
Abstract
Phosphoinositide 3-kinases (PI3K) and phosphoinositide 3-kinase-related protein kinases (PIKK) are two structurally related families of kinases that play vital roles in cell growth and DNA damage repair. Dysfunction of PIKK members and aberrant stimulation of the PI3K/AKT/mTOR signalling pathway are linked to a plethora of diseases including cancer. In recent decades, numerous inhibitors related to the PI3K/AKT/mTOR signalling have made great strides in cancer treatment, like copanlisib and sirolimus. Notably, most of the PIKK inhibitors (such as VX-970 and M3814) related to DNA damage response have also shown good efficacy in clinical trials. However, these drugs still require a suitable combination therapy to overcome drug resistance or improve antitumor activity. Based on the aforementioned facts, we summarised the efficacy of PIKK, PI3K, and AKT inhibitors in the therapy of human malignancies and the resistance mechanisms of targeted therapy, in order to provide deeper insights into cancer treatment.
Keywords:
Introduction
Phosphoinositide 3-kinase (PI3K) is a family of protein and lipid kinases, which phosphorylates the 3′-OH position on the inositol ring of phosphatidylinositol (4,5)-bisphosphate (PtdIns(4,5)P2) to generate PtdIns(3,4,5)P3Citation1. PtdIns(3,4,5)P3 (PIP3) serves as a docking point to recruit proteins to the vicinity of the plasma membrane by connecting to the protein’s pleckstrin homologous (PH) domain, with the most important protein being the AKT family of serine threonine kinasesCitation2. AKT, also referred to as protein kinase B (PKB), is a key signalling hub in the PI3K pathway. Its activation and overexpression can further regulate multiple downstream signalling targets and are closely correlated with resistance to cancer radiotherapy or chemotherapy treatmentsCitation3. PIKK and PI3K are two closely related families with similar kinase domains. PIKK is composed of six atypical serine/threonine protein kinases, including ataxia telangiectasia mutated (ATM), ataxia telangiectasia-and-rad3-related (ATR), DNA-dependent protein kinase (DNA-PK), mammalian target of rapamycin (mTOR), suppressor of morphogenesis in genitalia (SMG-1), and transformation/transcription associated protein (TRAAP)Citation4. PIKK members ATM, ATR, and DNA-PK associated with DNA damage response (DDR) are closely related to maintaining genomic stabilityCitation5. mTOR is the main mediator of PI3K/AKT signalling, which can regulate cell growth, survival, and metabolism. Therefore, its activation can promote tumour growth and metastasisCitation6. SMG-1 was first identified for RNA monitoring through nonsense-mediated mRNA decay (NMD), but it was later discovered that SMG-1 is independent of NMD itself and inhibits DNA repair through non-homologous end connectionsCitation7,Citation8. There is limited research on TRAAP, which is the only member of the PIKK kinase family that lacks all catalytic residues and can activate gene expression in important processesCitation9,Citation10. In summary, PI3K and PIKK family kinases play crucial roles in regulating various responses, including cell proliferation, metabolism, survival, and gene stability, making them important targets for research.
The PI3K/AKT/mTOR signalling pathway is one of the key intracellular signalling pathways that can undergo abnormal activation in most cancersCitation11–13. Mutations in genes such as PIK3CA, phosphatase and tension homologues (PTEN), AKT1, and tuberous sclerosis 1/2 (TSC1/2) can all lead to dysregulation of signalling pathways, which can drive important physiological processes such as tumour metabolism, proliferation, angiogenesis, and metastasisCitation14–17. Therefore, targeting key molecules in the PI3K/AKT/mTOR pathway is a promising cancer treatment strategy. PIKK members ATM, ATR, and DNA-PK are closely related to DNA damage repair and have complex effects on cancer progression and treatment. On the one hand, genomic instability is a basic characteristic of cancer, and defects in the DNA repair pathway can lead to the formation and development of cancer cellsCitation18. On the other hand, this also exposes the fragility of these tumour cells, making them more susceptible to DNA damage agentsCitation19. A method called "synthetic lethal" can selectively kill tumour cells by using drugs to inhibit the remaining DNA repair pathwaysCitation20. For example, in ATM-deficient xenograft models, ATR inhibition can induce synthesis lethality and exert chemical sensitisation effectsCitation21. It is worth noting that inhibiting DNA repair can lead to the accumulation of cytoplasmic DNA and induce type I interferon (IFN) in a STING (stimulator of interferon genes) -dependent manner, which is beneficial for activating the innate immune response and promoting tumour eradicationCitation22. Given the limited research of other members of PIKK and the prominent role of ATM, ATR, and DNA-PK in DNA damage reactions, this article mainly introduces the mechanisms of action of ATM, ATR, and DNA-PK. In summary, the abnormal activation of the PI3K/AKT/mTOR signalling pathway and the dysfunction of PIKK members in DNA damage repair are closely related to the occurrence of cancer and drug resistance during treatment. Therefore, the development of targeted drugs has become an effective approach for anti-tumour therapy.
In this review, we introduce the activation of the PI3K/AKT/mTOR signalling pathway in cancer and the mechanisms by which PIKK members ATM, ATR, and DNA-PK repair DNA damage. In addition, we summarise the efficacy and achievements of PIKK, PI3K, and AKT inhibitors in various human malignancies. We focused particularly on the drugs that entered clinical trials and were already approved for marketing, as well as the efficacy of these drugs in combination with other anticancer drugs applied in therapy of different types of cancers. We also discuss tumour resistance to the inhibitors and propose new approaches to improve antitumor activity by developing multi-targeted drugs, utilising drug delivery systems, and optimising combination regimens. Taken together, this article reviews the clinical research stages and efficacy of PIKK, PI3K, and AKT inhibitors as antitumor drugs in order to provide new perspectives and prospects for cancer treatment.
Phosphoinositide 3-kinase (PI3K)
PI3Ks are an evolutionarily conserved family of lipid kinases classified into three types according to their structure and functionCitation23. Class, I PI3Ks consist of a heterodimer of a regulatory and a catalytic subunit (p110α, p110β, p110δ, and p110γ), which is most associated with cancer cell growthCitation24,Citation25. Class I PI3Ks contain two categories, namely IA and IB. The catalysed subunits of category IA, p110α, p110β, and p110δ, are encoded by PIK3CA, PIK3CB, and PIK3CD genes, respectively, and the regulatory subunit is usually p85 type, including p85α (and its splicing variants p55α and p50α), p85β, p55γCitation26. The catalytic subunit p110γ of class IB is encoded by PIK3CG and its regulatory subunits are p101, p84 (p84 or p87)Citation26. However, the p110α subunit mutation in PIK3CA is considered one of the most prevalent mutations involved in cancer developmentCitation27. The most important mutation sites of p110α are the helical domain E542K and E545K sites, as well as the kinase domain H1047R, all of which have a synergistic effect on cellular transformationCitation28. PI3Kα and β isoforms can be found in many different tissues, while δ and γ are mainly present in the immune systemCitation29,Citation30. The other two PI3K classes have received less attention in terms of cancer research. Class II PI3K has three isomers, namely PI3KC2α, PI3KC2β and PI3KC2γCitation31. At present, the physiological effects of class II PI3K are not fully understood, but they are known to affect cellular activities such as glucose transport, endocytosis, cell migration, and survivalCitation32,Citation33. The member of Class III PI3K is vacuolar protein sorting 34 (VPS34), which plays a key role in regulating intracellular transport, autophagy, and heart and liver functionCitation34.
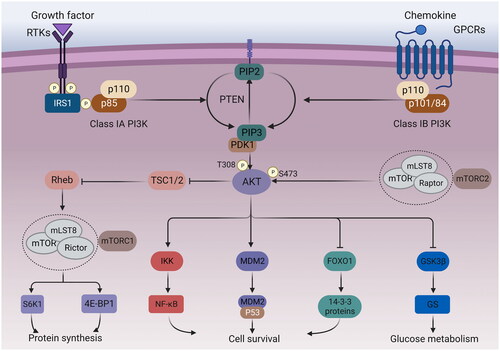
PI3K is a meaningful transduction target in signalling pathways. It is worth noting that class IA can be triggered by various receptor tyrosine kinases or the binding of Ras proteins to the p110 subunit, whereas Class IB can be activated by G protein-coupled receptorsCitation35,Citation36. After activation, the active PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate to PIP3Citation37. However, PTEN in turn can dephosphorylate PIP3Citation38. After binding to the PH domain of the AKT, PIP3 translocates the protein to the vicinity of the plasma membraneCitation39. Subsequently, the recruited AKT needs to be activated by the phosphorylation of two residues, T308 and S473, through phosphoinositol-dependent protein kinase 1 (PDK1) and mTOR complex 2 (mTORC2), respectivelyCitation39,Citation40. Activated AKT can activate and modulate a series of downstream targets, thereby significantly regulating tumour cell growth and proliferation, metastasis and invasion, and chemotherapy resistanceCitation41. The PI3K/AKT/mTOR signalling pathway is schematised in .
Figure 1. PI3K/AKT/mTOR signalling pathway. Growth factors bind to tyrosine kinase receptors (RTK) or G protein-coupled receptors (GPCR), activating PI3K heterodimers of the IA and IB families, respectively. Activated PI3K phosphorylates PIP2 into PIP3, while PTEN dephosphorylates PIP3. The activation of PIP3 attracts PDK1 and AKT to the plasma membrane. AKT can be phosphorylated by PDK1 at the T308 site, while mTORC2 phosphorylates the S473 site. Afterwards, the activated AKT can activate multiple downstream targets. The phosphorylation of tuberous sclerosis 2 (TSC2) by AKT can inhibit the TSC1/TSC2 complex, leading to indirect activation of mTORC1 by blocking the negative regulation of the Ras homolog (Rheb) by TSC1/2. S6 kinase 1 protein (S6K1) and eucaryotic initiation factor 4E-binding protein (4E-BP1) are downstream targets of mTORC1, which can control protein synthesis. In addition, AKT can also phosphorylate IKK, MDM2, FoxO1 to regulate cell survival, and GSK3β to regulate glucose metabolism.
PI3K inhibitors
In the early stages of research and development, PI3K-targeted drugs were mainly composed of a series of pan-PI3K (pan-PI3K) inhibitors, with Wortmannin and LY294002 being prototypes of PI3K inhibitorsCitation42. Non-selective pan-PI3K inhibitors target all PI3K subtypes and may produce strong side effects, making subtype-specific PI3K inhibitors one of the hotspots in drug developmentCitation43. In addition, dose limitations may also be a problem in the development process of PI3K inhibitors. Due to the fact that the PI3K signalling pathway is also indispensable in normal cells (with only low expression), high-dose medication is bound to affect the normal functions of other cells, leading to serious side effects that can be unbearable for patients and interrupting treatmentCitation44. At present, researchers are committed to developing a series of novel inhibitors with significant efficacy, low toxicity, and a few side effects. PI3K inhibitors can be subdivided into pan-PI3K inhibitors, isoform-specific inhibitors, and dual PI3K/mTOR inhibitors ().
Table 1. A summary of major PI3K inhibitors.
Pan-PI3K inhibitors
Pan-PI3K inhibitors are effective against all four isomers of Class I PI3K, leading to severe toxic side effects (). Therefore, the development of a large number of related inhibitors has been discontinued. The current research on Pan-PI3K inhibitors entering clinical practice is as follows.
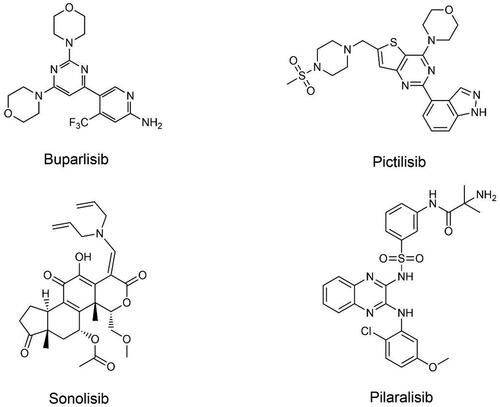
Figure 2. Structures of pan-PI3K inhibitors.
Buparlisib/NVP-BKM120/BKM120
Buparlisib is an oral pan-PI3K inhibitor belonging to a family of 2, 6-dimorpholinopyrimidine derivatives that inhibits 4 isoforms of PI3K via ATP competitionCitation45. Buparisib exhibits significant brain penetration in both in vitro and in vivo mouse models, indicating that it is a highly promising treatment option for intracranial tumorsCitation46. However, buparlisib showed low monotherapy efficacy in a phase II trial in patients with PI3K-activated glioblastoma (GBM), possibly due to insufficient inhibition of the PI3K pathway in the tumorCitation47. As a result, later combined treatment trials were conducted. Unfortunately, buparlisib combined with radiotherapy and temozolomide is not safe. Therefore, Novartis has decided not to develop buparlisib in newly diagnosed glioblastomaCitation48. At present, buparlisib appears to be a promising treatment for breast and head and HNSCC. In the phase III trial of buparlisib plus fulvestrant in hormone receptor-positive/epidermal growth factor receptor-2 negative (HR+/HER2-) advanced breast cancer, the combination of fulvestrant and buparlisib significantly prolonged progression-free survival, but the occurrence of severe side effects was higher than that of the placebo group, at 23% and 16%, respectivelyCitation49. Buparlisib monotherapy was found to be ineffective in a phase II trial in HNSCC, but combination therapy had an unexpected synergistic anti-tumour effect without significantly increasing treatment toxicityCitation50. At present, buparlisib has obtained the fast track approval granted by the FDA, and has been approved for a global multi-center phase III study (NCT04338399) of buparlisib plus paclitaxel for the treatment of HNSCC.
Pictilisib/GDC-0941
Pictilisib is an oral and potent PI3K inhibitor with significant activity against PI3K α/δ isoforms and moderate selectivity for PI3K β/γ, showing promising antitumor activity in preclinical xenograft modelsCitation51. When pictilisib is coupled with anastrozole for treating patients with HR+/HER2- early breast cancer, it can significantly inhibit tumour cell proliferationCitation52, but there is no superior benefit when pictilisib is combined with paclitaxel or fulvestrantCitation53,Citation54. Additionally, the combination of pictilisib with erlotinib or cobimetinib demonstrated only minimal anticancer effects and tolerability in patients with solid tumorsCitation55,Citation56. A study on pictilisib was conducted in JapanCitation57. Four of twelve patients achieved stable disease in the first stage of monotherapy for advanced solid tumours, and no patients were disengaged from the study due to adverse events. In the second phase of the experiment, pictilisib in combination with carboplatin-paclitaxel and bevacizumab was studied in individuals who had non-squamous non-small cell lung cancer (NSCLC), and tumour reduction has been witnessed in 6 patients. Therefore, pictilisib demonstrated preliminary antitumor activity as a single agent as well as in combination treatment in this studyCitation57. For pictilisib, multiple studies are still needed to explore the appropriate combination therapy for different patient groups.
Sonolisib/PX-866
Sonolisib is an oral PI3K inhibitor that can covalently bind to the ATP-binding site of the catalytic subunit of kinase p110 to effectively inhibit class IA PI3KCitation58. As a wortmannin derivative, it is more stable than wortmannin but exhibits lower liver toxicityCitation58. At present, clinical research is devoted to the efficacy of sonolisib in combination with other drugs, aiming to improve its antitumor activity, but the results are not satisfactory. In several phase II randomised trials, sonolisib failed to improve progression-free survival (PFS) as well as overall survival (OS) when combined with cetuximab or docetaxel in different types of cancer. As a result, the therapeutic potential of these combination therapies is restrictedCitation59–62. When raloxifene and sonolisib are combined, the expansion of breast cancer cells is significantly reduced, proving that the combination therapy has certain potential for patients with breast cancerCitation63. In recent studies, sonolisib has been shown to inhibit temozolomide-induced autophagy and promote apoptosis in GBM cells, thereby enhancing antitumor efficacy. This provides the rationale for sequential therapy with temozolomide and sonolisibCitation64. At present, the research progress of sonolisib is less published, and the use of sonolisib needs to be further studied.
Pilaralisib/XL147/SAR245408
Pilaralisib is a reversible, potent class I PI3K inhibitor. By inhibiting the formation of PIP3, pilaralisib inhibits the phosphorylation of downstream targets and is therefore useful in various cancer indicationsCitation65. Both the capsule and tablet formulations of pilaralisib have been demonstrated to have anticancer effectsCitation66,Citation67. In a clinical trial, patients with nonsteroidal aromatase inhibitor-refractory, recurrent, or metastatic HR+/HER2− breast cancer were given pilaralisib or voxtalisib in combination with letrozole tablets. The PFS rates at 6 months in the two groups were 17% and 8%, respectively, indicating that the pilaralisib group had a better pharmacodynamic effectCitation68. When pilaralisib was used in patients with chronic lymphocytic leukaemia (CLL) or lymphoma, PFS > 6 months was observed in 56.0% of patientsCitation69. Preliminary clinical activity has been demonstrated in studies, indicating that further development is warranted. A phase II experiment of pilaralisib in endometrial carcinoma patients observed an ORR of 6.0% and a PFS of more than 6 months in 11.9% of patients. As a result, pilaralisib has restricted clinical activityCitation70. Moreover, the combination of paclitaxel and carboplatin with pilaralisib did not enhance the antitumor efficacy of patients with solid tumorsCitation71. Therefore, further clinical studies are needed to evaluate effective combination strategies with other inhibitors. Interestingly, in in vitro settings, it was found that pilaralisib-treated tumour cells were rapidly damaged by blue-wavelength (430 nm) light irradiation, and the degree of cell damage/death had to be dependent on the dose of pilaralisib and the light power of the irradiation. Pilaralisib accumulates in tumour cells when exposed to blue light and can be photosensitised by blue light to generate reactive oxygen species (ROS), causing tumour cell destruction. This opens up new avenues for using anticancer medicines in photodynamic therapyCitation72.
Isoform-specific inhibitors
Isoform-specific inhibitors are more popular because of their ability to selectively restrict different PI3K isoforms with less off-target toxicity. At present, several inhibitors have entered clinical studies, among which alpelisib, idelalisib, and umbralisib have been approved. The structures of PI3K isoform-specific inhibitors are shown in .
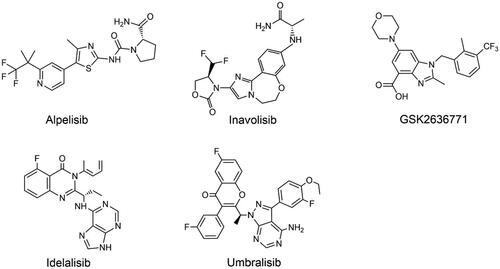
Figure 3. Structures of PI3K isoform-specific inhibitors.
Alpelisib/BYL719
Alpelisib belongs to the 2-aminothiazole family and is a PI3K inhibitor that can selectively inhibit the p110α subtype, which has great potential in breast cancer treatmentCitation73. A combination of alpelisib and fulvestrant was tested in the phase III trial to treat women with HR+/HER2- breast cancer. Patients in the PIK3CA mutation cohort who received alpelisib had significantly longer median PFS in comparison to placebo (11 months vs. 5.7 months, respectively), and median OS was 39.3 months vs. 31.4 months, respectively. In the cohort without PIK3CA-mutated cancers, no clinically relevant treatment benefit was observed with alpelisib plus fulvestrantCitation74. Hyperglycaemia, rash, and diarrhoea are the most frequently occurring side effects of alpelisib treatment. Because of the drug’s short half-life, the toxicity is reversible when the drug is interrupted or it can be controlled with early intervention and supportive therapyCitation75. Given the findings of this phase III trial, the FDA approved alpelisib plus fulvestrant in May 2019 for patients suffering from HR+/HER2- advanced breast cancer with PIK3CA mutationsCitation76. A phase 1b clinical study recently found that alpelisib plus olaparib was tolerable in patients suffering from advanced triple-negative breast cancer (TNBC), with 59% of patients achieving disease control and an ORR of 18%Citation77. Besides, given the bidirectional crosstalk of oestrogen receptor (ER) and HER2 signalling, and the role of PI3K in mediating resistance to anti-HER2 drugs and endocrine therapy, it is speculated that simultaneous inhibition of PI3K, HER2, and ER pathways may be a reasonable therapeutic modality. A phase III randomised trial was therefore planned to evaluate alpelisib plus trastuzumab with or without fulvestrant in patients with PIK3CA-mutated HER2+ advanced breast cancerCitation78.
