
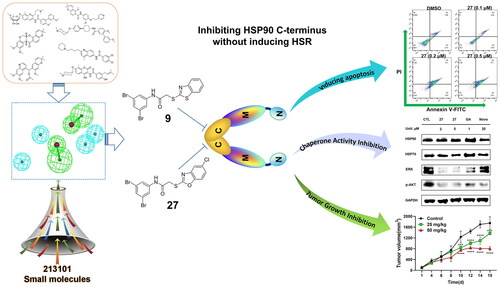
Abstract
Targeting Heat shock protein 90 (HSP90) C-terminus is an important strategy to develop HSP90 inhibitors without inducing heat shock response. The development of C-terminal inhibitors, however, is hampered by a lack of understanding regarding the interaction between the HSP90 C-terminus and the present inhibitors. We collected seven classical and structurally diverse HSP90 C-terminal inhibitors and constructed a ligand-based pharmacophore model. The subsequent virtual screening and structural optimisation led to the identification of 2-heteroarylthio-N-arylacetamides as novel HSP90 C-terminal inhibitors. 9 and 27 exhibited strong antitumour activity in vitro by inhibiting proliferation and inducing apoptosis in multiple cancer cell lines. These compounds disrupted the interaction between HSP90 C-terminus and peptidylprolyl isomerase D, exerting a stronger inhibitory effect than novobiocin. 27 significantly induced the degradation of HSP90 clients without triggering heat shock response. In an in vivo study using 4T1 mice breast cancer models, 9 showed a potent antitumour effect without obvious toxicity.
Graphical Abstract
Introduction
Heat shock protein 90 (HSP90) is a promising anticancer target because it is responsible for maintaining the homeostasis of a variety of oncoproteins, which are involved in all hallmarks of cancerCitation1,Citation2. HSP90 inhibition disrupts multiple signalling pathways and causes the apoptosis of cancer cellsCitation3,Citation4. As a result, it has been thought that modulating HSP90 activity is a promising strategy for treating cancerCitation5,Citation6. Because it regulates the maturation of many cancer-resistant proteins, HSP90 also plays a prominent role in mediating cancer resistanceCitation7,Citation8. Combining HSP90 inhibitors with various anticancer therapeutics has been extensively studied to overcome drug resistance in cancer therapyCitation9–12.
Traditional HSP90 inhibitors bind to the ATP binding site at the N-terminus of HSP90, halting the molecular chaperone cycle and leading to the degradation of HSP90 client proteins by the proteasomal systemCitation13,Citation14. Although numerous HSP90 N-terminal inhibitors have undergone clinical trial evaluation for cancer therapyCitation15–17, only pimitespib has received an approval for the treatment of GIST in Japan. The therapeutic effect of HSP90 N-terminal inhibitors is hampered by the compensatory expression of heat shock proteins, which is called the heat shock response (HSR). As an alternative way to modulate HSP90, inhibitors that target the HSP90 C-terminus prevent the induction of HSRCitation18,Citation19. In recent years, great efforts have been focused on the development of HSP90 C-terminal inhibitors, resulting in the identification of numerous HSP90 C-terminal inhibitors from both natural products and synthetic small molecules ()Citation20,Citation21.
Figure 1. Representative examples of HSP90 C-terminal inhibitors.
Despite the significant progress in the discovery of HSP90 C-terminal inhibitors, it should be emphasised that many of them are found randomly in natural products and the following structural optimisation. Furthermore, up to the present, there is still no resolved crystal structure of the inhibitor bound to HSP90 C-terminus. According to the literature, current HSP90 inhibitors may bind to a variety of binding sites at C-terminusCitation22–25. The lack of knowledge regarding the interaction between the HSP90 C-terminus and inhibitors significantly hinders the receptor-based development of HSP90 C-terminal inhibitors. In this context, pharmacophore modelling would be an attractive way to rationally discover new HSP90 C-terminal inhibitorsCitation26. In 2010, the Colombo group reported a receptor-based pharmacophore model for screening HSP90 inhibitorsCitation24. In 2020, the Tomasic group constructed both receptor-based and ligand-based pharmacophore models for virtual screeningCitation27. In their work, the ligands were limited to only two kinds of chemical scaffolds, coumarin and diphenyl, for creating pharmacophore models. In the present research, we developed a new ligand-based pharmacophore model using seven well-known HSP90 C-terminal inhibitors with various chemical scaffolds and successfully employed it to discover novel HSP90 C-terminal inhibitors.
Results and discussions
A pharmacophore model bearing six features was generated from seven ligands
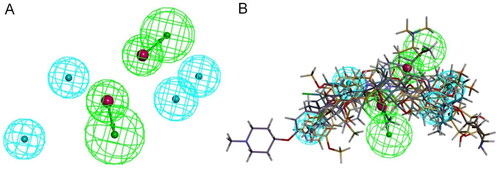
Ligand-based pharmacophore modelling technique extracts the common structural features of a number of inhibitors, which may be crucial for the inhibitors to interact with their biological target. To create the ligand-based pharmacophore model for identifying new HSP90 C-terminal inhibitors, seven well-known inhibitors carrying various structural scaffolds were used (). While inhibitors 1–5 are synthetic small molecules, 6 and 7 are natural compoundsCitation28–34. For each inhibitor, up to 255 conformations were calculated, and multiple alignments were performed based on the chemical features in 3D space of all conformations. The top ten pharmacophore models were generated and ranked based on alignment scores (Table S1). With a rank value of 111.122, the top-ranked pharmacophore_01 has six features, including four hydrophobic features (blue) and two hydrogen bond acceptor features (green) (). This model didn’t include either an aromatic ring feature or a hydrogen bond donor feature. displays the alignments of seven inhibitors with the pharmacophore_01 model. In order to assess the specificity of this pharmacophore model, four classical HSP90 N-terminal inhibitors were used as negative ligands for validation (Figure S1). None of these N-terminal inhibitors can map to pharmacophore_01, indicating that this pharmacophore model has good specificity.
