
Abstract
New aromatic O-alkyl pyridine derivatives were designed and synthesised as Proviral Integration Moloney (PIM)-1 kinase inhibitors. 4c and 4f showed potent in vitro anticancer activity against NFS-60, HepG-2, PC-3, and Caco-2 cell lines and low toxicity against normal human lung fibroblast Wi-38 cell line. Moreover, 4c and 4f induced apoptosis in the four tested cancer cell lines with high percentage. In addition, 4c and 4f significantly induced caspase 3/7 activation in HepG-2 cell line. Furthermore, 4c and 4f showed potent PIM-1 kinase inhibitory activity with IC50 = 0.110, 0.095 µM, respectively. Kinetic studies indicated that 4c and 4f were both competitive and non-competitive inhibitors for PIM-1 kinase enzyme. In addition, in silico prediction of physiochemical properties, pharmacokinetic profile, ligand efficiency, ligand lipophilic efficiency, and induced fit docking studies were consistent with the biological and kinetic studies, and predicted that 4c and 4f could act as PIM-1 kinase competitive non-adenosine triphosphate (ATP) mimetics with drug like properties.
Graphical Abstract
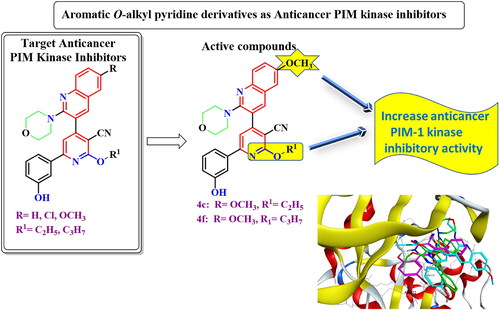
Introduction
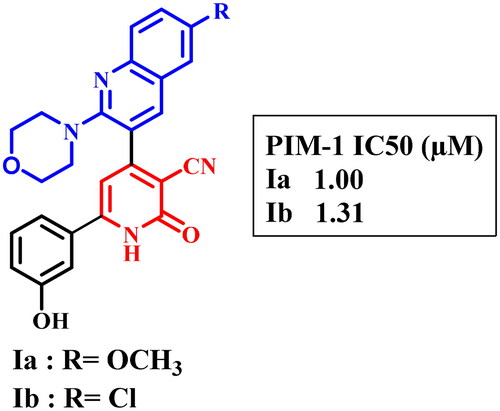
Proviral Integration Moloney (PIM)-1 kinase is a member of serine/threonine kinase enzymes and is encoded in chromosome 6Citation1. It has a crucial role in tumorigenesis because of its interactions with numerous signalling pathways which participate in drug resistance and cell proliferation of many tumoursCitation1,Citation2, such as leukaemia, lymphomaCitation3–6, prostateCitation7, colonCitation8, hepaticCitation9, and breast cancersCitation10. Based on previous studies, liver, colon, and prostate cancers as well as leukaemia are the most prevalent malignant tumours. PIM 1 is highly expressed and mediates the cell progression of the studied human cancer cell lines; HepG-2, Caco-2, and PC-3, as well as human leukaemia cell linesCitation11–14. Moreover, PIM-1 upregulates endonucleases which participate in DNA breaking and inhibition of DNA repair in several cell linesCitation15. In addition, PIM-1 has a role in cell cycle, apoptosisCitation16. Besides, PIM-1 kinase induced hypoxia in tumour cells resulted in chemotherapy resistanceCitation17. On the other hand, adenosine triphosphate (ATP) binding pocket of PIM-1 kinase contains proline 123 in the hinge region which imparts specificity to PIM-1 kinase among other kinases as it prevents ATP form forming a second hydrogen bond to the hinge regionCitation18–20. Furthermore, the majority of PIM-1 inhibitors were ATP-competitive inhibitors either ATP-mimetics (bind to hinge region via Glu121) or non-ATP mimetics (bind to Lys67)Citation21–23. In a previous study, new pyridone derivatives Ia,b were discovered as potent PIM-1 kinase inhibitorsCitation24 ().
Figure 1. Previously discovered pyridone derivatives as PIM-1 kinase inhibitors.
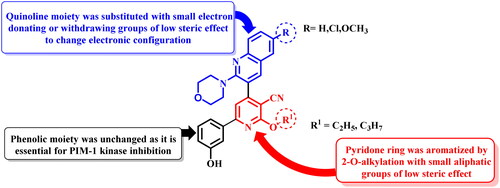
In order to increase the PIM-1 kinase inhibitory activity of the previously published compounds Ia,bCitation24, the pyridone moiety was aromatised by 2-alkylation by small aliphatic alkyl groups of low steric effect. In addition, the quinoline moiety was substituted at position 6 with electron donating or withdrawing groups while 3-hydroxyphenyl moiety was unchanged as it is essential for binding to PIM-1 kinase active site. The aromatisation of pyridone moiety and substitution on quinoline with electron donating or withdrawing groups could change the electronic configuration, conjugation, and possible conformations of the target compounds inside PIM-1 kinase active site aiming to increase binding affinity hence biological activity ().
Figure 2. Design of aromatic O-alkyl pyridine derivatives.
Experimental
Chemistry
General
Solvents and chemicals were bought from commercial suppliers. Griffin melting point apparatus was used to determine melting points in open-glass capillaries, which were uncorrected. The progress of the reactions was monitored by thin-layer chromatography (TLC) on commercially available precoated silica gel aluminium-backed plates, and the spots were visualised by UV-lamp using λ 254 nm wavelength for few seconds or exposure to iodine vapours. Infra-red (IR) spectra were recorded, for KBr discs, on a PerkinElmer RXIFT-IR spectrometer. Nuclear magnetic resonance spectra,1H NMR and 13C NMR were recorded on Bruker spectrometer (400 MHz) using DMSO-d6 as solvent using deuterated dimethyl sulphoxide as solvents. The results were calculated as chemical shifts or δ values (ppm) relative to internal standard (tetramethyl silane (TMS)). Signals were indicated by the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet, and br. = broad. Gas chromatograph/mass spectrophotometer Shimadzu GCMS/QP-2010 plus (70 eV) (Kyoto, Japan) was used to run electron impact mass spectra (EIMS), and relative intensity % corresponding to the most characteristic fragments was recorded. Elemental microanalyses were performed at the microanalytical unit, the Regional Center of Microbiology and Biotechnology, Al-Azhar University, Egypt.
Compounds 1a–c and 2a–c were prepared as reportedCitation24.
General experimental procedure for the synthesis of 3-(5-cyano-6-ethoxy or propoxy-4-(2-morpholin-4-yl-6-substituted quinolin-3-yl)pyridin-2-yl)phenyl acetate (3a–f)
A mixture of compounds 2a–c (1 mmol), anhydrous potassium carbonate (0.207 g, 1.5 mmol), and appropriate alkyl halides (1 mmol) in dry acetone (10 ml) was heated under reflux for 4 h. The reaction mixture was then cooled and triturated with ice-cold water. The reaction mixture was then filtered and the precipitate was crystallised from ethanol.
3-(5-Cyano-6-ethoxy-4-(2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)phenyl acetate (3a)
Yield, 90%; m.p. 158–160 °C; IR (KBr, cm−1): 2222 (C≡N), 1765 (COCH3), 1583 (C = N).
3-(4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-5-cyano-6-ethoxypyridin-2-yl)phenyl acetate (3b)
Yield, 90%; m.p. 179–180 °C; IR (KBr, cm−1): 2224 (C≡N), 1764 (COCH3), 1588 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 1.47 (t, J = 4,8 Hz, 3H, CH3); 2.34 (s, 3H, COCH3); 3.16–3.19 (m, 4H, morpholine-C2,6-H); 3.55–3.57 (m, 4H, morpholine-C3,5-H); 4.69 (q, J = 8 Hz, 2H, CH2); 7.34 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.64 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.75 (d, 1H, quinoline-C7-H); 7.84 (d, J = 8 Hz, 1H, quinoline-C8-H); 8.03–8.05 (m, 2H, phenyl-C2-H and quinoline-C5-H); 8.11 (s, 1H, pyridine-C5-H); 8.20 (d, J = 8 Hz, 1H, phenyl-C6-H); 8.39 (s, 1H, quinoline-C4-H).
3-(5-Cyano-6-ethoxy-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)phenyl acetate (3c)
Yield, 90%; m.p. 279–280 °C; IR (KBr, cm−1): 2223 (C≡N), 1762 (COCH3), 1600 (C═N).
