
Abstract
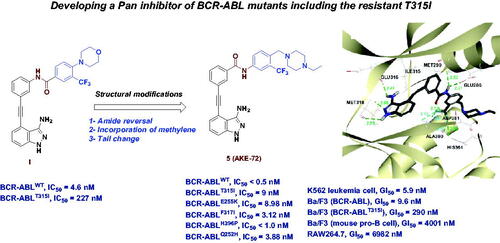
BCR-ABL inhibition is an effective therapeutic approach for the treatment of chronic myeloid leukaemia (CML). Herein, we report the discovery of AKE-72 (5), a diarylamide 3-aminoindazole, as a potent pan-BCR-ABL inhibitor, including the imatinib-resistant mutant T315I. A focussed array of compounds 4a, 4b, and 5 has been designed based on our previously reported indazole I to improve its BCR-ABLT315I inhibitory activity. Replacing the morpholine moiety of I with the privileged tail (4-ethylpiperazin-1-yl)methyl afforded 5 (AKE-72) with IC50 values of < 0.5 nM, and 9 nM against BCR-ABLWT and BCR-ABLT315I, respectively. Moreover, AKE-72 potently inhibited a panel of other clinically important mutants in single-digit nanomolar IC50 values. AKE-72 elicited remarkable anti-leukemic activity against K-562 cell line (GI50 < 10 nM, TGI = 154 nM). In addition, AKE-72 strongly inhibited the proliferation of Ba/F3 cells expressing native BCR-ABL or its T315I mutant. Overall, AKE-72 may serve as a promising candidate for the treatment of CML, including those harbouring T315I mutation.
GRAPHICAL ABSTRACT
Introduction
Chronic myeloid leukaemia (CML) is a myeloproliferative tumour of white blood cells in bone marrow and accounts for 15% of adult leukaemiaCitation1. Break-point cluster region-Abelson (BCR-ABL) is a fusion protein generated from the Philadelphia chromosome (Ph), which drives the pathogenesis of most of CML as well as a subset acute lymphoblastic leukaemia (Ph-positive ALL)Citation2,Citation3. Therefore, the design of kinase inhibitors targeting the BCR-ABL oncoprotein represents an effective strategy for the therapy of CML and/or ALL.
Imatinib, the first targeted therapy drug, is a first-line BCR-ABL inhibitor, which showed remarkable efficacy for most patients diagnosed with CML, with an overall 5-year survival rate of 89%Citation4,Citation5. Imatinib therapy for CML was estimated to have a long-term success rate of 83.3% after 10 yearsCitation6. Nevertheless, the dose intolerance and acquired resistance to imatinib, through the emergence of point mutations, hampered its efficacy for CML patients in accelerated and blastic phasesCitation5,Citation7. Nilotinib, bafetinib, dasatinib, and bosutinib have been developed as second-generation BCR-ABL inhibitors, with superior potency over imatinib, to treat adult CML patients with imatinib resistanceCitation8–11. However, these drugs are effective against most imatinib-resistant forms of BCR-ABL, except the most refractory gatekeeper T315I mutation, which arises in more than 20% of CML patientsCitation12,Citation13. The T315I mutation restrains the binding of first and second-generation BCR-ABL inhibitors to the ABL catalytic domain by either a direct steric hindrance or stabilising the active kinase conformation, which renders the design of new inhibitors targeting the open and active conformation of the T315I mutant as a major challengeCitation14.
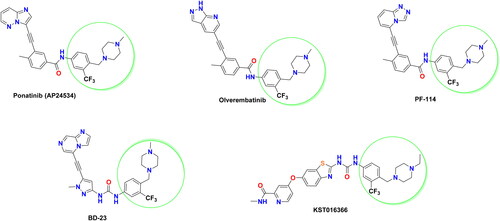
Ponatinib is the first FDA-approved third-generation BCR-ABL inhibitor for CML (), which displayed excellent potency to inhibit BCR-ABLWT, BCR-ABLT315I, and other clinically significant ABL mutants, like Q252H, Y253H, M351T, and H396PCitation15,Citation16. Most recently, asciminib has been approved by FDA in October 2021 for the treatment of CMLCitation17. Being different from the existing tyrosine kinase inhibitors (TKIs), asciminib is the first allosteric BCR-ABL1 inhibitor specifically targeting the ABL1 myristoyl pocket (STAMP)Citation18. It has demonstrated clinical activity in heavily pre-treated CML patients with or without mutations, with promising efficacy in patients with T315I mutationCitation19. In addition, a number of structurally diverse BCR-ABLT315I inhibitors have been identified (). Replacing imidazo[1,2-b]pyridazine, the hinge binding motif, of ponatinib with other bioisosteric heterocycles afforded several promising BCR-ABLT315I inhibitors. Among them, olverembatinib (HQP1351)Citation20,Citation21, and PF-114Citation22,Citation23 were developed as potent BCR-ABLT315I inhibitors with better safety profile than ponatinib. Apart from these diarylamide acetylene-containing inhibitors, certain heteroaryl ureides such as BD-23Citation24 and KST016366Citation25 () were also reported as potent ABLT315I inhibitors. As highlighted in , the presence of 4-((4-methyl(ethyl)piperazin-1-yl) methyl)-3-(trifluoromethyl)phenyl moiety is a common prerequisite structural feature in the above mentioned BCR-ABLT315I inhibitors. This privileged moiety serves as a tail, where the terminal nitrogen of piperazinyl methylene is predicted to be protonated at physiologic pH and then can engage in a hydrogen bond (HB) with the carbonyl oxygen of residue Ile360 in the activation loop of BCR-ABLT315ICitation15.
