
Abstract
Orthosiphon aristatus is a well-known folkloric medicine and herb for Guangdong soup for the treatment of rheumatism in China. Eight isopimarane-type and migrated pimarane-type diterpenoids (1-8), including a new one with a rarely occurring α,β-unsaturated diketone C-ring, were isolated from O. aristatus. Their structures were determined by spectroscopic methods and quantum chemical calculations. Furthermore, the most abundant compound, orthosiphol K, was structurally modified by modern synthetic techniques to give seven new derivatives (9-15). The anti-rheumatoid arthritis activity of these diterpenoids were evaluated on a TNF-α induced MH7A human rheumatoid fibroblast-like synoviocyte model. Compound 10 showed the most potent activity among these compounds. Based on their inhibitory effects on the release levels of IL-1β, the preliminary structure-activity relationships were concluded. Furthermore, western blot analysis revealed that 10 could increase the expression of IκBα and decrease the expression of NF-κB p65, and the expression levels of COX-2 and NLRP3 proteins were consequently down-regulated.
Graphical Abstract
Introduction
Rheumatoid arthritis (RA), a chronic autoimmune disease characterised by progressive inflammation, synovial hyperplasia, and joint destruction and deformity, affects up to 1% of the world’s population and over 5 million patients are suffering from it in ChinaCitation1. Inflammation is one of the most important factors throughout the courses of RA. Proinflammatory cytokines, such as TNF-α, IL-1β, IL-6, etc., not only recruit various immune cells that release more cytokines and cause persistent inflammation in joints, but also trigger the activation of resident fibroblast-like synoviocytes and contribute to cartilage and bone damageCitation2–4. Therefore, controlling the level of proinflammatory cytokines in synoviocytes has been a key stratagem in RA treatment. Inflammatory cytokines are involved in the pathogenesis of RA through activation of several signalling pathways, such as MAPK, NF-κB, PI3K/AKT, JAK/STAT, etcCitation5. The activation of NF-κB is a positive feedback process of proinflammatory cytokines, forming a vicious cycle and leading to abnormal fibroblast-like synoviocytes apoptosis in RACitation6,Citation7. Recently, the priming and activation of NLRP3 inflammasome have been revealed to play important role in the development of RA, in which the priming process is deeply associated with the NF-κB signalling pathway. Activation of NF-κB signalling pathway upregulates the expression of NLRP3 and pro-IL-1β. Under multiple stimuli, NLRP3 binds to the adaptor protein ASC and recruits pro-caspase-1 to finally form the NLRP3 inflammasome. Activated NLRP3 inflammasome activates caspase-1 to produce IL-1 and IL-18, which makes inflammasome an connector between innate and adaptive immunityCitation8,Citation9. The above mechanistic understanding is leading to potential therapeutics that target the cytokines, NF-κB pathway, and NLRP3 inflammasome.
Digging of therapeutical natural products from folkloric medicines is still a practical way for drug discovery. Orthosiphon aristatus (synonym: Clerodendranthus spicatus), also called “kidney tea” in Chinese, is a well-known folkloric medicine and herb for Guangdong soup for the treatment of rheumatism, nephritis, urolithiasis, diabetes, hypertension, etc.Citation10,Citation11. Previous phytochemical studies on this genus showed that flavonoidsCitation12, phenolic acidsCitation13,Citation14, diterpenoidsCitation15,Citation16, triterpenesCitation17, and lignansCitation18 are main components of these plants. Among them, the characteristic constituent is isopimarane-type diterpenoid, which has already been demonstrated to have various pharmacological activities, such as anti-inflammationCitation19, anti-hyperglycemiaCitation20, antitumorCitation21, and renoprotectionCitation22,Citation23.
In the present study, eight isopimarane-type and migrated pimarane-type diterpenoids (1–8) of O. aristatus were isolated and structurally characterised, including a new one (1) with a rarely occurring α,β-unsaturated diketone C-ring. The high content compound, orthosiphol K (4), was further chemically modified to obtain seven new derivatives (9–15). Their anti-inflammatory activity on TNF-α induced MH7A human rheumatoid fibroblast-like synoviocytes were evaluated by measuring the pro-inflammatory cytokines using ELISA, and the expression levels of several NF-κB signalling pathway related proteins were determined using Western blot technique.
Result and discussion
Structure identification of 1–8
Compound 1 was isolated as a light yellow powder and assigned a molecular formula of C20H28O6 on the basis of a positive HR-ESI-MS pseudo-ion peak at m/z 387.1776 [M + Na]+ (calcd for C20H28O6Na, 387.1778). The IR spectrum showed vibrational absorption peaks at 1674, 1649, and 1618 cm−Citation1 for carbonyl and double bonds. The 1H NMR spectrum () of 1 indicated a typical terminal monosubstituted double bond (vinyl group) [δH 6.16 (dd, J = 17.4, 10.8 Hz, H-15), 5.13 (d, J = 10.8 Hz, H-16a), δH 5.10 (d, J = 17.4 Hz, H-16b)] and four methyl singlets [δH 1.08 (s, H-17), 1.24 (s, H-18), 0.94 (s, H-19), δH 1.27 (s, H-20)]. The 13C NMR () and DEPT 135 spectra demonstrated resonances for 20 carbons, including 4 methyl groups, 3 methylene groups, 6 methine carbons, and 7 quaternary carbons. Two carbons resonated at δc 201.9 and 200.4 indicated two ketone carbonyl groups, similar to those of the isopimarane-type diterpenoid 3-O-deacetylrothosiphol I (7) and 2-O-deacetylorthosiphol J (8) ()Citation24. Compared with the NMR data of compounds 7 and 8, the absence of signals for benzoyl and acetoxy groups, combined with the presence of signals (δc 157.0 and 143.7) assignable to an additional tetrasubstituted double bond suggested that the CH(9)-C(8)-OH moiety might be dehydrated to form a double bond. The above elucidation was confirmed by 2D NMR experiments. Based on assignments of hydrogens to their bonding carbons by HMQC spectrum, the 1H-1H COSY spectrum indicated three main correlated fragments as shown in bold bonds in . Key HMBC correlations from H-7 to C-8, C-9, and C-14, from H2-12 to C-11 and C-9, and from H3-17 and H-15 to C-14 verified the rarely occurring C ring with α,β-unsaturated diketone structure. The relative configuration of 1 was determined by NOESY experiment. The NOESY spectrum showed correlations of H3-20 with H-1, H-2, and H3-19, H-2 with H-1, H-3, and H3-19, and H-5 with H3-18, indicating the relative configuration around ring A (). NOESY correlations of H3-19/H-6β, H-7/H-6β, H3-20/H-6β revealed the α-orientation of the hydroxy group on C-7. The C-13 position is too far away from the chiral centres of rings A and B, therefore, its configuration cannot be easily determined by NOESY spectrum. From biogenetic point of view, the configuration at C-13 of these compounds is conservative. Comparison of chemical shifts of C-13, and C-15 − C-17 of compound 1 with those of 7 showed high similarity, which indicated again that the C-13 configuration of 1 should be in consistent with its analogues. This deduction was further confirmed by quantum chemical calculations of NMR spectra on structures of the 17β-CH3 configuration 1a and 17α-CH3 configuration 1b (see Supplementary data). DP4+ probability analysis of the calculated NMR data of the two epimers revealed that the final score of 1a (100.00%) showed an absolute advantage over that of 1b, which again suggested 17β-CH3 configuration 1a as the correct relative configuration.