Inavolisib/GDC-0077
Inavolisib is an innovative PI3K inhibitor with over 300-fold selectivity for PI3Kα isoforms over other class I PI3K isoforms, thus showing remarkable selectivity for PI3Kα isoformsCitation79. PIK3CA-encoded p110α mediates most metabolic responses to insulin, so targeting it disrupts glucose metabolism across multiple tissuesCitation80. Insulin signalling inhibition, for example, promotes hepatic glycogenolysis while inhibiting glucose uptake in skeletal muscle and adipose tissue, leading to transitory hyperglycaemia following PI3K inhibition. The pancreas can release compensatory insulin (insulin feedback) to restore the body to normal glucose homeostasis, however this insulin feedback may reactivate PI3K/AKT/mTOR signalling in tumours, thereby affecting the effect of targeted therapyCitation80. The development of PI3Kα mutant-selective inhibitors to avoid wild-type PI3Kα enzymes is a potential solution to this problem. As a PI3Kα selective inhibitor and mutant PI3Kα degrader, inavolisib can degrade mutant p110α protein without significantly altering wild-type p110α protein, so it can effectively improve the efficacy of PIK3CA mutant tumour patientsCitation81. Inavolisib exhibits strong efficacy in breast cancer patients, and several clinical studies have already been performed in combination with other drugs. Inavolisib, in conjunction with palbociclib and fulvestrant, demonstrated preliminary antitumor activity and manageable safety in patients with PIK3CA-mutated breast cancerCitation82. Several clinical trials are currently underway for the treatment of breast cancer patients.
GSK2636771
GSK2636771 is a potent and orally selective PI3Kβ inhibitor that reduces off-target effects compared to pan-PI3K inhibitorsCitation83. The PI3Kβ isoform is an important lipid kinase that regulates the PI3K signalling pathway and the survival of PTEN-deficient tumour cells, so PI3Kβ inhibitors have a role in PTEN-deficient cancers and are expected to improve clinical efficacy through drug combinationCitation84. GSK2636771 demonstrated favourable clinical benefit and a manageable safety profile in PTEN-deficient and/or PIK3CB-abnormal cancer patients in a first-in-human clinical studyCitation83. β-subtype-specific inhibitors can significantly inhibit the migration and invasion of prostate cancer cells at lower concentrations, showing strong in vitro anti-tumour metastatic activityCitation85. Thus, in a phase, I study of GSK2636771 in combination with the androgen receptor antagonist enzalutamide in patients with metastatic castration-resistant prostate cancer (mCRPC), a 12-week non-progressive disease incidence of 50% was observedCitation86. Additionally, the clinical benefit rate of GSK2636771 in combination with pembrolizumab in patients with PTEN-deficient melanoma or other advanced solid tumours was 52%Citation87. Although the combination of GSK2636771 is well tolerated, the antitumor activity shown is still limited.
Idelalisib
Idelalisib, a PI3Kδ inhibitor for continuous oral use, was approved by the FDA in 2014 for the treatment of relapsed CLL, relapsed follicular B cell NHL, and relapsed small lymphocytic lymphoma (SLL)Citation88. The combination of idelalisib and rituximab demonstrates a significant advantage in patients with relapsed CLL, with ORRs of 81% in the idelalisib group and 13% in the control groups, respectivelyCitation89. The trial was terminated early due to the significant efficacy of the idelalisib arm, and an extension study was initiated, in which patients in the first two arms could participate in an extension study to receive idelalisib monotherapy. Results show that idelalisib significantly improves PFS and OS in patients with relapsed CLL compared with rituximab aloneCitation90. Given that targeting PI3Kδ leads to impairment of the T cell immune response, there have been some adverse events such as PJP, cytomegalovirus (CMV)-related death, and respiratory disease caused by infectionCitation91. In the extension study described above, 4 patients (3.6%) in the idelalisib group and 1 patient in the control group (0.9%) developed PJP infections. However, none of these five patients received anti-PJP prophylaxis, so the trial reinforces the PJP prophylaxis recommendation for idelalisib-treated patientsCitation90. Serious pulmonary complications including PJP infection and CMV reactivation may occur when idelalisib is used in combination with bendamustine. Therefore, PJP prevention and CMV surveillance are recommendedCitation92. Idelalisib frequently causes immune-mediated hepatotoxicity in first-line therapy, and younger subjects and those with immunoglobulin heavy chain (IGHV)-mutated disease are more likely to experience early hepatotoxicityCitation93. Idelalisib treatment is also accompanied by side effects such as severe diarrhoea, colitis, pneumonia, and intestinal perforation, making the clinical discontinuation rate as high as 50%Citation94. Several relevant clinical trials have been conducted, but the use of idelalisib is usually restricted because of its severe toxicity. In addition, Gilead announced the termination of the follow-up development plan and withdrew the indications of idelalisib for the treatment of FL and SLL in 2022, which is basically equivalent to withdrawing from the market.
Umbralisib
Umbralisib is an oral dual inhibitor of PI3Kδ and casein kinase 1ε (CK1ε) with improved selectivity for the PI3K isoform, while inhibiting CK1 signalling reduces the autoimmune toxicity associated with this class of drugsCitation95. From an analysis of comprehensive toxicity data, 13.7% of patients withdrew treatment for adverse effects caused by umbralisib treatment, while the rates of discontinuation owing to adverse effects in clinical studies of idelalisib, copanlisib, and duvelisib were 20.0%, 25%, and 52.0%, respectively. In contrast, umbralisib showed better tolerability and therapeutic potentialCitation96. In 2021, umbralisib was licenced in the United States for the treatment of adults with relapsed or refractory MZL and FLCitation97. Multiple clinical studies have explored umbralisib-related combination therapy effects. In a phase III trial, patients with relapsed/refractory CLL were given umbralisib plus ublituximab (U2) and obinuzumab plus chlorambucil (O + Chl). Both the U2 group and the O + Chl group significantly prolonged PFS, with ORRs of 83.3% and 68.7% respectively, and 34 (16.5%) and 16 (7.6%) patients discontinued treatment due to adverse effects in the two groups respectivelyCitation98. Therefore, it can be stated that the combination therapy has a high degree of feasibility. However, the efficacy of umbralisib combination therapy is still being explored, hoping to bring new breakthroughs to the treatment of patients with hematological malignancies.
Dual PI3K/mTOR or dual PI3K inhibitors
PI3K/mTOR or dual PI3K inhibitors can more completely inhibit the PI3K/AKT/mTOR signalling pathway and the occurrence of negative feedback regulation, thus possessing a wider range of activitiesCitation42,Citation99. On the other hand, dual targeting will result in more unpredicted clinically relevant negative consequences and adverse reactions, making their innovation more complex and difficult. Therefore, the development of a high-efficiency and low-toxic dual inhibitor has become the research and development goal of such drugs. The structures of dual PI3K/mTOR or dual PI3K inhibitors are shown in .
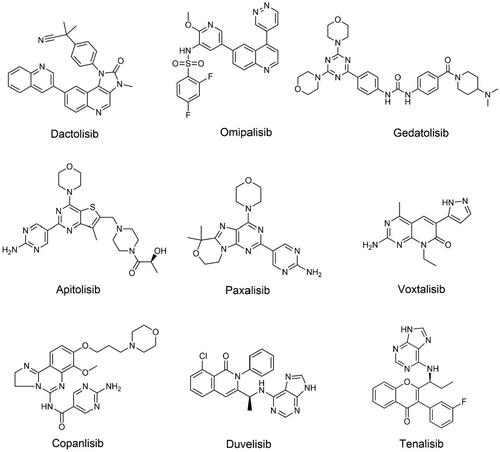
Figure 4. Structures of dual PI3K/mTOR or dual PI3K inhibitors.
Dactolisib/BEZ-235/NVP-BEZ235
Dactolisib, an imidazoquinoline derivative, is the first dual PI3K/mTOR inhibitor to enter clinical trials with IC50s of 4, 5, 7, 75, and 20.7 nmol/L for p110α/γ/δ/β and mTOR, respectivelyCitation42. Compared with first-generation mTOR inhibitors, dactolisib can simultaneously inhibit mTORC1 and mTORC2, thereby avoiding the activation of feedback regulatory pathwaysCitation100. In a phase II study of treatment-naïve patients with advanced pancreatic neuroendocrine tumours, the median PFS was 8.2 months with dactolisib compared with 10.8 months with everolimus. Dactolisib did not show higher efficacy than everolimus due to the poor tolerability of dactolisibCitation101. Similarly, tolerability issues have limited further clinical studies of dactolisib in renal cell carcinoma as well as prostate cancerCitation102,Citation103. Dactolisib is highly hydrophobic, so high-dose oral administration is often used in clinical trials. Due to the low bioavailability and high toxicity, the relevant clinical trial results are not satisfactory. Irreversible electroporation (IRE) is an emerging minimally invasive tumour ablation technique. When IRE is combined with an intratumorally injected liposomal formulation of dactolisib, IRE is able to release dactolisib from its liposomal encapsulation, thereby increasing its solubility in aqueous solutionsCitation104. This combined approach has significant antitumor effects and efficacy in limiting cancer cell proliferation, and is a promising combination therapy for cancer treatment. Unfortunately, BEZ-235 has now been withdrawn from development.
Omipalisib/GSK2126458/GSK458
Omipalisib, an oral and reversible dual inhibitor of PI3K/mTOR discovered by GlaxoSmithKline, showed broad antitumor activity in preclinical studiesCitation105. In a first-in-human phase I study, omipalisib treatment triggered durable objective responses in multiple tumour types, including endometrial, oropharyngeal, renal cell, and bladder cancerCitation105. Only one patient achieved a partial response when the mitogen-activated protein kinase (MEK) inhibitor trametinib was combined with omipalisib, instead 42% of patients had dose interruptions due to adverse eventsCitation106. Overlapping side effects are often seen in combination therapy, so omipalisib plus trametinib showed poor tolerance and poor clinical efficacy. Importantly, recent studies have shown that omipalisib can inhibit non-homologous end joining (NHEJ) and sensitise cancer cells to radiotherapy and chemotherapy, thus being a potential inhibitor of DNA double-strand break repairCitation107. Omipalisib has shown limited efficacy in cancer treatment. However, PI3K and mTOR are involved in the pathogenesis of idiopathic pulmonary fibrosis (IPF). It has been shown that omipalisib was well tolerated by subjects with IPF, supporting further evaluation of omipalisib as a novel therapy for IPFCitation108.
Gedatolisib/PF-05212384/PKI-587
Gedatolisib is an intravenous PI3Kα, PI3Kγ, and mTOR inhibitor developed by Pfizer. Gedatolisib can inhibit the activation of the PI3K/mTOR signalling pathway and induce G0/G1 cell cycle arrest, which has great potential for tumour therapyCitation109. Two PI3K/mTOR inhibitors, PF-04691502 and gedatolisib, were administered in combination with irinotecan or PD-0325901 in a phase I study. Both combinations of PF-04691502 were discontinued early because of poor tolerance, and no maximum tolerated dose (MTD) for either group was established. The ORRs of gedatolisib combined with irinotecan or PD-0325901 were 4.7% and 11.1%, respectively, and preliminary antitumor activity was observed. Tolerability remains a non-negligible issue, however, and both groups require frequent dose delays and dose reductions to mitigate the toxicity associated with combination therapyCitation110. In the phase II trial, PF-04691502 was compared with gedatolisib in subjects with recurrent endometrial carcinoma. Because of intolerable toxic effects, two PF-04691502 research groups were terminated soon. The clinical benefit response rates for the gedatolisib groups were 53% and 26%, respectivelyCitation111. At present, in a phase I study in cancer patients, gedatolisib was combined with paclitaxel and carboplatin, the ORR is 65%, and the ORR of clear cell ovarian cancer is 80%, showing good tolerability and preliminary efficacy. A follow-up trial is therefore plannedCitation112.
Apitolisib/GDC-0980
Apitolisib, a novel oral PI3K/mTOR inhibitor developed by Genentech, has shown anti-tumour activity in breast, prostate, endometrial, and other cancers by blocking the cell cycle and thereby inducing apoptosisCitation113. The phase I study demonstrated modest antitumor activity in patients with PIK3CA-mutated head and neck cancer, peritoneal mesothelioma, and pleural mesotheliomaCitation114. Furthermore, the most prevalent apitolisib-related toxicities were hyperglycaemia, pneumonia, rash, liver dysfunction, and diarrhoeaCitation114. Apitolisib and everolimus were assessed in a phase II study on people with renal cell carcinoma (mRCC). The results showed that apitolisib was not as effective as everolimus, with PFS of 3.7 months and 6.1 months, and ORR of 7.1% and 11.6%, respectivelyCitation115. Apitolisib monotherapy is often inefficient, and its combination therapy with chemotherapeutics should be further explored. Studies have shown that apitolisib and the MEK inhibitor refametinib can exert an anti-proliferative effect on hypopharyngeal squamous cell carcinoma cell linesCitation116. Apitolisib in combination with gemcitabine and/or cisplatin can improve chemotherapy sensitivity and antitumor activity in advanced cholangiocarcinomaCitation117. In addition, recent studies have shown that apitolisib can inhibit the proliferation of human GBM cells, thus apitolisib is expected to be further evaluated in clinical trials aimed at GBMCitation118. Therefore, apitolisib is a promising candidate for cancer treatment.
Paxalisib/GDC-0084
Paxalisib is an oral and selective PI3K/mTOR inhibitor that permeates the blood-brain barrier (BBB). A previously designed potent and BBB-penetrating PI3K inhibitor showed poor stability, and the purine-based inhibitor paxalisib was finally identified through continuous design optimizationCitation119. Paxalisib is a drug specifically designed to treat brain cancer. Paxalisib has been discovered in the latest research to dramatically reduce cell survival in PIK3CA-mutated breast cancer brain metastatic cell linesCitation120. Given that paxalisib has shown some activity in improving intracranial tumours, further clinical trials are scheduled to test its effectiveness in treating brain metastases. In the phase I study in patients with high-grade glioma, paxalisib efficiently crossed the BBB, and nineteen patients (40%) achieved stable disease. Therefore, paxalisib has obvious potential to treat brain cancerCitation121. According to interim results from a phase II study, the MTD for paxalisib in patients with newly diagnosed GBM was 60 mg/d. When all patients were included in the analysis, the PFS was 8.5 months, indicating a promising efficacy signalCitation122. To achieve the maximum efficacy of paxalisib, clinical studies in combination with other medications are still needed to meet the urgent need for the management of brain tumours.
Voxtalisib/XL765/SAR245409
Voxtalisib has the strongest effect on p110γ, while also inhibiting mTOR. There have been several human studies of voxtalisib with other chemotherapeutic agents. The phase I/II study compared pilaralisib and voxtalisib plus letrozole in breast cancer patients. PFS rates at 6 months were 17% in the pilarisib group and 8% in the voxtalisib group, and in the combination therapy cohort both demonstrated limited clinical activityCitation68. When voxtalisib was coupled with temozolomide in patients diagnosed with high-grade glioma, the partial response was estimated to be 4%, of which 68% had stable diseaseCitation123. When voxtalisib was combined with the MEK inhibitor pimatinib in cancer patients, 51 patients (46%) achieved stable diseaseCitation124. In recent years, voxtalisib monotherapy demonstrated promising efficacy in FL patients in a phase II trial, with an ORR of 41.3% and a complete response rate of 10.9%. However, in patients with mantle cell lymphoma, diffuse large B cell lymphoma or CLL/SLL, efficacy is limitedCitation125. Currently, the efficacy of voxtalisib in different tumour settings is being further explored.
Copanlisib/BAY 80–6946
Copanlisib, an intermittent intravenous inhibitor, inhibits four PI3K isoforms and significantly inhibits the activity of PI3Kα/δ isoformsCitation126. The FDA has authorised copanlisib monotherapy for the treatment of adult individuals with relapsed and refractory follicular lymphoma (FL), as well as for pre-treatment breakthrough designation for patients with marginal zone lymphoma (MZL)Citation127. FL is refractory, and most patients usually have disease recurrenceCitation128. At present, the optimisation scheme for FL treatment is constantly advancing.
Copanlisib exhibits significant efficacy and controllable safety in the therapy of indolent non-Hodgkin lymphoma (iNHL). CHRONOS-1 (phase II trial) for iNHL included parts A and B, and the objective response rates (ORR) were 43.8% and 59.2%, respectivelyCitation129. According to the findings of phase III clinical study CHRONOS-3, the PI3K inhibitor copanlisib + rituximab group diminished the probability of disease development/death by 48% when contrasted to the placebo group, proving that copanlisib combined with rituximab has good efficacy in the treatment of relapsed iNHL patients (including MZL)Citation130. The phase III clinical trial CHRONOS-4 was designed to compare copanlisib combined with rituximab plus bendamustine (RB) with copanlisib combined with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in patients with relapsed indolent B cell lymphoma. The ORRs of the copanlisib plus RB and R-CHOP groups were 90% and 100%, respectively. Due to the small number of patients, further trials are still needed to confirm these clinical dataCitation131. The most common adverse reactions to copanlisib treatment are temporary hyperglycaemia/hypertension, which can recover to baseline levels after reaching their peakCitation132,Citation133. There is a significant acute liver injury during copanlisib therapy, therefore it is recommended that patients receiving copanlisib are monitored for liver functionsCitation134. The most prevalent infection in treated patients is pneumonia, for which Pneumocystis jirovecii pneumonia (PJP) prophylaxis is recommendedCitation134. At present, the combination of copanlisib and afatinib for the treatment of head and neck squamous cell carcinoma (HNSCC) can markedly restrict the growth and survival of tumour cells in vitro, which would be a potential method to replace cisplatin-resistant therapyCitation135. Furthermore, PIK3CA is the most frequently mutated oncogene, and copanlisib was also tested in patients with PIK3CA mutations in the NCI-MATCH (EAY131) trial. The trial revealed encouraging therapeutic efficacy in refractory tumours with PIK3CA mutations, with an ORR of 16%Citation136. copanlisib shows greater safety and clinical potential for cancer treatment than other similar drugs, and further in-depth study of its clinical effects is needed.
Duvelisib
Duvelisib targets vital tumorigenic routes by inhibiting the γ and δ isoforms of PI3K, which is not only directly toxic to tumour cells but also abrogates pro-survival cytokine and chemokine signalling in the tumour microenvironmentCitation137. In the phase I trial, duvelisib demonstrated favourable efficacy in patients suffering from advanced malignant tumours, including those with high-risk biologics. The ORR was 46% in patients with 17p deletion or TP53 mutation and 51% in patients with unmutated IGHVCitation138. The ORR in the phase II DYNAMO trial of duvelisib monotherapy in patients with iNHL was 47.3%, with a median PFS of 9.5 months. As a result, monotherapy appears to have an encouraging clinical benefitCitation139. In the phase III DUO trial, duvelisib was contrasted to ofatumumab in patients with CLL/SLL. The results showed that the PFS was 13.3 months and 9.9 months respectively, and the ORR was 74% and 45%, respectivelyCitation140. Based on the significant efficacy of duvelisib in this trial, duvelisib received the first global approval in the United States for the treatment of CLL/SLL adult patientsCitation141. When duvelisib was combined with other drugs in patients with NHL and CLL, the ORR in group 1 (duvelisib plus rituximab) was 78.3%, and the ORR in group 2 (duvelisib plus bendamustine and rituximab) was 62.5%Citation142. Combination therapy showed no superimposed toxicity, so concomitant use is safe and feasible. Duvelisib is very similar to idelalisib, and expanded use will be limited by toxicities that fall into two broad categories: immune-mediated and infectiousCitation143. Measures should be taken to monitor, for example, in patients receiving duvelisib, CMV, and PJP prophylaxis is strongly suggested to lower the risk of some possibly deadly toxicitiesCitation144.
