
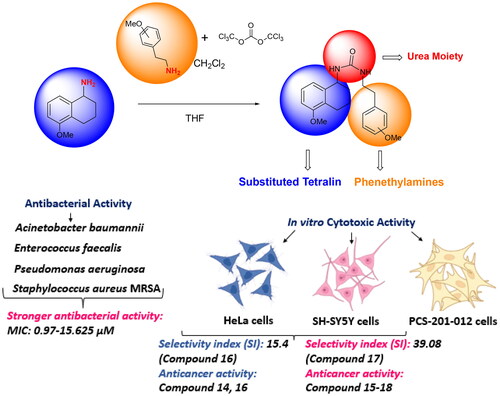
Abstract
Cancer and antibiotic-resistant bacterial infections are significant global health challenges. The resistance developed in cancer treatments intensifies therapeutic difficulties. In addressing these challenges, this study synthesised a series of N,N′-dialkyl urea derivatives containing methoxy substituents on phenethylamines. Using isocyanate for the efficient synthesis yielded target products 14–18 in 73–76% returns. Subsequently, their antibacterial and anticancer potentials were assessed. Cytotoxicity tests on cancer cell lines, bacterial strains, and a healthy fibroblast line revealed promising outcomes. All derivatives demonstrated robust antibacterial activity, with MIC values ranging from 0.97 to 15.82 µM. Notably, compounds 14 and 16 were particularly effective against the HeLa cell line, while compounds 14, 15, and 17 showed significant activity against the SH-SY5Y cell line. Importantly, these compounds had reduced toxicity to healthy fibroblast cells than to cancer cells, suggesting their potential as dual-functioning agents targeting both cancer and bacterial infections.
Graphical Abstract
Introduction
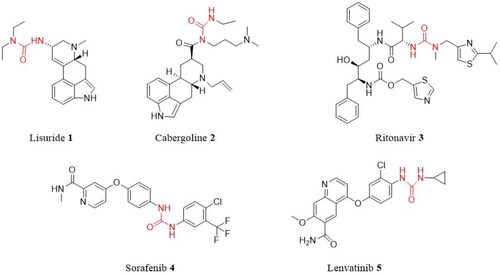
Urea derivatives are a large class of biologically active compounds, and molecules with the urea functional group are increasingly used in medicinal chemistry and drug development. Many studies have demonstrated that urea derivatives have a wide variety of biological activities, including antimalarialCitation1, antiviralCitation2, anti-cancerCitation3–5, anti-tubercularCitation5, and anti-microbialCitation6,Citation7. Also, extensive biological activity studies on urea compounds have led to the development of some urea-derived drugs. For instance, the urea medication lisuride (1), also known as dopargine, is a serotonin 5-HT2B receptor antagonistCitation7. Cabergoline (2) is used for the treatment of hyperprolactinaemia and Parkinson’s diseaseCitation8,Citation9. Ritonavir (3) (trade name Norvir) is an antiretroviral drug used as an HIV protease inhibitorCitation9. Sorafenib (4) (trade name Nexavar) is a kinase inhibitor drug used to treat thyroid, kidney, and advanced primary liver cancerCitation10. The anti-cancer medication lenvatinib (5) (brand name Lenvima) is used to treat thyroid cancer ()Citation11.
Figure 1. Examples of commercially available urea drugs 1–5.
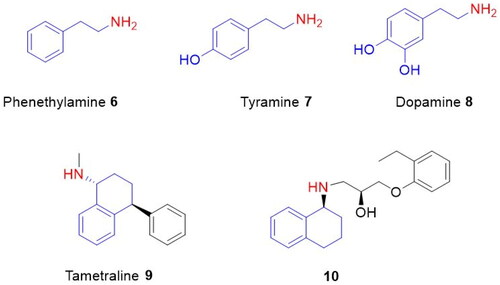
On the other hand, 2-phenylethylamines and 1-aminotetralins represent crucial building blocks for synthetic organic chemistry and medicinal organic compounds.
For example, phenylethylamine (6) is an endogenous trace amine that acts as neuromodulation in the central nervous system of mammalsCitation12. Tyramine (7) is a biogenic amine found in nature and formed from tyrosine by enzymatic decarboxylationCitation13. Dopamine (8), a hormone-like compound, has a very important role as a neurotransmitter in the brain and bodyCitation14,Citation15. Tametraline (9) is a norepinephrine-dopamine reuptake inhibitorCitation16. Aminotetralin 10 is used as a common β3-adrenoceptor antagonist ()Citation15.
Figure 2. Phenethylamine 6, tyramine 7, dopamine 8, tametraline 9, and 1-aminotetralin derivative 10.
In a previous study, we synthesised ureas with a skeletal structure consisting of 6- and 7-methoxy-1-aminotetralins and methoxy-substituted phenethylamines and investigated their antibacterial, anticancer, and toxicity properties. We have seen that some compounds can be used as anticancer and antibacterial agentsCitation17.
As can be seen from the information given above, ureas, phenethylamines, and 1-aminotetralins are important biologically active compounds. The presence of these three biologically active groups in one molecule and the high antibacterial and anticancer activity of similar molecules in our previous study led us to this study. Therefore, as an experience from our previous study, we aimed to synthesise a series of N,N-dialkyl ureas containing the urea functional group, 5-methoxy-1-aminotetralin, and substituted phenethylamines in their skeletal structures to investigate their biological properties, such as antibacterial and anticancer activities.
Materials and methods
The synthesis of 5-methoxy-1,2,3,4-tetrahydronaphthalen-1-amine (13)
5-Methoxy-1-aminotetralin 13Citation18 was synthesised as described previously.
General procedure for the synthesis of N,N′-dialkyl ureas (14–18)
A solution of triphosgene (1 mmol) in CH2Cl2 (15 ml) was cooled to 0 °C. Then, 2-phenethylamine 11(a–e) (3 mmol) dissolved in CH2Cl2 (5 ml) was added to it in a drop-wise manner. Following this, a NaOH solution (3 mmol) in 7 ml of water was introduced. The mixture was stirred at ambient temperature for 1 h post-addition. The aqueous layer was then separated. The remaining organic layer was rinsed with water three times, each with 20 ml, and dried using magnesium sulphate. Evaporating the filtrate yielded the corresponding isocyanate, which was used as-is without any additional purification. This isocyanate 12(a–e) (1 mmol) was either dissolved or suspended in dry THF (10 ml) and cooled using an ice bath. Separately, 1-aminotetralin 13 (1 mmol) was dissolved in dry THF (5 ml) and then gradually added to the earlier mixture. The combined mixture was stirred at room temperature for 6 h. After this, the solvent was removed by evaporation. The final products, N,N′-dialkyl ureas 14–18, were obtained through recrystallisation using a solvent mix of EtOAc/hexane at a ratio of 3:1.
