
Abstract
Butyrylcholinesterase (BuChE) and neuroinflammation have recently emerged as promising therapeutic directions for Alzheimer’s disease (AD). Herein, we synthesised 19 novel pyranone-carbamate derivatives and evaluated their activities against cholinesterases and neuroinflammation. The optimal compound 7p exhibited balanced BuChE inhibitory activity (eqBuChE IC50 = 4.68 nM; huBuChE IC50 = 9.12 nM) and anti-neuroinflammatory activity (NO inhibition = 28.82% at 10 μM, comparable to hydrocortisone). Enzyme kinetic and docking studies confirmed compound 7p was a mix-type BuChE inhibitor. Additionally, compound 7p displayed favourable drug-likeness properties in silico prediction, and exhibited high BBB permeability in the PAMPA-BBB assay. Compound 7p had good safety in vivo as verified by an acute toxicity assay (LD50 > 1000 mg/kg). Most importantly, compound 7p effectively mitigated cognitive and memory impairments in the scopolamine-induced mouse model, showing comparable effects to Rivastigmine. Therefore, we envisioned that compound 7p could serve as a promising lead compound for treating AD.
Graphical Abstract
Introduction
Alzheimer’s disease (AD) is a prevalent neurodegenerative disease that causes aberrant neurological function and irreparable brain damage, eventually leading to deathCitation1. More than 55 million people worldwide are affected by dementia, with AD accounting for 60–70% of dementia. By 2050, the number of patients will increase to 139 millionCitation2. As a consequence, searching for potential therapeutics, particularly safe and effective small molecule drugs, has been a great focus within the medicinal chemistry community.
The cholinergic hypothesis, a fundamental theory of AD, suggests that the decline of the neurotransmitter acetylcholine (ACh) is closely associated with cognitive dysfunctionCitation3–5. The current available drugs, such as donepezil, rivastigmine, and galantamine, are acetylcholinesterase (AChE) inhibitors that improve cognitive memory by reducing ACh hydrolysisCitation6. However, these drugs have limited therapeutic efficacy in halting the progression of AD, and long-term inhibition of AChE can also result in peripheral cholinergic side effects such as vomiting, diarrhoea, nausea, and tremorsCitation7,Citation8. Butyrylcholinesterase (BuChE) is another crucial enzyme responsible for ACh hydrolysisCitation9. With the progression of AD, BuChE functions as the predominant cholinesterase hydrolysing enzyme, with a broader substrate specificity compared with AChECitation10. Additionally, without causing serious cholinergic side effects, inhibiting BuChE is deemed a safer choice than AChECitation11–13. Therefore, searching for highly selective and safe BuChE inhibitors has attracted increasing interest in AD drug development.
Chronic neuroinflammation is another significant AD pathologyCitation14. Because of the development of inflammation, an imbalance between pro- and anti-inflammatory responses occurs owing to the continuous release of a variety of inflammatory cytokines that trigger physiological changesCitation15,Citation16. In AD pathology studies, the levels of inflammatory cytokines in the brains of AD patients were significantly higherCitation17. The growing inflammatory cytokines stimulate the central nervous system (CNS), which triggers neuroinflammation, damage to neuronal cells, and acceleration of AD progressionCitation18,Citation19. Thus, alleviating neuroinflammation might be an appealing strategy for treating AD.
Due to the complexity of AD, molecules having two or more concurrent activities are considered to be more favourable therapeutic agentsCitation20–22. In view of the significance of BuChE and neuroinflammation, dual-functional molecules are promising for treating AD. However, such molecules with concurrent anti-BuChE and anti-neuroinflammatory properties are uncommon.
Previously, we discovered the 3-hydroxylpyranone skeleton had a variety of anti-AD properties, including anti-inflammation, antioxidant, neuroprotection, and easy access to the brainCitation23,Citation24. Using 3-hydroxylpyranone as a scaffold, we herein introduced the privileged carbamate moiety, which has numerous advantages such as recognising and inactivating cholinesterases, stabilising structure, and enhancing pharmacokineticsCitation25–27. Styrene was chosen as the linker to unite pyranone and carbamate scaffolds. Based on the design strategy, we synthesised 19 novel pyranone-carbamate hybrids and assessed for anti-AChE/BuChE and anti-neuroinflammation properties in vitro. Among them, compound 7p showed selective and potent inhibition of BuChE with effective anti-neuroinflammatory activity, as well as the ability to ameliorate cognitive impairment in scopolamine-induced mouse model.
Results and discussion
Chemistry
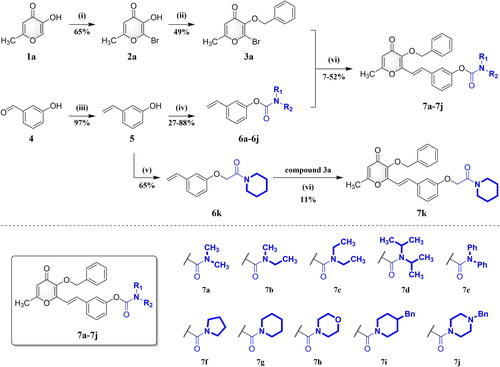
The pyranone-carbamate derivatives were synthesised using the precise synthetic procedures depicted in Schemes 1 and 2. Initially, bromoallomaltol (2a) was obtained by brominating allomaltol (1a) with N-bromosuccinimide (NBS). Compound 2a was then reacted with benzyl bromide and steadily converted into 3-(benzyloxy)-2-bromoallomaltol (3a). For the vinyl parts, m-hydroxybenzaldehyde (4) was transformed into m-hydroxystyrene (5) by the Wittig reaction, which was treated with various amino carbonyl chlorides to produce styrene-carbamate derivatives 6a–6j with the presence of 4-dimethylaminopyridine (4-DMAP) as a catalyst. Treatment of compound 5 with 2-chloro-1-(piperidin-1-yl)ethan-1-one in basic media at room temperature yielded chain-prolonged compound 6k. The Heck reaction was finally performed with Pd(OAc)2 as the catalyst to couple compound 3a and compounds 6a–6k to produce the corresponding target compounds 7a–7k (Scheme 1).
