
Abstract
In this study, a series of potential ligands for the treatment of AD were synthesised and characterised as novel harmine derivatives modified at position 9 with benzyl piperazinyl. In vitro studies revealed that the majority of the derivatives exhibited moderate to potent inhibition against hAChE and Aβ1 − 42 aggregation. Notably, compounds 13 and 17d displayed potent drug − likeness and ADMET properties, demonstrating remarkable inhibitory activities towards AChE (IC50 = 58.76 nM and 89.38 nM, respectively) as well as Aβ aggregation (IC50 = 9.31 μM and 13.82 μM, respectively). More importantly, compounds 13 and 17d showed exceptional neuroprotective effects against Aβ1 − 42−induced SH − SY5Y damage, while maintaining low toxicity in SH − SY5Y cells. Further exploration of the mechanism through kinetic studies and molecular modelling confirmed that compound 13 could interact with both the CAS and the PAS of AChE. These findings suggested that harmine derivatives hold great potential as dual − targeted candidates for treating AD.
Introduction
Alzheimer’s disease (AD) is a chronic and progressive neurodegenerative disorder, characterised by cognitive decline, encompassing memory impairment, language dysfunction, mood disturbances, and loss of motor functionCitation1,Citation2. The World Health Organisation (WHO) estimated that approximately 45 million individuals worldwide were affected by AD in 2015, with projections indicating a staggering increase to over 150 million cases by the year 2050Citation3. This imposes a significant socioeconomic burden. Currently, the Food and Drug Administration (FDA) has approved only six drugs for the treatment of AD, which include three acetylcholinesterase (AChE) inhibitors (donepezil, rivastigmine and galantamine), one N − methyl − D−aspartate receptor antagonist (memantine), and two monoclonal antibodies targeting β−amyloid (Aβ) peptide deposits (aduhelm and lecanemab)Citation4–6. Unfortunately, the efficacy of AChE inhibitors and NMDA receptor antagonists in alleviating AD symptoms is only partial and fails to provide a cure for the disease. Meanwhile, monoclonal antibodies are limited to treating mild − to − moderate AD but come with potent adverse effects that have sparked controversy regarding their usage Citation7,Citation8. The current situation is concerning and necessitates further endeavours in the development of more efficacious drugs for AD treatment.
To date, the precise aetiology of AD remains incompletely understood; however, deficits in acetylcholine (ACh), Aβ deposition, tau protein (τ) aggregation, neuroinflammation and oxidative stress have been implicated as significant contributors to its pathogenesisCitation9–12. Due to the intricate nature of AD and the interconnectedness of molecular events in its progression, single − targeted drugs may not be suitable for treating AD. Consequently, numerous researchers have shifted their focus towards designing multi − targeted ligands (MTDLs) that can simultaneously interact with two or more diseased targets as a superior strategy for treating AD compared to solely targeting one targetCitation1,Citation3–15.
In the amyloid cascade hypothesis, the deposition of Aβ plays a crucial role in the pathogenesis of AD. The aggregation of Aβ leads to neurotoxic effects on cerebral neurons, thereby initiating memory impairments and cognitive dysfunctionCitation16,Citation17. Considerable efforts have been devoted to exploiting efficient strategies for either reducing Aβ production or enhancing Aβ clearanceCitation18,Citation19. Another prominent hypothesis suggests that the decline of ACh is intricately associated with symptomatic cognitive impairment in patients with AD. The utilisation of AChE inhibitors, which enhance ACh levels by inhibiting its hydrolysis within the synaptic cleft, represents a pivotal strategy for alleviating cognitive deficitsCitation20. Additionally, more evidence suggests that the peripheral anionic site (PAS) of AChE contributes to Aβ aggregationCitation21. Furthermore, AChE has the ability to form a complex with Aβ, thereby exhibiting enhanced neurotoxicity compared to the effects of Aβ aloneCitation22. Consequently, it is plausible that a singular compound possessing inhibitory activity against both the dual site of AChE and Aβ aggregation could serve as a promising therapeutic agent for AD.
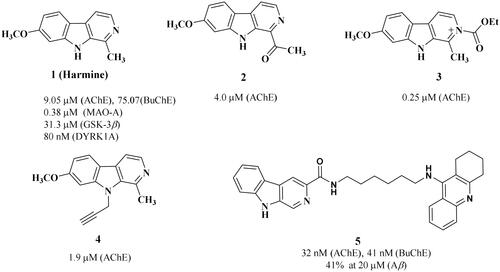
Harmine, a naturally occurring β−carboline alkaloid, is originally isolated from the seeds of Peganum harmala L. in the 1840sCitation23. A plethora of studies demonstrate that this compound exhibits an extensive range of biological, psychopharmacological, and toxicological effectsCitation24–26. Especially, harmine and its derivatives () have been identified as ligands for several biomolecular targets involved in the pathogenesis of Alzheimer’s disease (AD), including Aβ aggregation, AChE, monoamine oxidase (MAO) and NMDA receptorCitation27. Furthermore, harmine treatments have been demonstrated to enhance short − term memory in aged rats, improve spatial learning and memory ability in APP/PS1 transgenic mice, as well as alleviate cognitive dysfunction induced by scopolamine in miceCitation28–30. These findings suggest that harmine holds great promise as a multi − target therapeutic agent for AD.
Figure 1. Chemical structures of harmine and its derivatives.
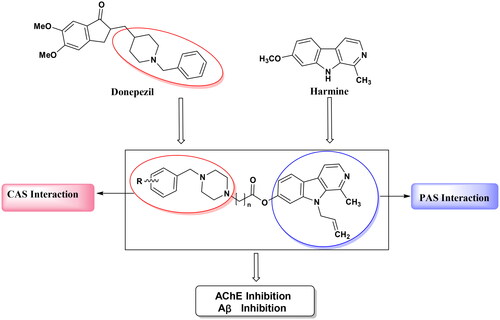
In this work, a pharmacophore combination strategy was employed, utilising harmine and donepezil as the lead compounds (). Furthermore, the piperidine fragment of donepezil was substituted with a piperazine moiety owing to the pronounced AChE inhibition exhibited by specific aryl piperazine − containing compoundsCitation31. The introduction of an allyl substituent at position 9 of harmine significantly augmented its activity against AChECitation27. In accordance with the aforementioned assumption, these novel derivatives may possess dual binding sites for AChE inhibition and exhibit inhibitory effects on Aβ aggregation simultaneously. The evaluation of the pharmacological profile for synthesised harmine derivatives involved determining (I) inhibitory activity against AChE/Aβ aggregation, (II) ADME computer predictions, (III) inhibition of AChE − induced Aβ1 − 42 peptide aggregation, (IV) in vitro cytotoxic effects and protection against cell damage induced by Aβ aggregation, (V) enzyme kinetic studies on AChE, and (VI) molecular docking studies.
Figure 2. Design strategy for the new series of harmine derivatives targeting AChE and Aβ1-42 aggregation.
Results and discussion
Synthesis
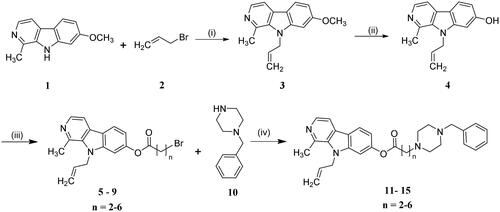
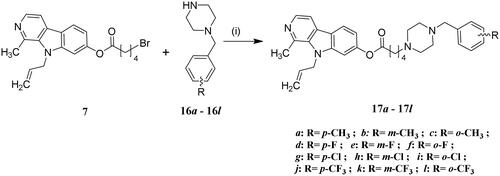
The synthesis of compounds 13 − 17 l was carried out following the procedures described in Schemes 1 and 2. The reaction commenced with harmine (1), which underwent allylation using allyl bromide as a catalyst in dry DMF, facilitated by NaH, resulting in the formation of the 9 − allyl harmine derivative (3). Subsequently, compound 3 was subjected to refluxing in AcOH and HBr for demethylation of methoxyl groups. Following this step, an esterification reaction between compound 4 and various acylbromides yielded key intermediates 5 − 9. Finally, the target compounds 13 − 17 l were obtained through nucleophilic substitution reactions between compounds 5 − 9 to different benzylpiperazine derivatives.
Scheme 1. Synthesis of compounds 11 − 15. Reagents and conditions: (i) NaH, DMF, rt; (ii) AcOH, HBr, reflux; (iii) Different acylbromides, EDCI, dry CH2Cl2, reflux; (iv) NaHCO3, EtOH, reflux.
Scheme 2. Synthesis of compounds 17a − 17l. Reagents and conditions: (i) NaHCO3, EtOH, reflux.
