
Abstract
Our previous studies have shown that the introduction of structurally diverse benzyl side chains at the C5-NH2 position of oseltamivir to occupy 150-cavity contributes to the binding affinity with neuraminidase and anti-influenza activity. To obtain broad-spectrum neuraminidase inhibitors, we designed and synthesised a series of novel oseltamivir derivatives bearing different N-heterocycles substituents that have been proved to induce opening of the 150-loop of group-2 neuraminidases. Among them, compound 6k bearing 4-((r)-2-methylpyrrolidin-1-yl) benzyl group exhibited antiviral activities similar to or weaker than those of oseltamivir carboxylate against H1N1, H3N2, H5N1, H5N6 and H5N1-H274Y mutant neuraminidases. More encouragingly, 6k displayed nearly 3-fold activity enhancement against H3N2 virus over oseltamivir carboxylate and 2-fold activity enhancement over zanamivir. Molecular docking studies provided insights into the explanation of its broad-spectrum potency against wild-type neuraminidases. Overall, as a promising lead compound, 6k deserves further optimisation by fully considering the ligand induced flexibility of the 150-loop.
Introduction
Influenza virus, the “silent terrorist” of the 21st century, endangers human health and social development. Influenza, mainly caused by influenza A virus, is still one of the most grievous global epidemics, with an estimated about 3–5 million cases of severe illness and 290,000–650,000 deaths annually all over the worldCitation1,Citation2. In the past, several influenza A (Flu A) pandemics have caused enormous effects on global human population (e.g. 1918HIN1 and 2009H7N9)Citation3. According to the latest reports, compared with SARS-CoV-2 mono-infection, co-infection with influenza viruses was associated with higher morbidity and mortalityCitation4.
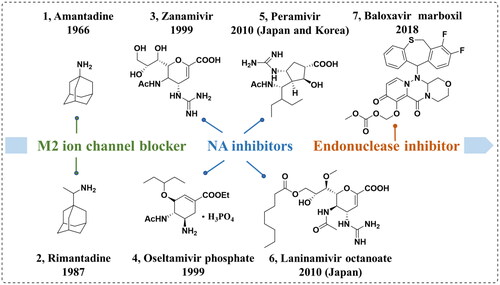
Influenza vaccines and antiviral drugs can be used to effectively prevent and treat influenza. Although the influenza vaccines are considered the most effective measure for the prophylaxis of influenza, there are many limitations because of high variability of influenza and rapid antigenic drift; hence, the influenza vaccines are only available for the current epidemic influenza virus strainsCitation5. Additionally, the influenza vaccine must be administered annually and its effectiveness is dependent on individual immunity. Thus, antiviral drugs remain an important weapon against influenza infections. There are currently three classes of anti-influenza agents that have been approved by the FDA (), namely polymerase acidic protein (PA) endonuclease inhibitor (baloxavir marboxil)Citation6, M2 ion-channel blockers (amantadine and rimantadine) and neuraminidase inhibitors (NAIs, oseltamivir, zanamivir, peramivir, and laninamivir octanoate)Citation7–10. However, some of these licenced drugs have several drawbacksCitation11.
Figure 1. Structures of approved agents for the treatment of influenza viral infection.
Neuraminidase (NA), a crucial surface antigenic glycoprotein, is responsible for viral penetration through mucosal secretions, helping the virus to access the target cells by mucus degradation. Moreover, NA allows the detachment of the virion from infected cells and avoids the self-aggregation of progeny virions at late stages of infection by disrupting haemagglutinin–sialic acid interactions, thus promoting the release and spread of influenza virus. Among NAIs, oseltamivir phosphate, with high efficiency, high bioavailability and low toxicity, can be administered orally. However, several NA variants acquired resistance to oseltamivir, such as H274Y mutant and I117T mutant, which have seriously limited its applicationCitation12,Citation13. Consequently, there is a pressing need to develop novel improved NAIs.
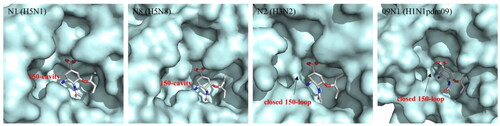
NA subtypes can be categorised into two groups: group-1 (N1, N4, N5 and N8), and group-2 (N2, N3, N6, N7 and N9) according to the phylogenetic treeCitation14. To the best of current crystal structure knowledge, the 150-loop, containing residues 147–152, adopts an open conformation among group-1 NAs, forming the 150-cavity ()Citation15. In contrast, in group-2 NAs, this loop is always closedCitation16. Further studies indicated that the conformation of group-2 NAs can be induced to form open 150-cavity by neuraminidase inhibitorsCitation17. Therefore, a deeper understanding of the relationship between the catalytic site and 150-loop can provide a new strategy for designing new influenza antiviral drugs that target both sites at the same time.
Figure 2. Comparison of the crystal structures of representative group-1 Nas (N1, PDB code: 2HU0, N8, PDB code 2HT7), group-2 Nas (N2, PDB code 4GZP), and 09N1 (PDB code 3TI6).
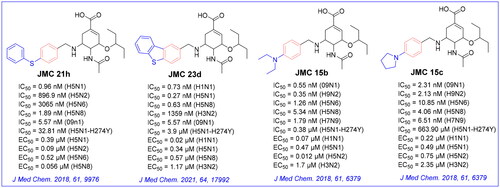
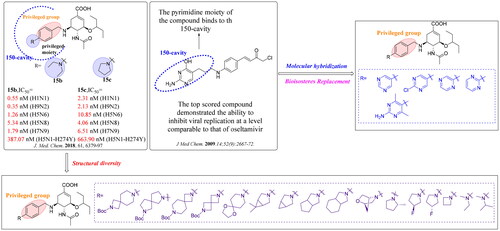
The crystal complex of NA-oseltamivir revealed that the C5-NH2 group of oseltamivir is directed towards the 150-cavity, making such an amino group a potential modification site. Previous research efforts from our laboratory have identified several oseltamivir derivatives with more potent inhibitory activity than oseltamivir carboxylate (OSC, the active form of oseltamivir) against several subtypes of influenza virus ()Citation18–23. The discovery of these oseltamivir derivatives bearing different substituted benzyl groups indicated that it is reasonable to occupy 150-cavity with an appropriate substituent. Accordingly, a substituted benzyl group that mediates the hydrophobic force is a privileged moiety of the 150-cavity. Additional hydrophobic interactions with the 150-cavity could improve the potency of these compounds against N1-H274Y mutants while retaining their outstanding potency against other group-1 NAs. In our recent screening, we identified compound EJMC43bCitation20 with the specific inhibition of group-1 NAs, which was a derivative of JMC21h. Although the inhibitory effect of these compounds was greatly improved, their inhibitory potency on group-2 NAs was robustly reduced. Unlike several compounds that were found to be selective group-1 NA inhibitors, compounds JMC15b and JMC15c showed potent inhibitory effects on both group-1 and group-2 NAsCitation19. These results suggested that nitrogen-containing substituents such as polar groups or aromatic rings at the terminal end of benzene ring are capable to induce the open 150-cavity conformation and retain the potent inhibitory activity against group-2 NA.
Figure 3. Structures of our previously reported group-1-specific influenza NA inhibitors (JMC23d and JMC21h) and non-specific group-1 and group-2 NA inhibitors (JMC15b and JMC15c).
Thus, to extend our previous work, we intended to introduce different N-substituted groups at the end of benzene ring to further explore the effects of substitutions on benzyl group. Herein, we designed and synthesised 21 novel oseltamivir analogues bearing enriched N-heterocycles substitutions (), anticipating that: (1) N-containing aryl groups, such as pyrimidine substitutions at the para-position of the benzyl group, would bind effectively to the 150-cavity of group-1 NAs because of sufficiently large size, sufficient length, relative flexibility and hydrophobic force driving their binding; (2) the diverse N-heterocycles substitutions such as spiro substituents, bridge-ring substituents and cycloalkane substituents can induce the open 150-cavity conformation; (3) the introduction of different N-heterocycles substitutions could increase the Fsp3 value to improve “drug-likeness” of the newly designed oseltamivir derivatives.
Figure 4. The novel designed oseltamivir analogues via targeting the 150-cavity.
Materials and methods
Chemistry
The key chemical reactant oseltamivir phosphate was provided by Shandong Qidu Pharmaceutical Co., Ltd (Zibo, China). Other chemical materials were obtained from Bidepharmatech. Ltd (Shanghai, China) and were at least 97% pure. Solvents were obtained from Fuyuqy Co., Ltd (Tianjin, China). Thin-layer chromatography (TLC) was performed using plates coated with Silica Gel GF254 (SKU: 1077301000) for TLC (Merck, Shanghai, China), and the product spots were visualised under irradiation with UV light (l ¼ 254 nm). Flash column chromatography was conducted on columns packed with Silica Gel (200e300 mesh, catalogue numbers: C0583530035), purchased from Qingdao Haiyang Chemical Company (Qingdao, China). Melting points (mp) of all compounds were measured with a micro melting point apparatus (RY-1G, Tianjin TianGuang Optical Instruments) without correction. 1H NMR and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer with TMS as an internal standard and CD3OD or DMSO‑d6 as a solvent. Coupling constants (J) were expressed in hertz (Hz), and chemical shifts were reported in values (ppm) from TMS. Mass spectra (MS) were registered with API 4000 LC/MS spectrometer (Applied Biosystems, USA).
General procedure for the preparation of compounds 1a∼1e
Compounds 1a∼1e were obtained by Suzuki reaction of 4-formylphenylboronic acid with corresponding substituted aryl bromides. The reaction was monitored by TLC. To a solution of 4-formylphenylboronic acid (1.04 g, 6.93 mmol, 1.1 equiv), K3PO4 (4.7 g, 22.05 mmol, 3.5 equiv), Pd(PPh3)4 (0.7 g, 0.63 mmol, 0.1 equiv), N2, in PhMe/H2O = 25/2 (30 ml) was added corresponding substituted aryl bromides (1 equiv). The reaction mixture was stirred under a nitrogen atmosphere at 100 °C for 12 h. Then, toluene was removed under reduced pressure, and brine (30 ml) was added. The mixture was extracted with ethyl acetate (3 × 30 ml), dried over anhydrous MgSO4, and concentrated in vacuo to give the crude product, which was purified by silica gel chromatography with ethyl acetate/petroleum ether (1:100) as the eluent to provide intermediates 1a∼1e.