3-(5-Cyano-4-(2-morpholin-4-yl-quinolin-3-yl)-6-propoxypyridin-2-yl)phenyl acetate (3d)
Yield, 90%; m.p. 180–182 °C; IR (KBr, cm−1): 2223 (C≡N), 1763 (COCH3), 1576 (C═N).
3-(4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-5-cyano-6-propoxypyridin-2-yl)phenyl acetate (3e)
Yield, 90%; m.p. 238–240 °C; IR (KBr, cm−1): 2223 (C≡N), 1763 (COCH3), 1585 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 1.07 (t, J = 8 Hz, 3H, CH3); 1.87 (sextet, J = 8 Hz, 2H, CH2); 2.34 (s, 3H, COCH3); 3.16–3.20 (m, 4H, morpholine-C2,6-H); 3.55–3.57 (m, 4H, morpholine-C3,5-H); 4.60 (t, J = 8 Hz, 2H, CH2); 7.34 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.64 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.75 (d, 1H, quinoline-C7-H); 7.84 (d, J = 8 Hz, 1H, quinoline-C8-H); 8.03–8.05 (m, 2H, phenyl-C2-H and quinoline-C5-H); 8.11 (s, 1H, pyridine-C5-H); 8.19 (d, J = 8 Hz, 1H, phenyl-C6-H); 8.40 (s, 1H, quinoline-C4-H).
3-(5-Cyano-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)-6-propoxypyridin-2-yl)phenyl acetate (3f)
Yield, 90%; m.p. 258–260 °C; IR (KBr, cm−1): 2223 (C≡N), 1763 (COCH3), 1600 (C═N).
General experimental procedure for the synthesis of 2-ethoxy or propoxy-6-(3-hydroxyphenyl)-4-(2-morpholin-4-yl-6-substituted quinolin-3-yl)pyridine-3-carbonitrile (4a–f)
Compounds 3a–f (1 mmol) and sodium bicarbonate (0.756 g, 9 mmol) were heated under reflux in methanol/water (1:1) for 1 h, cooled, diluted with water, and neutralised with acetic acid (pH = 7). The formed precipitate was filtered, washed with water, air dried, and crystallised from ethanol.
2-Ethoxy-6-(3-hydroxyphenyl)-4-(2-morpholin-4-yl-quinolin-3-yl)pyridine-3-carbonitrile (4a)
Yield, 90%; m.p. 258–260 °C; IR (KBr, cm−1): 3236 (OH), 2224 (C≡N), 1584 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 1.46 (t, J = 6.8,7.2 Hz, 3H, CH3); 3.12–1.16 (m, 4H, morpholine-C2,6-H); 3.53–3.56 (m, 4H, morpholine-C3,5-H); 4.66 (q, J = 5.2,8 Hz, 2H, CH2); 6.94 (d, J = 7.6 Hz, 1H, phenyl-C4-H); 7.35 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.47 (t, J = 7.2 Hz, 1H, quinoline-C6-H); 7.67–7.69 (m, 2H, phenyl-C2-H and quinoline-C8-H); 7.74 (t, J = 8 Hz, 1H, quinoline-C7-H); 7.82 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.92 (d, J = 8 Hz, 1H, quinoline-C5-H); 7.97 (s, 1H, pyridine-C5-H); 8.39 (s, 1H, quinoline-C4-H); 9.76 (s, 1H, OH, D2O-exchangeable). 13C NMR (400 MHz, DMSO-d6, δ ppm): 14.87, 49.66, 63.58, 66.24, 93.35, 100.00, 113.98, 114.42, 118.35, 118.64, 123.65, 124.63, 126.14, 127.37, 128.67, 130.54, 131.41, 138.36, 140.38, 147.26, 155.32, 157.70, 158.27, 158.37, 164.17. Elemental analysis Calcd for C27H24N4O3 (452.51): C 71.67, H 5.35, N 12.38. Found: C 71.50, H 5.49, N 12.61.
4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-2-ethoxy-6-(3-hydroxyphenyl)pyridine-3-carbonitrile (4b)
Yield, 90%; m.p. 228–230 °C; IR (KBr, cm−1): 3239 (OH), 2225 (C≡N), 1587 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 1.46–1.51 (m, 3H, CH3); 3.15–3.19 (m, 4H, morpholine-C2,6-H); 3.54–3.56 (m, 4H, morpholine-C3,5-H); 4.67 (q, J = 8 Hz, 2H, CH2); 6.96 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.36 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.67–7.69 (m, 2H, phenyl-C2-H and quinoline-C7-H); 7.74 (d, J = 8 Hz, 1H, quinoline-C8-H); 7.83 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.97 (s, 1H, quinoline-C5-H); 8.03 (s, 1H, pyridine-C5-H); 8.37 (s, 1H, quinoline-C4-H); 9.76 (s, 1H, OH, D2O-exchangeable). EI-MS m/z (% relative abundance): 486 [M+•] (85.94); 475 (85.76); 377 (100); 354 (78.30). Elemental analysis Calcd for C27H23ClN4O3 (486.96): C 66.60, H 4.76, N 11.51. Found: C 66.49, H 4.88, N 11.69.
2-Ethoxy-6-(3-hydroxyphenyl)-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)pyridine-3-carbonitrile (4c)
Yield, 90%; m.p. 280–281 °C; IR (KBr, cm−1): 3447 (OH), 2220 (C≡N), 1579 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 1.43 (t, J = 8 Hz, 3H, CH3); 3.01–3.05 (m, 4H, morpholine-C2,6-H); 3.49–3.52 (m, 4H, morpholine-C3,5-H); 3.75 (s, 3H, OCH3); 4.60–4.64 (m, 2H, CH2); 6.94 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.35–7.39 (m, 3H, quinoline-C5,7-H and phenyl-C5-H); 7.61–7.64 (m, 2H, phenyl-C2-H and quinoline-C8-H); 7.75 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.87 (s, 1H, pyridine-C5-H); 8.24 (s, 1H, quinoline-C4-H); 9.75 (s, 1H, OH, D2O-exchangeable). Elemental analysis Calcd for C28H26N4O4 (482.54): C 69.70, H 5.43, N 11.61. Found: C 69.87, H 5.68, N 11.79.
6-(3-Hydroxyphenyl)-4-(2-morpholin-4-yl-quinolin-3-yl)-2-propoxypyridine-3-carbonitrile (4d)
Yield, 88%; m.p. 270–272 °C; IR (KBr, cm−1): 3421 (OH), 2222 (C≡N), 1584 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 1.05–1.07 (m, 3H, –CH3); 1.86–1.88 (m, 2H, CH2CH3); 3.15 (s, 4H, morpholine-C2,6-H); 3.55 (s, 4H, morpholine-C3,5-H); 4.55–4.57 (m, 2H, O-CH2); 6.93–6.95 (m, 1H, phenyl-C4-H); 7.33–7.35 (m, 1H, phenyl-C5-H); 7.47–7.49 (m, 1H, quinoline-C6-H); 7.66–7.82 (m, 4H, quinoline-C7,8-H and phenyl-C2,6-H); 7.94–7.97 (m, 2H, pyridine-C5-H and quinoline-C5-H); 8.40 (s, 1H, quinoline-C4-H); 9.76 (s, 1H, OH, D2O-exchangeable). Elemental analysis Calcd for C28H26N4O3 (466.54): C 72.09, H 5.62, N 12.01. Found: C 71.88, H 5.78, N 12.23.
4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-6-(3-hydroxyphenyl)-2-propoxypyridine-3-carbonitrile (4e)
Yield, 88%; m.p. >300 °C; IR (KBr, cm−1): 3434 (OH), 2225 (C≡N), 1598 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 1.06 (t, J = 4 Hz, 3H, CH3); 1.85–1.89 (m, 2H, CH2CH3); 3.14–3.17 (m, 4H, morpholine-C2,6-H); 3.53–3.56 (m, 4H, morpholine-C3,5-H); 4.57 (t, J = 4 Hz 2H, OCH2); 6.95 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.36 (t, J = 8.8, 4.8 Hz, 1H, phenyl-C5-H); 7.64–7.66 (m, 2H, phenyl-C2-H and quinoline-C5-H); 7.72 (d, J = 8 Hz, 1H, quinoline-C7-H); 7.82 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.95 (s, 1H, pyridine-C5-H); 8.02 (s, 1H, quinoline-C8-H); 8.36 (s, 1H, quinoline-C4-H); 9.76 (s, 1H, OH, D2O-exchangeable). Elemental analysis Calcd for C28H25ClN4O3 (500.98): C 67.13, H 5.03, N 11.18. Found: C 67.40, H 5.28, N 11.37.