Figure 1. Representative examples of potent BCR-ABLT315I inhibitors bearing 4-((4-methyl(ethyl)piperazin-1-yl) methyl)-3-(trifluoromethyl)phenyl moiety.
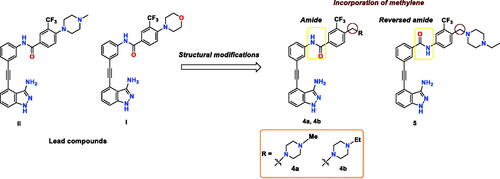
Recently, our group reported a series of 3-aminoindazole based ABL inhibitorsCitation26, where compound I () was identified as a potent ABLWT inhibitor (IC50 = 4.6 nM) with excellent anti-leukemic activity against K-562 cell line (GI50 < 10 nM). However, compound I elicited moderate potency against the imatinib-resistant T315I mutant (IC50 = 227 nM). As disclosed from the SAR study of this series, the nature of the substituent installed on the 3-trifluoromethylphenyl ring greatly affects the ABLT315I inhibitory potencyCitation26. In view of this finding, along with the substantial role of 4-((4-methyl(ethyl)piperazin-1-yl) methyl)-3-(trifluoromethyl)phenyl moiety in achieving favourable activity towards ABLT315I, we were motivated to further improve the activity of I against BCR-ABLT315I as a part of our ongoing endeavours to discover potent BCR-ABLT315I inhibitorsCitation27,Citation28. In the current study, we designed a concise library of three indazoles 4a, 4b, and 5, where the amide bond of I was conserved, as in 4a, 4b, while it was reversed in compound 5 (). Most importantly, the 4-morpholino moiety of I was replaced by the privileged 4-((4-methyl(ethyl)piperazin-1-yl) methylene in all three compounds. To investigate the impact of methylene insertion between the terminal phenyl and substituted piperazine on BCR-ABLT315I potency, we compared the activity of compounds 4a, 4b, and 5 with compound II, which lacks such methylene group (). The newly designed compounds were synthesised, and evaluated against both ABLWT and ABLT315I, and the most potent member 5 was further profiled against a set of clinically important ABL mutants.
Figure 2. The chemical structure of lead compounds II and I, and the newly designed target compounds.
Results and discussion
Chemistry
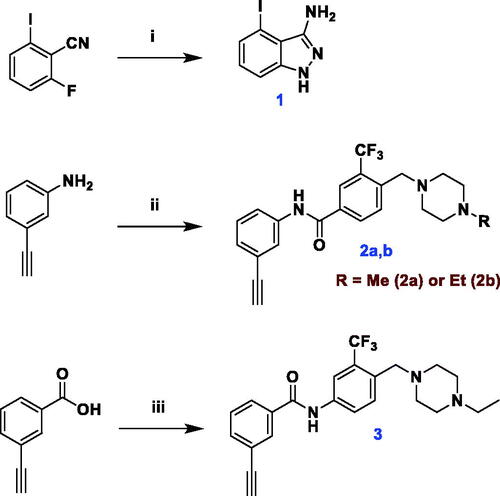
The key precursor for synthesis of target compounds, 4-iodoindazol-3-yl amine 1, as well as diarylamides intermediates 2a, 2b and 3 were prepared as depicted in Scheme 1. Treatment of 2-fluoro-6-iodobenzonitrile with hydrazine hydrate in n-butanol at 110 °C furnished the 3-aminoindazole 1Citation29. Amide coupling of 3-ethynylaniline or 3-ethylnylbenzoice acid with the proper 4-((4-methyl(ethyl)piperazin-1-yl)methyl)-3-(trifluoromethyl)benzoic acid or aniline was accomplished using hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) and N,N-diisopropylethylamine (DIPEA) in DMF to afford the corresponding diarylamides.
Scheme 1. Synthesis of 3-aminoindazole 1 and diarylamides 2a, 2b and 3. Reagents and reaction conditions: i) 2-fluoro-6-iodobenzonitrile, n-BuOH, hydrazine hydrate, 110 °C, 2 h, 99%; ii) 4-((4-methyl(ethyl)piperazin-1-yl)methyl)-3-(trifluoromethyl)benzoic acid, HATU, DIPEA, DMF, rt, overnight, 74% (2a), 66% (2b); iii) 4-((4-ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)aniline, HATU, DIPEA, DMF, 60 °C, 3 h, 31%.
