
Abstract
A novel library of human carbonic anhydrase (hCA) inhibitors based on the 2-sulfanilamido[1,2,4]triazolo[1,5-a]pyrimidine skeleton modified at its 7-position was prepared by an efficient convergent procedure. These derivatives were evaluated in vitro for their inhibition properties against a representative panel of hCA isoforms (hCA I, II, IV, IX, and XII). The target tumour-associated isoforms hCA IX and XII were potently inhibited with KIs in the low nanomolar range of 5–96 nM and 4–72 nM, respectively. Compounds 1d, 1j, 1v, and 1x were the most potent hCA IX inhibitors with KIs of 5.1, 8.6, 4.7, and 5.1 nM, respectively. Along with derivatives 1d and 1j, compounds 1r and 1ab potently inhibited hCA XII isoform with KIs in a single-digit nanomolar range of 8.8, 5.4, 4.3, and 9.0 nM, respectively. Compounds 1e, 1m, and 1p exhibited the best selectivity against hCA IX and hCA XII isoforms over off-target hCA II, with selectivity indexes ranging from 5 to 14.
Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are a ubiquitous superfamily of zinc-metalloenzymes responsible for the reversible catalytic hydration of carbon dioxide (CO2) to bicarbonate and a proton, facilitating various biochemical and physiological functions in both prokaryotes and eukaryotesCitation1–4. To date, eight genetically distinct CA families (α − ι) have been identified in nature, among which only the alpha class of carbonic anhydrases is expressed in humans (hCAs)Citation5,Citation6.
The human αCAs encompass 15 distinct isoforms (I–XV), 12 of which are catalytically active (CAs I–IV, CA VA–VB, CA VI, CA VII, CA IX, and CAs XII–XIV). These isoforms are differentiated by their unique catalytic activity, cellular localisation, and organ/tissue distributionCitation7,Citation8, whereas hCAs VIII, X, and XI are non-catalytically active CA-related proteinsCitation9,Citation10. The catalytically active isoforms can be further classified into four distinct classes based on localisation: cytosolic isoforms (hCAs I–III, VII, and XIII), transmembrane isoforms (hCAs IV, IX, XII, and XIV), mitochondrial isoforms (hCAs VA and VB), and the secreted isoenzyme hCA VI released in saliva and milkCitation11.
hCAs are widely distributed across various tissues and organs in our body, helping to maintain ion and pH cellular homeostasis. They play pivotal roles in numerous physiological and pathological conditionsCitation12,Citation13. As such, dysregulation of hCAs is linked with a broad spectrum of diseases, including glaucoma (hCA I, II, IV, and XII), epilepsy (hCA VII and XIV), cerebral and retinal oedema (hCA I and II), and tumours (hCA IX and hCA XII)Citation14–24.
Within the superfamily of α-CAs, isoforms CA IX and XII have been extensively studied due to their crucial role in the survival of hypoxic tumoursCitation25–27. These cancer-associated CAs exhibit limited expression in normal tissues whereas they are frequently overexpressed in various hypoxic solid tumours. This is likely due to the strong transcriptional activation of the hypoxia-inducible transcription factor 1 (HIF1)Citation28, where they can influence cell proliferation, cell adhesion, and malignant cell invasionCitation29.
The overexpression of hCA IX isoform in hypoxic tumours contributes to an acidic extracellular microenvironment. This acidity is essential to maintaining a slightly alkaline intracellular pH that supports cancer cell survival and proliferation, compromising the survival and proliferation of normal cellsCitation30–34. Concurrently, CA XII is overexpressed in numerous solid tumours, such as breast, lung, brain, and cervical cancerCitation35,Citation36. Consequently, the development of selective hCA IX and hCA XII inhibitors represents an encouraging approach for discovering potential antitumor/antimetastatic agentsCitation37–39.
However, achieving selectivity of hCA IX/XII inhibitors over the off-targets, the ubiquitous hCA I and II isoforms involved in several physiological processes, remains a challenging task. This selectivity is crucial to prevent side effectsCitation40,Citation41, despite some studies reporting the overexpression of intracellular CA II in certain malignant cellsCitation42–47. The development of selective and potent inhibitors of hCA IX/XII isoforms has been hindered due to the structural similarity between the off-target isoenzymes, CA I and II, and CA IXCitation48–50.
All catalytically active hCAs feature a Zn2+ ion at their highly conserved active site coordinated by three histidine residues (His94, His96, and His119) and a water molecule, playing a crucial role in the catalytic process. The majority of hCA inhibitors function as zinc-binders, with aromatic/heterocyclic primary sulphonamides (RSO2NH2) comprising the most significant class of hCA inhibitors (CAIs) due to their high affinity for numerous CA isoformsCitation51–53. In their deprotonated form (SO2NH–), these inhibitors interact with the Zn2+ ion in the active site and displace the Zn-coordinated water molecule, thereby inhibiting the enzyme.
Despite the efficacy of primary sulphonamide in developing potent CAIs, these molecules suffer from a lack of selectivity for a particular human isoform. This drawback is caused by the similarity in active site architecture among the human isoforms, leading to off-target side effectsCitation1,Citation13,Citation17.
To address this issue, the most effective adopted strategy has been the “tail approach”Citation54. Here, one or more different aryl or heteroaryl “tails” are appended to an aromatic or heterocyclic scaffold possessing the zinc-binding group (ZBG) such as sulphonamide or its bioisostere, such as sulfamate and sulfamideCitation53, through various functionalised linkersCitation55–58. This strategy optimises the interactions with specific hydrophobic/hydrophilic residues in the peripheral part of the active site cavity, which varies among the different hCA isoformsCitation59–62.
A successful example for the “tail approach” led to the discovery of the selective hCA IX inhibitor SLC-0111 ()Citation63. This ureido substituted sulphonamide is currently undergoing clinical trials for treating advanced hypoxic tumours with metastasesCitation64. Given its promise, significant research efforts are being devoted to developing potent and selective SLC-0111 analoguesCitation65–67.
Figure 1. Structure of SLC-0111 and molecular structures of 2-sulphanilamide-7-substituted [1,2,4]triazolo [1,5-a]pyrimidines 1a–ac reported in this article.
![Figure 1. Structure of SLC-0111 and molecular structures of 2-sulphanilamide-7-substituted [1,2,4]triazolo [1,5-a]pyrimidines 1a–ac reported in this article.](/cms/asset/d4c18182-2489-4f77-b9f9-790d56f6a06d/ienz_a_2270180_f0001_c.jpg)
The [1,2,4]triazolo[1,5-a]pyrimidine skeleton has been gained remarkable research attention due to its biological importance as a pharmacophore for designing anticancer agentsCitation68. Depending on the particular substitution pattern at the 2-, 5-, 6-, and 7-positions of the triazolopyrimidine scaffold, these agents can act on different targets, such as tubulinCitation69–74, BRD4Citation75, LSD1Citation76,Citation77, ABCB1Citation78, and CDK2Citation79. In our search for new selective hCA IX and hCA XII inhibitors, we identified the [1,2,4]triazolo[1,5-a]pyrimidine bicyclic system with appropriate substituents at its 2- and 7-positions as a suitable scaffold for producing compounds that inhibit both these tumour-associated hCA isoforms.
This new library of compounds, characterised by the general structure 1, incorporated a common p-sulphanilamide (4′-NH2SO2C6H4NH) group as the Zn-binding function (ZBG) at the 2-position of the [1,2,4]triazolo[1,5-a]pyrimidine nucleus. Different aryl or heteroaryl “tails” were inserted to the 7-position to interact with the variable amino acid residues within the active sites of different CA isoforms. For compounds 1b–e, we evaluated the effect of replacing the phenyl group of compound 1a with bioisosteric heterocyclic rings, such as 2′-thienyl, 4′-piridinyl, and 3′-piridinyl, to yield derivatives 1b–d, respectively. For compound 1e, the unsubstituted phenyl ring was substituted with the more lipophilic 2′-naphthyl group.
In preparing derivatives 1f–1ac, we explored the structure–activity relationship (SAR) by examining the impact of various substitutions with electron-withdrawing groups (EWGs) such as nitro (NO2) and halogens (F, Cl, Br, and I) or electron-releasing groups (ERGs), namely methyl (Me), methoxy (MeO), and ethoxy (EtO), on the phenyl ring at the 7-position of triazolopyrimidine scaffold.
A noteworthy point is that the preparation of these derivatives was accomplished via an efficient and flexible one-step procedure, starting from a common intermediate, 4-((5-amino-1H-1,2,4-triazol-3-yl)amino)benzenesulfonamide (compound 2). To the best of our knowledge, this is the first study of compounds characterised by the [1,2,4]triazolo[1,5-a]pyrimidine skeleton as the main scaffold for CA inhibitors. This represents unexplored territory, providing a potentially fruitful avenue for further research.
Materials and methods
Chemistry-general
1H and 13C NMR spectra were recorded on a Bruker AC 200 and Varian 400 Mercury Plus spectrometer, respectively. Chemical shifts (δ) are given in ppm upfield from tetramethylsilane as internal standard, and the spectra were recorded in appropriate deuterated solvents, as indicated. Mass spectra were recorded by an ESI single quadrupole mass spectrometer Waters ZQ 2000 (Waters Instruments, Wilmslow, UK) and the values are expressed as [M + 1]+. Melting points (mp) were determined on a Buchi-Tottoli apparatus and are uncorrected. All products reported showed 1H and 13C NMR spectra in agreement with the assigned structures. The purity of tested compounds was determined by combustion elemental analyses conducted by the Microanalytical Laboratory of the Chemistry Department of the University of Ferrara with a Yanagimoto MT-5 CHN recorder elemental analyser. All tested compounds yielded data consistent with a purity of at least 95% as compared with the theoretical values. All reactions were carried out under an inert atmosphere of dry nitrogen. Standard syringe techniques were used for transferring dry solvents. Reaction courses and product mixtures were routinely monitored by TLC on silica gel (precoated F254 Merck plates), and compounds were visualised with aqueous KMnO4. Flash chromatography was performed using 230–400 mesh silica gel and the indicated solvent system. Organic solutions were dried over anhydrous Na2SO4. 1H NMR, 13C NMR, UV–vis, and ESI-mass spectra of final compounds 1a–ac are included in the Supplementary Material.
Preparation of (Z)-phenyl N′-cyano-N-(4-sulfamoylphenyl)carbamimidate (2)
To a suspension of diphenyl cyanocarbonimidate (952 mg, 4 mmol) in THF (5 mL) was added 4-aminobenzenesulfonamide (688 mg, 4 mmol) and the mixture was refluxed for 24 h. After this time, the solvent was removed under reduced pressure, the resulting residue was washed CH2Cl2 (10 mL) and filtered to furnish the desired product 2 as a white solid. Yield: 79%, mp: 204–205 °C. 1H NMR (d6-DMSO) δ: 7.29–7.32 (m, 3H), 7.34 (s, 3H), 7.44 (t, J = 7.2 Hz, 2H), 7.62 (d, J = 8.8 Hz, 1H), 7.80 (d, J = 8.8 Hz, 1H), 11.1 (s, 1H). MS (ESI): [M + 1]+ = 317.3.
Preparation of 4-[(5-amino-1-benzoyl-1H-1,2,4-triazol-3-yl)amino]-benzenesulfonamide (3)
To a stirred suspension of compound 2 (1 g, 3.15 mmol) in THF (10 mL) was added hydrazine monohydrate (0.3 mL, 6.3 mmol, 2 equiv.) and the mixture was heated to reflux for 18 h. After this time, the reaction mixture was warmed to room temperature, the solid was collected by filtration and washed with tetrahydrofuran to afford compound 3 as a white solid. Yield 84%; mp 260 °C. 1H NMR (d6-DMSO) δ: 5.92 (s, 2H), 7.02 (bs, 2H), 7.55 (d, J = 9.2, 2H), 7.58 (d, J = 9.2 Hz, 2H), 9.18 (s, 1H), 11.1 (bs, 1H). MS (ESI): [M + 1]+ = 255.3.
General synthetic procedures
General procedure A for the preparation of compounds 5a–ac
A mixture of the appropriate 1-arylethanones 4a–ac (5 mmol) and DMF-DMA (2.7 mL, 2.38 g, 20 mmol, 4 equiv.) in DMF (2 mL) was stirred at reflux for 4 h. The reaction mixture was evaporated in vacuo, the crude residue was purified by flash chromatography on silica gel or suspended with diethyl ether, filtered and used for the next step without further purification, to afford enaminone derivatives 5a–ac, respectively.
Compounds (E)-3-(dimethylamino)-1-(thiophen-2-yl)prop-2-en-1-one (5b), (E)-3-(dimethylamino)-1-(4-nitrophenyl)prop-2-en-1-one (5q), (E)-3-(dimethylamino)-1-(p-tolyl)prop-2-en-1-one (5t), (E)-3-(dimethylamino)-1-(o-tolyl)prop-2-en-1-one (5v), (E)-3-(dimethylamino)-1-(4-methoxyphenyl)prop-2-en-1-one (5w), (E)-1-(3,4-dimethoxyphenyl)-3-(dimethylamino)prop-2-en-1-one (5ab), and (E)-3-(dimethylamino)-1-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (5ac) showed spectroscopic and analytical data in agreement with those previously published in the article: Huo et al. (Citation71).
General procedure B for the synthesis of compounds 1a–ac
To a solution of the appropriate enaminones 5a–ac (0.4 mmol, 2 equiv.) in glacial acetic acid (2 mL) was added 4-[(5-amino-1-benzoyl-1H-1,2,4-triazol-3-yl)amino]-benzenesulfonamide 3 (51 mg, 0.2 mmol), and the resulting mixtures were stirred for 4 h at 80 °C and then evaporated to dryness in vacuo. The crude residue was suspended in dichloromethane (5 mL) and filtered. The filtrate was rinsed with ethyl ether (2 × 3 mL) to afford the appropriate 2-(4′-aminobenzenesulfonamido)-7-substituted [1,2,4]triazolo[1,5-a]pyrimidines 1a–ac.
(E)-3-(Dimethylamino)-1-phenylprop-2-en-1-one (5a)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5a as a yellow solid. Yield 84%, mp 82–84 °C. 1H NMR (CDCl3) δ: 2.94 (s, 3H), 3.18 (s, 3H), 5.80 (d, J = 12.4 Hz, 1H), 7.41–7.46 (m, 3H), 7.80–7.88 (m, 2H), 7.90 (d, J = 12.4 Hz, 1H). MS (ESI): [M + 1]+ = 176.3.
(E)-3-(Dimethylamino)-1-(pyridin-4-yl)prop-2-en-1-one (5c)
Synthesised according to procedure A, the crude residue was taken up in ethyl ether and the suspension stirred at room temperature for 15 min. Filtration of the solid provided 5c as an orange solid. Yield: 82%, mp 115–117 °C. 1H NMR (CDCl3) δ: 2.96 (s, 3H), 3.19 (s, 3H), 5.63 (d, J = 12.4 Hz, 1H), 7.67 (d, J = 6.0 Hz, 2H), 7.83 (d, J = 12.4 Hz, 1H), 8.70 (d, J = 6.0 Hz, 2H). MS (ESI): [M + 1]+ = 177.3.
(E)-3-(Dimethylamino)-1-(pyridin-3-yl)prop-2-en-1-one (5d)
Synthesised according to procedure A, the crude residue was taken up in ethyl ether and the suspension stirred at room temperature for 15 min. Filtration of the solid provided 5d as an orange solid. Yield: 79%, p.f. 85–87 °C, 1H NMR (CDCl3) δ: 2.95 (s, 3H), 3.18 (s, 3H), 5.65 (d, J = 12.2 Hz, 1H), 7.39 (m, 1H), 7.83 (d, J = 12.2 Hz, 1H), 8.22 (d, J = 8.0 Hz, 1H), 8.66 (m, 1H), 9.08 (s, 1H). MS (ESI): [M + 1]+ = 177.2.
(E)-3-(Dimethylamino)-1-(naphthalen-2-yl)prop-2-en-1-one (5e)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5e as a yellow solid. Yield 92%, mp 66–67 °C. 1H NMR (CDCl3) δ: 2.96 (s, 3H), 3.20 (s, 3H), 5.86 (d, J = 12.4 Hz, 1H), 7.50–7.54 (m, 2H), 7.84–7.88 (m, 2H), 7.92 (d, J = 8.8 Hz, 1H), 7.96 (d, J = 8.8 Hz, 1H), 8.00 (d, J = 12.4 Hz, 1H), 8.41 (s, 1H). MS (ESI): [M + 1]+ = 225.9.