Figure 1. Chemical structures of diterpenoids 1 − 8 isolated from O. aristatus.
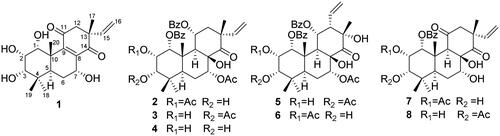
Figure 2. 1H-1H COSY (bold lines), HMBC (blue arrows), and NOESY (dash line arrows) correlations of compound 1.
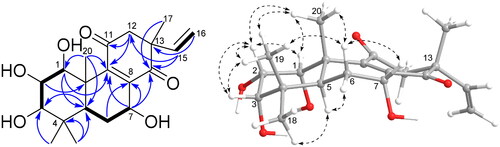
Table 1. 1H and 13C NMR spectral data (500 MHz for 1H and 125 MHz for 13C in methanol-d4) of compound 1 (δ in ppm, J in Hz).
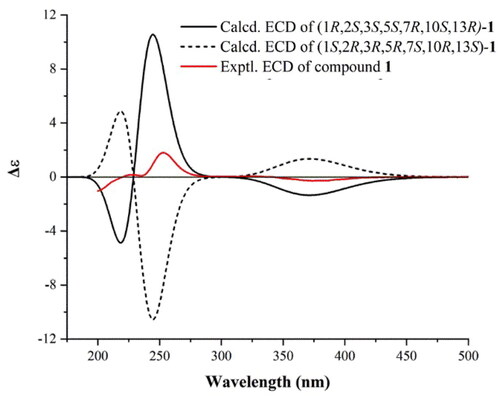
In order to further determine the absolute configuration of compound 1, the theoretical electronic circular dichroism (ECD) spectrum was obtained by quantum chemical calculations. As shown in , both the experimental ECD curve of compound 1 and the theoretical ECD curve of 1 R,2S,3S,5S,7R,10S,13R-1 showed a negative first Cotton effect in the 310–430 nm region, and a positive second Cotton effect in the 225–300 nm region, which allowed the determination of the absolute configuration of compound 1 as 1 R,2S,3S,5S,7R,10S,13R. According to literature survey, the α,β-unsaturated diketone structure on the C ring is very rare among the isopimarane-type diterpenoids, and compound 1 was named as clerodendranthin G according to the naming order.
Figure 3. Experimental ECD curve of compound 1 (solid red line), and M062X/TZVP//B3LYP/6-31G(d) calculated ECD spectra of 1 R,2S,3S,5S,7R,10S,13R-1 (solid black line) and 1S,2R,3R,5R,7S,10R,13S-1 (dash black line).
In addition to the new compound 1, seven known diterpenoids () were isolated from the EtOAc and CH2Cl2 extracts. By comparison of their spectroscopic data with those reported in literatures, the structures of them were identified as orthosiphol A (2)Citation25, orthosiphol B (3)Citation25, orthosiphol K (4)Citation26, neoorthosiphol A (5)Citation27, neoorthosiphol B (6)Citation27, 3-O-deacetylorthosiphol I (7)Citation24, 2-O-deacetylorthosiphol J (8)Citation24.
Structure modification of orthosiphol K (4)
To expand the structural diversity of the isolated diterpenoids, structure modification of the most abundant compound orthosiphol K (4) was carried out and seven new derivatives were obtained. (Scheme 1). Briefly, etherification reaction of 2-OH with 3-OMe benzyl bromide or ethyl bromide gave compounds 9 and 10, respectively. Esterification reaction of 2-OH with propionic acid and DMAP produced compound 11. Methylation of 2-OH with methyl iodide created compound 12, and further Jones reagent oxidised 3-OH secondary alcohol to ketone carbonyl group in 13. Directly oxidisation of 4 by Jones reagent gave compound 14. C-1 and C-11 esters were hydrolysed to alcohol in ammonium hydroxide-methanol system to give compound 15.
Scheme 1. Structures and reactions in modifications of orthosiphol K (4).a
a Reagents and conditions: (a) Cs2CO3, DMF, 3-OMePhCH2Br, 80 °C, reflux; (b) Cs2CO3, DMF, CH3CH2Br, 80 °C, reflux; (c) Propanoic acid, DMAP, EDCI, CH2Cl2, 0-25 °C,6h; (d) Cs2CO3, DMF, MeI, r.t., 24h; (e) Jones reagent, acetone, 0-25 °C, 1.5-2h; (f) NH4OH/MeOH, 110 °C, reflux, 0.5-1h.