Results of inhibition activity against hAChE, hBuChE and Aβ1 − 42 self − aggregation
The Ellman’s assay, a widely employed method for evaluating enzyme inhibition, was utilised to investigate the inhibitory potential of novel harmine derivatives against hAChE and hBuChE. Tacrine and donepezil were employed as reference compounds in this study. We initially screened all compounds at a concentration of 1 μM, and subsequently determined IC50 values for those exhibiting over 50% inhibitory activity against AChE in the primary screen. The resulting IC50 values and their corresponding selectivity indices are presented in . The tested target compounds demonstrated significant inhibitory activity against hAChE, with IC50 values below 1 μM. Most of these compounds exhibited selectivity for hAChE over hBuChE, except for compounds 11 and 17h. Among them, six compounds (13, 15, 17a, 17c, 17d and 17 l) displayed significantly higher inhibitory activity against hAChE compared to tacrine (IC50 = 81.36 nM). Additionally, compounds 17a and 17c (IC50 = 12.57 and 15.73 nM, respectively) demonstrated greater inhibitory activity against hAChE than donepezil with an IC50 of 17.86 nM. Additionally, eight compounds exhibited dual inhibitory activities against both hAChE and hBuChE, demonstrating IC50 values at the nanomolar level. In the human brain, BuChE is present in glial cells and neurons, as well as in plaques and tangles observed in patients with Alzheimer’s disease (AD). Additionally, it has been reported to exhibit associations with drug metabolism and detoxification processesCitation32,Citation33. Consequently, both AChE and BuChE have been proposed as potential therapeutic targets for AD treatment.
Table 1. Inhibition of hAChE, hBuChE, and Aβ1 − 42 aggregation by the synthesised compounds.
The inhibitory potency of derivatives on Aβ1 − 42 self − aggregation was assessed using the Thioflavin − T (ThT) fluorescence assay, with resveratrol and curcumin serving as controls (). As depicted in , compounds 13, 15, 17a, 17c, 17d and 17 l also demonstrated significant inhibition of Aβ1 − 42 aggregation (IC50 = 9.31, 18.33, 6.74, 16.02, 13.82 and 12.42 μM, respectively), which were comparable to or greater than that of resveratrol (IC50=11.51 μM) and curcumin (IC50 = 15.47 μM). Furthermore, a survey investigating key structure-activity relationships revealed a pronounced influence of alkyl spacer length on inhibitory activities. Compounds 12 (n = 3) and 13 (n = 4) exhibited superior AChE inhibitory activity with IC50 values of 49.69 and 58.76 nM, respectively, compared to compounds 11 (n = 2), 14 (n = 5), and 15 (n = 6). Furthermore, compounds 13 (n = 4) and 15 (n = 6) exhibited more potent inhibitory activity against Aβ1 − 42 aggregation with IC50 values of 9.31 and 18.33 μM, respectively, compared to other carbon chain lengths (compounds 11, 12, and 13). Thus, Lengthening or shortening the alkyl spacer resulted in reduced inhibition activities. Compound 13 (n = 4) demonstrated enhanced potency as a hybrid molecule against both AChE and Aβ1 − 42 aggregation compared to the other compounds tested. In addition, the methyl − substituted compounds 17a, 17b and 17c exhibited superior inhibitory activity when compared to their unsubstituted counterparts with identical chain linkers. Conversely, the presence of electron − withdrawing substituents (excluding compound 17 l) such as − F, −Cl and − CF3 resulted in a decrease in hAChE inhibitory activity. These findings suggest that augmenting the strength of electron − donating groups may potentially enhance hAChE inhibitory activity.
Predicted physicochemical properties and drug − likeness
To investigate the drug − like properties of compounds exhibiting potent inhibition against AChE and Aβ aggregation, the physicochemical properties of active compounds 13, 15, 17a, 17c, 17d and 17 l, such as the: molecular weight (MW), number of hydrogen acceptors (HBA), number of hydrogen donors (HBD), topological polar surface area (tPSA), log octanol/water partition coefficient (LogP) and the verification of Lipinski’s rule of five, were predicted. As shown in , it was evident that the molecular MW and logP values of compounds 17a, 17c, and 17d do not meet Lipinski’s rule criteria. To enhance their potential for future drug development, further optimisation of their chemical structures as well as reduction in both MW and logP values are necessary. Derivatives 13 and 17d were found to comply with the rule and were subsequently utilised in further studies.
Table 2. Physiochemical properties for some active compounds, tacrine, donepezil, resveratrol and curcumin.
ADMET prediction
The ADMET property of derivatives 13 and 17d was assessed utilising ADMETlab 2.0 (https://admetmesh.scbdd.com)Citation34and ProTox − II (http://tox.charite.de/protox_II)Citation35, with tacrine, donepezil, resveratrol, and curcumin serving as reference compounds. The results () showed that compounds 13 and 17d exhibited comparable ADMET properties. The Caco − 2 (Caucasian colon adenocarcinoma cell lines) permeability scores of derivatives 13 and 17d (−5.00 and −5.02 log units, respectively) were found to be slightly below the minimum optimal score of −5.15 log units, closely resembling those of the reference drugs. The MDCK (Madin–Darby canine kidney) permeability score of 1.8 × 1 0 −5 cm/s indicated a moderate level of permeability (0.2–2 × 1 0 −5 cm/s), which fell within the acceptable range. The HIA (Human intestinal absorption) predicted values for all tested compounds exceeded 30%. Derivatives 13 and 17d, as well as resveratrol, were anticipated to exhibit bioavailabilities greater than 30% (F30%, −−−).
Table 3. Predicted ADME and toxicity results for some active compounds, tacrine, donepezil, resveratrol and curcumin.
The predicted PPB (plasma proteinbinding) scores of derivatives 13 and 17d were 93.8% and 94.3% respectively, which closely approximated those of donepezil (87.7%), resveratroland (96.0%), and curcumin (89.7%). Moreover, the projected outcomes for the BBB (blood − brain barrier) permeability of these two derivatives suggested a likelihood similar to that observed with tacrine and donepezil. Additionally, the calculated volumes of distribution (VD) for derivatives 13 and 17d were determined to be 2.56 L/kg and 2.63 L/kg, respectively, surpassing those associated with the reference drugs in question. It is worth noting that both VD values fall within the optimal range of between 0.04 L/kg and 20 L/kg.
Metabolism plays a pivotal role in determining the plasma concentration of drugs. In this database, ligands are categorised as either inhibitors (category 1) or non-inhibitors (category 0), based on their ability to hinder the enzyme’s activity. As shown in , derivatives 13 and 17d exhibited inhibitory effects on CYP1A2, CYP2C19, and CYP2D6 isoforms, respectively. However, similar to donepezil and tacrine, they lacked the capability to inhibit the CYP2C9 isoform.
The clearance rates (CL) of derivatives 13 and 17d were calculated to be 9.75 and 9.52 ml/min/kg, respectively, indicating a moderate range of clearance rates (5 − 15 ml/min/kg). The half − life values (T1/2) for these derivatives were determined to be 0.04 and 0.01 respectively, indicating a high probability of short half − lives (< 3 h).
As presented in , compounds 13 and 17d demonstrated LD50 values that were 25 − times higher than tacrine and twice as high as donepezil. The toxicity classification of all tested compounds was elevated to level 4, surpassing the level 2 classification of tacrine. The ADMET profiles indicated that the majority of parameters fell within the optimal range, suggesting that derivatives 13 and 17d exhibited favourable pharmacological profiles and hold potential as orally bioavailable drug candidates for the central nervous system (CNS).
Inhibition of AChE − induced Aβ1 − 42 peptide aggregation assay
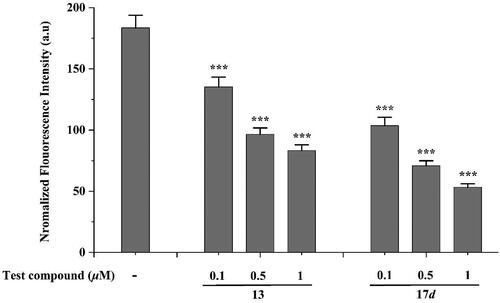
As research unfolds, it is increasingly evident that AChE plays a crucial role in the aggregation of Aβ, with accumulating evidence suggesting its active promotion of the aggregation of this toxic protein fragment beyond cholinergic functions. Several studies have demonstrated the effective inhibition of AChE − accelerated Aβ aggregation through the binding of compounds to the PAS of AChECitation36. Therefore, we evaluated the inhibitory effects of derivatives 13 and 17d on AChE-induced Aβ1 − 42 aggregation using the Th-T assay. The co-incubation of synthesised compounds and Aβ1-42 with AChE for 24h, as illustrated in , resulted in a significant reduction in Aβ1 − 42 aggregation, which was positively correlated with compound concentration. These findings clearly demonstrate that the two compounds have the ability to interfere with AChE − induced Aβ1 − 42 aggregation.
Figure 3. AChE induced Aβ1-42 aggregation inhibition by compounds 13 and 17d. *** p < 0.001 vs control group.
In vitro cytotoxicity and neuroprotective effects assay
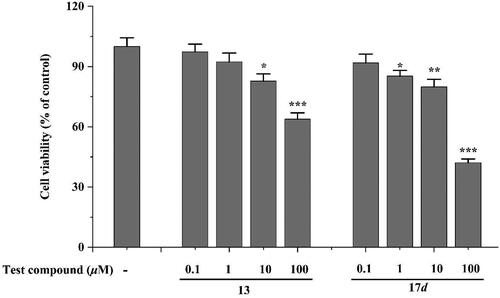
The compounds 13 and 17d, which possess the potential to inhibit AChE and Aβ1 − 42 aggregation, were selected for toxicity evaluation in the human neuronal cell line SHSY5Y. As illustrated in , compounds 13 exhibited negligible cytotoxicity at concentrations ranging from 0.1 to 1 μM following a 48h incubation period. Upon increasing the concentration to 10 μM, these compounds demonstrated significant effects on SH − SY5Y cell viability; however, the cell viability remained above 79%. These findings suggest that both compounds 13 and 17d exhibit weak toxicity towards SHSY5Y cells within the tested concentration range.
