
Abstract
Tumour associated carbonic anhydrases (CAs) IX and XII have been recognised as potential targets for the treatment of hypoxic tumours. Therefore, considering the high pharmacological potential of the chromene scaffold as selective ligand of the IX and XII isoforms, two libraries of compounds, namely 2H-chromene and 7H-furo-chromene derivatives, with diverse substitution patterns were designed and synthesised. The structure of the newly synthesised compounds was characterised and their inhibitory potency and selectivity towards human CA off target isoforms I, II and cancer-associated CA isoforms IX and XII were evaluated. Most of the compounds inhibit CA isoforms IX and XII with no activity against the I and II isozymes. Thus, while the potency was influenced by the substitution pattern along the chromene scaffold, the selectivity was conserved along the series, confirming the high potential of both 2H-chromene and 7H-furo-chromene scaffolds for the design of isozyme selective inhibitors.
Graphical Abstract
Introduction
Cancer, the second leading cause of death in most of the countries after cardiovascular diseases, is a major public health issue that affects all populations and the disease distribution and life expectancy among different social status, country, and sex, and has been investigated and discussed at lengthCitation1–3. In recent years, significant progress has been made in terms of diverse cancer such as non-small cell lung cancer, breast cancer, colon, and pancreaticCitation4–7 prevention and treatment options. However, despite this, cancer burden will continue to rise as life-expectancy and global population growCitation8,Citation9. Hence, it is of global interest to reduce this through early detection of cancer and appropriate treatment and care of cancer patients.
Carbonic anhydrases (CAs) are a class of metalloenzymes widely distributed in all living organisms encoded by eight different gene families or classes: α-, β-, γ-, δ-, ζ-, η-, θ-, and ι-CAsCitation10–16, that catalyse the reversible hydration reaction of carbon dioxide into bicarbonate and a proton (CO2 + H2O ↔ HCO3– + H+)Citation17,Citation18. Mammals encode only for α-CA classes that have 16 known isoforms with different subcellular localisation and catalytic activity. Accordingly, CA I, II, III, VII, and XIII are expressed in the cytoplasm; CA IX, XII, and XIV are membrane bound forms; CA IV and XV (this one not expressed in humansCitation19) are glycosylphosphatidylinositol (GPI)-anchored membrane forms; CA VA and CA VB are mitochondrial forms and CA VI is a saliva-secreted form. Three isoforms (VIII, X, and XI), called CA-related proteins (CARPs), are catalytically inactive as they lack one or more histidine residues coordinating the zinc ion in the active site. On the other hand, transmembrane isoforms are highly active enzymes and glycoproteinsCitation18,Citation20,Citation21.
Tumour-associated human carbonic anhydrase (hCA) IX and XII have been consistently validated as markers of disease progression in many solid tumours. Experimental evidence has indicated a strong link between pH regulation and tumour cell proliferation and survivalCitation22. These two isoforms are actively involved in carbon dioxide metabolism, and consequently play a role in pH control and tumour progression. Their overexpression in cancer and contribution to tumour physiology emphasise their relevance as both biomarkers and therapeutic targetsCitation23, therefore, attracting the research community attention on these isoforms in the last two decadesCitation23–25. The membrane-bound hCA isoforms hCA IX and hCA XII, as well as intracellular hCAs such as hCA I and hCA II, are the primarily hCA isoforms expressed in cancerCitation23,Citation26. Particularly, hCA IX is almost exclusively expressed in a broad range of tumours representing a more reliable indicator of malignant lesionsCitation23,Citation25,Citation27–30. hCA XII role in tumour biology is much less addressed in the literature when compared with hCA IX, primarily because it is expressed in both tumour and normal tissuesCitation31–33.
Not surprisingly, due to the hCA IX and XII overexpression in many tumours, in response to the hypoxia inducible factor pathway, the research on these isozymes role in cancer has significantly progressedCitation34. Studies have indicated that hCA XII is less strongly regulated by hypoxia when compared with hCA IXCitation35, but both are associated with tumour hypoxia, tumour propagation, malignant progression, and resistance to chemotherapy and radiotherapyCitation36. Hence, the two main strategies to target the tumour associated hCAs for cancer therapy include the development of monoclonal antibodies and the design and synthesis of small molecules that selectively inhibit hCA IX and XIICitation37.
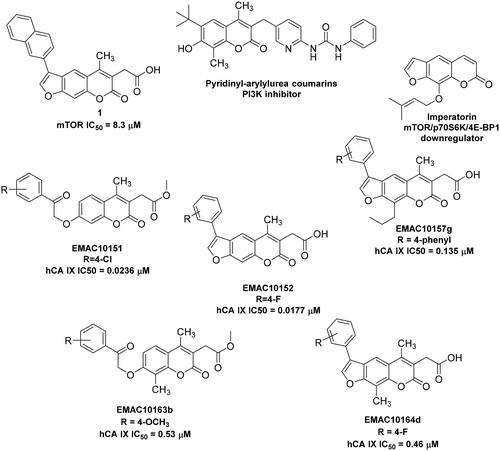
Coumarins (2H-chromen-2-one, 2H-1-benzopyran-2-one) are heterocycles, constituted by a pyrone ring fused with benzene ringCitation38. They belong to a wide family of secondary metabolites found in various species of plants, fungi, and microorganisms. According to structure differences, they are divided in different classes: simple coumarins, iso-coumarins, furanocoumarins, pyranocoumarins, and bis-coumarinsCitation39,Citation40. The type and/or location of substituents in the coumarin nucleus strongly influence the biological activity of the resulting derivativesCitation40. Coumarins exhibit several pharmacological activities including anticoagulantCitation41,Citation42, anti-inflammatoryCitation43, neuroprotectiveCitation44, antioxidantCitation45, antibacterialCitation46, anti-fungalCitation47, antiviralCitation48, and anticancerCitation38,Citation49,Citation50. Over the last 50 years, coumarin compounds have been widely used as pharmaceuticalsCitation40. Some coumarin derivatives capable to inhibit hCAs IX and XII have also been reportedCitation51–53. Very interestingly, similar coumarin derivatives have been identified as inhibitors of both mTOR and PI3K, suggesting an intriguingly potential of these molecules as poly-pharmacological agents in hypoxic tumours ()Citation54–57. Moreover, depending on the substitution pattern, coumarins could have several attractive features as scaffolds in drug discovery programs, such as low molecular weight, simple structure, high bioavailability, high solubility in most of the organic solvents and low toxicity. These features, together with their multifaceted biological activities, as well as their simple structural architecture and chemical stability, ensure them a prominent role as privileged scaffolds in drug research and developmentCitation39,Citation40.
Figure 1. Examples of 2H-chromene and 7H-furo-chromene-based potential anticancer derivativesCitation51,Citation52,Citation54,Citation55 and newly synthesised chromene series.
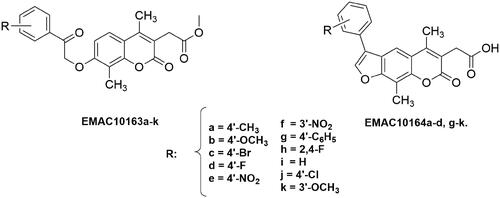
Pursuing on the research in the field of CA and anticancer agents, performed by our groupCitation51,Citation52,Citation58–62, we have designed and synthesised a new series of 2H-chromene and 7H-furo-chromene derivatives, namely compounds EMAC10163a-k and EMAC10164a-d, g-k, () to evaluate their activity and selectivity on hCA IX and XII with respect to the off-target isoforms hCA I and II. All the synthesised 2H-chromene derivatives are substituted in the positions 4 and 8 with two methyl moieties, while in the positions 3 and 7 a methylacetate and 2-oxo-2-arylethoxy groups have been introduced, respectively. To further explore the effects of structural modification on the inhibition potency and selectivity towards the diverse hCA isozymes, 7H-furo-chromene derivatives are characterised by the presence of two methyl substituents in the positions 4 and 9, a different substituted aromatic ring in the position 6 and a methylcarboxylic acid in the position 3.
Figure 2. Structure and substitution pattern of 2H-chromene EMAC10163a-k and 7H-furo-chromene EMAC10164a-d, g-k.
Results and discussion
Synthesis of 2H-chromene and 7H-furo-chromene derivatives
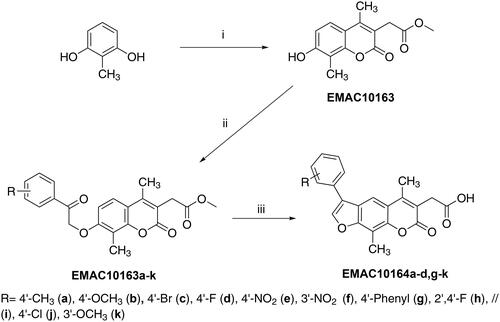
The synthetic procedures to obtain compounds EMAC10163a-k and EMAC10164a-d, g-k are represented in Scheme 1. Starting 2-(7-hydroxy-4,8-dimethyl-2-oxo-2H-chromen-3-yl)acetate, EMAC10163 was synthetised under the Pechmann condensation between 2-methylresorcinol and dimethyl 2-acetylsuccinateCitation51. EMAC10163 was further reacted under Williamson condition with differently substituted α-halogen ketones, to give derivatives EMAC10163a-k that were used as starting compounds to synthetise the furo-chromenes EMAC10164a-d, g-k. Thus, EMAC10163a-k were refluxed in a sodium hydroxide water solution and the resulting slurry was poured into iced water and acidified with a solution of HCl 6 M to give compounds EMAC10164a-d, g-k. All compounds were characterised by analytical and spectroscopic methods before being submitted to biological evaluation.
Scheme 1. Synthetic pathway to obtain compounds EMAC10163a-k and EMAC10164a-d, g-k. Reagents, and conditions: (i) dimethyl 2-acetylsuccinate, H2SO4 98%; (ii) acetone, K2CO3, α-haloketone, reflux, 6–24 h; (iii) propan-2-ol, NaOH, reflux 2–5 h.
Inhibitory activity of 2H-chromene and 7H-furo-chromene derivatives towards hCA I, II, IX, and XII
All the synthetised 2H-chromene and 7H-furo-chromene derivatives EMAC10163a-k and EMAC10164a-d, g-k were evaluated towards hCA I, II, IX, and XII isoforms to assess their inhibitory activity and selectivity towards the different isozymes. Acetazolamide (AAZ), a known potent but unselective inhibitor of hCA, was used as a reference standard. The results are summarised in .
Table 1. Inhibition data towards hCA I, II, IX, and XII of EMAC10163a-k and EMAC10164a-d, g-k.