(E)-3-Dimethylamino-1-(4-fluorophenyl)-prop-2-en-1-one (5f)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5f as a yellow solid. Yield 87%, mp 90–92 °C. 1H NMR (CDCl3) δ: 2.95 (s, 3H), 3.17 (s, 3H), 5.66 (d, J = 12.3 Hz, 1H), 7.05 (t, J = 9.0 Hz, 2H), 7.88–7.96 (m, 3H). MS (ESI): [M + 1]+ = 194.3.
(E)-3-(Dimethylamino)-1-(3-fluorophenyl)prop-2-en-1-one (5g)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5g as a yellow solid. Yield 95%, mp 86–88 °C. 1H NMR (CDCl3) δ: 2.95 (s, 3H), 3.18 (s, 3H), 5.65 (d, J = 12.4 Hz, 1H), 7.13–7.16 (m, 1H), 7.30–7.36 (m, 1H), 7.56 (d, J = 9.2 Hz, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.80 (d, J = 12.2 Hz, 1H). MS (ESI): [M + 1]+ = 194.2.
(E)-3-(Dimethylamino)-1-(2-fluorophenyl)prop-2-en-1-one (5h)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5h as a yellow oil. Yield 92%. 1H NMR (CDCl3) δ: 2.92 (s, 3H), 3.15 (s, 3H), 5.65 (d, J = 12.4 Hz, 1H), 7.06–7.10 (m, 1H), 7.21 (d, J = 9.2 Hz, 1H), 7.30–7.38 (m, 1H), 7.65 (m, 1H), 7.72 (d, J = 12.2 Hz, 1H). MS (ESI): [M + 1]+ = 194.1.
(E)-1-(2,6-Difluorophenyl)-3-(dimethylamino)prop-2-en-1-one (5i)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5i as a yellow solid. Yield 89%, mp 114–116 °C. 1H NMR (CDCl3) δ: 2.88 (s, 3H), 3.12 (s, 3H), 5.64 (d, J = 12.4 Hz, 1H), 6.88–6.92 (m, 2H), 7.28–7.32 (m, 2H). MS (ESI): [M + 1]+ = 211.3.
(E)-3-(Dimethylamino)-1-(2,3,6-trifluorophenyl)prop-2-en-1-one (5j)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc–petroleum ether 8–2 v/v as eluent, to furnish compound 5j as a yellow solid. Yield 82%, mp 110–112 °C. 1H NMR (CDCl3) δ: 2.96 (s, 3H), 3.18 (s, 3H), 5.62 (d, J = 12.4 Hz, 1H), 6.83–6.87 (m, 1H), 7.10–7.14 (m, 1H), 7.72 (d, J = 12.4 Hz, 1H). MS (ESI): [M + 1]+ = 229.9.
(E)-3-Dimethylamino-1-(4-chlorophenyl)-prop-2-en-1-one (5k)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc–petroleum ether 8–2 v/v as eluent, to furnish compound 5k as a yellow solid. Yield 94%, mp 96–98 °C. 1H NMR (CDCl3) δ: 2.94 (s, 3H), 3.06 (s, 3H), 5.65 (d, J = 12.4 Hz, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 12.4 Hz, 1H). MS (ESI): [M + 1]+ = 210.6.
(E)-3-Dimethylamino-1-(3-chlorophenyl)-prop-2-en-1-one (5l)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc–petroleum ether 8–2 v/v as eluent, to furnish compound 5l as a yellow solid. Yield 93%, mp 96–98 °C. 1H NMR (CDCl3) δ: 2.97 (s, 3H), 3.19 (s, 3H), 5.65 (d, J = 12.4 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.43 (ddd, J = 8.0, 2.2 and 1.2 Hz, 1H), 7.75 (dd, J = 8.0 and 1.2 Hz, 1H), 7.86 (d, J = 1.6 Hz, 1H), 7.96 (d, J = 12.4 Hz, 1H). MS (ESI): [M + 1]+ = 210.3.
(E)-3-Dimethylamino-1-(2-chlorophenyl)-prop-2-en-1-one (5m)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5m as a yellow oil. Yield 84%. 1H NMR (CDCl3) δ: 2.85 (s, 3H), 3.06 (s, 3H), 5.33 (d, J = 12.4 Hz, 1H), 7.24–7.26 (m, 2H), 7.34–7.38 (m, 3H). MS (ESI): [M + 1]+ = 210.6.
(E)-3-Dimethylamino-1-(4-bromophenyl)-prop-2-en-1-one (5n)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5n as a yellow solid. Yield 92%, mp 84–86 °C. 1H NMR (CDCl3) δ: 2.96 (s, 3H), 3.18 (s, 3H), 5.65 (d, J = 12.4 Hz, 1H), 7.53 (d, J = 9.2 Hz, 2H), 7.75 (d, J = 9.2 Hz, 2H), 7.92 (d, J = 12.4 Hz, 1H). MS (ESI): [M]+ = 254.2, [M + 2]+ = 256.2.
(E)-3-Dimethylamino-1-(3-bromophenyl)-prop-2-en-1-one (5o)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5o as a yellow solid. Yield 92%, mp 90–92 °C. 1H NMR (CDCl3) δ: 2.97 (s, 3H), 3.19 (s, 3H), 5.64 (d, J = 11.6 Hz, 1H), 7.59 (t, J = 8.0 Hz, 1H), 7.72 (dd, J = 8.0 and 1.6 Hz, 1H), 7.82 (dd, J = 8.0 and 1.6 Hz, 1H), 7.94 (d, J = 11.6 Hz, 1H), 8.01 (d, J = 1.6 Hz, 1H). MS (ESI): [M]+ = 254.2, [M + 2]+ = 256.2.
(E)-3-Dimethylamino-1-(4-iodophenyl)-prop-2-en-1-one (5p)
Synthesised according to procedure A, the crude residue was taken up in ethyl ether and the suspension was stirred at room temperature for 15 min. Filtration of the solid provided 5p as a yellow solid. Yield: 76%, mp 117–119 °C, 1H NMR (CDCl3) δ: 2.98 (s, 3H), 3.15 (s, 3H), 5.63 (d, J = 12.4 Hz, 1H), 7.60 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 12.4 Hz, 1H). MS (ESI): [M + 1]+ = 302.07.
(E)-3-(Dimethylamino)-1-(3-nitrophenyl)prop-2-en-1-one (5r)
Synthesised according to procedure A, the crude residue was taken up in ethyl ether and the suspension was stirred at room temperature for 15 min. Filtration of the solid provided 5r as a yellow solid. Yield: 87%, mp 112–114 °C, 1H NMR (CDCl3) δ: 2.99 (s, 3H), 3.21 (s, 3H), 5.7 (d, J = 12.4 Hz, 1H), 7.59 (t, J = 8.0 Hz, 1H), 7.98 (d, J = 12.4 Hz, 1H), 8.26 (m, 2H), 8.68 (s, 1H). MS (ESI): [M + 1]+ = 221.2.
(E)-3-(Dimethylamino)-1-(2-nitrophenyl)prop-2-en-1-one (5s)
Synthesised according to procedure A, the crude residue was suspended with diethyl ether and filtered to furnish compound 5s as a yellow solid. Yield 94%, mp 163–165 °C. 1H NMR (CDCl3) δ: 2.90 (s, 3H), 3.14 (s, 3H), 5.70 (d, J = 12.4 Hz, 1H), 7.62 (td, J = 7.8 and 2.0 Hz, 1H), 7.68 (dd, J = 7.8 and 2.0 Hz, 1H), 7.72 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 12.4 Hz, 1H). MS (ESI): [M + 1]+ = 221.3.
(E)-3-(Dimethylamino)-1-(m-tolyl)prop-2-en-1-one (5u)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5u as a yellow solid. Yield 88%, mp 90–92 °C. 1H NMR (CDCl3) δ: 2.39 (s, 3H), 2.93 (s, 3H), 3.12 (s, 3H), 5.68 (d, J = 12.4 Hz, 1H), 7.23–7.33 (m, 2H), 7.66 (d, J = 7.2 Hz, 1H), 7.70 (s, 1H), 7.77 (d, J = 12.4 Hz, 1H). MS (ESI): [M + 1]+ = 189.9.
(E)-3-(Dimethylamino)-1-(3-methoxyphenyl) prop-2-en-1-one (5x)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5x a yellow oil. Yield 78%. 1H NMR (400 MHz, CDCl3) δ: 2.86 (s, 3H), 2.93 (s, 3H). 3.84 (s, 3H), 5.70 (d, J = 12.4 Hz, 1H), 6.99–7.02 (m, 1H), 7.29–7.33 (m, 1H), 7.43–7.45 (m, 2H), 7.79 (d, J = 12.4 Hz, 1H). MS (ESI): [M + 1]+ = 206.2.
(E)-3-(Dimethylamino)-1-(2-methoxyphenyl) prop-2-en-1-one (5y)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc–MeOH 9.5–0.5 (v/v) as eluent, to furnish compound 5x a yellow oil. Yield: 70.6%, 1H NMR (CDCl3) δ: 2.87 (m, 2H), 2.95 (s, 3H), 3.84 (s, 3H) 5.58 (d, J = 12 Hz, 1H), 6.91 (m, 2H), 7.33 (m, 1H), 7.45–7.6 (bs, 2H). MS (ESI): [M + 1]+ = 206.14.
(E)-3-Dimethylamino-1-(4-ethoxyphenyl)-prop-2-en-1-one (5z)
Synthesised according to procedure A, the crude residue was suspended with diethyl ether and filtered to furnish compound 5z a yellow solid. Yield 68%, mp 113–115 °C. 1H NMR (CDCl3) δ: 1.43 (t, J = 7.0 Hz, 3H), 3.06 (s, 3H), 3.15 (s, 3H), 4.06 (q, J = 7.0 Hz, 2H), 5.70 (d, J = 12.2 Hz, 1H), 6.87 (d, J = 7.0 Hz, 2H), 7.87 (d, J = 7.0 Hz, 2H), 8.02 (d, J = 12.2 Hz, 1H). MS (ESI): [M + 1]+ = 220.3.
(E)-1-(3,5-Dimethoxyphenyl)-3-(dimethylamino)prop-2-en-1-one (5aa)
Synthesised according to procedure A, the crude residue was purified by flash chromatography, using EtOAc as eluent, to furnish compound 5aa as a yellow solid. Yield 67%, mp 134–135 °C. 1H NMR (CDCl3) δ: 2.92 (s, 3H), 3.14 (s, 3H), 3.83 (s, 6H), 5.72 (d, J = 12.4 Hz, 1H), 6.56 (t, J = 2.4 Hz, 1H), 7.03 (d, J = 2.4 Hz, 2H), 7.82 (d, J = 12.4 Hz, 1H). MS (ESI): [M + 1]+ = 236.3.
4-((7-Phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1a)
Following general procedure B, compound 1a was obtained as a yellow solid. Yield: 63%, mp 230–232 °C. UV (EtOH): λmax nm (log ε/M−1 cm−1): 355 (4.67). 1H NMR (DMSO-d6) δ: 7.14 (s, 2H), 7.45 (d, J = 4.8 Hz, 1H), 7.65–7.66 (m, 3H), 7.72 (d, J = 8.6 Hz, 2H), 7.81 (d, J = 8.6 Hz, 2H), 8.21–8.23 (m, 2H), 8.73 (d, J = 4.8 Hz, 1H), 10.37 (s, 1H). 13C NMR (DMSO-d6) δ: 109.05, 110.00, 116.60, 127.37 (2C), 129.15 (2C), 129.82 (2C), 130.38, 132.01, 136.00, 144.24, 146.11, 153.78, 155.65, 163.13. MS (ESI) m/z (%): 367.13 ([M + 1]+, 100); calcd for C17H14N6O2S [M]+: 366.09. Anal. calcd for C17H14N6O2S. C, 55.73; H, 3.85; N, 22.94; found: C, 55.51; H, 3.69; N, 22.69.
4-((7-(Thiophen-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1b)
Following general procedure B, compound 1b was obtained as a yellow solid. Yield: 74%, mp 244–246 °C. UV (EtOH): λmax nm (log ε/M−1 cm−1): 345 (4.82). 1H NMR (DMSO-d6) δ: 7.17 (s, 2H), 7.44 (t, J = 4.5 Hz, 1H), 7.74–7.88 (m, 3H), 7.96 (d, J = 8.5 Hz, 2H), 8.24 (d, J = 5.0 Hz, 1H), 8.54 (d, J = 3.9 Hz, 1H), 8.66 (d, J = 4.8 Hz, 1H), 10.48 (s, 1H). 13C NMR (DMSO-d6) δ: 105.12, 116.74 (2C), 127.50 (2C), 128.73 130.23, 133.15, 136.03, 136.17, 140.06, 143.94, 153.09, 155.18, 162.57. MS (ESI) m/z (%): 373.14 ([M + 1]+, 100), 353.35 (80), 331.48 (51); calcd for C15H12N6O2S [M]+: 372.05. Anal. calcd for C15H12N6O2S. C, 48.38; H, 3.25; N, 22.57; found: C, 48.27; H, 3.12; N, 22.35.
4-((7-(Pyridin-4-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1c)
Following general procedure B, compound 1c was obtained as a yellow solid. Yield: 65%, mp >300 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 373 (4.55). 1H NMR (DMSO-d6): 7.15 (s, 2H), 7.58 (d, J = 4.8 Hz, 1H), 7.74–7.85 (m, 4H), 8.17–8.21 (m, 2H), 8.79 (d, J = 4.8 Hz, 1H), 8.88–8.90 (m, 2H), 10.42 (s, 1H). 13C NMR (DMSO-d6) δ: 109.47, 116.69 (2C), 123.55 (2C), 127.43 (2C), 136.14, 137.74, 143.50, 144.09, 150.74 (2C), 153.95, 155.56, 163.24. MS (ESI) m/z (%): 381.38 (51), 368.18 ([M + 1]+, 90), 341.50 (100), 313.43 (57), 268.28 (51); calcd for C16H13N7O2S [M]+: 367.09. Anal. calcd for C16H13N7O2S. C, 52.31; H, 3.57; N, 26.69; found: C, 52.10; H, 3.46; N, 26.50.
4-((7-(Pyridin-3-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1d)
Following general procedure B, compound 1d was obtained as a yellow solid. Yield: 68%, mp 280–282 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 364 (4.51). 1H NMR (DMSO-d6) δ: 1H NMR (DMSO-d6): 7.15 (s, 2H), 7.56 (d, J = 4.8 Hz, 1H), 7.69–7.75 (m, 3H), 7.77–7.87 (m, 2H), 8.64 (dt, J = 8.1 and 2.0 Hz, 1H), 8.77 (d, J = 4.8 Hz, 1H), 8.81 (dd, J = 4.8 and 1.6 Hz, 1H), 9.35 (d, J = 2.3, 1H), 10.39 (s, 1H). 13C NMR (DMSO-d6) δ: 109.26, 116.66 (2C), 123.99, 127.38 (2C), 136.11, 137.44, 143.68, 144.15, 150.17, 152.36, 153.85, 155.53, 163.16, 172.44. MS (ESI) m/z (%): 381.41 (52), 368.26 ([M + 1]+, 100), 341.33 (63), 313.47 (50); calcd for C16H13N7O2S [M]+: 367.09. Anal. calcd for C16H13N7O2S. C, 52.31; H, 3.57; N, 26.69; found: C, 52.17; H, 3.44; N, 26.52.
4-((7-(Naphthalen-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamideide (1e)
Following general procedure B, compound 1e was obtained as a yellow solid. Yield: 59%, mp 230–232 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 325 (4.79). 1H NMR (DMSO-d6) δ: 7.15 (bs, 2H), 7.60 (d, J = 5.9 Hz, 1H), 7.64–7.70 (m, 2H), 7.71–7.76 (m, 2H), 7.81–7.89 (m, 2H), 8.03–8.14 (m, 2H), 8.17 (d, J = 8.8 Hz, 1H), 8.28 (dd, J = 8.8 and 1.8 Hz, 1H), 8.78 (d, J = 4.8 Hz, 1H), 8.92 (d, J = 1.8 Hz,1H), 10.40 (s, 1H). 13C NMR (DMSO-d6) δ: 109.29, 116.66 (2C), 126.00, 127.35 (2C), 127.61, 127.74, 128.23, 128.59, 128.76, 129.38, 130.67, 132.70, 134.44, 136.04, 144.23, 146.04, 153.81, 155.72, 163.16. MS (ESI) m/z (%): 313.41 (48), 353.46 (100), 381.55 (51), 417.38 ([M + 1]+, 50); calcd for C21H16N6O2S [M]+: 416.11. Anal. calcd for C21H16N6O2S. C, 60.56; H, 3.87; N, 20.18; found: C, 60.46; H, 3.70; N, 20.02.