Figure 4. Neurotoxicity of compounds in SH-SY5Y cells. *p < 0.05, **p < 0.01 and *** p < 0.001 vs control group (untreated cells).
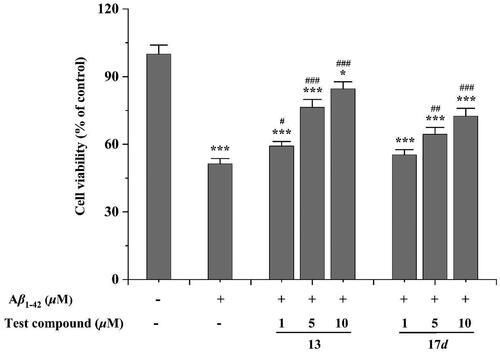
The neuroprotective effects of the test compounds against Aβ1 − 42−induced damage in SH − SY5Y cells were evaluated through a cell viability assay at three distinct concentrations: 1, 5, and 10 μM. clearly depicted the impact of Aβ1 − 42 and tested compounds on SH − SY5Y cells. The results showed a significant decrease in cell viability by 51.4% (***p < 0.001) after exposure to Aβ1 − 42 at a concentration of 20 μM for 48 h. However, the tested compounds exhibited good neuroprotective effects within the concentration range of 5 to 10 μM. Specifically, compound 13 demonstrated exceptional cellular recovery even at the lowest concentration of just 1 μM (#p < 0.05), compared to the group treated with Aβ1 − 42 alone. These results illustrate that the tested compounds have potential neuroprotective properties against Aβ1 − 42−induced neurotoxicity. This observation suggests their potential as promising therapeutic options for AD. Nevertheless, further investigation is necessary to comprehensively elucidate the underlying mechanisms responsible for these protective effects.
Figure 5. Neuroprotective effects of compounds 13 and 17d against Aβ1-42-induced cell death in SH-SY5Y cells. # p < 0.05 and ### p < 0.001 vs control group (untreated cells); *p < 0.05 and ***p < 0.001 vs Aβ1-42-treated cells.
Kinetic characterization of AChE inhibition
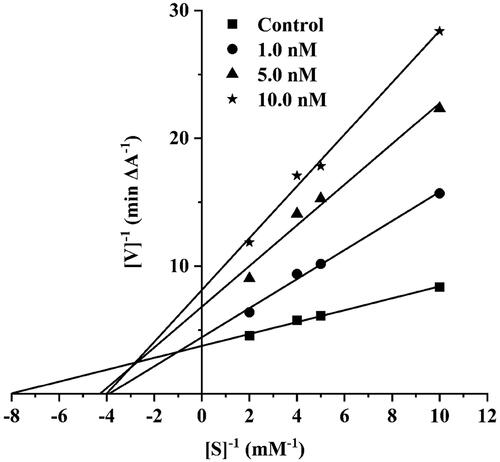
To further explore the intricacies of AChE activity inhibition by synthesised compounds, the specific compound 13 was chosen for an enzyme kinetic study due to its remarkable potency. Analysis of the Lineweaver–Burk plot () showed that the slope increased and the Vmax decreased as the concentration of compound 13 increased, which indicating a mixed − type inhibitionCitation34,Citation35. The results of this study imply that compound 13 exhibits binding affinity not only towards the CAS but also towards the PAS of AChE, thereby confirming the validity of our design.
Figure 6. The Lineweaver-Burk plot of hAChE exhibits competitive inhibition with compound 13.
Docking studies on AChE and Aβ1 − 42
To further investigate the binding mode of these compounds with their target enzymes, we carefully selected the potent compound 13 and conducted docking analysis within hAChE (PDB code: 4EY7) and Aβ1 − 42 (PDB code: 1IYT) active sites. The molecular docking simulations were performed using Surflex − Dock program integrated in Sybyl − X 2.0 software ( and ).
Figure 7. Proposed binding mode of compound 13 inside AChE active site, (A) 3D image, the blue dashed lines stand for π-π stacking, the red dashed lines represent hydrogen bondsnd and the black dashed lines mean hydrophobic interaction (B) 2D image.
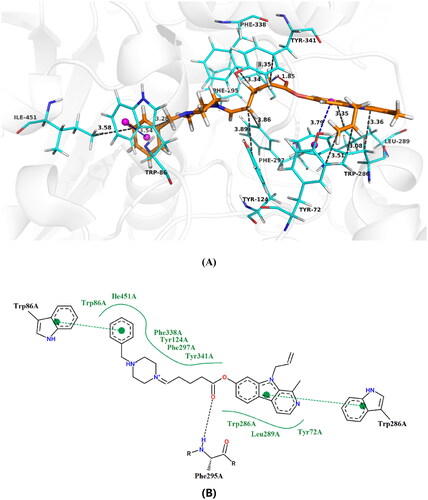
As illustrated in , the compound 13 demonstrated the ability to simultaneously bind to both the CAS and the PAS. Analysis of the binding sites revealed that the pyrrole ring of harmine group binds to the PAS via a π − π interaction with Trp286 at a distance of 3.8 Å. Additionally, an interaction with the residue Phe295 (1.85 Å) through a hydrogen bond was observed at mid-gorge sites, potentially augmenting its interaction with hAChE. Furthermore, the benzene ring of piperazine exhibited a π − π stacking interaction with Trp86 (3.5 Å), enabling binding to the CAS. This suggests that increasing the electron − donating group on the benzene ring could enhance hAChE inhibitory activity, consistent with enzyme kinetic studies. Moreover, the compound 13 exhibited hydrophobic interactions with residues Tyr72 (3.51 Å), Trp86 (3.86 Å), Tyr124 (3.59 Å), Trp286 (3.08 Å and 3.76 Å), Leu289 (3.35 Å), Phe297 (3.86 Å), Phe338 (3.34 Å), Tyr341 (3.35 Å) and He451(3.58 Å). These interactions may strengthen the binding affinity between small ligands and hAChE, ultimately resulting in potent inhibition.
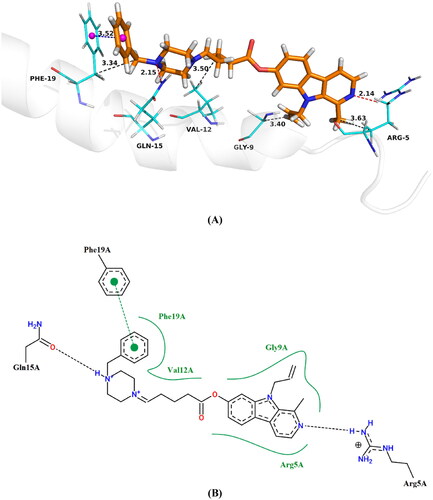
The examination of revealed that the benzene ring of piperazine was engaged in a π − π stacking interaction with the residue Phe19 at a distance of 3.5 Å. Additionally, the amino groups of pyridine and piperazine formed intriguing hydrogen bonds with Arg5 and Gln15 at distances of 2.1 and 2.2 Å, respectively. Furthermore, residues Arg5 (3.63 Å), Gly9 (3.40 Å), Val12 (3.50 Å) and Phe19 (3.34 Å) were found to engage in four hydrophobic interactions, potentially enhancing the inhibitory activity against Aβ1 − 42 aggregation.
Figure 8. Proposed binding mode of compound 13 inside Aβ1-42 active site, (A) 3D image, the blue dashed lines stand for π-π stacking, the red dashed lines represent hydrogen bondsnd and the black dashed lines mean hydrophobic interaction, (B) 2D image.
Conclusions
A novel series of harmine derivatives were designed and synthesised as potent inhibitors of AChE and Aβ aggregation, with the aim of developing therapeutic interventions for AD. All compounds underwent in vitro evaluation to assess their inhibitory activities against AChE, BuChE, and Aβ aggregation. The tested compounds, with the exception of compounds 11 and 17h, exhibited significant inhibitory activity against hAChE (IC50 <1 μM) and demonstrated selectivity for hAChE over hBuChE. Among these compounds, namely 12, 13, 17a, 17b, 17c, and 17 l showed significantly higher inhibitory activity against hAChE compared to tacrine (IC50 = 81.36 nM). Furthermore, compounds 17a and 17c displayed greater inhibitory activity against hAChE (IC50 = 12.57 and 15.73 nM respectively) than donepezil (IC50 = 17.86 nM). Meanwhile, six derivatives (13, 15, 17a, 17c, 17d and 17 l) demonstrated significant inhibition of Aβ1 − 42 aggregation with IC50 values ranging from 6.74 to 18.33 μM. These results were comparable to or surpassed the inhibitory effects observed for resveratrol (IC50 = 11.51 μM) and curcumin (IC50 = 15.47 μM). The drug − likeness and ADMET prediction results indicated that compounds 13 and 17d complied with Lipinski’s rule of five, demonstrating favourable characteristics as potential drugs for AD. Further investigation revealed that these compounds possessed significant neuroprotective activity against Aβ1 − 42−induced damage in SH − SY5Y cells, while exhibiting low toxicity towards neuroblastoma (SH − SY5Y). Kinetic profiling and molecular modelling studies demonstrated that compound 13 could bind to both the CAS and the PAS of AChE. The binding processes were primarily driven by hydrophobic, hydrogen bonding and π − π stacking interactions with key residues of AChE and Aβ. These findings offered valuable insights for the further development of harmine derivatives as promising lead compounds in research towards potential anti − AD drug candidates.