General procedure for the preparation of compounds 2a∼2e
Via Borch reduction reaction, compounds 2a∼2e were synthesised from oseltamivir phosphate and an aldehyde (1a∼1e). A mixture of oseltamivir phosphate (0.82 g, 2.0 mmol) and an aldehyde (1a∼1e) (2.0 mmol, 1 eq) in 30 ml methanol was stirred at room temperature for 0.5 h, NaBH3CN (0.15 g, 2.5 mmol, 2.5 equiv) was added slowly. The reaction mixture was stirred at room temperature for 6–7 h. After the reaction completed (judged by TLC), methanol was evaporated in vacuum, and then, brine (30 ml) and saturated sodium carbonate solution (10 ml) were added. This mixture was extracted with ethyl acetate (3 × 30 ml), dried over anhydrous MgSO4, and concentrated under reduced pressure to give the crude product, which was purified by silica gel column chromatography with petroleum ether/ethyl acetate (1/1) and ethyl acetate to obtain the intermediates 2a∼2e.
General procedure for the preparation of compounds 3a∼3e
Compounds 3a-3e were prepared by direct hydrolysis of the intermediates 2a-2e. Each intermediate of 2a∼2e (1.0 mmol) was dissolved in 30 ml of methanol, and 1 M NaOH aqueous solution (5 ml) was added. The mixture was stirred at room temperature for 2 h. After that, most methanol was removed under reduced pressure and the residue was dissolved with 30 ml water. The aqueous phase was acidified (pH to 2 − 3) by 3 M HCl to precipitate the target compounds 3a∼3e which were collected by filtration and dried in vacuo.
(3R,4R,5S)-4-acetamido-3-(pentan-3-yloxy)-5-((4-(pyrimidin-5-yl)benzyl)amino)cyclohex-1-ene-1-carboxylic acid (3a)
White power, 83% yield, mp: 150–151 °C. 1H NMR (600 MHz, CD3OD) δ: 9.16 (s, 1H, Pyrazine-H), 8.70 (d, J = 2.2 Hz, 1H, Pyrazine-H), 8.58 (s, 1H, Pyrazine-H), 8.20 (d, J = 8.0 Hz, 2H, 2Ph-H), 7.68 (d, J = 7.9 Hz, 2H, 2Ph-H), 6.89 (s, 1H, CH), 4.50 (d, J = 13.1 Hz, 1H, CH), 4.37 (d, J = 13.1 Hz, 1H, CH), 4.31 − 4.18 (m, 2H, 2CH), 3.74 − 3.64 (m, 1H, CH), 3.50 − 3.43 (m, 1H, CH), 3.08 (dd, J = 17.3, 5.3 Hz, 1H, CH), 2.75 − 2.66 (m, 1H, CH), 2.08 (s, 3H, CH3), 1.62 − 1.47 (m, 4H, 2CH2), 0.92 (dt, J = 12.4, 7.4 Hz, 6H, 2CH3). 13C NMR (150 MHz, CD3OD) δ: 151.87, 144.46, 143.19, 141.83, 137.37, 132.61, 130.30, 127.44, 82.36, 74.55, 68.71, 55.15, 51.59, 25.86, 25.74, 25.26, 22.08, 21.70, 8.41, 8.17. ESI-MS: m/z 453.15 [M + H]+, C25H32N4O4 (452.56).
(3R,4R,5S)-4-acetamido-3-(pentan-3-yloxy)-5-((4-(pyrimidin-5-yl)benzyl)amino)cyclohex-1-ene-1-carboxylic acid (3b)
White power, 81% yield, mp: 150–151 °C. 1H NMR (400 MHz, Methanol-d4) δ 8.87 (d, J = 4.8 Hz, 2H, 2 Pyrimidine-H), 8.50 (d, J = 7.7 Hz, 2H, 2Ph-H), 7.64 (d, J = 7.9 Hz, 2H, 2Ph-H), 7.40 (t, J = 4.8 Hz, 1H, Pyrimidine-H), 6.88 (s, 1H, CH), 4.49 (d, J = 12.7 Hz, 1H, CH), 4.41 − 4.19 (m, 3H, 3CH), 3.71 (q, J = 6.2, 5.8 Hz, 1H, CH), 3.47 (p, J = 5.5 Hz, 1H, CH), 3.08 (d, J = 18.3 Hz, 1H, CH), 2.77 − 2.63 (m, 1H, CH), 2.08 (s, 3H, CH3), 1.54 (dq, J = 13.5, 6.9 Hz, 4H, 2CH2), 0.91 (q, J = 7.6 Hz, 6H, 2CH3).13C NMR (150 MHz, CD3OD) δ: 163.47, 157.46, 138.66, 137.35, 133.37, 129.80, 128.55, 119.80, 82.36, 74.58, 55.16, 51.57, 25.90, 25.73, 25.25, 22.07, 8.41, 8.17. ESI-MS: m/z 453.15 [M + H]+, C25H32N4O4 (452.56).
(3R,4R,5S)-4-acetamido-3-(pentan-3-yloxy)-5-((4-(pyrimidin-5-yl)benzyl)amino)cyclohex-1-ene-1-carboxylic acid (3c)
White power, 72% yield, mp: 132–133 °C. 1H NMR (400 MHz, CD3OD) δ: 9.31 − 8.96 (m, J = 23.8, 9.8 Hz, 3H, 3Pyridine-H), 7.81 (d, J = 8.2 Hz, 2H, 2Ph-H), 7.64 (d, J = 8.2 Hz, 2H, 2Ph-H), 6.76 (s, 1H, CH), 4.30 (d, J = 13.3 Hz, 1H, CH), 4.23 − 4.00 (m, 3H, 3CH), 3.81 − 3.59 (m, 1H, CH), 3.45 (dt, J = 11.3, 5.5 Hz, 1H, CH), 2.97 (dd, J = 17.6, 5.2 Hz, 1H, CH), 2.59 − 2.45 (m, 1H, CH), 2.06 (s, 3H, CH3), 1.62 − 1.47 (m, 4H, 2CH2), 0.99 − 0.85 (m, 6H, 2CH3). 13C NMR (100 MHz, CD3OD) δ: 173.09, 169.72, 156.75, 154.72, 135.36, 134.83, 134.19, 133.95, 130.44, 130.10, 127.28, 82.08, 75.12, 68.71, 54.83, 52.82, 27.81, 25.78, 25.24, 21.69, 8.46, 8.19. ESI-MS: m/z 453.39 [M + H]+, C25H32N4O4 (452.56).
(3R,4R,5S)-4-acetamido-5-((4–(2-chloropyrimidin-5-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (3d)
Yellow power, 78% yield, mp: 162–163 °C. 1H NMR (400 MHz, CD3OD) δ: 8.83 (s, 2H, 2Pyridine-H), 7.67 (d, J = 8.0 Hz, 2H, 2Ph-H), 7.54 (d, J = 7.9 Hz, 2H, 2-Ph-H), 6.66 (s, 1H, CH), 4.18 (d, J = 13.2 Hz, 1H, CH), 4.11 − 4.06 (m, 2H, 2CH), 4.05 − 3.97 (m, 2H, 2CH), 3.41 (t, J = 5.6 Hz, 1H, CH), 2.92 (dd, J = 17.6, 5.3 Hz, 1H, CH), 2.44 (dd, J = 17.3, 9.3 Hz, 1H, CH), 2.02 (s, 3H, CH3), 1.57 − 1.47 (m, 4H, 2CH2), 0.90 (q, J = 7.5 Hz, 6H, 2CH3). 13C NMR (100 MHz, DMSO-d6) δ: 172.69, 164.13, 156.47, 135.86, 134.43, 130.62, 129.81, 129.73, 129.67, 126.61, 126.11, 81.49, 73.86, 67.91, 53.54, 51.00, 25.43, 24.93, 24.42, 20.88, 7.62, 7.37. ESI-MS: m/z 487.53 [M + H]+, C25H31ClN4O4 (487.00).
(3R,4R,5S)-4-acetamido-5-((4–(2-amino-4,6-dimethylpyrimidin-5-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (3e)
White power, 71% yield, mp: 188–190 °C. 1H NMR (400 MHz, CD3OD) δ: 7.46 (d, J = 7.9 Hz, 2H, 2Ph-H), 7.30 (d, J = 8.3 Hz, 2H, 2Ph-H), 6.47 (s, 1H, CH), 4.41 − 4.33 (m, 1H, CH), 3.90 − 3.80 (m, 3H, 3CH), 3.66 (m, 1H, CH), 3.43 − 3.32 (m, 1H), 2.92 (ddt, J = 12.3, 5.9, 0.9 Hz, 1H), 2.69 (s, 6H), 2.51 (ddt, J = 12.5, 3.1, 1.0 Hz, 1H), 1.95 (s, 3H), 1.61 − 1.48 (m, 4H, 2CH2), 0.93 (t, J = 7.2 Hz, 6H, 2CH3).13C NMR (100 MHz, DMSO-d6) δ: 173.50, 165.67, 139.89, 136.75, 135.03, 130.67, 130.15, 130.07, 129.56, 127.62, 127.55, 82.33, 74.69, 55.26, 51.67, 26.22, 25.75, 25.25, 22.08, 21.61, 21.32, 21.23, 8.44, 8.18. ESI-MS: m/z 496.22 [M + H]+, C27H37N5O4 (495.62).
General procedure for the preparation of compounds 4a∼4p
Compounds 4a∼4p were prepared by electrophilic substitution reaction of corresponding amines with 4-fluorobenzaldehyde. Corresponding amines (10 mmol, 1.0 equiv) and K2CO3 (2.76 g, 20 mmol, 2.0 equiv) were added to DMF (25 ml), and the mixture was heated at 100 °C for 0.5 h. Sequentially, 4-fluorobenzaldehyde (1.24 g, 10 mmol, 1.0 equiv) was added and the reaction was continued for 12 h at 100 °C. The mixture was poured into water and extracted with ethyl acetate (3 × 40 ml). The combined ethyl acetate solution was washed with brine (2 × 30 ml), dried over anhydrous MgSO4 and concentrated to give the crude product. Purification by column chromatography with a gradient of ethyl acetate/petroleum ether (1/50 to 1/10) as the eluent gave the corresponding compounds 4a∼4p.
General procedure for the preparation of compounds 5a∼5p
The synthetic procedures of 5a∼5p were similar to that of 2a∼2e, starting from intermediates 4a∼4p.
General procedure for the preparation of compounds 6a∼6p
The synthetic procedures of 6a∼6p were similar to that of 3a∼3e, starting from intermediates 5a∼5p.
