex of PD patients was significantly decreased, while the concentration of the neurotoxin 3-HK in the striatum and SNpc was significantly increased. At the same time, Beal et al.Citation37 found that the levels of KYNA in the cortical areas, caudate nucleus, and cerebellum of PD patients were significantly decreased. The direct consequence of this low endogenous KYNA level is a reduced ability to limit the excitotoxicity induced by QUIN and/or excess glutamate through NMDAR. It has been reported that the KYN/TRP ratio in the CSF and serum of PD patients is significantly increased compared to age- and gender-matched healthy controlsCitation38. Animal experiments have confirmed that the expression level of KP enzyme kynurenine aminotransferase-I (KAT-I), which leads to KYNA formation, is significantly decreased in the SNpc of MPTP-treated miceCitation39. According to data from post-mortem PD brain tissue and MPTP-treated mice, the activity of KAT-I and KAT-II is significantly decreased, consistent with lower plasma KYNA levelsCitation40. Subsequently, more and more research evidence has shown that KP is closely related to the occurrence and development of PDCitation14,Citation41. Heilman et al.Citation39 used a mass spectrometry-based targeted metabolomics analysis method, confirmed that plasma 3-HK levels were significantly elevated in PD patients, while the metabolic products 3-hydroxyanthranilic acid (3-HAA) and NAD+ (the final product of the KP pathway) were significantly reduced. The 5-HT/KYN ratio in the plasma of PD patients is also lower; the level of neuroprotective metabolite KYNA in the cerebrospinal fluid of PD patients is significantly decreased, the KYNA/KYN ratio is lower, and the ratio of neurotoxicity (QUIN/KYNA) is significantly increased. The plasma 3-HK level in PD patients is significantly correlated with the score reflecting the severity of PD symptoms (Unified Parkinson’s Disease Rating Scale, UPDRS I, II, and III), and the cerebrospinal fluid QUIN level is significantly correlated with higher UPDRS II and III scores. The higher KYN/TRP ratio is also significantly correlated with higher ADL and motor scores. The QUIN/PIC and QUIN/3-HK ratios are both closely related to the severity of ADL symptoms in PD patients (UPDRS II). The QUIN/KYNA ratio is related to higher non-motor daily life symptom scores (UPDRS I). In addition, it has been found that the increase in the QUIN/PIC ratio in the cerebrospinal fluid of PD patients is related to the increase in R2* value (a magnetic resonance imaging (MRI) index reflecting iron content) in the substantia nigra. C-reactive protein and serum amyloid protein α in the blood are associated with signs of increased KP activity in the cerebrospinal fluid of PD patients. These findings indicate that the levels of KP metabolites are altered in both the peripheral and central nervous systems in PD, and these changes are closely related to the severity of symptoms, consistent with previous research resultsCitation42–44. Sorgdrager et al.Citation44 used solid-phase extraction-liquid chromatography-tandem mass spectrometry to detect KP metabolites in the serum and cerebrospinal fluid of PD patients with clear clinical features and age-matched controls. The results showed that age was closely related to the increase in KYN, KYNA, and QUIN concentrations in the serum and cerebrospinal fluid, but the KYNA concentration in PD patients was significantly decreased compared to the age-matched control group. There was a strong positive correlation between KYN and QUIN levels in the serum and cerebrospinal fluid, and the concentrations of other metabolites, 3-HK, KYNA, and XA, were correlated with their corresponding concentrations in the cerebrospinal fluid. Bai et al.Citation45 studied the urinary KYN levels in early PD patients and found that urinary KYN levels could distinguish early PD patients from healthy controls. Urinary KYN levels were positively correlated with Hoehn-Yahr staging and disease duration, and negatively correlated with mini-mental state examination (MMSE) scores in PD patients. These results suggest that urinary KYN may be a new biomarker for detecting early PD and evaluating PD progression. Urinary KYN measurement may help clinicians identify early PD patients.
In summary, the levels of KP metabolites are altered in both the peripheral and central nervous systems in PD, and are closely related to the occurrence and development of PD. KP metabolites may become biomarkers for PD.