1-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)-3-phenethylurea (14)
The product 14 was obtained as a white powder, yield: 73%, mp 141–143 °C. IR spectrum, ѵ, cm−1: 3319, 2937, 2368, 1616, 1558, 1467, 1249, 1016, and 762 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.33–7.14 (m, 5H, ArH), 7.12 (t, J = 7.8 Hz, 1H, ArH), 6.92 (d, J = 7.8 Hz, 1H), 6.69 (d, J = 8.0 Hz, 1H, ArH), 4.91–4.83 (m, 1H, CH-N), 4.62 (br.d, J = 8.4 Hz, 1H, NH), 4.45 (t, J = 5.4 Hz, 1H, NH), 3.79 (s, 3H, OCH3), 3.41 (dd, J = 12.9, 6.6 Hz, 2H, CH2), 2.79 (t, J = 6.9 Hz, 2H, CH2), 2.69–2.50 (m, 2H, CH2), 1.96–1.84 (m, 1H, CH2), 1.83–1.66 (m, 3H, CH2). 13C NMR (101 MHz, CDCl3) δ 157.63 (CO), 156.94 (C), 139.22 (C), 138.93 (C), 128.84 (2CH), 128.56 (2CH), 126.50 (C), 126.37 (CH), 126.35 (CH), 120.74 (CH), 108.00 (CH), 55.30 (OCH3), 48.25 (CH), 41.70 (CH2), 36.41 (CH2), 30.26 (CH2), 22.96 (CH2), 19.29 (CH2). Analysis calculated for C20H24N2O2: C 74.05; H 7.46; N 8.63. Found, %: C 74.15; H 7.49; N 8.59. M: 336.39.
1-(5-Methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-methoxyphenethyl)urea (15)
The product 15 was obtained as a white powder, yield: 76%, mp 174–176 °C. IR spectrum, ѵ, cm−1: 3331, 3287, 2928, 2853, 1614, 1574, 1468, 1263, 1242, 1084, 1035, and 745 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.22–7.18 (m, 1H, ArH), 7.15–7.11 (m, 2H, ArH), 6.96 (d, J = 7.8 Hz, 1H, ArH), 6.88 (t, J = 7.3 Hz, 1H, ArH), 6.82 (d, J = 8.2 Hz, 1H, ArH), 6.71 (d, J = 8.0 Hz, 1H, ArH), 4.99–4.90 (m, 1H, CH-N), 4.68 (br.s, 1H, NH), 4.52 (br.s, 1H, NH), 3.81 (s, 3H, OCH3), 3.66 (s, 3H, OCH3), 3.35 (t, J = 6.6 Hz, 2H), 2.87–2.80 (m, 2H), 2.75–2.50 (m, 2H), 1.98–1.92 (m, 1H), and 1.82–1.74 (m, 3H).13C NMR (101 MHz, CDCl3) δ 157.69 (CO), 157.49 (C), 156.99 (C), 138.93 (C), 130.65 (CH), 127.84 (CH), 127.27 (C), 126.64 (C), 126.40 (CH), 120.87 (CH), 120.67 (CH), 110.36 (CH), 108.04 (CH), 55.33 (OCH3), 55.10 (OCH3), 48.33 (CH), 40.82 (CH2), 31.27 (CH2), 30.31 (CH2), 23.02 (CH2), and 19.27 (CH2). Analysis calculated for C21H26N2O3: C 71.16; H 7,39; N 7.90. Found, %: C 71.28; H 7.42; N 7.92. M: 354.45.
1-(5-Methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(3-methoxyphenethyl)urea (16)
The product 16 was obtained as a white powder, yield: 74%, mp 155–157 °C. IR spectrum, ѵ, cm−1: 3308, 2938, 2857, 1616, 1578, 1512, 1468, 1260, 1163, 1045, 849, and 779 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.21 (t, J = 7.7 Hz, 1H, ArH), 7.14 (t, J = 8.0 Hz, 1H, ArH), 6.94 (d, J = 7.9 Hz, 1H, ArH), 6.82–6.68 (m, 4H, ArH), 4.98–4.88 (m, 1H, CH-N), 4.42 (br.s, 2H, NH), 3.81 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.46 (t, J = 6.8 Hz, 2H, CH2), 2.81 (t, J = 6.8 Hz, 2H, CH2), 2.70–2.52 (m, 2H, CH2), 2.01–1.89 (m, 1H, CH2), and 1.78 (d, J = 5.8 Hz, 3H, CH2). 13C NMR (101 MHz, CDCl3) δ 159.76 (C), 157.67 (CO), 156.93 (C), 140.85 (C), 138.93 (C), 129.50 (CH), 126.47 (C), 126.31 (CH), 121.13 (CH), 120.72 (CH), 114.45 (CH), 111.79 (CH), 108.01 (CH), 55.26 (OCH3), 55.12 (OCH3), 48.23 (CH), 41.56 (CH2), 36.45 (CH2), 30.27 (CH2), 22.94 (CH2), and 19.27 (CH2). Analysis calculated for C21H26N2O3: C 71.16; H 7.39; N 7.90. Found, %: C 71.22; H 7.41; N 7.88. M: 354.45.
1-(3,4-Dimethoxyphenethyl)-3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)urea (17)
The product 17 was obtained as a white powder, yield: 76%, mp 168–170 °C. IR spectrum, ѵ, cm−1: 3346, 3310, 2928, 2351, 1732, 1616, 1587, 1516, 1259, 1140, 1029, and 794 cm–1; 1H NMR (400 MHz, CDCl3) δ 6.99 (t, J = 7.9 Hz, 1H, ArH), 6.81 (d, J = 7.7 Hz, 1H, ArH), 6.70–6.62 (m, 1H, ArH), 6.62–6.53 (m, 3H, ArH), 4.88 (d, J = 6.5 Hz, 1H, CH-N), 4.76 (br. d, J = 3.0 Hz, 2H, NH), 3.72 (s, 6H, OCH3), 3.69 (s, 3H, OCH3), 3.25 (t, J = 6.5 Hz, 2H, CH2), 2.60 (t, J = 6.9 Hz, 2H, CH2), 2.54–2.44 (m, 2H, CH2), 1.86–1.73 (m, 1H, CH2), and 1.72–1.53 (m, 3H, CH2). 13C NMR (101 MHz, CDCl3) δ 157.79 (CO), 156.92 (C), 148.91 (C), 147.50 (C), 138.99 (C), 131.82 (C), 126.43 (CH), 126.27 (CH), 120.69 (CH), 120.65 (CH), 112.00 (CH), 111.31 (CH), 107.96 (CH), 55.84 (OCH3), 55.75 (OCH3), 55.25 (OCH3), 48.16 (CH), 41.76 (CH2), 36.01 (CH2), 30.30 (CH2), 22.94 (CH2), and 19.28 (CH2). Analysis calculated for C22H28N2O4: C 68.73; H 7.34; N 7.29. Found, %: C 68.75; H 7.36; N 7.32. M: 384.48.