6-(3-Hydroxyphenyl)-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)-2-propoxypyridine-3-carbonitrile (4f)
Yield, 88%; m.p. 200–202 °C; IR (KBr, cm−1): 3444 (OH), 2225 (C≡N), 1601 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 1.07 (t, J = 7.2 Hz, 3H, –CH3); 1.87 (sextet, J = 7.2 Hz, 2H, CH2CH3); 3.07–3.09 (m, 4H, morpholine-C2,6-H); 3.54–3.56 (m, 4H, morpholine-C3,5-H); 3.88 (s, 3H, OCH3); 4.58 (t, J = 6.4 Hz, 2H, OCH2); 6.95 (d, J = 7.2 Hz, 1H, phenyl-C4-H); 7.34–7.42 (m, 3H, phenyl-C5-H and quinoline-C5,7-H); 7.67–7.68 (m, 2H, quinoline-C8-H and phenyl-C2-H); 7.77 (d, J = 9.2 Hz, 1H, phenyl-C6-H); 7.96 (s, 1H, pyridine-C5-H); 8.31 (s, 1H, quinoline-C4-H); 9.78 (s, 1H, OH, D2O-exchangeable). 13C NMR (400 MHz, DMSO-d6, δ ppm): 10.81, 22.24, 49.91, 55.96, 66.31, 68.92, 93.48, 106.98, 114.07, 114.39, 115.50, 118.35, 118.59, 123.20, 124.18, 125.61, 128.97, 130.56, 138.36, 139.58, 142.92, 155.27, 156.43, 156.60, 158.14, 158.38, 164.27. Elemental analysis Calcd for C29H28N4O4 (496.57): C 70.15, H 5.68, N 11.28. Found: C 70.38, H 5.84, N 11.50.
General experimental procedure for the synthesis of 3-(3-amino-4-(2-morpholin-4-yl-6-substitutedquinolin-3-yl)-1H-pyrazolo[3,4-b]pyridin-6-yl)phenol (6a–c)
A suspension of compounds 3a–c (1 mmol) and excess of hydrazine hydrate 99% (1 ml) in ethanol (10 ml) was heated under reflux for 4 h then cooled to room temperature and diluted with water. The precipitate was collected by filtration, washed with water, air dried, and crystallised from ethanol/DMF (9:1).
3-(3-Amino-4-(2-morpholin-4-yl-quinolin-3-yl)-1H-pyrazolo[3,4-b]pyridin-6-yl)phenol (6a)
Yield, 90%; m.p. 285–286 °C; IR (KBr, cm−1): 3440 (OH), 3400 (νasN–H), 3270 (νsN–H), 3200 (NH), 1630 (δ N–H), 1597 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 3.19–3.22 (m, 4H, morpholine-C2,6-H); 3.45–4.48 (m, 4H, morpholine-C3,5-H); 4.76 (s, 2H, NH2, D2O-exchangeable); 6.87 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.31 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.43 (t, J = 4,8 Hz, 1H, quinoline-C6-H); 7.59–7.62 (m, 2H, phenyl-C2-H and quinoline-C8-H); 7.65 (s, 1H, pyridine-C5-H); 7.70 (t, J = 8 Hz, 1H, quinoline-C7-H); 7.82 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.91 (d, J = 8 Hz, 1H, quinoline-C5-H); 8.24 (s, 1H, quinoline-C4-H); 9.64 (s, 1H, OH, D2O-exchangeable); 12.38 (s, 1H, NH, D2O-exchangeable). Elemental analysis Calcd for C25H22N6O2 (438.49): C 68.48, H 5.06, N 19.17. Found: C 68.71, H 5.24, N 19.38.
3-(3-Amino-4-(6-chloro-2-morpholin-4-yl-quinolin-3-yl)-1H-pyrazolo[3,4-b]pyridin-6-yl)phenol (6b)
Yield, 93%; m.p. >300 °C; IR (KBr, cm−1): 3417 (OH), 3300 (νasN–H), 3260 (νsN–H), 3200 (NH), 1650 (δ N–H), 1593 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 3.18–3.25 (under DMSO, 8H, morpholine-H); 4.74 (s, 2H, NH2, D2O-exchangeable); 6.87 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.31 (d, J = 8 Hz, 1H, quinoline-C7-H); 7.61–7.63 (m, 3H, quinoline-C5-H and phenyl-C2,5-H); 7.69 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.81 (d, J = 8 Hz, 1H, quinoline-C8-H); 8.00 (s, 1H, pyridine-C5-H); 8.21 (s, 1H, quinoline-C4-H); 9.65 (s, 1H, OH, D2O-exchangeable); 12.38 (s, 1H, NH, D2O-exchangeable). EI-MS m/z (% relative abundance): 472 [M+•] (32.07); 379 (100); 314 (70.05). Elemental analysis Calcd for C25H21ClN6O2 (472.93): C 63.49, H 4.48, N 17.77. Found: C 63.26, H 4.69, N 17.58.
3-(3-Amino-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)-1H-pyrazolo[3,4-b]pyridin-6-yl)phenol (6c)
Yield, 93%; m.p. 275 °C; IR (KBr, cm−1): 3419 (OH), 3320 (νasN–H), 3270 (νsN–H), 3200 (NH), 1660 (δ N–H), 1604 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 3.11–3.14 (m, 4H, morpholine-C2,6-H); 3.45–3.47 (m, 4H, morpholine-C3,5-H); 3.86 (s, 3H, OCH3); 4.77 (s, 2H, NH2, D2O-exchangeable); 6.87 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.32 (t, J = 8 Hz, 1H, phenyl-C5-H); 7.36–7.38 (m, 2H, quinoline-C5,7-H); 7.59–7.62 (m, 2H, phenyl-C2-H and quinoline-C8-H); 7.65 (s, 1H, pyridine-C5-H); 7.75–7.78 (m, 1H, phenyl-C6-H); 8.18 (s, 1H, quinoline-C4-H); 9.64 (s, 1H, OH, D2O-exchangeable); 12.37 (s, 1H, NH, D2O-exchangeable). 13C NMR (400 MHz, DMSO-d6, δ ppm): 49.93, 55.90, 66.21, 102.74, 106.96, 112.45, 114.15, 116.90, 118.35, 122.48, 124.63, 125.78, 128.80, 130.30, 140.01, 140.54, 142.49, 143.59, 148.24, 153.80, 156.15, 156.37, 156.48, 158.24. Elemental analysis Calcd for C26H24N6O3 (468.52): C 66.65, H 5.16, N 17.94. Found: C 66.54, H 5.37, N 17.83.
General experimental procedure for the synthesis of 3-(5-cyano-6-(cyanomethoxy)-(4-(2-morpholin-4-yl-6-substituted quinolin-3-yl)pyridin-2-yl)phenyl acetate (7a–c)
To a mixture of compounds 2a–c (1 mmol), anhydrous potassium carbonate (0.207 g, 1.5 mmol) in dry acetone (10 ml) chloroacetonitrile (0.063 ml, 1 mmol) was added. The mixture was heated under reflux for 4 h, cooled, and treated with ice-cold water. The formed precipitate (green colour) was filtered, washed with water, air dried, and crystallised from ethanol.
3-(5-Cyano-6-(cyanomethoxy)-4-(2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)phenyl acetate (7a)
Yield, 85%; m.p. 149–150 °C; IR (KBr, cm−1): 2226 (C≡N), 1765 (C═O), 1593 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 2.30 (s, 3H, COCH3); 3.20–3.21 (m, J = 7.6 Hz, 4H, morpholine-C2,6-H); 3.52–3.60 (m, 4H, morpholine-C3,5-H); 5.59 (s, 2H, CH2); 7.36 (d, J = 8 Hz, 1H, phenyl-C4-H); 7.49 (t, J = 6.8, 7.2 Hz, 1H, quinoline-C6-H); 7.65 (t, J = 7.6,8 Hz, 1H, phenyl-C5-H); 7.76 (t, J = 7.2,8 Hz, 1H, quinoline-C7-H); 7.84 (d, J = 8 Hz, 1H, quinoline-C8-H); 7.93 (d, J = 7.6 Hz, 1H, quinoline-C5-H); 8.12 (s, 1H, pyridine-C5-H); 8.26–8.28 (m, 2H, phenyl-C6-H and quinoline-C4-H); 8.45 (s, 1H, phenyl-C2-H).