The role of KP in the treatment of PD
The role of KP metabolites in PD treatment
In animal models of Parkinson’s disease (PD), many studies have shown that increasing endogenous KYNA or reducing QUIN production can prevent and/or delay PD progression. Miranda et al.Citation46 demonstrated that injection of QUIN (60 nmol) or N-methyl-D-aspartic acid (15 nmol) into the substantia nigra of rats significantly depleted tyrosine hydroxylase activity in the striatum. However, co-infusion of nicotinylalanine (5–6 nmol), an inhibitor of kynureninase and kynurenine hydroxylase activity, with kynurenine (the precursor of KYNA) (450 mg/kg i.p.) and probenecid (an inhibitor of organic acid transport) (200 mg/kg i.p.) into the lateral ventricle of rats for 3 h resulted in a 3.3-fold increase in KYNA in the whole brain and a 1.5-fold increase in the substantia nigra, and the elevation of endogenous KYNA prevented the decrease in tyrosine hydroxylase induced by QUIN. This suggests that increasing the level of endogenous KYNA can prevent the loss of dopaminergic neurons in the substantia nigra induced by focal infusion of quinolinic acid or N-methyl-D-aspartic acid. Silva-Adaya et al.Citation47 treated 6-OHDA-lesioned PD model rats with the precursor of KYNA, L-kynurenine (L-KYN), and probenecid (PROB), an inhibitor of organic acid transport, for 7 consecutive days. The results showed that systemic administration of L-KYN + PROB significantly increased KYNA and DA levels in the striatum of PD model rats, significantly decreased (hydroxycinnamic acid + dihydroxyphenylacetic acid)/DA and glial fibrillary acidic protein (GFAP) expression levels, significantly reduced Fluoro Jade staining fluorescence intensity, and significantly improved motor dysfunction (as evidenced by a significant decrease in apomorphine-induced rotations). In addition to regulating the release of glutamate from the cortex to the striatum, the elevation of KYNA can also directly antagonise NMDA receptors, thereby reducing Glu excitotoxicity. Studies have shown that zonisamide, a sulphonamide antiepileptic drug used to treat PD, can significantly improve the motor symptoms of PD patients with levodopa-induced dyskinesia and significantly increase KYNA productionCitation17; injection of KYNA (50 μg) into the bilateral internal pallidum of MPTP-induced PD model monkeys can significantly alleviate motor disorders, tremors, and muscle stiffnessCitation47. This further supports the potential role of KYNA in alleviating PD-related behavioural and functional disorders. Fukuyama et al.Citation48 further demonstrated that acute and chronic exposure of astrocytes to zonisamide not only resulted in the production of KYNA but also led to the production of other neuroactive metabolites in the KP pathway, such as xanthurenic acid and quinolinic acid. It has been shown that xanthurenic acid, quinolinic acid, and cinnamic acid are endogenous agonists of group II and III metabotropic glutamate receptorsCitation49. The activation of group II and III metabotropic glutamate receptors is a potential important drug target for PD symptom relief and neuroprotectionCitation50, in addition to KYNA, other neuroactive metabolites of KP may also have therapeutic importance in PDCitation43. Furthermore, long-term exposure to zonisamide can promote IFN-γ-mediated increase in quinolinic acid while reducing QUIN generationCitation17. This provides evidence for the enhancement of neuroprotection by KYNA, uric acid, and quinolinic acid, while limiting the excitotoxicity of QUIN at lower levels.