Anti-rheumatoid arthritis activity and SAR study
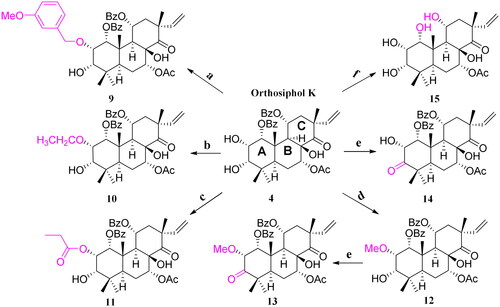
Since inflammation is one of the most important factors throughout the courses of rheumatoid arthritis, the anti-rheumatoid arthritis activity of all the diterpenoids were evaluated on a TNF-α induced MH7A human rheumatoid fibroblast-like synoviocyte model. MTT cell viability assay was firstly performed for the 15 diterpenoids on MH7A cell model induced by TNF-α. The results showed that, except compounds 10, 11, 12, and 14 showed cytotoxicities at 20 or 50 μM, other compounds exhibited no significant effects on cell viabilities in the range of 5–50 μM (See Supplementary data). From another point of view, inhibition of synovial cell proliferation is also of positive significance for the treatment of rheumatoid arthritis. In order to eliminate the influence of cytotoxicity on the subsequent anti-inflammation experiments, a concentration of 15 μM was selected according to the cell viability assay.
As shown in of ELISA tests results, the release content of pro-inflammatory cytokines IL-1β and IL-6 in the model group was very significantly increased after TNF-α (10 ng/mL) treatment, and compounds 1, 2, 3, 4, 10, 11, 12, 13, and 14 could effectively inhibit the release of both of the two pro-inflammatory cytokines. Among them, compounds 10 and 3 showed the most significance (p < 0.0001) vs. the model group, and their inhibitory efficacy at 15 μM were in the same level as that of positive control dexamethasone (5 μM). By comparing the release levels of IL-1β of the 15 compounds with their structures, preliminary SARs (structure-activity relationships) could be concluded. Briefly, substitutes on C-ring significantly influenced the activity. The vinyl migrated pimarane type diterpenoids showed no activity vs. the isopimarane type diterpenoids (5 vs. 3, 6 vs. 2). C-11 OBz group and C-7 OAc group are important to maintain the activity (7 vs. 2, 8 vs. 3, and 15 vs. 4). For substitutes on ring A, electron withdrawing group on 2-hydroxyl decreased the activity and electron donating group improved the activity (2 vs. 4, 2/9/11 vs. 4/12/10, 4 vs. 12/10). Oxidation of 3-OH into carbonyl will decrease the activity (12 vs. 13) and Ac substitutes on it may enhance the activity (3 vs. 4). Therefore, substituents on ring C and A significantly influenced the activity of the isopimarane type diterpenoids.
Figure 4. Cytokine release levels in TNF-α-induced MH7A cell model. (A) IL-1β release levels. (B) IL-6 release levels (####p < 0.0001, vs. control group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, vs. model group).
Compound 10 suppressed the activation of NF-κB pathway
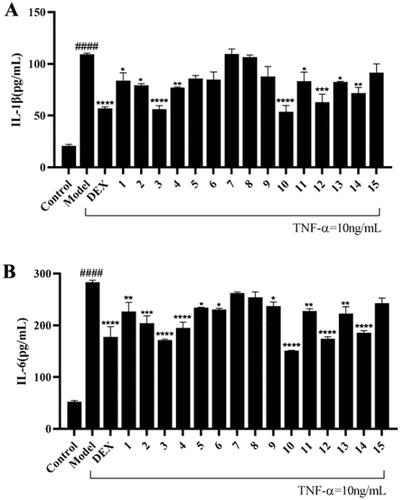
Compound 10 showed the most potent anti-inflammatory activity and was further studied for its regulation on the expression of NF-κB signalling pathway related proteins. As introduced in the previous part, IκBα and NF-κB p65 are important proteins in the activation of the NF-κB pathway. The induced intracellular NF-κB p65 protein is stimulated to transfer from the cytoplasm to the nucleus, thereby up-regulating the expression of COX-2. In the forming of the inflammasome, the activation of NF-κB pathway can stimulate the expression of NLRP3 protein, which is one of the important components of NLRP3 inflammasome. As shown in , the expression level of IκBα, an inhibitor of NF-κB, was down-regulated and NF-κB p65 protein in MH7A cells was up-regulated after the induction of TNF-α, indicating the activation of NF-κB pathway that leads to inflammation. Compound 10 can up-regulate the expression level of IκBα protein and down-regulate the expression level of p65 protein. The expression levels of COX-2 and NLRP3 was consequently increased after TNF-α stimulation, while they were decreased after dexamethasone and compound 10 treatment. These results suggested that the anti-inflammatory effect of compound 10 may be caused by inhibiting the activation of NF-κB signalling pathway and down-regulating the expression levels of COX-2 and NLRP3 proteins consequently.
Figure 5. Effect of compound 10 on TNF-α-induced IκBα, NF-κB p65, COX-2, and NLRP3 protein expression in MH7A cells. (A) Western blot analysis and (B) Quantified protein expression levels. (#p < 0.05, ##p < 0.01, ####p < 0.0001, vs control group; *p < 0.05, ***p < 0.001, ****p < 0.0001, vs. model group).
Conclusion
Rheumatoid arthritis is a severe chronic disease that needs continuous discovery of innovative drugs. Natural products are still important sources for the discovery of new drug lead compounds, especially those inspired by traditional applications. By modern phytochemical and synthetic techniques, 15 isopimarane type or migrated pimarane type diterpenoids were isolated and prepared. Some of them showed significant anti-inflammatory activity on a TNF-α induced MH7A human rheumatoid fibroblast-like synoviocyte model, and the primary SAR were discussed. These diterpenoids may inhibit the activation of NF-κB pathway and consequently down-regulate the expression levels of the key inflammation related COX-2 and NLRP3 proteins. The above findings not only revealed the chemical basis of O. aristatus for the treatment of RA, but also indicated the isopimarane diterpenoid maybe a good lead for anti-RA drug development.
Experimental section
General experimental procedures
Plant material and isolation
Spectroscopic data of new compound 1
Clerodendranthin G (1)
Yellow amorphous powder; [α]D20 = +7.7 (c 0.52, MeOH); UV (MeOH) λmax (log ε) 237 (3.67) nm; ECD (MeOH) λmax (Δε) 253 (+1.83), 375 (− 0.26) nm; IR (KBr) νmax 3366, 2962, 2925, 1674, 1649, 1618, 1381, 1266, 1065, 948, 920 cm−Citation1; 1H and 13C NMR data, see ; HR-ESI-MS m/z 387.1776 [M + Na]+ (calcd for C20H28O6Na, 387.1778).