Scheme 1. Synthesis of compounds 7a–7k. Reagents and conditions: (i) NBS, NH4OAc, CH3CN, RT; (ii) BnBr, K2CO3, CH3CN, 75 °C; (iii) CH3Ph3P-Br+, NaH, THF, N2, 80 °C; (iv) R1R2NCOCl, 4-DMAP, K2CO3, CH3CN, N2, 75 °C; (v) 2-chloro-1-(piperidin-1-yl)ethan-1-one, 4-DMAP, K2CO3, CH3CN, N2, 75 °C; (vi) Pd(OAc)2, TEA, DMF, N2, 100 °C.
Similar to the bromination and benzylation of compound 1a, pyromeconic acid (1b) and kojic acid (1c) could functionalise and yield compounds 3b and 3c. In addition, the 3-hydroxyl group of compound 2a was further transformed into compounds 3d–3f in the presence of 2-(bromomethyl)naphthalene, 4-(bromomethyl)-1,1′-biphenyl, and iodomethane, respectively. Subsequently, pyranone derivatives 3b–3f were reacted with 3-vinylphenyl piperidine-1-carboxylate (6g) to afford the matching products 7l–7p (Scheme 2(A)). Compounds 6l and 6m were synthesised through the Suzuki reaction of compound 3a with different hydroxyl arylboronic esters and catalysed by PdCl2(dppf), then reacted with piperidine-1-carbonyl chloride to yield compounds 7q and 7r, respectively (Scheme 2(B)). The double bond of compound 7g was hydrogenated using 10% Pt/C as the catalyst under H2 atmosphere to obtain compound 7s (Scheme 2(C)).
Scheme 2. Synthesis of compounds 7l–7s. Reagents and conditions: (A) (i) NBS, NH4OAc, CH3CN, RT; (ii) BnBr, K2CO3, CH3CN, 75 °C; (iii) 2-(bromomethyl)naphthalene, K2CO3, CH3CN, 75 °C; (iv) 4-(bromomethyl)-1,1’-biphenyl, K2CO3, CH3CN, 75 °C; (v) CH3I, K2CO3, DMF, RT; (vi) Pd(OAc)2, TEA, DMF, N2, 100 °C. (B) (i) PdCl2(dppf), K2CO3, THF: H2O (9:1, v/v), N2, 75 °C; (ii) Piperidine-1-carbonyl chloride, 4-DMAP, K2CO3, CH3CN, N2, 75 °C. (C) (i) H2, 10% Pt/C, THF, 50 °C. (D) X ray of compound 7p (CCDC Number 2299602).
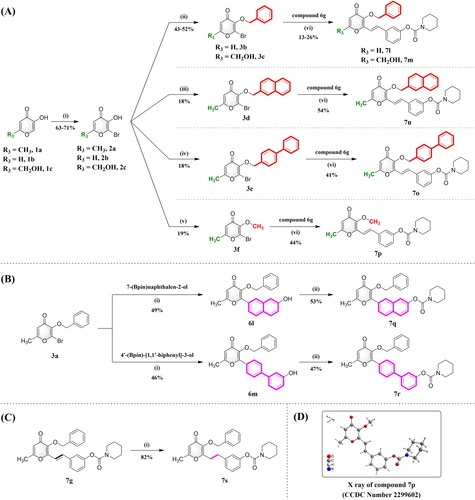
The structures of all target compounds were carefully examined by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectroscopy (HRMS), respectively. Their purities were determined by melting point (mp) and high-performance liquid chromatography (HPLC), ensuring the purities were higher than 95% prior to activity evaluations. Additionally, the absolute configuration of compound 7p was verified by X-ray analysis (Scheme 2(D), CCDC Number 2299602). We clearly observed the trans-configuration of the vinyl bond constructed by the Heck reaction. (For more detail, see the Experimental procedures and Supporting information.)
Biological evaluation
AChE and BuChE assays
The inhibitory activities of compounds 7a–7s against eeAChE (from electric eel), eqBuChE (from equine serum), and huBuChE (from human serum) were measured by the modified Ellman’s methodCitation28. Tacrine was used as a reference compound. The inhibitory potencies were initially tested at 20 μM (eeAChE) or 10 μM (eqBuChE and huBuChE). For compounds with inhibitory activities greater than 50%, IC50 values were subsequently determined. The results of the biological evaluations are shown in and .
Table 1. In vitro eeAChE, eqBuChE, and huBuChE inhibitory assay results (IC50 values)Table Footnotea of compounds 7a–7k.
Table 2. In vitro eeAChE, eqBuChE, and huBuChE inhibitory assay results (IC50 values)Table Footnotea of compounds 7l–7s.