The role of KP key enzyme regulators in the treatment of PD
Inhibitors of key KP enzymes are a potential method for management. KMO is a midstream enzyme in the KP metabolic pathway responsible for converting KYN to downstream neurotoxic metabolite 3-HK. Pharmacological inhibition of KMO has been widely shown to stimulate KYNA productionCitation51. Studies have shown that KMO inhibitors can significantly reduce the severity of hamster muscle tone disorders. Therefore, KMO inhibitors may be potential candidate drugs for managing movement disorders associated with striatal dysfunction in PDCitation52. Several studies have demonstrated that selective pharmacological inhibitors of KMO, such as Ro61-8048, not only promote dose-dependent increases in peripheral and central KYNA production but also alleviate levodopa-induced motor dysfunction in MPTP-treated non-human primate PD models without impairing the therapeutic effects of levodopaCitation53,Citation54. Another study showed that co-administration of KYNase, propentofylline (which prevents KYNA clearance from the brain), and KMO inhibitor nicotine propionyl-L-carnitine strongly promotes KYNA production, preventing NMDA and QUIN-induced excitotoxicity in dopaminergic neurons of the substantia nigraCitation55,Citation56. IDO is one of two rate-limiting enzymes (IDO-1 and IDO-2 and TDO) that convert TRP to KYN in the initial step of KP. When the body is in an immune response or stress state, IDO is overactivated. Studies have shown that treatment of 6-OHDA-induced PD model mice with IDO-1 inhibitor 1-methyltryptophan (1-MT) significantly reduces cortical and striatal lipid peroxidation markers and serum nitrite levels. Cortical and striatal reduced glutathione, catalase, superoxide dismutase, mitochondrial complex I and IV activity significantly increased. Expression levels of pro-inflammatory cytokines (IL-6, NF-κB, TNF-α, and IFN-ϒ) and apoptosis markers caspase-3 significantly decreased. Expression levels of antioxidant damage markers such as haem oxygenase-1 (HO-1) and neurotrophic factors such as brain-derived neurotrophic factor significantly increased. Striatal extracellular DA and homovanillic acid content significantly increased, while QUIN content significantly decreased. Motor coordination and spontaneous movement significantly improved, as evidenced by a significant increase in the latency period of falling off the rod, and an increase in the distance and number of standing in the open fieldCitation57. TDO, a tryptophan degradation enzyme, was identified as a potential drug target for PD in a genome-wide RNA interference screen for ageing-related α-synuclein toxicity regulators in C. elegansCitation58. Studies have shown that after 21 days of oral administration of TDO inhibitor (NTRC 3531–0) to PD model mice induced by rotenone, TDO activity was inhibited, and TRP levels in plasma and brain significantly increased. The number of TH-positive cells in the substantia nigra significantly increased, the volume of microglia significantly decreased, the number of branches significantly increased, the expression of glial fibrillary acidic protein, a marker of enteric glia, decreased, and the accumulation of α-synuclein in enteric plexus decreased. The latency period of falling off the rotating rod significantly increased, the time spent exploring shifted objects significantly increasedCitation59.
The role of analogs or precursor drugs of KP major neuroprotective metabolites in the treatment of PD
KYNA analogs or KYNA precursor drugs are another potential method for preventing and/or delaying PD progression. Since KYNA is difficult to penetrate the blood-brain barrier (BBB), its use as a neuroprotective agent is limited. However, KYNA precursor drugs or analogs can easily penetrate the BBB and be converted to KYNA in the brainCitation60. The precursor drug 4-chloro-KYN (a KYNA analog) of 7-chloro-KYNA (a synthetic KYNA precursor drug) overcomes the difficulty of KYNA penetrating the BBB and provides excitotoxic neuroprotection after systemic administrationCitation61. Conjugated compounds of 7-Cl-KYNA with D-glucose or D-galactose esters can promote the transport of 7-Cl-KYNA across the BBB. Intraperitoneal injection of these substances indicates their potential significance in experimental treatment of neurodegenerative diseasesCitation62. Studies have also shown that a newly synthesised KYNA analog, aminoglycoside-KYNA, is more easily penetrable through the BBB than KYNA and can more effectively modify the conformational site of NMDA receptorsCitation63. In addition, FK506, a ligand for pro-neurotrophins used clinically as an immunosuppressant, stands out in PD treatment. FK506 not only enhances KYNA formation in the cortex but also eliminates the inhibition of KYNA synthesis induced by 1 - methy l − 4 - phenylpyridinium and 3-nitropropionic acidCitation64.
In summary, the use of regulators of the main KP enzyme system (such as agonists or antagonists) and their main neuroprotective metabolites or analogs to increase KYNA levels in the brain may reduce the overactivation of excitatory amino acid receptors and may delay and/or alleviate the progression of PD neurodegenerative diseases, making it a potential therapeutic strategy for PD treatment ().
Figure 2. KP putative targets for drug development. TDO: tryptophan 2,3-dioxygenase; IDO1: indoleamine 2, 3 - dioxygenases; KYN: kynurenine; KYNA: kynurenic acid; AA: Anthranilic acid; KATs:kynurenine aminotransferase; KMO: kynurenine - 3 - monooxygenase; 3 - HAA: 3-hydroxyanthranilic acid; 3-HAO: 3,4-dioxygenase of 3-hydroxyanthranilic acid; 3-HK: 3-hydroxyuric acid; NAD +: nicotinamide adenine dinucleotide; PicA: picolinic acid; QUIN: quinone; XA: xanthine acid; ACMS: aminocarboxymuconate - semialdehyde; AMS: 2-aminomuconic 6-semialdehyde.