Tenalisib (RP6530)
Tenalisib is a potent dual PI3K δ/γ inhibitor with IC50s of 25 nM and 33 nM, respectivelyCitation145. Tenalisib, as a single drug, showed significant clinical activity in the treatment of T cell lymphoma (TCL) patients, with a total remission rate of 45.7%Citation146. Subsequently, the efficacy evaluation of tenalisib combined with romidepsin in the treatment of peripheral TCL and skin TCL was conducted, and the results showed that the ORR was 75% and 53.3%, respectivelyCitation147. Therefore, both monotherapy and combination therapy with tenalisib have shown high safety and excellent clinical activity. At present, Rhizon Pharmaceutical Company has announced that tenalisib has been approved by the FDA as a fast-track and Orphan drug for the treatment of PTCLCitation145. Besides, tenalisib also carried out a phase II study on metastatic breast cancer (NCT05021900)Citation148. Tenalisib is still being studied clinically for solid tumours and hematological malignancies.
Phosphoinositide 3-kinase-related protein kinases (PIKK)
PIKKs are a group of atypical serine/threonine kinases with conserved domain structures. From N-terminal to C-terminal, these are the FRAP-ATM-TRRAP (FAT) domain, the kinase domain (KD), the PIKK-regulatory domain (PRD), and the FAT-C-terminal (FATC) domain, with KD having significant sequence homology with PI3KCitation149. There are currently six PIKK members, including ATM, ATR, DNA-PK, mTOR, SMG-1, and TRAAP. Members closely related to DNA damage repair (ATM, ATR, and DNA-PK) can detect DNA damage, activate cell cycle checkpoints, and initiate DNA repair signalling cascadesCitation150. mTOR is capable of regulating nutrient-dependent signalling, SMG-1 mainly controls nonsense-mediated mRNA decay, and TRRAP is capable of regulating transcription but lacks kinase activityCitation151–153. Based on the correlation between the above proteins and cancer and the breadth of research, we mainly introduce the PIKK members ATM, ATR, and DNA-PK related to DRR, as well as the key target mTOR of the PI3K signalling pathway.
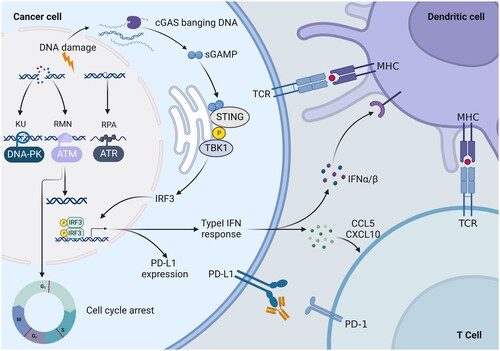
DNA is susceptible to damage by intrinsic and extrinsic toxic effects, including ROS, ultraviolet radiation, and ionising radiation, so the body develops a complex and organised DDR mechanism to preserve the genome’s integrityCitation154. Double-strand breaks (DSBs) are one of the most virulent types of DNA lesions, and their repair is accomplished by two distinct mechanisms: homologous recombination (HR) and NHEJCitation155. ATM is mainly engaged in HR repair and recruited to DNA ends, and it is activated by binding to the MRE11-RAD50-NBS1 complexCitation156. DNA-PK is a complex made up of a catalytic subunit (DNA-PKcs) and a Ku70/80 heterodimer (Ku70/80), and full activation of DNA-PKcs occurs only in the presence of DNA and Ku70/80Citation157. Single-strand breaks (SSBs) are repaired by ATR through homologous recombination. ATR is recruited to single-stranded DNA coated by replication protein A (RPA) by binding to ATR interacting protein (ATRIP), which is then activated by DNA topoisomerase 2-binding protein 1 (TOPBP1)Citation158,Citation159. ATM, ATR, and DNA-PK are situated at the upper end of the signalling pathway, activating multiple downstream targets. ATR and ATM kinases, respectively, regulate the activity of checkpoint kinase 1 (Chk1) and checkpoint kinase 2 (Chk2)Citation160. Subsequently, Chk1 and Chk2 can regulate certain substrates associated with cell cycle arrest, such as cell division cyclin25 family phosphatases, resulting in cell cycle stagnation at G2/M and S, respectivelyCitation160. DNA-PK phosphorylates different residues on the tumour suppressor gene p53, thereby inhibiting the ubiquitination and degradation of p53 by murine double microgene 2 (MDM2), leading to G1 cell cycle stagnationCitation161. In summary, the ATM, ATR, and DNA-PK signalling pathways are interconnected and intertwined, producing a complex signalling network that can promote DNA damage repair, trigger cell cycle checkpoints, and cause cellular senescence or apoptosis in cellsCitation5. Since the DNA repair mechanism within cancer cells can lead to resistance to DNA-damaging anti-cancer therapies, blocking DNA repair in cancer cells has the potential to boost sensitivity to chemotherapy and radiotherapyCitation162,Citation163. Moreover, there is a close relationship between the immune response and DNA repair pathways. The replication stress response (RSR) is defined as the activation of the DDR pathway and the inhibition of cell cycle progressionCitation164. Small-molecule inhibitors of RSR components such as ATM, ATR, and DNA-PK can enhance the instability of the tumour genome to stimulate innate immune responsesCitation165–167. DDR gene inactivation results in the accumulation of chromosome fragments in the cytoplasm, which can be bound by cytoplasmic cyclic GMP-AMP synthase (cGAS)Citation168. DNA binding causes conformational changes in cGAS that facilitate the synthesis of cGAMP, which activates the interferon gene stimulator (STING) and promotes its localisation to the Golgi apparatus. Subsequently, Tank-bound kinase 1 (TBK1) was recruited to phosphorylate interferon regulatory factor 3 (IRF3), and the activated IRF3 entered the nucleus to promote the transcription of type-I interferon (IFN) genesCitation168. Type I IFN signalling promotes the activation of dendritic cells, allowing them to present tumour antigens to CD8 T cellsCitation169. Type I IFN response up-regulates chemokine C-X-C motif chemokine ligand 10 (CXCL10) and chemokine ligand 5 (CCL5), which can effectively promote the infiltration of effector T cells into the tumorCitation164. However, the GAS-STING pathway can result in increased expression of programmed cell death ligand 1 (PDL-1)Citation170 (). The combination of DNA damage agents and immunotherapy seems to be a promising treatment, but its effectiveness still needs further study.
Figure 5. PIKK mediated DNA damage response (DDR) inactivation regulates the immune response. PIKK members ATM, ATR, and DNA-PK have the ability to promote cell cycle arrest and DNA repair. DNA-PK, via the Ku heterodimer, binds to double-strand broken DNA ends, promoting non-homologous end-joining (NHEJ). The MRN complex recruits ATM to single-strand broken DNA, which is then repaired via homologous recombination (HR). In response to HR, ATR is recruited to single-stranded DNA wrapped by replicating protein A (RPA) for DNA repair. DNA damage response (DDR) gene inactivation leads to the accumulation of chromosomal fragments in the cytoplasm, which activates the cyclic GMP-AMP synthase-interferon gene stimulator (cGAS-STING) pathway. STING activates interferon regulatory factor 3 (IRF3) transcription by binding to and activating tank-binding kinase 1 (TBK1). The type I IFN response is upregulated by the chemokine C-X-C motif, chemokine ligand 10 (CXCL10) and chemokine receptor 5 (CCL5), thereby promoting the infiltration of effector T cells into tumours. In addition, the expression of programmed cell death 1 ligand 1 (PD-L1) was upregulated. Secreted IFN-α and IFN-β promoted the antigen-presenting ability of dendritic cells.
mTOR can form two structurally distinct protein complexes, namely mTORC1 and mTORC2. mTORC1 is a rapamycin-sensitive protein kinase composed of three core components, namely mTOR, the regulatory-associated protein of mTOR (Raptor), and mammalian lethal with Sec13 protein 8 (mLST8, also known as GβL)Citation171,Citation172. In addition, it also contains two non-core components, namely proline-rich AKT1 substrate 1 (RAPTOR) and the DEP domain-containing mTOR-interacting protein (DEPTOR)Citation173,Citation174. Various growth factors can activate mTORC1 by stimulating signalling pathways involving PI3K and its downstream effector AKT. In this process, activated AKT mediates TSC2 phosphorylation and inhibits the TSC complex, thereby preventing negative regulation of the Ras homologue enriched in the brain (Rheb) by the complex, allowing for Rheb enrichment and activation of rapamycin-sensitive mTORC1Citation175. Ribosomal protein S6 kinase 1 protein (S6K1) and eukaryotic initiation factor 4E-binding protein (4E-BP1) are the major phosphorylation targets of mTORC1, both of which are involved in translation and protein synthesisCitation176. In addition, S6K1 can directly phosphorylate insulin receptor substrate 1 (IRS-1) as part of the negative feedback loop, thereby reducing insulin-based PI3K activationCitation177. mTORC2 also includes mTOR and mLST8. The difference is that there is no Raptor in mTORC2, but rather a rapamycin-insensitive companion of mTOR (Rictor) insteadCitation178,Citation179. In addition to its core components, mTORC2 also contains DEPTOR as well as regulatory subunits mSin1 and Protor1/2Citation174,Citation180,Citation181. mTORC2 can phosphorylate AKT to regulate signalling pathways, control cytoskeleton rearrangement, and regulate glucose metabolism, as well as participate in cell growth, apoptosis, and cell cycle controlCitation182. Collectively, mTOR is able to participate in intracellular and extracellular signalling, regulating multiple downstream pathways involved in a range of pathophysiological processes, such as cancer, metabolic disorders, neurodegeneration, and agingCitation183.
PIKK inhibitors
Members of the PIKK family engage in a wide range of physiological functions, and their functional abnormalities can lead to various diseases, including cancer. Therefore, targeted inhibition of PIKK family members has broad prospects for treating cancer. Due to the similar kinase domains of PI3K and PIKK, the chemical inhibitors of the two family kinases seem to share similar structural motifsCitation184. Currently, many studies on DNA damage repair and mTOR-related inhibitors are ongoing, and some drugs have entered clinical trials. However, only mTOR inhibitors have been approved.
ATR, ATM, and DNA-PK inhibitors
DDR is crucial for maintaining genomic stability in the body. However, inhibiting DNA repair in tumours can significantly improve the sensitivity of genotoxicity therapy (chemotherapy and radiotherapy)Citation162,Citation163. Moreover, the use of synthetic lethal relationships can selectively kill tumour cells with specific DDR pathway defectsCitation20. Therefore, DDR-related inhibitors have become cancer-specific therapeutic targets. DDR inhibitors are promising targets for breaking resistance to standard care treatments, with relatively mild and tolerable side effects. Currently, the anticancer therapy hotspots ATM, ATR, and DNA PK are members of the PIKK family, and several inhibitors are in preclinical and clinical development. However, no medications have yet received approval (). The structures of PIKK family members ATR, ATM, and DNA-PK inhibitors are shown in .
Figure 6. Structures of PIKK family members ATR, ATM, and DNA-PKcs inhibitors.
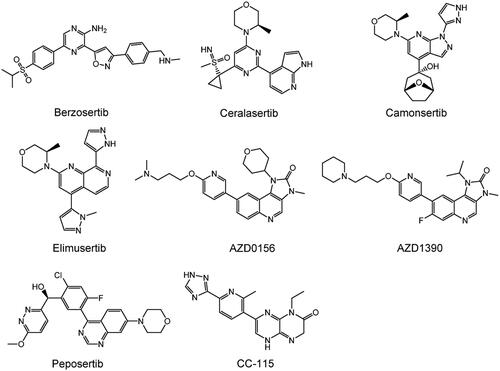
Table 2. A summary of major PIKK inhibitors.
Berzosertib/VX-970/VE-822/M6620
Berzosertib is a powerful ATR inhibitor administered intravenously and is currently being evaluated by Merck. To improve the targeted therapy of selective inhibitors, it is possible to target defective pathways in cancer cells, use synthetic lethal interactions to kill cancer cells or rely on synergy with other drugs to enhance anticancer effectsCitation185. In the phase I study, the berzosertib plus gemcitabine group had a partial response of 8.3% and a stable disease of 60.4%Citation186. In the berzosertib plus gemcitabine plus cisplatin group, partial response (PR) was 14% and stable disease was 57%. Additionally, berzosertib was well tolerated only in combination with cisplatin-free gemcitabine, so the suggested phase II dose (RP2D) for this combination was establishedCitation186. Following that, a phase II trial of berzosertib plus gemcitabine in high-grade serous ovarian cancer was conducted. Berzosertib combined with gemcitabine significantly prolonged PFS compared to gemcitabine alone (22.9 weeks vs. 14.7 weeks), and the combination therapy reduced the proportion of patients with severe adverse reactions (26% vs. 28%)Citation187. Berzosertib has good BBB penetration in preclinical studies of GBM. Studies have shown that berzosertib exhibits a radiosensitizing effect in preclinical NSCLC brain metastases models, so a clinical trial of berzosertib combined with whole brain irradiation for patients with NSCLC brain metastases is currently underwayCitation188.
Ceralasertib/AZD6738
Ceralasertib is a potent and selective oral ATR inhibitor currently being evaluated by AstraZeneca. Ceralasertib elicits promising antitumor activity in preclinical studies, so multiple clinical trials were conducted to evaluate the effectiveness of ceralasertib in combination with various chemotherapeutic drugs. Ceralasertib plus carboplatin or paclitaxel demonstrated excellent anticancer potential in phase I studies in cancer patients, providing a recommended RP2DCitation189,Citation190. Besides, ceralasertib plus durvalumab showed promising clinical effects in patients with metastatic melanoma and advanced gastric cancer. The ORRs were 31.0% and 22.6% respectively, and the disease control rates were 63.3% and 58.1% respectively. Common adverse events were mainly haemorrhagic and could be managed with dose interruptions and reductionsCitation191,Citation192. In contrast, the combination of ceralasertib and olaparib was less beneficial for treating genetically mutated ovarian cancer patients, with an ORR of 8.3%Citation193. The phase II study of this medication plan was then carried out, and the results showed that 9 patients had stable disease. Active signalling is especially observed in BRCA1-related diseases. Since no objective response has been received, further evaluation of this combination is requiredCitation194.
Camonsertib/RP-3500
Camonsertib is an oral ATR inhibitor that is currently undergoing clinical evaluation. In a clinical study related to camonsertib, it was shown that poly (adenosinediphosphate-ribose) polymerase (PARP) inhibitors not only block DNA break repair but also stabilise the PARP-DNA complex, thereby stopping the replication fork. Therefore, both effects can lead to the accumulation of DSB in DNA replication and its dependence on HRCitation195. The combination of ATR and PARP showed excellent antitumor activity in preclinical models. However, this combination results in overlapping toxicities, including myelosuppression, that limit tolerance to the combination. Therefore, the researchers used a short-term intermittent dosing regimen when combining camonsertib with a PARP inhibitor, and the results showed that this method can maximise the efficacy and alleviate the occurrence of anaemia, which is a potential method for cancer treatmentCitation195. Based on the good preclinical anticancer effect, the phase I study related to camonsertib is currently underway.
Elimusertib/BAY-1895344
Elimusertib is a potent, orally available, and selective ATR inhibitor. In preclinical models, elimusertib monotherapy showed anti-tumour activity in prostate cancer, colorectal cancer, and mantle cell lymphoma models, with the most sensitive response in lymphomaCitation196. Given the promising activity of elimusertib in preclinical testing, multiple clinical trials were conducted to evaluate its efficacy in humans. The MTD of the phase I clinical study was 40 mg. Among patients treated at the MTD or higher, the disease control rate was 69.2%, the ORR was 30.8%, and the median time to response was 78 dCitation197. Elimusertib has shown preliminary clinical activity in first-in-human trials, and multiple trials are currently recruiting.
AZD0156
AZD0156 is an imidazo[4,5-c]quinolin-2-one core-based selective ATM inhibitor and the first ATM inhibitor to be enrolled in clinical trialsCitation198. Compounds such as KU-55933 and KU-60019 have been shown to inhibit ATM in vitro, but their moderate potency, low water solubility, and low oral bioavailability have limited their further developmentCitation198. AZD0156 was discovered after the chemical optimisation of a series of novel ATM inhibitors. Due to its high selectivity and excellent physicochemical properties, AZD0156 has become a good candidate for clinical drug developmentCitation198. In preclinical trials, AZD0156 enhanced the usefulness of the PARP inhibitor olaparib in lung, gastric, and breast cancer cell linesCitation199. In addition, AZD0156 increased radiation sensitivity in a mouse model of gliomaCitation200. Based on good preclinical anti-tumour activity, phase I evaluation of AZD0156 is currently underway, and further evaluation of its efficacy in combination with other drugs is planned.
AZD1390
AZD1390 is an ATM inhibitor with an oral activity that can penetrate the central nervous system. Compared to other PIKK family-related enzymes, AZD1390 has significant selectivity for ATM202. Prior to this, AZD0156 was clinically developed for the treatment of systemic tumours. However, as a matrix for human efflux transporters, AZD0156 is unlikely to significantly cross the BBB. AZD1390 was discovered during the optimal design of compounds using in vitro MDCK efflux transporter assaysCitation201. As a weak substrate of the transporter, it has significant penetration across the BBB, thus showing good therapeutic potential for central nervous system malignancies. AZD1390 enhances radiosensitization in multiple preclinical cancer cell types, making it a promising antitumor therapy. GBM is the most universal and potentially fatal type of brain tumour caused by malignant glial cells, and AZD1390 is presently in phase I clinical research phase of combined therapy with radiotherapy for GBM (NCT03423628). In addition, BRCA1/2 is a gene involved in the HR pathway. Research has shown that the combination of AZD1390 and EZH2 inhibitors has a synthetic lethal effect in the treatment of BRCA1-deficient breast tumorsCitation202. Thus, synergistic interactions can serve as an effective model for cancer treatment.
Peposertib/M3814
Although the older generation of DNA-PK inhibitors showed some efficacy, their selectivity was limited due to their structural similarity to PI3Ks. Peposertib is a new generation of oral and highly potent DNA-PK inhibitorCitation203. In preclinical studies, peposertib effectively inhibited radiation-induced NHEJ repair when combined with radiotherapy, thereby enhancing the malignant cells’ reaction to radiationCitation204. Furthermore, it has shown synergistic antitumor effects in NSCLC models when used in combination with the chemotherapeutic drugs paclitaxel and etoposideCitation205. The good preclinical antitumor activity provides a theoretical basis for the follow-up clinical trials of peposertib. Peposertib was studied in a phase I study in combination with radiotherapy. There were 12 patients developing stable disease, and the RP2D was 400 mg twice daily (BID)Citation206. At present, the combination of capecitabine and radiotherapy in the treatment of rectal cancer has not entered the Phase II trial, and the Phase Ib part has been completed (NCT03770689). Anyway, other studies combining M3814 with radiotherapy and chemotherapy are still ongoing.