Although lacking significant anti-AChE activity (IC50 > 20 μM), compounds 7a–7k exhibited potent eqBuChE inhibitory activity. The substituents at the N-terminus had critical importance in influencing BuChE activity (). For example, from the methyl group (7a) to the isopropyl group (7d), the inhibition of eqBuChE decreased sharply, with the IC50 value increasing from 24.58 to 8525.33 nM. When alkyl chains were replaced by aromatic scaffolds, e.g. compound 7e, the anti-BuChE activity almost disappeared (IC50 > 10,000 nM). Notably, compared with alkane-chain compounds, the six-membered ring compound 7g exhibited the most appealing activities, with an IC50 value of 15.29 nM. When the ring size was reduced, for example, piperidine changed to pyrrolidine (7f, IC50 = 31.40 nM), the inhibitory activity was half that of compound 7g. Also, replacing piperidine with morpholine (7h, IC50 = 72.59 nM), benzylpiperidine (7i, IC50 = 75.19 nM), and benzylpiperazine (7j, IC50 = 31.75 nM) did not increase anti-BuChE activity. Additionally, extending the carbamate bond, even by one carbon atom, greatly reduced the efficacy. For instance, the IC50 value of compound 7k (IC50 = 1426.67 nM) was much greater than compound 7g.
In addition to the eqBuChE assay, we confirmed the inhibitory efficacy of compounds 7a–7k by the huBuChE assay (), and summarised their structure-activity relationships (SAR), which showed a trend similar to that of the eqBuChE assay.
Having found the optimal substituents on the N-terminus, we next conducted our structural modifications on the pyranone moiety and the vinyl linker. Despite significant structural dissimilarities compared to compound 7g, compounds 7l–7s remained inactive against AChE (IC50 > 20 μM, ).
When we removed the methyl group at the 6-position of pyranone, or switched it to the hydroxymethyl group, it had a positive effect on the BuChE inhibitory activity. For example, the IC50 values of compounds 7l (eqBuChE IC50 = 8.10 nM; huBuChE IC50 = 6.93 nM) and 7m (eqBuChE IC50 = 5.22 nM; huBuChE IC50 = 16.53 nM) were lower than compound 7g.
The substituents at the 3-position of pyranone were also essential to optimising its potency. When we substituted the 3-O-benzyl group with larger steric hindrance groups, e.g. compounds 7n (eqBuChE IC50 = 18.95 nM; huBuChE IC50 = 39.43 nM) and 7o (eqBuChE IC50 = 19.19 nM; huBuChE IC50 = 37.95 nM), the BuChE inhibitory effects were slightly weakened. However, adding a smaller substituent to the 3-hydroxyl group of pyranone improved the inhibition of BuChE, like compound 7p with a 3-O-methyl group, which showed a 3-fold and 4-fold increase in inhibiting eqBuChE (IC50 = 4.68 nM) and huBuChE (IC50 = 9.12 nM), respectively.
Furthermore, adjustments in the linker group exert directly affected the BuChE inhibitory activity. The effect was negative when styrene was substituted by naphthalene (7q, eqBuChE IC50 = 41.74 nM; huBuChE IC50 = 230.03 nM) or biphenyl (7r, eqBuChE IC50 = 94.30 nM; huBuChE IC50 = 651.43 nM), whereas the single-bond product 7s produced by hydrogenation of the vinyl was more effective in inhibiting BuChE (eqBuChE IC50 = 12.27 nM; huBuChE IC50 = 10.97 nM).
Anti-neuroinflammation
Nitric oxide (NO), a crucial pro-inflammatory mediator, is neurotoxic under specific circumstancesCitation29. We used the Griess assay to assess the anti-neuroinflammatory effects of compounds with the lipopolysaccharide (LPS)-induced NO production model of mouse microglia cells (BV-2)Citation30,Citation31. Hydrocortisone (HC) was used as a reference compound.
To avoid false-positive results, we initially used the 3–(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide (MTT) method to determine safe concentrations of tested compounds. There were no significant differences in cell viability after the addition of 10 μM hydrocortisone and compounds 7a–7s compared to the control group (Figure S1). Therefore, we chose 10 μM for the subsequent tests.
Compared with the control group, the LPS-activated group dramatically increased NO production, which indicated that the model was effective (). Consequently, we tested hydrocortisone and compounds 7a–7s, and found that the production of NO dropped differentially under the circumstances of LPS.
Figure 1. Inhibitory activity of hydrocortisone and compounds 7a–7s on nitric oxide production. The data are expressed as the mean ± SEM of at least three experiments and analysed with a one-way ANOVA followed by Dunnett’s post hoc analysis. ####p < 0.0001 compared to the control group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to the LPS model group. LPS: lipopolysaccharide; HC: Hydrocortisone.