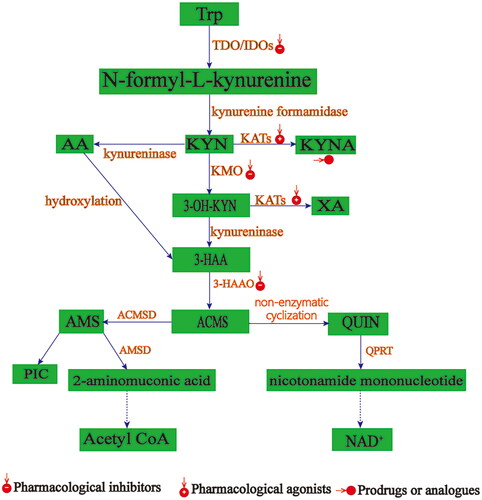
Summary
The levels of KP metabolites are altered in both the peripheral and central nervous systems in PD, and are closely related to the development of PD. KP metabolites may serve as biomarkers for PD. Modulation of the KP main enzyme system (such as agonists or antagonists) and its major neuroprotective metabolites or analogs may become a potential therapeutic strategy for PD.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Carrarini C, Russo M, Dono F, Di Pietro M, Rispoli MG, Di Stefano V, Ferri L, Barbone F, Vitale M, Thomas A, et al. A stage-based approach to therapy in Parkinson’s Disease. Biomolecules. 2019;9(8):388.
- Azeggagh S, Berwick DC. The development of inhibitors of leucine-rich repeat kinase 2 (LRRK2) as a therapeutic strategy for Parkinson’s disease: the current state of play. Br J Pharmacol. 2022;179(8):1478–1495.
- Li G, Ma J, Cui S, He Y, Xiao Q, Liu J, Chen S. Parkinson’s disease in China: a forty-year growing track of bedside work. Transl Neurodegener. 2019;8:22.
- Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V. Inflammation and immune dys function in Parkinson disease. Nat Rev Immunol. 2022;22(11):657–673.
- Chen Z, Zhang Z-Y, Zhang W, Xie T, Li Y, Xu X-H, Yao H. Direct and indirect pathway neurons in ventrolateral striatum differentially regulate licking movement and nigral responses. Cell Rep. 2021;37(3):109847.
- Feng Y-S, Yang S-D, Tan Z-X, Wang M-M, Xing Y, Dong F, Zhang F. The benefits and mechanisms of exercise training for Parkinson’s disease. Life Sci. 2020;245:117345.
- Knott C, Stern G, Wilkin GP. Inflammatory regulators in parkinsons disease: iNOS, Iipocortin-1, and cyclooxygenases-1 and 2. Mol Cell Neurosci. 2000;16(6):724–739.
- Koprich JB, Reske-Nielsen C, Mithal P, Isacson O. Neuroinflammation mediated by IL-1β increases susceptibility of dopamine neurons to degeneratiion in an animal model of parkinsons disease. J Neuroinflammation. 2008;5:8.
- Tan E-K, Chao Y-X, West A, Chan L-L, Poewe W, Jankovic J. Parkinson disease and the immune system - associations, mechanisms and therapeutics. Nat Rev Neurol. 2020;16(6):303–318.
- Mikita T, Campbell D, Wu P, Williamson K, Schindler U. Requirements for interleukin- 4 induced gene expression and function characterizaton of stat6. Mol Cell Biol. 1996;16(10):5811–5820.
- Fathi M, Vakili K, Yaghoobpoor S, Tavasol A, Jazi K, Hajibeygi R, Shool S, Sodeifian F, Klegeris A, McElhinney A, et al. Dynamic changes in metabolites of the kynurenine pathway in Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: a systematic review and meta-analysis. Front Immunol. 2022;13:997240.
- Mor A, Tankiewicz-Kwedlo A, Krupa A, Pawlak D. Role of kynurenine pathway in oxidative stress during neurodegenerative disorders. Cells. 2021;10(7):1603.
- Zhang Z, Zhang S, Fu P, Zhang Z, Lin K, Ko JK-S, Yung KK-L. Roles of glutamate receptors in Parkinson’s disease. IJMS. 2019;20(18):4391.