According to our previous observation, the majority of the new derivatives are selective towards the isoforms of interest, namely hCA IX and XII. Thus, although all the active compounds show lower potency than the reference AAZ, they exhibit a better profile in terms of anticancer potential. Furthermore, some conclusions must be highlighted about selectivity. None of the compounds were active against CA I and II up to 100 µM (highest tested concentration). In the case of 7H-furo-chromene derivatives, the introduction in the position 6 of the furo-chromene of a 4′-methylphenyl, 4′-methoxyphenyl, and a 4′-bromophenyl moiety (compounds EMAC10164a, b, and c, respectively) produced a complete loss of activity towards hCA I, II, and IX, leading to a selective inhibition of CA XII. Within the 2H-chromene derivatives, the most remarkable compound was EMAC10163b, with a 4′-methoxyphenyl-7-oxoethoxy moiety in the position 7 of the chromene. This compound inhibited tumour associated hCA IX and XII at a low micromolar concentration (Ki = 0.53 µM and 0.47 µM towards hCA IX and XII, respectively). Interestingly, when the 3′-methoxyphenyl moiety is introduced in the position 7 of the furo-chromene scaffold, a decrease of the inhibition potency towards CA IX and XII was observed (Ki = >100 µM and 4.86 µM, respectively). The compound with better inhibitory activity and selectivity towards CAs IX and XII, within the EMAC10164 series, was EMAC10164d, with a 4′-fluorophenyl moiety in the position 7 of the furo-chromene (Ki = 0.46 µM and 0.80 µM, respectively). The beneficial effect of the 4′-fluorophenyl moiety in the 7 position of furo-chromenes derivatives on both activity and selectivity towards hCA IX and XII was in accordance with the data previously reportedCitation51,Citation52. These results indicate that both chromene and furo-chromene derivatives are promising scaffolds to develop potent and selective inhibitors of tumour associated hCA IX and XII. In all probability, both series of derivatives EMAC10163 and EMAC10164 share the mechanism of action already observed for similar derivatives where the chromene is hydrolysed within the CA active site to the corresponding 2-hydroxycinnamic acidsCitation51,Citation63. However, the structural requirements for activity and selectivity are different between the chromene and furo-chromene scaffolds, suggesting that distinct optimisation strategies should be applied for the two families of compounds.
Molecular modelling studies
To gain more information that could be exploited for future compound optimisation, the two derivatives that showed the highest potency and selectivity towards hCA IX and XII isoforms of each series were selected and their binding mode was investigated in more detail by the application of a validated docking protocol.
Docking experiments on hCA IX isoform of EMAC10163a and EMAC10163b are depicted in . For each compound, two different proposed binding modes were obtained. The chromene ring is oriented towards the zinc in one of the proposed binding modes. In the second proposed mode, on the other hand, the substituted phenyl ring points towards the zinc.
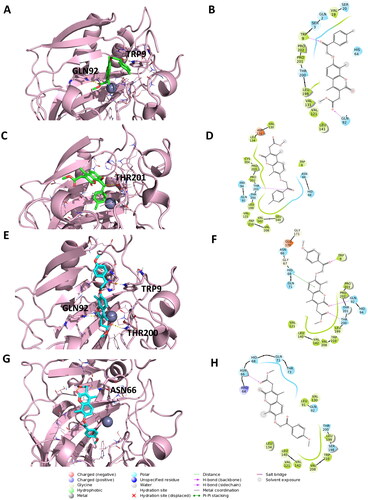
Figure 3. Representation of the putative binding mode of the EMAC10163a and EMAC10163b compounds obtained by docking experiments in complex with hCA IX. (A) 3D representation of EMAC10163a and its respective interactions with CA IX residues; (B) 2D depiction of interactions; (C) 3D depiction of EMAC10163a and its respective interactions with CA IX residues considering a different orientation of the compound; (D) 2D depiction of interactions; (E) 3D depiction of EMAC10163b and its respective interactions with CA IX residues; (F) 2D depiction of interactions; (G) 3D depiction of EMAC10163b and its respective interactions with CA IX residues considering a different orientation of the compound; (H) 2D depiction of interactions.
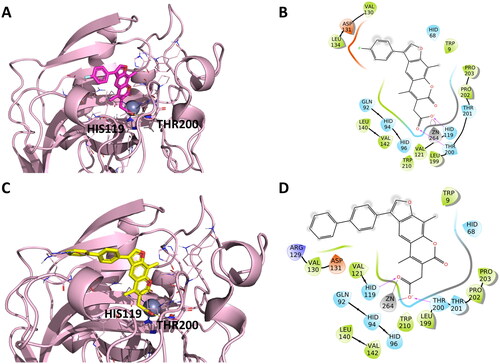
Conversely, docking experiments of EMAC10164d and EMAC10164g () always show the same orientation, with the chromene ring orientated towards the zinc. Docking experiments using the IX isoform revealed that the higher rigidity of the 7H-furo-chromene, EMAC10164 series, compared to the 2H-chromene series could help orienting the compounds in the binding pocket with the chromene portion pointing towards the Zn2+. The different orientation might be the reason why in general a better inhibitory activity of this series was observed. With respect to our previously reported series of compounds, EMAC10151 and EMAC10152 (), the introduction of a methyl moiety in the position 8 of the 2H-chromene and 9 of the 7H-furo-chromene was detrimental for the activity but conserved the selectivity. Conversely, the new derivatives exhibit a similar activity when compared with the previously reported 8-propyl EMAC10157 and 9-propyl EMAC10160Citation51,Citation52.
Figure 4. Representation of the putative binding mode of the EMAC10164 series most potent compounds obtained by docking experiments in complex with hCA IX. (A) 3D depiction of EMAC10164d and its respective interactions with CA IX residues; (B) 2D depiction of interactions; (C) 3D depiction of EMAC10164g and its respective interactions with CA IX residues; (D) 2D depiction of interactions.
To gain a deeper comprehension of the putative binding mode of the chromene derivative, and considering that, according to the reported mechanism, both chromene and furo-chromene are the prodrugs of the active corresponding inhibitor 2-hydroxycinnamic acidsCitation51–53,Citation63, the two most active chromene derivatives, EMAC10163a and EMAC10163b were submitted to docking experiments also in their open forms.
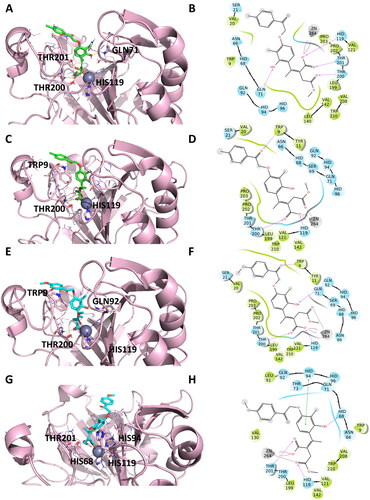
Hence, both the E and Z configurations of the 2-hydroxycinnamic acids were subjected to docking experiments to predict the putative binding mode of the hydrolysed forms (). The docking results of the open compounds revealed that Zn2+ chelation and interactions between the newly formed carboxylate moiety in the catalytic site were important for ligand–enzyme interactions. The complexes are further stabilised by interactions with residues near the Zn2+ ion, such as His119, Thr200, and Thr201 (Figures S1 and S2).
Figure 5. Representation of the putative binding mode of the most potent compounds from EMAC10163-open form series obtained by docking experiments in complex with hCA IX. (A) 3D depiction of EMAC10163a-open-E and its respective interactions with CA IX residues; (B) 2D depiction of interactions; (C) 3D depiction of EMAC10163a-open-Z and its respective interactions with CA IX residues; (D) 2D depiction of interactions; (E) 3D depiction of EMAC10163b-open-E and its respective interactions with CA IX residues; (F) 2D depiction of interactions; (G) 3D depiction of EMAC10163b-open-Z and its respective interactions with CA IX residues; (H) 2D depiction of interactions.
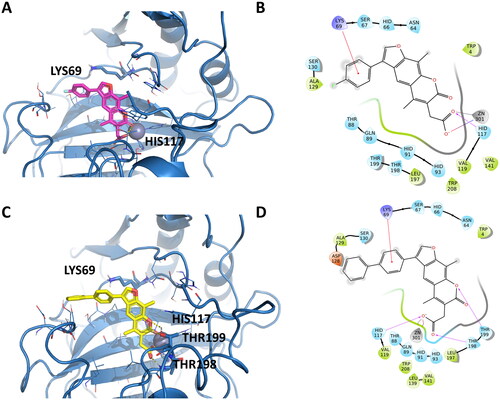
Docking experiments of the two best compounds of each series were also performed on hCA XII. EMAC10163a and EMAC10163b binding modes are represented in . Only one binding mode with the chromene ring oriented to the Zn2+ was obtained for EMAC10163a. In the case of EMAC10163b, however, two possible orientations were obtained. One has the chromene ring oriented towards the zinc, while the other has the substituted phenyl ring oriented towards the zinc. Docking experiments with EMAC10164d and EMAC10164g () show docking predicted binding modes with the chromene ring always oriented towards the Zn2+.
Figure 6. Representation of the putative binding mode of the EMAC10163 series most potent compounds obtained by docking experiments in complex with hCA XII. (A) 3D depiction of EMAC10163a and its respective interactions with CA XII residues; (B) 2D depiction of interactions; (C) 3D depiction of EMAC10163b and its respective interactions with CA XII residues; (D) 2D depiction of interactions; (E) 3D depiction of EMAC10164b and its respective interactions with CA XII residues considering a different orientation of the compound; (F) 2D depiction of interactions.
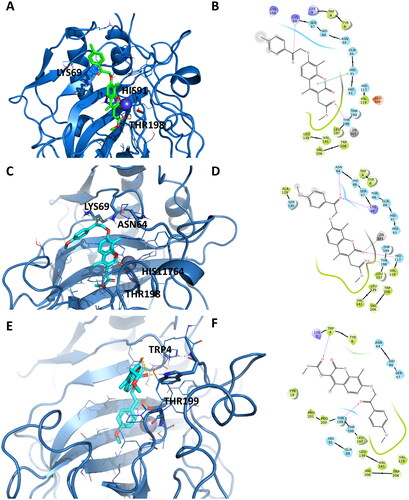
Figure 7. Representation of the putative binding mode of the EMAC10164 series most potent compounds obtained by docking experiments in complex with hCA XII. (A) 3D depiction of EMAC10164d and its respective interactions with CA XII residues; (B) 2D depiction of interactions; (C) 3D depiction of EMAC10164g and its respective interactions with CA XII residues; (D) 2D depiction of interactions.