1-(5-Methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(4-methoxyphenethyl)urea (18)
The product 18 was obtained as a white powder, yield: 75%, mp 150–152 °C. IR spectrum, ѵ, cm−1: 3296, 2935, 2323, 1614, 1568, 1513, 1467, 1250, 1083, 1039, and 821 cm–1; 1H NMR (400 MHz, CDCl3) δ 7.18–7.05 (m, 3H, ArH), 6.93 (d, J = 7.8 Hz, 1H, ArH), 6.83 (d, J = 8.4 Hz, 2H, ArH), 6.70 (d, J = 8.0 Hz, 1H, ArH), 4.97–4.89 (m, 1H, CH-N), 4.48 (br.s, 1H, NH), 4.30 (br.s, 1H, NH), 3.80 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.40 (t, J = 6.7 Hz, 2H, CH2), 2.75 (t, J = 6.8 Hz, 2H, CH2), 2.71–2.52 (m, 2H, CH2), 1.99–1.88 (m, 1H, CH2), and 1.84–1.70 (m, 3H, CH2). 13C NMR (101 MHz, CDCl3) δ 158.19 (C), 157.56 (CO), 156.96 (C), 138.87 (C), 131.11(C), 129.76 (2CH), 126.53 (C), 126.36 (CH), 120.72 (CH), 113.99 (2CH), 108.04 (CH), 55.31 (OCH3), 55.26 (OCH3), 48.28 (CH), 41.93 (CH2), 35.44 (CH2), 30.24 (CH2), 22.95 (CH2), and 19.26 (CH2). Analysis calculated for C21H26N2O3: C 71.16; H 7.39; N 7.90. Found, %: C 71.20; H 7.42; N 7.87. M: 354.45.
Bacterial strains
The bacteria employed for evaluation included Acinetobacter baumannii ATCC 1605, Enterococcus faecalis ATCC 49452, Pseudomonas aeruginosa ATCC 9027, and Staphylococcus aureus MRSA ATCC 43300. These strains were sourced from the Molecular Biology and Genetics Laboratory at Erzurum Technical University. They were immersed in Mueller-Hinton Broth (MHB) and then incubated at a temperature of 37 °C for a duration of 16–24 h.
Disc diffusion method
Bacteria were adjusted to a McFarland standardisation of 0.5, equivalent to a cell density of 1.5 × 108/ml, and were then introduced into MHB. Subsequently, a 100 μl volume of this bacterial mixture was spread onto Mueller-Hinton agar plates. Sterile empty discs were then treated with the formulated ureas at concentrations of 1000, 500, and 250 µM. Oflaxin (10 g/disc) and netilmicin (30 g/disc) were used as positive controls, while DMSO served as the negative control. The plates were then placed in an incubator at 37 °C for a period ranging from 24 to 72 h, depending on the optimal conditions. Post incubation, the diameter of the inhibition zones was gauged to determine the antibacterial efficacy. This entire process was replicated three timesCitation19.
Micro-well dilution assay
The synthesised ureas’ minimum inhibitory concentrations (MICs) against the bacterial samples were ascertained using the microdilution broth technique. Utilising a method of twofold serial dilutions, the ureas were diluted to concentrations spanning from 500 to 0.48 M. The bacterial turbidity was set to match the 0.5 McFarland standardCitation20. In the 96-well plates, each well received 100 μl of MHB and 5 μl of the bacteria being tested. For the negative control, 195 μl of MHB and 5 μl of the bacterial sample were added. The plate was then covered with a sterile seal and allowed to incubate for 24 h at a temperature of 37 °C. Post incubation, the growth of the bacteria was assessed at 600 nm using the Fluoroskan Ascent® FL. The MIC represented the smallest amount of the chemical that effectively halted bacterial growth. This procedure was conducted three timesCitation19.
Cell line and culture conditions
The cells in culture were frequently sub-cultured, about 2–3 times weekly, until they reached a confluence of 80–90%. The experiments were organised into seven cell groups as follows: group I served as the control, while groups II through VII were pre-treated with varying dosages of the synthesised ureas (ranging from 12.5 µM to 500 µM). The cytotoxic potential of these ureas was assessed in vitro using the Cell Viability Detection Kit-8 (CVDK-8, Ecotech Biotechnology, Erzurum, Turkey), a method based on WST-8 quantification for human primary dermal fibroblasts and cervical cancer cellsCitation21. Healthy fibroblast, HeLa, and SH-SY5Y cells were allocated in 96-well plates, with each well containing 5 × 103 cells in 100 µL of DMEM:F12 (supplemented with 1% penicillin/streptomycin and 10% FBS). After an overnight growth, these cells were exposed to specified concentrations of the synthesised ureas for a duration of 48 h at 37 °C in a 5% CO2 atmosphere. Cisplatin acted as the positive control with concentration gradients of 0–250 µM for HeLa cells and 0–20 µM for the SH-SY5Y cells. Post incubation, in sterile, dimly lit conditions, 10% WST-8 solution was added to every well and further incubated at 37 °C within 5% CO2 for another 3–4 h. The optical density of the 96-well plate was gauged at a 450 nm wavelength using a spectrophotometer (BioTek, EPOCH, Santa Clara, CA). The control group’s main purpose was to measure the absorbance of just the cells and the growth medium.
Selectivity index (SI)
This research denotes the selectivity level of the created compounds as follows: SI is calculated by dividing the IC50 value of the unadulterated compound in the standard cell line by the IC50 value of the same compound in the cancerous cell line. Here, IC50 represents the concentration required to eradicate 50% of the cell communityCitation22.
Statistical analysis
In analytical statistics, all of the experiments were carried out in triplets. All analyses were carried out using the GraphPad Prism program (GraphPad Software, La Jolla, CA), and the findings were given as mean ± standard deviation.
Results
In the initial phase of the synthesis, phenethylamines, which are a class of organic compounds that contain a phenyl ring attached to an amino group, were designated as 11a–e based on variations in their molecular structures or substituents. These were subjected to a reaction with triphosgene. Triphosgene is an organic compound often used as a more convenient substitute for phosgene, as it releases phosgene upon heating. In the context of this reaction, triphosgene serves as a reagent to convert the phenethylamine derivatives 11a–e into isocyanate intermediates. Isocyanates are functional groups containing the –N═C═O moiety. The resulting isocyanate intermediates were labelled as compounds 12a–e. Without undergoing any further purification, these isocyanate intermediates (12a–e) were immediately employed in the subsequent stage of the synthesis. 5-Methoxy-1-aminotetralin, which is labelled as compound 13, possesses an amine group (–NH2). This compound was reacted with the aforementioned isocyanate intermediates 12a–e. The choice of solvent for this reaction was tetrahydrofuran (THF), which is a polar aprotic solvent commonly used in organic synthesis. The reactions were carried out under specific temperature conditions, ranging from 0 °C to 25 °C. Under these conditions, the amine group from the 5-methoxy-1-aminotetralin 13 undergoes a nucleophilic attack on the electrophilic carbon of the isocyanate group, leading to the formation of N,N′-dialkyl ureas. Ureas are compounds with the functional group –NH–CO–NH–, and in this case, they are dialkylated, implying the presence of two alkyl groups attached to the nitrogen atoms. The resultant novel N,N′-dialkyl ureas were denoted as compounds 14–18. The overall efficiency of this synthesis is commendable, with high yields reported between 73% and 76% (Scheme 1).
Scheme 1. Synthesis of substituted tetrahydronaphthalen-1-yl-phenethyl ureas 14–18.