- Venkatesan D, Iyer M, Narayanasamy A, Siva K, Vellingiri B. Kynurenine pathway in Parkinson’s disease-An update. NeurologicalSci. 2020;21:100270.
- Behl T, Kaur I, Sehgal A, Singh S, Bhatia S, Al-Harrasi A, Zengin G, Bumbu AG, Andronie-Cioara FL, Nechifor AC, et al. The footprint of kynurenine pathway in neurodegeneration: janus-faced role in Parkinson’s disorder and therapeutic implications. IJMS. 2021;22(13):6737.
- Zhang S, Collier MEW, Heyes DJ, Giorgini F, Scrutton NS. Advantages of brain penetrating inhibitors of kynurenine-3-monooxygenase for treatment of neurodegenerative diseases. Arch Biochem Biophys. 2021;697:108702.
- Lim CK, Fernández-Gomez FJ, Braidy N, Estrada C, Costa C, Costa S, Bessede A, Fernandez-Villalba E, Zinger A, Herrero MT, et al. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog Neurobiol. 2017;155:76–95.
- Zádor F, Joca S, Nagy-Grócz G, Dvorácskó S, Szűcs E, Tömböly C, Benyhe S, Vécsei L. Pro-inflammatory cytokines: potential links between the endocannabinoid system and the kynurenine pathway in depression. IJMS. 2021;22(11):5903.
- Brown SJ, Huang XF, Newell KA. The kynurenine pathway in major depression: what we know and where to next. Neurosci Biobehav Rev. 2021;127:917–927.
- Qin W, Shi Y, Chen W, Jia X, Asakawa T. Can kynurenine pathway be considered as a next-generation therapeutic target for Parkinson’s disease? An update information. Biosci Trends. 2022;16(4):249–256.
- Morales-Puerto N, Giménez-Gómez P, Pérez-Hernández M, Abuin-Martínez C, Gil de Biedma-Elduayen L, Vidal R, Gutiérrez-López MD, O'Shea E, Colado MI. Addiction and the kynurenine pathway: a new dancing couple? Pharmacol Ther. 2021;223:107807.
- Ball HJ, Jusof FF, Bakmiwewa SM, Hunt NH, Yuasa HJ. Tryptophan-catabolizing enzymes - party of three. Front Immunol. 2014;5:485.
- O'Connor JC, André C, Wang Y, Lawson MA, Szegedi SS, Lestage J, Castanon N, Kelley KW, Dantzer R. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2, 3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette- Guerin. J Neurosci. 2009;29(13):4200–4209.
- Huang Y-S, Ogbechi J, Clanchy FI, Williams RO, Stone TW. IDO and kynurenine metabolites in peripheral and CNS disorders. Front Immunol. 2020;11:388.
- Savitz J. The kynurenine pathway: a finger in every pie. Mol Psychiatry. 2020;25(1):131–147.
- Ciapała K, Mika J, Rojewska E. The kynurenine pathway as a potential target for neuropathic pain therapy design: from basic research to clinical perspectives. IJMS. 2021;22(20):11055.
- Castellano-Gonzalez G, Jacobs KR, Don E, Cole NJ, Adams S, Lim CK, Lovejoy DB, Guillemin GJ. Kynurenine 3-Monooxygenase Activity in Human Primary Neurons and Effect on Cellular Bioenergetics Identifies New Neurotoxic Mechanisms. Neurotox Res. 2019;35(3):530–541.
- Bai MY, Lovejoy DB, Guillemin GJ, Kozak R, Stone TW, Koola MM. Galantamine-memantine combination and kynurenine pathway enzyme inhibitors in the treatment of neuropsychiatric disorders. Complex Psychiatry. 2021;7(1-2):19–33.
- Modoux M, Rolhion N, Mani S, Sokol H. Tryptophan Metabolism as a Pharmacological Target. Trends Pharmacol Sci. 2021;42(1):60–73.
- Platten M, Nollen EAA, Röhrig UF, Fallarino F, Opitz CA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. 2019;18(5):379–401.
- Lovelace MD, Varney B, Sundaram G, Franco NF, Ng ML, Pai S, Lim CK, Guillemin GJ, Brew BJ. Current Evidence for a Role of the Kynurenine Pathway of Tryptophan Metabolism in Multiple Sclerosis. Front Immunol. 2016;7:246.