(3R,4R,5S)-4-acetamido-5-((4–(2-(tert-butoxycarbonyl)-2,7-diazaspiro[3.5]nonan-7-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6a)
White power, 76% yield, mp: 156–158 °C. 1H NMR (400 MHz, CD3OD) δ: 7.31 (d, J = 8.5 Hz, 2H, 2Ph-H), 7.01 (d, J = 8.5 Hz, 2H, 2Ph-H), 6.71 (s, 1H, CH), 4.23 (d, J = 13.0 Hz, 1H, CH), 4.19 − 4.03 (m, 3H, 3CH), 3.67 (s, 4H, 2CH2), 3.49 − 3.38 (m, J = 10.8, 5.3 Hz, 2H, 2CH), 3.26 − 3.11 (m, 4H, 2CH2), 2.96 (dd, J = 17.8, 4.8 Hz, 1H, CH), 2.58 (dd, J = 17.5, 9.3 Hz, 1H, CH), 2.03 (s, 3H, CH3), 1.92 − 1.81 (m, 4H, 2CH2), 1.57 − 1.48 (m, 4H, 2CH2), 1.44 (s, 9H, 3CH3), 0.89 (dd, J = 13.3, 7.3 Hz, 6H, 2CH3). 13C NMR (100 MHz, CD3OD) δ: 173.29, 156.95, 152.11, 133.94, 130.53, 121.11, 116.22, 82.11, 79.61, 74.77, 54.48, 51.75, 47.90, 47.68, 45.83, 34.54, 33.15, 27.27, 25.76, 25.18, 21.91, 8.45, 8.16. ESI-MS: m/z 598.92 [M + H]+, C33H50N4O6 (598.79).
(3R,4R,5S)-4-acetamido-5-((4–(7-(tert-butoxycarbonyl)-2,7-diazaspiro[4.4]nonan-2-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6b)
White power, 74% yield, mp: 160–161 °C. 1H NMR (400 MHz, CD3OD) δ: 7.28 (d, J = 8.4 Hz, 2H, 2Ph-H), 6.80 (s, 1H, CH), 6.60 (d, J = 8.5 Hz, 2H, 2Ph-H), 4.25 (d, J = 13.0 Hz, 1H, CH), 4.21 − 4.07 (m, J = 19.0, 10.5 Hz, 3H, 3CH), 3.53 − 3.34 (m, 7H, 3CH2, CH), 3.27 − 3.20 (m, J = 6.6 Hz, 2H, CH2), 2.99 (dd, J = 17.4, 5.3 Hz, 1H, CH), 2.61 (dd, J = 17.1, 9.6 Hz, 1H, CH), 2.04 (s, 3H, CH3), 2.03 − 1.89 (m, 4H, 2CH2), 1.58 − 1.42 (m, 14H, 3CH3, 2CH2, CH), 0.90 (q, J = 7.3 Hz, 6H, 2CH3). 13C NMR (100 MHz, CD3OD) δ: 173.40, 155.10, 148.68, 136.61, 130.73, 127.90, 111.63, 82.28, 79.60, 74.49, 66.74, 54.05, 51.48, 47.68, 46.56, 34.26, 27.36, 25.96, 25.72, 25.22, 21.97, 8.41, 8.14. ESI-MS: m/z 599.21 [M + H]+, C33H50N4O6 (598.79).
(3R,4R,5S)-4-acetamido-5-((4–(7-(tert-butoxycarbonyl)-2,7-diazaspiro[3.5]nonan-2-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6c)
White power, 80% yield, mp: 157–158 °C. 1H NMR (400 MHz, CD3OD) δ: 7.28 (d, J = 8.4 Hz, 2H, 2Ph-H), 6.78 (s, 1H, CH), 6.50 (d, J = 8.4 Hz, 2H, 2Ph-H), 4.25 (d, J = 13.0 Hz, 1H, CH), 4.14 (dd, J = 18.2, 10.5 Hz, 3H, 3CH), 3.64 (s, 4H, 2CH2), 3.52 − 3.38 (m, 6H, 3CH2), 2.98 (dd, J = 17.5, 5.3 Hz, 1H, CH), 2.60 (dd, J = 17.2, 9.5 Hz, 1H, CH), 2.04 (s, 3H, CH3), 1.77 (dd, J = 11.7, 6.3 Hz, 4H, 2CH2), 1.53 (dq, J = 14.0, 7.1 Hz, 4H, 2CH2), 1.46 (s, 9H, 3CH3), 0.90 (q, J = 7.2 Hz, 6H, 2CH3).13C NMR (100 MHz, CD3OD) δ: 173.39, 168.88, 155.16, 152.59, 135.40, 130.57, 129.22, 118.39, 111.27, 82.22, 79.74, 74.71, 61.03, 54.25, 51.58, 47.90, 35.20, 34.40, 27.29, 26.32, 25.73, 25.19, 22.00, 8.45, 8.17.ESI-MS: m/z 598.80 [M + H]+, C33H50N4O6 (598.79).
(3R,4R,5S)-4-acetamido-5-((4–(6-(tert-butoxycarbonyl)-2,6-diazaspiro[3.3]heptan-2-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6d)
White power, 79% yield, mp: 155–156 °C. 1H NMR (400 MHz, Methanol-d4) δ: 7.28 (d, J = 7.9 Hz, 2H, 2Ph-H), 6.75 (s, 1H, CH), 6.51 (d, J = 7.8 Hz, 2H, 2Ph-H), 4.23 (d, J = 12.9 Hz, 1H, CH), 4.19 − 4.13 (m, 2H, 2CH), 4.13 − 4.03 (m, 5H, CH, 2CH2), 3.97 (s, 4H, 2CH2), 3.51 − 3.39 (m, 2H, 2CH), 2.96 (dd, J = 17.4, 4.7 Hz, 1H, CH), 2.59 (dd, J = 17.4, 9.1 Hz, 1H, CH), 2.03 (s, 3H, CH3), 1.54 (dq, J = 13.2, 6.5 Hz, 4H, 2CH2), 1.44 (s, 9H, 3CH3), 0.90 (q, J = 6.7 Hz, 6H, 2CH3).13C NMR (100 MHz, Methanol-d4) δ: 173.28, 169.33, 156.54, 152.31, 134.58, 130.47, 129.85, 119.40, 111.81, 82.14, 79.76, 74.60, 61.56, 54.37, 51.62, 48.10, 47.73, 33.34, 27.23, 26.44, 25.75, 25.19, 21.90, 8.42, 8.14.ESI-MS: m/z 570.94 [M + H]+, C31H46N4O6 (570.73).
(3R,4R,5S)-5-((4–(1,4-dioxa-8-azaspiro[4.5]decan-8-yl)benzyl)amino)-4-acetamido-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6e)
Brown power, 65% yield, mp: 131–133 °C. 1H NMR (400 MHz, CD3OD) δ: 7.30 (d, J = 8.6 Hz, 2H, 2Ph-H), 7.00 (d, J = 8.6 Hz, 2H, 2Ph-H), 6.62 (s, 1H, CH), 4.21 − 4.03 (m, 4H, 4CH), 3.97 (s, 4H, 2CH2), 3.42 (t, J = 5.6 Hz, 1H, CH), 3.38 − 3.33 (m, 4H, 2CH2), 3.31 (s, 1H, CH), 2.99 − 2.90 (m, 1H, CH), 2.59 − 2.48 (m, 1H, CH), 2.02 (s, 3H, CH3), 1.80 − 1.74 (m, 4H, 2CH2), 1.57 − 1.47 (m, 4H, 2CH2), 0.89 (q, J = 7.3 Hz, 6H, 2CH3). 13C NMR (100 MHz, DMSO-d6) δ: 173.44, 151.54, 136.43, 131.05 − 130.48 (m), 120.17, 115.91, 106.77, 82.28, 74.68, 63.97, 54.37, 51.52, 33.83, 26.11, 25.72, 25.21, 22.02, 8.45, 8.17. ESI-MS: m/z 516.6 [M + H]+, C28H41N3O6 (515.65).
(3R,4R,5S)-4-acetamido-5-((4–(6,6-dimethyl-3-azabicyclo[3.1.0]hexan-3-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6f)
White power, 65% yield, mp: 143–145 °C. 1H NMR (400 MHz, CD3OD) δ: 7.25 (d, J = 8.4 Hz, 2H, 2Ph-H), 6.62 (s, 1H, CH), 6.50 (d, J = 8.4 Hz, 2H, 2Ph-H), 4.26 − 3.98 (m, 4H, 4CH), 3.46 − 3.33 (m, 4H, 2CH2), 3.24 (d, J = 9.8 Hz, 2H, CH2), 2.96 (dd, J = 17.4, 5.0 Hz, 1H, CH), 2.57 (dd, J = 17.4, 9.4 Hz, 1H, CH), 2.02 (s, 3H, CH3), 1.59 − 1.44 (m, 6H, 2CH2, 2CH), 1.07 (s, 3H, CH3), 0.88 (dd, J = 13.5, 7.1 Hz, 6H, 2CH3), 0.84 (s, 3H, CH3).13C NMR (100 MHz, CD3OD) δ: 172.43, 171.00, 146.44, 131.75, 131.16, 129.81, 116.02, 110.52, 81.15, 74.14, 53.44, 50.94, 46.96, 26.755, 26.29, 24.95, 24.33, 21.06, 18.24, 10.27, 7.65, 7.35. ESI-MS: m/z 484.6 [M + H]+, C28H41N3O4 (483.65).
(3R,4R,5S)-5-((4–(3-azabicyclo[3.1.0]hexan-3-yl)benzyl)amino)-4-acetamido-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6 g)
White power, 72% yield, mp: 153–155 °C. 1H NMR (400 MHz, CD3OD) δ: 7.25 (d, J = 8.2 Hz, 2H, 2Ph-H), 6.69 (s, 1H, CH), 6.58 (d, J = 8.2 Hz, 2H, 2Ph-H), 4.28 − 4.02 (m, 4H, 4CH), 3.55 − 3.36 (m, 4H, 4CH), 3.26 − 3.12 (m, 2H, CH2), 2.97 − 2.92 (m, 1H, CH), 2.65 − 2.52 (m, 1H, CH), 2.03 (s, 3H, CH3), 1.68 (dt, J = 7.4, 3.4 Hz, 2H, 2CH), 1.59 − 1.46 (m, 4H, 2CH2), 0.89 (dd, J = 7.1 Hz, 6H, 2CH3), 0.81 − 0.66 (m, 1H, CH), 0.30 − 0.19 (m, 1H, CH).13C NMR (100 MHz, CD3OD) δ: 173.32, 170.60, 149.39, 133.51, 131.16, 130.63, 117.26, 112.26, 112.16, 82.08, 74.92, 54.22, 51.71, 49.80, 39.13, 26.79, 25.76, 25.16, 21.97, 15.61, 9.70, 8.47, 8.18. ESI-MS: m/z 456.69 [M + H]+, C26H37N3O4 (455.60).