Quantum chemical calculation of compound 1
Synthesis of compounds 9 − 15
The reaction conditions for the synthesis of above-mentioned compounds are shown in Scheme 1.
2–(3-Methoxybenzyloxy)-orthosiphol K (9)
To a solution of 4 (0.002 mmol) in DMF (1 ml), 3-methoxybenzyl bromide (0.004 mmol) and Cs2CO3 (0.004 mmol) were added, and then the mixture was refluxed at 80 °C for 1–2 h. The reaction process was monitored by TLC. The reaction mixture was then diluted with ethyl acetate and washed three times with pure water, 5% sodium chloride solution, and saturated sodium chloride solution, successively. The organic phase was concentrated and purified by preparative TLC (petroleum ether: EtOAc: acetone = 10:2:1) to give compound 9 (13.6 mg, 55% yield). White amorphous powder; ESI-MS m/z 755.6 [M + H]+; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 7.3 Hz, 2H, 1-OCOPh), 7.63 (t, J = 7.5 Hz, 1H, 1-OCOPh), 7.55 (d, J = 6.9 Hz, 2H, 11-OCOPh), 7.46 (t, J = 7.3 Hz, 1H, 11-OCOPh), 7.39 (t, J = 7.6 Hz, 2H, 11-OCOPh), 7.29 (t, J = 7.9 Hz, 1H, H-23), 7.13 (t, J = 7.8 Hz, 2H, 1-OCOPh), 6.86 (dd, J = 8.2, 2.5 Hz, 1H, H-26), 6.84 (d, J = 7.2 Hz, 1H, H-25), 6.78 (t, J = 2.1 Hz, 1H, H-27), 5.92 (d, J = 5.9 Hz, 1H, H-11), 5.87 (dd, J = 4.0, 1.9 Hz, 1H, H-2), 5.73 (dd, J = 17.6, 10.6 Hz, 1H, H-15), 5.02 (d, J = 3.0 Hz, 1H, H-1), 4.95 (d, J = 17.6 Hz, 1H, H-16 cis), 4.92 (s, 1H, H-7), 4.84 (d, J = 10.7 Hz, 1H, H-16 tran), 4.54 (d, J = 11.2 Hz, 1H, H-21α), 4.41 (dd, J = 3.7, 2.0 Hz, 1H, H-3), 4.14 (d, J = 11.2 Hz, 1H, H-21β), 3.82 (s, 3H, 25-OMe), 3.40 (d, J = 7.1 Hz, 1H, H-9), 2.50 (dd, J = 15.6, 5.5 Hz, 1H, H-12α), 2.45 (dd, J = 13.3, 2.3 Hz, 1H, H-5), 2.24 (s, 3H, 7-OCOCH3), 2.01 (dt, J = 14.7, 3.0 Hz, 2H, H-6), 1.83 (d, J = 7.7 Hz, 1H, H-12β), 1.39 (s, 3H, Me-20), 1.06 (d, J = 1.2 Hz, 6H, Me-17, Me-19), 0.86 (s, 3H, Me-18); 13C NMR (125 MHz, CDCl3) δ 205.0 (C-14), 168.8 (7-OCOCH3), 167.8 (1-OCOPh), 166.3 (11-OCOPh), 159.9 (C-24), 142.4 (C-15), 138.3 (C-22), 133.7, 130.5, 130.1, 128.3, (1-OCOPh), 132.9, 130.0, 129.7, 128.2 (11-OCOPh), 129.7 (C-26), 119.8 (C-27), 113.8 (C-16), 113.4 (C-23), 113.4 (C-25), 80.3 (C-2), 78.7 (C-8), 78.4 (C-7), 69.0 (C-11), 66.5 (C-3), 66.3 (C-1), 66.2 (C-21), 55.4 (24-OMe), 48.0 (C-13), 44.2 (C-10), 42.4 (C-9), 39.9 (C-12), 37.2 (C-4), 36.5 (C-5), 27.9 (C-18), 25.3 (C-17), 22.8 (C-19), 21.7 (C-6), 20.4 (7-OCOCH3), 16.9 (C-20).
2-Ethyloxy-orthosiphol K (10)
The prepare process of 10 was similar as that of 9, except ethyl bromide was applied instead of 3-methoxybenzyl bromide for 9. Compound 10 (5.5 mg, 53% yield) was purified by HPLC (methanol: water = 8: 2, tR= 15.5 min). White amorphous powder; ESI-MS (positive) m/z 685.4 [M + Na]+;1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 7.3 Hz, 2H, 1-OCOPh), 7.62 (t, J = 7.5 Hz, 1H, 1-OCOPh), 7.56 (d, J = 7.0 Hz, 2H, 11-OCOPh), 7.46 (t, J = 7.5 Hz, 1H, 11-OCOPh), 7.39 (t, J = 7.6 Hz, 2H, 11-OCOPh), 7.13 (t, J = 7.6 Hz, 2H, 1-OCOPh), 5.90 (t, J = 6.3 Hz, 1H, H-11), 5.72 (dd, J = 17.4, 10.5 Hz, 1H, H-15), 5.72 (s, 1H, H-7), 5.03 (d, J = 2.6 Hz, 1H, H-1), 4.91 (d, J = 18.0 Hz, 1H, H-16 cis), 4.82 (d, J = 10.3 Hz, 1H, H-16 tran), 4.41 (dd, J = 3.8, 2.1 Hz, 1H, H-2), 3.53 (d, J = 7.3 Hz, 2H, H-21), 3.32 (d, J = 7.1 Hz, 1H, H-9), 3.12 (d, J = 7.0 Hz, 1H, H-3), 2.53 (dd, J = 15.4, 5.3 Hz, 1H, H-12α), 2.41 (dd, J = 13.4, 2.3 Hz, 1H, H-5), 2.21 (s, 3H, 7-OCOCH3), 2.03–1.95 (m, 2H, H-6), 1.85 (d, J = 15.5 Hz, 1H, H-12β), 1.39 (s, 3H, Me-20), 1.25 (s, 3H, Me-22), 1.10 (s, 3H,Me-17), 1.08 (s, 3H, Me-19), 0.86 (s, 3H, Me-18); 13C NMR (125 MHz, CDCl3) δ 205.0 (C-14), 168.9 (7-OCOCH3), 167.9 (1-OCOPh), 166.3 (11-OCOPh), 142.5 (C-15), 133.6, 130.5, 130.1, 128.3, (1-OCOPh), 132.9, 130.1 129.7, 128.2 (11-OCOPh), 113.7 (C-16), 79.9 (C-2), 78.8 (C-8), 78.4 (C-7), 69.1 (C-11), 66.6 (C-3), 66.3 (C-1), 59.6 (C-21), 47.8 (C-13), 44.2 (C-10), 42.2 (C-9), 39.9 (C-12), 37.2 (C-4), 36.5 (C-5), 27.9 (C-18), 25.3 (C-17), 22.8 (C-19), 21.7 (C-6), 21.2 (7-OCOCH3), 16.7 (C-20), 15.8 (C-22).