Figure 2. Ligand-based pharmacophore model. (A) Top-ranked pharmacophore model (pharmacophore_01). Hydrophobic and hydrogen bond acceptor features are coloured blue and green, respectively. (B) Alignments of seven HSP90 C-terminal inhibitors with the pharmacophore_01 model.
Virtual screening identified a series of compounds with strong antiproliferative activity against the MCF-7 cell line
depicts the virtual screening flow used in this study. According to Lipinski and Verber’s Rules, 155 436 out of 213 101 molecules passed the drug-likeness screening (Table S2). Subsequent pharmacophore-based virtual screening found 5149 molecules that could map to pharmacophore_01. These molecules were ranked by FitValue. The top-ranked 100 molecules were subjected to the following manual inspection, which included chemical structure cluster analysis and novelty analysis. Twenty compounds were finally selected, purchased, and tested for their antiproliferative activity against the human breast cancer MCF-7 cell line via MTT assay (Table S3). Eight compounds (8–15) exhibited antiproliferative activity against MCF-7 cells with IC50 values less than 50 µM. It should be noted that none of these eight compounds have been reported as HSP90 inhibitors yet. Six compounds (8–13) interestingly share the structural features of two (hetero)aryl scaffolds and one short linker, the latter of which is helpful to provide good flexibility to molecules and maintain distance between two aromatic rings. Compounds 14 and 15 attracted our attention because of their symmetrical chemical structures ().
Figure 3. Virtual screening flow.
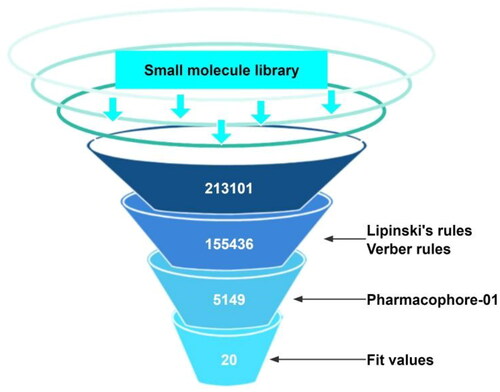
Figure 4. FitValue evaluation results and antiproliferative activity against MCF-7 cells of representative compounds. IC50 values (μM) were presented as the mean ± SD from three independent experiments.
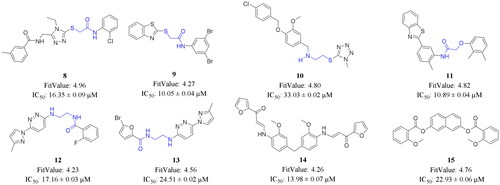
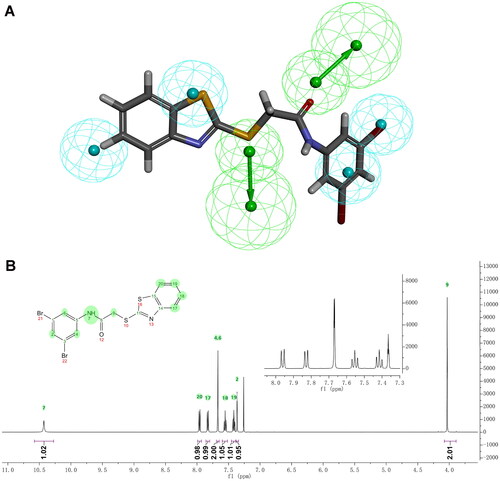
Compound 9, bearing a thioacetamide linker, manifested the strongest antiproliferative activity against MCF-7 cells (IC50 = 10.05 ± 0.04 μM). It is worthwhile noting that a series of analogues of 9 have been determined as antitubercular agents, possibly by inhibiting decaprenylphosphoryl-β-D-ribose oxidase (DprE1)Citation35. The alignment of 9 with pharmacophore model_01 is shown in . The carbonyl group and sulphur atom in the thioacetamide linker can act as two hydrogen bond acceptors. Hydrophobic features can be found in the two aromatic rings and bromo atoms. Compound 11, which has an oxyacetamide linker, showed comparable antiproliferative activity to 9. Since 9 displayed significant cytotoxicity against MCF-7 cells, it is worthwhile to take 9 as a starting point for further research. The chemical structure of 9 was verified using 1H NMR,13C NMR, and HRMS measurements (Figures S2–S4). displays the 1H NMR spectrum and the corresponding assignments.
Figure 5. Alignment of 9 with pharmacophore model_01 and characterisation of 9. (A) Alignment of 9 with pharmacophore model_01. (B) 1H NMR spectrum of 9 in CDCl3.