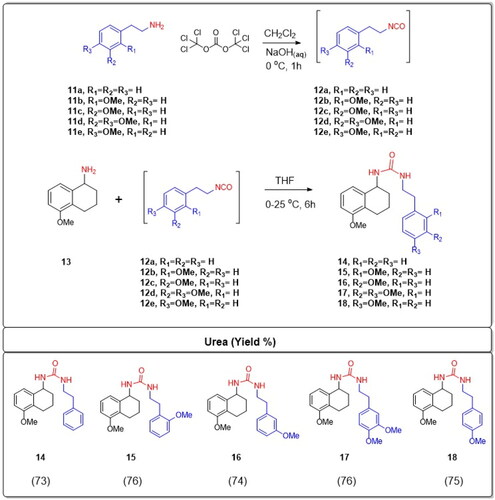
All the synthesised compounds had their chemical structures defined using elemental analysis, IR, 1H NMR, and 13C NMR. In the 1H NMR spectrum for N,N′-dialkyl ureas labelled 14–18, the protons related to the HN(CO)NH showed either as a broad singlet or broad doublet within the range of 4.76–4.30 ppm. For the same ureas, the OCH3 proton signals were observed as singlets between 3.81 and 3.66 ppm. Meanwhile, in the 13C NMR spectrum for these ureas, the –CO– signals were detected between 157.75 and 157.56 ppm, while the OCH3 signals ranged from 55.84 to 55.10 ppm.
The antibacterial properties of the formulated ureas, labelled 14–18, were evaluated against various microorganisms. This included two types of Gram-negative bacteria (namely Acinetobacter baumannii and Pseudomonas aeruginosa) and two strains of Gram-positive bacteria (specifically Enterococcus faecalis and Staphylococcus aureus MRSA) using the disc diffusion method. The outcomes of the antibacterial activity, along with the MIC values for these ureas, can be found in .
Table 1. Antibacterial activity of the synthesised ureas 14–18.
The WST-8 test was conducted on SH-SY5Y, HeLa, and PCS-201-012 cells after being treated with increasing concentrations of the produced compounds for 48 h to determine their cytotoxic effects. In this study, we used two different chemotherapeutic drugs as controls. One of them is cisplatin (cis-diammineplatinum(II) dichloride), which is a widely used antineoplastic drug for clinical cures and was used as a positive control (IC50 value for SH-SY5Y cell line: 10 µMCitation23, for HeLa cell line: >50 µMCitation24, and for fibroblast cells: >100 µMCitation25). Another control is melphalan, which is also a chemotherapy drug. It is used as a treatment for several different cancer types. The IC50 value for the HeLa cell is 61 µMCitation26, while it is 2.2 µMCitation27 for the SH-SY5Y cell. The synthesised ureas 14–18 were treated to SH-SY5Y, HeLa, and PCS-201-012 cells for 48 h at concentrations of 12.5, 25, 50, 100, 300, and 500 M. shows the IC50 values for several compounds.
Table 2. Cytotoxic activity (IC50, μM) of the synthesised ureas (n = 4).
Different concentrations of ureas (from 12.5 to 500 µM) were applied to SH-SY5Y, HeLa, and PCS-201-012 cells for 48 h. As shown in , SH-SY5Y cells exposed to compound 14 at concentrations of 12.5, 25, 50, 100, 300, and 500 µM showed the lowest cell viability rates of 25.48, 8.40, 4.68, 4.87, 3.77, and 1.67%, respectively. In addition, cell viability varies (58.3, 58, 31.1, 21.7, 6.4, and 6.4%) when HeLa cell lines were treated with newly synthesised ureas at concentrations of 12.5, 25, 50, 100, 300, and 500 µM ().
Figure 3. SH-SY5Y cells were exposed to produced ureas (12.5–500 M) for 48 h, and cytotoxicity was assessed using the WST-8 assay. GraphPad Prism 7.00 (GraphPad Software, La Jolla, CA) was used for statistical analysis, and ANOVA: Dunnett’s multiple comparison test calculated the results. *p < 0.01 when compared to the control group.
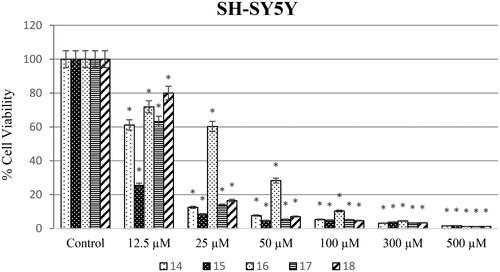
Figure 4. HeLa cells were exposed to produced ureas (12.5–500 M) for 48 h, and cytotoxicity was assessed using the WST-8 assay. GraphPad Prism 7.00 (GraphPad Software, La Jolla, CA) was used for statistical analysis, and ANOVA: Dunnett’s multiple comparison test calculated the results. *p < 0.01 when compared to the control group.
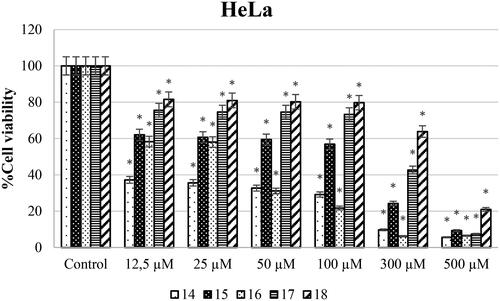
Anticarcinogenic analyses of the synthesised molecules on the SH-SY5Y cell line showed that compound 15 has the highest anticancer potential compared to other molecules. On the other hand, compound 16 was investigated to have the lowest anticarcinogenic property against the neuroblastoma cell line (). Besides, anticancer potential of synthesised molecules was found higher against the SH-SY5Y cell line compared to the HeLa cell line for all of the compounds. In parallel to the literature, it was shown in different studies that same anticarcinogen could exhibit diverse cytotoxic efficiency against different cancer cell typesCitation28,Citation29. Drug formulations should be chosen and modified according to tumour type and the molecular structures should be decided according to the target cell line.
Also, when examining the IC50 values of the synthesised compounds against the HeLa cell line, urea derivatives 14 and 16 had the best anticancer activity. However, urea derivative 18 had lower anticancer activity against the HeLa cell line. In our previous study, we reported that while urea derivative containing 7-methoxy-1-aminotetralin has the best activity with has 58.9 µM IC50 value against HeLa cell lineCitation17. On the other hand, we have shown that our compounds 14 (2.6 µM) and 16 (32.4 µM) have IC50 values against HeLa cell line in this study. When looking at this aspect, we can say that compounds containing 5-methoxy-1-aminotetralin have the best activity against cancer cell lines. In addition, Özgeris reported that urea derivatives containing substituted phenethylamine’s structure showed strong anticancer activity against HeLa cell linesCitation30, which supports our study.
On the other hand, the cytotoxic effect of compounds 14–18 against PCS201-012 cell lines was also assessed (). According to the results, compound 15 (IC50 on PCS201-012: 225.7 µM) was up to twofold more potent than cisplatin against the HeLa cell line while less toxic than cisplatin (IC50 on PCS201-012: 100 µM) against the PCS201-012 cell line. Also, compound 16's anticancer activity exhibited a low toxic effect against healthy cell lines compared to cisplatin treatment (IC50 on PCS201-012: 100 µM).