CC-115
CC-115 is a dual inhibitor of DNA-PK and mTOR with IC50s of 13 nM and 21 nM, respectivelyCitation207. Initial clinical performance of CC-115 was evidenced in 8 patients with ATM deletion/mutation with relapsed/refractory CLL/SLL, and these patients were then enrolled in a larger phase I studyCitation208. In this broader Phase I study, the efficacy of CC-115 in patients with hematological malignancies or solid tumours was evaluatedCitation209. CC-115 displayed surprising efficacy in cervical squamous cell carcinoma, Ewing sarcoma, GBM, and castration-resistant prostate cancer, with stable disease rates of 53%, 22%, 21%, and 64%, respectively. Furthermore, a group of CLL/SLL patients treated with CC-115 showed promising clinical outcomes. 38% of patients with CLL had a partial response, and 25% of patients with SLL had stable disease. CC-115 is well tolerated and has similar toxicity to mTOR inhibitors. Therefore, CC-115 (10 mg BID orally) is an excellent regimen for anti-cancer therapyCitation209. The II clinical trial of CC-115 combined with other drugs for GBM patients is currently underway (NCT02977780).
mTOR inhibitors
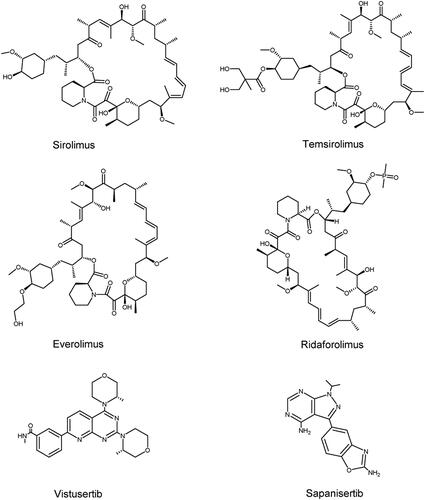
Drug resistance in targeted mTOR therapy can emerge through a range of mechanisms, including inadequate mTORC1 function suppression, negative feedback loop inhibition, and compensatory pathway activation due to mTORC1 inhibitionCitation210. Rapamycin and its analogs are the first generation of mTOR inhibitors that selectively inhibit mTORC1 activity. However, rapamycin can inhibit the mTORC1 substrate S6K1, but cannot completely inhibit the phosphorylation of 4E-BP1, resulting in the inability to effectively block the inhibition of tumour growthCitation211. Furthermore, S6K1 can phosphorylate insulin receptor substrate 1 (IRS-1), which is part of the negative feedback loopCitation179. S6K1 also phosphorylates mSIN1, a component of mTORC2, at T86 and T398 and has a certain inhibitory effect on MTORC2-mediated AKT activationCitation212. Since mTORC1 is the downstream target of several cancer-related pathways, inhibition of a single target can activate the compensation pathway, thereby decreasing the effectiveness of the cancerCitation210. Based on this, developers have developed a series of mTORC1/mTORC2 inhibitors and PI3K/mTOR inhibitors. While these drugs are effective in overcoming drug resistance, they can be detrimental to normal cells and lead to severe toxicity, which may restrict their clinical useCitation213. Due to the presence of multiple feedback loops and compensatory pathways, the effects of mTOR combined with other drugs are currently being explored. The structures of mTOR inhibitors are shown in .
Figure 7. Structures of PIKK family members mTOR inhibitors.
Rapamycin/sirolimus
Rapamycin, commonly known as sirolimus, has immunosuppressive and anti-proliferative effects, making it effective in the treatment of organ transplant rejection and cancerCitation214. Rapamycin does not directly suppress mTOR function, instead, it binds to the intracellular FK506-binding protein 12 (FKBP12) to create the FKBP12-rapamycin complex, which subsequently binds to the FKBP12-rapamycin-binding (FRB) structure of the mTOR regionCitation215. Rapamycin insertion into the cavity produced by the FKBP12 and FRB domains changes the conformation of mTOR, limiting mTORC1 action allostericallyCitation216. Due to the insensitivity of mTORC2 to rapamycin, prolonged exposure to rapamycin can reduce the level of mTORC2. Therefore, it is believed that rapamycin inhibits the phosphorylation of mTOR substrates by binding to mTORC1 rather than mTORC2Citation217. Rapamycin is limited in clinical practice due to its unfavourable physicochemical properties and pharmacokinetic characteristics. Hence, many structural changes and transformations of sirolimus have been carried out with the aim of finding more efficient rapamycin analogs. Furthermore, nab-sirolimus (ABI-009) is an innovative mTOR inhibitor, a suspension of sirolimus albumin-bound nanoparticles for injection. Due to its unique dosage form, nab-sirolimus has a superior pharmacokinetics profile, higher tumour tissue drug exposure, stronger target cell inhibition, and better safety profile than other marketed mTOR inhibitorsCitation218. In a phase II trial of advanced malignant PEComa, nab-sirolimus monotherapy resulted in an ORR of 39%, a median PFS of 10 months, and a median overall survival of 48 monthsCitation219. Based on the drug’s excellent efficacy in this trial, the FDA has approved it for intravenous injection in adult patients with malignant PEComaCitation220.
Temsirolimus/CCI-779
Temisirolimus, an ester analogue of rapamycin, was approved by the FDA in 2007 for the treatment of advanced renal cell carcinoma (RCC)Citation221. Temisirolimus’ efficacy in the treatment of various malignant cancers in combination with other chemotherapy medicines is still being evaluated. In patients with first-relapse rhabdomyosarcoma (RMS), a phase II trial was scheduled to estimate the feasibility of bevacizumab or temsirolimus in conjunction with chemotherapy. The ORR of the bevacizumab and temsirolimus groups was 28% and 47% respectively, and the 6-month event-free survival was 54.6% and 69.1%. Due to the superior efficacy of the temsirolimus group, temsirolimus will be recommended as the bioactive agent for RMSCitation222. Tesirolimus plus erlotinib was well tolerated in patients with solid tumours, of whom nine patients (82%) had stable disease, establishing the recommended RP2DCitation223. In one trial, temsirolimus was coupled with the AKT inhibitor perifosine to treat relapsed GBM. There have been no previous studies that combined these two kinds of medicines with the goal of efficiently inhibiting the PI3K/AKT signalling cascade. The MTD of the combination was established by the trial to be 115 mg of temsirolimus weekly and 100 mg of perifosine daily. Notably, the temsirolimus dose was more than four times higher than the FDA-approved amount for RCC (25 mg per week). The reasons why large dosages of temsirolimus were tolerated in this trial are unknown; nonetheless, additional pilot studies are plannedCitation224.
Everolimus/RAD-001
Everolimus, a 40-O-(2-hydroxyethyl) derivative of rapamycin, was granted FDA approval in 2019 for the treatment of advanced postmenopausal HR+/HER2− breast cancer in women, pancreatic-derived progressive neuroendocrine tumours, renal vascular smooth muscle lipoma, RCC, and other indicationsCitation225. In phase II randomised trial, everolimus plus letrozole was used to treat women with advanced premenopausal breast cancer who were HR+/HER2−. Patients in the everolimus plus letrozole group received longer median PFS than those in the letrozole alone group (19.4 months vs. 12.9 months), indicating that the combination was beneficialCitation226. Similarly, everolimus combined with an aromatase inhibitor (AI) has shown promising therapeutic activity in patients suffering from breast cancer. The median PFS in the everolimus plus AI group and the AI monotherapy group was 11.0 months and 7.2 months respectively, with ORRs of 22.4% and 19.2% respectivelyCitation227. When everolimus was combined with bevacizumab to treat patients with the RCC form of metastatic cancer, the median PFS was 13.7 months with an ORR of 35%. The combination’s robust activity supports this regimen as the standard choice for patients with RCCCitation228. Overall, everolimus has displayed considerable potential in various cancer treatments, and the efficacy of different combination regimens of everolimus is currently being further explored.
Ridaforolimus/deforolimus/MK-8669
Ridaforolimus is a sirolimus analog that was initially used to treat soft tissue and osteosarcoma before being extended to treat a wide range of solid and hematological malignanciesCitation229. A phase III trial evaluated the maintenance of disease control in advanced sarcoma with ridaforolimus. The median PFS was 17.7 weeks for ridaforolimus and 14.6 weeks for placebo. However, serious adverse effects (grade > 3) were more common with ridaforolimus (64.1% vs. 25.6%)Citation230. Due to its poor risk-benefit profile, the US FDA has not approved ridaforolimus for the treatment of sarcoma. Ridaforolimus underwent subsequent clinical trials for the treatment of various malignancies, but its safety still restricts its clinical application. In patients with advanced endometrial cancer, single-agent ridaforolimus displayed anticancer activity and tolerable tolerability, with 13 patients (29%) attaining clinical benefit and a 6-month PFS rate of 18%Citation231. Based on the positive clinical outcomes of this trial, a follow-up phase II trial was conducted in patients with endometrial cancerCitation232. The median PFS for the ridaforolimus group was 3.6 months and 1.9 months for the control group (progestin or chemotherapy). However, the proportion of patients in the ridaforolimus group who discontinued treatment due to AE was 33%, compared to 6% in the control group, with typical grade 3 toxicities being hyperglycaemia, anaemia, and diarrhoeaCitation232. Thus, oral ridaforolimus has strong toxicity in advanced endometrial cancer. Currently, ridaforolimus has stopped development.
Vistusertib/AZD2014
Vistusertib is an ATP-competitive mTOR inhibitor that inhibits the mTORC1 and mTORC2 complexesCitation233. Intermittent dosing regimens induce tumour regression when vistusertib is combined with fulvestrant, making the compound an ideal candidate for clinical combination with endocrine therapyCitation233. According to one study, when compared to single-agent anastrozole, combining vistusertib with anastrozole significantly improved clinical benefit in patients with endometrial cancer. The ORRs were 17.4% and 24.5% respectively, and the PFS were 1.9 and 5.2 months respectively, with manageable toxicityCitation234. A phase II randomised study compared vistusertib with everolimus in patients with mRCC. The PFS for AZD2014 and everolimus was 1.8 months and 4.6 months, respectively, with 35% and 48% of patients experiencing grade 3–4 adverse effects. As a result, while vistusertib is less hazardous, its anticancer activity is inferior to that of everolimusCitation235. When treating patients with polymorphic GBM, vistusertib plus temozolomide resulted in a 6-month PFS rate of 26.6%. The combination test demonstrates tentative efficacy and is relatively safeCitation236. The median PFS for both types of patients was 5.8 months when vistusertib plus paclitaxel was used in patients with ovarian cancer and NSCLC. Due to tolerability concerns, patients with NSCLC must adjust their dose accordingly, and the toxicity is manageableCitation237. A recent phase I/II study found that vistusertib was well tolerated in paediatric patients, indicating that it should be investigated in the appropriate paediatric therapy routeCitation238.
Sapanisertib/INK128/MLN0128/TAK-228
Sapanisertib is an oral, potent ATP-competitive mTOR inhibitorCitation239. In a phase I study of East Asian patients with advanced nonhematologic malignancies, sapanisertib revealed good tolerability and limited anticancer efficacy. Clinical benefit rates with once-daily (QD) and once-weekly doses were 45% and 67% respectively, with RP2D determined to be 3 mg QD240. Subsequently, clinical research was conducted on the combination of sapanisertib and other anticancer therapies. Both combinations of sapanisertib with alisertib or paclitaxel were well tolerated in patients with solid tumours, implying that further research into the combination’s efficacy is requiredCitation240,Citation241. A phase II trial looked into the effectiveness of sapanisertib plus fulvestrant in breast cancer patients. When compared to single-agent fulvestrant, sapanisertib plus fulvestrant significantly increased PFS by 3.5 months and 7.2 months, respectively. However, due to the increased toxicity produced by combination therapy, future research has been limitedCitation242. Moreover, research has indicated that combining tamoxifen and sapanisertib is a promising neoadjuvant modality for individuals with ER+ breast cancerCitation243. Currently, CB-839 HCl combined with sapanisertib treatment for advanced NSCLC in phase I/Ib trials is still recruiting patients(NSCLC).
AKT
AKT is a class of serine/threonine kinase that belongs to the AGC kinase family, also widely recognised as PKBCitation244. The Akt family consists of three members: Akt1 (PKB α), Akt2 (PKB β), and Akt3 (PKB γ). The three AKT isotypes have similar structures, including the PH domain, the central kinase domain, and the carboxyl-terminal regulatory domain containing hydrophobic motifsCitation245. Although the three subtypes have similar structures, they can perform different functions in cancer and physiology. AKT1, as a growth inducer, promotes tumour cell growth but damages metastasis, while AKT2 can increase tumour cell invasion and metastasisCitation246. Thus, AKT1 is hypothesised to act as an invasion suppressor in the early stages of the disease, while AKT2 promotes invasion in the advanced stages of the diseaseCitation247. AKT3 is the least studied and has been proven to be closely linked to the progression of triple-negative breast cancer (TNBC)Citation248. The subtype-specific function of AKT remains controversial, and the mechanisms by which it impacts cancer progression require further investigation. Nonetheless, AKT remains a key target for the treatment of cancer.
AKT is an indispensable part of PI3K-mediated signalling and its activation can phosphorylate a variety of kinases and transcription factors, thereby affecting cellular functions. For example, the phosphorylation of TSC2 by AKT can inhibit the GTPase activity of the TSC1/TSC2 complex, leading to the accumulation of Rheb-GTP and subsequent mTORC1 activationCitation179. AKT phosphorylates Glycogen synthase kinase-3β to facilitate glucose metabolismCitation249. AKT is able to activate the inhibitor of kappa B kinase (IKK), which is involved in the modulation of IκBα. Phosphorylated IκBα is rapidly degraded to release NF-κB, and the released NF-kB translocates to the nucleus and induces the expression of target genes, thus playing an important role in promoting tumour survivalCitation3. AKT phosphorylates forkhead box protein O1, inhibiting its nuclear translocation and preventing its transcriptional activationCitation250. AKT phosphorylates the ubiquitin ligase MDM2 (mouse double microgene 2) and translocates it to the nucleus to bind to p53, thereby affecting cell survival by increasing the degradation of the p53 proteinCitation251. AKT activation is closely related to the development and progression of various tumours, so suppressing AKT may be an excellent therapeutic approach.
AKT inhibitors
Overactivation of the PI3K/AKT pathway is common in many different types of cancer, and AKT is a critical hub of signalling pathways in cancer, making targeting AKT an appealing anticancer tacticCitation41. It can be challenging to develop specific inhibitors because the sequence identity of the kinase domains of the three isoforms is more than 80%, varying only at the PH structural domain and kinase structural domain junctionsCitation252. Drugs are classified as allosteric inhibitors or ATP-competitive inhibitors based on the structure of AKT and the difference in binding domain with the inhibitorCitation253 (). There are few reports of ATP-competitive inhibitors in early development, because the structural similarity of the ATP-binding site allows them to both target AKT and inhibit other kinases of the AGC kinase family with high AKT homology. Thus, lack of kinase selectivity makes them prone to off-target toxicityCitation254. Many AKT inhibitors exhibit limited anticancer activity as clinically indicated monotherapies; therefore, ongoing clinical trials of AKT inhibitors are focused on exploring their potential to improve standard cancer therapy. In addition, some AKT inhibitors such as miransertib have shown excellent efficacy in the treatment of cutaneous leishmaniasis or Proteus syndromeCitation255.
Table 3. A summary of major AKT inhibitors.
Allosteric inhibitors
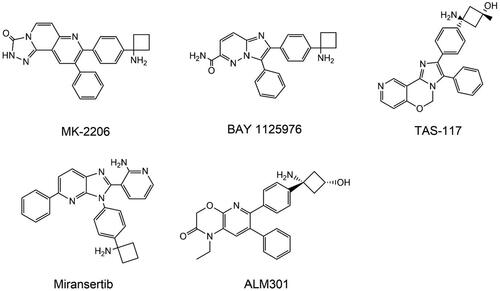
Allosteric inhibitors bind to a specific hydrophobic area at the intersection of the PH domain and the kinase domain, rather than the ATP binding site or the PH domain. As a result, a stable AKT "PH-in" conformation is formed, the phospholipid binding site is blocked, AKT activation is obstructed, and the selectivity of the three AKT isoforms is achieved by utilising the diversity and allosteric sites of the amino acid sequence of the AKT hinge regionCitation256. Allosteric modulators have several advantages, including greater specificity, fewer adverse effects, and lower toxicity. The structures of AKT allosteric inhibitors are shown in .
Figure 8. Structures of AKT allosteric Inhibitors.
MK-2206
MK-2206 is the first small molecule allosteric AKT inhibitor to enter clinical trials. MK-2206 inhibits AKT autophosphorylation at Thr 308 and Ser 473, which results in anticancer actionCitation257. In the phase II study, MK-2206 had little efficacy in advanced breast cancer patients with genetic alterations or deletionsCitation258. The ORR and 6-month PFS were 5.6% for the PIK3CA/AKT1 mutation group, while the ORR was 0% and the 6-month PFS was 11% for the PTEN deletion/mutation group. Toxicity and tumour-related features reduced dose delivery, leading to insufficient tumour target inhibition and ultimately limited anticancer efficacyCitation258. Likewise, MK-2206 monotherapy showed modest activity in renal cell carcinoma and in lymphomaCitation259,Citation260. When MK-2206 was combined with bendamustine and rituximab to treat patients with CLL, the ORR was 92% and the median PFS was 16 months, making it a promising novel therapeutic combination for CLLCitation261. In patients with HER2 amplified solid tumours, MK-2206 was coupled with paclitaxel and trastuzumab, and 10 patients (63%) responded with a median response duration of 6 monthsCitation262. Although some activity signals were observed in the combination therapy of MK-2206, no new research has been conducted on MK-2206 due to efficacy and tolerance issues.
BAY 1125976
BAY 1125976 is a novel, orally active, and allosteric AKT1/2 inhibitor. BAY 1125976 inhibits the expansion of multiple human cancer cells, with striking activity in breast and prostate cancer cell lines, paving the way for clinical developmentCitation263. In the phase I study, 30 of the 78 patients enrolled were stable. In 43 patients treated with RP2D, the clinical benefit rate was 27.9%Citation264. BAY 1125976 was well tolerated; however, clinical tumour responses were limited. At present, the development of BAY 1125976 is still in phase I testing.
TAS-117
TAS-117 is an oral allosteric AKT inhibitor. Preclinical evaluations show that TAS-117 can induce autophagy and apoptosis, and can significantly inhibit tumour growth in multiple myeloma cellsCitation265. AS-117 combined with the dual EZH2/1 inhibitor UNC1999 and the FGFR1-4 inhibitor futibatinib respectively has synergistic anti-tumour effects in cancer modelsCitation266,Citation267. At present, TAS-117 is in human clinical research. In a phase II trial in patients with advanced solid malignancies with PI3K/AKT pathway abnormalities, TAS-117 showed anti-tumour activity in patients with PIK3CA H1047R and AKT1E17K-mutated breast cancer and PIK3CA E545K-mutated ovarian cancer. The ORR was 8%, and the median PFS was 1.4 monthsCitation268. TAS-117 is being studied in a phase II trial in patients with advanced solid tumours possessing germline PTEN-inactivating mutations (NCT04770246).
Miransertib/ARQ-092
Miransertib is a novel oral AKT inhibitor that blocks membrane translocation of inactive AKT and dephosphorylates active AKT, thereby disrupting AKT activityCitation269. Miransertib was initially used to treat human malignant tumours, but in recent years it has primarily been used to treat a variety of rare disorders, including PIK3CA-related overgrowth spectrum caused by somatic activating mutations in PIK3CA and Proteus syndrome caused by somatic activation in AKT1Citation255,Citation269. As a result, targeting the PI3K pathway offers promise for treating such diseases, and several clinical studies have been performed with miransertib to treat related diseases. Furthermore, miransertib has a curative impact on visceral and cutaneous leishmaniasisCitation270.