2-Propionyloxy-orthosiphol K (11)
To a solution of orthosiphol K (4) (0.002 mmol) in DMAP (0.004 mmol), EDCI (0.004 mmol) and propanoic acid (20 μL) were added, and then 1 ml of anhydrous CH2Cl2 was added under ice bath. The reaction mixture was stirred for 6 h at room temperature and then diluted with ethyl acetate and washed three times with pure water, 5% sodium chloride solution, and saturated sodium chloride solution, successively. The organic phase was concentrated and purified by preparative TLC (petroleum ether: acetone = 2:1) to give compound 11 (3.59 mg, 33% yield). White amorphous powder; ESI-MS (positive) m/z 691.3 [M + H]+; 1H NMR (500 MHz, CDCl3) δ 8.12 (d, J = 7.3 Hz, 2H, 1-OCOPh), 7.74 (t, J = 7.5 Hz, 2H, 11-OCOPh), 7.53 (d, J = 7.5 Hz, 1H, 11-OCOPh), 7.40 (t, J = 7.5 Hz, 1H, 1-OCOPh), 7.36 (t, J = 7.6 Hz, 2H, 11-OCOPh), 7.26(t, J = 7.6 Hz, 2H, 1-OCOPh), 6.11 (dd, J = 17.5, 10.7 Hz, 1H, H-15), 5.82 (ddd, J = 9.3, 6.4, 2.9 Hz, 1H, H-11), 5.47 (d, J = 3.3 Hz, 1H, H-2), 5.32 (s, 1H, H-7), 5.29 (d, J = 3.4 Hz, 1H, H-3), 5.24 (d, J = 10.7 Hz, 1H, H-16 tran), 5.09 (d, J = 17.5 Hz, 1H, H-16 cis), 3.87 (d, J = 3.0 Hz, 1H, H-1), 3.42 (dd, J = 9.3 Hz, 1H, H-5), 3.13 (d, J = 7.0 Hz, 1H, H-9), 2.53 (dd, J = 15.6, 8.8 Hz, 1H, H-12α), 2.16 (s, 3H, 7-OCOCH3), 2.11 (dd, J = 12.1, 2.5 Hz, 2H, H-21), 1.99 (d, J = 15.3 Hz, 1H, H-12β), 1.92 (dt, J = 14.1, 2.9 Hz, 2H, H-6), 1.33 (s, 3H, Me-20), 1.33 (s, 3H, Me-22), 1.12 (s, 3H, Me-17), 0.88 (s, 3H, Me-19), 0.88 (s, 3H, Me-18); 13C NMR (125 MHz, CDCl3) δ 209.7 (C-14), 169.5 (3-COOCH2CH3), 169.3 (7-OCOCH3), 166.2 (1-OCOPh), 165.3 (11-OCOPh), 140.5(C-15), 133.2, 130.6, 129.8, 128.4, (1-OCOPh), 133.0, 130.6 129.6, 128.4 (11-OCOPh), 115.7 (C-16), 78.1 (C-2), 76.3 (C-1), 73.9 (C-8), 71.3 (C-7), 68.5 (C-11), 67.7 (C-3), 48.5 (C-13), 44.3 (C-10), 41.7 (C-9), 40.1 (C-12), 37.5 (C-4), 35.2 (C-5), 29.5 (C-18), 28.0 (3-COOCH2CH3), 26.3 (C-17), 22.8 (C-19), 21.5 (C-6), 21.1 (7-OCOCH3), 15.7 (C-20), 14.3 (3-COOCH2CH3).
2-Methyloxy-orthosiphol K (12)
Orthosiphol K (4) (0.002 mmol) and Cs2CO3 (0.004 mmol) were dissolved in DMF (1 ml), and MeI (0.004 mmol) were added in dropwise under ice bath. The reaction mixture was then heated to 80 °C and stirred for 24 h. Compound 12 (16.2 mg, 89% yield) was obtained by the same post-processing protocol as that of 11. White amorphous powder; ESI-MS (negative) m/z 647.4 [M − H]−;1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.0 Hz, 2H, 1-OCOPh), 7.62 (t, J = 7.3 Hz, 1H, 1-OCOPh), 7.56 (d, J = 7.6 Hz, 2H, 11-OCOPh), 7.46 (t, J = 7.4 Hz, 1H, 11-OCOPh), 7.38 (t, J = 7.6 Hz, 2H, 11-OCOPh), 7.13 (t, J = 7.6 Hz, 2H, 1-OCOPh), 5.88 (t, J = 5.8 Hz, 1H, H-11), 5.72 (dd, J = 17.7, 10.3 Hz, 1H, H-15), 5.72 (s, 1H, H-7), 5.02 (d, J = 3.5 Hz, 1H, H-1), 4.92 (d, J = 16.9 Hz, 1H, H-16 cis), 4.82 (d, J = 10.7 Hz, 1H, H-16 tran), 4.41 (dd, J = 3.5, 2.0 Hz, 1H, H-2), 3.32 (d, J = 7.2 Hz, 1H, H-9), 3.17 (s, 3H, OMe-2), 3.12 (d, J = 7.0 Hz, 1H, H-3), 2.48 (dd, J = 15.4, 5.4 Hz, 1H, H-12α), 2.41 (dd, J = 13.4, 2.3 Hz, 1H, H-5), 2.22 (s, 3H, 7-OCOCH3), 1.98 (dt, J = 14.6, 3.1 Hz, 2H, H-6), 1.86 (dd, J = 15.5, 1.4 Hz, 1H, H-12β), 1.49 (s, 3H, Me-20), 1.10 (s, 3H, Me-17), 1.08 (s, 3H, Me-19), 0.85 (s, 3H, Me-18); 13C NMR (125 MHz, CDCl3) δ 204.8 (C-14), 168.9 (7-OCOCH3), 167.8 (1-OCOPh), 166.3 (11-OCOPh), 142.5(C-15), 133.6, 130.5, 129.7, 128.3, (1-OCOPh), 132.9, 130.1 129.7, 128.2 (11-OCOPh), 113.3 (C-16), 79.9 (C-2), 78.8 (C-8), 78.4 (C-7), 69.0 (C-11), 66.3 (C-3), 66.1 (C-1), 51.5 (OMe-2), 47.9 (C-13), 44.2 (C-10), 42.2 (C-9), 39.9 (C-12), 37.2 (C-4), 36.5 (C-5), 27.9 (C-18), 25.3 (C-17), 22.8 (C-19), 21.5 (C-6), 21.1 (7-OCOCH3), 16.7 (C-20).