Figure 5. Percentage of cytotoxicity evaluated using the WST-8 test on PCS-201-012 cells after 48 h of exposure to produced ureas 14–18 (12.5–500 M). GraphPad Prism 7.00 (GraphPad Software, La Jolla, CA) was used for statistical analysis, and ANOVA: Dunnett’s multiple comparison test calculated the results. *p < 0.01 when compared to the control group.
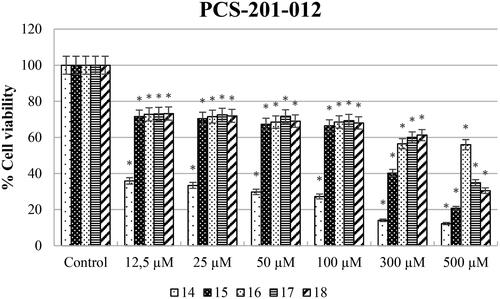
In addition, we can say that all synthesised compounds except compound 14 have fewer toxic effects on healthy cell lines when compared to cisplatin ().
Selectivity index value was calculated by dividing the IC50 value of non-cancerous human dermal fibroblast cells with the IC50 value of the cancer cell lines ().
Table 3. SI values of the synthesised molecules (n = 4).
According to the results, all compounds except 14 had at least 25-fold less cytotoxic effect for the SH-SY5Y cell line. The most effective compound is 18 with 36.72-fold. For the HeLa cell line, compound 14 up to threefold (SI: 3.04), compound 16 exhibited up to 15-fold less toxic effect (SI: 15.4). Suffness reported that derivatives exhibiting an SI value >2 were accepted as selectiveCitation31. This report supports that our results have the best toxicity profiles in the aspect of the SI. Therefore, all urea derivatives except for compound 14 can be considered as anticancer agents for SH-SY5Y and urea derivatives 14 and 16 can be considered against HeLa cancer.
Discussion
A number of experimental methodologies were developed to produce urea derivatives such as using phosgene or triphosgene functionalisationCitation32, addition of amine to isocyanateCitation32, 1,1-carbonyldiimidazole (CDI) couplingCitation33, carbonylimidazolium saltCitation34, dialkyl-based exchange processesCitation35, oxidative carbonylation of aminesCitation36, and derivatisation of chloroformateCitation37. Isocyanates play a pivotal role as precursor intermediates in the creation of biologically significant compounds. Alkyl isocyanates enable the straightforward production of unsymmetrical ureas. A predominant technique for isocyanate synthesis involves reacting amines with phosgene. Nonetheless, handling and storing phosgene poses challenges due to its toxic gaseous natureCitation38. Therefore, the use of phosgene in the laboratory is disadvantageous. Triphosgene (bis(trichloromethyl) carbonate) is easy to store and transport since it is a crystalline, stable solidCitation39. Therefore the most frequent alternative to phosgene for the production of isocyanates is triphosgeneCitation40. Hence, phenethylamines 11a–e were reacted with triphosgene to give isocyanate intermediates 12a–e. Compounds 12a–e were used in the next step without further purification. The reactions of 5-methoxy-1-aminotetralin 13 with isocyanates 12a–e in THF at 0–25 °C yielded novel N,N′-dialkyl ureas 14–18 in high yields (73–76%).
The antibacterial properties of the formulated ureas, particularly those labelled as 14–18, have shown significant potential in combating a range of bacterial pathogens. The Gram-negative bacteria, especially Acinetobacter baumannii and Pseudomonas aeruginosa, have historically been associated with various resistant infections, particularly in healthcare settingsCitation41. Similarly, the Gram-positive strains, Enterococcus faecalis and Staphylococcus aureus MRSA, are notorious for their virulence and multidrug resistance profilesCitation42. The evaluation of these ureas against such formidable bacterial adversaries using the disc diffusion method provides a comprehensive understanding of their potential applications in the therapeutic landscape. The inclusion of MIC values in further elucidates the potency of these compounds in inhibiting bacterial growth. With the global increase in antibiotic-resistant bacterial strains, the discovery and assessment of such compounds become invaluableCitation43.
Urea compound 14 does not have any modifications on its phenethylamine ring, while compound 18 is characterised by a methoxy group positioned at the para location on its phenethylamine ring. When evaluating their MIC values alongside their molecular structures, compound 18 stood out, showing values between 0.97 and 15.625 µM against all bacterial strains tested. On the other hand, urea compound 14 did not display any antibacterial properties against the studied bacteria. Interestingly, a prior study suggested that thiourea variants with methoxy groups on the phenethylamine ring often have diminished antibacterial capabilitiesCitation44. Contrary to that study, we found that urea derivatives with a methoxy group at the para position on the phenethylamine ring displayed significantly potent antibacterial properties. Furthermore, a recent study highlighted that thiourea compounds with a fluorine group in the same para position on the phenethylamine ring showed only moderate antibacterial effectsCitation30. Another study found that benzoyl thiourea compounds with a para methoxy aniline group have lower antibacterial activity (MIC: generally >1000 M) than standard drugsCitation45. As it is seen that, urea compounds having a methoxy group on the phenethylamine ring demonstrate strong antibacterial activity, while thiourea compounds containing a methoxy group on a similar structure demonstrate lower antibacterial activity. This may be explained by the presence of the CO group. The remaining urea compounds 15–18 showed strong to moderate antibacterial activity against all tested bacterial strains except compound 15.
The evaluation of ureas across varying concentrations provides essential insights into their potential as anticarcinogenic agents, particularly against specific cell lines. The SH-SY5Y cell line, which represents a model for neuroblastoma cells, demonstrated a significant reduction in cell viability upon exposure to compound 14, suggesting a potent cytotoxic effectCitation46. The viability rates observed in SH-SY5Y cells were substantially lower compared to those in the HeLa cell line when exposed to the same urea concentrations. This could hint at a targeted or selective cytotoxic action of these ureas towards neuroblastoma cells over cervical cancer cells (i.e. HeLa)Citation47. Interestingly, compound 15 stood out as the most potent anticarcinogenic molecule against SH-SY5Y, while compound 16 exhibited the least efficacy. Such variations in the anticancer potential of individual urea compounds might be attributed to differences in their chemical structures and how they interact with cellular mechanismsCitation48. The notable differential cytotoxic effects between the SH-SY5Y and HeLa cell lines underscore the importance of screening potential anticancer agents across diverse cell types to ascertain specificity and potency.
Cisplatin and melphalan are FDA-approved and are among the most crucial chemotherapy drugs used today for cancer treatment. For a drug to be approved by the FDA, it must meet specific safety criteria. Considering this information, we see that the effects of the newly synthesised urea derivatives on SH-SY5Y cells are generally in parallel with the IC50 value of cisplatin when compared with cisplatin. In addition, the effects of newly synthesised compounds on healthy cells should be examined to be predicted as a new candidate drug. For this, when the SI values are examined, it is seen that the others, except for compound number 14, have a very low cytotoxic effect against healthy cells. The IC50 values of cisplatin and melphalan used as controls were roughly the same on the HeLa cell line. Considering this value, it can be said that compounds 14 and 16, newly synthesised compounds, are more effective for HeLa.