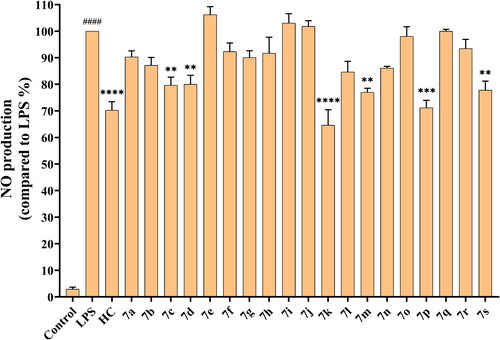
As shown in , we found similarities and differences between the SAR of NO inhibitory activity and BuChE inhibitory activity. As we lengthened the alkane-chain on the N-terminus, the NO inhibitory activity increased from compound 7a (NO production = 90.31%) to 7d (NO production = 80.01%). When the N-terminus was substituted by benzene rings, the activity of compound 7e (NO production = 106.16%) was completely disappeared. In addition, compound 7g (NO production = 90.09%) also exhibited the highest activity among compounds 7e–7j with different rigid rings. However, it is noteworthy that the NO inhibitory effect of compound 7k (NO production = 64.59%) was obviously enhanced when the carbamate bond was extended.
Moreover, we found the NO inhibitory effects were enhanced by three strategies of modifying compound 7g, such as converting the 6-methyl group into 6-hydroxymethyl group (7m, NO production = 76.97%), replacing the 3-O-benzyl group with 3-O-methyl group (7p, NO production = 71.18%), and hydrogenating the vinyl group (7s, NO production = 77.77%). In particular, the efficacy of compound 7p was equivalent to hydrocortisone (NO production = 70.25%).
In , we summarised preliminary SAR involving BuChE and NO inhibition, which could guide further structural optimizations. In addition, although compound 7k (NO inhibition = 35.41% at 10 μM) had the strongest NO inhibitory activity, its inhibitory effect on BuChE was modest. For comparison, compound 7p (NO inhibition = 28.82% at 10 μM), which possessed more balanced BuChE and NO inhibitory activities, had more potential for further studies.
Figure 2. Structure-activity relationships of compounds 7a–7s.
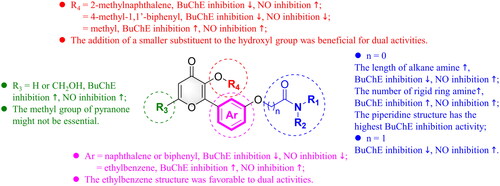
Kinetics of BuChE inhibition
We conducted an enzyme kinetic analysis with compound 7p to elucidate the mechanism of BuChE inhibition. The analysis of Dixon and Cornish-Bowden plots revealed that compound 7p exhibited characteristics consistent with mixed-type inhibition (). The inhibition constant for the enzyme (Kia) and the inhibition constant for the enzyme-substrate complex (Kib) of compound 7p were estimated to be 3.15 and 3.84 nM, respectively. These results suggested that compound 7p could bind to both the peripheral anionic site (PAS) and the catalytic active site (CAS) of BuChE, thus exerting an inhibitory effect on BuChE.
Figure 3. BuChE inhibition kinetics of compound 7p. (A) Dixon plot of compound 7p; (B) Cornish-Bowden plot of compound 7p.
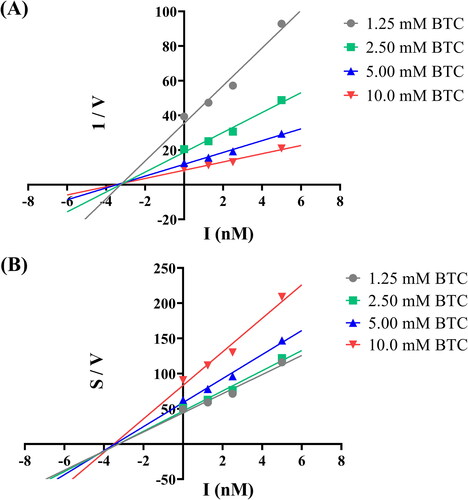
Docking studies
To explain the interaction pattern between compound 7p and BuChE, we performed docking studies using Autodock 4.2 softwareCitation32. We found that compound 7p stacked properly into the active site groove of the BuChE (). The carbamate of compound 7p was situated in the vicinity of CAS and interacted with Ser-198 and His-438 via hydrogen bonds. Moreover, the 4-carbonyl group and 1-oxygen atom of pyranone formed hydrogen bonds with Asp-70 of the PAS and Thr-120, respectively (). These results confirmed previous kinetic studies which showed compound 7p was a mixed-type inhibitor.
Figure 4. Docking studies of compound 7p. (A) The binding pocket of compound 7p with BuChE by the surface representation; (B) The interactions of compound 7p with BuChE (PDB code: 1P0I); (C) The binding pocket of compound 7p with AChE by the surface representation; (D) The interactions of compound 7p with AChE (PDB code: 4EY7).
Conversely, because of the narrow groove of the AChE, compound 7p could not completely bind to the active site of AChE, especially the CAS. Thus, compound 7p interacted with only the PAS, explaining its weaker activity against AChE (). In sum, the docking studies provided a reasonable explanation for the selective and powerful BuChE inhibitory activity of compound 7p.
SwissADME calculations
The characteristics of drug-likeness and the permeability of the blood-brain barrier (BBB) are crucial in developing drugs for treating AD. Table S1 displays the predicted results of the physiochemical properties, drug-likeness, and BBB permeation of compounds 7a–7s. Most of the compounds exhibited favourable drug-like characteristics, according to Lipinski’s rule of fiveCitation33. Considering that BBB permeability is a necessary condition for drugs affecting the effectiveness of CNS drugsCitation34, we calculated the BBB permeability of compounds 7a–7s and discovered that the optimal compound 7p had properties suitable for BBB penetration.