ALM301
Based on the Bicyclic pyridine and oxazidone core, which has demonstrated remarkable biochemical efficacy in drug development, a novel variant of the AKT inhibitor ALM301 has been developed in recent yearsCitation271. ALM301 effectively and selectively inhibits AKT1 and AKT2, with IC50 values of 125 nM and 95 nM, respectivelyCitation271. ALM301 has significant antiproliferative effects on sensitive cell lines and exhibits excellent efficacy in both single and combination therapy in rats, providing important clues for us to further explore the potential of ALM301 in clinical treatment.
Competitive inhibitors
ATP-competitive inhibitors compete with ATP for the AKT kinase domain active sites, preventing activated AKT from phosphorylating its downstream target proteinsCitation256. Since the catalytic domains are so similar, ATP-competitive inhibitors are ineffective against AKT isozymes and very marginally selective for closely related kinasesCitation254. A future goal is to create ATP-competitive inhibitors that specifically inhibit specific subtypes of AKT. The following is a list of some of the ATP-competitive inhibitors that have reached the clinical stage. The structures of AKT competitive inhibitors are shown in .
Figure 9. Structures of AKT competitive inhibitors.
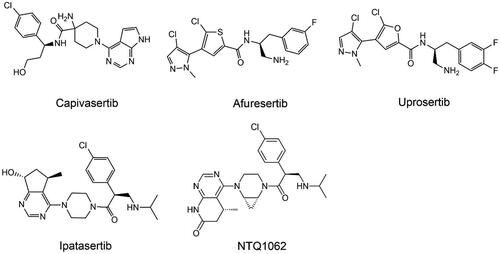
Capivasertib/AZD5363
Capivasertib is a potent oral ATP-competitive inhibitor developed by AstraZeneca that can effectively inhibit all isoforms of AKT. Capivasertib has a limited single-agent effect, but it is more effective when combined with traditional chemotherapy, and can be used as a sensitiser for tumour chemotherapyCitation272. Several trials have confirmed the effectiveness of combination drugs. Capivasertib has shown promising efficacy in breast cancer and has significant activity against AKT1-E17K-mutated cancers. The median PFS of TNBC patients receiving treatment with capivasertib and paclitaxel was 5.9 months. In PIK3CA/AKT1/PTEN-altered tumours, this regimen was significantly beneficial, with a median PFS of 9.3 monthsCitation273. When capivasertib was coupled with fulvestrant in patients with resistant breast cancer, the capivasertib group considerably surpassed the placebo group in terms of median PFS (10.3 months and 4.8 months, respectively). As a result, a relevant phase III trial is plannedCitation274. Furthermore, AKT signalling overexpression causes radioresistance in oral squamous cell carcinoma (OSCC) cells, and capivasertib encapsulated in new nanoparticles can sensitise radioresistant OSCC cells to IRCitation275. Therefore, combining Capivasertib with nanoparticles is an effective strategy for treating OSCC patients. Currently, research on the combination therapy of Capivasertib is ongoing.
Afuresertib/GSK2110183
Afuresertib is an oral ATP-competitive inhibitor that inhibits all isoforms of AKT. Afuresertib showed single-agent activity against hematological malignancies, particularly Multiple myeloma (MM), with 3 of 34 MM patients achieving PR (9%), and the others maintaining long-term disease stabilityCitation276. Afuresertib plus carboplatin and paclitaxel is an attractive tactic for treating ovarian cancer. The ORR and median PFS were 32% and 7.1 months, respectivelyCitation277. In patients with solid tumours, continuous daily dosing of afuresertib plus trametinib was poorly tolerated. Therefore, further study of intermittent dosing regimens is requiredCitation278. In the phase II study in patients with CLL, the ORR of afuresertib in combination with ofatumumab was 50%, with a median PFS of 8.5 monthsCitation279. Although combination therapy has certain activities and good tolerability, it cannot demonstrate greater benefits than single-drug treatment with afuresertib. At present, new drug combinations related to afuresertib are under intensive research.
Uprosertib/GSK2141795/GSK795
Uprosertib is an orally active N-alkylpyrazole AKT inhibitor that induces tumour cell growth arrest in vitro, enhances cisplatin-induced apoptosis, and reduces tumour volume in vivo when combined with platinum-based cytostaticsCitation280. Therefore, AKT inhibition has clinical therapeutic potential for platinum-resistant cancers. Clinical trials have used dual suppression of PI3K and RAS signalling. In patients with leukaemia, endometrial cancer, and melanoma, uprosertib in combination with trametinib displayed modest clinical activity, indicating the need for more effective therapeutic optionsCitation281–283. When trametinib was combined with uprosertib in patients with recurrent cervical cancer, 8 of 14 patients had stable disease, and 3 progressed to the best response. This combination is feasible in cervical cancer patients, but dose reductions are still required to reduce adverse eventsCitation284. The investigators will further explore more effective combination therapy strategies related to uprosertib.
Ipatasertib/GDC0068/RG7440
Ipatasertib is a highly selective oral ATP-competitive inhibitor that targets all subtypes of AKTCitation285. At present, ipatasertib is mainly developed for the treatment of solid tumours, and a number of clinical trials have been conducted to evaluate its efficacy. The combination of ipatasertib and abiraterone has entered a phase III clinical study in patients with PTEN-deficient mCRPC287. Ipatasertib inhibits PI3K-AKT-mTOR signalling, whereas abiraterone suppresses androgen signalling. The combination therapy increased radiographic PFS by up to 18.5 monthsCitation286. Patients with TNBC were enrolled in the phase II trial of ipatasertib plus paclitaxelCitation286. When contrasted to the placebo group, the ipatasertib group had a slightly longer median PFS (6.2 months vs. 4.9 months). This was also true in 48 patients with PTEN-low tumours (6.2 months vs. 3.7 months)Citation287. In a nutshell, ipatasertib has surprising potential in the treatment of breast and prostate cancers.
NTQ1062
NTQ1062 was discovered based on the ipatasertib structural skeleton by optimising hinge adhesive regions and connectionsCitation288. NTQ1062 showed good potency and pharmacokinetic properties with IC50 values of 0.4, 6.3, and 0.1 nM for Akt1/Akt2/Akt3, respectivelyCitation288. Given that NTQ1062 has demonstrated promising anticancer efficacy in vitro and in vivo, phase I clinical studies of NTQ1062 are currently underway (CTR20211999).
Conclusions and future outlooks
In the past few years, PI3K and AKT have become the research objects of targeted cancer therapy due to their crucial roles in the PI3K/AKT/mTOR signalling cascade. At present, a variety of inhibitors have entered clinical research, and they have shown good prospects in clinical trials. Among them, five drugs targeting PI3K have been approved by the FDA for marketing. PIKK and PI3K are two related kinase families with identical origins and very similar kinase structural domains but distinct characteristics. Extensive research on these kinases has demonstrated that inhibiting PIKK family kinases is useful in overcoming resistance to cancer therapy, and so the design and implementation of PIKK inhibitors could be a novel technique for effective cancer and other illness treatment. LY294002, the first-generation PI3K inhibitor, may efficiently inhibit PI3K, mTOR, DNA-PK, and ATM. Wortmannin can inhibit all four isoforms of PI3K, as well as mTOR and DNA-PK. Due to their poor physicochemical properties or adverse effects, both inhibitors have been excluded from further biological evaluation and clinical trials. They are, however, useful chemical tools for the development of other PI3K and PIKK inhibitors. In recent years, as the understanding of PIKK family kinases has deepened, inhibitors related to DNA damage response and mTOR inhibitors have started to appear prevalently in the public spotlight. A broad number of ATM, ATR, DNA-PK, and mTOR inhibitors have entered clinical trials, but only mTOR has an authorised medication on the market.
At present, safety and resistance concerns are still key obstacles to the development of this category of medications, making it difficult to make a splash in the oncology drug market. Cancers can rapidly adapt to a single chemotherapeutic drug due to the reactivation of blocked pathways or the activation of parallel signalling pathways caused by the initiation of compensatory mechanisms. Combining additional medications is thus regarded as a very interesting research avenue. These medications are typically administered in two ways: orally and intravenously. To improve anti-cancer efficacy, the most appropriate drug delivery method must be selected based on the pharmacokinetic characteristics of different drugs. Furthermore, nanodrug delivery systems (NDDS) can help overcome delivery barriers, reduce chemotherapy side effects, and achieve precision treatment, making NDDS an effective treatment strategy when combined with chemotherapy drugs. In addition, compared to continuous administration, intermittent administration of PI3K-related inhibitors is not only well tolerated but also more effective as an anticancer therapy compared to continuous dosing. Tumour invasion and progression are of considerable complexity. In recent years, multi-targeted therapy has been considered an attractive means of treating cancer to circumvent drug resistance and improve the safety of treatment. For example, PI3K/mTOR, PI3K/HDAC, PI3K/AKT/mTOR, and PDGFR/PI3K/EGFR/VEGFR2 multi-target inhibitors were developed with the hope of effectively improving anti-tumour activity. As approval of anticancer drugs becomes more difficult, developers are forced to reconsider safety issues, thus facilitating the re-optimisation of inhibitors. The safety and target selectivity of drugs can be enhanced through the development of combination therapies, optimisation of drug delivery routes and regimens, and the design of drug structures with the aim of improving the safety and resistance of drugs.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Noorolyai S, Shajari N, Baghbani E, Sadreddini S, Baradaran B. The relation between PI3K/AKT signalling pathway and cancer. Gene. 2019;698:120–128.
- Yudushkin I. Control of Akt activity and substrate phosphorylation in cells. IUBMB Life. 2020;72(6):1115–1125.
- Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z, Li W, Hu J, Lu C, Liu Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020;11(9):797.
- Smith GCM, Jackson SP. Chapter 77 – The PIKK family of protein kinases. In: Bradshaw RA, Dennis EA, editors. Handbook of cell signaling. 2nd ed. San Diego: Academic Press; 2010. p. 575–580.
- Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66(6):801–817.
- Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. 2019;12(1):71.
- Grimson A, O’Connor S, Newman CL, Anderson P. SMG-1 is a phosphatidylinositol kinase-related protein kinase required for nonsense-mediated mRNA Decay in Caenorhabditis elegans. Mol Cell Biol. 2004;24(17):7483–7490.
- Kamp JA, Lemmens BBLG, Romeijn RJ, González-Prieto R, Olsen JV, Vertegaal ACO, van Schendel R, Tijsterman M. THO complex deficiency impairs DNA double-strand break repair via the RNA surveillance kinase SMG-1. Nucleic Acids Res. 2022;50(11):6235–6250.
- Elías-Villalobos A, Fort P, Helmlinger D. New insights into the evolutionary conservation of the sole PIKK pseudokinase Tra1/TRRAP. Biochem Soc Trans. 2019;47(6):1597–1608.
- Knutson BA, Hahn S. Domains of Tra1 important for activator recruitment and transcription coactivator functions of SAGA and NuA4 complexes. Mol Cell Biol. 2011;31(4):818–831.
- Fattahi S, Amjadi-Moheb F, Tabaripour R, Ashrafi GH, Akhavan-Niaki H. PI3K/AKT/mTOR signaling in gastric cancer: epigenetics and beyond. Life Sci. 2020;262:118513.
- Papadimitrakopoulou V. Development of PI3K/AKT/mTOR pathway inhibitors and their application in personalized therapy for non–small-cell lung cancer. J Thorac Oncol. 2012;7(8):1315–1326.
- Ciruelos Gil EM. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat Rev. 2014;40(7):862–871.
- Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, Naing A, Falchook GS, Moroney JW, Piha-Paul SA, et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther. 2011;10(3):558–565.
- Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS, Tsimberidou AM, Stepanek VM, Moulder SL, Lee JJ, et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014;6(2):377–387.
- Hyman DM, Smyth LM, Donoghue MTA, Westin SN, Bedard PL, Dean EJ, Bando H, El-Khoueiry AB, Pérez-Fidalgo JA, Mita A, et al. AKT inhibition in solid tumors with AKT1 mutations. J Clin Oncol. 2017;35(20):2251–2259.
- Kwiatkowski DJ, Choueiri TK, Fay AP, Rini BI, Thorner AR, de Velasco G, Tyburczy ME, Hamieh L, Albiges L, Agarwal N, et al. Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2016;22(10):2445–2452.
- Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–724.
- Cheng B, Pan W, Xing Y, Xiao Y, Chen J, Xu Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur J Med Chem. 2022;230:114109.
- Kantidze OL, Velichko AK, Luzhin AV, Petrova NV, Razin SV. Synthetically lethal interactions of ATM, ATR, and DNA-PKcs. Trends Cancer. 2018;4(11):755–768.
- Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016;127(5):582–595.
- da Costa AABA, Chowdhury D, Shapiro GI, D’Andrea AD, Konstantinopoulos PA. Targeting replication stress in cancer therapy. Nat Rev Drug Discov. 2023;22(1):38–58.
- Djordjevic S, Driscoll PC. Structural insight into substrate specificity and regulatory mechanisms of phosphoinositide 3-kinases. Trends Biochem Sci. 2002;27(8):426–432.
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644.
- Goncalves MD, Hopkins BD, Cantley LC. Phosphatidylinositol 3-kinase, growth disorders, and cancer. N Engl J Med. 2018;379(21):2052–2062.
- Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15(1):7–24.
- Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F, Stivala F, McCubrey JA, Libra M. PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle. 2009;8(9):1352–1358.
- Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008;105(7):2652–2657.
- Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3(4):317–330.
- Cao S, Cao R, Liu X, Luo X, Zhong W. Design, synthesis and biological evaluation of novel benzothiazole derivatives as selective PI3Kβ inhibitors. Molecules. 2016;21(7):876.
- Bilanges B, Posor Y, Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol. 2019;20(9):515–534.
- Gulluni F, De Santis MC, Margaria JP, Martini M, Hirsch E. Class II PI3K functions in cell biology and disease. Trends Cell Biol. 2019;29(4):339–359.
- Margaria JP, Ratto E, Gozzelino L, Li H, Hirsch E. Class II PI3Ks at the intersection between signal transduction and membrane trafficking. Biomolecules. 2019;9(3)
- Jaber N, Zong WX. Class III PI3K Vps34: essential roles in autophagy, endocytosis, and heart and liver function. Ann N Y Acad Sci. 2013;1280:48–51.
- Kong D, Yamori T. Phosphatidylinositol 3-kinase inhibitors: promising drug candidates for cancer therapy. Cancer Sci. 2008;99(9):1734–1740.
- Brana I, Siu LL. Clinical development of phosphatidylinositol 3-kinase inhibitors for cancer treatment. BMC Med. 2012;10:161.
- Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell. 2017;170(4):605–635.
- Chen CY, Chen J, He L, Stiles BL. PTEN: tumor suppressor and metabolic regulator. Front Endocrinol. 2018;9:338.
- Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20(2):74–88.
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101.
- Shariati M, Meric-Bernstam F. Targeting AKT for cancer therapy. Expert Opin Investig Drugs. 2019;28(11):977–988.
- Mishra R, Patel H, Alanazi S, Kilroy MK, Garrett JT. PI3K inhibitors in cancer: clinical implications and adverse effects. Int J Mol Sci. 2021;22(7):3464.
- Wang X, Ding J, Meng LH. PI3K isoform-selective inhibitors: next-generation targeted cancer therapies. Acta Pharmacol Sin. 2015;36(10):1170–1176.
- Buchanan CM, Lee KL, Shepherd PR. For better or worse: the potential for dose limiting the on-target toxicity of PI 3-kinase inhibitors. Biomolecules. 2019;9(9)
- Xing J, Yang J, Gu Y, Yi J. Research update on the anticancer effects of buparlisib. Oncol Lett. 2021;21(4):266.
- de Gooijer MC, Zhang P, Buil LCM, Çitirikkaya CH, Thota N, Beijnen JH, van Tellingen O. Buparlisib is a brain penetrable pan-PI3K inhibitor. Sci Rep. 2018;8(1):10784.
- Wen PY, Touat M, Alexander BM, Mellinghoff IK, Ramkissoon S, McCluskey CS, Pelton K, Haidar S, Basu SS, Gaffey SC, et al. Buparlisib in patients with recurrent glioblastoma harboring phosphatidylinositol 3-kinase pathway activation: an open-label, multicenter, multi-arm, phase II trial. J Clin Oncol. 2019;37(9):741–750.
- Rosenthal M, Clement PM, Campone M, Gil-Gil MJ, DeGroot J, Chinot O, Idbaih A, Gan H, Raizer J, Wen PY, et al. Phase I, open-label, multicentre study of buparlisib in combination with temozolomide or with concomitant radiation therapy and temozolomide in patients with newly diagnosed glioblastoma. ESMO Open. 2020;5(4):e000673.
- Baselga J, Im S-A, Iwata H, Cortés J, De Laurentiis M, Jiang Z, Arteaga CL, Jonat W, Clemons M, Ito Y, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(7):904–916.
- Kim HR, Kang HN, Yun MR, Ju KY, Choi JW, Jung DM, Pyo KH, Hong MH, Ahn M-J, Sun J-M, et al. Mouse-human co-clinical trials demonstrate superior anti-tumour effects of buparlisib (BKM120) and cetuximab combination in squamous cell carcinoma of head and neck. Br J Cancer. 2020;123(12):1720–1729.
- O’Brien C, Wallin JJ, Sampath D, GuhaThakurta D, Savage H, Punnoose EA, Guan J, Berry L, Prior WW, Amler LC, et al. Predictive biomarkers of sensitivity to the phosphatidylinositol 3’ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin Cancer Res. 2010;16(14):3670–3683.
- Schmid P, Pinder SE, Wheatley D, Macaskill J, Zammit C, Hu J, Price R, Bundred N, Hadad S, Shia A, et al. Phase II randomized preoperative window-of-opportunity study of the PI3K Inhibitor pictilisib plus anastrozole compared with anastrozole alone in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2016;34(17):1987–1994.
- Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, Yardley DA, Melichar B, Forero-Torres A, Lee SC, et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17(6):811–821.
- Vuylsteke P, Huizing M, Petrakova K, Roylance R, Laing R, Chan S, Abell F, Gendreau S, Rooney I, Apt D, et al. Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3-kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor-positive, HER2-negative, locally recurrent, or metastatic breast cancer: interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann Oncol. 2016;27(11):2059–2066.
- Leong S, Moss RA, Bowles DW, Ware JA, Zhou J, Spoerke JM, Lackner MR, Shankar G, Schutzman JL, van der Noll R, et al. A phase I dose-escalation study of the safety and pharmacokinetics of pictilisib in combination with erlotinib in patients with advanced solid tumors. Oncologist. 2017;22(12):1491–1499.
- Shapiro GI, LoRusso P, Kwak E, Pandya S, Rudin CM, Kurkjian C, Cleary JM, Pilat MJ, Jones S, de Crespigny A, et al. Phase Ib study of the MEK inhibitor cobimetinib (GDC-0973) in combination with the PI3K inhibitor pictilisib (GDC-0941) in patients with advanced solid tumors. Invest New Drugs. 2020;38(2):419–432.
- Yamamoto N, Fujiwara Y, Tamura K, Kondo S, Iwasa S, Tanabe Y, Horiike A, Yanagitani N, Kitazono S, Inatani M, et al. Phase Ia/Ib study of the pan-class I PI3K inhibitor pictilisib (GDC-0941) administered as a single agent in Japanese patients with solid tumors and in combination in Japanese patients with non-squamous non-small cell lung cancer. Invest New Drugs. 2017;35(1):37–46.
- Howes AL, Chiang GG, Lang ES, Ho CB, Powis G, Vuori K, Abraham RT. The phosphatidylinositol 3-kinase inhibitor, PX-866, is a potent inhibitor of cancer cell motility and growth in three-dimensional cultures. Mol Cancer Ther. 2007;6(9):2505–2514.