Materials and methods
General remarks
All solvents, chemicals, and reagents were procured from commercial suppliers and utilised without any further refinement. Thin layer chromatography (TLC) was employed for reaction monitoring purposes. Flash chromatography purification was carried out using silica gel (200 − 300 mesh) sourced from Qingdao Marine Chemical Company. 1H NMR and 13C NMR spectra were acquired using a JEOL ECZ600R/S3 (JEOL RESONANCE Inc., Japan) operating at 600 MHz for 1H and 150 MHz for 13C. Chemical shifts were reported in parts per million (ppm, δ) with CDCl3 as the solvent and tetramethylsilane (TMS) as an internal standard. Multiplicities were denoted by singlet (s), doublet (d), triplet (t), doublet of doublet (dd), or multiplet (m). High resolution mass spectra (HRMS) were obtained on Agilent 6520 Q − TOF LC/MS.
Preparation of intermediates 3 − 9
The synthesis of compounds 3 and 4 was carried out using a previously reported methodCitation37. To a solution of acylbromides (1 equiv), DMAP (0.1 equiv) and EDCI (1.2 equiv) in dichloromethane, the mixture was stirred at 0 °C for 10 min. Subsequently, compound 4 (1.5 equiv) was added to the suspension, and the reaction proceeded at room temperature for 24 h under TLC monitoring. The resulting reaction solution was sequentially washed with water and brine, dried over anhydrous Na2SO4. The corresponding intermediates 5 − 9 were obtained by purifying through column chromatography.
9 − Allyl − 1 − methyl − 9H − pyrido[3,4 − b]indol − 7 − yl 3 − bromopropanoate (5)
Light yellow solid; 76%; 1H NMR (600 MHz, Chloroform−d) δ: 8.28 (d, J = 5.2 Hz, 1H), 7.99 (d, J = 8.6 Hz, 1H), 7.75 (d, J = 5.2 Hz, 1H), 6.96 (dd, J = 2.0, 8.6 Hz, 1H), 6.90 (d, J = 2.0 Hz, 1H), 6.02–6.09 (m, 1H), 5.83–5.79 (2 m, 2H), 4.48–4.44 (m, 2H), 3.76 (t, J = 5.6 Hz, 2H), 3.02 (t, J = 5.6 Hz, 2H), 2.90 (s, 3H).
9 − Allyl − 1 − methyl − 9H − pyrido[3,4 − b]indol − 7 − yl 4 − bromobutanoate (6)
Light yellow solid; 78%; 1H NMR (600 MHz, Chloroform−d) δ: 8.30 (d, J = 5.2 Hz, 1H), 8.00 (d, J = 8.6 Hz, 1H), 7.76 (d, J = 5.2 Hz, 1H), 6.90 (dd, J = 2.0, 8.6 Hz, 1H), 6.87 (d, J = 1.9 Hz, 1H), 6.03–6.09 (m, 1H), 5.81–5.78 (m, 1H), 4.52–4.46 (m, 1H), 3.68 (t, J = 6.0 Hz, 2H), 2.90 (s, 3H), 2.67 (t, J = 5.8 Hz, 2H), 2.24–2.18 (m, 2H)
9 − Allyl − 1 − methyl − 9H − pyrido[3,4 − b]indol − 7 − yl 5 − bromopentanoate (7)
Light yellow solid; 85%;1H NMR (600 MHz, Chloroform−d) δ: 8.27 (d, J = 5.2 Hz, 1H), 7.99 (d, J = 8.6 Hz, 1H), 7.76 (d, J = 5.1 Hz, 1H), 6.96 (dd, J = 2.0, 8.6 Hz, 1H), 6.91 (d, J = 2.0 Hz, 1H), 6.01 − 6.07 (m, 1H), 5.78 − 5.74 (m, 2H), 4.54 − 4.48 (m, 2H), 3.66 (t, J = 5.8 Hz, 2H), 2.88 (s, 3H), 2.65 (t, J = 5.8 Hz, 2H), 2.26–2.20 (m, 2H), 1.94–1.88 (m, 2H).
9 − Allyl − 1 − methyl − 9H − pyrido[3,4 − b]indol − 7 − yl 6 − bromohexanoate (8)
Light yellow solid; 89%; 1H NMR (600 MHz, Chloroform−d) δ: 8.27 (d, J = 5.2 Hz, 1H), 7.99 (d, J = 8.6 Hz, 1H), 7.75 (d, J = 5.2 Hz, 1H), 6.96 (dd, J = 2.0, 8.6 Hz, 1H), 6.90 (d, J = 2.0 Hz, 1H), 6.02 − 6.09 (m, 1H), 5.78 − 5.73 (m, 2H), 4.50 − 4.46 (m, 2H), 3.61 (t, J = 5.8 Hz, 2H), 2.90 (s, 3H), 2.63 (t, J = 5.6 Hz, 2H), 2.26–2.20 (m, 2H), 1.96–1.80 (m, 2H), 1.49 − 1.45 (m, 2H).
9 − Allyl − 1 − methyl − 9H − pyrido[3,4 − b]indol − 7 − yl 7 − bromoheptanoate (9)
Light yellow solid; 84%;1H NMR (600 MHz, Chloroform−d) δ: 8.28 (d, J = 5.2 Hz, 1H), 7.99 (d, J = 8.6 Hz, 1H), 7.75 (d, J = 5.2 Hz, 1H), 6.96 (dd, J = 2.0, 8.6 Hz, 1H), 6.90 (d, J = 2.0 Hz, 1H), 6.09–6.02 (m, 1H), 5.83–5.79 (m, 2H), 4.48–4.44 (m, 2H), 3.68 (t, J = 5.6 Hz, 2H), 2.88 (s, 3H), 2.66 (t, J = 5.6 Hz, 2H), 2.28–2.22 (m, 2H), 1.97–1.83 (m, 2H), 1.51 − 1.47 (m, 2H), 1.44 − 1.38 (m, 1H).
General procedure for the Preparation of compounds 11 − 17 l
A solution containing compounds 5 − 9 (1 mmol), K2CO3 (280 mg, 2 mmol), and benzylpiperazine derivatives (1.5 mmol) in acetonitrile was subjected to reflux with stirring. Following the disappearance of compounds 5 − 9, as detected by TLC, the solvents were evaporated under reduced pressure. Distilled water was then added and the resulting mixture underwent chloroform extraction. The organic solvent phase was subsequently washed with water and brine before being dried over anhydrous Na2SO4 and evaporated under vacuum. Compounds 11 − 17 l were purified using flash chromatography on silica gel.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 3–(4-benzylpiperazin-1-yl)propanoate (11)
White solid; 51%; 1H NMR (600 MHz, Chloroform−d) δ: 8.25 (d, J = 5.2 Hz, 1H), 7.95 (d, J = 8.5 Hz, 1H), 7.70 (d, J = 5.1 Hz, 1H), 7.36–7.30 (m, 4H), 7.10–7.04 (m, 1H), 6.93 (dd, J = 2.0, 8.5 Hz, 1H), 6.86 (d, J = 2.0 Hz, 1H), 6.10–6.03 (m, 1H), 5.80–5.74 (m, 2H), 4.45–4.40 (m, 2H), 4.11 (t, J = 5.8 Hz, 2H), 3.65 (s, 2H), 3.03 (t, J = 5.8 Hz, 2H), 2.85 (s, 3H), 2.70–2.51 (m, 8H); 13C NMR (150 MHz, Chloroform−d) δ: 176.3, 158.2, 143.2, 142.1, 139.0, 136.9, 135.3, 129.3, 128.8 (2 C), 127.9 (2 C), 127.3, 126.4, 125.1, 121.3, 116.3, 115.2, 109.4, 107.2, 62.5, 54.5 (2 C), 53.7 (2 C), 52.1, 48.4, 31.8, 15.6; HRMS (ESI): Calcd. for C29H33N4O2 [M + H]+ 469.2559, found 469.2567.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 4–(4-benzylpiperazin-1-yl)butanoate (12)
White solid; 59%; 1H NMR (600 MHz, Chloroform−d) δ: 8.26 (d, J = 5.2 Hz, 1H), 7.96 (d, J = 8.5 Hz, 1H), 7.71 (d, J = 5.1 Hz, 1H), 7.38–7.32 (m, 4H), 7.12–7.06 (m, 1H), 6.98 (dd, J = 2.0, 8.5 Hz, 1H), 6.87 (d, J = 2.0 Hz, 1H), 6.12–6.05 (m, 1H), 5.81–5.76 (m, 2H), 4.47–4.42 (m, 2H), 3.68 (s, 2H), 3.36 (t, J = 6.0 Hz, 2H), 2.88 (s, 3H), 2.79 (t, J = 5.8 Hz, 2H), 2.68–2.49 (m, 8H), 2.20–2.16 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 176.8, 158.5, 143.5, 142.7, 139.2, 137.3, 135.1, 129.5, 128.9 (2 C), 128.1 (2 C), 127.5, 126.7, 125.3, 121.0, 116.5, 115.6, 109.7, 107.0, 62.6, 58.4, 55.4 (2 C), 54.6 (2 C), 48.1, 29.2, 22.1, 15.8; HRMS (ESI): Calcd. for C30H35N4O2 [M + H]+ 483.2715, found 483.2740.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-benzylpiperazin-1-yl)pentanoate (13)