In vitro blood-brain barrier permeation assay
To confirm the BBB penetration potential of compound 7p, we used a parallel artificial membrane permeation assay (PAMPA-BBB) as described by Di et alCitation35,Citation36. As a validation, we selected six commercially available drugs as references. The experimental data was plotted against the reported values, resulting in a good linear correlation, Pe(exp.) = 0.9631 × Pe(ref.) + 0.1001, R2 = 0.9986 (Figure S2). Based on the measured Pe value (), compound 7p displayed high BBB permeability (Pe = 5.70 × 10−6 cm/s), indicating its ability to cross the BBB.
Table 3. Permeability Pe of six commercial drugs and compound 7p in the PAMPA-BBB assay.
Acute toxicity test
Considering the significance of safety, we chose ICR mice to evaluate the safety of compound 7p in an acute toxicity assay. The mice were randomised into a control group (0.5% CMC-Na) and a 7p-treated group (1000 mg/kg) and observed for 14 consecutive days. All the mice were in good health after the administration, with no abnormal behaviours or deaths. Additionally, the weights of mice varied within a certain range and showed an overall growth trend ().
Figure 5. Toxicity evaluation of compound 7p. (A) The trends of body weight changes in control and 7p-treated group (14 d). (B) The histomorphological appearance of heart, liver, spleen, and brain between control and 7p-treated group (40 ×, scale at 50 μm).
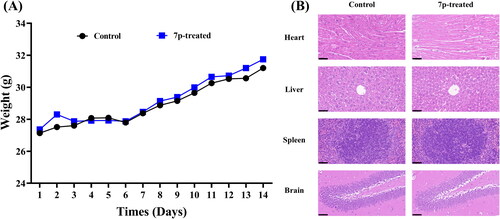
After a period of 14 d, the mice were sacrificed and dissected, and their organs were stained with hematoxylin-eosin (HE). We did not observe obvious pathological changes in the heart, liver, spleen, and brain in the 7p-treated group compared with the control group (). Therefore, compound 7p did not induce acute toxicity when given orally to mice at doses up to 1000 mg/kg, demonstrating a favourable in vivo safety profile (LD50 > 1000 mg/kg).
Behaviour studies
The BBB permeability and safety profile of compound 7p prompted us to investigate its potential for cognitive improvement. Scopolamine is a gold-standard tool for causing cholinergic deficits, resulting in memory and learning impairmentCitation37–39. Therefore, we used the Morris water maze (MWM) test to evaluate the effect of compound 7p in a scopolamine-induced cognitive impairment mouse model.
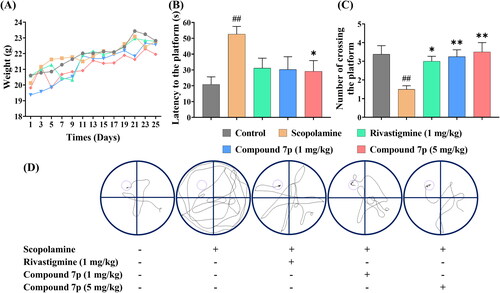
All animals showed an upward trend in weight during the entire trial, which confirmed that the mice were healthy (). The escape latency and trajectory to find the platform and the number of crossing times of the mice in each group are shown in . The scopolamine group (52.63 s, 1.5 times) exhibited a significantly longer latency to reach the platform and fewer traverses compared with the control group (20.82 s, 3.4 times), demonstrating that the scopolamine-induced model had been successfully established. In contrast, the latency period was considerably decreased in the groups treated with Rivastigmine (1 mg/kg, 31.15 s), compound 7p (1 mg/kg, 30.24 s), and compound 7p (5 mg/kg, 29.12 s). Furthermore, the number of crossings was enhanced as compound 7p concentration increased (1 mg/kg, 3.3 times; 5 mg/kg, 3.5 times), which was comparable to the effects of Rivastigmine (3.0 times). Collectively, the behavioural tests revealed that compound 7p effectively alleviated scopolamine-induced cognition and memory impairment in mice.
Figure 6. The effects of Rivastigmine and compound 7p on scopolamine-induced cognitive impairment in the Morris water maze test. (A) The daily weight of mice in different groups during the entire period. (B) The escape latency to the platform on the fifth day of training trials. (C) The number of crossing the platform on the last day without platform. (D) The representative tracks of mice on the fifth day of training trials. The data are expressed as the mean ± SEM and analysed with a one-way ANOVA followed by Dunnett’s post hoc analysis. ##p < 0.01 compared to the control group; *p < 0.05, **p < 0.01 compared to the scopolamine model group.
Conclusions
To develop effective drugs for treating AD, we designed and synthesised 19 novel pyranone-carbamate derivatives using concurrent inhibition of BuChE and neuroinflammation as a strategy. On the basis of bioactivity, we found that compound 7p was a mix-type potent BuChE inhibitor with anti-neuroinflammatory activity, high BBB permeability, favourable safety, and cognitive improvement properties. Therefore, compound 7p could be considered a potential agent for future studies.
Experimental procedures
Chemistry
Unless otherwise specified, all reagents and solvents were acquired commercially and utilised without additional purification. The reactions were monitored by thin-layer chromatography (TLC). NMR spectra were recorded on a Bruker IMPACT-II 400 or 600 spectrometer. HRMS spectra were obtained using an Agilent 6540 or Bruker FTICRMS 7TSOLARIX spectrometer (Electrospray ionisation, ESI). Melting points were measured using the MP100 melting-point apparatus. HPLC analysis was performed on a Shimadzu LC2A-MS2020 using a C18 column (2.1 mm × 150 mm, 3.5 µm) at 30 °C with gradient elution using the mobile phase 0.5% acetic acid in water and methanol; flow rate = 0.2 ml/min; detection wavelength = 254 nm.