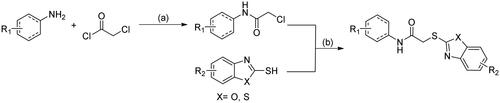
Scheme 1. Synthetic route of 2-heteroarylthio-N-arylacetamides 16–32. Reagents and conditions: (a) Et3N, CH2Cl2, r.t; (b) Et3N, THF, 60 °C.
2-Heteroarylthio-N-arylacetamides were synthesised through a two-step process
To understand the structure-activity relationships (SAR), many analogues of 9 were further synthesised. 2-Heteroarylthio-N-arylacetamides were readily prepared through a two-step synthetic route, as indicated in Scheme 1. 2-Chloroacetamides were obtained by the reaction of (hetero)aryl amine with chloroacetyl chloride in the presence of a base such as triethylamine. Afterwards, the nucleophilic substitution of 2-chloroacetamides with heteroaryl thiols afforded 2-heteroarylthio-N-arylacetamides 16–32.
Biological evaluation
9 and 27 Exhibited strong antiproliferative activity against multiple cancer cell lines
The cytotoxicity of 1, 2-heteroarylthio-N-arylacetamides 9 and 16–32 against cancer cell lines was investigated after a 48-h co-incubation. All compounds were evaluated against MCF-7 cells, and those with strong activity (IC50 < 50 µM) were further investigated against the mouse breast cancer 4T1 cell line (). It is notable that 9 inhibited the proliferation of 4T1 cells with an IC50 value of 0.61 ± 0.03 µM. The activity was dramatically decreased when other (hetero)aryl scaffolds were used in place of the 3,5-dibromophenyl. Only two compounds, 24 and 25, exhibited cytotoxicity against MCF-7 cells with IC50 values less than 50μM. We next studied the effect of benzothiazole on the activity. We were delighted to observe that compound 27 bearing a 5-chlorobenzoxazole scaffold inhibited the proliferation of MCF-7 cells at low micromolar concentrations (IC50 = 1.28 ± 0.04 µM). Replacing the 3,5-dibromophenyl in 27 resulted in the loss of cytotoxicity, suggesting that the 3,5-dibromophenyl scaffold may play an important role in maintaining the activity of this kind of compound.
Table 1. Antiproliferative activity of 2-heteroarylthio-N-arylacetamides. 
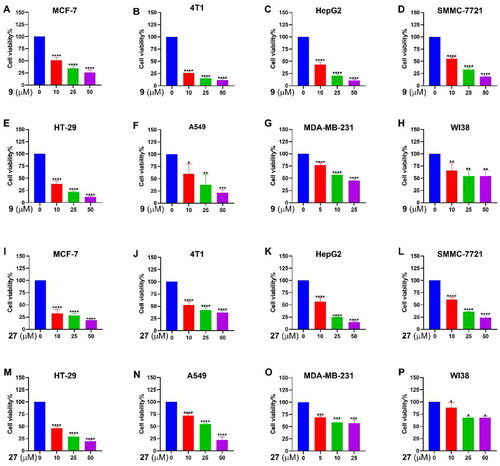
HSP90 inhibitors has been studied in clinical trials against a variety of cancers. To further explore the anticancer potential of 9 and 27, the antiproliferative activity was tested against a series of cancer cell lines. As shown in , 9 and 27 exerted dose-dependent, broad-spectrum antiproliferative effects. 9 and 27 demonstrated strong antiproliferative activities against MCF-7, 4T1, human liver cancer cell lines HepG2 and SMMC-7721, human colorectal adenocarcinoma cell line HT-29, and the human lung cancer cell line A549. Nevertheless, 9 and 27 had relatively mediocre activity against the human triple-negative breast cancer MDA-MB-231 cell line. It’s also noteworthy that 9 and 27 showed much weaker effects on the proliferation of human WI-38 cells, a normal fibroblast cell line, suggesting that 9 and 27 suppress cell proliferation preferentially in cancer cells over normal cells.
9 and 27 Induced apoptosis of MCF-7 cells
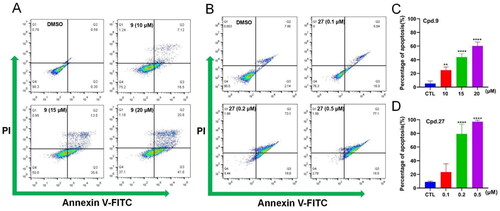
On the basis of the prominent antiproliferative activity of 9 and 27 against the MCF-7 cell line, the capacity of 9 and 27 to induce apoptosis of MCF-7 cells was examined by flow cytometry (). 9 and 27 caused the apoptosis of MCF-7 cells in a dose-dependent manner. For example, following the administration of 27, the percentage of early and late apoptotic cells rose from 23.7% (0.1 µM) to 93.6% (0.5 µM) (). These findings revealed that 9 and 27 inhibited the proliferation of MCF-7 cells through the effective induction of cell apoptosis.
Figure 6. Effects of 9 and 27 on the viability of cancer cells. Cancer cells were treated with 9 (A–H) or 27 (I-P) for 48 h at indicated concentrations. Cell viability was determined by the MTT assay. The data represent the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 by one-way ANOVA compared with vehicle-treated control group.