4-((7-(4-Fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1f)
Following general procedure B, compound 1f was obtained as a yellow solid. Yield: 63%, mp 212–214 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 355 (4.57). 1H NMR (DMSO-d6) δ: 7.14 (bs, 2H), 7.46 (d, J = 4.8 Hz, 1H), 7.52 (t, J = 8.9 Hz, 2H), 7.74 (d, J = 8.9 Hz, 2H), 7.80 (d, J = 8.8 Hz, 2H), 8.31–8.34 (m, 2H), 8.72 (d, J = 4.8 Hz, 1H), 10.38 (s, 1H). 13C NMR (DMSO-d6) δ: 108.97, 116.19 and 116.41 (J2CF = 22.2 Hz, 2C), 116.62 (2C), 126.84, 127.42 (2C), 132.55 and 132.64 (J3CF = 9.0 Hz, 2C), 136.03, 144.20, 145.12, 153.75, 155.64, 163.11 and 165.48 (J1CF = 237 Hz), 172.46. MS (ESI) m/z (%): 178.27 (72), 385.22 ([M + 1]+, 100); calcd for C17H13FN6O2S [M]+: 384.08. Anal. calcd for C17H13FN6O2S. C, 53.12; H, 3.41; N, 21.86; found: C, 53.01; H, 3.29; N, 21.70.
4-((7-(3-Fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1g)
Following general procedure B, compound 1g was obtained as a yellow solid. Yield: 68%, mp 218–220 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 361 (4.68). 1H NMR (DMSO-d6) δ: 7.15 (bs, 2H), 7.50–7.58 (m, 2H), 7.68–7.76 (m, 3H), 7.81 (d, J = 8.8 Hz, 2H), 8.06 (d, J = 8.0 Hz, 1H), 8.18 (d, J = 9.2 Hz, 1H), 8.75 (d, J = 4.8 Hz, 1H), 10.40 (s, 1H). 13C NMR (DMSO-d6) δ: 109.25, 116.64 (2C), 116.73 and 116.97 (J2CF = 24 Hz), 118.80 and 119.01 (J2CF = 21 Hz), 126.09, 127.36 (2C), 131.31 and 132.39 (J3CF = 8.0 Hz), 132.34 and 132.43 (J3CF = 9 Hz), 136.10, 144.18, 144.55, 153.78, 155.64, 163.14, 160.99 and 163.41 (J1CF = 242 Hz). MS (ESI) m/z (%): 353.49 (49), 385.15 ([M + 1]+, 100); calcd for C17H13FN6O2S [M]+: 384.08. Anal. calcd for C17H13FN6O2S. C, 53.12; H, 3.41; N, 21.86; found: C, 53.02; H, 3.26; N, 21.75.
4-((7-(2-Fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1h)
Following general procedure B, compound 1h was obtained as a yellow solid. Yield: 52%, mp 223–225 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 354 (4.55). 1H NMR (DMSO-d6) δ: 7.13 (bs, 2H), 7.38 (d, J = 4.8 Hz, 1H), 7.45–7.53 (m, 3H), 7.69 (d, J = 8.6 Hz, 2H), 7.76 (d, J = 8.4 Hz, 2H), 7.89 (t, J = 8.2 Hz, 1H), 8.76 (d, J = 4.8 Hz, 1H), 10.38 (s, 1H). 13C NMR (DMSO-d6) δ: 111.12, 116.59 (2C), 116.59 and 116.81 (J2CF = 21 Hz), 118.75, 125.29, 127.33 (2C), 131.95, 133.97 and 134.05 (J3CF = 8.4 Hz), 136.06, 141.69, 144.16, 153.56, 155.14, 158.48 and 160.98 (J1CF = 250 Hz), 163.15. MS (ESI) m/z (%): 280.38 (50), 385.24 ([M + 1]+, 100); calcd for C17H13FN6O2S [M]+: 384.08. Anal. calcd for C17H13FN6O2S. C, 53.12; H, 3.41; N, 21.86; found: C, 52.94; H, 3.18; N, 21.63.
4-((7-(2,6-Difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1i)
Following general procedure B, compound 1i was obtained as a yellow solid. Yield: 59%, mp 248–250 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 355 (4.64). 1H NMR (DMSO-d6) δ: 7.14 (bs, 2H), 7.43 (td, J = 8.5 and 4.5 Hz, 2H), 7.52–7.61 (m, 1H), 7.69–7.71 (m, 2H), 7.74–7.83 (m, 3H), 8.81 (d, J = 4.8 Hz, 1H), 10.43 (s, 1H). 13C NMR (DMSO-d6) δ: 112.49, 112.79 and 113.01 (J2CF = 22 Hz, 2C), 116.66 (2C), 127.33 (2C), 134.68, 134.78 and 134.88 (J3CF = 10.0 Hz), 135.92, 136.21, 144.01, 153.48, 155.00, 158.69 and 161.19 (J1CF = 250 Hz, 2C), 163.29. MS (ESI) m/z (%): 353.52 (51), 403.11 ([M + 1]+, 100); calcd for C17H12F2N6O2S [M]+: 402.07. Anal. calcd for C17H12F2N6O2S. C, 50.74; H, 3.01; N, 20.89; found: C, 50.55; H, 2.85; N, 20.71.
4-((7-(2,3,6-Trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1j)
Following general procedure B, compound 1j was obtained as a yellow solid. Yield: 52%, mp 263–265 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 359 (4.62). 1H NMR (DMSO-d6) δ: 7.14 (bs, 2H), 7.50 (tdd, J = 9.2, 3.7 and 2.0 Hz, 1H), 7.58 (d, J = 4.8 Hz, 1H), 7.69 (d, J = 9.2 Hz, 2H), 7.75 (d, J = 9.2 Hz, 2H), 7.89 (qd, J = 9.6 and 5.0 Hz, 1H), 8.84 (d, J = 4.8 Hz, 1H), 10.45 (s, 1H). 13C NMR (DMSO-d6) δ: 109.82 and 109.98 (J3CF = 16.0 Hz), 112.52, 112.98 and 113.22 (J2CF = 24 Hz), 114.99, 116.71 (2C), 121.31 and 121.50 (J2CF = 19 Hz), 121.41 and 121.60 (J2CF = 19 Hz), 127.34 (2C), 133.53, 134.69, 136.30, 143.96 and 146.42 (J1CF = 245 Hz), 148.15 145.86 and 148.27 (J1CF = 241 Hz), 153.59, 154.10 and 156.58 (J1CF = 248 Hz), 155.00, 163.34. MS (ESI) m/z (%): 421.29 ([M + 1]+, 100); calcd for C17H11F3N6O2S [M]+: 420.06. Anal. calcd for C17H11F3N6O2S. C, 48.57; H, 2.64; N, 19.99; found: C, 48.45; H, 2.48; N, 19.77.
4-((7-(4-Chlorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1k)
Following general procedure B, compound 1k was obtained as a yellow solid. Yield: 62%, mp 242–244 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 360 (4.63). 1H NMR (DMSO-d6) δ: 7.14 (bs, 2H), 7.47 (d, J = 4.8 Hz, 1H), 7.74 (d, J = 8.8 Hz, 4H), 7.80 (d, J = 8.8 Hz, 2H), 8.26 (d, J = 8.8 Hz, 2H), 8.73 (d, J = 4.8 Hz, 1H), 10.38 (s, 1H). 13C NMR (DMSO-d6) δ: 109.01, 116.65 (2C), 127.43 (2C), 129.16, 129.27 (2C), 131.71 (2C), 136.06, 136.81, 144.15, 144.92, 153.80, 155.61, 163.10. MS (ESI) m/z (%): 331.34 (35), 353.33 (100), 401.23 ([M + 1]+, 41), 402.63 ([M + 1]+, 18); calcd for C17H13ClN6O2S [M]+: 400.05. Anal. calcd for C17H13ClN6O2S. C, 50.94; H, 3.27; N, 20.97; found: C, 50.78; H, 3.12; N, 20.78.
4-((7-(3-Chlorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1l)
Following general procedure B, compound 1l was obtained as a yellow solid. Yield: 64%, mp 238–240 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 363 (4.60). 1H NMR (DMSO-d6) δ: 7.16 (bs, 2H), 7.52 (d, J = 4.4 Hz, 1H), 7.66–7.77 (m, 4H), 7.82 (d, J = 8.8 Hz, 2H), 8.13 (d, J = 7.6 Hz, 1H), 8.40 (s, 1H), 8.74 (d, J = 4.8 Hz, 1H), 10.41 (s, 1H). 13C NMR (DMSO-d6) δ: 109.23, 116.64 (2C), 127.35 (2C), 128.52, 129.65, 131.11, 131.76, 132.27, 133.72, 136.10, 144.18, 144.45, 153.83, 155.58, 163.09. MS (ESI) m/z (%): 331.42 (32), 353.47 (76), 381.56 (40), 401.14 ([M + 1]+, 100), 402.72 ([M + 1]+, 41); calcd for C17H13ClN6O2S [M]+: 400.05. Anal. calcd for C17H13ClN6O2S. C, 50.94; H, 3.27; N, 20.97; found: C, 50.72; H, 3.15; N, 20.76.
4-((7-(2-Chlorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1m)
Following general procedure B, compound 1m was obtained as a yellow solid. Yield: 67%, mp 242–243 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 349 (4.57). 1H NMR (DMSO-d6) δ: 7.12 (bs, 2H), 7.34 (d, J = 4.4 Hz, 1H), 7.58 (td, J = 7.5 and 1.3 Hz, 1H), 7.64 (dd, J = 8.8 and 1.8 Hz, 1H), 7.68 (q, J = 1.8 Hz, 1H), 7.70 (d, J = 2.2 Hz, 1H), 7.72–7.76 (m, 4H), 8.78 (d, J = 4.8 Hz, 1H), 10.40 (s, 1H). 13C NMR (DMSO-d6) δ: 111.16, 116.55 (2C), 127.32 (2C), 128.05, 130.19, 131.92 (2C), 132.67, 132.86, 136.06, 144.14, 144.23, 153.57, 155.00, 163.29. MS (ESI) m/z (%): 353.41 (78), 381.43 (40), 401.14 ([M + 1]+, 100), 403.25 ([M + 1]+, 40); calcd for C17H13ClN6O2S [M]+: 400.05. Anal. calcd for C17H13ClN6O2S. C, 50.94; H, 3.27; N, 20.97; found: C, 50.70; H, 3.12; N, 20.78.
4-((7-(4-Bromophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1n)
Following general procedure B, compound 1n was obtained as a yellow solid. Yield: 62%, mp 242–244 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 312 (4.79), 360 (4.64). 1H NMR (DMSO-d6) δ: 7.14 (bs, 2H), 7.47 (d, J = 4.8 Hz, 1H), 7.74 (d, J = 8.8 Hz, 2H), 7.80 (d, J = 8.8 Hz, 2H), 7.88 (d, J = 8.8 Hz, 2H), 8.18 (d, J = 8.4 Hz, 2H), 8.73 (d, J = 4.8 Hz, 1H), 10.38 (s, 1H). 13C NMR (DMSO-d6) δ: 108.97, 116.66 (2C), 125.75, 127.43 (2C), 129.53, 131.84 (2C), 132.22 (2C), 136.07, 144.15, 145.01, 153.82, 155.63, 163.11. MS (ESI) m/z (%): 313.39 (52), 331.36 (50), 353.39 (100), 381.57 (72), 445.19 ([M + 1]+, 70), 447.19 ([M + 1]+, 78); calcd for C17H13BrN6O2S. [M]+: 444.00, 446. Anal. calcd for C17H13BrN6O2S. C, 45.85; H, 2.94; N, 18.87; found: C, 45.69; H, 2.77; N, 18.73.
4-((7-(3-Bromophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1o)
Following general procedure B, compound 1o was obtained as a yellow solid. Yield: 65%, mp 252–254 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 363 (4.60). 1H NMR (DMSO-d6) δ: 7.16 (bs, 2H), 7.52 (d, J = 4.8 Hz, 1H), 7.62 (t, J = 8.0 Hz, 1H), 7.74 (d, J = 8.8 Hz, 2H), 7.82–7.87 (m, 3H), 8.15 (d, J = 8.0 Hz, 1H), 8.57 (s, 1H), 8.74 (d, J = 4.8 Hz, 1H), 10.41 (s, 1H). 13C NMR (DMSO-d6) δ: 109.17, 116.64 (2C), 122.12, 127.35 (2C), 128.86, 131.33, 132.49 (2C), 134.63, 136.10, 144.17, 144.37, 153.86, 155.56, 163.06. MS (ESI) m/z (%): 313.42 (54), 331.33 (50), 353.47 (100), 381.56 (52), 445.20 ([M + 1]+, 33), 447.22 ([M + 1]+, 35); calcd for C17H13BrN6O2S. [M]+: 444.00, 446. Anal. calcd for C17H13BrN6O2S. C, 45.85; H, 2.94; N, 18.87; found: C, 45.71; H, 2.74; N, 18.70.
4-((7-(4-Iodophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1p)
Following general procedure B, compound 1p was obtained as a yellow solid. Yield: 75%, mp 265–267 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 314 (4.87). 1H NMR (DMSO-d6) δ: 7.14 (s, 2H), 7.46 (d, J = 4.8 Hz, 1H), 7.74 (d, J = 8.8 Hz, 2H), 7.78 (d, J = 8.8 Hz, 2H), 8.01–8.07 (m, 4H), 8.72 (d, J = 4.8 Hz, 1H), 10.37 (s, 1H). 13C NMR (DMSO-d6) δ: 99.79, 108.78, 116.65 (2C), 127.43 (2C), 129.76, 131.56 (2C), 136.06, 138.05 (2C), 144.14, 145.26, 153.80, 158.96, 163.09. MS (ESI) m/z (%): 491.25 ([M–1]+, 100), 492.63 ([M–1]+, 38); calcd for C17H13IN6O2S. [M–1]+: 491.29. Anal. calcd for C17H13IN6O2S. C, 41.48; H, 2.66; N, 17.07; found: C, 41.33; H, 2.46; N, 16.88.
4-((7-(4-Nitrophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1q)
Following general procedure B, compound 1q was obtained as a yellow solid. Yield: 73%, mp 263–265 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 380 (4.76). 1H NMR (DMSO-d6) δ: 7.14 (bs, 2H), 7.55 (d, J = 4.8 Hz, 1H), 7.74 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 8.8, 2H), 8.45–8.50 (m, 4H), 8.79 (d, J = 4.8 Hz, 1H), 10.41 (s, 1H). 13C NMR (DMSO-d6) δ: 109.79, 115.01, 116.71 (2C), 124.13 (2C), 127.46 (2C), 131.39 (2C), 136.14, 136.40, 144.06, 149.31, 153.98, 155.52, 163.19. MS (ESI) m/z (%): 318.58 (100), 412.25 ([M + 1]+, 97); calcd for C17H13N7O4S [M]+: 411.07. Anal. calcd for C17H13N7O4S. C, 49.63; H, 3.19; N, 23.83; found: C, 49.50; H, 2.98; N, 23.71.