Sonogashira coupling of 1 with the proper acetylene diarylamide 2a, 2b or 3 was carried out utilising PdCl2(PPh3)2 and CuI as catalysts in Et3N/DMF (1:1) at 85 °C to yield the diarylamide tethered indazoles 4a, 4b and 5 as illustrated in Scheme 2.
Scheme 2. Synthesis of compounds 4a, 4b and 5. Reagents and reaction conditions: i) PdCl2(PPh3)2, CuI, Et3N, DMF, 85 °C, dark, overnight, 19–27%.
In vitro biochemical kinase screening
ABLWT and ABLT315I kinase evaluations
Compounds 4a, 4b and 5 were tested against the native BCR-ABLWT and its clinically relevant imatinib-resistant mutant BCR-ABLT315I to determine their IC50 values, using ponatinib as a reference compound, and the pan kinase inhibitor staurosporine, as a positive control (). As disclosed from the data, compounds 4a and 5 showed > 300 folds superior potency against BCR-ABLWT rather the lead indazole II with IC50 values less than 0.51 nM. Such finding points out the remarkable role of methylene linker incorporated between the terminal phenyl and substituted piperazine in improving BCR-ABLWT inhibition, which may stem from the ability of extended piperazine moiety to form tight interactions with the allosteric site of ABL. The N-methylpiperazine amide 4a elicited slightly better inhibitory activity (BCR-ABLT315I IC50 = 96 nM) than its corresponding N-ethylpiperazine analog 4b (BCR-ABLT315I IC50 = 142 nM). Meanwhile, both 4a and 4b surpassed the potency of lead indazoles II and I towards BCR-ABLT315I with 8–11, and about two times, respectively. Interestingly, compound 5, the reversed amide of 4b, showed robust potency towards BCR-ABLT315I with IC50 value of 9 nM, being 16 and 25 folds more potent than 4b and lead I, respectively. Accordingly, it could be inferred that both the amide bond direction, the DFG binding motif, and the substitution of distal phenyl with ethylpiperazine methylene moiety at para-position contribute significantly in achieving optimal ABLT315I inhibition. Compared with ponatinib, compound 5 showed better potency against ABLWT, and relatively lower potency for BCR-ABLT315I, yet both candidates possess single-digit nanomolar IC50 values. While comparing compound 5 with two ponatinib analogs, olverembatinib (HQP1351)Citation20 and PF-114Citation22, 5 showed comparable activity against BCR-ABLWT and lower potency over BCR-ABLT315I. On the other hand, compound 5 (AKE-72) exerted superior potency than the ureidobenzothiazole KST016366 (BCR-ABLWT and BCR-ABLT315I IC50 = 53 nM)Citation25.
Table 1. IC50 values (nM) of compounds II, I, 4a, 4b, and 5 against BCR-ABLWT and BCR-ABLT315I.a,b.
Compound 5 (AKE-72) was further profiled against a number of clinically relevant ABL mutants, including E255K, F317I, H396P, M351T, and Q252H (). This outcome confirmed the ability of compound 5 to potently suppress the activity of the clinically relevant ABL mutants in single-digit nanomolar IC50 values.
Table 2. In vitro enzymatic activity of 5 (AKE-72) over wild-ABL and a panel of its clinically resistant mutants.
Kinase profile of 5
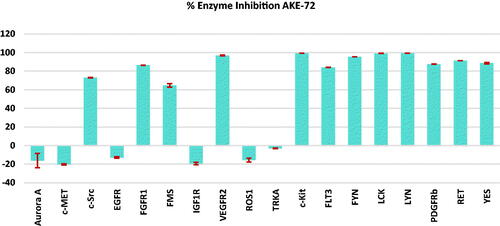
Furthermore, the most potent indazole member 5 was tested, at 50 nM concentration, over a panel of 18 major oncogenic kinases including the commonly kinases inhibited by ponatinib. As depicted in , compound 5 showed significant inhibition against c-Kit, FGFR1, FLT3, FYN, LCK, LYN, PDGFRβ, RET, VEGFR2, and YES kinases with (83.9–99.3% inhibition). While 5 displayed moderate suppressive activity (64.5–73% inhibition) over c-Src and FMS kinases, it exerted no inhibition against the other investigated kinases. This inhibition pattern presents compound 5 (AKE-72) as a multi-tyrosine kinase inhibitor in a similar fashion to that observed with ponatinibCitation16,Citation30.
Figure 3. Enzyme inhibition percentage (relative to DMSO controls) of compound 5 (0.05 µM) against a panel of 18 kinases.