2-Heteroarylthio-N-arylacetamides targeted HSP90 C-terminus
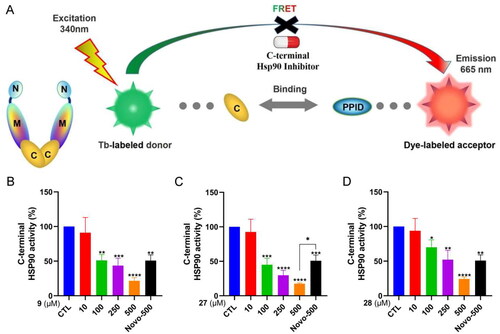
To further confirm the interplay between 2-heteroarylthio-N-arylacetamides and the C-terminus of HSP90, we conducted a specific HSP90α C-terminal domain time-resolved fluorescence resonance energy transfer (TR-FRET) experiment, which is designed to measure the inhibition of HSP90α binding ability to its protein target, peptidylprolyl isomerase D (PPID) (). As shown in , 2-heteroarylthio-N-arylacetamides (9, 27, and 28) significantly inhibited the binding between HSP90 C-terminus and PPID. These compounds manifested stronger activity than the positive control, novobiocin. Compound 27 was the strongest HSP90 C-terminal inhibitor with an IC50 value of 81.81 µM.
Figure 7. Effects of 9 and 27 on cell apoptosis determined by flow cytometry. (A, B) Apoptosis rate in MCF-7 cells treated with 9 (A) and 27 (B) at indicated concentrations. (C, D) Statistical analysis of the percentage of apoptosis rate of 9 (C) and 27 (D). Data represent the mean ± SD of three independent experiments. **p < 0.01 and ****p < 0.0001 compared to the control group.
27 Induced the degradation of HSP90 clients without triggering HSR
HSR is a pro-survival process in cancer cells that contributes to apoptosis inhibition, tumourigenicity, tumour resistance, and poor prognosisCitation36. Compelling evidence suggests that the HSR counteracts the benefit of HSP90 inhibition by N-terminal inhibitors in cancer therapyCitation37. A key feature of the HSR is the overexpression of HSPs, including HSP27, HSP70, and HSP90. To test whether these novel HSP90 C-terminal inhibitors induced HSR or not, we examined the effect of 27 on the expression of HSP70 and HSP90 in MCF-7 cells. As shown in , 27 did not induce the overexpression of HSP70 and HSP90 in a similar manner to novobiocin, a well-known HSP90 C-terminal inhibitor. Moreover, 27 even reduced the expression of HSP90 and HSP70 in MCF-7 cells. As a negative control, geldanamycin, a traditional HSP90 N-terminal inhibitor, significantly enhanced the expression of HSP90 and HSP70. We next investigated the effect of 27 on the expression of two HSP90 clients, ERK and p-Akt. 27 significantly decreased the expression of p-Akt and ERK in MCF-7 cells ()). Taken together, 27 inhibited HSP90 chaperone activity without triggering HSR, which was consistent with the characteristic of HSP90 C-terminal inhibitors.
Figure 8. HSP90α C-terminal domain TR-FRET assay. (A) The principle of TR-FRET assay. (B–D) The C-terminal HSP90 activity after the treatment with 9, 27, 28 and novobiocin at the indicated concentrations. Novobiocin was evaluated at a concentration of 500 μM. Data represent the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 by one-way ANOVA compared with vehicle-treated control group.
9 and 27 Fit into the binding site at the hexamer interface of HSP90
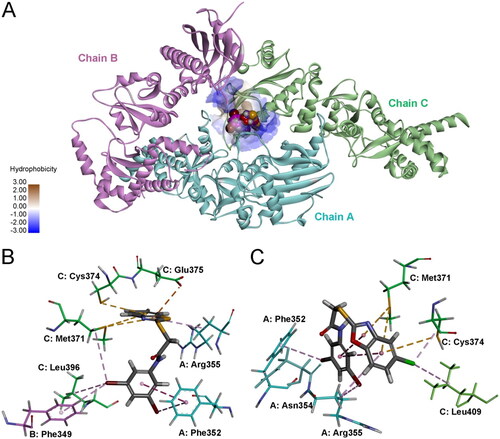
Previous studies have indicated that there are possibly at least 4 binding sites for inhibitors in the C-terminus of HSP90Citation38. Recently, the Matts and Deng group reported a cocrystal structure of the HSP90α and a coumarin derivative, 7-diethylamino-3-[N-(2-maleimidoethyl) carbamoyl]coumarin (MDCC)Citation39. MDCC is located in the binding site that is formed at the hexamer interface of HSP90α. Molecular docking was performed using the MDCC binding pocket to predict the plausible interactions between 2-heteroarylthio-N-arylacetamides and HSP90. As shown in , both 9 and 27 could fit into the binding pocket at the hexamer interface, with the top-CDOCKER energies of 18.80 and 24.28 kcal/mol, respectively. The favourable electrostatic and hydrophobic interactions that stabilise the complexes of 9 and 27 with HSP90 are shown in . The benzothiazole moiety of 9 and the 5-chlorobenzoxazole moiety of 27 are oriented towards inside of the binding site, which is formed by amino acid residues including Met371, Cys374, Glu375 and Leu409 in chain C, and Arg355 in chain A. The 3,5-dibromophenyl moieties of both molecules are pointing towards the opening of the binding site. Two bromine atoms form multiple interactions with neighbouring lipophilic amino acid residues.
Figure 9. Western blotting assays. (A) Expression of HSP90, HSP70, ERK, p-AKT in MCF-7 cells after 24 h treatment with 27; quantification analysis of detected proteins HSP90 (B), HSP70 (C), ERK (D), and p-AKT (E). Data represent the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 by one-way ANOVA compared with vehicle-treated control group.