General procedures for the synthesis of compounds 2a–2c
Compound 1a (2.00 g, 15.86 mmol) and ammonium acetate (1.22 g, 15.86 mmol) were added to acetonitrile (25 ml) with magnetic stirring. N-bromosuccinimide (3.39 g, 19.03 mmol) was then added under an ice bath. The resulting mixture was stirred overnight at room temperature. Once the starting materials (1a) were consumed, the mixture was concentrated under reduced pressure and extracted with ethyl acetate. The combined organic layers were washed with brine and dried over anhydrous sodium sulphate (Na2SO4). After the solvent evaporated, the crude product was purified using column chromatography (petroleum ether/ethyl acetate, 2:1, v/v) to obtain the desired product 2a. Compounds 2b and 2c were synthesised using a similar procedure as compound 2a.
2-Bromo-3-hydroxy-6-methyl-4H-pyran-4-one (2a)
Pale yellow solid; yield: 65% (2.10 g); Rf: 0.37 (petroleum ether/ethyl acetate, 1:2, v/v).1H NMR (600 MHz, DMSO-d6) δ 9.83 (s, 1H), 6.31 (s, 1H), 2.27 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.64, 167.27, 143.96, 127.88, 111.13, 20.10. HRMS (ESI) (m/z): calculated for C6H5BrO3 [M + H]+ 204.9500, found 204.9489.
2-Bromo-3-hydroxy-4H-pyran-4-one (2b)
Orange solid; yield: 63% (5.40 g); Rf: 0.47 (petroleum ether/ethyl acetate, 1:2, v/v). 1H NMR (600 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.15 (d, J = 5.5 Hz, 1H), 6.47 (d, J = 5.5 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 171.76, 156.40, 145.28, 130.06, 113.92. HRMS (ESI) (m/z): calculated for C5H3BrO3 [M + Na]+ 212.9163, found 212.9158.
2-Bromo-3-hydroxy-6-(hydroxymethyl)-4H-pyran-4-one (2c)
Yellow solid; yield: 71% (10.96 g); Rf: 0.21 (petroleum ether/ethyl acetate, 1:2, v/v). 1H NMR, 13C NMR and HRMS have been reported in our previous workCitation40.
General procedures for the synthesis of compounds 3a–3e
Compound 2a (2.00 g, 9.76 mmol) and potassium carbonate (2.02 g, 14.63 mmol) were dissolved in acetonitrile (25 ml). Then, benzyl bromide (1.84 g, 10.73 mmol) was added and the mixture was stirred at reflux temperature overnight. After cooling to room temperature, the mixture was filtered and the filter cake was washed with ethyl acetate. The combined organic solvents were concentrated under reduced pressure. The resulting residue was purified through column chromatography (petroleum ether/ethyl acetate, 5:1, v/v) to obtain product 3a. Compounds 3b-3e were synthesised using a similar procedure as compound 3a.
3-(Benzyloxy)-2-bromo-6-methyl-4H-pyran-4-one (3a)
Dark yellow oil; yield: 49% (1.40 g); Rf: 0.56 (petroleum ether/ethyl acetate, 1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 7.29 (dd, J = 5.0, 1.9 Hz, 3H), 7.19 (dd, J = 6.5, 2.9 Hz, 2H), 6.39 (s, 1H), 5.09 (s, 2H), 2.15 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 173.41, 166.49, 143.97, 139.69, 136.33, 128.60, 128.33, 114.26, 73.09, 18.79. HRMS (ESI) (m/z): calculated for C13H11BrO3 [M + H]+ 294.9970, found 294.9983.
3-(Benzyloxy)-2-bromo-4H-pyran-4-one (3b)
Yellow oil; yield: 43% (3.13 g); Rf: 0.62 (petroleum ether/ethyl acetate, 1:1, v/v). 1H NMR (600 MHz, DMSO-d6) δ 8.16 (d, J = 5.6 Hz, 1H), 7.44–7.41 (m, 2H), 7.40–7.34 (m, 3H), 6.51 (d, J = 5.6 Hz, 1H), 5.12 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.98, 156.56, 145.26, 140.66, 136.20, 128.56, 128.32, 128.28, 116.68, 73.11. HRMS (ESI) (m/z): calculated for C12H9BrO3 [M + H]+ 280.9813, found 280.9808.
3-(Benzyloxy)-2-bromo-6-(hydroxymethyl)-4H-pyran-4-one (3c)
Yellow oil; yield: 52% (7.31 g); Rf: 0.33 (petroleum ether/ethyl acetate, 1:1, v/v). 1H NMR, 13C NMR and HRMS have been reported in our previous workCitation40.