(3R,4R,5S)-4-acetamido-5-((4-(hexahydrocyclopenta[c]pyrrol-2(1H)-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6h)
White power, 63% yield, mp: 165–167 °C. 1H NMR (400 MHz, CD3OD) δ: 7.27 (d, J = 8.2 Hz, 2H, 2Ph-H), 6.76 (s, 1H, CH), 6.64 (d, J = 8.3 Hz, 2H, 2Ph-H), 4.29 − 4.06 (m, 4H, 4CH), 3.52 − 3.44 (m, 1H, CH), 3.45 − 3.37 (m, 3H, 3CH), 3.05 (dd, J = 9.7, 3.3 Hz, 2H, CH2), 2.99 (dd, J = 17.3, 5.2 Hz, 1H, CH), 2.85 − 2.74 (m, 2H, 2CH), 2.66 − 2.55 (m, 1H, CH), 2.04 (s, 3H, CH3), 1.95 − 1.83 (m, 2H, 2CH), 1.80 − 1.67 (m, 1H, CH), 1.66 − 1.57 (m, 1H, CH), 1.57 − 1.46 (m, 6H, 6CH), 0.94 − 0.85 (m, 6H, 2CH3).13C NMR (100 MHz, CD3OD) δ: 173.37, 169.32, 149.54, 134.93, 130.60, 129.71, 117.19, 112.89, 82.17, 74.78, 54.69, 54.14, 51.61, 42.68, 32.61, 26.43, 25.74, 25.29, 25.18, 21.99, 8.46, 8.17. ESI-MS: m/z 482.29 [M - H]-, C28H41N3O4 (483.65).
(3R,4R,5S)-4-acetamido-5-((4-(octahydro-2H-isoindol-2-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6i)
White power, 68% yield, mp: 147–148 °C. 1H NMR (400 MHz, CD3OD) δ: 7.26 (d, J = 8.5 Hz, 2H, 2Ph-H), 6.63 (s, 1H, CH), 6.56 (d, J = 8.6 Hz, 2H, 2Ph-H), 4.22 − 4.07 (m, J = 15.4, 10.3 Hz, 3H, 3CH), 4.04 (d, J = 12.9 Hz, 1H, CH), 3.48 − 3.40 (m, 1H, CH), 3.31 − 3.28 (m, 1H, CH), 3.19 (dd, J = 9.1, 4.9 Hz, 2H, CH2), 2.97 (dd, J = 17.6, 5.3 Hz, 1H, CH), 2.63 − 2.51 (m, 1H, CH), 2.37 (dp, J = 11.7, 6.0 Hz, 2H, CH2), 2.04 (s, 3H, CH3), 1.83 − 1.13 (m, 14H, 6CH2, 2CH), 0.91 (dd, J = 13.2, 7.3 Hz, 6H, 2CH3).13C NMR (100 MHz, CD3OD) δ: 173.16, 172.29, 148.90, 133.10, 131.67, 130.47, 129.40, 117.04, 111.08, 110.54, 81.94, 75.17, 54.28, 52.08, 51.38, 37.26, 27.53, 25.99, 25.79, 25.15, 22.74, 21.90, 8.46, 8.17. ESI-MS: m/z 496.32 [M - H]-, C29H43N3O4 (497.68).
(3R,4R,5S)-4-acetamido-5-((4-(octahydro-2H-isoindol-2-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6j)
Lavender power, 58% yield, mp: 147–148 °C. 1H NMR (400 MHz, CD3OD) δ: 7.31 (d, J = 8.5 Hz, 2H, 2Ph-H), 6.86 (s, 1H, CH), 6.70 (d, J = 8.5 Hz, 2H, 2Ph-H), 4.68 (s, 1H, CH), 4.57 (s, 1H, CH), 4.37 − 4.07 (m, 4H, overlapped, 4CH), 3.83 (dd, J = 23.6, 7.2 Hz, 2H, CH2), 3.61 − 3.42 (m, 3H, overlapped, 3CH), 3.10 (d, J = 9.5 Hz, 1H, CH), 3.03 (dd, J = 17.5, 5.1 Hz, 1H, CH), 2.64 (dd, J = 17.3, 9.2 Hz, 1H, CH), 2.07 (s, 3H, CH3), 2.04 − 1.94 (m, J = 21.9, 5.4 Hz, 2H, CH2), 1.62 − 1.48 (m, 4H, 2CH2), 0.98 − 0.86 (m, 6H, 2CH3). 13C NMR (100 MHz, CD3OD) δ: 172.60, 167.08, 147.48, 135.64, 130.04, 127.31, 116.84, 112.17, 81.48, 75.62, 73.79, 70.86, 56.97, 56.24, 53.44, 50.69, 47.08, 46.88, 35.42, 25.25, 24.91, 24.41, 21.18, 7.60, 7.34. ESI-MS: m/z 472.39 [M + H]+, C26H39N3O4 (471.60).
(3R,4R,5S)-4-acetamido-5-((4-((R)-2-methylpyrrolidin-1-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6k)
White power, 79% yield, mp: 160–161 °C. 1H NMR (400 MHz, CD3OD) δ: 7.27 (d, J = 7.9 Hz, 2H, 2Ph-H), 6.85 − 6.45 (m, J = 12.6 Hz, 3H, CH, 2Ph-H), 4.34 − 4.10 (m, 3H, 3CH), 4.07 (d, J = 12.8 Hz, 1H, CH), 3.95 − 3.86 (m, 1H, CH), 3.64 − 3.36 (m, 3H, CH, CH2), 3.17 (dd, J = 16.2, 8.0 Hz, 1H, CH), 3.08 − 2.87 (m, 1H, CH), 2.70 − 2.47 (m, 1H, CH), 2.24 − 1.88 (m, 6H, CH2, CH, CH3), 1.80 − 1.69 (m, 1H, CH), 1.64 − 1.44 (m, 4H, 2CH2), 1.16 (d, J = 6.1 Hz, 3H, CH3), 0.91 (dd, J = 12.0, 7.1 Hz, 6H, 2CH3).13C NMR (100 MHz, CD3OD) δ: 147.92, 130.62, 129.41, 127.95, 116.58, 111.82, 111.39, 81.99, 74.96, 54.30, 53.47, 51.83, 32.56, 25.77, 25.16, 22.71, 21.89, 17.86, 8.45, 8.16.ESI-MS: m/z 456.6 [M - H]-, C26H39N3O4 (457.62).
(3R,4R,5S)-4-acetamido-5-((4-((R)-2-methylpyrrolidin-1-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6 l)
White power, 58% yield, mp: 150–151 °C. 1H NMR (400 MHz, CD3OD) δ: 7.27 (d, J = 7.9 Hz, 2H, 2Ph-H), 6.85 − 6.45 (m, J = 12.6 Hz, 3H, CH, 2Ph-H), 4.34 − 4.10 (m, 3H, 3CH), 4.07 (d, J = 12.8 Hz, 1H, CH), 3.95 − 3.86 (m, 1H, CH), 3.64 − 3.36 (m, 3H, CH, CH2), 3.17 (dd, J = 16.2, 8.0 Hz, 1H, CH), 3.08 − 2.87 (m, 1H, CH), 2.70 − 2.47 (m, 1H, CH), 2.24 − 1.88 (m, 6H, CH2, CH, CH3), 1.80 − 1.69 (m, 1H, CH), 1.64 − 1.44 (m, 4H, 2CH2), 1.16 (d, J = 6.1 Hz, 3H, CH3), 0.91 (dd, J = 12.0, 7.1 Hz, 6H, 2CH3).13C NMR (100 MHz, CD3OD) δ: 170.68, 148.36, 133.11, 130.73, 111.80, 82.04, 74.78, 54.27, 51.65, 47.82, 45.00, 31.92, 26.77, 25.76, 25.15, 21.88, 8.43, 8.14. ESI-MS: m/z 460.7 [M - H]-, C25H36FN3O4 (461.59).
(3R,4R,5S)-4-acetamido-5-((4-((R)-2-fluoropyrrolidin-1-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6 m)
White power, 63% yield, mp: 145–147 °C. 1H NMR (400 MHz, CD3OD) δ: 7.30 (d, J = 8.6 Hz, 2H), 6.75 (s, 1H), 6.63 (d, J = 8.6 Hz, 2H), 5.38 (dt, J = 53.5, 3.8 Hz, 1H), 4.25 (d, J = 13.0 Hz, 1H), 4.20 − 4.07 (m, 3H), 3.65 − 3.53 (m, 1H), 3.51 (dd, J = 7.2, 3.5 Hz, 1H), 3.47 − 3.38 (m, 4H), 3.00 (dd, J = 17.5, 5.5 Hz, 1H), 2.65 − 2.55 (m, 1H), 2.33 (ddd, J = 18.6, 13.3, 5.4 Hz, 1H), 2.24 − 2.09 (m, 1H), 2.04 (s, 3H), 1.59 − 1.46 (m, 4H), 0.90 (q, J = 7.4 Hz, 6H).13C NMR (100 MHz, DMSO-d6) δ: 170.82, 167.73, 147.59, 137.65, 130.86, 128.96, 112.13, 81.48, 75.33, 54.09, 52.42, 45.67, 32.07, 26.04, 25.51, 23.76, 9.87, 9.35. ESI-MS: m/z 462.5 [M + H]+, C25H36FN3O4 (461.58).
(3R,4R,5S)-4-acetamido-5-((4-(azetidin-1-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6n)
White power, 69% yield, mp: 156–158 °C. 1H NMR (400 MHz, CD3OD) δ: 7.49 − 7.26 (m, 2H, 2Ph-H), 6.73 (s, 1H, CH), 6.61 − 6.38 (m, J = 8.4 Hz, 2H, 2Ph-H), 4.41 − 3.96 (m, J = 32.0, 29.2, 21.9 Hz, 4H, overlapped, 4CH), 3.92 − 3.83 (m, J = 11.5, 7.2 Hz, 3H, overlapped, 3CH), 3.82 − 3.64 (m, 1H, CH), 3.57 − 3.37 (m, 2H, CH2), 3.08 − 2.89 (m, 1H, CH), 2.57 (dd, J = 52.4, 21.8 Hz, 1H, CH), 2.47 − 2.24 (m, J = 14.9, 7.4 Hz, 2H, CH2), 2.05 (s, 3H, CH3), 1.62 − 1.45 (m, J = 15.4 Hz, 4H, 2CH2), 1.01 − 0.79 (m, J = 13.3, 7.3 Hz, 6H, 2CH3). 13C NMR (100 MHz, CD3OD) δ: 153.05, 130.40, 127.94, 111.34, 82.06, 74.83, 68.71, 54.47, 52.45, 51.9, 51.75, 25.76, 25.17, 21.88, 21.68, 16.31, 8.43, 8.14. ESI-MS: m/z 429.91 [M + H]+, C24H35N3O4 (429.56).