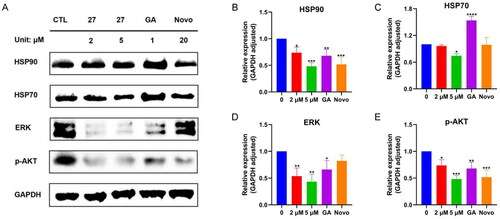
Figure 10. Predicted binding modes and interactions of 9 and 27 with HSP90 (PDB ID: 7RY1). (A) Binding pose of 9 and 27 in the HSP90 binding site. 9 and 27 are referred as CPK model. (B) Interactions between 9 and HSP90. (C) Interactions between 27 and HSP90.
9 Suppressed the tumour growth in the 4T1 tumour-bearing mice model
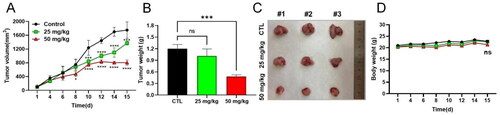
Because 9 exerted the strongest antiproliferative activity against 4T1 cells, we further investigated the in vivo anticancer activity of 9 in 4T1 tumour-bearing mice models. The tumour-bearing mice were treated intraperitoneally with 9 at 25 and 50 mg/kg for two weeks. As shown in , the growth of the tumour was significantly inhibited by the treatment with 9 in the high dose group (50 mg/kg), although there was no statistically significant change in the low dose group (25 mg/kg). Additionally, there was no significant decline in body weight in both groups, suggesting that 9 was well tolerated in this animal model.
Figure 11. In vivo assay of the antitumour activity of 9 in 4T1 tumour-bearing mice models. (A) tumour volume change after administration of 9 in two weeks. The tumour volumes were determined every other day. (B) Statistical analysis of the tumour weight in the indicated groups after two weeks administration of 9. (C) Photographs of tumours in the indicated groups after two weeks of administration of 9. (D) Statistical analysis of the body weights in the indicated groups in two weeks. The body weights were determined every other day. Data represent the mean ± SD. ns, p > 0.05, not significantly different from the control group. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 by one-way ANOVA compared with vehicle-treated control group.
Conclusion
In this study, a number of 2-heteroarylthio-N-arylacetamides were successfully identified as novel HSP90 C-terminal inhibitors using a ligand-based pharmacophore model. Virtual screening and a preliminary SAR study discovered 9 and 27 with potent HSP90 inhibition and anticancer activities. The inhibition activity against HSP90 C-terminus was validated by western blotting and the TR-FRET assay. An In vivo study revealed that 9 reduced tumour growth without significant body weight change. Our study indicates that 9 and 27 can serve as potent leads for further exploration of novel anticancer agents. The pharmacophore model can also be applied to screen other small molecule libraries for discovering other new HSP90 C-terminal inhibitors.
Experimental section
Pharmacophore modelling and virtual screening
The generation of pharmacophore models
The pharmacophore model for virtual screening was built using Discovery Studio 2016. The module “common feature pharmacophores” was used to generate the pharmacophore model. Seven previously reported HSP90 C-terminal inhibitors (1–7) were selected as ligands (). The conformation generation was set to “best”. Common Features were set as HB_ACCEPTOR 0–5, HB_DONOR 0–5, HYDROPHOBIC 0–5, RING_AROMATIC 0–5. Other parameters were set as default. Four HSP90 N-terminal inhibitors were used as negative controls to validate the pharmacophore model (Figure S1). The top-ranked model was used for virtual screening.
Virtual screening
A molecule library containing 213 101 molecules was obtained from Topscience (Shanghai, China). Drug-likeness prediction was performed using the module “Filter by Lipinski and Verber Rules”. The parameters were set as shown in Table S2. The passed molecules were further screened by the pharmacophore model. The pharmacophore-based virtual screening was performed using the module “Screen Library” in Discovery Studio 2016. The mapped molecules were ranked by “Fitvalue”. Higher fit scores represent a better fit to the model.
Chemistry
General method for chemistry
Unless otherwise noted, all chemical reagents were purchased from commercial sources and were used without further purification. All reactions were monitored by thin layer chromatography (TLC) on GF254 TLC plates. Silica gel (200–300 mesh) was used for column chromatography purification.1H and 13C NMR spectra were recorded on an AVANCE III HD at 500 MHz (Bruker, Billerica, MA, USA). Multiplicity of 1H NMR signals was reported as singlet (s), doublet (d), triplet (t), and multiplet (m). HRMS was carried out on a Q Exactive mass spectrometer (Thermo Fisher, Waltham, MA, USA) with electrospray ionisation (ESI) as the ionisation source. Analyst Software 1.6.3 was used for data acquisition and processing. The purity of target compounds was determined by 1H NMR. All target compounds achieved a minimum of 95% purity.
General procedure for 2-heteroarylthio-N-arylacetamides
To a solution of aniline (1 equiv.) in CH2Cl2, Et3N (2 equiv.), and chloroacetyl chloride (4 equiv.) was added. The reaction mixture was stirred at room temperature for 4 h and monitored by TLC. Water and CH2Cl2 were added for extraction. The organic layer was collected and washed with water and brine. The organic layer was then dried over anhydrous MgSO4, and concentrated under reduced pressure. The intermediate was purified using column chromatography.