3-(4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-5-cyano-6-(cyanomethoxy)pyridin-2-yl)phenyl acetate (7b)
Yield, 82%; m.p. 218–220 °C; IR (KBr, cm−1): 2225 (C≡N), 1764 (C═O), 1593 (C═N).
3-(5-Cyano-6-(cyanomethoxy)-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)phenyl acetate (7c)
Yield, 88%; m.p. 244–245 °C; IR (KBr, cm−1): 2225 (C≡N), 1762 (C═O), 1596 (C═N).
General procedure for synthesis of 2-((3-cyano-6-(3-hydroxyphenyl)-4-(2-morpholin-4-yl-6-substituted quinolin-3-yl)pyridin-2-yl)oxy)acetic acid (9a–c)
A mixture of compounds 7a–c (1 mmol), and sodium bicarbonate (0.756 g, 9 mmol) in methanol/water (1:1) was refluxed for 1 h. The reaction mixture was then cooled, triturated with water and neutralised with acetic acid (pH = 7). The precipitate of the product was collected, filtered, washed with water, air dried, and crystallised from ethanol.
2-((3-Cyano-6-(3-hydroxyphenyl)-4-(2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)acetic acid (9a)
Yield, 90%; m.p. 179–180 °C; IR (KBr, cm−1): 3403 (OH), 3350–3050 (COOH), 2224 (C≡N), 1732 (C═O), 1643 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 3.09–3.19 (m, 4H, morpholine-C2,6-H); 3.55–3.62 (m, 4H, morpholine-C3,5-H); 5.15 (s, 2H, CH2); 6.95 (d, J = 7.6 Hz, 1H, phenyl-C4-H); 7.34 (t, J = 7.6,8 Hz, 1H, phenyl-C5-H); 7.48 (t, J = 7.2,7.6 Hz, 1H, quinoline-C6-H); 7.60 (s, 1H, phenyl-C2-H); 7.65 (d, J = 7.6 Hz, 1H, quinoline-C8-H); 7.75 (t, J = 7.2, 7.6 Hz, 1H, quinoline-C7-H); 7.84 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.93 (d, J = 7.6 Hz, 1H, quinoline-C5-H); 8.02 (s, 1H, pyridine-C5-H); 8.43 (s, 1H, quinoline-C4-H); 9.77, 13.22 (2brs, each for 1H, 2OH, D2O-exchangeable). 13C NMR (400 MHz, DMSO-d6, δ ppm): 49.69, 63.77, 66.15, 93.33, 114.52, 114.78, 115.30, 118.44, 118.67, 123.70, 124.71, 125.21, 127.42, 128.71, 130.47, 131.43, 138.06, 140.78, 147.29, 155.38, 157.79, 158.08, 158.37, 163.38, 170.09. Elemental analysis Calcd for C27H22N4O5 (482.50): C 67.21, H 4.60, N 11.61. Found: C 67.49, H 4.83, N 11.88.
2-((4-(6-Chloro-2-morpholin-4-yl-quinolin-3-yl)-3-cyano-6-(3-hydroxyphenyl)pyridin-2-yl)oxy)acetic acid (9b)
Yield, 89%; m.p. 279–280 °C; IR (KBr, cm−1): 3448 (OH), 3600–3000 (COOH), 2225 (C≡N), 1626 (C═O), 1597 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 3.02–3.30 (m, 4H, morpholine-C2,6-H); 3.50–3.70 (4H, under DMSO, morpholine-C3,5-H); 4.96 (s, 2H, CH2); 6.89 (d, J = 7.6 Hz, 1H, phenyl-C4-H); 7.26 (t, J = 7.6,8 Hz, 1H, phenyl-C5-H); 7.58–7.60 (m, 2H, quinoline-C5-H and phenyl-C6-H); 7.73 (d, J = 9 Hz, 1H, quinoline-C7-H); 7.82 (d, J = 9 Hz, 1H, quinoline-C8-H); 7.88 (s, 1H, phenyl-C2-H); 8.03 (s, 1H, pyridine-C5-H); 8.32 (s, 1H, quinoline-C4-H); 10.09 (brs, 1H, OH, D2O-exchangeable). EI-MS m/z (% relative abundance): 518 [M+•+2] (21.67); 516 [M+•] (32.78); 413 (100); 391 (86.16). Elemental analysis Calcd for C27H21ClN4O5 (516.94): C 62.73, H 4.09, N 10.84. Found: C 62.96, H 4.13, N 11.06.
2-((3-Cyano-6-(3-hydroxyphenyl)-4-(6-methoxy-2-morpholin-4-yl-quinolin-3-yl)pyridin-2-yl)oxy)acetic acid (9c)
Yield, 92%; m.p. 274–275 °C; IR (KBr, cm−1): 3422 (OH), 3600–3050 (COOH), 2224 (C≡N), 1598 (C═O), 1549 (C═N). 1H NMR (400 MHz, DMSO-d6, δ ppm); 3.02–3.12 (m, 4H, morpholine-C2,6-H); 3.55–3.60 (m, 4H, morpholine-C3,5-H); 3.87 (s, 3H, OCH3); 4.89 (s, 2H, –CH2); 6.92 (d, J = 8.4 Hz, 1H, phenyl-C4-H); 7.31 (t, J = 8,8.4 Hz, 1H, phenyl-C5-H); 7.38–7.41 (m, 2H, quinoline-C5,7-H); 7.60–7.67 (m, 2H, phenyl-C2-H and quinoline-C8-H); 7.77 (d, J = 8 Hz, 1H, phenyl-C6-H); 7.90 (s, 1H, pyridine-C5-H); 8.30 (s, 1H, quinoline-C4-H); 9.84, 13.10 (2brs, each for 1H, 2OH, D2O-exchangeable). Elemental analysis Calcd for C28H24N4O6 (512.52): C 65.62, H 4.72, N 10.93. Found: C 65.90, H 4.89, N 11.24.
Biological evaluationCitation25
All solvents, chemicals, kits, and media were purchased from commercial suppliers. Biological evaluation procedures were performed in Medical Biotechnology Department, Genetic Engineering and Biotechnology Research Institute, City of Scientific Research and Technological Applications (SRTA-City), Egypt.
Cytotoxicity screening
Cytotoxicity of the tested compounds was evaluated using normal human lung fibroblast Wi-38 cell line compared to currently used anticancer drug (doxorubicin (Dox)). MTT method was used for assaying cell viabilityCitation26 (Supp. Page S17) (approved by Ethics CommitteeCitation25).
Determination of the anticancer activity
Anticancer effect was evaluated using colon cancer cells (Caco-2), myeloid leukaemia cell line (NFS-60), liver cancer cell line (HepG-2), and prostate cancer cell line (PC-3) utilising MTT method for assaying cell viabilityCitation26 (Supp. Page S17) (approved by Ethics CommitteeCitation25).
Flow cytometric analysis of apoptosis
The apoptosis-dependent anticancer effect of the most active compounds was determined by quantification of annexin-stained apoptotic cells using the FITC signal detector (FL1) against the phycoerythrin emission signal detector (FL2) (Supp. Page S17) (approved by Ethics CommitteeCitation25).
Caspase 3/7 activation assay
The percentage of caspase 3/7 activation was quantified using the Apo-ONE® caspase 3/7 kit following the manufacturer’s instructionsCitation27 (Supp. Page S18) (approved by Ethics CommitteeCitation25).
Statistical analysis
Results were calculated as mean ± standard error of the mean (SEM). Multiple comparisons Tukey post hoc analysis of variance (ANOVA) was used to estimate statistical significance utilising the SPSS16 programCitation28 (SPSS Inc., Chicago, IL) (Supp. Page S18).
PIM-1 kinase inhibitory activity
The most active anticancer compounds were tested for their ability to in vitro inhibit PIM-1 kinase utilising PIM-1 Kinase Assay Kit – Promega Corporation (Madison, WI) catalogue #V4032, following the manufacturer’s instructionsCitation29.
Molecular modelling
Induced fit docking
The molecular modelling studies were performed using the Molecular Operating Environment (MOE 2016.08) software (Chemical Computing Group, Montreal, Canada)Citation30 and the crystal structures of the proteins were downloaded from the Protein Data Bank (PDB) website (Page: S18, Supplementary file).
In silico predication of physiochemical properties, pharmacokinetic profile (Page: S19, Supplementary file).