In vitro evaluation of the anti-leukemic activity by SRB assay
Motivated by the promising biochemical assay data, the anti-leukemic activities of compounds 4a, 4b, and 5 were evaluated against a panel of six human leukaemia cell lines -using sulforhodamine B (SRB) assay- at National Cancer Institute (NCI, Developmental Therapeutics Program, Bethesda, MD, USA). The three compounds were tested initially at a single dose (10 μM), and their corresponding GI50 values were further determined by advancing to five-dose testing mode (), and compared with the lead indazole I ().
Figure 4. Dose-response curves of compounds I, 4a, 4b, and 5 against a panel of six human leukaemia cell lines.
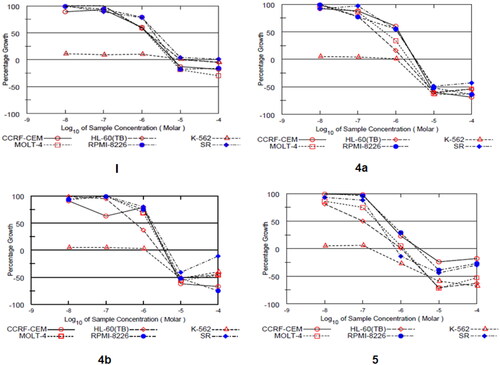
Table 3. The antiproliferative activities (% Growth inhibition (GI) at 10 µM and GI50 (µM)) of compounds I, 4a, 4b, and 5 against a panel of six human leukaemia cell lines.a,b.
The single-dose assay results pointed out the sound antiproliferative activity of compound 5 (GI > 100%) against all tested leukaemia cell lines. Most importantly, the five dose testing findings highlighted the remarkable selective anti-leukemic activities of all three compounds towards the ABL overexpressing leukaemia cell K562 with half-maximal growth inhibition concentration (GI50) values of less than 10 nM. This observation emphasises the excellent ability of compounds 4a, 4b, and 5 to suppress ABL activity on the cellular level. Over other leukaemia cells, both amides 4a and 4b showed comparable antiproliferative activity, with a special preference for HL-60 and MOLT-4 cell lines. For example, compound 4a displayed GI50 values of 0.278 and 0.494 µM against HL-60 and MOLT-4 cells, respectively. Of special importance, the indazole reversed amide 5 (AKE-72) elicited the highest cellular potency over all cell lines with sub-micromolar GI50 values. However, based on the activity order, 5 possessed particular selectivity towards the K-562 cell line. The noticed differential cellular potency of compounds 4a, 4b, and 5 may refer to their potential enzymatic selectivity towards ABL rather than other cancer-related kinases.
Apart from the potency parameter GI50, two additional measures; TGI (total growth inhibition) and LC50 (half-maximal lethal concentration) were calculated from the five dose assay and used as efficacy indices (). All three compounds exerted selective superior efficacy against K-562 cell line rather the lead indazole I. Particularly, indazole 5 induced total growth inhibition of K-562 cells at 154 nM, with 87-fold improved efficacy than I (TGI = 13500 nM). In addition, compound 5 triggered 50% cytotoxic lethal activity towards K-562 cells at 5.25 µM, while the lead I could not achieve same effect up to 100 µM.
Table 4. TGI and LC50 (µM) of compounds I, 4a, 4b and 5 against a panel of six human leukaemia cell lines.
Further cell-based evaluation of the antiproliferative activity of compound 5 (AKE-72)
Further cellular proliferation assays were carried out for compound 5 (AKE-72) with normal RAW264.7 macrophage cells, Ba/F3 mouse pro-B cells and Ba/F3 cells expressing native BCR-ABL or BCR-ABLT315I kinases following the reported protocolCitation31 and using ponatinib as a reference compound (). AKE-72 potently suppressed the proliferation of Ba/F3 cells expressing wild-type BCR-ABL (GI50 = 9.6 nM). In addition, the BCR-ABLT315I mutant showed sensitivity to AKE-72 (GI50 = 290 nM). Growth of Ba/F3 mouse pro-B cells and normal RAW264.7 macrophage cells was inhibited only at significantly higher GI50 of 4001 nM and 6982 nM, respectively. These cellular outcomes underscore the favourable differential selectivity of compound AKE-72 for inhibiting BCR-ABL-dependent cells with minimal impact on normal cells. We also tested AKE-72 against BCR-ABL-positive and BCR-ABL-negative cell lines. Compound AKE-72 strongly inhibited the growth of K562 (BCR-ABL-positive, GI50 = 5.9 nM), however, had no significant activity against U937 (BCR-ABL-negative, GI50 = 1900 nM) leukaemia cell line ().