The intermediate (1 equiv.) was dissolved in THF, and triethylamine was added (2 equiv.). The corresponding heteroaryl thiol (1 equiv.) was added to the reaction mixture, and the solution was stirred at 60 °C for 6 h. The reaction was monitored by TLC. Upon completion, the reaction was terminated, and THF was removed by vacuum concentration. Water and ethyl acetate were added to extract the product. The organic layer was collected, washed with water and brine, dried over anhydrous magnesium sulphate, and concentrated under vacuum. The residue was purified by column chromatography (petroleum ether/ethyl acetate) to give 2-heteroarylthio-N-arylacetamides 16–32.
Biological evaluation
Cell culture, antiproliferative activity assay and western blotting assay in this study were performed according to the previous reportCitation40.
Cell culture
Cells in this study were purchased from the Institute of Basic Medicine, Chinese Academy of Medical Sciences (Beijing, China). MCF-7, MDA-MB-231, HepG2, SMMC-7721, HT-29, A549 and WI38 cells were cultured in DMEM and 4T1 cells in RPMI-1640 using 10% foetal bovine serum (FBS) and 1% penicillin–streptomycin (PS) solution in humidified air containing 5% CO2 at 37 °C.
Antiproliferative activity assay
Cell proliferation assay of target compounds against cancer cell lines MCF-7, MDA-MB-231, 4T1, and A549 was determined using the MTT assay (Beyotime, Shanghai, China). The compounds were dissolved in DMSO, diluted with cell culture medium to the desired concentrations. Cells were seeded into a 96-well plate at a density of 7000 cells/well (200 μL/well) and incubated overnight. Compound solutions at different concentrations were added and incubated for 48 h or 72 h. MTT solution was added and further incubated for 4 h in the dark. Absorbance was measured at a wavelength of 490 nm using Synergy H1 (BioTek Instruments, Inc., Winooski, VT, USA). IC50 values were calculated by GraphPad Prism 8 (GraphPad Software, Inc., San Diego, CA, USA). Three parallel experiments were performed.
Western blotting assay
Cells were incubated in a 6-well plate at a density of 106 cells/well (4 ml/well) at 37 °C for 12 h. The cells were then treated with target compounds, geldanamycin, and novobiocin at the indicated concentrations for 24 h. The 6-well plate was placed on ice for 15 min. Cells were collected into a centrifuge tube and lysed in a radioimmunoprecipitation (RIPA) solution containing 1% phenylmethanesulphonyl fluoride (PMSF). The lysates were cleared by centrifugation at a speed of 14 000 r/min for 10 min. Protein concentrations were determined using the BCA assay. Equal amounts of protein were separated by 10% Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). The protein on the separating gel was transferred to the polyvinylidene fluoride (PVDF) membrane. The membrane was blocked in skim milk for 80 min and incubated with the primary antibodies against HSP70 (Abclonal, A12948), HSP90 (Abclonal, A5027), ERK (Beyotime, AF1051), p-AKT (Beyotime, AF5734) and GAPDH (Bioworld, AP0063), respectively. After further incubation with the HRP anti-rabbit IgG (H + L) (Abbkine, A21020) at room temperature for 1 h, blots were imaged with a gel imager (ProteinSimple, San Jose, CA, USA). The density of proteins was determined using AlphaView SA (Alpha Innotech Corp., version 3.4.0.0, San Leandro, CA, USA).
Annexin V/PI double staining
MCF-7 cells were plated into a 6-well plate at a density of 3.0 × 105 cells and incubated for 24 h. Cells were treated with vehicle, 9 (10, 15, 20 μM), and 27 (0.1, 0.2, 0.5 μM) for additional two days, respectively. Cells were harvested by trypsinisation, stained with Annexin V-FITC or PI, and analysed by flow cytometry according to the manufacturer’s recommended procedure (BD Biosciences).
Fluorescence resonance energy transfer assay
HSP90α C-Terminal Domain TR-FRET Assay Kit (#50289) was obtained from BPS Bioscience (CA, USA). All assays were measured using a SpectraMax M2 microplate reader. Diluted HSP90α (2 ng/µL) and PPID (3 ng/µL) with novobiocin, 9, and 27, were loaded in the Optiplate along with Tb donor (10 µL) and Dye-labelled acceptor (10 µL) in accordance with the manufacturer’s guide. HSP90α:PPID binding activity with each HSP90 inhibitor was analysed with calculation formula provided by the manufacturer. Two sequential measurements were conducted. Tb-donor emission should be measured at 620 nm, followed by dye-acceptor emission at 665 nm. Data analysis is performed using the TR-FRET ratio (665 nm emission/620 nm emission). When percentage activity is calculated, the FRET value from the negative control can be set to 0% activity and the FRET value from the positive control can be set to 100% activity.
Animal experiments
Female Balb/c mice were fed freely, with standard mouse chow and water, and housed in a temperature- and humidity-controlled facility with a 12-h light/12-h dark cycle. For the allograft experiments, 2 × 106 4T1 cells were subcutaneously injected into the six- to eight-week-old mice. After the tumour volume had reached 50–150 mm3, the mice were randomly assigned to different experimental groups (n = 3 per group). Once a day for two weeks, the animals were administered with vehicle (10% DMSO, 40% PEG300, 5% Tween 80, and normal saline) or 9 (25 or 50 mg/kg, dissolved in corn oil with 10% DMSO). Tumour growth was determined by measuring the short and long diameter of the tumour tissue using a calliper. The tumour volume was calculated using the following formula: tumour volume (mm3) = (small diameter)Citation2 × (large diameter) × 0.5. The body weight of mice was measured at the beginning and end of administration. All mice were killed after two weeks of treatment. The tumour tissue was collected and weighed.