- Gorecki AM, Anyaegbu CC, Anderton RS. TLR2 and TLR4 in Parkinson’s disease pathogenesis: the environment takes a toll on the gut. Transl Neurodegener. 2021;10(1):47.
- Iovino L, Tremblay ME, Civiero L. Glutamate-induced excitotoxicity in Parkinson’s disease: the role of glial cells. J Pharmacol Sci. 2020;144(3):151–164.
- Latif S, Jahangeer M, Maknoon Razia D, Ashiq M, Ghaffar A, Akram M, El Allam A, Bouyahya A, Garipova L, Ali Shariati M, et al. Dopamine in Parkinson’s disease. Clin Chim Acta. 2021;522:114–126.
- Joisten N, Ruas JL, Braidy N, Guillemin GJ, Zimmer P. The kynurenine pathway in chronic diseases: a compensatory mechanism or a driving force?. Trends Mol Med. 2021;27(10):946–954.
- Ogawa T, Matson WR, Beal MF, Myers RH, Bird ED, Milbury P, Saso S. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology. 1992;42(9):1702–1706.
- Beal MF, Matson WR, Storey E, Milbury P, Ryan EA, Ogawa T, Bird ED. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J Neurol Sci. 1992;108(1):80–87.
- Widner B, Leblhuber F, Fuchs D. Increased neopterin production and tryptophan degradation in advanced Parkinson’s disease. J Neural Transm (Vienna)). 2002;109(2):181–189.
- Knyihár-Csillik E, Csillik B, Pákáski M, Krisztin-Péva B, Dobó E, Okuno E, Vécsei L. Decreased expression of kynurenine aminotransferase-I (KAT-I) in the substantia nigra of mice after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment. Neuroscience. 2004;126(4):899–914.
- Hartai Z, Klivenyi P, Janaky T, Penke B, Dux L, Vecsei L. Kynurenine metabolism in plasma and in red blood cells in Parkinson’s disease. J Neurol Sci. 2005;239(1):31–35.
- Heilman PL, Wang EW, Lewis MM, Krzyzanowski S, Capan CD, Burmeister AR, Du G, Escobar Galvis ML, Brundin P, Huang X, et al. Tryptophan metabolites are associated with symptoms and nigral pathology in Parkinson’s disease. Mov Disord. 2020;35(11):2028–2037.
- Heyes MP, Saito K, Crowley JS, Davis LE, Demitrack MA, DER M, Dilling LA, Elia J, Kruesi MJP, Lackner A, et al. Quinolinic acid and kynurenine pathway metabolism in inflflammatory and noninflammatory neurological disease. Brain. 1992;115(5):1249–1273.
- Chang K-H, Cheng M-L, Tang H-Y, Huang C-Y, Wu Y-R, Chen C-M. Alternations of metabolic profifile and kynurenine metabolism in the plasma of Parkinson’s disease. Mol Neurobiol. 2018;55(8):6319–6328.
- Sorgdrager FJH, Vermeiren Y, Van Faassen M, van der Ley C, Nollen EAA, Kema IP, De Deyn PP. Age- and disease-specific changes of the kynurenine pathway in Parkinson’s and Alzheimer’s disease. J Neurochem. 2019;151(5):656–668.
- Bai JH, Zheng YL, Yu YP. Urinary kynurenine as a biomarker for Parkinson’s disease. Neurol Sci. 2021;42(2):697–703.
- Miranda AF, Boegman RJ, Beninger RJ, Jhamandas K. Protection against quinolinic acid-mediated excitotoxicity in nigrostriatal dopaminergic neurons by endogenous kynurenic acid. Neuroscience. 1997;78(4):967–975.
- Silva-Adaya D, Pérez-De La Cruz V, Villeda-Hernández J, Carrillo-Mora P, González-Herrera IG, García E, Colín-Barenque L, Pedraza-Chaverrí J, Santamaría A. Protective effect of L-kynurenine and probenecid on 6-hydroxydopamine-induced striatal toxicity in rats: implications of modulating kynurenate as a protective strategy. Neurotoxicol Teratol. 2011;33(2):303–312.
- Fukuyama K, Tanahashi S, Hoshikawa M, Shinagawa R, Okada M. Zonisamide regulates basal ganglia transmission via astroglial kynurenine pathway. Neuropharmacology. 2014;76:137–145.