Results and discussion
Chemistry
The target compounds were synthesised according to the steps outlined in Scheme 1. Herein, we illustrated the study for the synthesis of O-substituted pyridine derivatives via treatment of intermediated 2a–c with alkyl halides in presence of potassium carbonate to give compounds 3a–f. IR spectra of the target compounds 3a–f lacked the characteristic absorption bands corresponding to C═O and NH functions present in the precursor and exhibited stretching absorption band for C═O of acetyl function group at 1762–1765 cm−1. 1H NMR spectra for compounds 3b and 3e lacked the D2O exchangeable signal assigned for NH found in the precursor. Compound 3b demonstrated up field triplet at δ 1.47 ppm and quartet signals corresponding to OCH2CH3 protons at δ 4.69 ppm, while compound 3e showed up field triplet at δ 1.07 ppm, sextet at δ 1.87 ppm and triplet signal at δ 4.60 ppm corresponding to OCH2CH2CH3. Compounds 3a–f were hydrolysed using sodium bicarbonate in equivalent amounts of methanol and water to prepare the final compounds 4a–f. IR spectra of compounds 4a–f lacked the characteristic absorption band due to acetyl C═O function and appearance of a characteristic absorption band due to OH function at 3236–3447 cm−1. 1H NMR spectra for compounds 4a–f lacked the upfield singlet signal corresponding to CH3 of acetyl group. In addition, the spectra displayed D2O exchangeable signal corresponding to OH at a range of δ 9.75–9.78 ppm. Besides, other signals corresponding for the molecule were detected at their expected chemical shifts.13C NMR spectrum for compound 4a demonstrated shielded signal corresponding to CH3 and OCH2 at δ 14.87 and 63.58 ppm, respectively. While spectrum for compound 4f showed shielded signal corresponding to CH3, CH2, and OCH2 at δ 10.81, 22.24, and 68.92 ppm, respectively. EI/MS for 4b showed the molecular ion peak [M+•] at m/z 486 and the base peak at m/z 377. Furthermore, the intermediates 3a–f were heated under reflux with excess hydrazine hydrate to afford the corresponding hydrazine derivatives (5a–c). But, in the present investigation, trials to synthesis hydrazine derivatives were unsuccessful, and resulted in the formation of 3-amino pyrazolo[3,4-b]pyridine derivatives (6a–c). Formation of compounds (6a–c) was proposed to occur through nucleophilic attack of hydrazine on pyridine-C2 to eliminate O-alkyl group to form 2-hydrazino derivatives. After that, the 2-hydrazino group attacked the neighbouring 3-CN which resulted in cyclisation to the 3-aminopyrazolo[3,4-b]pyridine derivativesCitation31 ().
Scheme 1. Synthesis of O-alkylpyridine derivatives.
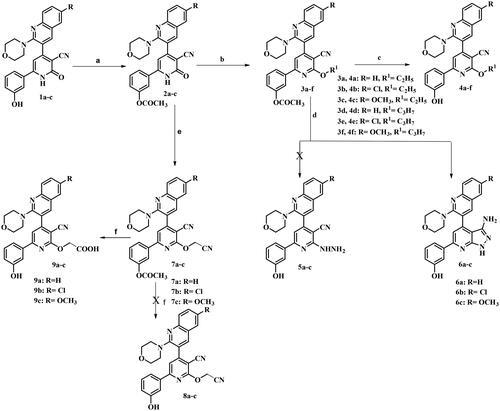
Figure 3. Proposed mechanism for formation of 3-aminopyrazolo[3,4-b]pyridine derivatives 6a–c.
![Figure 3. Proposed mechanism for formation of 3-aminopyrazolo[3,4-b]pyridine derivatives 6a–c.](/cms/asset/cbfdd290-ddd9-48c6-8bd8-340981719deb/ienz_a_2304044_f0003_b.jpg)
IR spectra of compounds 6a–c lacked the characteristic absorption bands due to acetyl C═O and C≡N functions, and appearance of characteristic absorption bands due to OH and NH at 3417–3440 and 3200 cm−1, respectively. In addition, appearance of asymmetric and symmetric absorption bands corresponding to NH2 functions at 3300–3400 and 3260–3270 cm−1, respectively. 1H NMR spectra for compounds 6a–c lacked the upfield singlet signal corresponding to CH3 of acetyl group. In addition, the spectra displayed three D2O exchangeable signals corresponding to NH2, OH, and NH, and at range of δ 4.74–4.77, 9.64–9.65, and 12.37–12.38 ppm, respectively. 13C NMR spectrum for compound 6c showed disappearance of a signal corresponding to C≡N. It demonstrated shielded signal corresponding to OCH3 at δ 55.90 ppm signal, and deshielded signal corresponding to phenyl-C3 at 158.24. EI/MS for 6b showed the molecular ion peak [M+.] at m/z 472 and base peak at m/z 379. Furthermore, the intermediates 2a–c were subjected to react with chloro acetonitrile in the presence of anhydrous potassium carbonate to give the corresponding compounds 7a–c. IR spectra of the target compounds 7a–c lacked the characteristic absorption bands for the amide C═O and NH functions present in the precursor. They showed stretching an absorption band due to C≡N function at 2225–2226 cm−1 and an absorption band due to C═O of the ester at 1762–1765 cm−1. 1H NMR spectrum for compound 7a lacked a D2O exchangeable signal corresponding to NH function. It revealed appearance of up field singlet signal integrated for two protons corresponding to CH2 at δ 5.59 ppm and singlet signal integrated for three protons assigned for COCH3 at δ 2.30 ppm. However, trials to deacetylate compounds 7a–c via refluxing with sodium bicarbonate in methanol/water (1:1) as a solvent to get compounds 8a–c went in vein. Under these reaction conditions, hydrolysis of CN function group to COOH group was observed and hence, compounds 9a–c were separated instead. The obtained products were verified using melting point.
Biological evaluationCitation25
Cytotoxicity screening
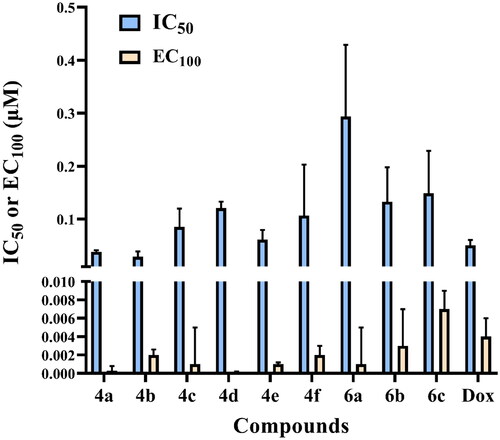
Compounds 4a–f and 6a–c were tested for their cytotoxic activity against normal human lung fibroblast (Wi-38) using Dox as standard anticancer drug utilising microculture MTT-based cytotoxicity methodCitation26. All the tested compounds showed significant noncytotoxic effect on human lung fibroblasts (Wi-38) as they showed IC50 values higher than Dox with IC50 ranging from 0.029 to 0.294 µM compared to Dox (IC50 = 0.0266 µM) () (Table 1 in Supp.) (approved by Ethics CommitteeCitation25).
Figure 4. IC50 and EC100 (μM) of the tested compounds against normal human cells (wi-38) after 72 h incubation.
In vitro anticancer screening
Compounds 4a–f and 6a–c were evaluated against liver cancer (HepG2) (ATCC® HB-8065™, Manassas, VA), leukaemia cancer (NFS-60) (ATCC® CRL-1838™, Manassas, VA), colon cancer (Caco-2) (ATCC® HTB-37™, Manassas, VA), and prostate cancer (PC-3) (ATCC® CRL-1435™, Manassas, VA) using Dox as the reference drug following reported procedureCitation26 (Table 1 in Supp., and ). Most of the tested compounds elicited broad antitumor activity against the four cancer cell lines. The obtained results revealed that, O-alkylation of the 2-oxo group as in compounds 4a–f enhanced the anticancer activity. Compound 4c with 6-methoxy quinoline and O-ethyl moieties was discovered to be the most active among this series as it comprised a remarkable increase in the potency more than the reference drug Dox against HepG-2, Caco-2, PC-3, and NFS-60 cell lines with IC50 = 0.0132, 0.007, 0.005, and 0.006 µM, respectively. Similarly, compound 4f with 6-methoxy quinoline and O-propyl moieties showed significant improvement in the anticancer activity against HepG-2 cell line with IC50 = 0.0164 µM with promising activity against Caco-2, PC-3, and NFS-60 cell lines. Furthermore, compound 4b observed approximately equipotent activity to that produced by the standard drug against HepG-2 cell line with IC50 = 0.059 µM. Besides, fusion of pyrazole moiety to pyridine ring as compounds 6a–c decreased the anticancer activity, which confirms the importance of free pyridine ring for the activity. Compound 6a showed slight anticancer activity against HepG-2 cell line with IC50 = 0.0667 µM.