As with CA IX, the hCA esterase-mediated inhibition mechanism of 2-hydroxicinnamic acids was considered for CA XII. As a result, docking experiments involving the compounds in their pyran-2-one open forms were submitted. Also in this case, the putative binding mode of the hydrolysed forms was predicted using the two possible originated diastereoisomers, E and Z. Thus, four compounds were docked, and two distinct potential binding modes, for the E and Z diastereoisomers, were observed. Only the binding modes obtained for the E configuration appeared to be reasonable (Figures S3 and S4), because the proposed binding modes for Z configurations showed the compounds oriented with the substituted phenyl ring facing the Zn2+. This latter position appeared to be less preferred, because the pyran-2-one ring must be oriented towards the Zn2+ to allow the formation of the active 2-hydroxicinnamic acid and coordinate the bivalent ion.
Docking poses for hCA IX revealed several interactions with common residues such as Lys69, His117, Thr198, and Thr199. A similar pattern was observed in the case of the hCA XII isoform, where an array of interactions involving the catalytic cavity residues was observed.
In silico prediction of physicochemical properties, ADMET parameters, and drug-likeness
Finally, considering the high relevance of early determination of drug like properties, all the compounds under investigation were submitted to ADME calculations by means of the QikProp Schrodinger softwareCitation64 (Tables S2–S7). In most cases, 2H-chromene and 7H-furo-chromene exhibit a good drug-like profile.
Conclusions
We have designed and synthesised 2H-chromene and 7H-furo-chromene derivatives and evaluate their activity on hCA I, II, IX, and XII isozymes based on our previous work aiming to the identification of selective tumour associated hCA isozymes inhibitors. The new derivatives exhibited higher isozyme selectivity when compared with the reference AAZ. In particular, the majority of the compounds were selective towards the tumour associated hCA isoforms IX and XII. Our data suggest that, when compared with less complex sulphonamides derivatives, such as AAZ, 2H-chromene, and 7H-furo-chromene derivatives generally exhibit a better selectivity profile towards tumour associated hCA isoforms IX and XII. Furthermore, our results indicate that the substitution pattern surrounding the 2H-chromene and 7H-furo-chromene scaffolds can affect the potency of these derivatives. In line with our previous findings on similar derivatives, we found that introducing a fluorine in the position 4 of the aromatic ring in 7H-furo-chromene derivatives improves activity and selectivity towards hCA IX and XII isozymes. According to literature data, in most cases, 2H-chromene and 7H-furo-chromene exhibit good pharmacokinetic properties and, accordingly, a preliminary computational study has shown that these new derivatives might possess a good drug-like profile. All these findings encouraged us to further investigate these scaffolds to optimise both the activity and to further increase the isozyme selectivity.
Materials and methods
Materials and apparatus
2-Methylresorcinol was purchased from Thermo Fisher GmbH (Kandel, Germany). Dimethylsuccinate was purchased from Sigma-Aldrich, Co. (St. Louis, MO). The differently substituted α-halogen ketones were purchased from Fluorochem Ltd (Hadfield, UK). All solvents were pro analysis grade from Carlo Erba Reagents (Val-de-Reuil, France) and Thermo Fisher GmbH (Kandel, Germany). NMR spectra were registered on a Bruker AMX (400 MHz) spectrometer (chemical shifts in δ values). Chemical shifts are reported referenced to the solvent in which they were measured. Coupling constants J are expressed in hertz (Hz). For the DEPT sequence, the width of the 90° pulse for 13C was 4 μs, and that of the 90° pulse for 1H was 9.5 μs; the delay 2JC,H-1 was set to 3.5 ms (underlined values). TLC chromatography was performed using silica gel plates (Merck F 254, Darmstadt, Germany), and spots were visualised by UV light. Mass spectra were acquired on an Orbitrap Exploris mass spectrometer (Thermo Fisher Scientific, Bremen, Germany).
Full MS spectra were acquired at a resolution of 240 000, in the m/z range 180–1000 (Table S1). For that, compounds were initially dissolved in dimethylsulphoxide (DMSO) at 5 mg/ml concentration. Stock solutions were then diluted 50-fold in acetonitrile and diluted further 10-fold in 70% acetonitrile containing 0.1% of formic acid. Solutions were directly infused in the mass spectrometer at 5 µl/min. Mass spectra were acquired in positive ion mode (3200 V). Ion transfer tube temperature was 320 °C, whereas S-lens value was 20 units. Melting points were determined by the capillary method on a Stuart Scientific melting point apparatus and are uncorrected. The SD contains NMR spectra and HRMS data.
Synthetic procedures
Methyl 2-(7-hydroxy-4,8-dimethyl-2-oxo-2H-chromen-3-yl)acetate (EMAC10163)
To a mixture of 2-methylresorcinol (0.5 g, 4.03 mmol) and dimethylsuccinate (650 µl, 4.03 mmol), 3 ml of H2SO4 98% were added. The mixture was vigorously stirred at room temperature for 8 h. The homogeneous mixture was poured into iced water and stirred until the melting of the ice. The precipitate was kept in the fridge overnight and filtered in the next day. The desired compound was obtained as a white solid after drying in a vacuum oven. Yield: 62.6%; m.p. 179–180 °C; Rf (DCM/EtOAc = 8/2): 0.58; 1H NMR (400 MHz, DMSO): δ = 2.16 (s, 3H, CH3), 2.34 (s, 3H, CH3), 3.62 (s, 3H, CH3), 3.65 (s, 2H, CH2), 6.88 (d, J = 8.8 Hz, 1H, H(Ar)), 7.51 (d, J = 8.8, 1H, H(Ar)), 10.38 (s, 1H, OH); 13C NMR (100 MHz, DMSO): δ = 8.41 (CH3), 15.51 (CH3), 32.80 (CH2), 52.27 (CH3), 110.92 (C(Ar)), 112.40 (C(Ar)), 112.62 (C(Ar)), 115.07 (C(Ar)), 123.92 (C(Ar)), 150.33 (C(Ar)), 151.94 (C(Ar)), 158.94 (C7), 161.57 (C2), 171.22 (COOCH3).
Methyl 2‐{4,8‐dimethyl‐7‐[2‐(4‐methylphenyl)‐2‐oxoethoxy]‐2‐oxo‐2H‐chromen‐3‐yl}acetate (EMAC10163a)
EMAC10163 (1.00 g, 3.81 mmol), acetone (20 ml), and K2CO3 (1.31 g, 9.5 mmol) were mixed and 2-bromo-4′-methylacetophenone (0.89 g, 4.18 mmol) was added. After 6 h, the mixture was poured into 100 ml of H2SO4 0.5 M. For the purification by chromatographic column, DCM/EtOAc 88:12 to 100% of EtOAc were used as eluents. The white solid was obtained by recrystallisation with DCM/n-hexane. Yield: 86.1%; m.p. 178–181 °C; Rf (DCM/EtOAc = 5:5): 0.86; 1H NMR (400 MHz, CDCl3): δ = 2.35 (s, 3H, CH3), 2.38 (s, 3H, CH3), 2.43 (s, 3H, CH3), 3.70 (s, 3H, OCH3), 3.72 (s, 2H, CH2), 5.36 (s, 2H, CH2), 6.71 (d, J = 8.9 Hz, 1H, H(Ar)), 7.28–7.34 (m, 2H, 2 x H(Ar′)), 7.39 (d, J = 9.0 Hz, 1H, H(Ar)), 7.85–7.94 (m, 2H, 2 x H(Ar′)); 13C NMR (100 MHz, CDCl3): δ = 8.42 (CH3), 15.38 (CH3), 21.80 (CH3), 32.75 (CH2), 52.21 (CH3), 71.17 (CH2), 107.90 (C(Ar)), 114.68 (C(Ar)), 114.73 (C(Ar)), 116.62 (C(Ar)), 122.64 (C(Ar)), 128.26 (2 x C(Ar′)), 129.59 (2 x C(Ar′)), 131.86 (C(Ar)), 145.16 (C(Ar)), 149.04 (C(Ar)), 151.68 (C(Ar)), 158.36 (C(Ar)), 161.84 (C(Ar)), 170.88 (COOCH3) 193.71 (CO); MS (ESI) m/z (%, fragment): 395 (100).
Methyl 2-(7-(2-(4-methoxyphenyl)-2-oxoethoxy)-4,8-dimethyl-2-oxo-2H-chromen-3-yl)acetate (EMAC10163b)
EMAC10163 (0.50 g, 1.91 mmol), acetone (10 ml), and K2CO3 (0.66 g, 4.8 mmol) were mixed and 2-bromo-4′-methoxyacetophenone (0.48 g, 2.10 mmol) was added. After 6 h, the mixture was poured into 50 ml of H2SO4 0.5 M. For the purification by chromatographic column, DCM/EtOAc 88:12 to 100% of EtOAc were used as eluents. The white solid was obtained by recrystallisation with DCM/n-hexane. Yield: 27.8%; m.p. 173–175 °C; Rf (DCM/EtOAc 5:5): 0.80; 1H NMR (400 MHz, CDCl3): δ = 2.35 (s, 3H, CH3), 2.38 (s, 3H, CH3), 3.70 (s, 3H, CH3), 3.72 (s, 2H, CH2), 3.89 (s, 3H, CH3), 5.33 (s, 2H, CH2), 6.73 (d, J = 8.9 Hz, 1H, H(Ar)), 6.94–7.00 (m, 2H, 2 x H(Ar′)), 7.39 (d, J = 8.7 Hz, 1H, H(Ar)), 7.96–8.02 (m, 2H, 2 x H(Ar′)). 13C NMR (100 MHz, CDCl3): δ = 8.43 (CH3), 15.38 (CH3), 32.75 (CH2), 52.21 (CH3), 55.57 (CH3), 71.15 (CH2), 107.91 (C(Ar)), 114.10 (2 x C(Ar′)), 114.67 (C(Ar)), 114.68 (C(Ar)), 116.60 (C(Ar)), 122.65 (C(Ar)), 127.37 (C(Ar)), 130.57 (2 x C(Ar′)), 149.04 (C(Ar)), 151.68 (C(Ar)), 158.40 (C(Ar)), 161.84 (C(Ar)), 164.25 (C(Ar)), 170.88 (COOCH3) 192.64 (CO). MS (ESI) m/z (%, fragment): 411 (100).