- Kubicova L, Chobot V. Potential of kynurenine metabolites in drug development against neurodegenerative diseases. Neural Regen Res. 2021;16(2):308–309.
- Chen P, Li X. Study on effect of striatal mglur2/3 in alleviating motor dysfun -ction in rat pd model treated by exercise therapy. Front Aging Neurosci. 2019;11:255.
- Phillips RS, Iradukunda EC, Hughes T, Bowen JP. Modulation of enzyme activity in the kynurenine pathway by kynurenine monooxygenase inhibition. Front Mol Biosci. 2019;6:3.
- Beaumont V, Mrzljak L, Dijkman U, Freije R, Heins M, Rassoulpour A, Tombaugh G, Gelman S, Bradaia A, Steidl E, et al. The novel KMO inhibitor CHDI-340246 leads to a restoration of electrophysiological alterations in mouse models of Huntington’s disease. Exp Neurol. 2016;282:99–118.
- Abdel-Magid AF. Kynurenine Monooxygenase (KMO) Inhibitors for the Treatment of Acute Pancreatitis and Neurodegenerative Disorders. ACS Med Chem Lett. 2015;6(9):954–955.
- Lu Y, Shao M, Wu T. Kynurenine-3-monooxygenase: a new direction for the treatment in different diseases. Food Sci Nutr. 2020;8(2):711–719.
- Fukushima T, Umino M, Sakamoto T, et al. A review of chromatographic methods for bioactive tryptophan metabolites, kynurenine, kynurenic acid, quinolinic acid, and others, in biological fluids. Biomed Chromatogr. 2022;36:e5308.
- Harris CA, Miranda AF, Tanguay JJ, Boegman RJ, Beninger RJ, Jhamandas K. Modulation of striatal quinolinate neurotoxicity by elevation of endogenous brain kynurenic acid. Br J Pharmacol. 1998;124(2):391–399.
- Sodhi RK, Bansal Y, Singh R, Saroj P, Bhandari R, Kumar B, Kuhad A. IDO-1 inhibition protects against neuroinflammation, oxidative stress and mitochondrial dysfunction in 6-OHDA induced murine model of Parkinson’s disease. Neurotoxicology. 2021;84:184–197.
- van der Goot AT, Zhu W, Vázquez-Manrique RP, Seinstra RI, Dettmer K, Michels H, Farina F, Krijnen J, Melki R, Buijsman RC, et al. Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc Natl Acad Sci U S A. 2012;109(37):14912–14917.
- Perez-Pardo P, Grobben Y, Willemsen-Seegers N, Hartog M, Tutone M, Muller M, Adolfs Y, Pasterkamp RJ, Vu-Pham D, van Doornmalen AM, et al. Pharmacological validation of TDO as a target for Parkinson’s disease. Febs J. 2021;288(14):4311–4331.
- Nagy K, Plangár I, Tuka B, Gellért L, Varga D, Demeter I, Farkas T, Kis Z, Marosi M, Zádori D, et al. Synthesis and biological effects of some kynurenic acid analogs. Bioorg Med Chem. 2011;19(24):7590–7596.
- Wu HQ, Lee SC, Schwarcz R. Systemic administration of 4-chlorokynurenine prevents quinolinate neurotoxicity in the rat hippocampus. Eur J Pharmacol. 2000;390(3):267–274.
- Battaglia G, La Russa M, Bruno V, Arenare L, Ippolito R, Copani A, Bonina F, Nicoletti F. Systemically administered D-glucose conjugates of 7-chlorokynurenic acid are centrally available and exert anticonvulsant activity in rodents. Brain Res. 2000;860(1-2):149–156.
- Füvesi J, Somlai C, Németh H, Varga H, Kis Z, Farkas T, Károly N, Dobszay M, Penke Z, Penke B, et al. Comparative study on the effects of kynurenic acid and glucosamine-kynurenic acid. Pharmacol Biochem Behav. 2004;77(1):95–102.
- Luchowska E, Luchowski P, Wielosz M, et al. FK506 attenuates 1 - methy l - 4 - phenylpyridinium - and 3 - nitropropionic acid-evoked inhibition of kynurenic acid synthesis in rat cortical slices. Acta Neurobiol Exp (Wars). 2003;63(2):101–108.