Recent studies have converged on the therapeutic potential of urea and phenethylamine derivatives, positioning them as potent dual inhibitors of enzymes pivotal in the fight against cancer and bacterial infections. Urea derivatives have shown promising results as topoisomerase inhibitors, with compounds like benzimidazole urea derivatives emerging as dual inhibitors capable of targeting DNA topoisomerase II and IV, essential in cancer cell replication. Notably, the development of hexacyclic camptothecin analogues containing urea groups has highlighted their strong antitumor activities, challenging the efficacy of established chemotherapeutic agentsCitation49. Concurrently, tetrahydrobenzothieno[2,3-d]pyrimidine urea derivatives have demonstrated superior anticancer efficacy, offering new avenues for oncological drug developmentCitation50,Citation51. In parallel, phenethylamine derivatives, traditionally linked to neurotransmitter modulation, are being explored for their inhibitory action on DNA gyrase, an enzyme integral to bacterial DNA replication. Although research is in the early stages, the application of structure–activity relationship analysis and molecular docking suggests that phenethylamine derivatives could serve as a novel class of antimicrobial agentsCitation52,Citation53. This exploration is especially critical given the rise of antimicrobial resistance, which presents similar challenges to those of drug-resistant cancersCitation54,Citation55. Integrating insights from recent research, both urea and phenethylamine derivatives are emerging as multifaceted molecules with the potential to disrupt DNA processes critical to cancerous and bacterial cell proliferation, marking a significant stride in the development of new anticancer and antibacterial therapies. The convergence of anticancer and antibacterial properties within the same therapeutic agent is an area of significant interest in drug development. The synthesised molecules, which have been designed to possibly target critical enzymes involved in DNA replication and repair, showcase this duality of therapeutic action. These compounds likely owe their efficacy to a mechanism involving the inhibition of topoisomerase enzymes. By inhibiting these enzymes, our molecules induce DNA damage that ultimately leads to cell death in both bacterial and cancer cells. In bacterial cells, the synthesised molecules appear to target DNA gyrase and topoisomerase IV – key enzymes required for bacterial DNA replication. The inhibition of these enzymes prevents the supercoiling necessary for proper replication, leading to bacterial cell death. This mode of action is reminiscent of that utilised by fluoroquinolones, a well-established class of antibioticsCitation56. Conversely, in cancer cells, the same molecules exhibit the ability to interfere with topoisomerase II, a pivotal enzyme in DNA replication and repair in eukaryotic cells. The inhibition of topoisomerase II disrupts the replication process and can trigger apoptosis in rapidly dividing cancer cells, similar to the action of chemotherapeutic agents such as etoposideCitation57. Notably, this topoisomerase II inhibition characteristic has been evidenced in our molecular compounds, which have demonstrated significant anti-proliferative activity against a range of cancer cell lines. This dual inhibition underscores the potential versatility and wide-ranging applicability of our synthesised compounds. The anticancer and antibacterial activities of our synthesised molecules, stemming from their topoisomerase inhibition mechanism, present a unique therapeutic opportunity. They could potentially streamline treatment protocols by addressing both bacterial infections and cancerous growths concurrently, particularly in clinical scenarios where bacterial infections complicate cancer progression. Furthermore, these dual-action inhibitors may provide an alternative pathway to combat antibiotic resistance in bacteria and resistance to chemotherapy in cancer cells, a significant concern in both fieldsCitation58,Citation59. In essence, the dual pharmacological profiles of our molecules not only position them as promising candidates for further development but also underscore the importance of topoisomerase as a target for designing versatile and potent new drugs for complex diseasesCitation60,Citation61.
Conclusions
As a result, the resistance developed by cancer patients against chemotherapeutic drugs and the resistance developed by people against antibiotics have led scientists to find new drugs. It has been shown by previous studies that urea derivatives are the best candidates for this. Although the cytotoxic and antibacterial activities of many urea compounds have been investigated, the biological activities of our newly synthesised urea compounds against cancer cells or bacteria have not been investigated before. A series of substituted phenethylamine-based N,N′-dialkyl urea derivatives containing 1-aminotetralin and carrying methoxy substituents at various sites of phenethylamine were synthesised within the scope of this project. Synthesis of N,N′-dialkyl urea derivatives 14–18 was performed for the first time in good yields (73–76%). According to the antibacterial results, all tested compounds showed potent activity against all tested bacterial strains, except for compound 14 (MIC values: 0.97–15.82 µM). In anticancer activity against the HeLa cell line, urea derivatives 14 (IC50: 2.6 µM) and 16 (IC50: 32.4 µM) exhibited the best activity. In addition, the anticancer activities of the synthesised compounds showed a highly effective anticarcinogenic potential on the SH-SY5Y cell line. In particular, compound 15 exhibited the highest anticancer activity with an IC50 of 8.93 µM.
Moreover, the tested urea derivatives showed reasonable safety against the normal human dermal fibroblast cell line PCS201-012. On the other hand, it was observed that compounds 16, 17, and 18 were the most potent antimicrobial and anticarcinogenic candidate molecules. Also, among these compounds, molecule 16 has been shown to have the highest anticancer activity against the HeLa cell line, and compounds 17 and 18 exhibit the highest anticarcinogenic activity against the SH-SY5Y cell line. All newly synthesised compounds for the SH-SY5Y cell line were found to have the same cytotoxic effect as the average cisplatin compared to cisplatin used as a positive control. When the HeLa cell line is examined, it can be said that compounds 14 and 16 are more effective than cisplatin. Finally, looking at the cytotoxic effect on the healthy cell line, all seem to be less toxic than cisplatin, except for compound 14. Considering the results, it is predicted that the compounds can be used as anticancer and antibacterial drug candidate, but it needs to be developed with additional pharmacokinetic studies.
Author contributions
Conceptualisation, Y.A., A.M., and F.N.K.; methodology, Y.A. and F.N.K.; software, M.E.A.; validation, M.E.A., S.G., and H.T.; formal analysis, S.G.; investigation, H.T.; resources, A.M.; data curation, S.G.; writing – original draft preparation, Y.A. and M.E.A.; writing – review and editing, F.N.K. and Y.A.; visualisation, F.N.K.; supervision, H.T.; project administration, Y.A.; funding acquisition, S.G. All authors have read and agreed to the published version of the manuscript.
Acknowledgements
The authors would like to thank The Knut and Alice Wallenberg Foundation.
Disclosure statement
The authors declare no conflict of interest.
Data availability statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy.
Additional information
Funding
References
- Zhang Y, Anderson M, Weisman JL, Lu M, Choy CJ, Boyd VA, Price J, Sigal M, Clark J, Connelly M, et al. Evaluation of diarylureas for activity against Plasmodium falciparum. ACS Med Chem Lett. 2010;1(9):460–465.