4-((7-(3-Nitrophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1r)
Following general procedure B, compound 1r was obtained as a yellow solid. Yield: 56%, mp >300 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 370 (4.48). 1H NMR (DMSO-d6) δ: 7.17 (s, 2H), 7.64 (d, J = 4.0 Hz, 1H), 7.74 (d, J = 8.0 Hz, 2H), 7.84 (d, J = 8.0 Hz, 2H), 7.95 (t, J = 7.6 Hz, 1H), 8.48 (d, J = 7.6 Hz, 1H), 8.57 (d, J = 7.6 Hz, 1H), 8.79 (d, J = 4.4 Hz, 1H), 9.36 (s, 1H), 10.43 (s, 1H). 13C NMR (DMSO-d6) δ: 109.18, 116.72 (2C), 124.95, 126.52, 127.42 (2C), 130.91, 131.65, 136.07, 136.17, 143.60, 144.03, 148.13, 154.06, 155.52, 163.04. MS (ESI) m/z (%): 313.51 (50), 341.47 (100), 359.43 (58), 381.66 (70), 412.25 ([M + 1]+, 15); calcd for C17H13N7O4S [M]+: 411.07. Anal. calcd for C17H13N7O4S. C, 49.63; H, 3.19; N, 23.83; found: C, 49.46; H, 3.05; N, 23.70.
4-((7-(2-Nitrophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1s)
Following general procedure B, compound 1s was obtained as a yellow solid. Yield: 62%, mp 263–264 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 323 (4.58). 1H NMR (DMSO-d6) δ: 7.14 (bs, 2H), 7.49 (d, J = 4.4 Hz, 1H), 7.64–7.70 (m, 4H), 7.87 (dd, J = 7.6 and 1.6 Hz, 1H), 7.95 (t, J = 7.6 Hz, 1H), 8.04 (td, J = 7.6 and 1.6 Hz, 1H), 8.39 (d, J = 8.0 Hz, 1H), 8.83 (d, J = 4.8 Hz, 1H), 10.31 (s, 1H). 13C NMR (DMSO-d6) δ: 109.70, 116.59 (2C), 125.18, 125.53, 127.25 (2C), 133.17 (2C), 135.42, 136.21, 143.83, 144.47, 147.95, 154.45 (2C), 163.05. MS (ESI) m/z (%): 313.41 (50), 331.38 (45), 341.49 (25), 353.39 (100), 381.23 (48), 412.33 ([M + 1]+, 100); calcd for C17H13N7O4S [M]+: 411.07. Anal. calcd for C17H13N7O4S. C, 49.63; H, 3.19; N, 23.83; found: C, 49.48; H, 3.03; N, 23.68.
4-((7-(p-Tolyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1t)
Following general procedure B, compound 1t was obtained as a white solid. Yield: 54%, mp 234–236 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 315 (4.79). 1H NMR (DMSO-d6) δ: 2.44 (s, 3H), 7.14 (bs, 2H), 7.44 (d, J = 4.8 Hz, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.8 Hz, 2H), 7.81 (d, J = 8.8 Hz, 2H), 8.16 (d, J = 8.0 Hz, 2H), 8.70 (d, J = 4.8 Hz, 1H), 10.36 (s, 1H). 13C NMR (DMSO-d6) δ: 21.59, 108.59 (2C), 116.60 (2C), 127.38 (2C), 124.43, 129.73, 129.77, 135.99, 142.33, 144.24, 146.12, 153.70 (2C), 155.69, 163.07. MS (ESI) m/z (%): 381.30 ([M + 1]+, 100); calcd for C18H16N6O2S [M]+: 380.11. Anal. calcd for C18H16N6O2S. C, 56.83; H, 4.24; N, 22.09; found: C, 56.67; H, 4.13; N, 21.88.
4-((7-(m-Tolyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1u)
Following general procedure B, compound 1u was obtained as a white solid. Yield: 73%, mp 224–226 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 355 (4.63). 1H NMR (DMSO-d6) δ: 2.45 (s, 3H), 7.15 (bs, 2H), 7.44 (d, J = 4.8 Hz, 1H), 7.45–7.49 (m, 1H), 7.54 (t, J = 7.6 Hz, 1H), 7.72 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H), 8.01 (d, J = 7.6 Hz, 1H), 8.08 (s, 1H), 8.71 (d, J = 4.8 Hz, 1H), 10.37 (s, 1H). 13C NMR (DMSO-d6) δ: 21.50, 108.92, 116.61 (2C), 126.98, 127.33 (2C), 129.08 (2C), 130.24, 132.65, 136.02, 138.43, 144.25, 146.20, 153.78, 155.65, 163.09. MS (ESI) m/z (%): 313.41 (50), 331.45 (47), 353.46 (52), 381.23 [M + 1]+, 100); calcd for C18H16N6O2S [M]+: 380.11. Anal. calcd for C18H16N6O2S. C, 56.83; H, 4.24; N, 22.09; found: C, 56.62; H, 4.15; N, 21.81.
4-((7-(o-Tolyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1v)
Following general procedure B, compound 1v was obtained as a white solid. Yield: 69%, mp 220–222 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 344 (4.66). 1H NMR (DMSO-d6) δ: 2.18 (s, 3H), 7.12 (bs, 2H), 7.23 (d, J = 4.4 Hz, 1H), 7.37–7.45 (m, 3H), 7.49 (t, J = 7.2 Hz, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 8.4 Hz, 2H), 8.74 (d, J = 4.8 Hz, 1H), 10.36 (s, 1H). 13C NMR (DMSO-d6) δ: 19.81, 110.95, 116.46 (2C), 126.41, 127.33 (2C), 129.96, 130.80 (2C), 131.00, 135.95, 137.15, 144.23, 147.07, 153.48, 155.18, 163.30. MS (ESI) m/z (%): 381.23 ([M + 1]+, 100); calcd for C18H16N6O2S [M]+: 380.11. Anal. calcd for C18H16N6O2S. C, 56.83; H, 4.24; N, 22.09; found: C, 56.60; H, 4.11; N, 21.85.
4-((7-(4-Methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1w)
Following general procedure B, compound 1w was obtained as a yellow solid. Yield: 68%, mp 236–238 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 331 (4.91). 1H NMR (DMSO-d6) δ: 3.89 (s, 3H), 7.14 (bs, 2H), 7.20 (d, J = 8.8 Hz, 2H), 7.43 (d, J = 4.8 Hz, 1H), 7.74 (d, J = 8.8 Hz, 2H), 7.82 (d, J = 8.8 Hz, 2H), 8.30 (d, J = 8.8 Hz, 2H), 8.67 (d, J = 4.8 Hz, 1H), 10.36 (s, 1H). 13C NMR (DMSO-d6) δ: 56.05, 108.07, 114.65 (2C), 116.58 (2C), 122.25, 127.42 (2C), 131.75 (2C), 135.94, 144.27, 145.84, 153.52, 155.77, 162.32, 163.02. MS (ESI) m/z (%): 353.21 (33), 381.36 (24), 397.23 ([M + 1]+, 100); calcd for C18H16N6O3S [M]+: 396.10. Anal. calcd for C18H16N6O3S. C, 54.54; H, 4.07; N, 21.20; found: C, 54.42; H, 3.98; N, 21.07.
4-((7-(3-Methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1x)
Following general procedure B, compound 1x was obtained as a yellow solid. Yield: 63%, mp 267–269 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 324 (4.65). 1H NMR (DMSO-d6) δ: 3.87 (s, 3H), 7.16 (bs, 2H), 7.21 (dd, J = 8.4 and 2.6 Hz, 1H), 7.48 (d, J = 4.8 Hz, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.72–7.77 (m, 3H), 7.82-7.87 (m, 3H), 8.72 (d, J = 4.8 Hz, 1H), 10.38 (s, 1H), 13C NMR (DMSO-d6) δ: 55.88, 109.09, 115.13, 116.60 (2C), 117.91, 122.06, 127.33 (2C), 130.37, 131.54, 136.04, 144.22, 145.89, 153.79, 155.66, 159.56, 163.09. MS (ESI) m/z (%): 313.40 (24), 341.49 (34), 397.35 ([M + 1]+, 100); calcd for C18H16N6O3S [M]+: 396.10. Anal. calcd for C18H16N6O3S. C, 54.54; H, 4.07; N, 21.20; found: C, 54.38; H, 3.88; N, 21.10.
4-((7-(2-Methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1y)
Following general procedure B, compound 1y was obtained as a yellow solid. Yield: 52%, mp 252–254 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 343 (4.64). 1H NMR (DMSO-d6) δ: 3.78 (s, 3H), 7.12–7.21 (m, 3H), 7.26–7.29 (m, 2H), 7.58–7.64 (m, 2H), 7.68 (d, J = 9.2 Hz, 2H), 7.75 (d, J = 9.2 Hz, 2H), 8.70 (d, J = 4.4 Hz, 1H), 10.33 (s, 1H). 13C NMR (DMSO-d6) δ: 56.25, 111.14, 112.40, 116.46 (2C), 119.65, 120.88, 127.30 (2C), 131.08, 133.04, 135.89, 144.30, 144.96, 153.24, 155.04, 157.38, 162.99. MS (ESI) m/z (%): 313.40 (15), 341.41 (17), 397.18 ([M + 1]+, 100); calcd for C18H16N6O3S [M]+: 396.10. Anal. calcd for C18H16N6O3S. C, 54.54; H, 4.07; N, 21.20; found: C, 54.40; H, 3.90; N, 21.03.
4-((7-(4-Ethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1z)
Following general procedure B, compound 1z was obtained as a white solid. Yield: 60%, mp 280–282 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 334 (4.99). 1H NMR (DMSO-d6) δ: 1.37 (t, J = 7.2 Hz, 3H), 4.16 (q, J = 7.2 Hz, 2H), 7.14 (s, 2H), 7.18 (d, J = 8.8 Hz, 2H), 7.43 (d, J = 5.2 Hz, 1H), 7.74 (d, J = 9.2 Hz, 2H), 7.82 (d, J = 9.2 Hz, 2H), 8.28 (d, J = 8.8 Hz, 2H), 8.66 (d, J = 4.8 Hz, 1H), 10.35 (s, 1H), 13C NMR (DMSO-d6) δ: 14.99, 64.04, 108.01, 115.01 (2C), 116.59 (2C), 122.07, 127.41 (2C), 131.76 (2C), 135.94, 144.28, 145.84, 153.49, 153.78, 161.63, 163.02. MS (ESI) m/z (%): 313.52 (22), 341.46 (46), 411.24 ([M + 1]+, 100); calcd for C19H18N6O3 [M]+: 410.12. Anal. calcd for C19H18N6O3S. C, 55.60; H, 4.42; N, 20.48; found: C, 55.50; H, 4.33; N, 20.36.
4-((7-(3,5-Dimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1aa)
Following general procedure B, compound 1aa was obtained as a yellow solid. Yield: 53%, mp 245–246 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 317 (4.73). 1H NMR (DMSO-d6) δ: 3.85 (s, 6H), 6.79 (t, J = 2.3 Hz, 1H), 7.16 (bs, 2H), 7.42 (d, J = 2.4 Hz, 2H), 7.50 (d, J = 4.8 Hz, 1H), 7.71 (d, J = 8.8 Hz, 2H), 7.82 (d, J = 8.8 Hz, 2H), 8.72 (d, J = 4.8 Hz, 1H), 10.39 (s, 1H). 13C NMR (DMSO-d6) δ: 56.05 (2C), 104.00, 107.87 (2C), 109.11, 116.59 (2C), 127.28 (2C), 131.97, 136.07, 144.20, 145.82, 153.79, 155.62, 160.85 (2C), 163.05. MS (ESI) m/z (%): 353.51 (61), 381.44 (51), 427.35 ([M + 1]+, 100); calcd for C19H18N6O4S [M]+: 426.11. Anal. calcd for C19H18N6O4S. C, 53.51; H, 4.25; N, 19.71; found: C, 53.38; H, 4.13; N, 19.59.
4-((7-(3,4-Dimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino)benzenesulfonamide (1ab)
Following general procedure B, compound 1ab was obtained as a yellow solid. Yield: 61%, mp 286–288 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 344 (4.95). 1H NMR (DMSO-d6) δ: 3.89 (s, 3H), 3.90 (s, 3H), 7.16 (s, 2H), 7.22 (d, J = 8.6 Hz, 1H), 7.49 (d, J = 5.2 Hz, 1H), 7.72 (d, J = 8.8 Hz, 2H), 7.82 (d, J = 8.8 Hz, 2H), 7.92 (dd, J = 8.4 and 2.4 Hz, 1H), 7.99 (d, J = 2.4 Hz, 1H), 8.67 (d, J = 4.8 Hz, 1H), 10.36 (s, 1H), 13C NMR (DMSO-d6) δ: 56.15, 56.25, 108.15, 112.01, 113.04, 116.60 (2C), 122.24, 123.67, 127.33 (2C), 130.03, 144.21, 145.97, 148.79, 152.12, 153.99, 155.74, 162.96. MS (ESI) m/z (%): 313.49 (36), 341.48 (51), 427.22 ([M + 1]+, 100); calcd for C19H18N6O4S [M]+: 426.11. Anal. calcd for C19H18N6O4S. C, 53.51; H, 4.25; N, 19.71; found: C, 53.33; H, 4.12; N, 19.54.
4-((7-(3,4,5-Trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)amino) benzenesulfonamide (1ac)
Following general procedure B, compound 1ac was obtained as a yellow solid. Yield: 53%, mp 223–224 °C. UV (EtOH): λmax nm (log ε/M–1 cm–1): 3.38 (4.64). 1H NMR (DMSO-d6) δ: 3.79 (s, 3H), 3.91 (s, 6H), 7.17 (bs, 2H), 7.55 (d, J = 4.8 Hz, 1H), 7.65 (s, 2H), 7.71 (d, J = 8.8 Hz, 2H), 7.83 (d, J = 8.8 Hz, 2H), 8.71 (d, J = 4.8 Hz, 1H), 10.37 (s, 1H). 13C NMR (DMSO-d6) δ: 56.65 (2C), 60.74, 107.75 (2C), 108.76, 116.64 (2C), 125.28, 127.26 (2C), 136.14, 140.75, 144.14, 145.93, 153.20 (2C), 153.79, 155.63, 162.98. MS (ESI) m/z (%): 266.32 (100), 457.34 ([M + 1]+, 100), 595.55 (61); calcd for C20H20N6O5S [M]+: 456.12. Anal. calcd for C20H20N6O5S. C, 52.62; H, 4.42; N, 18.41; found: C, 52.52; H, 4.28; N, 18.30.
Ultraviolet–visible spectrophotometry assay
Stock solutions of compounds 1a–ac (10 mM) dissolved in DMSO were diluted in EtOH at the final concentration of 50 µM. UV–vis spectra (290–500 nm) were recorded in a Spark spectrophotomer (Tecan, Männedorf, Switzerland).
Biological assays
In vitro carbonic anhydrase inhibition
An SX.18MV-R Applied Photophysics stopped-flow instrument (Oxford, UK) has been used for the CO2 hydration activity measurements of various CA isoenzymes. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.5) as buffer, 0.1 M Na2SO4 (for maintaining constant the ionic strength), following the CA-catalysed CO2 hydration reaction for a period of 10–100 s. Saturated CO2 solutions in water at 20 °C were used as substrate. Stock solutions of inhibitors were prepared at a concentration of 1–3 mM (in DMSO–water 1:1, v/v) and dilutions up to 0.1 nM done with the assay buffer mentioned above. At least seven different inhibitor concentrations were used for measuring the inhibition constant. Inhibitor and enzyme solutions were preincubated together for 10 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. IC50 values were obtained from dose response curves working at seven different concentrations of the test compound (from 0.1 nM to 50 µM) by fitting the curves using PRISM (www.graphpad.com) and non-linear least squares methods. Triplicate experiments were done for each inhibitor concentration, and the values reported throughout the paper are the mean of such results. The inhibition constants (KI) were derived from the IC50 values by using the Cheng–Prusoff equation as follows: KI = IC50/(1 + [S]/KM) where [S] represents the CO2 concentration at which the measurement was carried out, and KM represents the concentration of the substrate at which the enzyme activity was at half maximal. All CA isoforms were recombinant proteins obtained in house, as described in the Supplementary data. The concentrations of enzymes used in the assay system were: hCA I, 11.9 nM; hCA II, 7.7 nM; hCA IV, 8.2 nM; hCA IX, 9.1 nM, and hCA XII, 11.5 nM.