White solid; 65%; 1H NMR (600 MHz, Chloroform−d) δ: 8.30 (d, J = 5.2 Hz, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.75 (d, J = 5.2 Hz, 1H), 7.40–7.34 (m, 4H), 7.28–7.23 (m, 1H), 7.03 (dd, J = 2.0, 8.5 Hz, 1H), 6.92 (d, J = 2.0 Hz, 1H), 6.10–6.05 (m, 1H), 5.86–5.79 (m, 2H), 4.50–4.45 (m, 2H), 3.70 (s, 2H), 3.40 (t, J = 6.0 Hz, 2H), 2.90 (s, 3H), 2.82 (t, J = 5.8 Hz, 2H), 2.74–2.55 (m, 8H), 2.16–2.12 (m, 2H), 2.04–1.99 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 178.2, 159.4, 144.7, 143.2, 139.9, 138.6, 136.2, 130.2, 129.4 (2 C), 128.5 (2 C), 128.1, 127.3, 125.9, 121.8, 117.3, 116.5, 110.5, 107.6, 63.0, 56.2, 55.8 (2 C), 55.0 (2 C), 48.8, 31.2, 24.1, 20.8, 16.4; HRMS (ESI): Calcd. for C31H37N4O2 [M + H]+ 497.2872, found 497.2898.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 6–(4-benzylpiperazin-1-yl)hexanoate (14)
White solid; 73%; 1H NMR (600 MHz, Chloroform−d) δ: 8.25 (d, J = 5.2 Hz, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.69 (d, J = 5.2 Hz, 1H), 7.36–7.30 (m, 4H), 7.24–7.18 (m, 1H), 6.96 (dd, J = 2.0, 8.5 Hz, 1H), 6.87 (d, J = 2.0 Hz, 1H), 6.06–6.01 (m, 1H), 5.82–5.76 (m, 2H), 4.48–4.42 (m, 2H), 3.66 (s, 2H), 3.35 (t, J = 6.0 Hz, 2H), 2.86 (s, 3H), 2.78 (t, J = 5.8 Hz, 2H), 2.72–2.53 (m, 8H), 2.14–2.08 (m, 2H), 1.96–1.88 (m, 4H); 13C NMR (150 MHz, Chloroform−d) δ: 177.8, 159.1, 144.3, 142.9,139.6, 138.6, 136.0, 129.8, 129.1 (2 C), 128.3 (2 C), 127.8, 127.1, 125.6, 121.5, 117.1, 116.2, 110.1, 107.3, 62.8, 56.3, 55.4 (2 C), 54.6 (2 C), 48.6, 31.1, 24.0, 22.5, 21.2, 16.1; HRMS (ESI): Calcd. for C32H39N4O2 [M + H]+ 511.3028, found 511.3062.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 7–(4-benzylpiperazin-1-yl)heptanoate (15)
White solid; 80%; 1H NMR (600 MHz, Chloroform−d) δ: 8.28 (d, J = 5.2 Hz, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.76 (d, J = 5.2 Hz, 1H), 7.38–7.33 (m, 4H), 7.26–7.20 (m, 1H), 6.92 (dd, J = 2.0, 8.5 Hz, 1H), 6.86 (d, J = 2.0 Hz, 1H), 6.08–6.02 (m, 1H), 5.83–5.78 (m, 2H), 4.48–4.44 (m, 2H), 3.68 (s, 2H), 3.36 (t, J = 6.0 Hz, 2H), 2.90 (s, 3H), 2.74 (t, J = 5.8 Hz, 2H), 2.70–2.48 (m, 8H), 2.16–2.11 (m, 2H), 1.98–1.87 (m, 6H); 13C NMR (150 MHz, Chloroform−d) δ: 178.6, 159.6, 144.8, 143.5, 140.1, 138.9, 136.3, 130.2, 129.5 (2 C), 128.8 (2 C), 128.2, 127.4, 125.8, 121.6, 117.5, 116.8, 110.6, 107.5, 62.9, 56.6, 55.6 (2 C), 54.7 (2 C), 48.9, 31.5, 24.7, 23.9, 22.9, 20.8, 16.5; HRMS (ESI): Calcd. for C33H41N4O2 [M + H]+ 525.3185, found 525.3216.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(4-methylbenzyl)piperazin-1-yl)pentanoate (17a)
White solid; 68%; 1H NMR (600 MHz, Chloroform−d) δ: 8.29 (d, J = 5.2 Hz, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.74 (d, J = 5.2 Hz, 1H), 7.28 (d, J = 8.0 Hz, 2H), 7.16 (d, J = 8.0 Hz, 2H), 7.01 (dd, J = 2.0, 8.5 Hz, 1H), 6.91 (d, J = 2.0 Hz, 1H), 6.10–6.04 (m, 1H), 5.85–5.78 (m, 2H), 4.50–4.44 (m, 2H), 3.70 (s, 2H), 3.38 (t, J = 6.0 Hz, 2H), 2.90 (s, 3H), 2.82 (t, J = 5.8 Hz, 2H), 2.76–2.57 (m, 8H), 2.27 (s, 3H), 2.16–2.12 (m, 2H), 2.01–1.93 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 178.4, 159.6, 144.9, 143.4, 140.1, 136.5, 130.8, 135.4, 132.7, 129.8 (2 C), 128.6 (2 C), 127.6, 126.1, 122.2, 117.6, 116.7, 110.8, 107.8, 63.1, 56.7, 55.8 (2 C), 54.8 (2 C), 49.3, 31.4, 24.3, 23.0, 20.5, 16.7; HRMS (ESI): Calcd. for C32H39N4O2 [M + H]+ 511.3028, found 511.3054.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(3-methylbenzyl)piperazin-1-yl)pentanoate (17b)
White solid; 72%; 1H NMR (600 MHz, Chloroform−d) δ: 8.30 (d, J = 5.2 Hz, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.75 (d, J = 5.2 Hz, 1H), 7.32 (d, J = 7.5 Hz, 1H), 7.20–7.14 (m, 2H), 7.09 (d, J = 7.4 Hz, 1H), 7.03 (dd, J = 2.0, 8.5 Hz, 1H), 6.94 (d, J = 2.0 Hz, 1H), 6.12–6.06 (m, 1H), 5.88–5.82 (m, 2H), 4.53–4.47 (m, 2H), 3.73 (s, 2H), 3.40 (t, J = 6.0 Hz, 2H), 2.90 (s, 3H), 2.82 (t, J = 5.8 Hz, 2H), 2.74–2.54 (m, 8H), 2.35 (s, 3H), 2.18–2.12 (m, 2H), 2.04–1.99 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 178.0, 159.4, 144.7, 143.2,139.7, 138.2, 136.5, 135.1, 130.5, 129.2, 127.4, 127.0, 126.1, 125.0, 123.8, 122.0, 117.3, 116.5, 110.5, 107.6, 63.6, 56.4, 55.6 (2 C), 54.5 (2 C), 49.1, 31.2, 24.1, 22.8, 21.2, 16.4; HRMS (ESI): Calcd. for C32H39N4O2 [M + H]+ 511.3028, found 511.3046.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(2-methylbenzyl)piperazin-1-yl)pentanoate (17c)
White solid; 63%; 1H NMR (600 MHz, Chloroform−d) δ: 8.28 (d, J = 5.2 Hz, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.73 (d, J = 5.2 Hz, 1H), 7.27 (d, J = 7.2 Hz, 1H), 7.20–7.14 (m, 2H), 7.09 (d, J = 7.4 Hz, 1H), 7.02 (dd, J = 2.0, 8.5 Hz, 1H), 6.90 (d, J = 2.0 Hz, 1H), 6.09–6.03 (m, 1H), 5.84–5.78 (m, 2H), 4.42–4.38 (m, 2H), 3.68 (s, 2H), 3.36 (t, J = 6.0 Hz, 2H), 2.88 (s, 3H), 2.80 (t, J = 5.8 Hz, 2H), 2.74–2.54 (m, 8H), 2.39 (s, 3H), 2.14–2.08 (m, 2H), 2.00–1.94 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 179.2, 160.6, 145.6, 144.2, 140.9, 137.5, 137.1, 136.2, 131.6, 130.8, 129.5, 128.4, 127.6, 126.8, 124.8, 122.9, 118.3, 117.6, 111.3, 108.4, 63.7, 57.2, 56.2 (2 C), 55.4 (2 C), 49.8, 31.9, 24.8, 23.6, 17.6, 17.1. HRMS (ESI): Calcd. for C32H39N4O2 [M + H]+ 511.3028, found 511.3047.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(4-fluorobenzyl)piperazin-1-yl)pentanoate (17d)
White solid; 76%;1H NMR (600 MHz, Chloroform−d) δ: 8.32 (d, J = 5.2 Hz, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.76 (d, J = 5.2 Hz, 1H), 7.29 (dd, J = 8.6, 5.5 Hz, 2H), 7.12 (t, J = 8.6 Hz, 2H), 7.04 (dd, J = 2.0, 8.5 Hz, 1H), 6.93 (d, J = 2.0 Hz, 1H), 6.12–6.08 (m, 1H), 5.89–5.83 (m, 2H), 4.55–4.49 (m, 2H), 3.72 (s, 2H), 3.40 (t, J = 6.0 Hz, 2H), 2.91 (s, 3H), 2.85 (t, J = 5.8 Hz, 2H), 2.78–2.2.53 (m, 8H), 2.20–2.14 (m, 2H), 2.07–2.01 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 176.8, 162.1 (d, J = 245.0 Hz), 158.2, 143.2, 142.0, 138.5, 135.1, 131.8, 130.1 (2 C), 129.5, 126.2, 125.5, 120.2, 117.9 (2 C), 116.1, 115.0, 108.2, 105.3, 61.8, 57.3, 55.2 (2 C), 53.4 (2 C), 48.3, 30.9, 22.8, 20.5, 15.9; HRMS (ESI): Calcd. for C31H36FN4O2 [M + H]+ 515.2778, found 515.2793.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(3-fluorobenzyl)piperazin-1-yl)pentanoate (17e)