Table 5. GI50 (µM) of compound 5 (AKE-72) and ponatinib against RAW264.7, Ba/F3 (Ba/F3 mouse pro-B cells), Ba/F3 (Bcr-Abl), Ba/F3 (Bcr-AblT315I), K562, and U937 cells.a
CYP450 assays and ADME-Tox predictions
Azole derivatives are well known for their ability to inhibit cytochrome P450 isozymes. Since our target compounds feature azole moiety as a part of the indazole scaffold, we tested the affinity of AKE-72 against four of the major cytochrome P450 isozymes at 10 µM; CYP1A2, CYP2D6, CYP2C9, and CYP3A4 (). As disclosed from the results, compound 5 (AKE-72) showed weak inhibitory activity towards both CYP1A2 and CYP2D6 with 9.88% and 13.92% inhibition (IC50 > 10 µM), being less potent than furafylline and ketoconazole, respectively. In contrast, 5 elicited significant affinity against CYP2C9 (50.67% inhibition) and CYP3A4 (90.57% inhibition). The strong inhibitory effect with CYP3A4 is a common feature for many tyrosine kinase inhibitors, including PonatinibCitation32. Therefore, AKE-72 has a reasonable CYP450-profile, which could be further improved in our next plan to offer merit for avoiding undesirable drug-drug interactions.
Table 6. CYP inhibition profile for compound 5 and ketoconazole/furafylline as reference standards.Table Footnotea
Furthermore, the pharmacokinetics ADME-Tox properties for compound 5 (AKE-72) were calculated by pkCSM, a web-based tool that can predict small-molecule pharmacokinetic and toxicity properties using graph-based signaturesCitation33. The ADME-Tox properties of compound 5 (data added to the supporting information) showed that it could be well absorbed from GIT. Compound 5 is predicted to be both a substrate and inhibitor for P-glycoprotein, which may offer the advantage of increasing bioavailability and efficacy, by inhibiting its own efflux from cells. In addition, compound 5 is predicted to be a non-substrate for hERG, indicating a low probability of causing Torsade de points cardiac adverse effects.
Molecular docking study
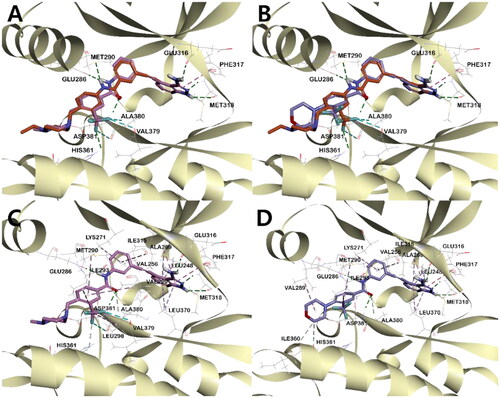
In order to rationalise the observed ABL kinase inhibitory results from a 3D structural perspective, the lead compounds I and II, and the newly designed derivatives 4a, 4b, and 5 were docked in the catalytic kinase domains of BCR-ABLWT (PDB code: 3OXZ) and BCR-ABLT315I (PDB code: 3OY3)Citation34. The docking study revealed the existence of two major hydrogen bonds (HB) between all compounds and ABL kinases. One HB is between the carbonyl oxygen in the central amide bond of the inhibitor and the hydrogen in the amide side chain of Asp381, and the other is between the amidic hydrogen in the inhibitor and the carboxyl oxygen on the side chain of Glu286 (, Figures S3 and S4). In addition, all inhibitors shared π-alkyl interactions between the phenyl group next to the indazole and Ile315 in BCR-ABLT315I. However, the main binding distance between compound 5 and the protein was relatively shorter in BCR-ABLWT than in BCR-ABLT315I, suggesting that 5 binds more tightly to ABLWT and exhibits a better inhibitory effect in ABLWT (Figure S2-4).
Figure 5. The binding mode of compound 5 in the kinase domain of (A) BCR-ABLWT and (B) BCR-ABLT315I. For clarity, only the major residues with hydrogen bonding and halogen interactions are shown. Compound 5 is shown in the stick model, and surrounding key interaction residues were shown in the line model. Hydrogen bonding and halogen interactions are shown in dashes green and sky blue, respectively. The number near the dashes indicates the bonding distance in Å.
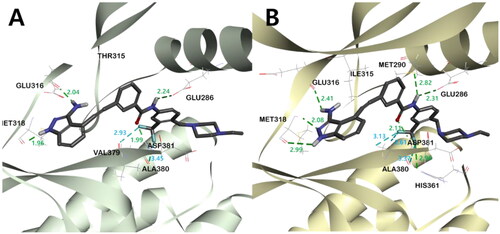
In all inhibitors, the indazole ring showed a very stable binding to both BCR-ABLWT and BCR-ABLT315I kinases. The indazole NH and 3-NH2 hydrogens were engaged in critical HB with the carboxyl group of Glu316 and Met318, respectively. The binding of indazole moiety to the ATP binding site of the ABL kinase was further stabilised by π- π interactions between Phe317 and the pyrazole moiety, as well as the hydrophobic interaction of the indazole moiety with hydrophobic residues such as Leu248, Val256, Ala269 and Leu370 ((), Figures S2–S4, S5C, S5D). Among all indazoles, compound 5 has the lowest binding energy and good inhibitory efficacy. The major difference between 5 and the other derivatives is that the central amide bond is reversed. This difference shifts the positions of the two phenyl groups slightly, resulting in different bonding modes and binding energies (Table S1). The phenyl group next to the stably bound indazole moiety exists on the same plane, but the plane of the opposite m-trifluoromethylphenyl group has different degrees of distortion. Compared to 5, 4a showed the smallest twist angle, and the m-trifluoromethylphenyl group planes of the remaining three inhibitors (4b, I, and II) were calculated to be bound to the protein by twisting at a relatively larger angle (, Figure S6). The trifloromethyl group of compounds 5 and 4a, which has the best potency against ABLT315I, was engaged in a halogen interaction with the Val379 and Ala380 residues, which is lacking in the other indazole derivatives ( and Figure S5 (C-D)).