2-Bromo-6-methyl-3-(naphthalen-2-ylmethoxy)-4H-pyran-4-one (3d)
White solid; yield: 18% (0.60 g); Rf: 0.35 (petroleum ether/ethyl acetate, 1:2, v/v). 1H NMR (600 MHz, DMSO-d6) δ 7.95–7.91 (m, 4H), 7.61 (dd, J = 8.4, 1.6 Hz, 1H), 7.55–7.51 (m, 2H), 6.36 (d, J = 0.8 Hz, 1H), 5.28 (s, 2H), 2.26 (d, J = 0.7 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 173.36, 166.46, 143.96, 139.65, 133.91, 132.70, 132.58, 127.91, 127.88, 127.55, 127.37, 126.45, 126.31, 126.28, 114.22, 73.14, 18.74. HRMS (ESI) (m/z): calculated for C17H13BrO3 [M + Na]+ 366.9946, found 366.9941.
3-([1,1′-Biphenyl]-4-ylmethoxy)-2-bromo-6-methyl-4H-pyran-4-one (3e)
White solid; yield: 18% (1.00 g); Rf: 0.36 (petroleum ether/ethyl acetate, 1:2, v/v). 1H NMR (600 MHz, DMSO-d6) δ 7.70–7.67 (m, 4H), 7.53–7.51 (m, 2H), 7.49–7.45 (m, 2H), 7.39–7.35 (m, 1H), 6.37 (d, J = 0.8 Hz, 1H), 5.15 (s, 2H), 2.28 (d, J = 0.7 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 173.37, 166.48, 144.01, 140.03, 139.64, 139.62, 135.52, 129.14, 128.93, 127.56, 126.64, 126.55, 114.25, 72.76, 18.74. HRMS (ESI) (m/z): calculated for C19H15BrO3 [M + Na]+ 393.0102, found 393.0098.
Preparation of 2-Bromo-3-methoxy-6-methyl-4H-pyran-4-one (3f)
Iodomethane (3.81 g, 26.83 mmol) was added to a solution of compound 2a (5.00 g, 24.39 mmol) and potassium carbonate (5.06 g, 36.58 mmol) in N,N-dimethylformamide (30 ml). The reaction mixture was stirred at room temperature overnight. After the completion of the reaction, the mixture was diluted with water and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over Na2SO4, concentrated, and finally purified using column chromatography (petroleum ether/ethyl acetate, 3:1, v/v) to obtain product 3f.
2-Bromo-3-methoxy-6-methyl-4H-pyran-4-one (3f)
Yellow solid; yield: 19% (1.00 g); Rf: 0.44 (petroleum ether/ethyl acetate, 1:1, v/v). 1H NMR (600 MHz, DMSO-d6) δ 6.32 (s, 1H), 3.75 (s, 3H), 2.27 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 174.24, 165.80, 146.20, 139.49, 115.05, 60.26, 19.57. HRMS (ESI) (m/z): calculated for C7H7BrO3 [M + H]+ 218.9657, found 218.9644.
Preparation of 3-Vinylphenol (5)
To a solution of anhydrous tetrahydrofuran (250 ml), methyltriphenylphosphonium bromide (58.50 g, 163.76 mmol) was added. The suspension was stirred under an N2 atmosphere, and sodium hydride (6.55 g, 163.76 mmol) was added portionwise. The resulting yellow suspension was stirred at room temperature for 2 h. 3-hydroxybenzaldehyde (4, 10.00 g, 81.89 mmol) was added dropwise under an ice bath. The reaction was then stirred for 5 h at 80 °C. TLC confirmed the completion of the reaction. After removing the solvent, the mixture was extracted with ethyl acetate and water. The combined organic phases were washed with brine and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified by column chromatography (petroleum ether/ethyl acetate, 10:1, v/v) to obtain product 5.
3-Vinylphenol (5)
Pale yellow oil; yield: 97% (9.50 g); Rf: 0.39 (petroleum ether/ethyl acetate, 5:1, v/v). 1H NMR (400 MHz, CDCl3) δ 9.41 (s, 1H), 7.13 (t, J = 7.8 Hz, 1H), 6.90–6.86 (m, 1H), 6.85–6.82 (m, 1H), 6.70–6.59 (m, 2H), 5.72 (dd, J = 17.6, 1.1 Hz, 1H), 5.21 (dd, J = 10.9, 1.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 154.71, 131.77, 128.78, 126.22, 123.91, 119.09, 115.72, 113.47. HRMS (ESI) (m/z): calculated for C8H8O [M-H]+ 119.0497, found 119.0489.
General procedures for the synthesis of compounds 6a–6k
To a mixture of dimethylcarbamoyl chloride (1.34 g, 12.48 mmol), anhydrous potassium carbonate (2.30 g, 16.65 mmol), and 4-dimethylaminopyridine (0.20 g, 1.66 mmol) in anhydrous acetonitrile (20 ml), 3-vinylphenol (5, 1.00 g, 8.32 mmol) was added dropwise at 0 °C. The mixture was heated overnight at 75 °C under N2 atmosphere. After the completion of the reaction, the solvent was evaporated under reduced pressure. Water was added to the residue, and the resulting mixture was extracted with ethyl acetate. The combined organic solvents were washed with brine and dried over Na2SO4. The solvent was evaporated under vacuum to give the crude product, which was purified by column chromatography (petroleum ether/ethyl acetate, 10:1, v/v) to obtain the desired product 6a. Compounds 6b–6k were synthesised using a similar procedure as compound 6a.