Methyl 2-(7-(2-(4-bromophenyl)-2-oxoethoxy)-4,8-dimethyl-2-oxo-2H-chromen-3-yl)acetate (EMAC10163c)
EMAC10163 (1.00 g, 3.81 mmol), acetone (20 ml), and K2CO3 (1.31 g, 9.51 mmol) were mixed and, 2,4′-dibromoacetophenone (1.17 g, 4.21 mmol) was added. After 24 h, the mixture was poured into 100 ml of H2SO4. For the purification by chromatographic column, DCM/EtOAc 88:12 to 100% of EtOAc were used as eluents. The white solid was obtained by recrystallisation with DCM/n-hexane. Yield: 46.1%; m.p. 199–201 °C; Rf (DCM/EtOAc 5:5): 0.89; 1H NMR (400 MHz, CDCl3): δ = 2.35 (s, 3H, CH3), 2.37 (s, 3H, CH3), 3.70 (s, 3H, CH3), 3.72 (s, 2H, CH2), 5.33 (s, 2H, CH2), 6.71 (d, J = 8.9 Hz, 1H, H(Ar)), 7.40 (d, J = 8.7 Hz, 1H, H(Ar)), 7.63–7.68 (m, 2H, 2 x H(Ar′)), 7.85–7.89 (m, 2H, 2 x H(Ar′)); 13C NMR (100 MHz, CDCl3): δ = 8.43 (CH3), 15.40 (CH3), 32.75 (CH2), 52.24 (OCH3), 71.23 (CH2), 107.76 (C(Ar)), 114.79 (C(Ar)), 114.88 (C(Ar)), 116.83 (C(Ar)), 122.70 (C(Ar)), 129.45 (C(Ar)), 129.71 (2 x C(Ar′)), 132.28 (2 x C(Ar′)), 133.05 (C(Ar)), 148.96 (C(Ar)), 151.71 (C(Ar)), 158.07 (C(Ar)), 161.75 (C(Ar)), 170.85 (COOCH3), 193.43 (CO); MS (ESI) m/z (%, fragment): 459 (44).
Methyl 2-(7-(2-(4-fluorophenyl)-2-oxoethoxy)-4,8-dimethyl-2-oxo-2H-chromen-3-yl)acetate (EMAC10163d)
EMAC10163 (1.00 g, 3.82 mmol), acetone (20 ml), and K2CO3 (1.32 g, 9.59 mmol) were mixed and, 2-bromo-4′-fluoroacetophenone (0.91 g, 4.22 mmol) was added. After 6 h, the mixture was poured into 100 ml of H2SO4. For the purification by chromatographic column, DCM/MeOH 98:2 to 20% of MeOH were used as eluents. The very light-yellow solid was obtained by recrystallisation with DCM/n-hexane. Yield: 63.0%; m.p. 155–157 °C; Rf (DCM/EtOAc 5:5): 0.89; 1H NMR (400 MHz, CDCl3): δ = 2.35 (s, 3H, CH3), 2.37 (s, 3H, CH3), 3.70 (s, 3H, OCH3), 3.72 (s, 2H, CH2), 5.34 (s, 2H, CH2), 6.72 (d, J = 8.9 Hz, 1H, H(Ar)), 7.13–7.22 (m, 2H, 2 x H(Ar′)), 7.40 (d, J = 8.9 Hz, 1H, H(Ar)), 8.02–8.08 (m, 2H, 2 x H(Ar′)); 13C NMR (100 MHz, CDCl3): δ = 8.42 (CH3), 15.39 (CH3), 32.75 (CH2), 52.23 (OCH3), 71.22 (CH2), 107.81 (C(Ar)), 114.76 (C(Ar)), 114.84 (C(Ar)), 116.17 (d, JCF = 22.0 Hz, C3′, C5′), 116.78 (C(Ar)), 122.70 (C(Ar)), 130.81 (d, JCF = 3.1 Hz, C1′), 131.00 (d, JCF = 9.5 Hz, C2′, C6′), 148.99 (C(Ar)), 151.70 (C(Ar)), 158.15 (C(Ar)), 161.77 (C(Ar)), 166.26 (d, JCF = 256.7 Hz, C4′), 170.86 (COOCH3), 192.75 (CO); MS (ESI) m/z (%, fragment): 399 (100).
Methyl 2-(4,8-dimethyl-7-(2-(4-nitrophenyl)-2-oxoethoxy)-2-oxo-2H-chromen-3-yl)acetate (EMAC10163e)
EMAC10163 (0.50 g, 1.91 mmol), acetone (10 ml), and K2CO3 (0.66 g, 4.80 mmol) were mixed and, 2-bromo-4′-nitroacetophenone (0.51 g, 2.10 mmol) was added. After 8 h, the mixture was poured into 50 ml of H2SO4. For the purification by chromatographic column, DCM/EtOAc 88:12 to 100% of EtOAc were used as eluents. The light brown solid was obtained by recrystallisation with DCM/n-hexane. Yield: 37.9%; m.p. 180–183 °C; Rf (DCM/EtOAc 5:5): 0.80; 1H NMR (400 MHz, CDCl3): δ = 2.27 (s, 3H, CH3), 2.29 (s, 3H, CH3), 3.64 (s, 3H, CH3), 3.65 (s, 2H, CH2), 5.31 (s, 2H, CH2), 6.67 (d, J = 8.9 Hz, 1H, H(Ar)), 7.35 (d, J = 8.7 Hz, 1H, H(Ar)), 8.09–8.12 (m, 2H, 2 x H(Ar′)), 8.27–8.29 (m, 2H, 2 x H(Ar)′); 13C NMR (100 MHz, CDCl3): δ = 8.43 (CH3), 15.41 (CH3), 32.73 (CH2), 52.26 (CH3), 71.53 (CH2), 107.64 (C(Ar)), 114.83 (C(Ar)), 115.10 (C(Ar)), 117.05 (C(Ar)), 122.79 (C(Ar)), 124.08 (2 x C(Ar′)), 129.44 (2 x C(Ar′)), 138.77 (C(Ar)), 148.90 (C(Ar)), 150.82 (C(Ar)), 151.71 (C(Ar)), 157.74 (C(Ar)), 161.65 (C(Ar)), 170.82 (COOCH3), 193.36 (CO); MS (ESI) m/z (%, fragment): 426 (100).
Methyl 2-(4,8-dimethyl-7-(2-(3-nitrophenyl)-2-oxoethoxy)-2-oxo-2H-chromen-3-yl)acetate (EMAC10163f)
EMAC10163 (1.00 g, 3.82 mmol), acetone (20 ml), and K2CO3 (1.31 g, 9.49 mmol) were mixed and, 3-nitrophenacyl bromide (1.02 g, 4.18 mmol) was added. After 7 h, the mixture was poured into 100 ml of H2SO4. For the purification by chromatographic column, DCM/EtOAc 88:12 to 100% of EtOAc were used as eluents. The yellow solid was obtained by recrystallisation with DCM/n-hexane. Yield: 19.6%; m.p. 165–167 °C; Rf (DCM/EtOAc 5:5): 0.78; 1H NMR (400 MHz, CDCl3): δ = 2.35 (s, 3H, CH3), 2.36 (s, 3H, CH3), 3.71 (s, 3H, CH3), 3.72 (s, 2H, CH2), 5.40 (s, 2H, CH2), 6.76 (d, J = 8.9 Hz, 1H, H(Ar)), 7.42 (d, J = 8.6 Hz, 1H, H(Ar)), 7.74 (t, J = 8.6 Hz, 1H, H5′), 8.34–8.36 (m, 1H, H(Ar′)), 8.47–8.49 (m, 1H, H(Ar′)), 8.85–8.86 (m, 1H, H(Ar′)); 13C NMR (100 MHz, CDCl3): δ = 8.40 (CH3), 15.41 (CH3), 32.74 (CH2), 52.25 (CH3), 71.53 (CH2), 107.67 (C(Ar)), 114.84 (C(Ar)), 115.10 (C(Ar)), 117.03 (C(Ar)), 122.82 (C(Ar)), 123.30 (C(Ar)), 128.27 (C(Ar)), 130.24 (C(Ar)), 133.91 (C(Ar)), 135.54 (C(Ar)), 148.49 (C(Ar)), 148.92 (C(Ar)), 151.71 (C(Ar)), 157.74 (C(Ar)), 161.65 (C(Ar)), 170.82 (COOCH3), 192.82 (CO); MS (ESI) m/z (%, fragment): 426 (51).
Methyl 2-(7-(2-([1,1′-biphenyl]-4-yl)-2-oxoethoxy)-4,8-dimethyl-2-oxo-2H-chromen-3-yl)acetate (EMAC10163g)
EMAC10163 (1.00 g, 3.83 mmol), acetone (20 ml), and K2CO3 (1.31 g, 9.49 mmol) were mixed and, 2-bromo-1-(4-phenylphenyl)ethan-1-one (1.15 g, 4.18 mmol) was added. After 6 h, the mixture was poured into 100 ml of H2SO4. For the purification by chromatographic column, DCM/EtOAc 88:12 to 100% of EtOAc were used as solvents. The white solid was obtained by recrystallisation with DCM/n-hexane. Yield: 73.9%; m.p. 168–169 °C; Rf (DCM/EtOAc 5:5): 0.94; 1H NMR (400 MHz, CDCl3): δ = 2.35 (s, 3H, CH3), 2.40 (s, 3H, CH3), 3.70 (s, 3H, CH3), 3.72 (s, 2H, CH2), 5.41 (s, 2H, CH2), 6.74 (d, J = 8.9 Hz, 1H, H(Ar)), 7.39–7.44 (m, 2H, 2 x H(Ar)), 7.46–7.50 (m, 2H, 2 x H(Ar)), 7.61–7.64 (m, 2H, 2 x H(Ar)); 7.71–7.74 (m, 2H, 2 x H(Ar)), 8.06–8.09 (m, 2H, 2 x H(Ar)); 13C NMR (100 MHz, CDCl3): δ = 8.45 (CH3), 15.39 (CH3), 32.75 (CH2), 52.22 (OCH3), 71.28 (CH2), 107.90 (C(Ar)), 114.75 (C(Ar)), 114.77 (C(Ar)), 116.68 (C(Ar)), 122.68 (C(Ar)), 127.29 (2 x C(Ar)), 127.50 (2 x C(Ar)), 128.54 (C(Ar)), 128.77 (2 x C(Ar)), 129.05 (2 x C(Ar)), 132.99 (C(Ar)), 139.53 (C(Ar)), 146.81 (C(Ar)), 149.03 (C(Ar)), 151.70 (C(Ar)), 158.32 (C(Ar)), 161.82 (C(Ar)), 170.87 (COOCH3), 193.70 (CO); MS (ESI) m/z (%, fragment): 457 (88).