- Paget CJ, Kisner K, Stone RL, DeLong DC. Heterocyclic substituted ureas. II. Immunosuppressive and antiviral activity of benzothiazolyl- and benzoxazolylureas. J Med Chem. 1969;12(6):1016–1018.
- Schroeder MC, Hamby JM, Connolly CJ, Grohar PJ, Winters RT, Barvian MR, Moore CW, Boushelle SL, Crean SM, Kraker AJ, et al. Soluble 2-substituted aminopyrido[2,3-d]pyrimidin-7-yl ureas. Structure–activity relationships against selected tyrosine kinases and exploration of in vitro and in vivo anticancer activity. J Med Chem. 2001;44(12):1915–1926.
- Wróbel TM, Kiełbus M, Kaczor AA, Kryštof V, Karczmarzyk Z, Wysocki W, Fruziński A, Król SK, Grabarska A, Stepulak A, et al. Discovery of nitroaryl urea derivatives with antiproliferative properties. J Enzyme Inhib Med Chem. 2016;31(4):608–618.
- North EJ, Scherman MS, Bruhn DF, Scarborough JS, Maddox MM, Jones V, Grzegorzewicz A, Yang L, Hess T, Morisseau C, et al. Design, synthesis and anti-tuberculosis activity of 1-adamantyl-3-heteroaryl ureas with improved in vitro pharmacokinetic properties. Bioorg Med Chem. 2013;21(9):2587–2599.
- Pandurangan K, Kitchen JA, Blasco S, Paradisi F, Gunnlaugsson T. Supramolecular pyridyl urea gels as soft matter with antibacterial properties against MRSA and/or E. coli. Chem Commun. 2014;50(74):10819–10822.
- Hofmann C, Penner U, Dorow R, Pertz HH, Jähnichen S, Horowski R, Latté KP, Palla D, Schurad B. Lisuride, a dopamine receptor agonist with 5-HT2B receptor antagonist properties: absence of cardiac valvulopathy adverse drug reaction reports supports the concept of a crucial role for 5-HT2B receptor agonism in cardiac valvular fibrosis. Clin Neuropharmacol. 2006;29(2):80–86.
- Reichmann H. Long-term treatment with dopamine agonists in idiopathic in Parkinson’s disease. J Neurol. 2000;247(4):IV17–IV19.
- Zeldin RK, Petruschke RA. Pharmacological and therapeutic properties of ritonavir-boosted protease inhibitor therapy in HIV-infected patients. J Antimicrob Chemother. 2004;53(1):4–9.
- Igwe KK, Ikpeazu OV, Otuokere IE, Department of Veterinary Biochemistry and Animal Production, Michael Okpara University of Agriculture, Umudike, Nigeria. Global energy and molecular interactions between pazopanib, axitinib and sorafenib anticancer drugs with vascular endothelial growth factor. J Biosci Biotechnol Discov. 2017;2(4):91–96.
- Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14(17):5459–5465.
- Sabelli HC, Mosnaim AD, Vazquez AJ, Giardina WJ, Borison RL, Pedemonte WA. Biochemical plasticity of synaptic transmission: a critical review of Dale’s principle. Biol Psychiatry. 1976;11(4):481–524.
- Finberg JPM, Gillman K. Selective inhibitors of monoamine oxidase type B and the “cheese effect”. Int Rev Neurobiol. 2011;100:169–190.
- Carlsson A. Biochemical and pharmacological aspects of Parkinsonism. Acta Neurol Scand Suppl. 1972;51:11–42.
- Bredikhina ZA, Kurenkov AV, Krivolapov DB, Bredikhin AA. Synthesis of all of the stereoisomers of β3-adrenoceptor antagonist SR 59230 based on the spontaneous resolution of 3-(2-ethylphenoxy) propane-1,2-diol. Tetrahedron Asymmetry. 2016;27(11–12):467–474.
- Welch WM, Kraska AR, Sarges R, Koe BK. Nontricyclic antidepressant agents derived from cis- and trans-1-amino-4-aryltetralins. J Med Chem. 1984;27(11):1508–1515.
- Akbaba Y, Kacı FN, Göksu S. Substituted tetrahydronaphthalen‐1‐yl‐phenethyl ureas: synthesis, characterization, and biological evaluations. ChemistrySelect. 2022;7(15):e202200450.
- Özgeriş B, Akbaba Y, Özdemir Ö, Türkez H, Göksu S. Synthesis and anticancer activity of novel ureas and sulfamides incorporating 1-aminotetralins. Arch Med Res. 2017;48(6):513–519.
- Gormez A, Bozari S, Yanmis D, Gulluce M, Sahin F, Agar G. Chemical composition and antibacterial activity of essential oils of two species of Lamiaceae against phytopathogenic bacteria. Pol J Microbiol. 2015;64(2):121–127.
- Wiegand I, Hilpert K, Hancock REW. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc. 2008;3(2):163–175.
- Ishiyama M, Miyazono Y, Sasamoto K, Ohkura Y, Ueno K. A highly water-soluble disulfonated tetrazolium salt as a chromogenic indicator for NADH as well as cell viability. Talanta. 1997;44(7):1299–1305.
- Koch A, Tamez P, Pezzuto J, Soejarto D. Evaluation of plants used for antimalarial treatment by the Maasai of Kenya. J Ethnopharmacol. 2005;101(1–3):95–99.
- Yang C, Tan J, Zhu J, Wang S, Wei G. YAP promotes tumorigenesis and cisplatin resistance in neuroblastoma. Oncotarget. 2017;8(23):37154–37163.
- Sasaki-Kudoh E, Kudo I, Kakizaki Y, Hosaka M, Ikeda S-I, Uemura S, Grave E, Togashi S, Sugawara T, Shimizu H, et al. Cisplatin inhibits AhR activation. Am J Mol Biol. 2018;8(1):69–82.
- Czarnomysy R, Bielawski K, Muszynska A, Bielawska A, Gornowicz A. Biological evaluation of dimethylpyridine–platinum complexes with potent antiproliferative activity. J Enzyme Inhib Med Chem. 2016;31(Suppl. 3):150–165.
- Mphahlele MJ, Gildenhuys S, Parbhoo N. Synthesis, cytotoxicity and molecular docking studies of the 9-substituted 5-styryltetrazolo [1, 5-c]quinazoline derivatives. Molecules. 2017;22(11):1719.
- Xu H, Cheung IY, Wei XX, Tran H, Gao X, Cheung NV. Checkpoint kinase inhibitor synergizes with DNA-damaging agents in G 1 checkpoint-defective neuroblastoma. Int J Cancer. 2011;129(8):1953–1962.
- Shao JW, Dai YC, Xue JP, Wang JC, Lin FP, Guo YH. In vitro and in vivo anticancer activity evaluation of ursolic acid derivatives. Eur J Med Chem. 2011;46(7):2652–2661.
- Zargan J, Sajad M, Umar S, Naime M, Ali S, Khan HA. Scorpion (Androctonus crassicauda) venom limits growth of transformed cells (SH-SY5Y and MCF-7) by cytotoxicity and cell cycle arrest. Exp Mol Pathol. 2011;91(1):447–454.