Molecular docking studies
The X-ray crystal structure of human carbonic anhydrases (hCAs) in complex with inhibitors were downloaded from the Protein Data Bank (CA I: 7q0d, CA II: 6ypw, CA IV: 3fw3, CA IX: 6g9u, CA XII: 6r6y) and prepared with Prep-Wizard module (Schrodinger Release 2022-3). Protonation states of protein residues were calculated considering a temperature of 300 K and a pH of 7, waters, and other co-crystallised molecules were removed, except for the ligand and Zn ions. The molecules were prepared using Lig-Prep (Schrodinger Release 2022-3) considering the ionisation states at pH 7 ± 2. A 12 Å docking grid (inner-box 10 Å and outer-box 20 Å) was prepared using as centroid the co-crystallised ligand. The docking studies were performed using Glide SP precision (Schrodinger Release 2022-3) keeping the default parameters and setting, and it was combined with molecular mechanics generalised Born surface area (MMGBSA) calculations, implemented in the Prime module from Maestro, to re-score the three output docking poses of each compound. Molecular Operating Environment (MOE-2022.2) was used to visualise the structures and acquire the images.
Cell lines and primary GBM cell isolation and culturing
In this study, we take advantage of patient derived glioblastoma cell lines already established in our laboratory. Primary glioblastoma cells (GBM) were isolated from GBM tumours (HuTu-109 and HuTu-192) at surgery and cultured as previously describedCitation80. Informed written consent was obtained from adult patients to acquire brain tissue samples for research purposes, in accordance with the ethical guidelines approved by the Ethical Committee of the Padova University-Hospital (Protocol 2462P). Freshly resected GBM samples were subjected to mechanical dissociation using sterile scalpels and enzymatic digestions with collagenase and dispase (Roche, Basel, Switzerland), resulting in the generation of single-cell suspensions. Cells were then seeded onto fibronectin-coated plates and cultured as monolayers using a "primary GBM medium” consisting of DMEM/F12 supplemented with 10% BIT9500 (StemCell Technologies Inc., Vancouver, Canada), 20 ng/mL basic fibroblast growth factor (bFGF), and 20 ng/mL epidermal growth factor (EGF) (Cell Guidance Systems Ltd, Cambridge, UK). After isolation, cells were plated at the density of 25 000 cells/cm2.
Two human medulloblastoma (MB) cell lines, HD-MB03 (CVCL_S506) and DAOY (CVCL_1167), were purchased from ATCC (Manassas, VA) and cultured in RPMI 1640 and MEM-ALPHA (Thermo Fisher Scientific, Waltham, MA), respectively, both supplemented with 10% foetal bovine serum (FBS), 1% glutamine and 1% penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA).
To mimic the hypoxic microenvironment characteristic of GBM and MB, cells were maintained in a H35 hypoxic cabinet (Don Whitley Scientific Ltd, Shipley, UK) with an atmosphere comprising 2% oxygen, 5% CO2, and balanced nitrogenCitation81.
Cell proliferation analysis
Drug stock solutions were prepared for each compound by dissolving in DMSO at the final concentration of 10 mM. Cells were seeded in 50 µL of medium in a 96-well plate at the concentration of 10 000 cells/well for HD-MB03, HuTuP108, and HuTuP192 and 1250 cells/well for DAOY. After 24 h, MDB cells were treated with the test compounds at the concentration of 10 μM, 1.8 µM cisplatin or with their combination meanwhile GBM cells were treated with the test compounds at the concentration of 10 μM, 500 µM temozolomide (TMZ) or with their combination. After 72 h of incubation, 10 μL of 100 μg/mL resazurin solution was added to each well and the plate was re-incubated for 3–4 h. The fluorescence of the wells in each plate was monitored using a Spark 10 M spectrophotometer (Tecan Group Ltd., Mannedorf, Switzerland) with a 535 nm excitation wavelength and a 600 nm emission wavelength.
Apoptosis assay
To assess the apoptotic effects of the test compounds, flow cytometric analysis was employed using the Annexin-V Fluos kit (Roche Diagnostics, Rotkreuz, Switzerland), following the manufacturer’s instructions. Cells treated with the test compounds, cisplatin for MB cells and TMZ for GBM cells as described above, were subsequently labelled with Annexin V/FITC and propidium iodide (PI). In particular, each sample was incubated for 15 min in the dark at room temperature with 2 µL of Annexin V reagent and 2 µL of PI solution in 100 µL of staining buffer and then analysed by flow cytometry. The flow cytometric analysis was conducted using a Coulter Cytomics FC500 instrument (Beckman Coulter, Brea, CA), with annexin V/FITC detected in the FL1 channel (525 nm/40) and PI fluorescence detected in the FL3 channel (620 nm/20). The whole cell population was considered in the analysis.
Antiproliferative activity in PBLs
Further experiments were conducted using peripheral blood lymphocytes (PBLs) obtained from human peripheral blood (leucocyte rich plasma-buffy coats) from healthy volunteers using a Lymphoprep™ (Serumwerk Bernburg AG, Bernburg, Germany) gradient and were used for the evaluation of the cytotoxic potential of compounds under study in normal human cellsCitation82.
Buffy coats were obtained from the Blood Transfusion Service, Azienda Ospedaliera of Padova and provided at this institution for research purposes without identifier. The samples were not obtained specifically for this study, and for this reason ethical approval was not required. Informed consent was obtained from blood donors according to Italian law no. 219 (21 October 2005). Data have been treated by the Blood Transfusion Service according to Italian law on personal management “Codice in materia di protezione dati personali” (Testo Unico D.L. giugno 30, 2003 196). The experimental procedures were carried out in strict accordance with approved guidelines.
After extensive washing with saline solution (BioConcept, Hank’s buffer saline solution), quiescent PBLs were resuspended (1.0 × 106 cells/mL) in RPMI-1640 medium supplemented with 10% FBS. Cytotoxicity evaluations were conducted also in cultures of proliferating PBLs, stimulated with 2.5 mg/mL PHA (Irvine Scientific, Irvine, CA).
To evaluate cytotoxicity in proliferating PBL cultures, non-adherent cells were resuspended at a concentration of 5 × 105 cells/mL in a growth medium containing 2.5 g/mL of PHA (Irvine Scientific, Irvine, CA). The same cellular density was utilised for resting PBL cultures, but without the addition of PHA. The test compounds were added at different concentrations, and after a 72-h incubation period, cell viability was determined using the Resazurin test.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, La Jolla, CA). The data presented in bar graphs are represented as mean ± standard error of the mean (SEM). Statistical comparisons among three or more experimental groups were performed using one-way ANOVA followed by Newman–Keuls post-test multiple comparison. Statistical significance is indicated by asterisks placed above the bars to denote a significant difference compared to control cells or specific experimental groups (indicated within brackets, if applicable). The significance levels were defined as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results and discussion
Synthesis
The novel designed 2-(4′-aminobenzenesulfonamido)-7-substituted-[1,2,4]triazolo[1,5-a]pyrimidines 1a–ac were synthesised using a three-step synthetic procedure described in Scheme 1.
Scheme 1. Reagents. (a) THF, sulphanilamide, reflux, 18 h; (b) NH2NH2·H2O, THF, rx, 18 h; (c) DMF-DMA, DMF, reflux, 4 h; (d) AcOH, 80 °C, 4 h.
The condensation of diphenyl cyanocarbonimidate with 4-aminobenzensulfonamide resulted in the formation of intermediate 2, which was heated to reflux with hydrazine hydrate in tetrahydrofuran to form the 4-((5-amino-1H-1,2,4-triazol-3-yl)amino)benzenesulfonamide derivative 3. This latter compound was the key intermediate for the preparation of final compounds 1a–ac via a parallel synthesis. The best cyclisation reaction conditions to synthesise 2-(4′-aminobenzenesulfonamido)-7-substituted-[1,2,4]triazolo[1,5-a]pyrimidine derivatives 1a–ac were treating triazole 3 (1 equiv.) and enaminones 5a–ac (2 equiv.) in glacial acetic acid at 80 °C for 4 h. Through this procedure, the insoluble triazole derivative 3 was completely consumed. Enaminones 5a–ac were generated by the condensation of commercially available ketones 4a–ac with an excess of N,N-dimethylformamide dimethyl acetal (DMF-DMA) at reflux for 4 h.
As far as we know, all the prepared compounds reported in this study are novel and were structurally characterised and confirmed by means of spectral analysis (1H NMR, 13C NMR, ESI-MS, and UV–vis) as well as elemental analysis. The synthetic details were described in “Materials and methods” section and the original spectra of these compounds were provided in the supplementary materials.
The 1H NMR spectra of the target compounds 1a–ac confirmed the success of the cyclisation reaction, revealing increased integration of the aromatic protons corresponding to the additional aryl or heteroaryl ring at the C-7 position of [1,2,4]triazolo[1,5-a]pyrimidine scaffold.
In the 1H NMR spectra of all synthesised molecules 1a–ac, the aromatic protons at the C-5 and C-6 positions of pyrimidine portion of triazolopyrimidine core can be identified as an AX system. A down-field doublet signal around δ 8.70–8.90 ppm with coupling constant of 4.8 Hz can be attributed to the proton (H-5) at the C-5 position.
In the 1H NMR spectra of compounds 1a–ac, the proton of anilinic nitrogen (NH) of p-sulphanilamide moiety was attributed to the singlet signal around δ 10.3–10.4 ppm, while both the hydrogens of sulphonamide moiety (SO2NH2) were identified by a signal as a sharp singlet around δ 7.14 ppm.
1H NMR spectra of compounds 1t, 1u, and 1v showed a characteristic up-field singlet peak at δ 2.44, 2.45, and 2.18 ppm, respectively, corresponding to the methyl protons on the phenyl ring at the 7-position of the triazolopyrimidine scaffold. For compounds 1w, 1x, and 1y, the presence of methoxy group on C-7 phenyl ring of the triazolopyrimidine nucleus was detected by the singlet signals at δ 3.89, 3.87, and 3.78 ppm, respectively.
The 1H NMR spectra of compound 1z revealed the presence of triplet signal at 1.37 ppm and a quartet signal around 4.17 ppm due to the ethoxy group at p-position of C-7 phenyl ring. The 13C NMR spectra of compounds 1z revealed two signals at 14.99 and 64.04 ppm assigned for the two carbon atoms of the ethoxy moiety.
13C NMR spectral analysis for tolyl derivatives 1t, 1u, and 1v displayed the typical absorptions for methyl carbons in the aliphatic region at δ 21.59, 21.50, and 19.81 ppm, respectively. 13C NMR spectra exhibit the signal referring to methoxy carbon of compounds 1w, 1x, and 1y at δ 56.05, 55.88, and 56.25, respectively. 13C NMR spectra for compound 1ab exhibited the signals with δ of 56.15 and 56.25 ppm assigned to the two methoxy groups at the 3′- and 4′-position of phenyl ring, confirmed by 1H NMR spectra with two peaks at δ 3.89 and 3.90 ppm. 13C NMR spectra for 1aa revealed the signal at δ 56.05 ppm assigned to magnetic equivalent 3′ and 5′-methoxy groups, verified by 1H NMR spectra with peak at δ 3.85 ppm.
The UV–vis spectra of compounds 1a–ac showed a single absorption with a λmax ranging from 312 to 380 nm, with the exception of compound 1n that displayed two absorption bands with λmax at 312 and 360 nM. Compound 1r showed an absorption band shoulder at 370 nM. Absorption data for compounds 1a–ac are presented in Table 1s as Supplementary data.
Carbonic anhydrase in vitro inhibition activity
The inhibition profiles for all the triazolopyrimidine derivatives 1a–ac against a panel of five different hCA isoforms involved in crucial physiologic/pathologic processes, namely hCAs I, II, IV, IX, and XII, are reported in . All inhibition constants (KIs) were determined by means of the stopped-flow CO2 hydrase assayCitation83. Results are compared with the sulphonamide CA inhibitors acetazolamide (AAZ) and SLC-0111 used as reference compounds. All synthesised compounds showed remarkable variation among the different CA isoenzymes and the following SARs might be drawn from the inhibition data reported in .
Table 1. Inhibition data of human CA isoforms I, II, IV, IX, and XII with novel synthesised triazolopyrimidine derivatives 1a–ac using acetazolamide (AAZ) and SLC-0111 as standard inhibitors.
The off-target cytosolic ubiquitous hCA I isoform was weakly inhibited by all the synthesised compounds reported in this manuscript, with KIs ranging between 0.50 and 9.2 µM, resulting less efficient inhibitors than reference drug AAZ (KI: 250 nM). In particular, compound 1j that incorporated the 2′,3′,6′-trifluorophenyl substitution displayed the highest inhibition profile among other compounds with a KI of 0.50 µM, while derivatives 1m (2′-chlorophenyl), 1o (3′-bromophenyl), and 1s (2′-nitrophenyl) showed less effective inhibition properties, with KIs of 9.1, 9.2, and 8.4 µM, respectively.
The in vitro kinetic data listed in revealed that all synthesised triazolopyrimidine derivatives 1a–ac displayed moderate to potent inhibitory activity towards the ubiquitous and physiologically dominant cytosolic off-target isoform hCA II (KI values ranging between 14 and 560 nM). Out of 29 evaluated compounds, eight molecules displayed the weakest hCA II inhibitory activity with KI values superior to 100 nM. In particular, compounds 1e (napht-2′-yl), 1l (3′-chlorophenyl), 1m (2′-chlorophenyl), 1n (4′-bromophenyl), 1o (3′-bromophenyl), 1p (4′-iodophenyl), 1aa (3′,5′-dimethoxyphenyl), and 1ac (3′,4′,5′-trimethoxyphenyl) showed the weakest inhibition profile with KI values of 110–559 nM. Compounds 1i (2′,6′-difluorophenyl) and its homologue 1j (2′,3′,6′-trifluorophenyl) emerged as the most efficient hCA II inhibitors with KIs of 18 and 14 nM, respectively, compared with the reference drug AAZ (KI: 12 nM), while all remaining compounds are weaker inhibitors than AAZ with no relevant SAR considerations to report. Noteworthy, replacement of the phenyl ring in the triazolopyrimidine derivative 1a (KI: 75.3 nM) with the lipophilic bulky 2′-napthyl (1e, KI: 326 nM) or the insertion of a sterically bulky iodo group at the 4′-position of the phenyl group (1p, KI: 559 nM) led to about four- and sevenfold reduced hCA II inhibitory effect, respectively.
As for the slow cytosolic isoform hCA I, also the membrane-bound isoform hCA IV was weakly inhibited by all of the reported compounds within the inhibition constants in the range of 0.33–8.8 µM. hCA IV inhibitory profiles of all evaluated compounds were found to be lower than the reference compound AAZ. Among them, compounds 1d (pyridin-3-yl) and 1x (3′-methoxyphenyl) showed slightly better activity as compared to other compounds from the series, with KIs of 0.33 and 0.56 µM, respectively.
Based on the in vitro kinetic data displayed in , all triazolopyrimidine derivatives reported here potently inhibited the target transmembrane tumour-associated hCA IX isoform with single/two-digit KI values ranging between 4.7 and 96 nM. Compounds 1d (pyridin-3-yl), 1j (2′,4′,6′-trifluoro), 1v (2′-methyl), and 1x (3′-methoxy) showed better inhibitory efficacy against hCA IX, as compared to other derivatives with KI in the single-digit nanomolar range, with derivative 1v (KI: 4.7 nM) as the best inhibitor in this series, which showed a selectivity index (SI) of 9.1 on off-target hCA II (). The shift of the 2-methyl group in compound 1v to the 3′-position, to yield compound 1u, resulted in a 5.5-fold decrease in activity. The reduction in activity was more dramatic (8.8-fold) for the 4′-tolyl derivative 1t. Another interesting result was for compound 1m (2′-chloro) that had best selectivity (10-fold) against hCA II, although it has threefold reduced potency as compared to the one of 1v (4.7 and 14.8 nM, respectively) for inhibiting hCA IX.
Table 2. Selectivity indexes (SIs) for the inhibition of transmembrane human CA isoforms hCA IX and XII over off-targets isoforms hCA I, IV, and II for target 2-sulfanilamido triazolopyrimidines 1a–ac, acetazolamide (AAZ) and SLC-0111.