White solid; 65%;1H NMR (600 MHz, Chloroform−d) δ: 8.29 (d, J = 5.2 Hz, 1H), 7.98 (d, J = 8.5 Hz, 1H), 7.73 (d, J = 5.2 Hz, 1H), 7.31–7.26 (m, 1H), 7.16–7.09 (m, 2H), 7.01 (dd, J = 2.0, 8.5 Hz, 1H), 6.92 − 6.88 (m, 2H), 6.10–6.06 (m, 1H), 5.86–5.80 (m, 2H), 4.52–4.46 (m, 2H), 3.70 (s, 2H), 3.38 (t, J = 6.0 Hz, 2H), 2.89 (s, 3H), 2.82 (t, J = 5.8 Hz, 2H), 2.76–2.57 (m, 8H), 2.22–2.16(m, 2H), 2.05–1.98 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 178.6, 162.9 (d, J = 245.6 Hz), 159.8, 145.0, 143.7,139.8, 136.5, 135.3, 131.3, 127.8, 127.1, 124.8, 122.4, 121.8, 118.0, 117.6, 116.5, 112.5, 109.7, 106.8, 62.6, 57.7, 55.9 (2 C), 54.9 (2 C), 49.6, 32.4, 24.6, 21.9, 17.2; HRMS (ESI): Calcd. for C31H36FN4O2 [M + H]+ 515.2778, found 515.2805.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(2-fluorobenzyl)piperazin-1-yl)pentanoate (17f)
White solid; 79%;1H NMR (600 MHz, Chloroform−d) δ: 8.30 (d, J = 5.2 Hz, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.74 (d, J = 5.2 Hz, 1H), 7.60 (d, J = 8.8 Hz, 1H), 7.42 (td, J = 7.5, 1.8 Hz, 1H), 7.15 (td, J = 7.5, 1.2 Hz, 1H), 7.12–7.06 (m, 1H), 7.02 (dd, J = 2.0, 8.5 Hz, 1H), 6.91 (d, J = 2.0 Hz, 1H), 6.10–6.05 (m, 1H), 5.87–5.80 (m, 2H), 4.53–4.48 (m, 2H), 3.70 (s, 2H), 3.38 (t, J = 6.0 Hz, 2H), 2.80 (s, 3H), 2.83 (t, J = 5.8 Hz, 2H), 2.76–2.57 (m, 8H), 2.18–2.12 (m, 2H), 2.02–1.96 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 178.0, 162.6 (d, J = 246.2 Hz), 159.6, 144.4, 143.2, 139.0, 136.3, 130.7, 129.6, 127.3, 126.6, 126.1, 124.6, 123.2, 121.4, 117.3, 116.1, 110.9, 109.3, 106.4, 62.9, 58.1, 56.3 (2 C), 54.5 (2 C), 49.5, 32.0, 22.8, 21.6, 16.8; HRMS (ESI): Calcd. for C31H36FN4O2 [M + H]+ 515.2778, found 515.2814.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(4-chlorobenzyl)piperazin-1-yl)pentanoate (17 g)
White solid; 80%; 1H NMR (600 MHz, Chloroform−d) δ: 8.28 (d, J = 5.2 Hz, 1H), 7.98 (d, J = 8.5 Hz, 1H), 7.72 (d, J = 5.2 Hz, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 6.99 (dd, J = 2.0, 8.5 Hz, 1H), 6.89 (d, J = 2.0 Hz, 1H), 6.08–6.04 (m, 1H), 5.85–5.80 (m, 2H), 4.51–4.45 (m, 2H), 3.68 (s, 2H), 3.36 (t, J = 6.0 Hz, 2H), 2.86 (s, 3H), 2.81 (t, J = 5.8 Hz, 2H), 2.78–2.59 (m, 8H), 2.20–2.14 (m, 2H), 2.05–1.98 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 177.6, 159.3, 144.5, 143.1, 139.3, 136.2, 135.9, 131.5, 130.1, 129.5 (2 C), 127.5, 126.3 (2 C), 126.3, 121.1, 117.0, 116.3, 109.5, 106.4, 62.1, 56.9 (2 C), 56.1, 54.6 (2 C), 48.9, 31.2, 24.0, 21.2, 16.3; HRMS (ESI): Calcd. for C31H36ClN4O2 [M + H]+ 531.2482, found 531.2506.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(3-chlorobenzyl)piperazin-1-yl)pentanoate (17h)
White solid; 73%; 1H NMR (600 MHz, Chloroform−d) δ: 8.24 (d, J = 5.2 Hz, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.68 (d, J = 5.2 Hz, 1H), 7.58 (s, 1H), 7.30–7.26 (m, 2H), 7.14 − 7.08 (m, 1H), 6.94 (dd, J = 2.0, 8.5 Hz, 1H), 6.85 (d, J = 2.0 Hz, 1H), 6.04–5.98 (m, 1H), 5.80–5.74 (m, 2H), 4.48–4.42 (m, 2H), 3.64 (s, 2H), 3.32 (t, J = 6.0 Hz, 2H), 2.82 (s, 3H), 2.76 (t, J = 5.8 Hz, 2H), 2.72–2.53 (m, 8H), 2.17–2.11 (m, 2H), 2.00–1.94 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 178.4, 159.9, 145.3, 143.8, 140.5, 137.1, 136.9, 133.6, 131.8, 130.7, 128.2, 127.1, 126.4, 125.8, 124.3, 121.8, 117.8, 117.0, 110.3, 107.2, 61.7, 57.1 (2 C), 56.8, 55.3 (2 C), 49.2, 31.7, 24.3, 22.1, 16.8; HRMS (ESI): Calcd. for C31H36ClN4O2 [M + H]+ 531.2482, found 531.2506.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(2-chlorobenzyl)piperazin-1-yl)pentanoate (17i)
White solid; 61%; 1H NMR (600 MHz, Chloroform−d) δ: 8.28 (d, J = 5.2 Hz, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.74 (d, J = 5.2 Hz, 1H), 7.42 (d, J = 7.5, 1H), 7.26–7.23 (m, 1H), 7.22 − 7.18 (m, 2H), 6.99 (dd, J = 2.0, 8.5 Hz, 1H), 6.89 (d, J = 2.0 Hz, 1H), 6.08–6.04 (m, 1H), 5.85–5.80 (m, 2H), 4.52–4.46 (m, 2H), 3.66 (s, 2H), 3.35 (t, J = 6.0 Hz, 2H), 2.85 (s, 3H), 2.80 (t, J = 5.8 Hz, 2H), 2.74–2.55 (m, 8H), 2.19–2.15 (m, 2H), 2.03–1.98 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 179.2, 160.6, 145.9, 144.6, 141.2, 137.9, 137.3, 134.3, 131.4, 130.5, 128.8, 128.2, 127.9, 127.2, 125.9, 122.4, 118.5, 117.6, 110.9, 107.8, 62.1, 58.6 (2 C), 57.2, 55.8 (2 C), 49.7, 32.5, 25.0, 22.7, 17.3; HRMS (ESI): Calcd. for C31H36ClN4O2 [M + H]+ 531.2482, found 531.2523.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)pentanoate (17j)
White solid; 82%; 1H NMR (600 MHz, Chloroform−d) δ: 8.30 (d, J = 5.2 Hz, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.74 (d, J = 5.2 Hz, 1H), 7.62–7.56 (m, J = 7.6 Hz, 2H), 7.18 (d, J = 7.6 Hz, 2H), 7.01 (dd, J = 2.0, 8.5 Hz, 1H), 6.92 (d, J = 2.0 Hz, 1H), 6.10–6.06 (m, 1H), 5.88–5.82 (m, 2H), 4.53–4.48 (m, 2H), 3.69 (s, 2H), 3.38 (t, J = 6.0 Hz, 2H), 2.88 (s, 3H), 2.84 (t, J = 5.8 Hz, 2H), 2.76–2.57 (m, 8H), 2.22–2.18 (m, 2H), 2.12–2.04 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 178.3, 159.8, 145.1, 143.7, 139.9, 138.6, 136.8, 130.7, 128.8 (2 C), 128.2, 126.9, 125.7, 124.6 (2 C), 121.8 (q, J = 229 Hz), 121.1, 117.6, 116.8, 110.2, 107.1, 62.7, 57.6 (2 C), 56.8, 55.1(2 C), 49.5, 31.5, 24.6, 21.7, 16.8; HRMS (ESI): Calcd. for C32H36F3N4O2 [M + H]+ 565.2746, found 565.2772.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(3-(trifluoromethyl)benzyl)piperazin-1-yl)pentanoate (17k)
White solid; 73%; 1H NMR (600 MHz, Chloroform−d) δ: 8.34 (d, J = 5.2 Hz, 1H), 8.03 (d, J = 8.5 Hz, 1H), 7.78 (d, J = 5.2 Hz, 1H), 7.64 (s, 1H), 7.48 (dd, J = 14.0, 7.7 Hz, 2H), 7.35 (t, J = 7.7 Hz, 1H), 7.05 (dd, J = 2.0, 8.5 Hz, 1H), 6.96 (d, J = 2.0 Hz, 1H), 6.12–6.08 (m, 1H), 5.94–5.88 (m, 2H), 4.58–4.52 (m, 2H), 3.74 (s, 2H), 3.42 (t, J = 6.0 Hz, 2H), 2.93 (s, 3H), 2.88 (t, J = 5.8 Hz, 2H), 2.82–2.63 (m, 8H), 2.23–2.18 (m, 2H), 2.14–2.06 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 176.2, 157.5, 142.8, 141.4, 137.6, 135.7, 134.2, 131.5, 128.6, 127.2, 125.8, 125.2, 124.5, 123.2, 120.7 (q, J = 231.9 Hz), 119.5, 118.6, 115.4, 114.5, 108.1, 106.7, 61.9, 56.6 (2 C), 55.2, 53.4 (2 C), 47.4, 30.2, 22.8, 20.5, 15.2; HRMS (ESI): Calcd. for C32H36F3N4O2 [M + H]+ 565.2746, found 565.2786.