- Bowles DW, Kochenderfer M, Cohn A, Sideris L, Nguyen N, Cline-Burkhardt V, Schnadig I, Choi M, Nabell L, Chaudhry A, et al. A randomized, phase II trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with metastatic colorectal carcinoma. Clin Colorectal Cancer. 2016;15(4):337.e2–344.e2.
- Jimeno A, Bauman JE, Weissman C, Adkins D, Schnadig I, Beauregard P, Bowles DW, Spira A, Levy B, Seetharamu N, et al. A randomized, phase 2 trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Oral Oncol. 2015;51(4):383–388.
- Jimeno A, Shirai K, Choi M, Laskin J, Kochenderfer M, Spira A, Cline-Burkhardt V, Winquist E, Hausman D, Walker L, et al. A randomized, phase II trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Ann Oncol. 2015;26(3):556–561.
- Levy B, Spira A, Becker D, Evans T, Schnadig I, Camidge DR, Bauman JE, Hausman D, Walker L, Nemunaitis J, et al. A randomized, phase 2 trial of Docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic non-small-cell lung cancer. J Thorac Oncol. 2014;9(7):1031–1035.
- Peek GW, Tollefsbol TO. Combinatorial PX-866 and raloxifene decrease rb phosphorylation, cyclin E2 transcription, and proliferation of MCF-7 breast cancer cells. J Cell Biochem. 2016;117(7):1688–1696.
- Harder BG, Peng S, Sereduk CP, Sodoma AM, Kitange GJ, Loftus JC, Sarkaria JN, Tran NL. Inhibition of phosphatidylinositol 3-kinase by PX-866 suppresses temozolomide-induced autophagy and promotes apoptosis in glioblastoma cells. Mol Med. 2019;25(1):49.
- Foster P, Yamaguchi K, Hsu PP, Qian F, Du X, Wu J, Won K-A, Yu P, Jaeger CT, Zhang W, et al. The selective PI3K inhibitor XL147 (SAR245408) inhibits tumor growth and survival and potentiates the activity of chemotherapeutic agents in preclinical tumor models. Mol Cancer Ther. 2015;14(4):931–940.
- Edelman G, Rodon J, Lager J, Castell C, Jiang J, Van Allen EM, Wagle N, Lindeman NI, Sholl LM, Shapiro GI. Phase I trial of a tablet formulation of pilaralisib, a pan-class I PI3K inhibitor, in patients with advanced solid tumors. Oncologist. 2018;23(4):401–438.
- Shapiro GI, Rodon J, Bedell C, Kwak EL, Baselga J, Braña I, Pandya SS, Scheffold C, Laird AD, Nguyen LT, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2014;20(1):233–245.
- Blackwell K, Burris H, Gomez P, Lynn Henry N, Isakoff S, Campana F, Gao L, Jiang J, Macé S, Tolaney SM. Phase I/II dose-escalation study of PI3K inhibitors pilaralisib or voxtalisib in combination with letrozole in patients with hormone-receptor-positive and HER2-negative metastatic breast cancer refractory to a non-steroidal aromatase inhibitor. Breast Cancer Res Treat. 2015;154(2):287–297.
- Brown JR, Davids MS, Rodon J, Abrisqueta P, Kasar SN, Lager J, Jiang J, Egile C, Awan FT. Phase I trial of the pan-pi3k inhibitor pilaralisib (SAR245408/XL147) in patients with chronic lymphocytic leukemia (CLL) or relapsed/refractory lymphoma. J Clin Cancer Res. 2015;21(14):3160–3169.
- Matulonis U, Vergote I, Backes F, Martin LP, McMeekin S, Birrer M, Campana F, Xu Y, Egile C, Ghamande S. Phase II study of the PI3K inhibitor pilaralisib (SAR245408; XL147) in patients with advanced or recurrent endometrial carcinoma. Gynecol Oncol. 2015;136(2):246–253.
- Wheler J, Mutch D, Lager J, Castell C, Liu L, Jiang J, Traynor AM. Phase I dose-escalation study of pilaralisib (SAR245408, XL147) in combination with paclitaxel and carboplatin in patients with solid tumors. Oncologist. 2017;22(4):377–337.
- Hayashida Y, Ikeda Y, Sawada K, Kawai K, Kato T, Kakehi Y, Araki N. Invention of a novel photodynamic therapy for tumors using a photosensitizing PI3K inhibitor. Int J Cancer. 2016;139(3):700–711.
- Alizadeh SR, Hashemi SM. Development and therapeutic potential of 2-aminothiazole derivatives in anticancer drug discovery. Med Chem Res. 2021;30(4):771–806.
- André F, Ciruelos EM, Juric D, Loibl S, Campone M, Mayer IA, Rubovszky G, Yamashita T, Kaufman B, Lu Y-S, et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: final overall survival results from SOLAR-1. Ann Oncol. 2021;32(2):208–217.
- Nunnery SE, Mayer IA. Management of toxicity to isoform α-specific PI3K inhibitors. Ann Oncol. 2019;30(Suppl 10):x21–x26.
- Markham A. Alpelisib: first global approval. Drugs. 2019;79(11):1249–1253.
- Batalini F, Xiong N, Tayob N, Polak M, Eismann J, Cantley LC, Shapiro GI, Adalsteinsson V, Winer EP, Konstantinopoulos PA, et al. Phase 1b clinical trial with alpelisib plus olaparib for patients with advanced triple-negative breast cancer. Clin Cancer Res. 2022;28(8):1493–1499.
- Pérez-Fidalgo JA, Criscitiello C, Carrasco E, Regan MM, Di Leo A, Ribi K, Adam V, Bedard PL. A phase III trial of alpelisib + trastuzumab ± fulvestrant versus trastuzumab + chemotherapy in HER2+ PIK3CA-mutated breast cancer. Future Oncol. 2022;18(19):2339–2349.
- Hanan EJ, Braun M-G, Heald RA, MacLeod C, Chan C, Clausen S, Edgar KA, Eigenbrot C, Elliott R, Endres N, et al. Discovery of GDC-0077 (Inavolisib), a highly selective inhibitor and degrader of mutant PI3Kα. J Med Chem. 2022;65(24):16589–16621.
- Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature. 2018;560(7719):499–503.
- Song KW, Edgar KA, Hanan EJ, Hafner M, Oeh J, Merchant M, Sampath D, Nannini MA, Hong R, Phu L, et al. RTK-dependent inducible degradation of mutant PI3Kα drives GDC-0077 (Inavolisib) efficacy. Cancer Discov. 2022;12(1):204–219.
- Schmid P, Bedard P, Jhaveri K, Cervantes A, Gambardella V, Hamilton E, Italiano A, Kalinsky K, Krop IE, Oliveira M, et al. A phase I/Ib study evaluating GDC-0077 (inavolisib) + palbociclib (palbo) + fulvestrant in patients (pts) with PIK3CA-mutant (mut), hormone receptor-positive/HER2-negative metastatic breast cancer (HR+/HER2− mBC). Senologie – Zeitschrift für Mammadiagnostik und -therapie. 2021;18(02): e36–e37.
- Mateo J, Ganji G, Lemech C, Burris HA, Han S-W, Swales K, Decordova S, DeYoung MP, Smith DA, Kalyana-Sundaram S, et al. A first-time-in-human study of GSK2636771, a phosphoinositide 3 kinase beta-selective inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2017;23(19):5981–5992.
- Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A. 2008;105(35):13057–13062.
- Zhang Z, Liu J, Wang Y, Tan X, Zhao W, Xing X, Qiu Y, Wang R, Jin M, Fan G, et al. Phosphatidylinositol 3-kinase β and δ isoforms play key roles in metastasis of prostate cancer DU145 cells. FASEB J. 2018;32(11):5967–5975.
- Sarker D, Dawson NA, Aparicio AM, Dorff TB, Pantuck AJ, Vaishampayan UN, Henson L, Vasist L, Roy-Ghanta S, Gorczyca M, et al. A phase I, open-label, dose-finding study of GSK2636771, a PI3Kβ inhibitor, administered with enzalutamide in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2021;27(19):5248–5257.
- Burton E, Ileana Dumbrava EE, Peng W, Milton D, Amaria R, Mcquade G, Glitza IC, Hong DS, Patel S, Rodon J, et al. 1085P Ph I/II study of PI3K-β inhibitor GSK2636771 (G771) in combination with pembrolizumab (P) in patients (pts) with PTEN loss and melanoma or other advanced solid tumors. Ann Oncol. 2021;32(S5):S899.
- Markham A. Idelalisib: first global approval. Drugs. 2014;74(14):1701–1707.
- Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997–1007.
- Sharman JP, Coutre SE, Furman RR, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn IW, et al. Final results of a randomized, phase III study of rituximab with or without idelalisib followed by open-label idelalisib in patients with relapsed chronic lymphocytic leukemia. J Clin Oncol. 2019;37(16):1391–1402.
- Tarantelli C, Argnani L, Zinzani PL, Bertoni F. PI3Kδ inhibitors as immunomodulatory agents for the treatment of lymphoma patients. Cancers. 2021;13(21):5535.
- Ghia P, Pluta A, Wach M, Lysak D, Kozak T, Simkovic M, Kaplan P, Kraychok I, Illes A, de la Serna J, et al. ASCEND: phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol. 2020;38(25):2849–2861.
- Lampson BL, Kasar SN, Matos TR, Morgan EA, Rassenti L, Davids MS, Fisher DC, Freedman AS, Jacobson CA, Armand P, et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood. 2016;128(2):195–203.
- Coutré SE, Barrientos JC, Brown JR, de Vos S, Furman RR, Keating MJ, Li D, O’Brien SM, Pagel JM, Poleski MH, et al. Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma. 2015;56(10):2779–2786.
- Deng C, Lipstein MR, Scotto L, Jirau Serrano XO, Mangone MA, Li S, Vendome J, Hao Y, Xu X, Deng S-X, et al. Silencing c-Myc translation as a therapeutic strategy through targeting PI3Kδ and CK1ε in hematological malignancies. Blood. 2017;129(1):88–99.
- Davids MS, O’Connor OA, Jurczak W, Samaniego F, Fenske TS, Zinzani PL, Patel MR, Ghosh N, Cheson BD, Derenzini E, et al. Integrated safety analysis of umbralisib, a dual PI3Kδ/CK1ε inhibitor, in relapsed/refractory lymphoid malignancies. Blood Adv. 2021;5(23):5332–5343.
- Dhillon S, Keam SJ. Umbralisib. Drugs. 2021;81(7):857–866.
- Ryan J, Wojciech J, Flinn IW, Grosicki S, Giannopoulos K, Wróbel T, Zafar SF, Cultrera JL, Kambhampati S, Danilov AV, et al. Efficacy and safety of Ublituximab in combination with umbralisib (U2) in patients with chronic lymphocytic leukemia (CLL) By treatment status: a sub-analysis of the phase 3 unity-CLL study. Blood. 2021;138(S1):3726.
- Wu X, Xu Y, Liang Q, Yang X, Huang J, Wang J, Zhang H, Shi J. Recent advances in dual PI3K/mTOR inhibitors for tumour treatment. Front Pharmacol. 2022;13:875372.
- Zhou HY, Huang SL. Current development of the second generation of mTOR inhibitors as anticancer agents. Chin J Cancer. 2012;31(1):8–18.
- Salazar R, Garcia-Carbonero R, Libutti SK, Hendifar AE, Custodio A, Guimbaud R, Lombard-Bohas C, Ricci S, Klümpen H-J, Capdevila J, et al. Phase II study of BEZ235 versus everolimus in patients with mammalian target of rapamycin inhibitor-naïve advanced pancreatic neuroendocrine tumors. Oncologist. 2018;23(7):766–e790.
- Carlo MI, Molina AM, Lakhman Y, Patil S, Woo K, DeLuca J, Lee C-H, Hsieh JJ, Feldman DR, Motzer RJ, et al. A phase Ib study of BEZ235, a dual inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin (mTOR), in patients with advanced renal cell carcinoma. Oncologist. 2016;21(7):787–788.
- Wei XX, Hsieh AC, Kim W, Friedlander T, Lin AM, Louttit M, Ryan CJ. A phase I study of abiraterone acetate combined with BEZ235, a dual PI3K/mTOR inhibitor, in metastatic castration resistant prostate cancer. Oncologist. 2017;22(5):503–543.
- Tian L, Qiao Y, Lee P, Wang L, Chang A, Ravi S, Rogers TA, Lu L, Singhana B, Zhao J, et al. Antitumor efficacy of liposome-encapsulated NVP-BEZ 235 in combination with irreversible electroporation. Drug Deliv. 2018;25(1):668–678.
- Munster P, Aggarwal R, Hong D, Schellens JHM, van der Noll R, Specht J, Witteveen PO, Werner TL, Dees EC, Bergsland E, et al. First-in-human phase I study of GSK2126458, an oral pan-class I phosphatidylinositol-3-kinase inhibitor, in patients with advanced solid tumor malignancies. Clin Cancer Res. 2016;22(8):1932–1939.
- Grilley-Olson JE, Bedard PL, Fasolo A, Cornfeld M, Cartee L, Razak ARA, Stayner L-A, Wu Y, Greenwood R, Singh R, et al. Phase Ib dose-escalation study of the MEK inhibitor trametinib in combination with the PI3K/mTOR inhibitor GSK2126458 in patients with advanced solid tumors. Invest New Drugs. 2016;34(6):740–749.
- Du J, Chen F, Yu J, Jiang L, Zhou M. The PI3K/mTOR inhibitor ompalisib suppresses nonhomologous end joining and sensitizes cancer cells to radio- and chemotherapy. Mol Cancer Res. 2021;19(11):1889–1899.
- Lukey PT, Harrison SA, Yang S, Man Y, Holman BF, Rashidnasab A, Azzopardi G, Grayer M, Simpson JK, Bareille P, et al. A randomised, placebo-controlled study of omipalisib (PI3K/mTOR) in idiopathic pulmonary fibrosis. Eur Respir J. 2019;53(3):1801992.
- Murase Y, Hosoya K, Sato T, Kim S, Okumura M. Antitumor activity of the dual PI3K/mTOR inhibitor gedatolisib and the involvement of ABCB1 in gedatolisib resistance in canine tumor cells. Oncol Rep. 2022;47(4):61.
- Wainberg ZA, Alsina M, Soares HP, Braña I, Britten CD, Del Conte G, Ezeh P, Houk B, Kern KA, Leong S, et al. A multi-arm phase I study of the PI3K/mTOR inhibitors PF-04691502 and gedatolisib (PF-05212384) plus irinotecan or the MEK inhibitor PD-0325901 in advanced cancer. Target Oncol. 2017;12(6):775–785.
- Del Campo JM, Birrer M, Davis C, Fujiwara K, Gollerkeri A, Gore M, Houk B, Lau S, Poveda A, González-Martín A, et al. A randomized phase II non-comparative study of PF-04691502 and gedatolisib (PF-05212384) in patients with recurrent endometrial cancer. Gynecol Oncol. 2016;142(1):62–69.
- Colombo I, Genta S, Martorana F, Guidi M, Frattini M, Samartzis EP, Brandt S, Gaggetta S, Moser L, Pascale M, et al. Phase I dose-escalation study of the dual PI3K-mTORC1/2 inhibitor gedatolisib in combination with paclitaxel and carboplatin in patients with advanced solid tumors. Clin Cancer Res. 2021;27(18):5012–5019.
- Wallin JJ, Edgar KA, Guan J, Berry M, Prior WW, Lee L, Lesnick JD, Lewis C, Nonomiya J, Pang J, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011;10(12):2426–2436.
- Dolly SO, Wagner AJ, Bendell JC, Kindler HL, Krug LM, Seiwert TY, Zauderer MG, Lolkema MP, Apt D, Yeh R-F, et al. Phase I study of apitolisib (GDC-0980), dual phosphatidylinositol-3-kinase and mammalian target of rapamycin kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2016;22(12):2874–2884.
- Powles T, Lackner MR, Oudard S, Escudier B, Ralph C, Brown JE, Hawkins RE, Castellano D, Rini BI, Staehler MD, et al. Randomized open-label phase II trial of apitolisib (GDC-0980), a novel inhibitor of the PI3K/mammalian target of rapamycin pathway, versus everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. 2016;34(14):1660–1668.
- Peng X, Liu Y, Zhu S, Peng X, Li H, Jiao W, Lin P, Zhang Z, Qiu Y, Jin M, et al. Co-targeting PI3K/Akt and MAPK/ERK pathways leads to an enhanced antitumor effect on human hypopharyngeal squamous cell carcinoma. J Cancer Res Clin Oncol. 2019;145(12):2921–2936.
- Jang DK, Lee YG, Chan Chae Y, Lee JK, Paik WH, Lee SH, Kim Y-T, Ryu JK. GDC-0980 (apitolisib) treatment with gemcitabine and/or cisplatin synergistically reduces cholangiocarcinoma cell growth by suppressing the PI3K/Akt/mTOR pathway. Biochem Biophys Res Commun. 2020;529(4):1242–1248.
- Omeljaniuk WJ, Krętowski R, Ratajczak-Wrona W, Jabłońska E, Cechowska-Pasko M. Novel dual PI3K/mTOR inhibitor, apitolisib (GDC-0980), inhibits growth and induces apoptosis in human glioblastoma cells. Int J Mol Sci. 2021;22(21):11511.
- Heffron TP, Ndubaku CO, Salphati L, Alicke B, Cheong J, Drobnick J, Edgar K, Gould SE, Lee LB, Lesnick JD, et al. Discovery of clinical development candidate GDC-0084, a brain penetrant inhibitor of PI3K and mTOR. ACS Med Chem Lett. 2016;7(4):351–356.
- Ippen FM, Alvarez-Breckenridge CA, Kuter BM, Fink AL, Bihun IV, Lastrapes M, Penson T, Schmidt SP, Wojtkiewicz GR, Ning J, et al. The dual PI3K/mTOR pathway inhibitor GDC-0084 achieves antitumor activity in PIK3CA-mutant breast cancer brain metastases. Clin Cancer Res. 2019;25(11):3374–3383.
- Wen PY, Cloughesy TF, Olivero AG, Morrissey KM, Wilson TR, Lu X, Mueller LU, Coimbra AF, Ellingson BM, Gerstner E, et al. First-in-human phase i study to evaluate the brain-penetrant PI3K/mTOR inhibitor GDC-0084 in patients with progressive or recurrent high-grade glioma. Clin Cancer Res. 2020;26(8):1820–1828.
- Wen P, de Groot J, Battiste J, Goldlust S, Damek D, Garner J, Simpson J, Olivero A, Cloughesy T. CTNI-44. Interim results of phase 2 study to evaluate PI3K/mTOR inhibitor paxalisib (GDC-0084) given to newly diagnosed glioblastoma patients with unmethylated O6-methylguanine-methyltransferase promoter. Neuro Oncol. 2020;22(Suppl 2):ii52–ii52.
- Wen PY, Omuro A, Ahluwalia MS, Fathallah-Shaykh HM, Mohile N, Lager JJ, Laird AD, Tang J, Jiang J, Egile C, et al. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro Oncol. 2015;17(9):1275–1283.
- Schram AM, Gandhi L, Mita MM, Damstrup L, Campana F, Hidalgo M, Grande E, Hyman DM, Heist RS. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br J Cancer. 2018;119(12):1471–1476.
- Brown JR, Hamadani M, Hayslip J, Janssens A, Wagner-Johnston N, Ottmann O, Arnason J, Tilly H, Millenson M, Offner F, et al. Voxtalisib (XL765) in patients with relapsed or refractory non-Hodgkin lymphoma or chronic lymphocytic leukaemia: an open-label, phase 2 trial. Lancet Haematol. 2018;5(4):e170–e180.