The protocol for this study was reviewed and approved by the Institutional Animal Care and Use Committee of the Dalian University of Technology (Approval number: DUTSLPS230301-01).
Molecular docking
Molecular docking was performed using CDOCKER in BIOVIA Discovery Studio (Dassault Systemes, San Diego, USA). The CHARMm force filed was used for protein preparation and molecular docking. The crystal structure of the middle and C-terminal domains of Hsp90α labelled with MDCC was downloaded from the Protein Data Bank database (https://www.rcsb.org/structure/7RY1). The protein structure was prepared before docking by procedures including building loops, protonation, and removing water molecules and ligands. The MDCC binding site was used as active site for docking. The coordinates of the active site sphere were as follows: x = −15.176, y = −52.776, z = 27.813. The radius of the active site sphere was set to 15. The protein-ligand complex with the highest-CDOCKER_EMERGY score was analysed.
Statistical analysis
The data are presented as the mean ± SD. The IC50 value represents the concentration of inhibitor that reduces cell viability or enzyme activity by 50% and is calculated by nonlinear regression using GraphPad Prism 8.0 software (GraphPad Software, Inc., La Jolla, USA). A p values less than 0.05 was statistically significant.
Supplemental Material
Download PDF (2.8 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Bagatell R, Whitesell L. Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther. 2004;3(8):1021–1030.
- Birbo B, Madu EE, Madu CO, Jain A, Lu Y. Role of HSP90 in cancer. Int J Mol Sci. 2021;22(19):10317.
- Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des. 2013;19(3):347–365.
- Serwetnyk MA, Blagg BSJ. The disruption of protein-protein interactions with co-chaperones and client substrates as a strategy towards Hsp90 inhibition. Acta Pharm Sin B. 2021;11(6):1446–1468.
- Ortiz AG, Salcedo JM. Heat shock proteins as targets in oncology. Clin Transl Oncol. 2010;12(3):166–173.
- Liu Q, Tu G, Hu Y, Jiang Q, Liu J, Lin S, Yu Z, Li G, Wu X, Tang Y, et al. Discovery of BP3 as an efficacious proteolysis targeting chimera (PROTAC) degrader of HSP90 for treating breast cancer. Eur J Med Chem. 2022;228:114013.
- Solárová Z, Mojžiš J, Solár P. Hsp90 inhibitor as a sensitizer of cancer cells to different therapies (review). Int J Oncol. 2015;46(3):907–926.
- Li ZN, Luo Y. HSP90 inhibitors and cancer: prospects for use in targeted therapies (review). Oncol Rep. 2023;49(1):6.
- Smyth T, Van Looy T, Curry JE, Rodriguez-Lopez AM, Wozniak A, Zhu M, Donsky R, Morgan JG, Mayeda M, Fletcher JA, et al. The HSP90 inhibitor, AT13387, is effective against imatinib-sensitive and -resistant gastrointestinal stromal tumour models. Mol Cancer Ther. 2012;11(8):1799–1808.
- Ray-Coquard I, Braicu I, Berger R, Mahner S, Sehouli J, Pujade-Lauraine E, Cassier PA, Moll UM, Ulmer H, Leunen K, et al. Part I of GANNET53: a European multicenter phase I/II trial of the Hsp90 inhibitor ganetespib combined with weekly paclitaxel in women with high-grade, platinum-resistant epithelial ovarian cancer-a study of the GANNET53 consortium. Front Oncol. 2019;9:832.
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93.
- Ewers KM, Patil S, Kopp W, Thomale J, Quilitz T, Magerhans A, Wang X, Hessmann E, Dobbelstein M. HSP90 inhibition synergizes with cisplatin to eliminate basal-like pancreatic ductal adenocarcinoma cells. Cancers. 2021;13(24):6163.
- Park JM, Kim YJ, Park S, Park M, Farrand L, Nguyen CT, Ann J, Nam G, Park HJ, Lee J, et al. A novel HSP90 inhibitor targeting the C-terminal domain attenuates trastuzumab resistance in HER2-positive breast cancer. Mol Cancer. 2020;19(1):161.
- Eroglu Z, Chen YA, Gibney GT, Weber JS, Kudchadkar RR, Khushalani NI, Markowitz J, Brohl AS, Tetteh LF, Ramadan H, et al. Combined BRAF and HSP90 inhibition in patients with unresectable BRAF (V600E)-mutant melanoma. Clin Cancer Res. 2018;24(22):5516–5524.
- Li L, Chen NN, You QD, Xu XL. An updated patent review of anticancer Hsp90 inhibitors (2013-present). Expert Opin Ther Pat. 2021;31(1):67–80.
- Xiao Y, Liu Y. Recent advances in the discovery of novel HSP90 inhibitors: an update from 2014. Curr Drug Targets. 2020;21(3):302–317.
- Koren J, Blagg BSJ. The right tool for the job: an overview of Hsp90 Inhibitors. Adv Exp Med Biol. 2020;1243:135–146.