9-allyl-1-methyl-9H-pyrido[3,4-b]indol-7-yl 5–(4-(2-(trifluoromethyl)benzyl)piperazin-1-yl)pentanoate (17 l)
White solid; 54%; 1H NMR (600 MHz, Chloroform−d) δ: 8.31 (d, J = 5.2 Hz, 1H), 8.00 (d, J = 8.5 Hz, 1H), 7.75 (d, J = 5.2 Hz, 1H), 7.61 (t, J = 7.9 Hz, 2H), 7.50 (t, J = 7.5 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 7.02 (dd, J = 2.0, 8.5 Hz, 1H), 6.92 (d, J = 2.0 Hz, 1H), 6.09–6.05 (m, 1H), 5.91–5.84 (m, 2H), 4.55–4.49 (m, 2H), 3.70 (s, 2H), 3.39 (t, J = 6.0 Hz, 2H), 2.90 (s, 3H), 2.85 (t, J = 5.8 Hz, 2H), 2.79–2.60 (m, 8H), 2.21–2.14 (m, 2H), 2.01–1.97 (m, 2H); 13C NMR (150 MHz, Chloroform−d) δ: 178.6, 159.8, 145.1, 143.5, 139.9, 136.5, 130.9, 130.4, 128.6, 128.1, 127.3, 126.7, 125.8, 125.1, 123.5, 122.6, 121.2 (q, J = 227.5 Hz), 117.7, 116.8, 110.3, 109.1, 63.1, 58.2 (2 C), 57.5, 54.8 (2 C), 49.1, 32.1, 24.7, 21.9, 17.3; HRMS (ESI): Calcd. for C32H36F3N4O2 [M + H]+ 565.2746, found 565.2771.
In vitro inhibition studies on AChE and BuChE
The Ellman’s assay was employed to evaluate the inhibitory potential of novel harmine derivatives against hAChE and hBuChE. 50 μL of hAChE (0.02 unit/mL) or hBChE (0.02 unit/mL) were incubated with 10 μL of the compound in 96 − well plates at 37 °C for 6 min. Subsequently, 30 μL of a 0.01 M substrate solution acetylthiocholine iodide (ATCI) or butyrylthiocholine iodide (BTCI) was added and the mixture was further incubated at 37 °C for an additional 12 min. Finally, the activity was measured by adding 150 μL of a 5,5′−dithiobis(2 − nitrobenzoic acid) (DTNB) solution (0.01 M) and measuring absorbance at a wavelength of 415 nm using an Evolution 300 PC UV − Vis Spectrophotometer. At least five different concentrations ranging from1 × 1 0 −5 to10−9 M were tested for each compound.
Aβ1 − 42 self − aggregation inhibition assay
The inhibition of Aβ1 − 42 self − aggregation was assessed by means of the thioflavin T (Th-T) fluorescence assay. 1,1,1,3,3,3 − hexafluoro − 2 − propanol (HFIP) pre-treated Aβ1 − 42 samples were dissolved in PBS (pH 7.4) to obtain a solution of 40 mM concentration. Meanwhile, Th-T solution was prepared in PBS (pH 7.4) at a concentration of 10 μM. Subsequently, each well containing different concentrations of test compounds received an addition of 10 μL of the aforementioned Aβ1 − 42 solution with a concentration of 40 μM. After incubation for 24 h at a temperature of 30 °C, the samples were diluted to a final volume of 200 μL using Th-T solution. Finally, a 300 s time scan was conducted to measure fluorescence intensity (λexc = 446 nm; λem = 490 nm). The plateau values were obtained by subtracting the background fluorescence of the thioflavin T solution and then averaged.
ADMET study
The ADMETlab 2.0 (https://admetmesh.scbdd.com)Citation34 was utilised to predict molecular properties, drug − likeness and ADME properties. The prediction of small molecule toxicity was conducted through ProTox − II (http://tox.charite.de/protox_II)Citation35, a webserver dedicated to this purpose.
Inhibition of AChE induced Aβ1 − 42 aggregation assay
Inhibition of AChE − induced aggregation of Aβ1 − 42 peptide was achieved through co − incubation with synthesised compounds (at concentrations of 0.1 μM and 1 μM) and AChE (at a concentration of 10 μM). Control experiments were conducted in the absence of test compounds. Aggregation of Aβ1 − 42 peptide was monitored using Th-T at 37 °C for a period of 24 h, with excitation wavelength set at 446 nm and emission ranging between 490 nm.
Assessment of in vitro cytotoxicity
The human neuroblastoma SH − SY5Y cell line (RRID: CVCL_0019) with the catalogue number of CL-0208, was obtained from Procell Life Science & Technology Co., Ltd. (Wuhan, China). All of the other chemicals and reagents were from Macklin Biochemical Co., Ltd (Shanghai, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 50 U/mL penicillin, and 50 mg/mL streptomycin at a temperature of 37 °C under a humidified atmosphere containing 5% CO2.
The cytotoxicity of the compounds was evaluated using an MTT assay as previously described. Briefly, SH − SY5Y cells were seeded at a density of 1 × 104 per well in 96 − well plates and incubated at 37 °C for 24 h. Subsequently, cells were treated with 100 μL of compounds 13 and 17d at various concentrations (ranging from 0.1 to 100 μM) for a duration of 48 h. The final concentration of DMSO in the culture media did not exceed 0.05% (v/v), and no cellular alterations were observed. Each experiment should include a control group consisting of complete medium without any cells. After incubation, the medium was aspirated and washed with phosphate-buffered saline (PBS), followed by addition of 50 ml of MTT solution. The plates were then incubated for two hours at a temperature of 37 °C in a 5% CO2 environment. Subsequently, the MTT solution was discarded and replaced with 100 ml of dimethyl sulfoxide (DMSO). The plates were further incubated for 10 min at room temperature before adding 5 ml of Sorensen Buffer. Absorbance at a wavelength of 570 nm was measured using a microplate reader. The cell viability was quantified as a percentage relative to the control values (blank control). The experiments were performed in triplicate.
Protection of SH − SY5Y cells against damage induced by Aβ1 − 42
SH − SY5Y cells were seeded at a density of 1 × 104 per well in 96 − well plates and cultured at 37 °C for 24 h. Aβ1 − 42 (20 μM) was diluted with medium and added to individual wells, along with test compounds 13, and 17d (at concentrations of 1 μM, 5 μM, and 10 μM). The plates were then incubated for an additional period of 48 h at the same temperature. Cell viability was assessed using the MTT assay protocol and expressed as a percentage relative to control cells.
Kinetic profiling of AChE inhibition
To elucidate the mechanism of AChE inhibition by synthesised compounds, varying concentrations (1 − 10 nM) of compound 13 were subjected to different concentrations of substrate (AChE) ranging from 0.1 to 0.5 mM. The resulting Lineweaver − Burk plot allowed for determination of Vm and Km values pertaining to the inhibition.