Methyl 2-(7-(2-(2,4-difluorophenyl)-2-oxoethoxy)-4,8-dimethyl-2-oxo-2H-chromen-3-yl)acetate (EMAC10163h)
EMAC10163 (0.50 g, 1.91 mmol), acetone (10 ml), and K2CO3 (0.66 g, 4.80 mmol) were mixed and, 2-chloro-1-(2,4-difluorophenyl)ethanone (0.41 g, 2.13 mmol) was added. The mixture was kept reacting overnight, and then poured into 50 ml of H2SO4. The resulting precipitate was recrystallised with DCM/n-hexane giving a light-yellow solid. Yield: 49.0%; m.p. 188–190; Rf (DCM/EtOAc 5:5): 0.87; 1H NMR (400 MHz, CDCl3): δ = 2.36 (s, 3H, CH3), 2.38 (s, 3H, CH3), 3.71 (s, 3H, CH3), 3.73 (s, 2H, CH2), 5.30 (m, 2H, CH2), 6.68 (d, J = 8.9 Hz, 1H, H(Ar)), 6.92–6.98 (m, 1H, H(Ar)), 7.02–7.07 (m, 1H, H(Ar)), 7.40 (d, J = 8.9 Hz, 1H, H(Ar)), 8.00–8.06 (m, 1H, H(Ar)); 13C NMR (100 MHz, CDCl3): δ = 8.35 (CH3), 15.40 (CH3), 32.76 (C3CH2), 52.23 (CH3), 73.71 (d, J = 12.3 Hz, CH2), 104.82 (dd, JCF = 27.5, 25.7 Hz, C3′), 107.84 (C(Ar)), 113.00 (dd, JCF = 21.6, 3.2 Hz, C5′), 114.78 (C(Ar)), 114.88 (C(Ar)), 116.73 (C(Ar)), 119.41 (dd, JCF = 15.3, 3.6 Hz, C1′), 122.59 (C(Ar)), 132.91 (dd, JCF = 10.8, 4.9 Hz, C6′), 149.00 (C(Ar)), 151.75 (C(Ar)), 158.28 (C(Ar)), 163.03 (dd, JCF = 256.1, 12.8 Hz, C2′), 166.54 (dd, JCF = 259.1, 12.5 Hz, C4′), 170.88 (COOCH3), 190.86 (d, JCF = 5.6 Hz, CO); MS (ESI) m/z (%, fragment): 417 (100).
Methyl 2-(4,8-dimethyl-2-oxo-7-(2-oxo-2-phenylethoxy)-2H-chromen-3-yl)acetate (EMAC10163i)
EMAC10163 (1.00 g, 3.82 mmol), acetone (20 ml), and K2CO3 (1.32 g, 9.56 mmol) were mixed and, 2-bromoacetophenone (0.8344 g, 4.19 mmol) was added. After 7 h, the mixture was poured into 100 ml of H2SO4 0.5 M. The light-yellow solid was obtained by recrystallisation with DCM/n-hexane. Yield: 88.6%; m.p. 157–159 °C; Rf (DCM/EtOAc 5:5): 0.84; 1H NMR (400 MHz, CDCl3): δ = 2.35 (s, 3H, CH3), 2.38 (s, 3H, CH3), 3.70 (s, 3H, CH3), 3.72 (s, 2H, CH2), 5.39 (s, 2H, CH2), 6.72 (d, J = 8.9 Hz, 1H, H(6)), 7.38–7.40 (m, 1H, H(Ar)), 7.49–7.53 (m, 2H, 2 x H(Ar′)), 7.61–7.65 (m, 1H, H(Ar′)); 7.99–8.01 (m, 2H, 2 x H(Ar′)). 13C NMR (100 MHz, CDCl3): δ = 8.42 (CH3), 15.39 (CH3), 32.76 (CH2), 52.22 (CH3), 71.20 (CH2), 107.88 (C(Ar)), 114.75 (C(Ar)), 114.80 (C(Ar)), 116.69 (C(Ar)), 122.65 (C(Ar)), 128.14 (2 x C(Ar′)), 128.93 (2 x C(Ar′)), 134.11 (C(Ar)), 134.36 (C(Ar)), 149.01 (C(Ar)), 151.71 (C(Ar)), 158.30 (C(Ar)), 161.81 (C(Ar)), 170.87 (COOCH3), 194.08 (CO); MS (ESI) m/z (%, fragment): 381 (100).
Methyl 2-(7-(2-(4-chlorophenyl)-2-oxoethoxy)-4,8-dimethyl-2-oxo-2H-chromen-3-yl)acetate (EMAC10163j)
EMAC10163 (0.50 g, 1.91 mmol), acetone (10 ml), and K2CO3 (0.66 g, 4.80 mmol) were mixed and, 2-bromo-4′-chloroacetophenone (0.49 g, 2.12 mmol) was added. After 6 h, the mixture was poured into 50 ml of H2SO4 0.5 M. For the purification by chromatographic column, DCM/EtOAc 80:20 to 30% of EtOAc were used as eluents. The white solid was obtained by recrystallisation with DCM/n-hexane. Yield: 46.0%; m.p. 182–184 °C; Rf (DCM/EtOAc 5:5): 0.80; 1H NMR (400 MHz, CDCl3): δ = 2.37 (s, 3H, CH3), 2.38 (s, 3H, CH3), 3.72 (s, 3H, OCH3), 3.74 (s, 2H, CH2), 5.35 (s, 2H, CH2), 6.73 (d, J = 8.6 Hz, 1H, H(Ar)), 7.41 (d, J = 8.9 Hz, 1H, H(Ar)), 7.48–7.52 (m, 2H, 2 x H(Ar′)), 7.95–7.98 (m, 2H, 2 x H(Ar′)); 13C NMR (100 MHz, CDCl3): δ = 8.42 (CH3), 15.39 (CH3), 32.74 (CH2), 52.23 (OCH3), 71.24 (CH2), 107.77 (C(Ar)), 114.76 (C(Ar)), 114.86 (C(Ar)), 116.80 (C(Ar)), 122.70 (C(Ar)), 129.28 (2 x C(Ar′)), 129.64 (2 x C(Ar′)), 132.64 (C(Ar)), 140.67 (C(Ar)), 148.97 (C(Ar)), 151.69 (C(Ar)), 158.08 (C(Ar)), 161.75 (C(Ar)), 170.85 (COOCH3) 193.20 (CO); MS (ESI) m/z (%, fragment): 415 (96).
Methyl 2-(7-(2-(3-methoxyphenyl)-2-oxoethoxy)-4,8-dimethyl-2-oxo-2H-chromen-3-yl)acetate (EMAC10163k)
EMAC10163 (1.00 g, 3.82 mmol), acetone (20 ml), and K2CO3 (1.32 g, 9.57 mmol) were mixed and, 2-bromo-3′-methoxyacetophenone (0.96 g, 4.19 mmol) was added. After 6 h, the mixture was poured into 100 ml of H2SO4 0.5 M. For the purification by chromatographic column, DCM/EtOAc 88:12 to 100% of EtOAc were used as eluents. The white solid was obtained by recrystallisation with DCM/n-hexane. Yield: 55.3%; m.p. 157–160 °C; Rf (DCM/EtOAc 5:5): 0.80; 1H NMR (400 MHz, CDCl3): δ = 2.35 (s, 3H, CH3), 2.39 (s, 3H, CH3), 3.70 (s, 3H, CH3), 3.72 (s, 2H, CH2), 3.86 (s, 3H, OCH3), 5.38 (s, 2H, CH2), 6.71 (d, J = 8.9 Hz, 1H, H(Ar)), 7.16–7.19 (m, 1H, H(Ar)), 7.38–7.44 (m, 2H, 2 x H(Ar′)), 7.51–7.52 (m, 1H, H(Ar)), 7.56–7.58 (m, 1H, H(Ar)); 13C NMR (100 MHz, CDCl3): δ = 8.43 (CH3), 15.39 (CH3), 32.76 (CH2), 52.22 (CH3), 52.52 (OCH3), 71.22 (CH2), 107.89 (C(Ar)), 112.51 (C(Ar)), 114.75 (C(Ar)), 114.82 (C(Ar)), 116.69 (C(Ar)), 120.52 (2 x C(Ar′)), 122.64 (C(Ar)), 129.93 (C(Ar)), 135.59 (C(Ar)), 149.02 (C(Ar)), 151.70 (C(Ar)), 158.31 (C(Ar)), 160.05 (C(Ar)), 161.82 (C(Ar)), 170.88 (COOCH3), 193.86 (CO); MS (ESI) m/z (%, fragment): 411 (98).
2-(5,9-Dimethyl-7-oxo-3-(p-tolyl)-7H-furo[3,2-g]chromen-6-yl)acetic acid (EMAC10164a)
EMAC10163a (0.50 g, 1.27 mmol), 5 ml of propan-2-ol and an aqueous solution of NaOH 1 M (5.1 ml) were mixed and stirred for 4 h. After filtration of the resulting precipitate, a light brown solid was obtained. Yield: 96.0%; m.p. 228–231 °C; Rf (DCM/EtOAc 5:5): 0.15; 1H NMR (400 MHz, DMSO): δ = 2.38 (s, 3H, CH3), 2.48 (s, 3H, CH3), 2.50 (s, 3H, CH3), 3.64 (s, 2H, CH2), 7.33–7.35 (m, 2H, 2 x H(Ar′)), 7.65–7.67 (m, 2H, 2 x H(Ar′)), 7.96 (s, 1H, H5), 8.39 (s, 1H, H7). 13C NMR (100 MHz, DMSO): δ = 8.66 (CH3), 16.07 (CH3), 21.29 (CH3), 33.44 (CH2), 108.96 (C(Ar)), 114.18 (C(Ar)), 117.05 (C(Ar)), 118.33 (C(Ar)), 121.94 (C(Ar)), 122.47 (C(Ar)), 127.52 (2 x C(Ar′)), 128.34 (C(Ar)), 130.19 (2 x C(Ar′)), 137.56 (C(Ar)), 144.07 (C(Ar)), 148.01 (C(Ar)), 149.92 (C(Ar)), 155.44 (C(Ar)), 161.21 (C(Ar)), 171.99 (COOH); MS (ESI) m/z (%, fragment): 363 (100).
2-(3-(4-Methoxyphenyl)-5,9-dimethyl-7-oxo-7H-furo[3,2-g]chromen-6-yl)acetic acid (EMAC10164b)
EMAC10163b (0.35 g, 0.86 mmol), 3 ml of propan-2-ol and an aqueous solution of NaOH 1 M (3.5 ml) were mixed and stirred for 4 h. After filtration of the resulting precipitate, a light brown solid was obtained. Yield: 67.3%; m.p. 226–227 °C; Rf (DCM/EtOAc 5:5): 0.35;c1H NMR (400 MHz, DMSO): δ = 2.52 (s, 3H, CH3), 2.54 (s, 3H, CH3), 3.66 (s, 2H, CH2), 3.83 (s, 3H, C4′OCH3), 7.10–7.12 (m, 2 x H(Ar′)), 7.73–7.75 (m, 2 x H(Ar′)), 8.01 (s, 1H, H5), 8.39 (s, 1H, H7), 12.50 (s, 1H, COOH); 13C NMR (100 MHz, DMSO): δ = 8.71 (CH3), 16.13 (CH3), 33.42 (CH2), 55.70 (C4′OCH3), 108.99 (C(Ar)), 114.28 (C(Ar)), 115.13 (2 x C(Ar′)), 117.07 (C(Ar)), 118.31 (C(Ar)), 121.75 (C(Ar)), 122.65 (C(Ar)), 123.54 (C(Ar)), 128.95 (2 x C(Ar′)), 143.70 (C(Ar)), 148.06 (C(Ar)), 150.04 (C(Ar)), 155.46 (C(Ar)), 159.44 (C(Ar)), 161.25 (C(Ar)), 171.98 (COOH); MS (ESI) m/z (%, fragment): 379 (100).