Further analysis of the results revealed that bioisosteric replacement of the phenyl ring in the derivative 1a (KI: 33.2 nM) with the heterocyclic 3′-pyridinyl ring (1d) led to about sevenfold enhanced hCA IX inhibition potency (KI: 5.1 nM), while there were only minor differences in activity between unsubstituted phenyl and 4′-pyridinyl derivatives 1a and 1c, respectively. Also, the incorporation of the electron-donating methoxy group on the phenyl ring (3′-methoxy derivative 1x; KI: 5.1 nM) resulted in sevenfold enhanced activity compared to the unsubstituted analogues 1a (KI: 38.1 nM), while grafting three lipophilic and electron-withdrawing fluorine atoms at the 2′, 3′, and 6′-positions on the phenyl ring led to fourfold enhanced activity (1j, KI: 8.6 nM). In contrast, the incorporation of the large electron-withdrawing iodine substituent at the 4′-position of phenyl ring (compound 1p, KI: 96 nM) elicited a worsening of effectiveness towards hCA IX in comparison to its unsubstituted counterpart 1a, with a threefold reduced potency and recording the least inhibiting activity in the group. Comparison of the halogenated derivatives at the 4′-position of the phenyl ring indicated that the order of activity was F (1f) ≫ Br (1n) > Cl (1k) > I (1p), with KIs of 28.8, 52.7, 74.1, and 95.7 nM, respectively. Turning specifically to the para-substituted phenyl derivatives, these showed highly variable potencies. With the exception of 4′-fuoro derivative 1f, it was found that electron-withdrawing chlorine (1k), bromine (1n) and nitro (1q) substituents in the 4′-position resulted in lower activity against hCA IX as compared to the unsubstituted parent compound 1a, with the following order of activity F > Br > Cl = NO2 > I. The insertion of a weak electron-releasing methyl group at the 4′-position of phenyl group (1t) was tolerated and had minor effects on activity, while its replacement with a stronger electron-releasing methoxy group (1w) slightly reduced the potency (KI: 41.5 and 50.8 nM, respectively). Moreover, by shifting the electron-withdrawing chlorine and nitro or the electron releasing methyl or methoxy groups from the para- to the meta- or ortho-position of the phenyl ring, an improved inhibitory activity towards hCA IX isoform (1k vs. 1l and 1m, 1q vs. 1r and 1s, 1t vs. 1u and 1v, 1w vs. 1x and 1y, respectively) was observed. The best results were obtained with the chloro (1m), nitro (1s), or methyl (1v) groups at the ortho-position. Only in the case of fluorine substituent on the phenyl ring, no appreciable differences were observed between the three regioisomers 1f (4′-F), 1g (3′-F), and 1h (2′-F), which showed very close inhibition potencies with KIs of 28.8, 22.4, and 19.5 nM, respectively. On the contrary, a progressive reduction of the KI values and increased activity were observed when the nitro group was shifted from the para- to the meta- and ortho positions on the phenyl ring in compounds 1q, 1r, and 1s, respectively, with KI values of 69.4, 49.9, and 20.4 nM. The same effect was observed for the three chlorine isomers 1k, 1l, and 1m, as well as for the ortho-, meta-, and para-toluidine analogues 1v, 1u, and 1t, respectively. Starting from the 3′-methoxy derivative 1x, moving the methoxy group from the meta- to the ortho- or para-positions (compounds 1y and 1w, respectively) led to a drop in potency, which resulted in twofold for 1y and 10-fold for 1w. Although the 3′-methoxy group of 1x proved beneficial in enhancing potency against hCA IX isoform (KI: 5.1 nM), introducing an additional methoxy group, to yield the corresponding 3′,5′-dimethoxy (1y) and 3′,4′-dimethoxy (1ab) analogues, led to a significant decrease in activity. Specifically, these alterations resulted in a 9.2-fold reduction in activity for 1y and a 6.9-fold reduction for 1ab, respectively, compared to 1x. These findings further illustrate the delicate balance in the placement and number of methoxy groups for optimal activity against the hCA IX isoform. The addition of a third methoxy group in the 3′,4′,5′-trimethoxy derivative 1ac, led to an even greater reduction in hCA IX activity compared to both the 3′,4′-dimethoxy and 3′,5′-dimethoxy analogues. Meanwhile, the p-ethoxyphenyl homologue 1z showed equivalent potency to its methoxy counterpart 1w. This information is vital for understanding the SAR of these inhibitors and for further refinement of their design.
It is worth highlighting that 10 out of the 29 prepared compounds (1d, 1g–j, 1m, 1s, 1v, and 1x-y) demonstrated a potent inhibitory activity (KIs: 5.1–22.4) against CA IX surpassing the reference drug AAZ, which had a KI of 25 nM. These data highlight not only the promising potential of these newly developed compounds but also the potential for improvement and refinement in their future design for even greater inhibitory activity against CA IX.
A similar situation to the one previously described for hCA IX was observed in the inhibition of the second transmembrane isoform under investigation, hCA XII. Much like the hCA IX inhibition profile, all the compounds investigated turned out to be potent inhibitors of the second tumour-associated isoform, hCA XII, with KI values ranging from 4.3 to 79 nM. Four analogues, 1d (pyridin-3′-yl), 1j (2′,4′,6′-trifluorophenyl), 1r (3′-nitrophenyl), and 1ab (3′, 4′-dimethoxyphenyl), emerged as the most potent hCA XII inhibitors with KI values of 8.8, 5.4, 4.3, and 9.0 nM, respectively. In particular, derivatives 1d and 1j demonstrated excellent inhibitory activity against hCA IX isoform as well (Ki < 10 nM). Moreover, only compounds 1j and 1r showed comparable (KI: 5.4 nM) and higher (KI: 4.3 nM) potency, respectively, in comparison to AAZ (Ki: 5.7 nM) and SLC-0111 (KI: 4.5 nM) against the tumour-associated isoform CA XII.
The position of the nitro substituent on the phenyl ring influenced activity against the hCA XII isoform. There was no significant difference between the isomeric 2′- and 4′-nitro analogues 1s and 1q (KIs of 48.9 and 42.1 nM for 1s and 1q, respectively), whereas both were up to 10-fold less potent than the 3′-nitro counterpart 1r (KI: 4.3 nM).
When the unsubstituted phenyl ring in compound 1a was replaced with its bioisosteric thien-2-yl or the more lipophilic naphth-2-yl, to afford derivatives 1b and 1e, respectively, the inhibitory effect was retained (KIs: 71.7, 66.9, and 62.5 nM). On the contrary, the isomeric pyridin-4′-yl (1c) and pyridin-3′-yl (1d) derivatives displayed enhanced activity by 4.7- and 12.8-fold, respectively, compared to the phenyl compound 1a.
The introduction of substituents with different electronic and steric properties on the phenyl ring allows to maintain, such as 4′-F (1f), 3′-Br (1o), 4′-CH3 (1t), or 3′-CH3 (1u), or improve the inhibition potency against the hCA XII isozyme, with activity increased by up to 16.6-fold for the 3′-nitrophenyl derivative 1r. In particular, ortho substitutions on the phenyl ring with both electron-releasing and ERGs, such as fluorine (1h, KI: 25.8 nM), chlorine (1m, KI: 16.9 nM), nitro (1s, KI: 48.9 nM), methyl (1v, KI: 30.4 nM), or methoxy (1y, KI: 42.6 nM) led to an increase in activity compared to the unsubstituted phenyl derivative 1a (KI: 71.7 nM). For the 2′-fluorophenyl derivative 1h, a double substitution introducing an additional fluorine atom in the ortho position, to furnish the 2′,6′-difluoro derivative 1i, maintained the activity (KI: 22.5 nM). The activity was enhanced up to fourfold by introducing a third fluorine atom at the meta-position, as in the 2′.3′,6′-trifluorophenyl analogue 1j (KI: 5.4 nM).
Most of the potent compounds against both tumour-associated CA IX and XII isoforms had one or more fluorine atoms on the phenyl ring (compounds 1f–j with KIs spanning from 5.4 to 62 nM), proving to be key moieties to achieve potent inhibitors against both these isoforms. The replacement of 4′-fluorine atom with a chlorine (compounds 1f and 1k, respectively) produced a twofold increase in activity on hCA XII isoform. Moving the chlorine atom into 3′-position (compound 1l) caused a slight reduction (1.5-fold) in potency, while the shift of the chlorine atom from the 3′-to the 2′-position (compound 1m) improved about threefold the activity. The replacement of 4′-chlorine with a bromine atom (compound 1n) maintained the activity, slightly worsening shifting the bromine into 3′-position (1o). Increasing the size of the halogen from bromine to iodine (compound 1p) caused about a 1.5-fold increase in activity. The replacement of the bromine atom with an a weak electron-releasing methyl group (compound 1t) did not affect the activity, which was maintained moving the methyl group from 4′- to 3′-position (compounds 1t and 1u, respectively). The shift of the methyl group into 2′-position (compound 1v) produced a twofold increased activity as compared to the isomeric 4′-methyl analogue 1t. For compound 1p, the replacement of the iodine atom with a strong electron-releasing methoxy group (compound 1w) retained the activity, which was slightly improved shifting the methoxy group from the 4′- to 2′-position (compound 1y). Among the three isomeric methoxy derivatives 1w–y, the 3′-methoxy derivative 1x resulted in twofold more potent than 2′-methoxy 1y and 4′-methoxy 1w analogues. The 4′-ethoxy homologue 1z was slightly less active (1.3-fold) than methoxy counterpart 1w, while the introduction of a second methoxy group at the 3′-position (compound 1ab) produced a 4.4-fold increase in hCA XII activity when compared with the derivative 1w. For the 3′,4′-dimethoxy compound 1ab, the shift of methoxy group into the 5′-position to furnish the isomeric 3′,5′-dimethoxy derivative 1aa, reduced the activity (threefold), which was similar to that of 3′,4′,5′-trimethoxy analogue 1ac (KIs: 27.1, 9.0, and 31.9 nM for 1aa, 1ab, and 1ac, respectively).
Comparing the effects of ERGs and EWGs on the phenyl at the C-7 position of the triazolopyrimidine core, no clear influence on inhibition of tumour-associated hCA IX and XII isoforms was observed. Several compounds with substituents showing opposite electronic effects displayed the same potency. For instance, compound 1g containing the electron-withdrawing fluoro group showed the same potency on hCA IX as compound 1u containing the electron-donating methyl group. Similarly, for hCA XII isoform, compound 1n containing the electron-withdrawing bromo group displayed the same potency as compound 1z containing the electron-donating methoxy group.
In conclusion, SAR analysis demonstrated that all the compounds 1a–ac were weaker hCAI and hCA IV inhibitors than AAZ. The slow cytosolic hCA I and the membrane-bound hCA IV were the least inhibited among the isoforms that were tested in the current work, with Kis in the range of 0.5–9.2 µM and 0.33–8.8 µM, respectively. Sixteen of the 29 synthesised compounds (1e, 1k–q, 1s–v, 1w, 1y–z, and 1ac) showed Kis superior to 1 µM against both these isoforms. With only few exceptions, hCA IX and XII inhibitory effects of compounds 1a–ac were superior than that on hCA I, II, and IV isoenzymes. The best inhibition for compounds 1a–ac was observed for the transmembrane tumour-associated isoforms hCA IX and hCA XII, with Kis ranging between 5–96 nM and 4–72 nM, respectively. Noteworthy, compounds 1e (2′-naphthyl), 1h (2′-fluorophenyl), 1i (2′,6′-difluorophenyl), 1j (2′,3′,6′-trifluorophenyl), 1m (2′-chlorophenyl), and 1n (4′-bromophenyl) were equally potent on both the tumour-associated isoforms hCA IX and XII.
Selectivity parameters of the target compounds 1a–ac
Given that hCA IX and XII isoforms are recognised targets for the treatment of human malignancies, designing selective inhibitors for these isoforms can be challenging due to the high level of amino acid conservation among the different hCA isoforms. This has often resulted in inhibitors that also impact the physiologically relevant off-target isoforms hCA I, II, and IV, which can lead to unwanted side effects.
To understand and quantify the selectivity of the synthesised compounds, a SI is typically used. The SI is calculated as the ratio of the KI (inhibition constant) values for the off-target isoforms (hCA I, II, and IV) to the KI values for the target isoforms (hCA IX and XII). The SI can be used as a measure of how selective a given inhibitor is for the target isoforms. Inhibitors with a higher SI have a higher degree of selectivity for hCA IX and XII, meaning they are more likely to inhibit these isoforms specifically and less likely to inhibit hCA I, II, and IV. Conversely, inhibitors with a lower SI are less selective and more likely to inhibit the off-target isoforms. The SI values for each of the synthesised compounds discussed are listed in .
These values can provide insight into which compounds might be the most promising as selective hCA IX and XII inhibitors. It is important to note that while SI can provide a helpful metric in early stages of drug development, it is not the only factor to consider. Other aspects like the drug’s pharmacokinetic properties, toxicity profile, and overall efficacy must also be taken into account.
According to the calculated off-target/target SIs displayed in , all the tested compounds possessed good selectivity towards hCA IX and XII over hCA I and hCA IV. In detail, all the series of synthesised compounds were characterised by an excellent selectivity towards hCA IX and XII over hCA I (SIs in the range 17–618 and 7.5–806, respectively) and over hCA IV (SIs ranges 17–1877 and 18–384, respectively). In relation to selectivity towards hCA IX and XII over the off-target isoenzyme hCA II, most of the synthesised molecules displayed interesting selectivity profile with SI II/IX and II/XII in the range 0.8–10.5 and 0.8–14.3, respectively, regardless of their efficient inhibition of hCA IX and XII isoforms.
Of note, only compounds 1e (2′-naphtyl), 1m (2′-chlorophenyl), and 1p (4′-iodophenyl) were capable of achieving the highest SIs (ranging from 5.2 to 14.3) towards both hCA IX and XII isoforms over hCA II isoform. Regarding selectivity towards hCA IX over the off-target isoform hCA II, compounds 1m and 1v (2′-tolyl) showed the highest SI values (10.5 and 9.1, respectively), while derivative 1m along with derivatives 1p, 1ab (3′.4′-dimethoxyphenyl) and 1r (3′-nitrophenyl) displayed the best selectivity towards hCA XII over the isoform hCA II, with SIs of 9.2, 14.3, 10.6, and 11.2, respectively.
Molecular docking
In an effort to elucidate the interactions between the novel inhibitors identified in this study and various CAs, we executed a series of molecular docking simulations. These were carried out utilising the available X-ray structures of hCAs complexed with known inhibitors. As depicted in , most of the compounds displayed a consistent binding mode across the different CAs.
Figure 2. Binding mode of compound 1a in the active site of CA isoforms I (A). Binding modes of compound 1j in the active site of CA isoforms II (B), IV(C), and XII (E). Binding mode of compound 1v in the active site of CA isoforms IX (D).
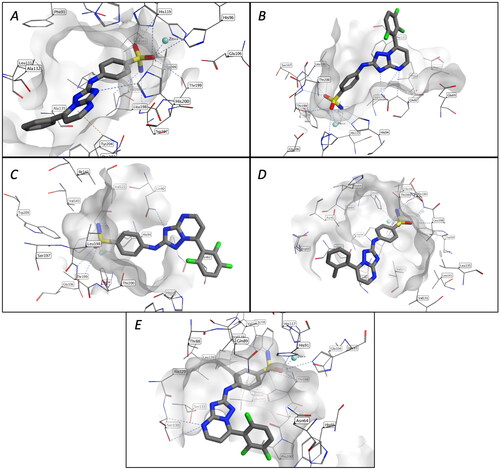
The sulphonamide moiety is located deep in the active site, showing the typical well-known coordination cluster that includes the zinc ion and the three histidine residues (His94, His96, and His119). The triazolopyrimidine core can be well accommodated within the binding pocket of CA I, II, IX, and XII, interacting with the surrounding hydrophobic and polar residues. However, the CA IV shows a smaller binding site, and despite an H-bond interaction with Gln92 and a hydrophobic interaction with the backbone of Ans62, the triazolopyrimidine core remains largely exposed to the solvent in this scenario. The aromatic ring, situated in 7-position of the triazolopyrimidine ring, forms additional interaction with the neighbouring hydrophobic residues, resulting to a general stabilisation of the binding.
Nonetheless, considering the inherent rigidity of the heterocyclic moiety, the different substituents on the phenyl group could potentially drive the selectivity of the different compounds. For instance, the methoxy group in ortho, para, or meta-position (compounds 1y, 1x, and 1w, respectively) might contribute towards the selectivity for the anhydrase IX, as illustrated in .
Figure 3. Binding modes of 1x (A), 1y (B), and 1w (C) in the active site of CA isoforms IX, and the superposition of the binding poses of the three analogues 1w–y (D).