Molecular docking
Docking was conducted using the Surflex − Dock program within Sybyl − X 2.0 Software, while the 3D structures of ligands were sketched in the Sybyl package. Atom types were verified, hydrogen atoms were appended, and Gasteiger − Marsili charges were assigned with Sybyl − X 2.0 Software. The protein structures of hAChE (PDB code: 4EY7) and Aβ1 − 42 (PDB code: 1IYT) were obtained from the RCSB Protein Data Bank (https://www.rcsb.org/). To facilitate molecular docking studies, the ligand was extracted from the crystal structure, hydrogen atoms were added, water molecules were removed, and side − chain amides were verified. Subsequently, a protomold was generated for further analysis. PyMOL and PoseView software tools were utilised to visualise the results of docking.
Author contribution
H.D. and Y.Y. were responsible for overseeing the research project. F. M. and J. S. synthesised the compounds under investigation. X. H. helped in conducting the structural analysis of the compounds. X.Z., H.G. and R.M. conducted biological activity tests to evaluate their effectiveness. The manuscript was written by H.D. and Y.Y., with contributions from all authors who have given their approval to the final version of the manuscript.
Supplemental Material
Download PDF (1.8 MB)Acknowledgement
The authors express our gratitude to Dr. Qiuju Zhou (Analysis and Testing Center, Xinyang Normal University) for her invaluable assistance in conducting compound structure analyses.
Disclosure statement
The authors report there are no competing interests to declare.
Additional information
Funding
References
- Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314(5800):777–781.
- Mendiola–Precoma J, Berumen LC, Padilla K, Garcia–Alcocer G. Therapies for prevention and treatment of Alzheimer’s disease. Biomed Res Int. 2016; 2016:2589276.
- Alzheimer’s Disease International. World Alzheimer Report. 2016. https://www.alz.co.uk/research/WorldAlzheimerReport2016.pdf.
- Tan CC, Yu JT, Wang HF, Tan MS, Meng XF, Wang C, Jiang T, Zhu XC, Tan L. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: a systematic review and meta–analysis. J Alzheimers Dis. 2014;41(2):615–631.
- Dal–Re R. Approval of aducanumab for Alzheimer’s disease in the United States: the surrender of science. Rev Neurologia. 2021; 73(8):296–297.
- Larkin HDD. Lecanemab gains FDA approval for early Alzheimer disease. Jama–J Am Med Associ. 2023;329(5):363.
- Zissimopoulos J, Jacobson M, Chen Y, Borson S. Knowledge and attitudes concerning aducanumab among older americans after FDA approval for treatment of Alzheimer disease. JAMA Netw Open. 2022;5(2):e2148355.
- Lythgoe MP, Jenei K, Prasad V. Regulatory decisions diverge over aducanumab for Alzheimer’s disease. BMJ. 2022; 376(8326):e069780.
- Bartus RT, Dean RL, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217(4558):408–414.
- Selkoe DJ. Alzheimer’s disease: Genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766.
- Querfurth HW, LaFerla FM. MECHANISMS OF DISEASE Alzheimer’s Disease. N Engl J Med. 2010;362(4):329–344. Review.
- Nasb M, Tao W, Chen N. Alzheimer’s disease puzzle: delving into pathogenesis hypotheses. Aging Dis. 2023;15(1):0608. doi:10.14336/AD.2023.0608.
- Savelieff MG, Nam G, Kang J, Lee HJ, Lee M, Lim MH. Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis in the last decade. Chem Rev. 2019;119(2):1221–1322.
- Ramesh M, Govindaraju T. Multipronged diagnostic and therapeutic strategies for Alzheimer’s disease. Chem Sci. 2022;13(46):13657–13689.
- Sang Z, Wang K, Dong J, Tang L. Alzheimer’s disease: Updated multi–targets therapeutics are in clinical and in progress. Eur J Med Chem. 2022;238:114464.
- Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, Fowler C, Li Q-X, Martins R, Rowe C, et al. High performance plasma amyloid–beta biomarkers for Alzheimer’s disease. Nature. 2018;554(7691):249–254.
- John A, Reddy PH. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P–tau and mitochondria. Ageing Res Rev. 2021;65:101208.
- Jeremic D, Jimenez–Diaz L, Navarro–Lopez JD. Past, present and future of therapeutic strategies against amyloid–beta peptides in Alzheimer’s disease: a systematic review. Ageing Res Rev. 2021;72:101496. doi:10.1016/j.arr.2021.101496.
- Zhang Y, Chen H, Li R, Sterling K, Song W. Amyloid beta–based therapy for Alzheimer’s disease: challenges, successes and future. Sig Transduct Target Ther. 2023;8(1):248–248.
- Marucci G, Buccioni M, Dal Ben D, Lambertucci C, Volpini R, Amenta F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology. 2021;190:108352.
- Tonelli M, Catto M, Sabaté R, Francesconi V, Laurini E, Pricl S, Pisani L, Miniero DV, Liuzzi GM, Gatta E, et al. Thioxanthenone–based derivatives as multitarget therapeutic leads for Alzheimer’s disease. Eur J Med Chem. 2023;250:115169.
- Ahmed S, Khan ST, Zargaham MK, Khan AU, Khan S, Hussain A, Uddin J, Khan A, Al –, Harrasi A. Potential therapeutic natural products against Alzheimer’s disease with reference of acetylcholinesterase. Biomed Pharmacother. 2021;139:111609. doi:10.1016/j.biopha.2021.111609.
- Patel K, Gadewar M, Tripathi R, Prasad SK, Patel DK. A review on medicinal importance, pharmacological activity and bioanalytical aspects of beta–carboline alkaloid ''Harmine. Asian Pac J Trop Biomed. 2012;2(8):660–664.
- Zhang L, Li D, Yu S. Pharmacological effects of harmine and its derivatives: a review. Arch Pharm Res. 2020;43(12):1259–1275.
- Brito–da–Costa AM, Dias–da–Silva D, Gomes NGM, Dinis–Oliveira RJ, Madureira–Carvalho A. Toxicokinetics and toxicodynamics of ayahuasca alkaloids N,N–dimethyltryptamine (DMT), harmine, harmaline and tetrahydroharmine: Clinical and forensic impact. Pharmaceuticals. 2020;13(11):334.
- Javeed M, Rasul A, Hussain G, Jabeen F, Rasool B, Shafiq N, Riaz A, Kaukab G, Ali M. Harmine and its derivatives: Biological activities and therapeutic potential in human diseases. Bangladesh J Pharmacol. 2018;13(3):203–213.
- Beato A, Gori A, Boucherle B, Peuchmaur M, Haudecoeur R. Beta–carboline as a privileged scaffold for multitarget strategies in Alzheimer’s disease therapy. J Med Chem. 2021;64(3):1392–1422.
- Mennenga SE, Gerson JE, Dunckley T, Bimonte–Nelson HA. Harmine treatment enhances short–term memory in old rats: Dissociation of cognition and the ability to perform the procedural requirements of maze testing. Physiol Behav. 2015;138:260–265.
- He D, Wu H, Wei Y, Liu W, Huang F, Shi H, Zhang B, Wu X, Wang C. Effects of harmine, an acetylcholinesterase inhibitor, on spatial learning and memory of APP/PS1 transgenic mice and scopolamine–induced memory impairment mice. Eur J Pharmacol. 2015;768:96–107.
- Li S-P, Wang Y-W, Qi S-L, Zhang Y-P, Deng G, Ding W-Z, Ma C, Lin Q-Y, Guan H-D, Liu W, et al. Analogous beta–carboline alkaloids harmaline and harmine ameliorate scopolamine–induced cognition dysfunction by attenuating acetylcholinesterase activity, oxidative stress, and inflammation in mice. Front Pharmacol. 2018;9:346.
- Iraji A, Firuzi O, Khoshneviszadeh M, Tavakkoli M, Mahdavi M, Nadri H, Edraki N, Miri R. Multifunctional iminochromene–2H–carboxamide derivatives containing different aminomethylene triazole with BACE1 inhibitory, neuroprotective and metal chelating properties targeting Alzheimer’s disease. Eur J Med Chem. 2017;141:690–702.
- Zhao YF, Ye F, Xu J, Liao QH, Chen L, Zhang WJ, Sun HP, Liu WY, Feng F, Qu W. Design, synthesis and evaluation of novel bivalent β -carboline derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg Med Chem. 2018;26(13):3812–3824.
- Lamie PF, Abdel-Fattah MM, Philoppes JN. Design and synthesis of new indole drug candidates to treat Alzheimer’s disease and targeting neuro-inflammation using a multi-target-directed ligand (MTDL) strategy. J Enzyme Inhib Med Chem. 2022;37(1):2660–2678.
- Xiong GL, Wu ZX, Yi JC, Fu L, Yang ZJ, Hsieh CY, Yin MZ, Zeng XX, Wu CK, Chen X, et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49(W1):W5–W14.
- Banerjee P, Eckert OA, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–W263.
- Liu Y, Uras G, Onuwaje I, Li W, Yao H, Xu S, Li X, Li X, Phillips J, Allen S, et al. Novel inhibitors of AChE and Aβ aggregation with neuroprotective properties as lead compounds for the treatment of Alzheimer’s disease. Eur J Med Chem. 2022;235:114305.
- Vaaland IC, López Ó, Puerta A, Fernandes MX, Padrón JM, Fernández-Bolaños JG, Sydnes MO, Lindbäck E. Investigation of the enantioselectivity of acetylcholinesterase and butyrylcholinesterase upon inhibition by tacrine-iminosugar heterodimers. J Enzyme Inhib Med Chem. 2023;38(1):349–360.