2-(3-(4-Bromophenyl)-5,9-dimethyl-7-oxo-7H-furo[3,2-g]chromen-6-yl)acetic acid (EMAC10164c)
EMAC10163c (0.50 g, 1.10 mmol), 5 ml of propan-2-ol and an aqueous solution of NaOH 1 M (4.4 ml) were mixed and stirred for 3 h. After filtration of the resulting precipitate, a light brown solid was obtained. Yield: 90.5%; m.p. 263–267 °C; Rf (DCM/EtOAc 5:5): 0.26; 1H NMR (400 MHz, DMSO): δ = 2.49 (s, 3H, CH3), 2.50 (s, 3H, CH3), 3.64 (s, 2H, CH2), 7.69–7.76 (m, 4H, 4 x H(Ar′)), 7.96 (s, 1H, H5), 8.50 (s, 1H, H7), 12.49 (s, 1H, COOH); 13C NMR (100 MHz, DMSO): δ = 8.68 (CH3), 16.13 (CH3), 33.40 (CH2), 109.11 (C(Ar)), 114.25 (C(Ar)), 117.23 (C(Ar)), 118.42 (C(Ar)), 120.99 (C(Ar)), 121.28 (C(Ar)), 121.98 (C(Ar)), 129.66 (2 x C(Ar′)), 130.56 (C(Ar)), 132.53 (2 x C(Ar′)), 144.93 (C(Ar)), 148.13 (C(Ar)), 150.00 (C(Ar)), 155.46 (C(Ar)), 161.16 (C(Ar)), 171.95 (COOH); MS (ESI) m/z (%, fragment): 427 (28).
2-(3-(4-Fluorophenyl)-5,9-dimethyl-7-oxo-7H-furo[3,2-g]chromen-6-yl)acetic acid (EMAC10164d)
EMAC10163d (0.50 g, 1.26 mmol), 5 ml of propan-2-ol and an aqueous solution of NaOH 1 M (5.0 ml) were mixed and stirred for 4 h. After filtration of the resulting precipitate, a light brown solid was obtained. Yield: 89.7%; m.p. 277–279 °C; Rf (DCM/EtOAc 5:5): 0.28; 1H NMR (400 MHz, DMSO): δ = 2.50 (s, 3H, CH3), 2.50 (s, 3H, CH3), 3.64 (s, 2H, CH2), 7.34–7.38 (m, 2H, 2 x H(Ar′)), 7.82–7.85 (m, 2H, 2 x H(Ar′)), 7.97 (s, 1H, H5), 8.44 (s, 1H, H7), 12.55 (s, 1H, COOH). 13C NMR (100 MHz, DMSO): δ = 8.66 (CH3), 16.11 (CH3), 33.39 (CH2), 109.04 (C(Ar)), 114.17 (C(Ar)), 116.52 (d, JCF = 21.5 Hz, C3′, C5′), 117.15 (C(Ar)), 118.36 (C(Ar)), 121.09 (C(Ar)), 122.24 (C(Ar)), 127.71 (d, JCF = 3.1 Hz, C1′), 129.67 (d, JCF = 8.1 Hz, C2′, C6′), 144.51 (C(Ar)), 148.08 (C(Ar)), 150.00 (C(Ar)), 155.41 (C(Ar)), 160.99 (C(Ar)), 162.30 (d, JCF = 225 Hz, C4′), 171.97 (COOH); MS (ESI) m/z (%, fragment): 367 (88).
2-(3-([1,1′-Biphenyl]-4-yl)-5,9-dimethyl-7-oxo-7H-furo[3,2-g]chromen-6-yl)acetic acid (EMAC10164g)
EMAC10163g (0.50 g, 1.20 mmol), 5 ml of propan-2-ol and an aqueous solution of KOH 1 M (5.0 ml) were mixed and stirred for 4 h. After filtration of the resulting precipitate, a light brown solid was obtained. Yield: 50.1%; m.p. 248–250 °C; Rf (DCM/EtOAc 5:5): 0.15; 1H NMR (400 MHz, DMSO): δ = 2.48 (s, 3H, CH3), 2.51 (s, 3H, CH3), 3.59 (s, 2H, CH2), 7.38–7.41 (m, 1H, H(Ar)), 7.47–7.51 (m, 2H, 2 x H(Ar)), 7.72–7.74 (m, 2H, 2 x H(Ar)), 7.79–7.81 (m, 2H, 2 x H(Ar)), 7.86–7.88 (m, 2H, 2 x H(Ar)), 8.01 (s, 1H, H5), 8.49 (s, 1H, H7); 13C NMR (100 MHz, DMSO): δ = 8.69 (CH3), 16.10 (CH3), 34.03 (CH2), 108.99 (C(Ar)), 114.17 (C5), 117.30 (C(Ar)), 119.11 (C(Ar)), 121.60 (C(Ar)), 122.23 (C(Ar)), 127.01 (2 x C(Ar)), 127.81 (2 x C(Ar)), 128.06 (C(Ar)), 128.11 (2 x C(Ar)), 129.49 (2 x C(Ar)), 130.47 (C(Ar)), 139.83 (C(Ar)), 140.04 (C(Ar)), 144.60 (C7), 148.07 (C(Ar)), 149.43 (C(Ar)), 155.44 (C(Ar)), 161.27 (CO), 172.01 (COOH); MS (ESI) m/z (%, fragment): 425 (57).
2-(3-(2,4-Difluorophenyl)-5,9-dimethyl-7-oxo-7H-furo[3,2-g]chromen-6-yl)acetic acid (EMAC10164h)
EMAC10163h (0.50 g, 1.20 mmol), 5 ml of propan-2-ol and an aqueous solution of NaOH 1 M (4.8 ml) were mixed and stirred for 2 h. After filtration of the resulting precipitate, a brown solid was obtained. Yield: 93.2%; m.p. 227–230 °C; Rf (DCM/EtOAc 5:5): 0.14; 1H NMR (400 MHz, DMSO): δ = 2.47 (s, 3H, CH3), 2.54 (s, 3H, CH3), 3.65 (s, 2H, CH2), 7.25–7.30 (m, 1H, H(Ar)), 7.45–7.51 (m, 1H, H(Ar)), 7.85–7.91 (m, 2H, 2 x H(Ar)), 8.41 (s, 1H, H7); 13C NMR (100 MHz, DMSO): δ = 8.68 (CH3), 16.07 (CH3), 33.42 (CH2), 105.30 (t, JCF = 26.2 Hz, C3′), 109.08 (C(Ar)), 112.74 (dd, JCF = 21.3, 3.5 Hz, C5′), 114.48 (d, JCF = 3.1 Hz, C5), 115.36 (dd, JCF = 14.6, 3.2 Hz, C1′), 117.23 (C(Ar)), 118.55 (C(Ar)), 122.44 (C(Ar)), 131.96 (dd, JCF = 9.8, 5.3 Hz, C6′), 146.16 (d, JCF = 5.5 Hz, C7), 148.15 (C(Ar)), 149.85 (C(Ar)), 154.91 (C(Ar)), 158.60 (C2), 159.91 (d, JCF = 237.2 Hz, C2′), 161.16 (C(Ar)), 162.38 (d, JCF = 235.7 Hz, C4′), 171.93 (COOH); MS (ESI) m/z (%, fragment): 385 (100).
2-(5,9-Dimethyl-7-oxo-3-phenyl-7H-furo[3,2-g]chromen-6-yl)acetic acid (EMAC10164i)
EMAC10163i (0.50 g, 1.32 mmol), 5 ml of propan-2-ol and an aqueous solution of NaOH 1 M (5.3 ml) were mixed and stirred for 5 h. After filtration of the resulting precipitate, a light brown solid was obtained. Yield: 87.1%; m.p. 242–245 °C; Rf (DCM/EtOAc 5:5): 0.39; 1H NMR (400 MHz, DMSO): δ = 2.49 (s, 3H, CH3), 2.50 (s, 3H, CH3), 3.63 (s, 2H, CH2), 7.41–7.45 (m, 1H, H4′), 7.52–7.56 (m, 2H, 2 x H(Ar′)), 7.77–7.80 (m, 2H, 2 x H(Ar′)), 7.99 (s, 1H, H5), 8.45 (s, 1H, H7); 13C NMR (100 MHz, DMSO): δ = 8.66 (CH3), 16.08 (CH3), 33.47 (CH2), 109.01 (C(Ar)), 114.19 (C(Ar)), 117.12 (C(Ar)), 118.41 (C(Ar)), 122.01 (C(Ar)), 122.30 (C(Ar)), 127.61 (2 x C(Ar′)), 128.22 (C(Ar)), 129.65 (2 x C(Ar)), 131.29 (C(Ar)), 144.50 (C(Ar)), 148.04 (C(Ar)), 149.90 (C(Ar)), 155.47 (C(Ar)), 161.20 (C(Ar)), 172.00 (COOH). MS (ESI) m/z (%, fragment): 349 (100).
2-(3-(4-Chlorophenyl)-5,9-dimethyl-7-oxo-7H-furo[3,2-g]chromen-6-yl)acetic acid (EMAC10164j)
EMAC10163j (0.35 g, 0.84 mmol), 4 ml of propan-2-ol and an aqueous solution of NaOH 1 M (3.4 ml) were mixed and stirred for 4 h. After filtration of the resulting precipitate, a light brown solid was obtained. Yield: 67.9%; m.p. 266–269 °C; Rf (DCM/EtOAc 5:5): 0.11; 1H NMR (400 MHz, DMSO): δ = 2.51 (s, 3H, CH3), 2.51 (s, 3H, CH3), 3.65 (s, 2H, CH2), 7.56–7.60 m, 2H, 2 x H(Ar)), 7.81–7.84 (m, 2H, 2 x H(Ar′)), 7.99 (s, 1H, H5), 8.51 (s, 1H, H7), 12.55 (s, 1H, COOH); 13C NMR (100 MHz, DMSO): δ = 8.68 (CH3), 16.12 (CH3), 33.41 (CH2), 109.10 (C(Ar)), 114.24 (C(Ar)), 117.22 (C(Ar)), 118.43 (C(Ar)), 120.94 (C(Ar)), 122.02 (C(Ar)), 129.36 (2 x C(Ar′)), 129.61 (2 x C(Ar′)), 130.20 (C(Ar)), 132.77 (C(Ar)), 144.95 (C(Ar)), 148.12 (C(Ar)), 149.99 (C(Ar)), 155.46 (C(Ar)), 161.16 (C(Ar)), 171.95 (COOH); MS (ESI) m/z (%, fragment): 383 (32).