Figure 6. Superposed model of inhibitors inside BCR-ABLT315I Kinase. (A) 5 (orange), 4a (pink), and 4b (green), (B) 5, I (lavender), and II (yellow).
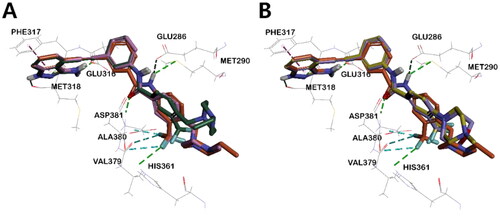
Figure 7. 3D structural overlay of compounds (A) 5 (orange) and 4a (pink), (B) 5 and I (lavender) in the ABLT315I Kinase. Inhibitors are shown in stick model and surrounding key interaction residues were shown in line model. The key interaction mode between 5 and BCR-ABLT315I indicated by a dash line. The binding mode of compounds (C) 4a, and (D) I in the BCR-ABLT315I Kinase. Various interactions are shown in dashes.
Importantly, in compound 5, the m-trifluoromethylphenyl group and N-ethylpiperazine moiety are connected by a methylene linker. Such bulky piperazine moiety adopts a sterically more stable conformation that binds tightly with the protein, where the hydrogens in N-ethylpiperazine form carbon-hydrogen bonds with Ile360, His361, and Asp381, respectively. However, in the lead compound I, the morpholine moiety was directly bonded to trifluoromethylphenyl, thus both groups were positioned perpendicular to each other and bound to the protein in a form that reduced steric hindrance. Only one hydrogen of morpholine in compound I formed a carbon-hydrogen bond with Ile360 and His361, while alkyl interaction was noticed between morpholine and Val289. Overall, compound I showed fewer binding interactions with ABL compared to compound 5. Therefore, the docking score of compound I was calculated to be lower than that of compound 5, which is in agreement with the observed inhibitory effect (,D), Figures S4A, S4E, CDocker interaction energy in ). Similarly, in the case of compound 4a, the presence of methylene linker allowed the bulky piperazine moiety to adopt a sterically more stable conformation with protein, and hydrogens in N-methylpiperazine could share carbon-hydrogen bonding with Ile360, His361, and Asp381, respectively. In contrast, in compound II, the phenyl group and bulky N-methyl piperazine were perpendicular to each other to reduce steric hindrance, which makes them bind to the ABL kinase in a different direction from that of N-methylpiperazine in 4a. (Figure S4B, S4D, S5B, S5D). Compounds 4a and 4b possess methyl and ethyl substituents in their piperazine terminal NH, respectively. Rather than the effect of this, as suggested earlier, since the twist angle of the trifluoromethyl phenyl group increased in 4b, it was unable to interact tightly with BCR-ABLT315I and halogen (fluorine) and could not interact properly with other residues, as evidenced by the docking score (Figure S4).
Table 7. CDocker interaction energy of compounds I, II, 4a, 4b, and 5 with BCR-ABLWT and BCR-ABLT315I kinases.
Experimental
The detailed experimental section is included in the supplementary data associated with this article.
Conclusion
In this short communication, we report the design and synthesis of new indazole amides 4a, 4b and reversed amide 5 in attempt to improve the BCR-ABL inhibitory profile of our previously reported indazole lead compound I. The target compounds have been designed based on changing the morpholine motif of indazole I with the substantial (4-methyl(ethyl)piperazin-1-yl)methyl moiety, that exist in several potent BCR-ABLT315I inhibitors. In vitro cell-free assays disclosed the excellent potency of all three compounds against BCR-ABLWT as evident by their IC50 values of < 1 nM. Interestingly, compound 5 (AKE-72) exerted superior potency over the indazole I towards the most refractory T315I mutant with IC50 value of 9 nM. In addition, several forms of BCR-ABL mutants such as were greatly suppressed by AKE-72 at single digit nanomolar IC50 values. Cellular screening of all compounds over a set of six human leukaemia cell lines, at NCI, pointed out their distinct and selective anti-leukemic potency towards K562 cell line, with GI50 less than 10 nM. In addition, AKE-72 selectively inhibited the proliferation of Ba/F3 cells expressing native BCR-ABL or its T315I mutant. The putative binding mode of AKE-72 in kinase domain of BCR-ABLWT, BCR-ABLT315I was investigated by molecular docking study and revealed the important role of 4-((4-methyl(ethyl)piperazin-1-yl) methyl)-3-(trifluoromethyl)phenyl tail moiety in achieving tight binding within BCR-ABL kinase domain. Taken together, our current report offers compound 5 (AKE-72) as a promising candidate for potential treatment of CML.