2-Methyloxy-3-oxo-orthosiphol K (13)
Compound 12 (0.002 mmol) was dissolved in acetone (0.6 ml), and Jones reagent (0.3 ml) was slowly added in dropwise under ice bath. The reaction mixture was stirred for 2 h at room temperature. The excess Jones reagent was quenched with large amount of methanol. The mixture was concentrated and re-dissolved in ethyl acetate and washed three times with pure water, 5% sodium chloride solution, and saturated sodium chloride solution, successively. Compound 13 (10.8 mg, 68% yield) was obtained by preparative TLC (petroleum ether: EtOAc = 2:1). White amorphous powder; ESI-MS (positive) m/z 685.4 [M + K]+; 1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 8.0 Hz, 2H, 1-OCOPh), 7.64 (t, J = 7.3 Hz, 1H, 11-OCOPh), 7.56 (d, J = 8.0 Hz, 2H, 11-OCOPh), 7.43 (t, J = 7.3 Hz, 2H, 1-OCOPh), 7.35 (t, J = 7.5 Hz, 1H, 1-OCOPh), 6.96 (t, J = 7.5 Hz, 2H, 11-OCOPh), 5.88 (dd, J = 17.6, 10.7 Hz, 1H, H-15), 5.79 (t, J = 5.9 Hz, 1H, H-11), 5.74 (s, 1H, H-7), 5.11 (d, J = 10.4 Hz, 1H, H-16 tran), 5.06 (d, J = 17.0 Hz, 1H, H-16 cis), 4.90 (s, 1H, H-2), 4.39 (s, 1H, H-1), 3.59 (d, J = 7.4 Hz, 1H, H-9), 3.18 (s, 3H, OMe-2), 2.68 (d, J = 12.9 Hz, 1H, H-5), 2.45 (dd, J = 15.6 5.5 Hz, 1H, H-12α), 2.21 (s, 3H, 7-COCH3), 2.00–1.96 (m, 2H, H-6), 1.86 (t, J = 13.9 Hz, 1H, H-12β), 1.33 (s, 3H, Me-20), 1.16 (s, 3H, Me-17), 0.98 (s, 3H, Me-19), 0.88 (s, 3H, Me-18); 13C NMR (125 MHz, CDCl3) δ 204.9 (C-14), 200.1 (C-3), 168.9 (7-OCOCH3), 165.7 (1-OCOPh), 164.6 (11-OCOPh), 142.5(C-15), 133.7, 130.0, 129.6, 128.6, (1-OCOPh), 133.0, 129.8 129.6, 128.1 (11-OCOPh), 113.8 (C-16), 82.4 (C-2), 81.1 (C-1), 79.9 (C-8), 68.5 (C-11), 66.1 (C-7), 51.6 (OMe-2), 48.1 (C-13), 46.2 (C-10), 42.5 (C-9), 40.9 (C-12), 39.3 (C-4), 37.4 (C-5), 26.9 (C-18), 25.3 (C-17), 21.8 (C-19), 21.7 (C-6), 21.1 (7-OCOCH3), 19.9 (C-20).
3-Oxo-orthosiphol K (14)
By the same protocol as that of 13, compound 4 (0.002 mmol) as applied instead of 12. 14 (11.8 mg, 94% yield) was obtained by HPLC (ACN: H2O = 70:30). White amorphous powder; ESI-MS (positive) m/z 671.2 [M + K]+; 1H NMR (500 MHz, CDCl3) δ 7.94 (d, J = 7.4 Hz, 2H, 1-OCOPh), 7.66 (t, J = 7.4 Hz, 1H, 1-OCOPh), 7.58 (d, J = 8.0 Hz, 2H, 11-OCOPh), 7.46 (t, J = 7.7 Hz, 2H, 11-OCOPh), 7.36 (t, J = 7.5 Hz, 1H, 11-OCOPh), 6.98 (t, J = 7.7 Hz, 2H, 1-OCOPh), 5.97 (dd, J = 17.6, 10.7 Hz, 1H, H-15), 5.84 (t, J = 6.8 Hz, 1H, H-11), 5.50 (s, 1H, H-7), 5.15 (d, J = 10.7 Hz, 1H, H-16 tran), 5.06 (d, J = 17.6 Hz, 1H, H-16 cis), 4.91 (s, 1H, H-1), 4.41 (s, 1H, H-2), 3.59 (d, J = 7.8 Hz, 1H, H-9), 2.69 (dd, J = 13.2, 2.8 Hz, 1H, H-5), 2.58 (dd, J = 15.7, 5.9 Hz, 1H, H-12α), 2.20 (s, 3H, 7-OCOCH3), 1.86 (d, J = 13.0 Hz, 1H, H-12β), 2.03–1.98 (m, 2H, H-6), 1.32 (s, 3H, Me-20), 1.17 (s, 3H, Me-17), 1.10 (s, 3H, Me-19), 0.89 (s, 3H, Me-18); 13C NMR (125 MHz, CDCl3) δ 209.1 (C-14), 200.1 (C-3), 168.8 (7-OCOCH3), 165.7 (1-OCOPh), 164.6 (11-OCOPh), 141.5(C-15), 133.7, 130.0, 129.7, 128.6, (1-OCOPh), 133.0, 129.8 129.6, 128.2 (11-OCOPh), 114.6 (C-16), 82.5 (C-2), 81.2 (C-1), 75.6 (C-8), 70.9 (C-7), 68.3 (C-11), 48.3 (C-13), 46.1 (C-10), 41.5 (C-9), 41.0 (C-12), 39.7 (C-4), 37.6 (C-5), 26.9 (C-18), 25.9 (C-17), 21.9 (C-19), 21.6 (C-6), 21.1 (7-OCOCH3), 19.9 (C-20).