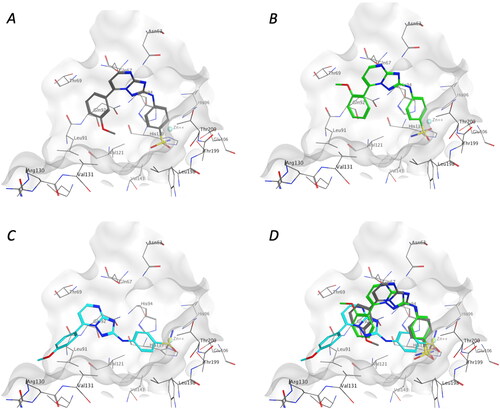
Compound 1x, featuring the meta-methoxy group, demonstrated higher activity against this isoform compared to its ortho- and para-isomers 1y and 1w, respectively. Notably, with the latter analogue 1w, the substitution changes the binding mode such that the substituted phenyl moiety becomes more exposed to the solvent, justifying the observed approximately 10-fold drop in inhibitory activity compared to the meta-isomer 1x. However, we could not observe an apparent distinction between the binding of the 4-pyridyl and the 3-pyridyl analogues 1c and 1d, respectively, towards isoform IX. On the other hand, the corresponding fluorinated analogues demonstrated reduced selectivity towards the different anhydrases, with no observable specificity in the docking results for these compounds. However, fluorination increases the overall activity against all the anhydrases, which could be related to an increase in lipophilicity for these compounds. These studies show that these compounds are promising starting points for the identification of highly selective inhibitors. As suggested from our results, modification of the aromatic ring can modulate not only the activities of compounds against CAs but also their selectivity towards the different isoforms.
Evaluation of cytotoxicity in peripheral blood lymphocytes
To get an indication of the potential toxicity of the synthesised compounds in human healthy cells, the most active compounds were tested on human lymphocytes isolated from healthy volunteers. As summarised in , as the reference compound SLC-0111, the tested compounds were inactive with a GI50 > 10 µM both in quiescent and in proliferating lymphocytes stimulated with phytohaemagglutinin (PHA). The only exception was the compound 1x that presents a GI50 of 6.0 µM in PHA stimulated lymphocytes. Altogether, these results indicate that the compounds potentially exhibit a good toxicity profile.
Table 3. Cytotoxicity of selected compound in PBL.
Biological evaluation on brain tumour cells under hypoxic conditions
hCA IX expression is controlled by the transcriptional activity of hypoxia-inducible factor 1 alpha (HIF-1a), the master regulator of the cellular response to hypoxia. hCA IX has been reported to be overexpressed in malignant brain tumours as results of the hypoxic intratumoural conditions, and have been described to be implicated in invasiveness, and correlated with therapeutic resistanceCitation84.
For these reasons, to evaluate the biological activity of those derivatives (1d, 1j, 1m, 1x, and 1y) that showed the best inhibitory activity on the hCA IX enzyme, we used two MB lines, HD-MB03 and DAOY, and two patient derived glioblastoma multiforme (GBM) cell lines, HuTuP-108 and HuTuP-192. To ensure the activation of HIF1 and the expression of CAIX, these brain tumour cell lines were grown in hypoxic conditions (1.5% oxygen tension)Citation28,Citation29.
As depicted in , in the two MB lines, at the concentration of 10 µM, the derivatives 1d, 1j, 1m, and 1y induced a decrease in cell viability in the range of about 20–35%, while compound 1x showed the best activity causing a remarkable decrease in viability around 50–60% in both cell lines. Of note, SLC-0111 taken as reference drugs and used at a concentration of 10 µM, induced only a modest decrease in viability (about 10–15%) compared to untreated cells. In order to evaluate the ability of these compounds to improve the cytotoxic activity of chemotherapeutics commonly used in clinical practice for the treatment of MB, we tested the new derivatives in combination with cisplatin (CDDP or cis-Pt). CDDP was used at concentration that induced approximately 50% viability reduction to which we added the test compounds at a concentration of 10 µM. As shown in , compounds 1j, 1m, and 1x significantly improved the efficacy of CDDP in reducing cell viability in both cell lines, while compound 1d and SLC-0111 did not.
Figure 4. Antiproliferative activity of the indicated compounds in medulloblastoma cell lines (DAOY, upper panels; HDMB lower panels). Cells were treated with the compounds at the concentration of 10 µM alone or in combination with cis-Pt (1.8 µM) for 72 h. SLC-0111 was used as reference compounds at the concentration of 10 µM. Data are expressed as mean ± SE of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
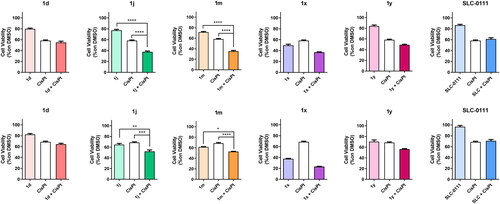
As far as the primary glioblastoma cell lines are concerned, the response of the new hCA IX inhibitors, various compounds including also the reference compound SLC-0111, was quite variable between the two lines used. In particular, as shown in , in HuTuP192, the hCA IX inhibitors induced a decrease in viability in a range from 20 to 40% (where compounds 1d and 1x are the most potent), while in HuTuP108 their antiproliferative activity is reduced. It is worthy to note that HuTuP108 cells were resistant also towards the TMZ, the chemotherapeutics commonly used for the clinical treatment of glioblastoma. In both cell lines, the treatment with the new compounds 1d, 1j, 1m, and 1y did not significantly improve the effect of TMZ.
Figure 5. Antiproliferative activity of the indicated compounds in glioblastoma primary cell lines (HuTuP192, upper panels; HuTuP108 lower panels). Cells were treated with the compounds at the concentration of 10 µM alone or in combination with TMZ (500 µM) for 72 h. SLC-0111 was used as reference compounds at the concentration of 10 µM. Data are expressed as mean ± SE of three independent experiments. *p < 0.05; ****p < 0.0001.
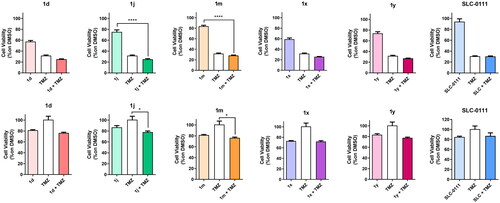
Compound 1j induced apoptosis in brain tumour cells
One of the best compounds resulting from the previous experiments was further studied for its ability to induce apoptosis both alone and in combination with cis-Pt and TMZ in MB and glioblastoma cells, respectively. As depicted in , at the concentration of 10 µM, the compound 1j induced apoptosis (about 25%) in HD-MB03 cells (, panel A), whereas, it was ineffective in HuTuP108 GBM primary cells (, panel C). Interestingly, when both HD-MB03 and HuTuP108 cell lines were treated with 1j in combination with cis-Pt and TMZ, respectively, we observed a significant increase in the percentage of apoptotic cells compared to both the single treatment with 1j and the chemotherapeutic drug.
Figure 6. Induction of apoptosis in medulloblastoma (HDMB, panels A and C) and GBM cells (HuTu-108, panels B and D) by 1j and SLC-0111 as reference compounds. Cells were treated with 1j (10 µM) or SLC-0111 (10 µM) both alone or in combination with cis-Pt (1.8 µM) or TMZ (500 µM) for 72 h. The cells were then labelled with Annexin-V and PI as described in “Materials and methods” section and analysed by flow cytometry. Data are expressed as mean ± SE of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
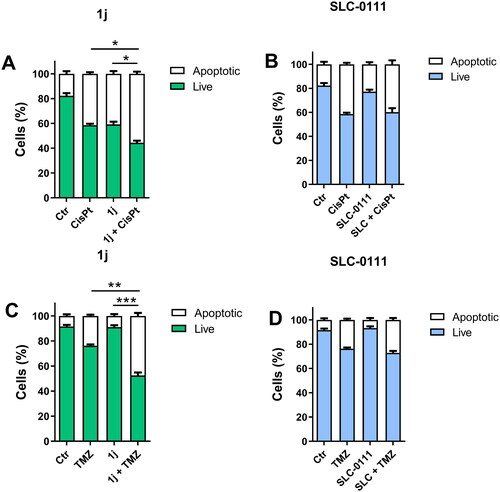
On the contrary, the reference drug SLC-0111 did not significantly induce apoptosis and did not cause any increase in cytotoxicity neither alone nor in combination with cis-Pt or TMZ in both cell lines (, panels B and D).
Conclusions
In this study, we have detailed the design, synthesis and evaluation of [1,2,4]triazolo [1,5-a]pyrimidine-based hCA inhibitors. We investigated the influence of the substitution pattern at the 7-position, while maintaining the para-sulphanilamide moiety at its 2-position. The synthesised compounds, product of an efficient three-step procedure, were evaluated for their inhibitory profiles against five therapeutically important hCA isoforms, specifically hCA I, II, IV, IX, and XII. The hCA inhibitory activity demonstrated by the synthesised analogues was highly dependent on the structural modifications at the 7-position of the triazolopyrimidine nucleus. The results unveiled their capabilities to variably influence the investigated isoforms, with KI ranges: 500–9200 nM for CA I, 14–560 nM for CA II, 330–8800 nM for CA IV, 5–96 nM for CA IX, and 4.3–79 nM for CA XII. All synthesised molecules powerfully inhibited both the transmembrane tumour-associated target isoforms hCA IX and XII, with single/two-digit KI ranging from 5 to 96 nM, with notable selectivity against the tumour isoforms hCA IX and XII over the off-target isoforms hCA I and IV. Notably, the 2′,3′.6′-trifluorophenyl (1j), 3′-nitrophenyl (1r), and 3′,5′-dimethoxyphenyl (1ab) analogues displayed superior potency towards hCA XII isoform with Ki values <10 nM (KI = 5.4, 9.0, and 4.3 nM, respectively), in comparison with the reference compound AAZ. Moreover, only the pyridin-3′-yl and 2′,3′,6′-trifluorophenyl derivatives 1d and 1j, respectively, emerged as single-digit nanomolar inhibitors against both the tumour-associated isoforms CA IX and CA XII. Compounds 1e (2′-naphtyl), 1m (2′-chlorophenyl), and 1p (4′-iodophenyl) also emerged as effective hCA IX (KIs: 58, 15, and 96 nM, respectively) and hCA XII (KIs: 62, 17, and 39 nM, respectively) inhibitors and displayed notable hCA IX/XII selectivity over the off-target hCA I, II, and IV isoforms.
Bioisosteric replacement of the phenyl with pyridin-3-yl (1d) ring considerably enhanced inhibitory activity against both hCA IX and hCA XII. The isomeric pyridin-4-yl (1c), on the other hand, yielded a fourfold increase of potency only against hCA-XII, suggesting that the substitution of phenyl with pyridine ring is advantageous for the inhibitory activity towards hCA XII. The three isomeric mono-fluorine compounds 1f–h were equally effective against hCA IX isoform, whereas their inhibitory activity against hCA XII isoform followed the order: 2′-F (1h) > 3′-F (1g) > 4′-F (1f). For the 2′-fluorophenyl derivative 1h, the insertion of an additional fluorine atom at the ortho-position on the phenyl ring, resulting in the 2′,6′-difluoro analogue 1i, sustained the activity against both hCA IX and XII isoforms. An additional fluorine at the meta-position of 1i, yielding the 2′,3′,6′-trifluorophenyl derivative 1j, led to a 2.5- and 4-fold increase in activity against hCA IX and XII isoforms, respectively.
The in vitro activity on human brain tumour cell lines showed that the compounds demonstrating the best inhibitory towards CA IX enzyme effectively suppressed the proliferation in comparison to the reference molecule SLC-0111, especially when paired with chemotherapeutics currently in clinical use. Preliminary cytotoxicity experiments further demonstrate that these compounds do not induce cytotoxicity in human non-tumour cells.
Overall, these results indicate that through small structural changes to the phenyl tail portion at the 7-position of triazolopyrimidine scaffold, allow modulation of both inhibitory activity and hCA isoform selectivity of this novel series of synthesised molecules.
Author contributions
Conceptualisation and supervision: Romeo Romagnoli, Stefano Manfredini, and Claudiu T. Supuran; writing-review and editing: Romeo Romagnoli, Stefano Manfredini, and Giampietro Viola; project administration and funding acquisition: Stefano Manfredini; software: Andrea Brancale; conducted experiments: Tiziano De Ventura, Erika Baldini, Alessio Nocentini, Roberta Bortolozzi, and Lorenzo Manfreda. All authors have read and agreed to the published version of the manuscript.
Supplemental Material
Download PDF (6 MB)Acknowledgements
R.R. and S.M. acknowledge the support of the PRIN 2017 under Grant 2017E84AA4_002.
Disclosure statement
All authors except CTS declare no competing financial interest. CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper.
Data availability statement
1H NMR, 13C NMR, ESI-mass, and UV–vis spectra of compounds 1a–ac. Preparation and purification CA isoforms. Supplementary data associated with this article can be found in the online version.
Additional information
Funding
References
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J. 2016;473(14):2023–2032.
- Krishnamurthy VM, Kaufman GK, Urbach AR, Gitlin I, Gudiksen KL, Weibel DB, Whitesides GM. Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein–ligand binding. Chem Rev. 2008;108(3):946–1051.
- Jensen EL, Clement R, Kosta A, Maberly SC, Gontero B. A new widespread subclass of carbonic anhydrase in marine phytoplankton. ISME J. 2019;13(8):2094–2106.
- Aspatwar A, Haapanen S, Parkkila S. An update on the metabolic roles of carbonic anhydrases in the model alga Chlamydomonas reinhardtii. Metabolites. 2018;8(1):22.
- Kupriyanova E, Pronina N, Los D. Carbonic anhydrase—a universal enzyme of the carbon-based life. Photosynthetica. 2017;55(1):3–19.
- Supuran CT, De Simone G. Carbonic anhydrases: an overview. In: Supuran CT, De Simone G, editors. Carbonic anhydrases as biocatalysts. From theory to medical and industrial applications. Amsterdam: Elsevier; 2015.
- Henry RP. Multiple roles of carbonic anhydrase in cellular transport and metabolism. Annu Rev Physiol. 1996;58(1):523–538.
- Geers C, Gros G. Carbon dioxide transport and carbonic anhydrase in blood and muscle. Physiol Rev. 2000;80(2):681–715.
- Alterio V, Di Fiore A, D'Ambrosio K, Supuran CT, De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev. 2012;112(8):4421–4468.
- Supuran CT, Capasso C. Chapter 13 – a catalytic carbonic anhydrases (CAs VIII, X, XI). In: Supuran CT, De Simone G, editors. Carbonic anhydrases as biocatalysts. Amsterdam: Elsevier; 2015. p. 239–245.
- D’Ambrosio K, De Simone G, Supuran CT. Chapter 2 - Human carbonic anhydrases: catalytic properties, structural features, and tissue distribution. In: Supuran CT, De Simone G, editors. Carbonic anhydrases as biocatalysts. Amsterdam: Elsevier; 2015. p. 17–30.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem. 2016;31(3):345–360.
- Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov. 2019;14(11):1175–1197.
- Supuran CT. Emerging role of carbonic anhydrase inhibitors. Clin Sci. 2021;135(10):1233–1249.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7(2):168–181.
- Aspatwar A, Tolvanen MEE, Barker H, Syrjänen L, Valanne S, Purmonen S, Waheed A, Sly WS, Parkkila S. Carbonic anhydrases in metazoan model organisms: molecules, mechanisms, and physiology. Physiol Rev. 2022;102(3):1327–1383.
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2012;27(6):759–772.
- Nocentini A, Supuran CT, Capasso C. An overview on the recently discovered iota-carbonic anhydrases. J Enzyme Inhib Med Chem. 2021;36(1):1988–1995.
- Buabeng ER, Henary M. Developments of small molecules as inhibitors for carbonic anhydrase isoforms. Bioorg Med Chem. 2021;39:116140.
- Supuran CT. Novel carbonic anhydrase inhibitors. Future Med Chem. 2021;13:1935–1937.
- Eldehna WM, Fares M, Ceruso M, Ghabbour HA, Abou-Seri SM, Abdel-Aziz HA, Abou El Ella DA, Supuran CT. Amido/ureidosubstituted benzenesulfonamides-isatin conjugates as low nanomolar/subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform XII. Eur J Med Chem. 2016;110:259–266.
- Thiry A, Dogné J-M, Supuran CT, Masereel B. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr Top Med Chem. 2007;7(9):855–864.