(3R,4R,5S)-4-acetamido-5-((4-(ethyl(methyl)amino)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6o)
Yellow power, 66% yield, mp: 153–155 °C. 1H NMR (400 MHz, DMSO-d6) δ: 7.26 (d, J = 8.5 Hz, 2H, 2Ph-H), 6.74 (d, J = 8.5 Hz, 2H, 2-Ph-H), 6.60 (s, 1H, CH), 4.21 − 3.98 (m, 4H, 4CH), 3.47 − 3.38 (m, 3H, 3CH), 3.36 − 3.32 (m, 1H, CH), 2.99 − 2.93 (m, 1H, CH), 2.91 (s, 3H, CH3), 2.54 (dd, J = 17.7, 9.2 Hz, 1H, CH), 2.02 (s, 3H, CH3), 1.58 − 1.44 (m, 4H, 2CH2), 1.09 (t, J = 7.0 Hz, 3H, CH3), 0.89 (q, J = 7.1 Hz, 6H, 2CH3). 13C NMR (100 MHz, CD3OD) δ: 172.62, 149.09, 135.26, 129.95, 128.53, 115.86, 111.25, 81.44, 73.88, 53.32, 50.70, 35.82, 35.49, 25.38, 25.00, 24.91, 24.46, 24.38, 21.18, 9.10, 7.62, 7.35. ESI-MS: m/z 430.61 [M - H]-, C24H37N3O4 (431.58).
(3R,4R,5S)-4-acetamido-5-((4-((R)-2-fluoropyrrolidin-1-yl)benzyl)amino)-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylic acid (6p)
Yellow power, 65% yield, mp: 141–143 °C. 1H NMR (400 MHz, DMSO-d6) δ: 7.27 (d, J = 8.5 Hz, 2H, 2Ph-H), 6.82 (d, J = 8.4 Hz, 2H, 2Ph-H), 6.62 (s, 1H, CH), 4.25 − 3.98 (m, 5H, 5CH), 3.41 (p, J = 5.6 Hz, 1H, CH), 3.38 − 3.32 (m, 1H, CH), 2.96 (dd, J = 17.5, 5.4 Hz, 1H, CH), 2.73 (s, 3H, CH3), 2.63 − 2.51 (m, 1H, CH), 2.02 (s, 3H, CH3), 1.52 (td, J = 9.6, 8.9, 6.6 Hz, 4H, 2CH2), 1.15 (d, J = 6.6 Hz, 6H, 2CH3), 0.89 (q, J = 7.1 Hz, 6H, 2CH3). 13C NMR (100 MHz, CD3OD) δ: 171.34, 169.13, 167.25, 132.03, 131.25, 128.02, 125.38, 81.57, 74.87, 54.41, 51.04, 45.31, 30.89, 26.04, 25.93, 25.47, 23.99, 9.86, 9.31. ESI-MS: m/z 446.11 [M + H]+, C25H39N3O4 (445.60).
Biological evaluation
Neuraminidase enzyme inhibitory assay in vitro
The influenza NA inhibition assay was performed as previously describedCitation21,Citation24,Citation26. Influenza viral suspensions of A/Goose/Guangdong/SH7/2013 (H5N1) and A/Duck/Guangdong/674/2014 (H5N6) were harvested from the allantoic fluid of influenza virus-infected chicken embryo layer, and A/PuertoRico/8/1934 (H1N1), A/Babol/36/2005 (H3N2) and A/Anhui/1/2005 (H5N1-H274Y) were obtained from Sino Biological Inc (Catalogue numbers: 40909-V08B, 40017-VNAHC and 11676-VNAHC1, respectively). NA inhibitory activities were examined by using a chemiluminescence-based assay with 2′-(4-methylumbelliferyl)-α-D-N-acetylneuraminic acid (MUNANA) as the substrate, which was cleaved by NA to provide a quantifiable fluorescent product. The tested compounds were dissolved in DMSO and diluted to the corresponding concentrations in MES buffer (1.27 g 2-(N-morpholino)-ethanesulphonic acid and 0.09 g CaCl2 in 200 ml Milli-Q water). Subsequently, 10 μL of diluted virus supernatant or NA assay diluent, along with 70 μL of MES buffer and 10 μL of test compounds at varying concentrations, were sequentially added to a 96-well plate and incubated for 10 min at 37 °C. To start the reaction, 10 μL of substrate was added to each well. Following a subsequent incubation period of 40 min, the reaction was stopped by adding 150 μL of stopping solution (3 g glycine and 1.6 g NaOH in 200 ml Milli-Q water). At Ex = 365 nm and Em = 460 nm, fluorescence was measured with a microplate reader (Molecular Devices, SpectraMax iD5). The values of IC50 (50%-inhibitory concentration) were obtained from the dose-response curves by plotting the percent inhibition of NA activity versus the concentration of the compounds.
In vitro anti-influenza virus assay and cytotoxicity assay in chicken embryo fibroblast (CEF)
The anti-influenza activity (EC50) and cytotoxicity (CC50) of the novel synthesised oseltamivir derivatives were determined with H5N1 and H5N6 strains in Chicken Embryo Fibroblasts (CEFs) (Extracted from 9-day-old chicken embryo eggs, provided by Institute of Poultry Science, Shandong Academy of Agricultural Sciences) utilising Cell Counting Kit-8 (CCK-8, Dojindo Laboratories) method as our previously describedCitation19,Citation20,Citation22,Citation26. Chicken embryo fibroblasts (CEFs) were infected with H5N1 and H5N6 as an antiviral detection model to determine the inhibitory effect of these compounds on the cytopathic effect (CPE) induced by IFV. The tested compounds and positive control were dissolved in DMSO and diluted to the corresponding concentrations by 3-fold dilution in medium (1% FBS in DMEM). Aliquots of 50 μL of diluted influenza viral suspension (H5N1, H5N6) were mixed with equal volumes of solutions of the novel synthesised compounds. In 96-well plates, the mixtures were added to CEFs and then incubated at 37 °C under 5% CO2 for 48 h. Then, 100 μL per well of CCK-8 reagent solution (10 μL of CCK-8 in 90 μL of media) was added. After incubating at 37 °C for 90 min, the absorbance at 450 nm was read on a microplate reader. The EC50 values were obtained by fitting the curve of percent CPE (cytopathic effect) versus inhibitor concentration. The values of CC50 (50% cytotoxic concentration) of NA inhibitors to CEFs were determined similarly to the EC50 but without virus infection.
Plaque reduction assay (PRA) in MDCK cells
The antiviral activity of selected compounds towards the H1N1 and H3N2 viruses was determined by PRA (plaque reduction assay). The procedure of this assay adhered to our recently published research and was conducted at the Department of Molecular Medicine, University of PadovaCitation20,Citation27,Citation28. Briefly, a confluent monolayer of MDCK cells (Madin-Darby Canine Kidney cells, from Department of Molecular Medicine, University of Padova) was seeded in 12-well plates. After incubating for 24 h at 37 °C, cells were infected with FluA virus (PR8 or WSN strain) at approximately 40 PFU/well in DMEM supplemented with 1 µg/mL of TPCK-treated trypsin (Worthington Biochemical Corporation, catalogue numbers: LS02120) and 0.14% BSA (Sigma, catalogue numbers: A7906) for 1 h at 37 °C. After virus adsorption, the cells were overlaid with medium containing 1 µg/mL TPCK-treated trypsin, 0.14% BSA, 1.2% Avicel cellulose and different concentrations of test compounds. Oseltamivir carboxylic acid (OSC) and zanamivir (ZAN) were included as reference compounds in each experiment. At 2 days post-infection, cells monolayers were fixed with 4% (v/v) formaldehyde and stained with 0.1% toluidine blue. Viral plaques were counted and the mean plaque number of DMSO-treated control group was set at 100% of plaque formation.
Cytotoxicity assay in MDCK cells
Cytotoxicity of representative compounds against MDCK cells was assessed by the 3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method as our previously reportedCitation29,Citation30. MDCK cells (seeded at density of 2 × 104 cells per well) were grown in 96-well plates for 24 h and then treated with serial dilutions of test compounds, or DMSO as a control, in DMEM supplemented with 10% FBS and 1% P/S. After incubation for 48h at 37 °C, MTT solution (5 mg/ml in PBS) was added to each well, and plates were incubated at 37 °C for 4 h in a CO2 incubator. Successively, a solubilisation solution (10% SDS, 0.01 N HCl) was added to lyse cells and incubated overnight at 37 °C. Finally, absorbance was read at the wavelength of 570 nm on a microtiter plate reader (MultiSkan FC, Thermo Scientific).
In silico studies
Computational methods
The molecular docking was performed utilising Schrödinger Maestro 11.8 software. The molecular structures of compounds 6k was optimised using LigPrep module. The stable conformer with minimum potential energy was generated via OPLS force fieldCitation31.
The crystal structures of N1 (H1N1, PDB code: 3BEQ), N1 (2HU0), and N2 (H2N2, PDB code: 4K1K) were performed from the Protein Data Bank. The addition of hydrogen atoms and removal of unwanted water molecules were implemented using the “Protein Preparation Wizard” tool of Maestro 11.8Citation32. Molecular docking of 6k was carried out with standard Glide protocol (Schrödinger Maestro 11.8)Citation33. Docking results were visualised using the software of PyMOL version 1.5.
The system builder module was used to build the molecular dynamic simulation structure. The atomic framework had been solvated with Transferable Interatomic Potential with Three Points (TIP3P) crystallographic water particles with orthorhombic intermittent limit conditions. The minimum distance was set as 10 Å. After eliminating the overlapping water molecules, Na+ or Cl- were added to neutralise the entire framework of atoms, and an extra 0.15 M NaCl was added into the system. With the NPT ensemble class of Nose-Hoover thermostat and barostat to maintain the constant temperature of 300 K and pressure of 1.013 bar in the system, the 200 ns productive simulation was performed. The protein backbone RMSD, ligand heavy atom RMSD, and RMSF plots were generated by analysing the resultant trajectory with Maestro.
Results and discussion
Chemistry
The C-5 modified oseltamivir derivatives (3a-3e, 6a-6p) were prepared via a concise and well-established synthetic route as outlined in Scheme 1 and Scheme 2. As shown in Scheme 1, a Suzuki reaction of the (4-formylphenyl) boronic acid with the corresponding bromo-substituted aromatic heterocycle gave five substituted biphenyl benzaldehydes (1a − 1e), which were used in synthesising intermediates 2a − 2e. Finally, the target compounds (3a-3e) were prepared by direct hydrolysis of the intermediates 2a-2e. Compounds 6a-6p were synthesised according to the synthetic protocols represented in Scheme 2. Compounds 4a-4p were prepared by the reaction of corresponding amines with 4-fluorobenzaldehyde in the presence of potassium carbonate. Then, following a previously reported procedure, 4a-4p were reacted with commercial oseltamivir phosphate in the presence of NaBH3CN to obtain the key intermediates 5a − 5p, which was followed by direct hydrolysis to afford the target compounds 6a-6p.