Figure 5. In vitro anticancer activity, IC50 (μM), of the tested compounds against human cancer cells after 72 h incubation.
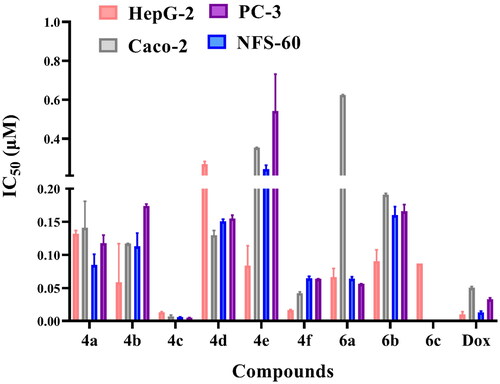
Figure 6. Morphological alterations of the most active compounds (0.06 µM)-treated cancer cells lines in comparison with the untreated cancer cells after 72 h incubation.
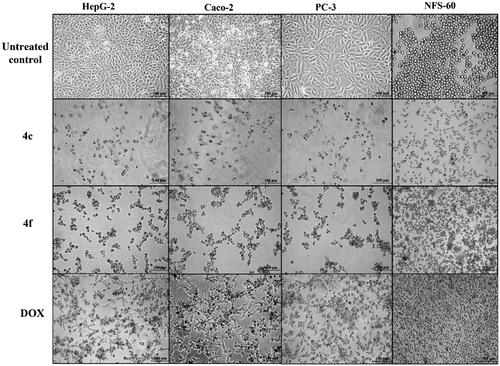
The estimated selectivity index (SI) declared that compounds 4c showed the highest selective cancer growth inhibition (SI ≥ 6) against all tested cancer cell lines, followed by compounds 4f (SI ≥ 1.6). Also, 6a revealed high SI values against all cancer cell lines, excluding Caco-2, liver cancer HepG-2 with SI values 6.439, 6.524, respectively (Table 1 in Supp.). However, compound 4c was the most selective compound towards all cancer cell lines. Additionally, morphological changes on the four cancer cell lines (HepG-2, Caco-2, NFS-60, and PC-3) were investigated before and after treatment with the most active compounds 4c and 4f in comparison with cells treated with the reference Dox. Compounds 4c and 4f resulted in severe collapsing and shrinking in the normal shape of the tested cancer cell lines () (approved by Ethics CommitteeCitation25)
Flow cytometric analysis of apoptosis
The apoptotic effects of compounds 4c and 4f were determined utilising flow cytometric annexin V/propidium iodide analysisCitation32. The induction of apoptosis was investigated after treatment with the lowest IC50 value that corresponds to each cancer cell line. Compounds 4c and 4f showed high percentages of annexin-stained population cells in the four cancer cell lines compared with the control untreated cancer cells ( and ). Compound 4f induced apoptosis-dependent death by more than 49% in the treated HepG-2, Caco-2, PC-3, and NFS-60 cancer cell lines compared to less than 39% apoptotic cell in case of Dox (approved by Ethics CommitteeCitation25).
Figure 7. Flowcharts of Annexin-PI analysis of 4c and 4f – treated cancer cell lines in comparison with the untreated cancer cells after 72 h incubation at concentrations equivalent to 0.013, 0.007, 0.005, and 0.006 µM, respectively.
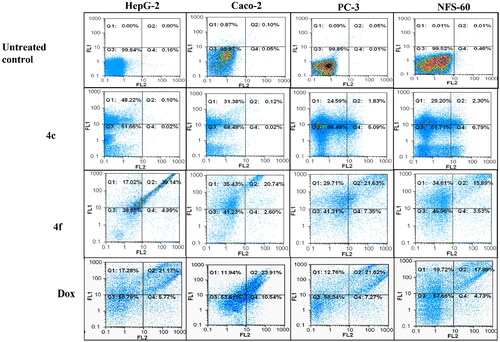
Table 1. The total percentage of the apoptotic cell population in the most effective compounds-treated cancer cells lines.
Caspase 3/7 activation assay
The apoptotic induction mechanism of compounds 4c and 4f was explained by estimating the fold increase in caspase 3/7 activation in the treated cancer cell line relative to the untreated HepG-2 cells. Compounds 4c and 4f exhibited high caspase 3/7 up regulation more than Dox treated HepG-2 cancer cell line () (approved by Ethics CommitteeCitation25).
Table 2. Relative fold increase in caspase activity by the most effective compounds relative to untreated HepG2 cancer cells.
In vitro PIM-1 kinase inhibitory activity
Compounds 4c and 4f were evaluated for their PIM-1 kinase inhibitory activities using quercetin (Qur) as reference drug (). Compounds 4c and 4f showed remarkable PIM-1 kinase inhibitory activity comparable to the reference Qur.
Table 3. In vitro PIM-1 kinase inhibition data of the most active compounds.
Mechanism of PIM-1 kinase inhibition
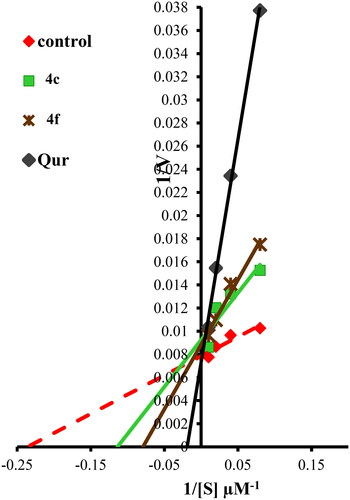
The mechanism of PIM-1 kinase inhibition in the presence of ATP for compounds 4c and 4f was investigated using enzyme kinetic parameters (maximum activity “Vmax” and substrate concentration at half Vmax “Km”), which were assessed by Lineweaver–Burk double-reciprocal plot (). Compounds 4c and 4f revealed Vmax at a higher position on the Y-axis (reduced in Vmax value) also has the negative sign on the X-intercept (right to control) which indicated increase in Km value. Therefore, compounds 4c and 4f are both ATP competitive and non-competitive inhibitors for PIM-1 kinase enzyme, which means they exhibit hybrid (mixed non-competitive) inhibition form ().
Figure 8. Lineweaver–Burk double-reciprocal plot for PIM-1 kinase inhibition by 4c and 4f in comparison with quercetin as reference inhibitor in the presence of ATP.
Structure activity relationship
The aim of the present investigation is to impart aromaticity to pyridone moiety by O-alkylation to increase PIM-1 kinase inhibitory activity. All O-alkyl derivatives 4a–f showed enhanced anticancer activity. While, fusion of pyridine with pyrazole with changing cyano moiety to amino methine as in 6a–c decreased the anticancer activity which confirms the importance of free pyridine ring and cyano moiety for the activity. On the other hand, substitution at position 6 of quinoline with methoxy group, 4c and 4f revealed higher activity than chloro and unsubstituted derivatives. Compounds 4c and 4f with aromatic O-alkylpyridine and 6-methoxy substitution on quinoline were the most active compounds with remarkable in vitro anticancer activity against HepG-2, Caco-2, PC-3, and NFS-60 cell lines, and in vitro PIM-1 kinase inhibitory activities.