2-(3-(3-Methoxyphenyl)-5,9-dimethyl-7-oxo-7H-furo[3,2-g]chromen-6-yl)acetic acid (EMAC10164k)
EMAC10163k (0.50 g, 1.22 mmol), 5 ml of propan-2-ol and an aqueous solution of NaOH 1 M (4.9 ml) were mixed and stirred for 4 h. After filtration of the resulting precipitate, a light brown solid was obtained. Yield: 91.7%; m.p. 220–224 °C; Rf (DCM/EtOAc 5:5): 0.41;1H NMR (400 MHz, DMSO): δ = 2.51 (s, 3H, CH3), 2.53 (s, 3H, CH3), 3.65 (s, 2H, CH2), 3.86 (s, 3H, OCH3), 6.99–7.02 (m, 1H, H(Ar)), 7.30 (m, 1H, H6′), 7.37–7.39 (m, 1H, H(Ar)), 7.44–7.49 (m, 1H, H(Ar)), 8.01 (s, 1H, H5), 8.48 (s, 1H, H7); 13C NMR (100 MHz, DMSO): δ = 8.69 (CH3), 16.08 (CH3), 33.42 (CH2), 55.67 (OCH3), 109.06 (C(Ar)), 112.99 (C(Ar)), 113.99 (C(Ar)), 114.29 (C(Ar)), 117.15 (C(Ar)), 118.39 (C(Ar)), 119.94 (C(Ar)), 121.99 (C(Ar)), 122.35 (C(Ar)), 130.79 (C(Ar)), 132.59 (C(Ar)), 144.77 (C(Ar)), 148.08 (C(Ar)), 149.96 (C(Ar)), 155.48 (C(Ar)), 160.28 (C(Ar)), 161.21 (C(Ar)), 171.97 (COOH). MS (ESI) m/z (%, fragment): 379 (100).
Molecular modelling
Ligand preparation
Maestro GUI softwareCitation57 was used to create theoretical 3D models of the compounds. Coumarin’s opening was also considered, and the compounds were constructed in both E and Z configurations, considering the same mechanism proposed by Maresca et al.Citation63,Citation65. The ligands most stable conformation has been determined by molecular mechanics conformational analysis performed by Macromodel software version 9.2Citation66, considering Merck Molecular Force Fields (MMFFs)Citation67 as force field and solvent effects by adopting the generalised Born/surface area (GB/SA) water implicit solvation modelCitation68. The simulations were performed allowing 5000 steps Monte Carlo analysis with Polak–Ribier Conjugate Gradient (PRCG) method and a convergence criterion of 0.05 kcal/(mol Å) was used. All the other parameters were left as default.
Protein preparation
The coordinates for hCA isoforms enzymes were taken from the RCSB Protein Data BankCitation69 (PDB codes 5FL4Citation70, for isoform IX and 5MSACitation71, for isoform XII). These 3D structures are high resolution X-ray models and the alignment with the other 3D structure did not highlight significative difference to justify the use of an ensemble docking approach. Maestro Protein Preparation Wizard protocol was used to prepare the proteins. The original water molecules and ligands were removed. The Gln and Asn residues were analysed and orientated with the best terminal amide position. Likewise, the best His tautomer was chosen according to the best orientation.
Docking experiments
Quantum Mechanics-Polarised Ligand (QMPL) Docking was used for the molecular docking studies applying the validated protocol, which was extended to more complexes (Table S8)Citation51,Citation52,Citation72. Grids were defined around the refined structure by centring on crystallised ligands. The other settings were left as default.
Post-docking experiments
The best pose complexes were then minimised to consider the induced fit phenomena and used to analyse the ligand binding mode. Ten thousand steps of the PRCG minimisation method were conducted on the top ranked theoretical complexes using OPLS_2005 force field. The optimisation process was performed up to the derivative convergence criterion equal to 0.1 kcal/(mol Å).
Drug-like properties prediction
Drug-like properties of compounds were theoretically predicted using QikProp software and reported in Tables S2–S7.
Biological activity
Carbonic anhydrase inhibition assay
The CA catalysed CO2 hydration/inhibition was measured by using a stopped-flow instrument as the method previously describedCitation73. Initial rates of the CA-catalysed CO2 hydration reaction were followed for 10–100 s. The CO2 concentrations were ranged from 1.7 to 17 mM for the determination of the inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction were used for assessing the initial velocity. The uncatalysed rates were subtracted from the total observed rates. Stock solutions of inhibitors (10 mM) and dilutions up to 0.01 nM were prepared in distilled-deionised water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 as reported earlier and represent the mean from at least three different determinations. hCA I, hCA II, hCA IX, and hCA XII (catalytic domain) were recombinant proteins produced inhouse using our standardised protocol and their concentration in the assay system was in the range of 3–10 nM. AAZ was used as reference CA inhibitorCitation64,Citation74,Citation75.
Author contributions
Lisa Sequeira: investigation, data curation, formal analysis, and writing – original draft. Simona Distinto: investigation, data curation, formal analysis, methodology, supervision, and writing – review and editing. Rita Meleddu: methodology, supervision, and writing – review and editing. Marco Gaspari: investigation. Andrea Angeli: investigation. Filippo Cottiglia: supervision and writing – review and editing. Daniela Secci: investigation. Alessia Onali: investigation. Erica Sanna: investigation. Fernanda Borges: funding acquisition, resourses, supervision, writing – review & editing. Eugenio Uriarte: resources, supervision, writing – review & editing. Stefano Alcaro: conceptualization, resources. Claudiu T. Supuran: conceptualization, resources. Elias Maccioni: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing.
Supplemental Material
Download PDF (4 MB)Disclosure statement
All authors declare no conflict of interest except CTS. CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30.
- Whiteman DC, Wilson LF. The fractions of cancer attributable to modifiable factors: a global review. Cancer Epidemiol. 2016;44:203–221.
- Mihor A, Tomsic S, Zagar T, Lokar K, Zadnik V. Socioeconomic inequalities in cancer incidence in Europe: a comprehensive review of population-based epidemiological studies. Radiol Oncol. 2020;54(1):1–13.
- Wu HX, Zhuo KQ, Wang K. Efficacy of targeted therapy in patients with HER2-positive non-small cell lung cancer: a systematic review and meta-analysis. Br J Clin Pharmacol. 2022;88(5):2019–2034.
- Modi S, Jacot W, Yamashita T, Sohn J, Vidal M, Tokunaga E, Tsurutani J, Ueno NT, Prat A, Chae YS, et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N Engl J Med. 2022;387(1):9–20.
- Zhang M, Fan Y, Nie L, Wang G, Sun K, Cheng Y. Clinical outcomes of immune checkpoint inhibitor therapy in patients with advanced non-small cell lung cancer and preexisting interstitial lung diseases: a systematic review and meta-analysis. Chest. 2022;161(6):1675–1686.
- Jaaks P, Coker EA, Vis DJ, Edwards O, Carpenter EF, Leto SM, Dwane L, Sassi F, Lightfoot H, Barthorpe S, et al. Effective drug combinations in breast, colon and pancreatic cancer cells. Nature. 2022;603(7899):166–173.
- Global Burden of Disease Cancer Collaboration. The global burden of cancer 2013. JAMA Oncol. 2015;1(4):505–527.
- de Martel C, Georges D, Bray F, Ferlay J, Clifford GM. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health. 2020;8(2):e180–e190.
- Supuran C. Carbonic anhydrases: an overview. Curr Pharm Des. 2008;14(7):603–614.
- Supuran CT. Carbonic anhydrases. Bioorg Med Chem. 2013;21(6):1377–1378.
- Nocentini A, Supuran CT, Capasso C. An overview on the recently discovered iota-carbonic anhydrases. J Enzyme Inhib Med Chem. 2021;36(1):1988–1995.
- Supuran CT, Capasso C. An overview of the bacterial carbonic anhydrases. Metabolites. 2017;7(4):56–74.
- Angeli A, Pinteala M, Maier SS, Del Prete S, Capasso C, Simionescu BC, Supuran CT. Inhibition of α-, β-, γ-, δ-, ζ- and η-class carbonic anhydrases from bacteria, fungi, algae, diatoms and protozoans with famotidine. J Enzyme Inhib Med Chem. 2019;34(1):644–650.
- Angeli A, Del Prete S, Alasmary FAS, Alqahtani LS, AlOthman Z, Donald WA, Capasso C, Supuran CT. The first activation studies of the η-carbonic anhydrase from the malaria parasite Plasmodium falciparum with amines and amino acids. Bioorg Chem. 2018;80:94–98.
- Schlicker C, Hall RA, Vullo D, Middelhaufe S, Gertz M, Supuran CT, Mühlschlegel FA, Steegborn C. Structure and inhibition of the CO2-sensing carbonic anhydrase Can2 from the pathogenic fungus Cryptococcus neoformans. J Mol Biol. 2009;385(4):1207–1220.
- Maren TH. Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol Rev. 1967;47(4):595–781.
- Singh S, Lomelino CL, Mboge MY, Frost SC, McKenna R. Cancer drug development of carbonic anhydrase inhibitors beyond the active site. Molecules. 2018;23(5):1045–1066.
- Hilvo M, Tolvanen M, Clark A, Shen B, Shah GN, Waheed A, Halmi P, Hänninen M, Hämäläinen JM, Vihinen M, et al. Characterization of CA XV, a new GPI-anchored form of carbonic anhydrase. Biochem J. 2005;392(Pt 1):83–92.
- Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem. 2016;31(5):689–694.
- Frost SC. Physiological functions of the alpha class of carbonic anhydrases. In: Frost SC, McKenna R, editors. Carbonic anhydrase: mechanism, regulation, links to disease, and industrial applications. Dordrecht: Springer Netherlands; 2014. p. 9–30.
- Mboge MY, Mahon BP, McKenna R, Frost SC. Carbonic anhydrases: role in pH control and cancer. Metabolites. 2018;8(1):19–50.
- Pastorekova S, Parkkila S, Závada J. Tumor‐associated carbonic anhydrases and their clinical significance. Adv Clin Chem. 2006;42:167–216.