- Bickel D, Gohlke H. C-terminal modulators of heat shock protein of 90 kDa (HSP90): state of development and modes of action. Bioorg Med Chem. 2019;27(21):115080.
- Byrd JC. HSP90 inhibition without heat shock response. Blood. 2018;132(3):241–242.
- Li L, Wang L, You QD, Xu XL. Heat shock protein 90 inhibitors: an update on achievements, challenges, and future directions. J Med Chem. 2020;63(5):1798–1822.
- Liew HY, Tan XY, Chan HH, Khaw KY, Ong YS. Natural HSP90 inhibitors as a potential therapeutic intervention in treating cancers: a comprehensive review. Pharmacol Res. 2022;181:106260.
- Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson SD, Verkhivker GM, Blagg BS. Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chem Biol. 2011;6(8):800–807.
- Khalid S, Paul S. Identifying a C-terminal ATP binding sites-based novel Hsp90-Inhibitor in silico: a plausible therapeutic approach in Alzheimer’s disease. Med Hypotheses. 2014;83(1):39–46.
- Morra G, Neves MAC, Plescia CJ, Tsustsumi S, Neckers L, Verkhivker G, Altieri DC, Colombo G. Dynamics-based discovery of allosteric inhibitors: selection of new ligands for the C-terminal domain of Hsp90. J Chem Theory Comput. 2010;6(9):2978–2989.
- Sgobba M, Degliesposti G, Ferrari AM, Rastelli G. Structural models and binding site prediction of the C-terminal domain of human Hsp90: a new target for anticancer drugs. Chem Biol Drug Des. 2008;71(5):420–433.
- Giordano D, Biancaniello C, Argenio MA, Facchiano A. Drug design by pharmacophore and virtual screening approach. Pharmaceuticals. 2022;15(5):646.
- Tomašič T, Durcik M, Keegan BM, Skledar DG, Zajec Ž, Blagg BSJ, Bryant SD. Discovery of novel Hsp90 C-terminal inhibitors using 3D-pharmacophores derived from molecular dynamics simulations. Int J Mol Sci. 2020;21(18):6898.
- Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem. 2000;275(47):37181–37186.
- Kusuma BR, Khandelwal A, Gu W, Brown D, Liu W, Vielhauer G, Holzbeierlein J, Blagg BS. Synthesis and biological evaluation of coumarin replacements of novobiocin as Hsp90 inhibitors. Bioorg Med Chem. 2014;22(4):1441–1449.
- Garg G, Forsberg LK, Zhao H, Blagg BSJ. Development of phenyl cyclohexylcarboxamides as a novel class of Hsp90 C-terminal inhibitors. Chemistry. 2017;23(65):16574–16585.
- Hyun SY, Le HT, Nguyen CT, Yong YS, Boo HJ, Lee HJ, Lee JS, Min HY, Ann J, Chen J, et al. Development of a novel Hsp90 inhibitor NCT-50 as a potential anticancer agent for the treatment of non-small cell lung cancer. Sci Rep. 2018;8(1):13924.
- Jiang F, Guo AP, Xu JC, Wang HJ, Mo XF, You QD, Xu XL. Identification and optimization of novel 6-acylamino-2-aminoquinolines as potent Hsp90 C-terminal inhibitors. Eur J Med Chem. 2017;141:1–14.
- Chang DJ, An H, Kim KS, Kim HH, Jung J, Lee JM, Kim NJ, Han YT, Yun H, Lee S, et al. Design, synthesis, and biological evaluation of novel deguelin-based heat shock protein 90 (HSP90) inhibitors targeting proliferation and angiogenesis. J Med Chem. 2012;55(24):10863–10884.
- Kim HS, Hong M, Ann J, Yoon S, Nguyen CT, Lee SC, Lee HY, Suh YG, Seo JH, Choi H, et al. Synthesis and biological evaluation of C-ring truncated deguelin derivatives as heat shock protein 90 (HSP90) inhibitors. Bioorg Med Chem. 2016;24(22):6082–6093.
- Mir F, Shafi S, Zaman MS, Kalia NP, Rajput VS, Mulakayala C, Mulakayala N, Khan IA, Alam MS. Sulfur rich 2-mercaptobenzothiazole and 1,2,3-triazole conjugates as novel antitubercular agents. Eur J Med Chem. 2014;76:274–283.
- Kurop MK, Huyen CM, Kelly JH, Blagg BSJ. The heat shock response and small molecule regulators. Eur J Med Chem. 2021;226:113846.
- Garcia-Carbonero R, Carnero A, Paz-Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol. 2013;14(9):e358–e369.
- Amatya E, Blagg BSJ. Recent advances toward the development of Hsp90 C-terminal inhibitors. Bioorg Med Chem Lett. 2023;80:129111.
- Peng S, Woodruff J, Pathak PK, Matts RL, Deng J. Crystal structure of the middle and C-terminal domains of Hsp90α labeled with a coumarin derivative reveals a potential allosteric binding site as a drug target. Acta Crystallogr D Struct Biol. 2022;78(Pt 5):571–585.
- Yang M, Li C, Li Y, Cheng C, Shi M, Yin L, Xue H, Liu Y. Design, synthesis, biological evaluation and molecular docking study of 2, 4-diarylimidazoles and 2, 4-bis (benzyloxy)-5-arylpyrimidines as novel HSP90 N-terminal inhibitors. J Enzyme Inhib Med Chem. 2022;37(1):2551–2565.