Molecular modelling
Induced fit docking to pim-1 kinase active site
Compounds 4c and 4f together with the co-crystallised ligand were subjected to induced fit docking into the active site of PIM-1 kinase (PDB: 2OBJ)Citation33 using MOE version 2016.0802 softwareCitation30. On the other hand, protein flexibility has essential role in the protein–ligand interaction. Induced fit docking predicts the protein–ligand interaction based on flexibility of both ligand and active site residues which leads to increased accuracy of predicted binding interactions and binding affinityCitation34–36. In the present investigation, MOE induced fit docking was used as a docking method. This method continuously sample ligand–receptor conformations as the docking proceeds, allowing for a best fit of the ligand within a potential binding pocket. In addition, five different compound-receptor fit complexes were obtained for each compound, and the best score was chosen. The docking solutions of the compounds 4c, 4f, and co-crystallised ligand ( and ) confirmed the potential activities against PIM-1 kinase as the mode of interactions of the best poses were comparable to the co-crystallised ligand. For validation of the method, the co-crystallised ligand was subjected to induced fit docking using the same parameters used for other compounds. The 2D interaction and overlay of the best fit pose to that of the co-crystallised conformer with RMSD = 1.431 ( and ) predicted success of the docking method. The mode of binding of 4c ( and ) was like that of co-crystallised ligand. It formed H-bond with the key amino acid Lys67, and two pi–H interactions with Val52. Also, 4c produced polar and hydrophobic interactions with all amino acids interacted with the co-crystallised ligand. Additionally, 4c interacted by polar and hydrophobic interactions with other amino acids which add to the potential binding affinity of the compound. 4c interacted with additional pi–H with Leu174 and polar interaction with Gly47, Gly50, Ser51, Lys169, Glu171, Asn172, and Asp128. Furthermore, 4f interacted with most of amino acids interacted with co-crystallised ligand ( and ) especially the key amino acid Lys67. 4f additionally interacted with pi–H with Leu174 beside additional polar interactions with Gly47, Gly50, Ser51, Asp131, Glu171, Asn172, and Asp128 which add to the potential binding affinity of compound 4f. Moreover, overlay of the best fit pose of 4c and 4f to co-crystallised conformer () predicted that the pyridine moiety of 4c and 4f was slightly shifted from the pyridone moiety of the co-crystallised ligand. In conclusion, aromatisation of the pyridine moiety of 4c and 4f by O-alkylation resulted in better binding affinity to the active site by additional pi–H interactions, and by directing the phenolic and quinoline moieties to other positions which increased the polar and hydrophobic interactions with more residues in active site. Besides, 4c and 4f could act as PIM-1 kinase competitive non-ATP mimetics as they interacted with Lys67.
Figure 9. Left: Co-crystallised ligand, ligand–enzyme interaction (2D). Right: Overlay of crystallised ligand (magenta) and docked ligand (yellow) with RMSD = 1.431 (3D) inside PIM-1 kinase active site.
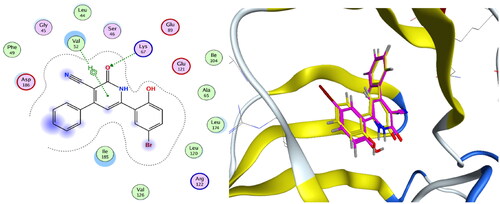
Figure 10. 4c, ligand–enzyme interaction 2D (left) and 3D (right) inside PIM-1 kinase active site.
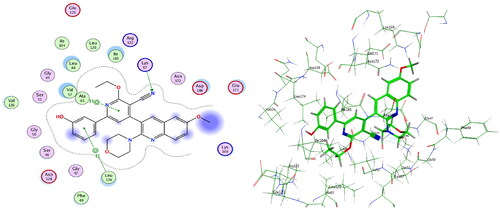
Figure 11. 4f, ligand–enzyme interaction 2D (left) and 3D (right) inside PIM-1 kinase active site.
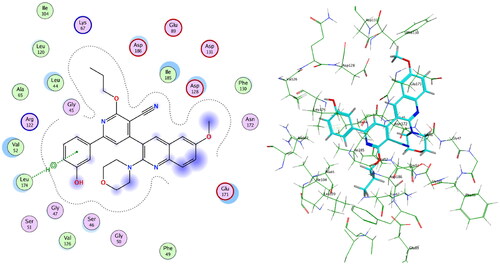
Figure 12. Overlay of 4c (green), 4f (cyan), and ligand (magenta) inside PIM-1 kinase active site.
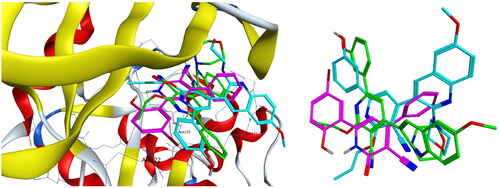
Table 4. Induced fit docking results of the active compounds inside PIM-1 kinase active site.
In silico predication of physiochemical properties, pharmacokinetic profile, and optimisation measures
Compounds 4c and 4f were subjected to molecular properties and bioactivity prediction by Molinspiration online property calculation toolkitCitation37, drug-likeness and solubility parameter calculation by Mol-Soft softwareCitation38, ADME profiling by PreADMET calculatorCitation39. Compounds 4c and 4f were predicted to possess considerable ranges of H-bond donors (1–3) and H-bond acceptors (5–10) (Table 2, Supp.). Moreover, compound 4c obeyed Lipinski’s rule of five. In addition, compounds 4c and 4f possessed rotatable bonds in the range (6 and 7, respectively) therefore, fulfilling the criterion and exhibiting high conformational flexibility. Furthermore, the calculated TPSA and % ABS values for 4c and 4f were within acceptable range (100.74 and 74.24%, respectively) for good oral bioavailability. Both compounds fulfilled the requirements of solubility of more than 0.0001 mg/l. Therefore, they could be considered as drug candidates with good oral absorption (Table 3, Supp.). Furthermore, the predicted drug-likeness scores for 4c and 4f were positive value (0.11–0.32) so they were considered drug-like candidates. 4c and 4f were calculated to have high human intestinal absorption (HIA) values, medium cell permeability in the Caco-2 cell model, low cell permeability in the MDCK cell model, strong plasma protein binding (PPB), and were found to be non-inhibitors of CYP2D6 enzyme. Therefore, they might be metabolised and excreted successfully. From the above-mentioned results, it can be concluded that compounds 4c and 4f were predicted to exert reasonable physicochemical properties, obey Lipinski’s rule of five with slight violations and exhibit acceptable pharmacokinetic parameters. Consequently, these predicted properties have pointed out that these compounds could be drug-like candidates.
Optimisation methods
Ligand lipophilic efficiency (LLE) and ligand efficiency (LE)
LE is the measurement of the binding energy which is related to the potency of the compound (pIC50) and its molecular sizeCitation40. The LE value for lead-like compound should be in the range of 0.3 while the acceptable LE value for drug-like compound should be higher than 0.3. On the other hand, LLE measures the effect of ligand lipophilicity on the binding to a given targetCitation40. LLE value for lead-like compound should be ≥3 and for drug-like candidate should be ≥5. LE and LLE were measured for 4c and 4f using reported methodCitation40 (). According to the predicted results, 4c and 4f obeyed acceptable ranges of LE (nearly 0.3), so they could act as lead-like compounds. On the other hand, LLE values for 4c and 4f were lower than 3, so their lipophilic properties did not significantly affect their binding to the PIM-1 kinase active site.
Table 5. LE and LLE values for the most active anticancer compounds against PIM-1 kinase enzyme.
Conclusions
The objective of the present investigation was to design and synthesise new O-alkyl pyridine derivatives to increase anticancer PIM-1 kinase inhibitory activity. Success was achieved and all O-alkyl derivatives 4a–f showed in vitro anticancer activity against myeloid leukaemia (NFS-60), liver cancer (HepG-2), prostate cancer (PC-3), and colon cancer (Caco-2) cell lines, and low toxicity against normal human lung fibroblast Wi-38 cell line. In addition, compounds 4c and 4f induced apoptosis with high percentage of annexin-stained apoptotic cells in the four tested cancer cell lines. Moreover, compounds 4c and 4f significantly induced caspase 3/7 activation in HepG-2 cell line. Consequently, the morphology of the treated cancer cells was substantially altered and cell shrinkage (confirming apoptosis incidence), compared to the untreated cells. Furthermore, 4c and 4f showed potent PIM-1 kinase inhibitory activity with IC50 = 0.110, 0.095 µM, respectively. On the other hand, kinetic studies proved that, 4c and 4f are both competitive and non-competitive inhibitors for PIM-1 kinase enzyme. In addition, in silico prediction of physiochemical properties, pharmacokinetic have pointed out that these compounds could be drug-like candidates. In addition, LE predicted that, 4c and 4f obeyed acceptable ranges of to act as lead-like compounds. Besides, LLE predicted that, 4c and 4f lipophilic properties did not significantly affect their binding to the PIM-1 kinase active site. Moreover, induced fit docking studies predicted that, aromatisation of the pyridine moiety in 4c and 4f by O-alkylation resulted in better binding affinity to the active site by additional pi–H interactions, and by directing the phenolic and quinoline moieties to other positions which increased the polar and hydrophobic interactions with more residues in active site. Besides, 4c and 4f could act as PIM-1 kinase competitive non-ATP mimetics as they interacted with Lys67.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supplemental Material
Download MS Word (4.3 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Wang BW, Huang CH, Liu LC, Cheng FJ, Wei YL, Lin YM, Wang YF, Wei CT, Chen Y, Chen YJ, et al. PIM1 kinase inhibitors exert anti-cancer activity against HER2-positive breast cancer cells through downregulation of HER2. Front Pharmacol. 2021;12:614673.