Supplemental Material
Download PDF (2.7 MB)Acknowledgements
The authors express our appreciation to the National Cancer Institute (NCI, Bethesda, Maryland, USA) for performing the in vitro anticancer evaluation of the new compounds.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Siegel RL, Miller Kimberly D, Fuchs Hannah E, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33.
- Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia specific P210-Protein is the product of the Bcr/Abl hybrid gene. Science. 1986;233(4760):212–214.
- Faderl S, Garcia‐Manero G, Thomas DA, Kantarjian HM. Philadelphia chromosome‐positive acute lymphoblastic leukemia–current concepts and future perspectives. Rev Clin Exp Hematol. 2002;6(2):142–160.
- Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1(7):493–502.
- Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MWN, Silver RT, Goldman JM, Stone RM, IRIS Investigators, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417.
- Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, Baccarani M, Deininger MW, Cervantes F, Fujihara S, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376(10):917–927.
- Pandrala M, Bruyneel AAN, Hnatiuk AP, Mercola M, Malhotra SV. Designing novel BCR-ABL inhibitors for chronic myeloid leukemia with improved cardiac safety. J Med Chem. 2022;65(16):10898–10919.
- Le Coutre P, Ottmann OG, Giles F, Kim D-W, Cortes J, Gattermann N, Apperley JF, Larson RA, Abruzzese E, O'Brien SG, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood. 2008;111(4):1834–1839.
- Santos FPS, Kantarjian H, Cortes J. Quintas-Cardama A. Bafetinib, a dual Bcr-Abl/Lyn tyrosine kinase inhibitor for the potential treatment of leukemia. Curr Opin Invest Dr. 2010;11(12):1450–1465.
- Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius LAM, Das J, Doweyko AM, et al. Discovery of N-(2-chloro-6-methylphenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47(27):6658–6661.
- Puttini M, Coluccia AML, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, Ahmed S, Redaelli S, Piazza R, Magistroni V, et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl(+) neoplastic cells. Cancer Res. 2006;66(23):11314–11322.
- Roychowdhury S, Talpaz M. Managing resistance in chronic myeloid leukemia. Blood Rev. 2011;25(6):279–290.
- Quintas-Cardama A, Cortes J. Therapeutic options against BCR-ABL1 T315I-positive chronic myelogenous leukemia. Clin Cancer Res. 2008;14(14):4392–4399.
- Zhang D, Li P, Gao Y, Song Y, Zhu Y, Su H, Yang B, Li L, Li G, Gong N, et al. Discovery of a Candidate Containing an (S)-3,3-Difluoro-1-(4-methylpiperazin-1-yl)-2,3-dihydro-1H-inden Scaffold as a Highly Potent Pan-Inhibitor of the BCR-ABL Kinase Including the T315I-Resistant Mutant for the Treatment of Chronic Myeloid Leukemia. J Med Chem. 2021;64(11):7434–7452.
- Huang W-S, Metcalf CA, Sundaramoorthi R, Wang Y, Zou D, Thomas RM, Zhu X, Cai L, Wen D, Liu S, et al. Discovery of 3-[2-(Imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a Potent, Orally Active Pan-Inhibitor of Breakpoint Cluster Region-Abelson (BCR-ABL) Kinase Including the T315I Gatekeeper Mutant. J Med Chem. 2010;53(12):4701–4719.
- O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang W-S, Xu Q, et al. AP24534, a Pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412.
- Yeung DT, Shanmuganathan N, Hughes TP. Asciminib: a new therapeutic option in chronic-phase CML with treatment failure. Blood. 2022;139(24):3474–3479.
- Schoepfer J, Jahnke W, Berellini G, Buonamici S, Cotesta S, Cowan-Jacob SW, Dodd S, Drueckes P, Fabbro D, Gabriel T, et al. Discovery of asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1. J Med Chem. 2018;61(18):8120–8135.
- Cortes JE, Hughes TP, Mauro MJ, Hochhaus A, Rea D, Goh YT, Janssen J, Steegmann JL, Heinrich MC, Talpaz M, et al. Asciminib, a First-in-Class STAMP inhibitor, provides durable molecular response in Patients (pts) with Chronic Myeloid Leukemia (CML) Harboring the T315I mutation: primary efficacy and safety results from a phase 1 trial. Blood. 2020;136(Supplement 1):47–50.