- Liu N, Rowley BR, Bull CO, Schneider C, Haegebarth A, Schatz CA, Fracasso PR, Wilkie DP, Hentemann M, Wilhelm SM, et al. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol Cancer Ther. 2013;12(11):2319–2330.
- Munoz J, Follows GA, Nastoupil LJ. Copanlisib for the treatment of malignant lymphoma: clinical experience and future perspectives. Target Oncol. 2021;16(3):295–308.
- Magagnoli M, Carlo-Stella C, Santoro A. Copanlisib for the treatment of adults with relapsed follicular lymphoma. Expert Rev Clin Pharmacol. 2020;13(8):813–823.
- Dreyling M, Santoro A, Mollica L, Leppä S, Follows G, Lenz G, Kim WS, Nagler A, Dimou M, Demeter J, et al. Long-term safety and efficacy of the PI3K inhibitor copanlisib in patients with relapsed or refractory indolent lymphoma: 2-year follow-up of the CHRONOS-1 study. Am J Hematol. 2020;95(4):362–371.
- Matasar MJ, Capra M, Özcan M, Lv F, Li W, Yañez E, Sapunarova K, Lin T, Jin J, Jurczak W, et al. Copanlisib plus rituximab versus placebo plus rituximab in patients with relapsed indolent non-Hodgkin lymphoma (CHRONOS-3): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021;22(5):678–689.
- Matasar MJ, Dreyling M, Leppä S, Santoro A, Pedersen M, Buvaylo V, Fletcher M, Childs BH, Zinzani PL. Feasibility of combining the phosphatidylinositol 3-kinase inhibitor copanlisib with rituximab-based immunochemotherapy in patients with relapsed indolent B-cell lymphoma. Clin Lymphoma Myeloma Leuk. 2021;21(11):e886–e894.
- Dreyling M, Santoro A, Mollica L, Leppä S, Follows GA, Lenz G, Kim WS, Nagler A, Panayiotidis P, Demeter J, et al. Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J Clin Oncol. 2017;35(35):3898–3905.
- Dreyling M, Morschhauser F, Bouabdallah K, Bron D, Cunningham D, Assouline SE, Verhoef G, Linton K, Thieblemont C, Vitolo U, et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann Oncol. 2017;28(9):2169–2178.
- Le T, Jerel D, Bryan LJ. Update on the role of copanlisib in hematologic malignancies. Ther Adv Hematol. 2021;12:20406207211006027.
- Yang Z, Liao J, Schumaker L, Carter-Cooper B, Lapidus RG, Fan X, Gaykalova DA, Mehra R, Cullen KJ, Dan H. Simultaneously targeting ErbB family kinases and PI3K in HPV-positive head and neck squamous cell carcinoma. Oral Oncol. 2022;131:105939.
- Damodaran S, Zhao F, Deming DA, Mitchell EP, Wright JJ, Gray RJ, Wang V, McShane LM, Rubinstein LV, Patton DR, et al. Phase II study of copanlisib in patients with tumors with PIK3CA mutations: results from the NCI-MATCH ECOG-ACRIN Trial (EAY131) subprotocol Z1F. J Clin Oncol. 2022;40(14):1552–1561.
- Vangapandu HV, Jain N, Gandhi V. Duvelisib: a phosphoinositide-3 kinase δ/γ inhibitor for chronic lymphocytic leukemia. Expert Opin Investig Drugs. 2017;26(5):625–632.
- O’Brien S, Patel M, Kahl BS, Horwitz SM, Foss FM, Porcu P, Jones J, Burger J, Jain N, Allen K, et al. Duvelisib, an oral dual PI3K-δ,γ inhibitor, shows clinical and pharmacodynamic activity in chronic lymphocytic leukemia and small lymphocytic lymphoma in a phase 1 study. Am J Hematol. 2018;93(11):1318–1326.
- Flinn IW, Miller CB, Ardeshna KM, Tetreault S, Assouline SE, Mayer J, Merli M, Lunin SD, Pettitt AR, Nagy Z, et al. DYNAMO: a phase II study of duvelisib (IPI-145) in patients with refractory indolent non-hodgkin lymphoma. J Clin Oncol. 2019;37(11):912–922.
- Flinn IW, Hillmen P, Montillo M, Nagy Z, Illés Á, Etienne G, Delgado J, Kuss BJ, Tam CS, Gasztonyi Z, et al. The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood. 2018;132(23):2446–2455.
- Blair HA. Duvelisib: first global approval. Drugs. 2018;78(17):1847–1853.
- Flinn IW, Cherry MA, Maris MB, Matous JV, Berdeja JG, Patel M. Combination trial of duvelisib (IPI-145) with rituximab or bendamustine/rituximab in patients with non-Hodgkin lymphoma or chronic lymphocytic leukemia. Am J Hematol. 2019;94(12):1325–1334.
- Lampson BL, Brown JR. The evolving use of phosphatidylinositol 3-kinase inhibitors for the treatment of chronic lymphocytic leukemia. Hematol Oncol Clin North Am. 2021;35(4):807–826.
- Iskierka-Jażdżewska E, Obracaj A, Urbaniak M, Robak T. New treatment options for newly-diagnosed and relapsed chronic lymphocytic leukemia. Curr Treat Options Oncol. 2022;23(6):775–795.
- Sabbah DA, Hajjo R, Bardaweel SK, Zhong HA. Phosphatidylinositol 3-kinase (PI3K) inhibitors: a recent update on inhibitor design and clinical trials (2016–2020). Expert Opin Ther Pat. 2021;31(10):877–892.
- Huen A, Haverkos BM, Zain J, Radhakrishnan R, Lechowicz MJ, Devata S, Korman NJ, Pinter-Brown L, Oki Y, Barde PJ, et al. Phase I/Ib study of tenalisib (RP6530), a dual PI3K δ/γ inhibitor in patients with relapsed/refractory T-cell lymphoma. J Cancers. 2020;12(8):2293.
- Iyer SP, Huen A, Ai WZ, Jagadeesh D, Lechowicz MJ, Okada C, Feldman TA, Ghione P, Alderuccio JP, Stevens DA, Kuzel TM, et al. Safety and efficacy of tenalisib given in combination with romidepsin in patients with relapsed/refractory T-cell lymphoma: final results from a phase I/II OPEN label multi-center study. J Blood. 2021;138(S1):1365.
- Makharadze T, Kiladze IZ, Dzagnidze G, Semionova-Peskova N, Katselashvili L, Vekua N, Routhu K, Barde P, Nair A. 224P Efficacy and safety of tenalisib, a PI3K delta/gamma and SIK3 inhibitor in patients with locally advanced or metastatic breast cancer: Results from a phase II study. Annals of Oncology. 2022;33:S640.
- Lempiäinen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. Embo J. 2009;28(20):3067–3073.
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434(7033):605–611.
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–484.
- Yamashita A, Kashima I, Ohno S. The role of SMG-1 in nonsense-mediated mRNA decay. Biochim Biophys Acta. 2005;1754(1–2):305–315.
- McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000;20(2):556–562.
- O’Connor MJ. Targeting the DNA Damage Response in Cancer. Molecular Cell. 2015;60(4):547–560.
- Rassool FV, Tomkinson AE. Targeting abnormal DNA double strand break repair in cancer. Cell Mol Life Sci. 2010;67(21):3699–3710.
- Qiu S, Huang J. MRN complex is an essential effector of DNA damage repair. J Zhejiang Univ Sci B. 2021;22(1):31–37.
- Zahid S, Seif El Dahan M, Iehl F, Fernandez-Varela P, Le Du MH, Ropars V, Charbonnier JB. The Multifaceted Roles of Ku70/80. Int J Mol Sci. 2021;22(8)
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300(5625):1542–1548.
- Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124(5):943–955.
- Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112.
- Yue X, Bai C, Xie D, Ma T, Zhou PK. DNA-PKcs: a multi-faceted player in DNA Damage response. Front Genet. 2020;11:607428.
- Deng S, Vlatkovic T, Li M, Zhan T, Veldwijk MR, Herskind C. Targeting the DNA damage response and DNA repair pathways to enhance radiosensitivity in colorectal cancer. Cancers. 2022;14(19):4874.
- ATM Inhibition Sensitizes ALT Neuroblastomas to Chemotherapy. Cancer Discov. 2021;11(10):2368.
- Ngoi NYL, Pham MM, Tan DSP, Yap TA. Targeting the replication stress response through synthetic lethal strategies in cancer medicine. Trends Cancer. 2021;7(10):930–957.
- Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife. 2012;1:e00047.
- Liu C, Wang X, Qin W, Tu J, Li C, Zhao W, Ma L, Liu B, Qiu H, Yuan X. Combining radiation and the ATR inhibitor berzosertib activates STING signaling and enhances immunotherapy via inhibiting SHP1 function in colorectal cancer. Cancer Commun. 2023;43(4):435–454.
- Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015;42(2):332–343.
- Reisländer T, Groelly FJ, Tarsounas M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol Cell. 2020;80(1):21–28.
- Lopez-Pelaez M, Young L, Vazquez-Chantada M, Nelson N, Durant S, Wilkinson RW, Poon E, Gaspar M, Valge-Archer V, Smith P, et al. Targeting DNA damage response components induces enhanced STING-dependent type-I IFN response in ATM deficient cancer cells and drives dendritic cell activation. Oncoimmunology. 2022;11(1):2117321.
- Mouw KW, Konstantinopoulos PA. From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD-L1 immune checkpoint activation. Br J Cancer. 2018;118(7):933–935.
- Kim D-H, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–175.
- Kim D-H, Sarbassov DD, Ali SM, Latek RR, Guntur KVP, Erdjument-Bromage H, Tempst P, Sabatini DM. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11(4):895–904.
- Wang L, Harris TE, Roth RA, Lawrence JC. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. 2007;282(27):20036–20044.
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–886.
- Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, Manning BD. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014;156(4):771–785.
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–1945.
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166(2):213–223.
- Sarbassov DD, Ali SM, Kim D-H, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–1302.
- Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976.
- Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, Sabatini DM. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16(18):1865–1870.
- Pearce LR, Huang X, Boudeau J, Pawłowski R, Wullschleger S, Deak M, Ibrahim AFM, Gourlay R, Magnuson MA, Alessi DR. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. 2007;405(3):513–522.
- Murugan AK. mTOR: role in cancer, metastasis and drug resistance. Semin Cancer Biol. 2019;59:92–111.
- Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10:31.
- Andrs M, Korabecny J, Jun D, Hodny Z, Bartek J, Kuca K. Phosphatidylinositol 3-kinase (PI3K) and phosphatidylinositol 3-kinase-related kinase (PIKK) inhibitors: importance of the morpholine ring. J Med Chem. 2015;58(1):41–71.
- Gorecki L, Andrs M, Rezacova M, Korabecny J. Discovery of ATR kinase inhibitor berzosertib (VX-970, M6620): Clinical candidate for cancer therapy. Pharmacol Ther. 2020;210:107518.
- Yap TA, O’Carrigan B, Penney MS, Lim JS, Brown JS, de Miguel Luken MJ, Tunariu N, Perez-Lopez R, Rodrigues DN, Riisnaes R, et al. Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol. 2020;38(27):3195–3204.
- Konstantinopoulos PA, Cheng S-C, Wahner Hendrickson AE, Penson RT, Schumer ST, Doyle LA, Lee EK, Kohn EC, Duska LR, Crispens MA, et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21(7):957–968.
- Baschnagel AM, Elnaggar JH, VanBeek HJ, Kromke AC, Skiba JH, Kaushik S, Abel L, Clark PA, Longhurst CA, Nickel KP, et al. ATR inhibitor M6620 (VX-970) enhances the effect of radiation in non-small cell lung cancer brain metastasis patient-derived xenografts. Mol Cancer Ther. 2021;20(11):2129–2139.
- Kim ST, Smith SA, Mortimer P, Loembé A-B, Cho H, Kim K-M, Smith C, Willis S, Irurzun-Arana I, Berges A, et al. Phase I study of ceralasertib (AZD6738), a novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer. Clin Cancer Res. 2021;27(17):4700–4709.
- Yap TA, Krebs MG, Postel-Vinay S, El-Khouiery A, Soria J-C, Lopez J, Berges A, Cheung SYA, Irurzun-Arana I, Goldwin A, et al. Ceralasertib (AZD6738), an oral ATR kinase inhibitor, in combination with carboplatin in patients with advanced solid tumors: a phase I study. Clin Cancer Res. 2021;27(19):5213–5224.
- Kim R, Kwon M, An M, Kim ST, Smith SA, Loembé AB, Mortimer PGS, Armenia J, Lukashchuk N, Shah N, et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Ann Oncol. 2022;33(2):193–203.
- Kwon M, Kim G, Kim R, Kim KT, Kim ST, Smith S, Mortimer PGS, Hong JY, Loembé AB, Irurzun-Arana I, et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced gastric cancer. J Immunother Cancer. 2022;10(7):e005041.
- Mahdi H, Hafez N, Doroshow D, Sohal D, Keedy V, Do KT, LoRusso P, Jürgensmeier J, Avedissian M, Sklar J, et al. Ceralasertib-mediated ATR inhibition combined with olaparib in advanced cancers harboring DNA damage response and repair alterations (olaparib combinations). JCO Precis Oncol. 2021;5:PO.20.00439.
- Shah PD, Wethington SL, Pagan C, Latif N, Tanyi J, Martin LP, Morgan M, Burger RA, Haggerty A, Zarrin H, et al. Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol Oncol. 2021;163(2):246–253.
- Roulston A, Zimmermann M, Papp R, Skeldon A, Pellerin C, Dumas-Bérube É, Dumais V, Dorich S, Fader LD, Fournier S, et al. RP-3500: a novel, potent, and selective ATR inhibitor that is effective in preclinical models as a monotherapy and in combination with PARP inhibitors. Mol Cancer Ther. 2022;21(2):245–256.
- Wengner AM, Siemeister G, Lücking U, Lefranc J, Wortmann L, Lienau P, Bader B, Bömer U, Moosmayer D, Eberspächer U, et al. The novel ATR inhibitor BAY 1895344 is efficacious as monotherapy and combined with DNA damage-inducing or repair-compromising therapies in preclinical cancer models. Mol Cancer Ther. 2020;19(1):26–38.
- Yap TA, Tan DSP, Terbuch A, Caldwell R, Guo C, Goh BC, Heong V, Haris NRM, Bashir S, Drew Y, et al. First-in-human trial of the oral ataxia telangiectasia and RAD3-related (ATR) inhibitor BAY 1895344 in patients with advanced solid tumors. Cancer Discov. 2021;11(1):80–91.
- Pike KG, Barlaam B, Cadogan E, Campbell A, Chen Y, Colclough N, Davies NL, de-Almeida C, Degorce SL, Didelot M, et al. The identification of potent, selective, and orally available inhibitors of ataxia telangiectasia mutated (ATM) kinase: the discovery of AZD0156 (8-{6-[3-(Dimethylamino)propoxy]pyridin-3-yl}-3-methyl-1-(tetrahydro-2 H-pyran-4-yl)-1,3-dihydro-2 H-imidazo[4,5- c]quinolin-2-one). J Med Chem. 2018;61(9):3823–3841.
- Riches LC, Trinidad AG, Hughes G, Jones GN, Hughes AM, Thomason AG, Gavine P, Cui A, Ling S, Stott J, et al. Pharmacology of the ATM inhibitor AZD0156: potentiation of irradiation and olaparib responses preclinically. Mol Cancer Ther. 2020;19(1):13–25.
- Qin T, Mullan B, Ravindran R, Messinger D, Siada R, Cummings JR, Harris M, Muruganand A, Pyaram K, Miklja Z, et al. ATRX loss in glioma results in dysregulation of cell-cycle phase transition and ATM inhibitor radio-sensitization. Cell Rep. 2022;38(2):110216.
- Durant ST, Zheng L, Wang Y, Chen K, Zhang L, Zhang T, Yang Z, Riches L, Trinidad AG, Fok JHL, et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci Adv. 2018;4(6):eaat1719.
- Ratz L, Brambillasca C, Bartke L, Huetzen MA, Goergens J, Leidecker O, Jachimowicz RD, van de Ven M, Proost N, Siteur B, et al. Combined inhibition of EZH2 and ATM is synthetic lethal in BRCA1-deficient breast cancer. Breast Cancer Res. 2022;24(1):41.
- Zenke FT, Zimmermann A, Sirrenberg C, Dahmen H, Kirkin V, Pehl U, Grombacher T, Wilm C, Fuchss T, Amendt C, et al. Pharmacologic inhibitor of DNA-PK, M3814, potentiates radiotherapy and regresses human tumors in mouse models. Mol Cancer Ther. 2020;19(5):1091–1101.
- Sun Q, Guo Y, Liu X, Czauderna F, Carr MI, Zenke FT, Blaukat A, Vassilev LT. Therapeutic implications of p53 status on cancer cell fate following exposure to ionizing radiation and the DNA-PK inhibitor M3814. Mol Cancer Res. 2019;17(12):2457–2468.
- Wang M, Chen S, Wei Y, Wei X. DNA-PK inhibition by M3814 enhances chemosensitivity in non-small cell lung cancer. Acta Pharm Sin B. 2021;11(12):3935–3949.
- van Bussel MTJ, Awada A, de Jonge MJA, Mau-Sørensen M, Nielsen D, Schöffski P, Verheul HMW, Sarholz B, Berghoff K, El Bawab S, et al. A first-in-man phase 1 study of the DNA-dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br J Cancer. 2021;124(4):728–735.
- Tsuji T, Sapinoso LM, Tran T, Gaffney B, Wong L, Sankar S, Raymon HK, Mortensen DS, Xu S. CC-115, a dual inhibitor of mTOR kinase and DNA-PK, blocks DNA damage repair pathways and selectively inhibits ATM-deficient cell growth in vitro. Oncotarget. 2017;8(43):74688–74702.
- Thijssen R, Ter Burg J, Garrick B, van Bochove GGW, Brown JR, Fernandes SM, Rodríguez MS, Michot J-M, Hallek M, Eichhorst B, et al. Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood. 2016;128(4):574–583.
- Munster P, Mita M, Mahipal A, Nemunaitis J, Massard C, Mikkelsen T, Cruz C, Paz-Ares L, Hidalgo M, Rathkopf D, et al. First-in-human phase I study of a dual mTOR kinase and DNA-PK inhibitor (CC-115) in advanced malignancy. Cancer Manag Res. 2019;11:10463–10476.
- Formisano L, Napolitano F, Rosa R, D’Amato V, Servetto A, Marciano R, De Placido P, Bianco C, Bianco R. Mechanisms of resistance to mTOR inhibitors. Crit Rev Oncol Hematol. 2020;147:102886.
- Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105(45):17414–17419.
- Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O, Liu D, Wan L, Zhai B, Yu Y, et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol. 2013;15(11):1340–1350.
- Popova NV, Jücker M. The role of mTOR signaling as a therapeutic target in cancer. Int J Mol Sci. 2021;22(4):1743.
- Mohamed MA, Elkhateeb WA, Daba GM. Rapamycin golden jubilee and still the miraculous drug: a potent immunosuppressant, antitumor, rejuvenative agent, and potential contributor in COVID-19 treatment. Bioresour Bioprocess. 2022;9(1):65.
- Oleksak P, Nepovimova E, Chrienova Z, Musilek K, Patocka J, Kuca K. Contemporary mTOR inhibitor scaffolds to diseases breakdown: a patent review (2015–2021). Eur J Med Chem. 2022;238:114498.
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369(6483):756–758.
- Sarbassov DD, Ali SM, Sengupta S, Sheen J-H, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–168.