Scheme 1. Reagents and conditions: (i) Corresponding bromo-substituted aromatic heterocycle, K3PO4, Pd(PPh3)4, N2, toluene/H2O = 25/2, 100 °C, 12 h; (ii) NaBH3CN, CH3OH, r.t., 6–7 h; (iii) 1 M NaOH, CH3OH, r.t., then 3 M HCl.
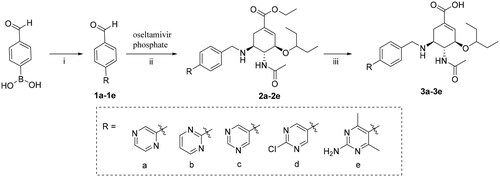
Scheme 2. Reagents and conditions: (i) Corresponding amine, K2CO3, DMF, 100 °C, 12 h; (ii) NaBH3CN, CH3OH, r.t., 6–7 h; (iii) 1 M NaOH, CH3OH, r.t., then 3 M HCl.
Biological evaluation
In vitro activities of influenza neuraminidase inhibitors
All the synthesised compounds were screened for their NA inhibitory activities using a chemiluminescence-based assay with 2′-(4-methylumbelliferyl)-α-D-N-acetylneuraminic acid (MUNANA) as the substrateCitation19–23,Citation25,Citation34. We chose wild-type N1 (H1N1, H5N1) or mutated N1 (H5N1-H274Y) from group-1, N2 (H5N2) and N6 (H5N6) from group-2 as representatives for testing. OSC (Oseltamivir acid) and ZNA (Zanamivir) was selected as positive control drug. The inhibitory activities of the synthesised compounds and OSC are reported in . As a broad-spectrum neuraminidase inhibitor, OSC showed high inhibitory potency against all selected wild-type NAs with IC50 values of 15 nM, 28 nM, 50 nM, and 16 nM against H1N1, H3N2, H5N1, and H5N6, respectively.
Table 1. Neuraminidase (NA) inhibition of oseltamivir derivatives in chemiluminescence-based assay.  .
.
In the tested series, all oseltamivir derivatives showed decreased inhibitory activities against selected wild-type NAs. Among them, 3a-3e with R group of pyrazinyl and pyrimidinyl exhibited a great decrease in inhibitory activity against H1N1 and H3N2 (IC50 = 4.74–12.14 μM). This confirmed our previous hypothesis that when electron-withdrawing substituents with higher electron cloud density were introduced into the dominant benzyl group targeting the 150-cavity hydrogen bond region, the activity of the compound decreased. In particular, when the nitrogen atom of the pyrimidine ring was at 3-position and 5-position (3c-3e), the absence of electron-donating conjugated effect of nitrogen atom on benzyl results in a more significant decrease in the electron cloud density of the benzyl group, which further reduced the inhibitory activity of compounds compared to 3a and 3b. Although the activity of 3a-3e decreased, they displayed similar inhibitory activity against group-1 NAs and group-2 NAs, without selectivity.
Replacing terminal aromatic nitrogen-containing heterocycles with nitrogen-containing aliphatic heterocycles greatly improved the inhibitory activity of compounds 6a-6p against NAs. Among them, 6k with R group of (r)-2-methylpyrrole (IC50 = 0.084 μM) exhibited similar activities towards H1N1 compared to OSC (IC50 = 0.015 μM). As for the other wild-type NAs, 6k was proved to have a strong inhibitory effect on both group-1 (H5N1, IC50 = 0.23 μM) and group-2 NAs (H3N2, IC50 = 0.12 μM; H5N6, IC50 = 0.15 μM) with similar activity or slightly weaker than that of OSC. The introduction of electron-withdrawing substituents on pyrrolidine groups was not conducive to an enhancement of activity, as seen with compounds 6 l and 6 m bearing 3-fluoropyrrole (H1N1, IC50 = 0.23 μM and 0.37 μM, respectively; H3N2, IC50 = 0.40 μM and 0.43 μM, respectively; H5N1, IC50 = 0.69 μM and 0.84 μM, respectively; H5N6, IC50 = 0.24 μM and 0.18 μM, respectively) and showing lower potency than 6k.
Further strengthening the flexibility and size of R group is detrimental to the improvement of the inhibitory activity of compounds, such as 6a-6e with R group of nitrogen-containing spiro-heterocycles. More extensive hydrophobic interactions brought by relatively large size did not contribute to the activity, probably because the large size of R group may make the OSC nucleus deflect to a certain extent. In addition, by replacing spiro substituents with bicyclic or bridge-ring substituents, we obtained compounds 6f-6j with more potent inhibitory activity than 6a-6e. This result demonstrated that conformational restriction by increasing the rigidity of substituents is beneficial to reinforce the compatibility of substituents with 150-cavity. Compounds 6f-6j exhibited similar potency towards group-1 and group-2 NAs with IC50 values of 0.14–0.59 μM. In nitrogen-containing bicyclic derivatives (6f-6i), the size of the terminal cycloalkane substituent has little effect on the inhibitory activity.
Inspired by the result of 6k and to further examine the size effects of N-substituted fragments, we performed ring contraction and ring opening for the pyrrole ring contained in 6k to obtain compounds 6n-6p. The activity results indicated that the more flexible conformation caused by the size reduction of substituents did not improve the inhibitory potency of compounds, which may be due to the fact that too small substituents cannot establish suitable hydrophobic interactions with surrounding amino acids to fully occupy 150-cavity. Among them, 6o bearing N, N-dimethylethylamine substituent displayed the lowest inhibitory activity (IC50 = 0.36–0.89 μM) against wild-type NAs compared with 6n (IC50 = 0.14–0.42 μM) and 6p (IC50 = 0.17–0.52 μM). Compound 6p showed improved activities by introducing methyl on N, N-dimethylethylamine substituent.
Overall, N-heterocycles substituted oseltamivir derivatives have been shown to have broad-spectrum inhibitory activity on group-1 and group-2 NAs. Consistently with our previous results, the NA inhibitory activity of novel N-heterocycles substituted oseltamivir derivatives is very sensitive to the flexibility, size and hydrophobicity of R groupCitation19. Small substituent changes have great influence on the inhibitory activity (activity cliff). In summary, 6k, endowed with moderate flexibility, size, and hydrophobicity of R groups, proved to be the most potent NA inhibitor and displayed similar or slightly weaker inhibitory activities than that of OSC towards selected wild-type NAs.
The presence of mutant NA-H274Y has severely limited the clinical applications of OSC and is presently of a great concernCitation35,Citation36. Thus, we investigated the inhibitory activity of all compounds against mutant H5N1-H274Y NA. In agreement with our previous data, OSC showed sharply reduced potency (IC50 = 2.10 μM), over 42-fold weaker than that of wild-type H5N1 NA. Among these novel oseltamivir derivatives, 3a-6e appeared to lack activity against the H5N1-H274Y NA with IC50 values of over 40 μM. It is noteworthy that 6k and 6i showed robust activity with IC50 values of 3.05 and 3.76 μM, respectively. Therefore, these compounds could use as lead compounds for further optimisation studies.
In vitro anti-influenza viral activity
In view of the potent inhibitory activity of several compounds in vitro, we further examined their antiviral activity and toxicity in Chicken Embryo Fibroblast cells (CEFs) via cell-based assays that detect the CPE of IAV infection using A/Goose/Guangdong/SH7/2013 (H5N1) and A/Duck/Guangdong/674/2014 (H5N6) strains, consistently with the above enzymatic assays. As previously performed in the enzyme inhibition assays, OSC was selected as a reference compound in parallel. The values of EC50 (anti-IAV activity) and CC50 (cytotoxicity) are outlined in . Notably, none of the tested compounds displayed significant cytotoxicity in CEFs at the highest tested concentration (CC50 > 200 μM).
Table 2. Anti-influenza virus activity and cytotoxicity of selected compounds in CEFs.
In the case of H5N1 virus, 6 g, 6i and 6j showed lower activities (EC50 = 2.81 μM, 2.99 μM, and 2.34 μM, respectiyely) compared to OSC (EC50 = 0.85 μM). In contrast, the activity of 6k (EC50 = 0.98 μM) was comparable to that of OSC, consistently with results obtained in the enzymatic assay. As for the case of H5N6 virus, 6 g and 6i-6k displayed reduced antiviral activity (EC50 = 4.06 μM, 5.14 μM, 5.41 μM, and 9.29 μM, respectively). Among them, 6 g exhibited the most potent inhibitory activity, albeit 3-fold weaker than OSC (EC50 = 1.44 μM), which is not in line with their activities in the enzymatic assay.
Next, we examined compounds’ activity against A/PR/8/34 (H1N1) and A/Wisconsin/67/05 (H3N2) strains in MDCK cells by plaque reduction assays (PRA). OSC and ZAN were selected as positive controls for inhibition. The EC50 and CC50 (determined via MTT assays) values are summarised in . No cytotoxicity was observed at the highest tested concentration (250 μM) in MDCK cells.
Table 3. Anti-influenza virus activity and cytotoxicity of selected compounds in MDCK cells.
As for H1N1 strain, all the tested oseltamivir derivatives 6 g and 6i-k exhibited nearly equivalent potency, with EC50 values of 0.03 μM, 0.03 μM, 0.03 μM, 0.02 μM, respectively, compared to OSC (EC50 = 0.02 μM) and ZAN (EC50 = 0.009 μM). Against H3N2 virus, 6j showed moderate activity (EC50 = 0.80 μM), which is 2-fold lower than OSC and 3-fold lower than ZA. Compounds 6 g, 6i, and 6k displayed more potent activity against the H3N2 (EC50 = 0.16 μM, 0.32 μM, and 0.12 μM, respectively) compared to OSC (EC50 = 0.36 μM). More encouragingly, 6 g and 6k exerted nearly 2-fold activity enhancement over ZA (EC50 = 0.27 μM).
Molecular modeling
In order to gather further insight into the binding mechanisms of novel oseltamivir derivatives in the 150-cavity of NAs, molecular docking studies of representative compound 6k bound to the crystal structures of N1 (PDB ID: 3BEQ), N1 (PDB ID: 2HU0), and N2 (PDB ID: 4K1K) were performed by utilising Schrödinger Maestro 12.9 software. The PyMOL 1.3 software was used to visualise the docking results.