- Supuran CT, Alterio V, Di Fiore A, D' Ambrosio K, Carta F, Monti SM, De Simone G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev. 2018;38(6):1799–1836.
- Nocentini A, Donald WA, Supuran CT. Chapter 8 – human carbonic anhydrases: tissue distribution, physiological role, and druggability. In: Supuran CT, Nocentini A, editors. Carbonic anhydrases. Cham, Switzerland: Academic Press; 2019. p. 151–185.
- Becker HM. Carbonic anhydrase IX and acid transport in cancer. Br J Cancer. 2020;122(2):157–167.
- Ondriskova E, Debreova M, Pastorekova S. Chapter 10 – tumor-associated carbonic anhydrases IX and XII. In: Supuran CT, De Simone G, editors. Carbonic anhydrases as biocatalysts. Amsterdam: Elsevier; 2015. p. 169–205.
- Pastorekova S, Gillies RJ. The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019;38(1–2):65–77.
- Aldera AP, Govender D. Carbonic anhydrase IX: a regulator of pH and participant in carcinogenesis. J Clin Pathol. 2021;74(6):350–354.
- Wingo T, Tu C, Laipis PJ, Silverman DN. The catalytic properties of human carbonic anhydrase IX. Biochem Biophys Res Commun. 2001;288(3):666–669.
- Türeci Ö, Sahin U, Vollmar E, Siemer S, Göttert E, Seitz G, Parkkila A-K, Shah GN, Grubb JH, Pfreundschuh M, et al. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc Natl Acad Sci U S A. 1998;95(13):7608–7613.
- Parkkila S, Parkkila A-K, Saarnio J, Kivelä J, Karttunen TJ, Kaunisto K, Waheed A, Sly WS, Türeci Ö, Virtanen I, et al. Expression of the membrane-associated carbonic anhydrase isozyme XII in the human kidney and renal tumors. J Histochem Cytochem. 2000;48(12):1601–1608.
- Waheed A, Sly WS. Carbonic anhydrase XII functions in health and disease. Gene. 2017;623:33–40.
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem. 2011;3(9):1165–1180.
- Mishra CB, Tiwari M, Supuran CT. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev. 2020;40(6):2485–2565.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7(2):168–181.
- Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008–2018). Expert Opin Ther Pat. 2018;28(10):729–740.
- Al-Warhi T, Sabt A, Elkaeed EB, Eldehna WM. Recent advancements of coumarin-based anticancer agents: an up-to-date review. Bioorg Chem. 2020;103:104163–104178.
- Annunziata F, Pinna C, Dallavalle S, Tamborini L, Pinto A. An overview of coumarin as a versatile and readily accessible scaffold with broad-ranging biological activities. Int J Mol Sci. 2020;21(13):4618–4699.
- Riveiro EM, De Kimpe N, Moglioni A, Vazquez R, Monczor F, Shayo C, Davio C. Coumarins: old compounds with novel promising therapeutic perspectives. Curr Med Chem. 2010;17(13):1325–1338.
- Wardrop D, Keeling D. The story of the discovery of heparin and warfarin. Br J Haematol. 2008;141(6):757–763.
- Au N, Rettie AE. Pharmacogenomics of 4-hydroxycoumarin anticoagulants. Drug Metab Rev. 2008;40(2):355–375.
- Dawood D, Batran R, Farghaly T, Khedr M, Abdalla M. New coumarin derivatives as potent selective COX-2 inhibitors: synthesis, anti-inflammatory, QSAR, and molecular modeling studies. Arch Pharm. 2015;348:878–888.
- de Souza LG, Rennã MN, Figueroa-Villar JD. Coumarins as cholinesterase inhibitors: a review. Chem Biol Interact. 2016;254:11–23.
- Salar U, Khan KM, Jabeen A, Faheem A, Naqvi F, Ahmed S, Iqbal E, Ali F, Kanwal , Perveen S. ROS inhibitory activity and cytotoxicity evaluation of benzoyl, acetyl, alkyl ester, and sulfonate ester substituted coumarin derivatives. Med Chem. 2020;16(8):1099–1111.
- Gormley NA, Orphanides G, Meyer A, Cullis PM, Maxwell A. The interaction of coumarin antibiotics with fragments of the DNA gyrase B protein. Biochemistry. 1996;35(15):5083–5092.
- Shaikh M, Subhedar D, Khan F, Sangshetti J, Shingate B. 1,2,3-Triazole incorporated coumarin derivatives as potential antifungal and antioxidant agents. Chin Chem Lett. 2016;27(2):295–301.
- Campagna M, Rivas C. Antiviral activity of resveratrol. Biochem Soc Trans. 2010;38(Pt 1):50–53.
- Goud NS, Kumar P, Bharath R. Recent developments of target based coumarin derivatives as potential anticancer agents. Mini Rev Med Chem. 2020;20(17):1754–1766.
- Küpeli Akkol E, Genç Y, Karpuz B, Sobarzo-Sánchez E, Capasso R. Coumarins and coumarin-related compounds in pharmacotherapy of cancer. Cancers. 2020;12(7):1959–1984.
- Melis C, Distinto S, Bianco G, Meleddu R, Cottiglia F, Fois B, Taverna D, Angius R, Alcaro S, Ortuso F, et al. Targeting tumor associated carbonic anhydrases IX and XII: highly isozyme selective coumarin and psoralen inhibitors. ACS Med Chem Lett. 2018;9(7):725–729.
- Meleddu R, Deplano S, Maccioni E, Ortuso F, Cottiglia F, Secci D, Onali A, Sanna E, Angeli A, Angius R, et al. Selective inhibition of carbonic anhydrase IX and XII by coumarin and psoralen derivatives. J Enzyme Inhib Med Chem. 2021;36(1):685–692.
- Fois B, Distinto S, Meleddu R, Deplano S, Maccioni E, Floris C, Rosa A, Nieddu M, Caboni P, Sissi C, et al. Coumarins from Magydaris pastinacea as inhibitors of the tumour-associated carbonic anhydrases IX and XII: isolation, biological studies and in silico evaluation. J Enzyme Inhib Med Chem. 2020;35(1):539–548.
- Park H, Choe H, Hong S. Virtual screening and biochemical evaluation to identify new inhibitors of mammalian target of rapamycin (mTOR). Bioorg Med Chem Lett. 2014;24(3):835–838.
- Mi C, Ma J, Wang KS, Zuo HX, Wang Z, Li MY, Piao LX, Xu GH, Li X, Quan ZS, et al. Imperatorin suppresses proliferation and angiogenesis of human colon cancer cell by targeting HIF-1α via the mTOR/p70S6K/4E-BP1 and MAPK pathways. J Ethnopharmacol. 2017;203:27–38.
- Ma C-C, Liu Z-P. Design and synthesis of coumarin derivatives as novel PI3K inhibitors. Anticancer Agents Med Chem. 2017;17(3):395–403.
- Menezes JC, Diederich M. Translational role of natural coumarins and their derivatives as anticancer agents. Future Med Chem. 2019;11(9):1057–1082.
- Meleddu R, Maccioni E, Distinto S, Bianco G, Melis C, Alcaro S, Cottiglia F, Ceruso M, Supuran CT. New 4-[(3-cyclohexyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzene-1-sulfonamides, synthesis and inhibitory activity toward carbonic anhydrase I, II, IX, XII. Bioorg Med Chem Lett. 2015;25(16):3281–3284.
- Melis C, Meleddu R, Angeli A, Distinto S, Bianco G, Capasso C, Cottiglia F, Angius R, Supuran CT, Maccioni E. Isatin: a privileged scaffold for the design of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2017;32(1):68–73.
- Distinto S, Meleddu R, Ortuso F, Cottiglia F, Deplano S, Sequeira L, Melis C, Fois B, Angeli A, Capasso C, et al. Exploring new structural features of the 4-[(3-methyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzenesulphonamide scaffold for the inhibition of human carbonic anhydrases. J Enzyme Inhib Med Chem. 2019;34(1):1526–1533.
- Skaraitė I, Maccioni E, Petrikaitė V. Anticancer activity of sunitinib analogues in human pancreatic cancer cell cultures under normoxia and hypoxia. Int J Mol Sci. 2023;24(6):5422.
- Di Maria S, Picarazzi F, Mori M, Cianciusi A, Carbone A, Crespan E, Perini C, Sabetta S, Deplano S, Poggialini F, et al. Novel pyrazolo[3,4-d]pyrimidines as dual Src/Bcr-Abl kinase inhibitors: synthesis and biological evaluation for chronic myeloid leukemia treatment. Bioorg Chem. 2022;128:106071.
- Maresca A, Temperini C, Pochet L, Masereel B, Scozzafava A, Supuran CT. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem. 2010;53(1):335–344.
- Bilginer S, Gonder B, Gul HI, Kaya R, Gulcin I, Anil B, Supuran CT. Novel sulphonamides incorporating triazene moieties show powerful carbonic anhydrase I and II inhibitory properties. J Enzyme Inhib Med Chem. 2020;35(1):325–329.
- Maresca A, Temperini C, Vu H, Pham NB, Poulsen S-A, Scozzafava A, Quinn RJ, Supuran CT. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc. 2009;131(8):3057–3062.
- Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. Macromodel—an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J Comput Chem. 1990;11(4):440–467.
- Halgren TA. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J Comput Chem. 1996;17(5–6):520–552.
- Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33(12):889–897.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242.
- Leitans J, Kazaks A, Balode A, Ivanova J, Zalubovskis R, Supuran CT, Tars K. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J Med Chem. 2015;58(22):9004–9009.
- Smirnov A, Zubrienė A, Manakova E, Gražulis S, Matulis D. Crystal structure correlations with the intrinsic thermodynamics of human carbonic anhydrase inhibitor binding. PeerJ. 2018;6:e4412.
- Chung JY, Hah J-M, Cho AE. Correlation between performance of QM/MM docking and simple classification of binding sites. J Chem Inf Model. 2009;49(10):2382–2387.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase: I. Stop-flow kinetic studies on the native human isoenzymes B And C. J Biol Chem. 1971;246(8):2561–2573.
- Berrino E, Angeli A, Zhdanov DD, Kiryukhina AP, Milaneschi A, De Luca A, Bozdag M, Carradori S, Selleri S, Bartolucci G, et al. Azidothymidine “Clicked” into 1,2,3-triazoles: first report on carbonic anhydrase–telomerase dual-hybrid inhibitors. J Med Chem. 2020;63(13):7392–7409.
- Pacchiano F, Carta F, McDonald PC, Lou Y, Vullo D, Scozzafava A, Dedhar S, Supuran CT. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem. 2011;54(6):1896–1902.