- Puente-Moncada N, Costales P, Antolín I, Núñez L-E, Oro P, Hermosilla MA, Pérez-Escuredo J, Ríos-Lombardía N, Sanchez-Sanchez AM, Luño E, et al. Inhibition of FLT3 and PIM kinases by EC-70124 exerts potent activity in preclinical models of acute myeloid leukemia. Mol Cancer Ther. 2018;17(3):614–624.
- Ogawa N, Yuki H, Tanaka A. Insights from PIM1 structure for anti-cancer drug design. Expert Opin Drug Discov. 2012;7(12):1177–1192.
- Schenone S, Tintori C, Botta M. Using insights into PIM1 structure to design new anticancer drugs. Curr Pharm Des. 2010;16(35):3964–3978.
- Blanco-Aparicio C, Carnero A. Pim kinases in cancer: diagnostic, prognostic and treatment opportunities. Biochem Pharmacol. 2013;85(5):629–643.
- Mohamed EA, Ismail NSM, Hagras M, Refaat H. Medicinal attributes of pyridine scaffold as anticancer targeting agents. Futur J Pharm Sci. 2021;7(1):24.
- Ye C, Zhang C, Huang H, Yang B, Xiao G, Kong D, Tian Q, Song Q, Song Y, Tan H, et al. The natural compound myricetin effectively represses the malignant progression of prostate cancer by inhibiting PIM1 and disrupting the PIM1/CXCR4 interaction. Cell Physiol Biochem. 2018;48(3):1230–1244.
- Liu K, Gao H, Wang Q, Wang L, Zhang B, Han Z, Chen X, Han M, Gao M. Hispidulin suppresses cell growth and metastasis by targeting PIM1 through JAK2/STAT3 signaling in colorectal cancer. Cancer Sci. 2018;109(5):1369–1381.
- Stafman LL, Mruthyunjayappa S, Waters AM, Garner EF, Aye JM, Stewart JE, Yoon KJ, Whelan K, Mroczek-Musulman E, Beierle EA. Targeting PIM kinase as a therapeutic strategy in human hepatoblastoma. Oncotarget. 2018;9(32):22665–22679.
- Brasó-Maristany F, Filosto S, Catchpole S, Marlow R, Quist J, Francesch-Domenech E, Plumb DA, Zakka L, Gazinska P, Liccardi G, et al. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat Med. 2016;22(11):1303–1313.
- Kronschnabl P, Grünweller A, Hartmann RK, Aigner A, Weirauch U. Inhibition of PIM2 in liver cancer decreases tumor cell proliferation in vitro and in vivo primarily through the modulation of cell cycle progression. Int J Oncol. 2020;56(2):448–459.
- Zhang M, Liu T, Sun H, Weng W, Zhang Q, Liu C, Han Y, Sheng W. PIM1 supports human colorectal cancer growth during glucose deprivation by enhancing the Warburg effect. Cancer Sci. 2018;109(5):1468–1479.
- Hu XF, Li J, Vandervalk S, Wang Z, Magnuson NS, Xing PX. PIM-1-specific mAb suppresses human and mouse tumor growth by decreasing PIM-1 levels, reducing Akt phosphorylation, and activating apoptosis. J Clin Invest. 2009;119(2):362–375.
- Li J, Hu XF, Loveland BE, Xing PX. PIM-1 expression and monoclonal antibody targeting in human leukemia cell lines. Exp Hematol. 2009;37(11):1284–1294.
- Liu Z, Zhang Y, Guo Y, Wang H, Fu R. An overview of PIM kinase as a target in multiple myeloma. Cancer Med. 2023;12(10):11746–11759.
- Abnous K, Manavi H, Mehri S, Alibolandi M, Kamali H, Ghandadi M, Hadizadeh F. In vitro evaluation of dihydropyridine-3-carbonitriles as potential cytotoxic agents through PIM-1 protein kinase inhibition. Res Pharm Sci. 2017;12(3):196–203.
- Chen J, Kobayashi M, Darmanin S, Qiao Y, Gully C, Zhao R, Kondo S, Wang H, Wang H, Yeung S-CJ, et al. Hypoxia-mediated up-regulation of PIM-1 contributes to solid tumor formation. Am J Pathol. 2009;175(1):400–411.
- Walhekar V, Bagul C, Kumar D, Muthal A, Achaiah G, Kulkarni R. Topical advances in PIM kinases and their inhibitors: medicinal chemistry perspectives. Biochim Biophys Acta Rev Cancer. 2022;1877(3):188725.
- Mori M, Tintori C, Christopher RSA, Radi M, Schenone S, Musumeci F, Brullo C, Sanità P, Delle Monache S, Angelucci A, et al. A combination strategy to inhibit PIM-1: synergism between noncompetitive and ATP-competitive inhibitors. ChemMedChem. 2013;8(3):484–496.
- Morwick T. PIM kinase inhibitors: a survey of the patent literature. Expert Opin Ther Pat. 2010;20(2):193–212.
- Nair JR, Caserta J, Belko K, Howell T, Fetterly G, Baldino C, Lee KP. Novel inhibition of PIM2 kinase has significant anti-tumor efficacy in multiple myeloma. Leukemia. 2017;31(8):1715–1726.
- Wähler K, Kräling K, Steuber H, Meggers E. Non-ATP-mimetic organometallic protein kinase inhibitor. ChemistryOpen. 2013;2(5–6):180–185.
- Lee SJ, Han B-G, Cho J-W, Choi J-S, Lee J, Song H-J, Koh JS, Lee BI. Crystal structure of PIM1 kinase in complex with a pyrido[4,3-d]pyrimidine derivative suggests a unique binding mode. PLOS One. 2013;8(7):e70358.
- El-Miligy MMM, Abdelaziz ME, Fahmy SM, Ibrahim TM, Abu-Serie MM, Mahran MA, Hazzaa AA. Discovery of new pyridine–quinoline hybrids as competitive and non-competitive PIM-1 kinase inhibitors with apoptosis induction and caspase 3/7 activation capabilities. J Enzyme Inhib Med Chem. 2023;38(1):2152810.
- Ethics Committee. Faculty of Medicine, Alexandria University, Member of ICLAS, IRB No.: 00018699, SN: 0201797(9-4-2023); 2023.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63.
- Xu T, Niu C, Zhang X, Dong M. B-ecdysterone protects SH-SY5Y cells against β-amyloid-induced apoptosis via c-Jun N-terminal kinase- and Akt-associated complementary pathways. Lab Invest. 2018;98(4):489–499.
- Brown AM. A new software for carrying out one-way ANOVA post hoc tests. Comput Methods Prog Biomed. 2005;79(1):89–95.
- Promega; 2023. Available from: https://www.promega.com/-/media/files/resources/protocols/kinase-enzyme, appnotes/pim1-kinase-assay.pdf?la=en
- Arrouchi H, Lakhlili W, Ibrahimi A. A review on PIM kinases in tumors. Bioinformation. 2019;15(1):40–45.
- Hassan AS, Hafez TS, Osman SA. Synthesis, characterization, and cytotoxicity of some new 5-aminopyrazole and pyrazolo[1,5-a]pyrimidine derivatives. Sci Pharm. 2015;83(1):27–39.
- Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1(3):1458–1461.
- Cheney IW, Yan S, Appleby T, Walker H, Vo T, Yao N, Hamatake R, Hong Z, Wu JZ. Identification and structure–activity relationships of substituted pyridones as inhibitors of PIM-1 kinase. Bioorg Med Chem Lett. 2007;17(6):1679–1683.
- Sotriffer Accounting AC. For induced-fit effects in docking: what is possible and what is not? Curr Top Med Chem. 2011;11(2):179–191.
- Barreca ML, Iraci N, De Luca L, Chimirri A. Induced‐fit docking approach provides insight into the binding mode and mechanism of action of HIV‐1 integrase inhibitors. ChemMedChem. 2009;4(9):1446–1456.
- Nirwan S, Chahal V, Kakkar R. A comparative study of different docking methodologies to assess the protein–ligand interaction for the E. coli murb enzyme. J Biomol Struct Dyn. 2022;40(21):11229–11238.
- Molinspiration; 2023. Available from: http://www.molinspiration.com/
- Molsoft; 2023. Available from: http://molsoft.com/mprop/
- Preadmet; 2023. Available from: http://preadmet.bmdrc.org/
- Hopkins AL, Keserü GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13(2):105–121.