As shown in , it can be observed that 4-((r)-2-methylpyrrolidin-1-yl)benzyl group of compound 6k was projected towards the 150-cavity of N1 () and N2 (), while the oseltamivir carboxylic acid part of this compound was well anchored to the active site of NAs, consistent with the binding pattern of OSC. In developed multiple hydrogen bonds with Arg371, Arg292 and Asp151 in the active site (H1N1 NA), in line with OSC binding. Meanwhile, the 4-((R)-2-methylpyrrolidin-1-yl)benzyl group extended into the 150-cavity and occupied a hydrophobic pocket consisting of Val116, Thr439, Gly147, Gln136 and Val149. However, compared with OSC, 6k lost the hydrogen bond between the amide group and Arg152, which may be due to the slight conformational deviation of 6k caused by the introduction of the amino side chain. Furthermore, the slight conformational deflection made the amide group of 6k twisting towards Arg156 and established an additional hydrogen bond with it. Although the formation of additional hydrogen bond helped to compensate for the energy loss caused by the destruction of the original hydrogen bond, the distorted conformation might not be optimal for the interaction of NA inhibitors with the active site. This might account for the reduction in the inhibitory activity of 6k, that was slightly weaker than that of OSC against H1N1 NA.
Figure 5. Docking of compound 6k in the binding site on NA of N1 (PDB ID: 3BEQ), N1 (PDB ID: 2HU0) and N2 (PDB ID: 4K1K). (A, B, C) Superposition of 6k and OSC with N1 NA and N2 NA, respectively; (D, E, F) The key interactions formed by OSC with N1 and N2 NA, respectively; (G, H, I) The key interactions formed by 6k with N1 and N2 NA, respectively. Hydrogen bonds are shown as dashed lines (yellow).
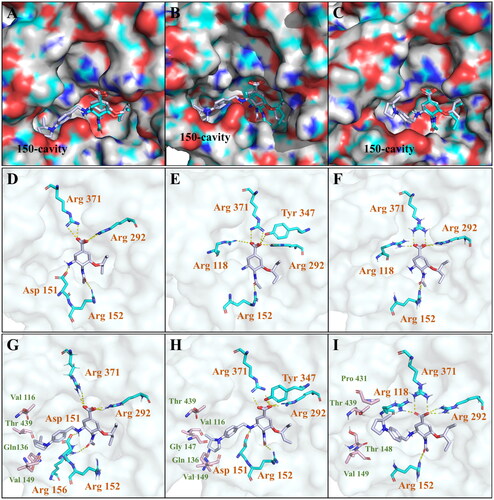
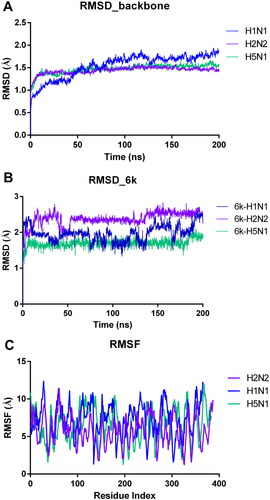
Figure 6. The result plots of molecular dynamics simulations. (A) The protein backbone RMSD plot of the NA with 6k binding throughout the MD trajectory of 200 ns; (B) The ligand heavy atom RMSD plot when bound to the NA; (C) RMSF plot of the protein chain in 6k bound state.
As for H5N1 NA (), 6k established similar interactions with OSC at the active site. Multiple hydrogen bonds between carboxylic of 6k and Arg371, Arg292, Tyr347 and Asp151 were retained as same as OSC. It’s a pity that 6k lost the hydrogen bond with Arg118. The amino group of 6k formed an additional hydrogen bond with the carbonyl group of Asp151. Compared with the conformation of 6k binding to H1N1 NA, the 2-methylpyrrolidinyl of 6k binding to H5N1 NA was positioned outward and also formed wide range of hydrophobic interactions with the amino acid residues that make up 150-cavitie.
As shown in , it was suggested that 6k retained the key interactions of carboxyl group with residues Arg292, Arg371 and Arg118, respectively, which was consistent with the action mode of OSC. Moreover, the conformation of 6k at the active site was highly overlapped with that of OSC cyclohexene, so that the hydrogen bond between the amide group and Arg152 could still be retained. Although the 150-cavity induced in group-2 NA was flatter and shallower, the 4-((R)-2-methylpyrrolidin-1-yl)benzyl group could still stretched into the 150-cavity due to the flexibility of the substituted benzylidene. Compound 6k might induce opening of the rigid N2 150-loop and was highly compatible with the active site and 150-cavity. Since 6k was only incubated with NA for 10 min in the enzyme activity test, it was difficult to induce the completely opening of the rigid N2 150-loop in such a short time. In in vitro anti-influenza virus activity assays, 6k was co-incubated with the H3N2 influenza virus for two days, in which the rigid N2 150-loop could actually be opened by 6k. This could explain why 6k had a weaker inhibitory activity against H3N2 NA than OSC in the enzyme activity assay, but it displayed nearly 3-fold activity enhancement over OSC against H3N2 strain in PRA.
Molecular dynamics simulations
To further examine the binding stability of 6k with NA, we performed MD simulation studies of 100 ns initiating from the docked states in Desmond software (Schrödinger Suite version 2022–1). The relevant RMSD (root mean square deviation) and RMSF (root mean square fluctuations) were displayed in . The illustrates the peaks of the RMSD probability density, indicating the stable conformational variations in the protein structure. The ligand heavy atom RMSD analysis in revealed the dynamic nature of 6k binding pose. RMSD values remained quite stable at around 2.0 Å, indicating the strong binding between 6k and NA. According to the dynamic results, the side chain of 6k oscillated slightly in the 150-cavity due to its own flexibility. In the RMSF plot (), the protein residues fluctuated greatly, further demonstrating that 6k is tightly bound to NA. These results confirm that the side chain of 6k can bind closely to the 150-cavity and establish stable interactions with the surrounding residues.
In vitro neuraminidase inhibitory activity assay of 6k with different preincubation time with NA
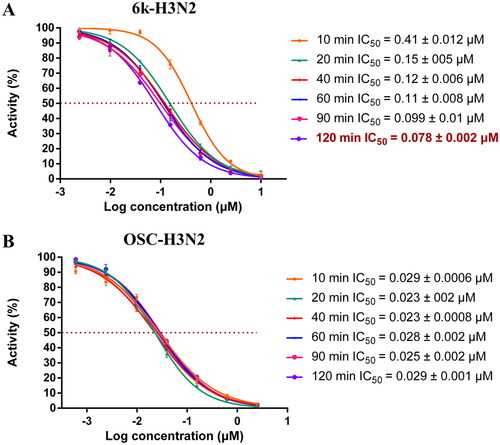
In order to further verify that compound 6k may induce the opening of N2 150-loop, we carried out neuraminidase inhibitory activity assay of 6k with different preincubation time with NA. The IC50 data can be seen in . As shown in , preincubation with different time of H3N2 NA with the OSC did not lead to significant changes of the IC50 value (0.0 2 3 ∼ 0.029 μM). However, according to , the inhibitory activity of 6k against H3N2 NA was significantly improved with the prolongation of preincubation time. It can be found that longer preincubation time gave lower IC50 values. When the incubation time was 10 min, the IC50 value of 6k was 0.41 μM. When the incubation time was extended to 120 min, the inhibitory activity was greatly improved (IC50 = 0.078 μM), being 5.3-fold more potent than 6k at 10 min incubation time. We also tried to extend the preincubation time to 4 h and 8 h. However, when the enzyme was placed for too long, its activity would be affected, so reliable data were not obtained. Combined with the antiviral activity results of 6k against H3N2 strain, there is no doubt that 6k can induce the opening of 150-loop and establish additional interactions, which explained its superior inhibitory activity against H3N2 strain comparable with OSC.
Figure 7. The neuraminidase inhibitory activity assay of 6k and OSC with different incubation time with NA. (A) The dose–response–inhibition curve of 6k at different incubation time; (B) The dose-response–inhibition curve of OSC at different incubation time.
In silico studies
In silico prediction of physicochemical properties
As shown in , all the selected compounds displayed gratifying Fsp3 valuesCitation36. To further investigate the drug-like properties of representative compounds 6 g, 6i, 6j, 6k, we utilised a free software (http://www.molinspiration.com/) to comprehensively address their physicochemical properties. The results confirmed that the parameters of the selected compounds didn’t exceed the normal scope and could meet the requirements for Lipinski’s “rule of five” concerning MW, nON, nOHNH, nrotb and miLog P. More remarkably, topological polar surface area (TPSA) is an important parameter related to the blood-brain barrier penetration, Caco-2 monolayers permeability and human intestinal absorption of compound. As seen from , TPSA values of all the compounds were in the range of 9 0 ∼ 100.13 Å2 (<140 Å2), confirming their advantages for intestinal absorption (<140 Å2) and inability to cross the blood-brain barrier, avoiding detrimental effects on the central nervous system (>60 Å2).
Table 4. Physicochemical properties of some representative compounds.
Conclusion
In summary, based on our previous studies, we designed and synthesized a series of novel N-heterocycles substituted oseltamivir derivatives to explore the chemical space of 150-cavity in NAs. Consistent with our previous findings, the introduction of nitrogen-containing heterocyclic substituents could induce the 150-cavity formation and maintain the inhibitory activity against group-2 NAs. As expected, all the tested novel compounds exhibited potent inhibitory activities against group-1 and group-2 NAs. Among them, compounds 6 g, 6i-6k displayed moderate inhibitory activities against N1 (H1N1), N2 (H3N2), N1 (H5N1), N6 (H5N6) and mutant N1 (H5N1-H274Y) compared to OSC. Moreover, compounds 6 g, 6i-6k with low cytotoxicity showed robust anti-IAV potencies against H5N1, H5N6, H1N1, and H3N2 strains in cellular assay. In particular, 6k exerted the most potent and broad-spectrum anti-IAV activity, with EC50 of 0.98 μM, 0.02 μM, 0.12 μM towards H5N1, H1N1, and H3N2, respectively, which were equivalent or more potent than that of OSC. Additionally, longer preincubation time gave lower IC50 values of 6k, further proving that N-heterocycles contribute to the opening of the N2 150-loop. The results of docking studies and molecular dynamics simulation studies clearly demonstrated that the side chain of 6k stably occupies the 150-cavity and the scaffold retains other key interactions in the active site of NAs. Therefore, compound 6k deserves to be further studied as a valuable lead compound.