- Świtaj T, Sobiborowicz A, Teterycz P, Klimczak A, Makuła D, Wągrodzki M, Szumera-Ciećkiewicz A, Rutkowski P, Czarnecka AM. Efficacy of sirolimus treatment in PEComa-10 years of practice perspective. J Clin Med. 2021;10(16):3705.
- Wagner AJ, Ravi V, Riedel RF, Ganjoo K, Van Tine BA, Chugh R, Cranmer L, Gordon EM, Hornick JL, Du H, et al. nab-sirolimus for patients with malignant perivascular epithelioid cell tumors. J Clin Oncol. 2021;39(33):3660–3670.
- Mann JE. Sirolimus protein-bound particles (Fyarro™). J Oncol Times. 2022;44(14):13, 19.
- Denise W, Eh J. Mechanistic target of rapamycin (mTOR) inhibitors. Handb Exp Pharmacol. 2022;272:53–72.
- Mascarenhas L, Chi Y-Y, Hingorani P, Anderson JR, Lyden ER, Rodeberg DA, Indelicato DJ, Kao SC, Dasgupta R, Spunt SL, et al. Randomized phase II trial of bevacizumab or temsirolimus in combination with chemotherapy for first relapse rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol. 2019;37(31):2866–2874.
- Park H, Williams K, Trikalinos NA, Larson S, Tan B, Waqar S, Suresh R, Morgensztern D, Van Tine BA, Govindan R, et al. A phase I trial of temsirolimus and erlotinib in patients with refractory solid tumors. Cancer Chemother Pharmacol. 2021;87(3):337–347.
- Kaley TJ, Panageas KS, Pentsova EI, Mellinghoff IK, Nolan C, Gavrilovic I, DeAngelis LM, Abrey LE, Holland EC, Omuro A, et al. Phase I clinical trial of temsirolimus and perifosine for recurrent glioblastoma. Ann Clin Transl Neurol. 2020;7(4):429–436.
- Everolimus HJ. Recent results in cancer research: Fortschritte Der Krebsforschung. Progres Dans Les Recherches Sur le Cancer. 2018;211:101–123.
- Fan Y, Sun T, Shao Z, Zhang Q, Ouyang Q, Tong Z, Wang S, Luo Y, Teng Y, Wang X, et al. Effectiveness of adding everolimus to the first-line treatment of advanced breast cancer in premenopausal women who experienced disease progression while receiving selective estrogen receptor modulators: a phase 2 randomized clinical trial. JAMA Oncol. 2021;7(10):e213428.
- Guarneri V, Giorgi CA, Cinieri S, Bengala C, Mariani G, Bisagni G, Frassoldati A, Zamagni C, De Rossi C, Amoroso V, et al. Everolimus plus aromatase inhibitors as maintenance therapy after first-line chemotherapy: final results of the phase III randomised MAIN-A (MAINtenance Afinitor) trial. Eur J Cancer. 2021;154:21–29.
- Feldman DR, Ged Y, Lee C-H, Knezevic A, Molina AM, Chen Y-B, Chaim J, Coskey DT, Murray S, Tickoo SK, et al. Everolimus plus bevacizumab is an effective first-line treatment for patients with advanced papillary variant renal cell carcinoma: final results from a phase II trial. Cancer. 2020;126(24):5247–5255.
- Mita MM, Gong J, Chawla SP. Ridaforolimus in advanced or metastatic soft tissue and bone sarcomas. Expert Rev Clin Pharmacol. 2013;6(5):465–482.
- Demetri GD, Chawla SP, Ray-Coquard I, Le Cesne A, Staddon AP, Milhem MM, Penel N, Riedel RF, Bui-Nguyen B, Cranmer LD, et al. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol. 2013;31(19):2485–2492.
- Colombo N, McMeekin DS, Schwartz PE, Sessa C, Gehrig PA, Holloway R, Braly P, Matei D, Morosky A, Dodion PF, et al. Ridaforolimus as a single agent in advanced endometrial cancer: results of a single-arm, phase 2 trial. Br J Cancer. 2013;108(5):1021–1026.
- Oza AM, Pignata S, Poveda A, McCormack M, Clamp A, Schwartz B, Cheng J, Li X, Campbell K, Dodion P, et al. Randomized phase II trial of ridaforolimus in advanced endometrial carcinoma. J Clin Oncol. 2015;33(31):3576–3582.
- Guichard SM, Curwen J, Bihani T, D’Cruz CM, Yates JWT, Grondine M, Howard Z, Davies BR, Bigley G, Klinowska T, et al. AZD2014, an inhibitor of mTORC1 and mTORC2, Is highly effective in ER + breast cancer when administered using intermittent or continuous schedules. Mol Cancer Ther. 2015;14(11):2508–2518.
- Heudel P, Frenel J-S, Dalban C, Bazan F, Joly F, Arnaud A, Abdeddaim C, Chevalier-Place A, Augereau P, Pautier P, et al. Safety and efficacy of the mTOR inhibitor, vistusertib, combined with anastrozole in patients with hormone receptor-positive recurrent or metastatic endometrial cancer: the VICTORIA multicenter, open-label, phase 1/2 randomized clinical trial. JAMA Oncol. 2022;8(7):1001–1009.
- Powles T, Wheater M, Din O, Geldart T, Boleti E, Stockdale A, Sundar S, Robinson A, Ahmed I, Wimalasingham A, et al. A randomised phase 2 study of AZD2014 versus everolimus in patients with VEGF-refractory metastatic clear cell renal cancer. Eur Urol. 2016;69(3):450–456.
- Lapointe S, Mason W, MacNeil M, Harlos C, Tsang R, Sederias J, Luchman HA, Weiss S, Rossiter JP, Tu D, et al. A phase I study of vistusertib (dual mTORC1/2 inhibitor) in patients with previously treated glioblastoma multiforme: a CCTG study. Invest New Drugs. 2020;38(4):1137–1144.
- Basu B, Krebs MG, Sundar R, Wilson RH, Spicer J, Jones R, Brada M, Talbot DC, Steele N, Ingles Garces AH, et al. Vistusertib (dual m-TORC1/2 inhibitor) in combination with paclitaxel in patients with high-grade serous ovarian and squamous non-small-cell lung cancer. Ann Oncol. 2018;29(9):1918–1925.
- Morscher RJ, Brard C, Berlanga P, Marshall LV, André N, Rubino J, Aerts I, De Carli E, Corradini N, Nebchi S, et al. First-in-child phase I/II study of the dual mTORC1/2 inhibitor vistusertib (AZD2014) as monotherapy and in combination with topotecan-temozolomide in children with advanced malignancies: arms E and F of the AcSé-ESMART trial. Eur J Cancer. 2021;157:268–277.
- Shimizu T, Kuboki Y, Lin C-C, Yonemori K, Yanai T, Faller DV, Dobler L, Gupta N, Sedarati F, Kim K-P. A phase 1 study of sapanisertib (TAK-228) in East Asian patients with advanced nonhematological malignancies. Target Oncol. 2022;17(1):15–24.
- Burris HA, Kurkjian CD, Hart L, Pant S, Murphy PB, Jones SF, Neuwirth R, Patel CG, Zohren F, Infante JR. TAK-228 (formerly MLN0128), an investigational dual TORC1/2 inhibitor plus paclitaxel, with/without trastuzumab, in patients with advanced solid malignancies. Cancer Chemother Pharmacol. 2017;80(2):261–273.
- Davis SL, Ionkina AA, Bagby SM, Orth JD, Gittleman B, Marcus JM, Lam ET, Corr BR, O’Bryant CL, Glode AE, et al. Preclinical and dose-finding phase I trial results of combined treatment with a TORC1/2 Inhibitor (TAK-228) and Aurora A kinase inhibitor (Alisertib) in solid tumors. Clin Cancer Res. 2020;26(17):4633–4642.
- García-Sáenz JÁ, Martínez-Jáñez N, Cubedo R, Jerez Y, Lahuerta A, González-Santiago S, Ferrer N, Ramos M, Alonso-Romero JL, Antón A, et al. Sapanisertib plus fulvestrant in postmenopausal women with estrogen receptor-positive/HER2-negative advanced breast cancer after progression on aromatase inhibitor. Clin Cancer Res. 2022;28(6):1107–1116.
- Koca E, Niravath PA, Ensor J, Patel TA, Li X, Hemati P, Wong H, Qian W, Boone T, Zhao J, et al. ANETT: phase II trial of neoadjuvant TAK-228 plus Tamoxifen in patients with hormone receptor-positive breast cancer. Breast Cancer Res Treat. 2021;188(2):433–439.
- Basu A, Lambring CB. Akt isoforms: a family affair in breast cancer. Cancers. 2021;13(14):3445.
- Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT–a major therapeutic target. Biochim Biophys Acta. 2004;1697(1–2):3–16.
- Dillon RL, Marcotte R, Hennessy BT, Woodgett JR, Mills GB, Muller WJ. Akt1 and akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Res. 2009;69(12):5057–5064.
- Riggio M, Perrone MC, Polo ML, Rodriguez MJ, May M, Abba M, Lanari C, Novaro V. AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins. Sci Rep. 2017;7:44244.
- Chin YR, Yoshida T, Marusyk A, Beck AH, Polyak K, Toker A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014;74(3):964–973.
- Mora A, Sakamoto K, McManus EJ, Alessi DR. Role of the PDK1-PKB-GSK3 pathway in regulating glycogen synthase and glucose uptake in the heart. FEBS Lett. 2005;579(17):3632–3638.
- Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813(11):1978–1986.
- Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277(24):21843–21850.
- Brown JS, Banerji U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol Ther. 2017;172:101–115.
- Dong C, Wu J, Chen Y, Nie J, Chen C. Activation of PI3K/AKT/mtor pathway causes drug resistance in breast cancer. Front Pharmacol. 2021;12:628690.
- Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Semin Cancer Biol. 2019;59:125–132.
- Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, Darling TN, Burton-Akright J, Bagheri M, Dombi E, Gruber A, Jarosinski PF, Martin S, et al. Pharmacodynamic study of miransertib in individuals with proteus syndrome. Am J Hum Genet. 2019;104(3):484–491.
- Lazaro G, Kostaras E, Vivanco I. Inhibitors in AKTion: ATP-competitive vs allosteric. Biochem Soc Trans. 2020;48(3):933–943.
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan B-S, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9(7):1956–1967.
- Xing Y, Lin NU, Maurer MA, Chen H, Mahvash A, Sahin A, Akcakanat A, Li Y, Abramson V, Litton J, et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019;21(1):78.
- Jonasch E, Hasanov E, Corn PG, Moss T, Shaw KR, Stovall S, Marcott V, Gan B, Bird S, Wang X, et al. A randomized phase 2 study of MK-2206 versus everolimus in refractory renal cell carcinoma. Ann Oncol. 2017;28(4):804–808.
- Oki Y, Fanale M, Romaguera J, Fayad L, Fowler N, Copeland A, Samaniego F, Kwak LW, Neelapu S, Wang M, et al. Phase II study of an AKT inhibitor MK2206 in patients with relapsed or refractory lymphoma. Br J Haematol. 2015;171(4):463–470.
- Larsen JT, Shanafelt TD, Leis JF, LaPlant B, Call T, Pettinger A, Hanson C, Erlichman C, Habermann TM, Reeder C, et al. Akt inhibitor MK-2206 in combination with bendamustine and rituximab in relapsed or refractory chronic lymphocytic leukemia: results from the N1087 alliance study. Am J Hematol. 2017;92(8):759–763.
- Chien AJ, Cockerill A, Fancourt C, Schmidt E, Moasser MM, Rugo HS, Melisko ME, Ko AH, Kelley RK, Korn WM, et al. A phase 1b study of the Akt-inhibitor MK-2206 in combination with weekly paclitaxel and trastuzumab in patients with advanced HER2-amplified solid tumor malignancies. Breast Cancer Res Treat. 2016;155(3):521–530.
- Politz O, Siegel F, Bärfacker L, Bömer U, Hägebarth A, Scott WJ, Michels M, Ince S, Neuhaus R, Meyer K, et al. BAY 1125976, a selective allosteric AKT1/2 inhibitor, exhibits high efficacy on AKT signaling-dependent tumor growth in mouse models. Int J Cancer. 2017;140(2):449–459.
- Schneeweiss A, Hess D, Joerger M, Varga A, Moulder S, Tsimberidou AM, Ma C, Hurvitz SA, Rentzsch C, Rudolph M, et al. Phase 1 dose escalation study of the allosteric AKT inhibitor BAY 1125976 in advanced solid cancer-lack of association between activating AKT mutation and AKT inhibition-derived efficacy. Cancers. 2019;11(12):1987.
- Naoya M, Hiroto O, Diana C, Francesca C, Gullu Topal G, Jiro M, Rikio S, Toshiyasu S, Teruhiro U, Teru H, et al. TAS-117, a novel selective Akt inhibitor demonstrates significant growth inhibition in multiple myeloma cells in vitro and in vivo. Blood. 2012;120(21):942.
- Junya I, Takuya K, Toshiharu K, Hitoshi S, Hiroshi H. Synergistic antitumor activity of futibatinib, an FGFR1-4 inhibitor, and TAS-117, a selective AKT inhibitor, in FGFR-deregulated cancer models. Cancer Res. 2020;80(16):661.
- Rizk M, Rizq O, Oshima M, Nakajima-Takagi Y, Koide S, Saraya A, Isshiki Y, Chiba T, Yamazaki S, Ma A, et al. Akt inhibition differently controls PRC2 components and synergizes with dual EZH2/1 inhibitor in the treatment of multiple myeloma. Blood. 2019;134(Suppl 1):4400–4400.
- Lee JB, Jung M, Beom SH, Kim GM, Kim HR, Choi HJ, Sohn JH, Ahn JB, Rha SY, Chung HC. Phase 2 study of TAS-117, an allosteric akt inhibitor in advanced solid tumors harboring phosphatidylinositol 3-kinase/v-akt murine thymoma viral oncogene homolog gene mutations. Invest New Drugs. 2021;39(5):1366–1374.
- Forde K, Resta N, Ranieri C, Rea D, Kubassova O, Hinton M, Andrews KA, Semple R, Irvine AD, Dvorakova V. Clinical experience with the AKT1 inhibitor miransertib in two children with PIK3CA-related overgrowth syndrome. Orphanet J Rare Dis. 2021;16(1):109.
- Devki N, Naixin Z, Yi Y, Schwartz B, Chen S, Kima PE, Reiner NE. Miransertib (ARQ 092), an orally-available, selective Akt inhibitor is effective against Leishmania. PLOS One. 2018;13(11):e0206920.
- Page N, Wappett M, O’Dowd CR, O’Rourke M, Gavory G, Zhang L, Rountree JSS, Jordan L, Barker O, Gibson H, et al. Identification and development of a subtype-selective allosteric AKT inhibitor suitable for clinical development. Sci Rep. 2022;12(1):15715.
- Andrikopoulou A, Chatzinikolaou S, Panourgias E, Kaparelou M, Liontos M, Dimopoulos M-A, Zagouri F. The emerging role of capivasertib in breast cancer. Breast. 2022;63:157–167.
- Schmid P, Abraham J, Chan S, Wheatley D, Brunt AM, Nemsadze G, Baird RD, Park YH, Hall PS, Perren T, et al. Capivasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer: the PAKT trial. J Clin Oncol. 2020;38(5):423–433.
- Howell SJ, Casbard A, Carucci M, Ingarfield K, Butler R, Morgan S, Meissner M, Bale C, Bezecny P, Moon S, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2022;23 (7):851– 864.
- Lang L, Lam T, Chen A, Jensen C, Duncan L, Kong FC, Kurago ZB, Shay C, Teng Y. Circumventing AKT-associated radioresistance in oral cancer by novel nanoparticle-encapsulated capivasertib. Cells. 2020;9(3):533.
- Spencer A, Yoon S-S, Harrison SJ, Morris SR, Smith DA, Brigandi RA, Gauvin J, Kumar R, Opalinska JB, Chen C, et al. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood. 2014;124(14):2190–2195.
- Blagden SP, Hamilton AL, Mileshkin L, Wong S, Michael A, Hall M, Goh JC, Lisyanskaya AS, DeSilvio M, Frangou E, et al. Phase IB dose escalation and expansion study of akt inhibitor afuresertib with carboplatin and paclitaxel in recurrent platinum-resistant ovarian cancer. Clin Cancer Res. 2019;25(5):1472–1478.
- Tolcher AW, Patnaik A, Papadopoulos KP, Rasco DW, Becerra CR, Allred AJ, Orford K, Aktan G, Ferron-Brady G, Ibrahim N, et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother Pharmacol. 2015;75(1):183–189.
- Chen CI, Paul H, Le LW, Wei EN, Snitzler S, Wang T, Levina O, Kakar S, Lau A, Queau M, et al. A phase 2 study of ofatumumab (Arzerra(®)) in combination with a pan-AKT inhibitor (afuresertib) in previously treated patients with chronic lymphocytic leukemia (CLL). Leuk Lymphoma. 2019;60(1):92–100.
- Cheraghchi-Bashi A, Parker CA, Curry E, Salazar J-F, Gungor H, Saleem A, Cunnea P, Rama N, Salinas C, Mills GB, et al. A putative biomarker signature for clinically effective AKT inhibition: correlation of in vitro, in vivo and clinical data identifies the importance of modulation of the mTORC1 pathway. Oncotarget. 2015;6(39):41736–41749.
- Algazi AP, Esteve-Puig R, Nosrati A, Hinds B, Hobbs-Muthukumar A, Nandoskar P, Ortiz-Urda S, Chapman PB, Daud A. Dual MEK/AKT inhibition with trametinib and GSK2141795 does not yield clinical benefit in metastatic NRAS-mutant and wild-type melanoma. Pigment Cell Melanoma Res. 2018;31(1):110–114.
- Ragon BK, Odenike O, Baer MR, Stock W, Borthakur G, Patel K, Han L, Chen H, Ma H, Joseph L, et al. Oral MEK 1/2 inhibitor trametinib in combination with AKT inhibitor GSK2141795 in patients with acute myeloid leukemia with RAS mutations: a phase II study. Clin Lymphoma Myeloma Leuk. 2019;19(7):431.e13–440.e13.
- Westin SN, Sill MW, Coleman RL, Waggoner S, Moore KN, Mathews CA, Martin LP, Modesitt SC, Lee S, Ju Z, et al. Safety lead-in of the MEK inhibitor trametinib in combination with GSK2141795, an AKT inhibitor, in patients with recurrent endometrial cancer: an NRG oncology/GOG study. Gynecol Oncol. 2019;155(3):420–428.
- Liu JF, Gray KP, Wright AA, Campos S, Konstantinopoulos PA, Peralta A, MacNeill K, Morrissey S, Whalen C, Dillon D, et al. Results from a single arm, single stage phase II trial of trametinib and GSK2141795 in persistent or recurrent cervical cancer. Gynecol Oncol. 2019;154(1):95–101.
- Yoshida K, Wilkins J, Winkler J, Wade JR, Kotani N, Wang N, Sane R, Chanu P. Population pharmacokinetics of ipatasertib and its metabolite in cancer patients. J Clin Pharmacol. 2021;61(12):1579–1591.
- Sweeney C, Bracarda S, Sternberg CN, Chi KN, Olmos D, Sandhu S, Massard C, Matsubara N, Alekseev B, Parnis F, et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet. 2021;398(10295):131–142.
- Kim S-B, Dent R, Im S-A, Espié M, Blau S, Tan AR, Isakoff SJ, Oliveira M, Saura C, Wongchenko MJ, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360–1372.
- Ma C, Wu J, Wang L, Ji X, Wu Y, Miao L, Chen D, Zhang L, Wu Y, Feng H, et al. Discovery of clinical candidate NTQ1062 as a potent and bioavailable Akt inhibitor for the treatment of human tumors. J Med Chem. 2022;65(12):8144–8168.