- Ren X, Pan X, Zhang Z, Wang D, Lu X, Li Y, Wen D, Long H, Luo J, Feng Y, et al. Identification of GZD824 as an orally bioavailable inhibitor that targets phosphorylated and nonphosphorylated Breakpoint Cluster Region-Abelson (Bcr-Abl) kinase and overcomes clinically acquired mutation-induced resistance against imatinib. J Med Chem. 2013;56(3):879–894.
- Jiang Q, Li Z, Qin Y, Li W, Xu N, Liu B, Zhang Y, Meng L, Zhu H, Du X, et al. Olverembatinib (HQP1351), a well-tolerated and effective tyrosine kinase inhibitor for patients with T315I-mutated chronic myeloid leukemia: results of an open-label, multicenter phase 1/2 trial. J Hematol Oncol. 2022;15(1):113.
- Ivanova ES, Tatarskiy VV, Yastrebova MA, Khamidullina AI, Shunaev AV, Kalinina AA, Zeifman AA, Novikov FN, Dutikova YV, Chilov GG, et al. PF-114, a novel selective inhibitor of BCR-ABL tyrosine kinase, is a potent inducer of apoptosis in chronic myelogenous leukemia cells. Int J Oncol. 2019;55(1):289–297.
- Mian AA, Rafiei A, Haberbosch I, Zeifman A, Titov I, Stroylov V, Metodieva A, Stroganov O, Novikov F, Brill B, et al. PF-114, a potent and selective inhibitor of native and mutated BCR/ABL is active against Philadelphia chromosome-positive (Ph plus) leukemias harboring the T315I mutation. Leukemia. 2015;29(5):1104–1114.
- Desai B, Dixon K, Farrant E, Feng Q, Gibson KR, van Hoorn WP, Mills J, Morgan T, Parry DM, Ramjee MK, et al. Rapid discovery of a novel series of Abl Kinase inhibitors by application of an integrated microfluidic synthesis and screening platform. J Med Chem. 2013;56(7):3033–3047.
- El-Damasy AK, Cho NC, Nam G, Pae AN, Keum G. Discovery of a nanomolar multikinase inhibitor (KST016366): a new benzothiazole derivative with remarkable broad-spectrum antiproliferative activity. Chemmedchem. 2016;11(15):1587–1595.
- El-Damasy AK, Jin H, Seo SH, Bang EK, Keum G. Design, synthesis, and biological evaluations of novel 3-amino-4-ethynyl indazole derivatives as Bcr-Abl kinase inhibitors with potent cellular antileukemic activity. Eur J Med Chem. 2020;207:112710
- El-Damasy AK, Cho NC, Kang SB, Pae AN, Keum G. ABL kinase inhibitory and antiproliferative activity of novel picolinamide based benzothiazoles. Bioorg Med Chem Lett. 2015;25(10):2162–2168.
- El-Damasy AK, Jin HW, Park JW, Kim HJ, Khojah H, Seo SH, et al. Overcoming the imatinib-resistant BCR-ABL mutants with new ureidobenzothiazole chemotypes endowed with potent and broad-spectrum anticancer activity. J Enzym Inhib Med Ch. 2023;38(1):2189097.
- Dai Y, Hartandi K, Ji Z, Ahmed AA, Albert DH, Bauch JL, Bouska JJ, Bousquet PF, Cunha GA, Glaser KB, et al. Discovery of N-(4-(3-amino-1H-indazol-4-yl)phenyl)-N'-(2-fluoro-5-methylphenyl)urea (ABT-869), a 3-aminoindazole-based orally active multitargeted receptor tyrosine kinase inhibitor. J Med Chem. 2007;50(7):1584–1597.
- Tan FH, Putoczki TL, Stylli SS, Luwor RB. Ponatinib: a novel multi-tyrosine kinase inhibitor against human malignancies. Onco Targets Ther. 2019;12:635–645.
- Kuchukulla RR, Hwang I, Park SW, Moon S, Kim SH, Kim S, Chung HW, Ji M-J, Park H-M, Kong G, et al. Novel 2,6,9-Trisubstituted Purines as Potent CDK Inhibitors Alleviating Trastuzumab-Resistance of HER2-Positive Breast Cancers. Pharmaceuticals-Base. 2022;15(9):1041.
- Teo YL, Ho HK, Chan A. Metabolism-related pharmacokinetic drug-drug interactions with tyrosine kinase inhibitors: current understanding, challenges and recommendations. Br J Clin Pharmacol. 2015;79(2):241–253.
- Pires DEV, Blundell TL, Ascher DB. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J Med Chem. 2015;58(9):4066–4072.
- Zhou T, Commodore L, Huang W-S, Wang Y, Thomas M, Keats J, Xu Q, Rivera VM, Shakespeare WC, Clackson T, et al. Structural Mechanism of the Pan-BCR-ABL Inhibitor Ponatinib (AP24534): Lessons for Overcoming Kinase Inhibitor Resistance. Chem Biol Drug Des. 2011;77(1):1–11.