- Özgeriş B. Synthesis of substituted phenethylamine-based thioureas and their antimicrobial and antioxidant properties. Russ J Org Chem. 2021;57(3):422–429.
- Suffness M, Pezzuto JM. Assays related to cancer drug discovery. In: Hostettmann, editor. Methods in plant biochemistry: assays for bioactivity. Vol. 6. London (UK): Academic Press; 1990. p. 71–133.
- McMorris TC, Chimmani R, Alisala K, Staake MD, Banda G, Kelner MJ. Structure–activity studies of urea, carbamate, and sulfonamide derivatives of acylfulvene. J Med Chem. 2010;53(3):1109–1116.
- Duspara PA, Islam MS, Lough AJ, Batey RA. Synthesis and reactivity of N-alkyl carbamoylimidazoles: development of N-methyl carbamoylimidazole as a methyl isocyanate equivalent. J Org Chem. 2012;77(22):10362–10368.
- Grzyb JA, Shen M, Yoshina-Ishii C, Chi W, Brown RS, Batey RA. Carbamoylimidazolium and thiocarbamoylimidazolium salts: novel reagents for the synthesis of ureas, thioureas, carbamates, thiocarbamates and amides. Tetrahedron. 2005;61(30):7153–7175.
- Tundo P, Selva M. The chemistry of dimethyl carbonate. Acc Chem Res. 2002;35(9):706–716.
- Diaz DJ, Darko AK, McElwee‐White L. Transition metal‐catalyzed oxidative carbonylation of amines to ureas. Eur J Org Chem. 2007;2007(27):4453–4465.
- Ready JM, Nijhawan D, Gonzales SS, Theodoropoulos P. Preparation of benzothiophenes and other compounds and methods and compositions for selective and targeted cancer therapy. PCT Int Appl. 2015; WO2015035051A1. 307 pp.
- Eckert H, Forster B. Triphosgene, a crystalline phosgene substitute. Angew Chem Int Ed Engl. 1987;26(9):894–895.
- Shi H, Hu W, Sun Y. Preparation of chloroformates using bis(trichloromethyl)carbonate. J Chem Res. 2004;2004(10):708–709.
- Rose TE, Morisseau C, Liu J-Y, Inceoglu B, Jones PD, Sanborn JR, Hammock BD. 1-Aryl-3-(1-acylpiperidin-4-yl) urea inhibitors of human and murine soluble epoxide hydrolase: structure–activity relationships, pharmacokinetics, and reduction of inflammatory pain. J Med Chem. 2010;53(19):7067–7075.
- Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother. 2007;51(10):3471–3484.
- Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK, Participating National Healthcare Safety Network Facilities. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect Control Hosp Epidemiol. 2008;29(11):996–1011.
- Prestinaci F, Pezzotti P, Pantosti A. Antimicrobial resistance: a global multifaceted phenomenon. Pathog Glob Health. 2015;109(7):309–318.
- Rauf MK, Talib A, Badshah A, Zaib S, Shoaib K, Shahid M, Flörke U, Imtiaz-Ud-Din Iqbal J. Solution-phase microwave assisted parallel synthesis of N,N′-disubstituted thioureas derived from benzoic acid: biological evaluation and molecular docking studies. Eur J Med Chem. 2013;70:487–496.
- Obradović D, Nikolić S, Milenković I, Milenković M, Jovanović P, Savić V, Roller A, Đorđić Crnogorac M, Stanojković T, Grgurić-Šipka S, et al. Synthesis, characterization, antimicrobial and cytotoxic activity of novel half-sandwich Ru (II) arene complexes with benzoylthiourea derivatives. J Inorg Biochem. 2020;210:111164.
- Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Ceña V, Gallego C, Comella JX. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J Neurochem. 2000;75(3):991–1003.
- Masters JR. HeLa cells 50 years on: the good, the bad and the ugly. Nat Rev Cancer. 2002;2(4):315–319.
- Reers M, Smith TW, Chen LB. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 1991;30(18):4480–4486.
- Abdelhaleem EF, Abdelhameid MK, Kassab AE, Kandeel MM. Design and synthesis of thienopyrimidine urea derivatives with potential cytotoxic and pro-apoptotic activity against breast cancer cell line MCF-7. Eur J Med Chem. 2018;143:1807–1825.
- Esteves-Souza A, Pissinate K, Graça Nascimento MD, Grynberg NF, Echevarria A. Synthesis, cytotoxicity, and DNA-topoisomerase inhibitory activity of new asymmetric ureas and thioureas. Bioorg Med Chem. 2006;14(2):492–499.
- Charifson PS, Grillot A-L, Grossman TH, Parsons JD, Badia M, Bellon S, Deininger DD, Drumm JE, Gross CH, LeTiran A, et al. Novel dual-targeting benzimidazole urea inhibitors of DNA gyrase and topoisomerase IV possessing potent antibacterial activity: intelligent design and evolution through the judicious use of structure-guided design and structure–activity relationships. J Med Chem. 2008;51(17):5243–5263.
- Alfonso EE, Deng Z, Boaretto D, Hood BL, Vasile S, Smith LH, Chambers JW, Chapagain P, Leng F. Novel and structurally diversified bacterial DNA gyrase inhibitors discovered through a fluorescence-based high-throughput screening assay. ACS Pharmacol Transl Sci. 2022;5(10):932–944.
- Kundu D, Zhu A, Kim E, Paudel S, Jang C-G, Lee YS, Kim K-M. Potential functional role of phenethylamine derivatives in inhibiting dopamine reuptake: structure–activity relationship. Biomol Ther. 2023;31(1):108–115.
- Khan T, Sankhe K. DNA gyrase inhibitors. In: Offermanns, editor. Encyclopedia of molecular pharmacology. Cham: Springer International Publishing; 2021. p. 1–8.
- Dighe SN, Collet TA. Recent advances in DNA gyrase-targeted antimicrobial agents. Eur J Med Chem. 2020;199:112326.
- Morgan H, Lipka-Lloyd M, Warren AJ, Hughes N, Holmes J, Burton NP, Mahenthiralingam E, Bax BD. A 2.8 Å structure of zoliflodacin in a DNA cleavage complex with Staphylococcus aureus DNA gyrase. Int J Mol Sci. 2023;24(2):1634.
- Shyian M, Shore D. Approaching protein barriers: emerging mechanisms of replication pausing in eukaryotes. Front Cell Dev Biol. 2021;9:672510.
- Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9(5):338–350.
- Francisco GD, Li Z, Albright JD, Eudy NH, Katz AH, Petersen PJ, Labthavikul P, Singh G, Yang Y, Rasmussen BA, et al. Phenyl thiazolyl urea and carbamate derivatives as new inhibitors of bacterial cell-wall biosynthesis. Bioorg Med Chem Lett. 2004;14(1):235–238.
- Webster J, Piscitelli G, Polli A, Ferrari CI, Ismail I, Scanlon MF. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. N Engl J Med. 1994;331(14):904–909.
- Molinoff PB, Axelrod J. Biochemistry of catecholamines. Annu Rev Biochem. 1971;40(1):465–500.