- Mishra CB, Tiwari M, Supuran CT. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev. 2020;40(6):2485–2565.
- Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov. 2017;12(1):61–88.
- Supuran CT. Carbonic anhydrase inhibitors: an update on experimental agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs. 2021;30(12):1197–1208.
- McDonald PC, Chafe SC, Supuran CT, Dedhar S. Cancer therapeutic targeting of hypoxia induced carbonic anhydrase IX: from bench to bedside. Cancers. 2022;14(14):3297.
- Supuran CT. Experimental carbonic anhydrase inhibitors for the treatment of hypoxic tumors. J Exp Pharmacol. 2020;12:603–617.
- Potter C, Harris AL. Hypoxia inducible carbonic anhydrase IX, marker of tumour hypoxia, survival pathway and therapy target. Cell Cycle. 2004;3(2):159–162.
- Pastorekova S, Zatovicova M, Pastorek J. Cancer-associated carbonic anhydrases and their inhibition. Curr Pharm Des. 2008;14(7):685–698.
- Lee SH, McIntyre D, Honess D, Hulikova A, Pacheco-Torres J, Cerdan S, Swietach P, Harris AL, Griffiths JR. Carbonic anhydrase IX is a pH-stat that sets an acidic tumour extracellular pH in vivo. Br J Cancer. 2018;119(5):622–630.
- Chiche J, Ilc K, Laferrière J, Trottier E, Dayan F, Mazure NM, Brahimi-Horn MC, Pouysségur J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009;69(1):358–368.
- De Simone G, Supuran CT. Carbonic anhydrase IX: biochemical and crystallographic characterization of a novel antitumor target. Biochim Biophys Acta. 2010;1804(2):404–409.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov. 2011;10(10):767–777.
- Pastorek J, Pastorekova S. Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: from biology to clinical use. Semin Cancer Biol. 2015;31:52–64.
- Kopecka J, Campia I, Jacobs A, Frei AP, Ghigo D, Wollscheid B, Riganti C. Carbonic anhydrase XII is a new therapeutic target to overcome chemoresistance in cancer cells. Oncotarget. 2015;6(9):6776–6793.
- Ulmasov B, Waheed A, Shah GN, Grubb JH, Sly WS, Tu C, Silverman DN. Purification and kinetic analysis of recombinant CA XII, a membrane carbonic anhydrase overexpressed in certain cancers. Proc Natl Acad Sci U S A. 2000;97(26):14212–14217.
- Wichert M, Krall N. Targeting carbonic anhydrase IX with small organic ligands. Curr Opin Chem Biol. 2015;26:48–54.
- Supuran CT, Alterio V, Di Fiore A, D' Ambrosio K, Carta F, Monti SM, De Simone G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev. 2018;38(6):1799–1836.
- Pacchiano F, Carta F, McDonald PC, Lou Y, Vullo D, Scozzafava A, Dedhar S, Supuran CT. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem. 2011;54(6):1896–1902.
- Dar’in D, Kantin G, Kalinin S, Sharonova T, Bunev A, Ostapenko GI, Nocentini A, Sharoyko V, Supuran CT, Krasavin M. Investigation of 3-sulfamoyl coumarins against cancer-related IX and XII isoforms of human carbonic anhydrase as well as cancer cells leads to the discovery of 2-oxo-2H-benzo[h]chromene-3-sulfonamide – a new caspase-activating proapoptotic agent. Eur J Med Chem. 2021;222:113589.
- McDonald PC, Winum JY, Supuran CT, Dedhar S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget. 2012;3(1):84–97.
- Noor SI, Jamali S, Ames S, Langer S, Deitmer JW, Becker HM. A surface proton antenna in carbonic anhydrase II supports lactate transport in cancer cells. eLife. 2018;7:e35176.
- Leppilampi M, Koistinen P, Savolainen ER, Hannuksela J, Parkkila AK, Niemela O, Pastorekova S, Pastorek J, Waheed A, Sly WS, et al. The expression of carbonic anhydrase II in hematological malignancies. Clin Cancer Res. 2002;8:2240–2245.
- Zhou R, Huang W, Yao Y, Wang Y, Li Z, Shao B, Zhong J, Tang M, Liang S, Zhao X, et al. CA II, a potential biomarker by proteomic analysis, exerts significant inhibitory effect on the growth of colorectal cancer cells. Int J Oncol. 2013;43(2):611–621.
- Parkkila S, Lasota J, Fletcher JA, Ou WB, Kivela AJ, Nuorva K, Parkkila AK, Ollikainen J, Sly WS, Waheed A, et al. Carbonic anhydrase II. A novel biomarker for gastrointestinal stromal tumors. Mod Pathol. 2010;23(5):743–750.
- Haapasalo J, Nordfors K, Haapasalo H, Parkkila S. The expression of carbonic anhydrases II, IX and XII in brain tumors. Cancers. 2020;12(7):1723.
- Zhang H, Zhuo C, Zhou D, Zhang F, Chen M, Xu S, Chen Z. Association between the expression of carbonic anhydrase II and clinicopathological features of hepatocellular carcinoma. Oncol Lett. 2019;17(6):5721–5728.
- Abdelrahman MA, Ibrahim HS, Nocentini A, Eldehna WM, Bonardi A, Abdel-Aziz HA, Gratteri P, Abou-Seri SM, Supuran CT. Novel 3-substituted coumarins as selective human carbonic anhydrase IX and XII inhibitors: synthesis, biological and molecular dynamics analysis. Eur J Med Chem. 2021;209:112897.
- Mboge MY, McKenna R, Frost SC. Advances in anti-cancer drug development targeting carbonic anhydrase IX and XII. Top Anticancer Res. 2015;5:3–42.
- Swenson ER. Safety of carbonic anhydrase inhibitors. Expert Opin Drug Saf. 2014;13:459–472.
- Akocak S, Ilies MA. Next-generation primary sulfonamide carbonic anhydrase inhibitors. In: Supuran CT, Cappasso C, editors. Targeting carbonic anhydrases. London: Future Science; 2014. p. 35–51.
- Vats L, Kumar R, Bua S, Nocentini A, Gratteri P, Supuran CT, Sharma PK. Continued exploration and tail approach synthesis of benzenesulfonamides containing triazole and dual triazole moieties as carbonic anhydrase I, II, IV and IX inhibitors. Eur J Med Chem. 2019;183:111698.
- Carta F, Supuran CT, Scozzafava A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med Chem. 2014;6(10):1149–1165.
- Kumar A, Siwach K, Supuran CT, Sharma PK. A decade of tail-approach based design of selective as well as potent tumor associated carbonic anhydrase inhibitors. Bioorg Chem. 2022;126:105920.
- Bozdag M, Ferraroni M, Nuti E, Vullo D, Rossello A, Carta F, Scozzafava A, Supuran CT. Combining the tail and the ring approaches for obtaining potent and isoform-selective carbonic anhydrase inhibitors: solution and X-ray crystallographic studies. Bioorg Med Chem. 2014;22(1):334–340.
- Liguori F, Carradori S, Ronca R, Rezzola S, Filiberti S, Carta F, Turati M, Supuran CT. Benzenesulfonamides with different rigidity-conferring linkers as carbonic anhydrase inhibitors: an insight into the antiproliferative effect on glioblastoma, pancreatic, and breast cancer cells. J Enzyme Inhib Med Chem. 2022;37(1):1857–1869.
- Wilkinson BL, Bornaghi LF, Houston TA, Innocenti A, Supuran CT, Poulsen SA. A novel class of carbonic anhydrase inhibitors: glycoconjugate benzene sulfonamides prepared by "click-tailing". J Med Chem. 2006;49(22):6539–6548.
- Fares M, Eldehna WM, Bua S, Lanzi C, Lucarini L, Masini E, Peat TS, Abdel-Aziz HA, Nocentini A, Keller PA, et al. Discovery of potent dual-tailed benzenesulfonamide inhibitors of human carbonic anhydrases implicated in glaucoma and in vivo profiling of their intraocular pressure-lowering action. J Med Chem. 2020;63(6):3317–3326.
- Allam HA, Fahim SH, Abo-Ashour MF, Nocentini A, Elbakry ME, Abdelrahman MA, Eldehna WM, Ibrahim HS, Supuran CT. Application of hydrazino and hydrazido linkers to connect benzenesulfonamides with hydrophilic/phobic tails for targeting the middle region of human carbonic anhydrases active site: selective inhibitors of hCA IX. Eur J Med Chem. 2019;179:547–556.
- Ibrahim HS, Allam HA, Mahmoud WR, Bonardi A, Nocentini A, Gratteri P, Ibrahim ES, Abdel-Aziz HA, Supuran CT. Dual-tail arylsulfone-based benzenesulfonamides differently match the hydrophobic and hydrophilic halves of human carbonic anhydrases active sites: selective inhibitors for the tumor-associated hCA IX isoform. Eur J Med Chem. 2018;152:1–9.
- Tanpure RP, Ren B, Peat TS, Bornaghi LF, Vullo D, Supuran CT, Poulsen SA. Carbonic anhydrase inhibitors with dual-tail moieties to match the hydrophobic and hydrophilic halves of the carbonic anhydrase active site. J Med Chem. 2015;58(3):1494–1501.
- Stams T, Christianson DW. X-ray crystallographic studies of mammalian carbonic anhydrase isozymes. In: Chegwidden WR, Carter ND, Edwards YH, editors. The carbonic anhydrases. EXS 90. Vol. 90. Basel: Birkhäuser; 2000. p. 159–174.
- Carta F, Vullo D, Osman SM, Alothman ZA, Supuran CT. Synthesis and carbonic anhydrase inhibition of a series of SLC-0111 analogs. Bioorg Med Chem. 2017;25(9):2569–2576.
- McDonald PC, Chia S, Bedard PL, Chu Q, Lyle M, Tang L, Singh M, Zhang Z, Supuran CT, Renouf DJ, et al. A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am J Clin Oncol. 2020;43(7):484–490.
- Eldehna WM, Abo-Ashour MF, Berrino E, Vullo D, Ghabbour HA, Rashood ST, Hassan GS, Alkahtani HM, Almehizia AA, Alharbi A, Abdel-Aziz HA, et al. SLC-0111 enaminone analogues, 3/4-(3-aryl-3-oxopropenyl)aminobenzenesulfonamides, as novel selective subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform IX. Bioorg Chem. 2019;83:549–558.
- Elbadawi MM, Eldehna WM, Nocentini A, Abo-Ashour MF, Elkaeed EB, Abdelgawad MA, Alharbi KS, Abdel-Aziz HA, Supuran CT, Gratteri P, et al. Identification of N-phenyl-2-(phenylsulfonyl) acetamides/propanamides as new SLC-0111 analogues: synthesis and evaluation of the carbonic anhydrase inhibitory activities. Eur J Med Chem. 2021;218:113360.
- Eldehna WM, Abo-Ashour MF, Nocentini A, El-Haggar RS, Bua S, Bonardi A, Al-Rashood ST, Hassan GS, Gratteri P, Abdel-Aziz HA, et al. Enhancement of the tail hydrophobic interactions within the carbonic anhydrase IX active site via structural extension: design and synthesis of novel N-substituted isatins-SLC-0111 hybrids as carbonic anhydrase inhibitors and antitumor agents. Eur J Med Chem. 2019;162:147–160.
- Dai X-J, Xue L-P, Ji S-K, Zhou Y, Gao Y, Zheng Y-C, Liu H-M, Liu H-M. Triazole-fused pyrimidines in target-based anticancer drug discovery. Eur J Med Chem. 2023;249:115101.
- Mohamed HS, Amin NH, El-Saadi MT, Abdel-Rahman HM. Design, synthesis, biological assessment, and in-silico studies of 1,2,4-triazolo[1,5-a]pyrimidine derivatives as tubulin polymerization inhibitors. Bioorg Chem. 2022;121:105687.
- Yang F, Yu LZ, Diao PC, Jian XE, Zhou MF, Jiang CS, You WW, Ma WF, Zhao PL. Novel [1,2,4]triazolo[1,5-a]pyrimidine derivatives as potent antitubulin agents: design, multicomponent synthesis and antiproliferative activities. Bioorg Chem. 2019;92:103260.
- Huo XS, Jian XE, Ou-Yang J, Chen L, Yang F, Lv DX, You WW, Rao JJ, Zhao PL. Discovery of highly potent tubulin polymerization inhibitors: design, synthesis, and structure–activity relationships of novel 2,7-diaryl-[1,2,4]triazolo[1,5-a]pyrimidines. Eur J Med Chem. 2021;220:113449.
- Alle T, Varricchio C, Yao Y, Lucero B, Nzou G, Demuro S, Muench M, Vuong KD, Oukoloff K, Cornec A-S, et al. Microtubule-stabilizing 1,2,4-triazolo[1,5‑a]pyrimidines as candidate therapeutics for neurodegenerative disease: matched molecular pair analyses and computational studies reveal new structure–activity insights. J Med Chem. 2023;66(1):435–459.
- Oliva P, Romagnoli R, Cacciari B, Manfredini S, Padroni C, Brancale A, Ferla S, Hamel E, Corallo D, Aveic S, et al. Synthesis and biological evaluation of highly active 7-anilino triazolopyrimidines as potent antimicrotubule agents. Pharmaceutics. 2022;14(6):1191.
- Romagnoli R, Oliva P, Prencipe F, Manfredini S, Budassi F, Brancale A, Ferla S, Hamel E, Corallo D, Aveic S, et al. Design, synthesis and biological investigation of 2-anilino triazolopyrimidines as tubulin polymerization inhibitors with anticancer activities. Pharmaceuticals. 2022;15(8):1031.
- Wang S, Shen DD, Zhao LJ, Yuan XH, Cheng JL, Yu B, Zheng YC, Liu HM. Discovery of [1,2,4]triazolo[1,5-a]pyrimidine derivatives as new bromodomain containing protein 4 (BRD4) inhibitors. Chin Chem Lett. 2020;31(2):418–422.
- Wang S, Li ZR, Suo FZ, Yuan XH, Yu B, Liu HM. Synthesis, structure–activity relationship studies and biological characterization of new [1,2,4]triazolo[1,5-a]pyrimidine-based LSD1/KDM1A inhibitors. Eur J Med Chem. 2019;167:388–401.
- Wang S, Zhao LJ, Zheng YC, Shen DD, Miao EF, Qiao XP, Zhao LJ, Liu Y, Huang R, Yu B, et al. Design, synthesis and biological evaluation of [1,2,4]triazolo[1,5-a]pyrimidines as potent lysine specific demethylase 1 (LSD1/KDM1A) inhibitors. Eur J Med Chem. 2017;125:940–951.
- Wang S, Wang SQ, Teng QX, Yang L, Lei ZN, Yuan XH, Huo JF, Chen XB, Wang M, Yu B, et al. Structure-based design, synthesis, and biological evaluation of new triazolo[1,5-a]pyrimidine derivatives as highly potent and orally active ABCB1 modulators. J Med Chem. 2020;63(24):15979–15996.
- Richardson CM, Williamson DS, Parratt MJ, Borgognoni J, Cansfield AD, Dokurno P, Francis GL, Howes R, Moore JD, Murray JB, et al. Triazolo[1,5-a]pyrimidines as novel CDK2 inhibitors: protein structure-guided design and SAR. Bioorg Med Chem Lett. 2006;16(5):1353–1357.
- Pistollato F, Rampazzo E, Abbadi S, Della Puppa A, Scienza R, D'Avella D, Denaro L, Te Kronnie G, Panchision DM, Basso G. Molecular mechanisms of HIF-1alpha modulation induced by oxygen tension and BMP2 in glioblastoma derived cells. PLOS One. 2009;4(7):e6206.
- Pistollato F, Abbadi S, Rampazzo E, Persano L, Della Puppa A, Frasson C, Sarto E, Scienza R, D'Avella D, Basso G. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells. 2010;28(5):851–862.
- Romagnoli R, Baraldi PG, Prencipe F, Oliva P, Baraldi S, Salvador MK, Lopez-Cara LC, Brancale A, Ferla S, Hamel E, et al. Synthesis and biological evaluation of 2-methyl-4,5-disubstituted oxazoles as a novel class of highly potent antitubulin agents. Sci Rep. 2017;7(1):46356.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.
- Pastorekova S, Gillies RJ. The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019;38(1–2):65–77.