1,11-Didebenzoxyl-orthosiphol K (15)
Orthosiphol K (4) (0.002 mmol) was dissolved in a mixture of ammonium hydroxide 0.6 ml and MeOH 0.5 ml and refluxed at 110 °C for 1 h. The mixture was diluted by ethyl acetate and washed three times with pure water, 5% sodium chloride solution, and saturated sodium chloride solution, successively. The organic phase was concentrated and purified by preparative TLC (CH2Cl2: MeOH = 15:1) to obtain compound 15 (8.6 mg, 74% yield). White amorphous powder; ESI-MS (positive) m/z 449.2 [M + Na]+; 1H NMR (500 MHz, CD3OD) δ 6.27 (dd, J = 17.6, 10.7 Hz, 1H, H-15), 5.28 (dd, J = 3.4, 1.6 Hz, 1H, H-7), 5.04 (d, J = 10.7 Hz, 1H, H-16 tran), 4.93 (dd, J = 17.7, 1.2 Hz, 1H, H-16 cis), 4.34 (d, J = 7.1 Hz, 1H, H-11), 3.84 (d, J = 3.1 Hz, 1H, H-2), 3.67 (d, J = 4.3 Hz, 1H, H-1), 2.50 (d, J = 7.3 Hz, 1H, H-9), 2.38 (dd, J = 14.8, 5.5 Hz, 1H, H-12α), 1.99 (dd, J = 11.2, 1.5 Hz, 1H, H-5), 1.97–1.94 (m, 2H, H-6), 1.93 (s, 3H, 7-OCOCH3), 1.85–1.78 (m, 1H, H-12β), 1.17 (s, 6H, Me-20, Me-17), 0.92 (s, 3H, Me-19), 0.91 (s, 3H, Me-18); 13C NMR (125 MHz, MeOD) δ 210.3 (C-14), 171.4 (7-COCH3), 143.0 (C-15), 114.5 (C-16), 81.4 (C-3), 77.9 (C-1), 76.3 (C-8), 72.9 (C-7), 65.6 (C-2), 65.5 (C-11), 46.7 (C-13), 45.0 (C-10), 43.0 (C-9), 39.3 (C-12), 36.1 (C-4), 33.0 (C-5), 29.0 (C-18), 26.7 (C-17), 23.0 (C-19), 22.1 (C-6), 21.1 (7-OCOCH3), 16.8 (C-20).
Cell culture and treatments
MH7A cells (Guangzhou Jennio Biotech Co., Ltd, P. R. China) were maintained in DMEM supplied with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin with a humidified atmosphere 5% CO2 at 37 °C. Passages between 4 and 7 of MH7A cells were used in the current study. All the compounds and other agents were dissolved in DMSO as stock solutions. MH7A cells were seeded in 96-well culture plates (Citation5 × 103 cell/well) for 24 h and then pre-treated or untreated with 15 μM tested compounds for 2 h, then incubated for another 24 h with or without stimulation of 10 ng/mL of TNF-α.
Cell viability assay
MH7A cell viability was detected using the MTT assay (BioFroxx, Guangzhou saiguo biotech Co., Ltd, Guangzhou, China). After incubation with compounds and TNF-α (Shanghai Chamot Biotech Co., Ltd, P. R. China.), the medium was replaced with fresh medium containing 0.5 mg/mL MTT. After incubation for 4 h, the medium was removed and 100 μL DMSO was added and incubated at 37 °C for 15 min on a plate shaker. The optical density (OD) values were measured using a microplate reader (Thermo Scientific Multiskan GO) at 550 nm. Each group had 5 duplicate wells and repeated 3 times.
Enzyme-linked immunosorbent assay (ELISA)
After incubation with compounds and TNF-α, the IL-1β and IL-6 released into the conditioned media were detected using specific ELISA Ready-SET-Go kits (Enzyme linked Biotech Co., Ltd, Shanghai, P. R. China). The release levels were quantified according to manufacturer’s protocols.
Western blot analysis
RIPA cell lysate buffer was mixed with phosphatase and protease inhibitors cocktail to lyse and extract total proteins. Total protein concentration was detected with the bicinchoninic acid (BCA) protein detection kit (Biosharp, Beijing, China). All protein samples were electrophoretic separated in equal amounts in 10% sodium dodecyl sulphate polyacrylamide (SDS-PAGE) gel and electrophoretic blot in polyvinylidene difluoride membrane. These blots were blocked with 5% skim milk for 1 h at room temperature, then incubated with corresponding primary antibodies IκBα, NF-κB p65, COX-2, and NLPR3 (all from CST, Massachusetts, USA) overnight at 4 °C. After that, it was then incubated with the HRP-labeled Goat Anti-Rabbit IgG (H + L) (Beyotime, Shanghai, P. R. China) for 2 h. Finally, after 5 min of reaction with the chemiluminescence solution (ECL, Amersham Biosciences Corp, NJ, USA), the target protein was observed using a gel imaging system (Tanon 5200; Shanghai Tianneng Technology Co., Ltd., Shanghai, China).
Statistical analysis
All experiment data are analysed by GraphPad Prism 8.0 and shown as the mean ± standard error. Statistical results were analysed by One-Way ANOVA test to express difference groups when p ˂ 0.5 was considered statistically significant. Western blotting results were analysed using Image J for gray scale.
Supplemental Material
Download PDF (2.9 MB)Disclosure statement
The authors declare no conflicts of interest.
Supplementary data
Supplementary data associated with this article can be accessed online at.