Author contributions
Jiwei Zhang and Chuanfeng Liu: Conceptualisation, Methodology, Formal analysis, Investigation, Resources, Data Curation, Writing - Original Draft, Visualisation
Ruifang Jia and Jian Zhang: Supervision, Writing - Review & Editing, Methodology
Xujie Zhang: Software
Chiara Bertagnin, Anna Bonomini, Laura Guizzo: Resource (Provision of study materials for the in vitro anti-influenza virus activity in MDCK cells)
Yuanmin Jiang, Huinan Jia, Shuzhen Jia: Resource, Data Curation
Xiuli Ma: Resource (Provision of study materials)
Arianna Loregian: Resource (Provision of study materials for the in vitro anti-influenza virus activity in MDCK cells), Writing - Review & Editing, Supervision, Project administration
Bing Huange, Peng Zhan, Xinyong Liu: Writing - Review & Editing, Supervision, Project administration, Funding acquisition
Supplemental Material
Download PDF (2.5 MB)Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (NSFC no. 81773574); Science Foundation for Outstanding Young Scholars of Shandong Province (ZR2020JQ31); Foreign cultural and educational experts Project (GXL20200015001); Shandong modern agricultural technology and industry system (SDAIT-21–06); Key Research and Development Program of Shandong Province (2022CXGC010606); China Postdoctoral Science Foundation (2022M711938); Natural Science Foundation of Jiangsu Province (SBK2023041680); Associazione Italiana per la Ricerca sul Cancro, AIRC, grants IG 2016 - ID. 18855 and IG 2021 - ID. 25899 (to A.L.); Ministero dell’Istruzione, dell’Università e della Ricerca, PRIN 2017-cod. 2017KM79NN (to A.L.); Fondazione Cassa di Risparmio di Padova e Rovigo-Bando Ricerca Covid-2019 No. 55777 2020.0162-ARREST-COV: AntiviRal PROTAC-Enhanced Small-molecule Therapeutics against COronaViruses (to A.L.).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Tang L, Yan H, Wu W, Chen D, Gao Z, Hou J, Zhang C, Jiang Y. Synthesis and anti-influenza virus effects of novel substituted polycyclic pyridone derivatives modified from baloxavir. J Med Chem. 2021;64(19):14465–14476.
- Wang Z, Zalloum WA, Wang W, Jiang X, De Clercq E, Pannecouque C, Kang D, Zhan P, Liu X. Discovery of novel dihydrothiopyrano[4,3-d]pyrimidine derivatives as potent HIV-1 NNRTIs with significantly reduced hERG inhibitory activity and improved resistance profiles. J Med Chem. 2021;64(18):13658–13675.
- Fineberg HV. Pandemic preparedness and response–lessons from the H1N1 influenza of 2009. N Engl J Med. 2014;370(14):1335–1342.
- Swets MC, Russell CD, Harrison EM, Docherty AB, Lone N, Girvan M, Hardwick HE, Visser LG, Openshaw PJM, Groeneveld GH, et al. SARS-CoV-2 co-infection with influenza viruses, respiratory syncytial virus, or adenoviruses. Lancet. 2022;399(10334):1463–1464.
- Chen JR, Liu YM, Tseng YC, Ma C. Better influenza vaccines: an industry perspective. J Biomed Sci. 2020;27(1):33.
- Shirley M. Baloxavir marboxil: a review in acute uncomplicated influenza. Drugs. 2020;80(11):1109–1118.
- Dunn CJ, Goa KL. Zanamivir: a review of its use in influenza. Drugs. 1999;58(4):761–784.
- McClellan K, Perry CM. Oseltamivir: a review of its use in influenza. Drugs. 2001;61(2):263–283.
- Scott LJ. Peramivir: a review in uncomplicated influenza. Drugs. 2018;78(14):1525.
- Kubo S, Tomozawa T, Kakuta M, Tokumitsu A, Yamashita M. Laninamivir prodrug CS-8958, a long-acting neuraminidase inhibitor, shows superior anti-influenza virus activity after a single administration. Antimicrob Agents Chemother. 2010;54(3):1256–1264.
- Loregian A, Mercorelli B, Nannetti G, Compagnin C, Palù G. Antiviral strategies against influenza virus: towards new therapeutic approaches. Cell Mol Life Sci. 2014;71(19):3659–3683.
- Baz M, Abed Y, Simon P, Hamelin ME, Boivin G. Effect of the neuraminidase mutation H274Y conferring resistance to oseltamivir on the replicative capacity and virulence of old and recent human influenza A(H1N1) viruses. J Infect Dis. 2010;201(5):740–745.
- Kode SS, Pawar SD, Tare DS, Keng SS, Hurt AC, Mullick J. A novel I117T substitution in neuraminidase of highly pathogenic avian influenza H5N1 virus conferring reduced susceptibility to oseltamivir and zanamivir. Vet Microbiol. 2019;235:21–24.
- Wu Y, Wu Y, Tefsen B, Shi Y, Gao GF. Bat-derived influenza-like viruses H17N10 and H18N11. Trends Microbiol. 2014;22(4):183–191.
- Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, Blackburn GM, Hay AJ, Gamblin SJ, Skehel JJ. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443(7107):45–49.
- Han N, Mu Y. Plasticity of 150-loop in influenza neuraminidase explored by Hamiltonian replica exchange molecular dynamics simulations. PLoS One. 2013;8(4):e60995.
- Wu Y, Qin G, Gao F, Liu Y, Vavricka CJ, Qi J, Jiang H, Yu K, Gao GF. Induced opening of influenza virus neuraminidase N2 150-loop suggests an important role in inhibitor binding. Sci Rep. 2013;3(1):1551.
- Ju H, Zhang J, Sun Z, Huang Z, Qi W, Huang B, Zhan P, Liu X. Discovery of C-1 modified oseltamivir derivatives as potent influenza neuraminidase inhibitors. Eur J Med Chem. 2018;146:220–231.
- Zhang J, Poongavanam V, Kang D, Bertagnin C, Lu H, Kong X, Ju H, Lu X, Gao P, Tian Y, et al. Optimization of N-substituted oseltamivir derivatives as potent inhibitors of group-1 and -2 influenza a neuraminidases, including a drug-resistant variant. J Med Chem. 2018;61(14):6379–6397.
- Jia R, Zhang J, Shi F, Bonomini A, Lucca C, Bertagnin C, Zhang J, Liu C, Jia H, Jiang Y, et al. Discovery of N-substituted oseltamivir derivatives as novel neuraminidase inhibitors with improved drug resistance profiles and favorable drug-like properties. Eur J Med Chem. 2023;252:115275.
- Zhang J, Murugan NA, Tian Y, Bertagnin C, Fang Z, Kang D, Kong X, Jia H, Sun Z, Jia R, et al. Structure-based optimization of n-substituted oseltamivir derivatives as potent anti-influenza a virus agents with significantly improved potency against oseltamivir-resistant N1-H274Y variant, E. J Med Chem. 2018;61(22):9976–9999.
- Jia R, Zhang J, Ju H, Kang D, Fang Z, Liu X, Zhan P. Discovery of novel anti-influenza agents via contemporary medicinal chemistry strategies (2014-2018 update. Future Med Chem. 2019;11(5):375–378.),
- Jia R, Zhang J, Bertagnin C, Cherukupalli S, Ai W, Ding X, Li Z, Zhang J, Ju H, Ma X, et al. Discovery of highly potent and selective influenza virus neuraminidase inhibitors targeting 150-cavity. Eur J Med Chem. 2021;212:113097.
- Jia R, Zhang J, Zhang J, Bertagnin C, Bonomini A, Guizzo L, Gao Z, Ji X, Li Z, Liu C, et al. Discovery of novel boron-containing N-substituted oseltamivir derivatives as anti-influenza A virus agents for overcoming N1-H274Y oseltamivir-resistant. Molecules. 2022;27(19):6426.
- Xie Y, Xu D, Huang B, Ma X, Qi W, Shi F, Liu X, Zhang Y, Xu W. Discovery of N-substituted oseltamivir derivatives as potent and selective inhibitors of H5N1 influenza neuraminidase. J Med Chem. 2014;57(20):8445–8458.
- Du J, Guo J, Kang D, Li Z, Wang G, Wu J, Zhang Z, Fang H, Hou X, Huang Z, et al. New techniques and strategies in drug discovery. Chin Chem Lett. 2020;31(7):1695–1708.
- Muratore G, Mercorelli B, Goracci L, Cruciani G, Digard P, Palù G, Loregian A. Human cytomegalovirus inhibitor AL18 also possesses activity against influenza A and B viruses. Antimicrob Agents Chemother. 2012;56(11):6009–6013.
- Nannetti G, Massari S, Mercorelli B, Bertagnin C, Desantis J, Palù G, Tabarrini O, Loregian A. Potent and broad-spectrum cycloheptathiophene-3-carboxamide compounds that target the PA-PB1 interaction of influenza virus RNA polymerase and possess a high barrier to drug resistance. Antiviral Res. 2019;165:55–64.
- D’Agostino I, Giacchello I, Nannetti G, Fallacara AL, Deodato D, Musumeci F, Grossi G, Palù G, Cau Y, Trist IM, et al. Synthesis and biological evaluation of a library of hybrid derivatives as inhibitors of influenza virus PA-PB1 interaction. Eur J Med Chem. 2018;157:743–758.
- Massari S, Bertagnin C, Pismataro MC, Donnadio A, Nannetti G, Felicetti T, Di Bona S, Nizi MG, Tensi L, Manfroni G, et al. Synthesis and characterization of 1,2,4-triazolo[1,5-a]pyrimidine-2-carboxamide-based compounds targeting the PA-PB1 interface of influenza A virus polymerase. Eur J Med Chem. 2021;209:112944.
- Lu C, Wu C, Ghoreishi D, Chen W, Wang L, Damm W, Ross GA, Dahlgren MK, Russell E, Von Bargen CD, et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput. 2021;17(7):4291–4300.
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221–234.
- Sinha SK, Shakya A, Prasad SK, Singh S, Gurav NS, Prasad RS, Gurav SS. An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. J Biomol Struct Dyn. 2021;39(9):3244–3255.
- Ju H, Murugan NA, Hou L, Li P, Guizzo L, Zhang Y, Bertagnin C, Kong X, Kang D, Jia R, et al. Identification of C5-NH(2) modified oseltamivir derivatives as novel influenza neuraminidase inhibitors with highly improved antiviral activities and favorable druggability. J Med Chem. 2021;64(24):17992–18009.
- Bialy D, Shelton H. Functional neuraminidase inhibitor resistance motifs in avian influenza A(H5Nx) viruses. Antiviral Res. 2020;182:104886.
- Samson M, Pizzorno A, Abed Y, Boivin G. Influenza virus resistance to neuraminidase inhibitors. Antiviral Res. 2013;98(2):174–185.