Additional information
Funding
References
- Aletaha D, Smolen JS. Diagnosis and management of rheumatoid arthritis: a review. JAMA. 2018;320(13):1360–1372.
- Chen Z, Bozec A, Ramming A, Schett G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat Rev Rheumatol. 2019;15(1):9–17.
- An Q, Yan W, Zhao Y, Yu K. Enhanced neutrophil autophagy and increased concentrations of IL-6, IL-8, IL-10 and MCP-1 in rheumatoid arthritis. Int Immunopharmacol. 2018;65:119–128.
- Robert M, Hot A, Mifsud F, Ndongo-Thiam N, Miossec P. Synergistic interaction between high bioactive IL-17A and joint destruction for the occurrence of cardiovascular events in rheumatoid arthritis. Front Immunol. 2020;11:1998.
- Liu S, Ma H, Zhang H, Deng C, Xin P. Recent advances on signaling pathways and their inhibitors in rheumatoid arthritis. Clin Immunol. 2021;230:108793.
- Noort AR, Tak PP, Tas SW. Non-canonical NF-κB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde? Arthritis Res Ther. 2015;17(1):15.
- Jimi E, Fei H, Nakatomi C. NF-κB signaling regulates physiological and pathological chondrogenesis. Int J Mol Sci. 2019;20(24):6275.
- Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489.
- Yin H, Liu N, Sigdel KR, Duan L. Role of NLRP3 inflammasome in rheumatoid arthritis. Front Immunol. 2022;13:931690.
- Yam MF, Asmawi MZ, Basir R. An investigation of the anti-inflammatory and analgesic effects of Orthosiphon stamineus leaf extract. J Med Food. 2008;11(2):362–368.
- Jia MR, Li XW. Chinese Ethnic Materia Medica. Y.X. Zhao (Ed.). 1st ed. Beijing, China: China Medical Science and Technology Press, 2005; p. 166–167.
- Malterud KE, Hanche-Olsen IM, Smith-Kielland I. Flavonoids from Orthosiphon spicatus. Planta Med. 1989;55(6):569–570.
- Sumaryono W, Proksch P, Wray V, Witte L, Hartmann T. Qualitative and quantitative analysis of the phenolic constituents from Orthosiphon aristatus. Planta Med. 1991;57(2):176–180.
- Ma GX, Zhang XP, Li PF, Sun ZH, Zhu NL, Zhu YD, Yang JS, Chen DL, Wu HF, Xu XD. Four new phenolic acid with unusual bicycle [2.2.2] octane moiety from Clerodendranthus spicatus and their anti-inflammatory activity. Fitoterapia. 2015;105:61–65.
- Awale S, Tezuka Y, Banskota AH, Kadota S. Inhibition of NO production by highly-oxygenated diterpenes of Orthosiphon stamineus and their structure-activity relationship. Biol Pharm Bull. 2003;26(4):468–473.
- Nguyen MT, Awale S, Tezuka Y, Chien-Hsiung C, Kadota S. Staminane- and isopimarane-type diterpenes from Orthosiphon stamineus of Taiwan and their nitric oxide inhibitory activity. J Nat Prod. 2004;67(4):654–658.
- Di XX, Wang SQ, Zhang XL, Wang B, Lou HX, Wang XN. Diterpenoids from the aerial parts of Orthosiphon aristatus var. aristatus. Phytochem Lett. 2013;6(3):412–417.
- Chen YL, Tan JJ, Lu LL, Tan CH, Jiang SH, Zhu DY. Water-soluble constituents of Clerodendranthus spicatus. Chin Tradit Herbal Drugs. 2009;40(5):689–693.
- Awale S, Tezuka Y, Kobayashi M, Ueda JY, Kadota S. Neoorthosiphonone A; a nitric oxide (NO) inhibitory diterpene with new carbon skeleton from Orthosiphon stamineus. Tetrahedron Lett. 2004;45(7):1359–1362.
- Nguyen PH, Tuan HN, Hoang DT, Vu QT, Pham MQ, Tran MH, To DC. Glucose uptake stimulatory and PTP1B inhibitory activities of pimarane diterpenes from Orthosiphon stamineus Benth. Biomolecules. 2019;9(12):859–869.
- Maheswari C, Sajna V, Venkatnarayanan R. In silico docking analysis of the compounds of Orthosiphon stamineus for the anticancer activity. Int Res J Pharm. 2016;7(4):17–23.
- Yoshimura H, Sugawara K, Saito M, Saito S, Murakami S, Miyata N, Kawashima A, Morimoto S, Gao N, Zhang X, et al. In vitro TGF-beta1 antagonistic activity of ursolic and oleanolic acids isolated from Clerodendranthus spicatus. Planta Med. 2003;69(7):673–675.
- Chen WD, Zhao YL, Sun WJ, He YJ, Liu YP, Jin Q, Yang XW, Luo XD. Kidney tea” and its bioactive secondary metabolites for treatment of gout. J Agric Food Chem. 2020;68(34):9131–9138.
- Awale S, Tezuka Y, Banskota AH, Adnyana IK, Kadota S. Highly-oxygenated isopimarane-type diterpenes from Orthosiphon stamineus of Indonesia and their nitric oxide inhibitory activity. Chem Pharm Bull (Tokyo)). 2003;51(3):268–275.
- Masuda T, Masuda K, Shiragami S, Jitoe A, Nakatani N. Orthosiphol A and B, novel diterpenoid inhibitors of TPA (12-O-tetradecanoylphorbol-13-acetate)-induced inflammation, from Orthosiphon stamineus. Tetrahedron. 1992;48(33):6787–6792.
- Awale S, Tezuka Y, Banskota AH, Kouda K, Tun KM, Kadota S. Five novel highly oxygenated diterpenes of Orthosiphon stamineus from Myanmar. J Nat Prod. 2001;64(5):592–596.
- Kazuyoshi O, Takako B, Toshiyuki M, Hirotaka S. Indonesian medicinal plants. XXIII.1) Chemical structures of two new migrated pimarane-type diterpenes, neoorthsiphols A and B, and suppressive effects on rat thoracic aorta of chemical constituents isolated from the leaves of Orthosiphon aristatus (Lamiaceae). Chem Pharm Bull. 2000;48(3):433–435.