3-Vinylphenyl dimethylcarbamate (6a)
White oil; yield: 82% (1.30 g); Rf: 0.29 (petroleum ether/ethyl acetate, 5:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 7.37–7.29 (m, 2H), 7.24–7.22 (m, 1H), 7.03–7.00 (m, 1H), 6.72 (dd, J = 17.7, 11.0 Hz, 1H), 5.89–5.84 (m, 1H), 5.31–5.28 (m, 1H), 3.04 (s, 3H), 2.91 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 153.95, 151.62, 138.42, 135.86, 129.29, 123.03, 121.37, 119.30, 115.10, 36.30, 36.07. HRMS (ESI) (m/z): calculated for C11H13NO2 [M + H]+ 192.1025, found 192.1026.
3-Vinylphenyl ethyl(methyl)carbamate (6b)
Pale yellow oil; yield: 73% (2.50 g); Rf: 0.25 (petroleum ether/ethyl acetate, 10:1, v/v). 1H NMR (600 MHz, DMSO-d6) δ 7.36–7.31 (m, 2H), 7.23 (s, 1H), 7.01 (d, J = 7.7 Hz, 1H), 6.73 (dd, J = 17.7, 10.9 Hz, 1H), 5.87 (d, J = 17.8 Hz, 1H), 5.29 (d, J = 11.0 Hz, 1H), 3.44–3.40 (q, J = 6.9 Hz, 1H), 3.30 (q, J = 6.9 Hz, 1H), 2.95 (d, J = 76.1 Hz, 3H), 1.14 (dt, J = 52.6, 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 153.60, 151.60, 138.43, 135.86, 129.28, 122.98, 121.36, 119.32, 115.10, 43.50, 33.90 (1/2 C), 33.54 (1/2 C), 12.97 (1/2 C), 12.18 (1/2 C). HRMS (ESI) (m/z): calculated for C12H15NO2 [M + H]+ 206.1181, found 206.1185.
3-Vinylphenyl diethylcarbamate (6c)
White oil; yield: 75% (1.37 g); Rf: 0.33 (petroleum ether/ethyl acetate, 10:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 7.37–7.29 (m, 2H), 7.23–7.21 (m, 1H), 7.03–7.00 (m, 1H), 6.73 (dd, J = 17.7, 11.0 Hz, 1H), 5.89–5.85 (m, 1H), 5.31 − 5.28 (m, 1H), 3.40 (q, J = 6.9 Hz, 2H), 3.29 (q, J = 6.9 Hz, 2H), 1.15 (dt, J = 35.7, 6.9 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 153.22, 151.57, 138.43, 135.86, 129.28, 122.92, 121.35, 119.33, 115.10, 41.76, 41.49, 14.08, 13.17. HRMS (ESI) (m/z): calculated for C13H17NO2 [M + H]+ 220.1338, found 220.1342.
3-Vinylphenyl diisopropylcarbamate (6d)
White oil; yield: 36% (1.50 g); Rf: 0.45 (petroleum ether/ethyl acetate, 10:1, v/v). 1H NMR (600 MHz, CDCl3) δ 7.33–7.29 (m, 1H), 7.23–7.21 (m, 1H), 7.17 − 7.15 (m, 1H), 7.02–7.00 (m, 1H), 6.69 (dd, J = 17.6, 10.9 Hz, 1H), 5.76–5.73 (m, 1H), 5.27–5.25 (m, 1H), 4.13 (s, 1H), 3.94 (s, 1H), 1.32 (d, J = 38.7 Hz, 12H). 13C NMR (100 MHz, DMSO-d6) δ 152.67, 151.87, 151.45, 138.43, 135.89, 130.89, 129.27, 122.69, 121.29, 119.31, 115.05, 46.19, 45.98, 21.10, 20.09. HRMS (ESI) (m/z): calculated for C15H21NO2 [M + H]+ 248.1651, found 248.1654.
3-Vinylphenyl diphenylcarbamate (6e)
White solid; yield: 48% (1.90 g); Rf: 0.43 (petroleum ether/ethyl acetate, 10:1, v/v). 1H NMR (600 MHz, DMSO-d6) δ 7.44–7.41 (m, 8H), 7.38–7.32 (m, 3H), 7.30–7.27 (m, 2H), 7.12–7.10 (m, 1H), 6.73 (dd, J = 17.7, 11.0 Hz, 1H), 5.92–5.87 (m, 1H), 5.32–5.29 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 152.46, 151.18, 142.17, 138.60, 135.72, 129.45, 129.11, 127.15, 126.65, 123.55, 121.24, 119.19, 115.38. HRMS (ESI) (m/z): calculated for C21H17NO2 [M + H]+ 316.1338, found 316.1340.
3-Vinylphenyl pyrrolidine-1-carboxylate (6f)
Yellow oil; yield: 62% (1.11 g); Rf: 0.26 (petroleum ether/ethyl acetate, 5:1, v/v). 1H NMR (600 MHz, CDCl3) δ 7.31–7.29 (m, 1H), 7.23–7.21 (m, 1H), 7.20–7.18 (m, 1H), 7.04–7.02 (m, 1H), 6.68 (dd, J = 17.6, 10.9 Hz, 1H), 5.75–5.72 (m, 1H), 5.27–5.25 (m, 1H), 3.57 (t, J = 6.7 Hz, 2H), 3.48 (t, J = 6.7 Hz, 2H), 2.00–1.90 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 152.14, 151.49, 138.41, 135.89, 129.27, 122.96, 121.38, 119.32, 115.06, 46.18, 45.98, 25.27, 24.42. HRMS (ESI) (m/z): calculated for C13H15NO2 [M + H]+ 218.1181, found 218.1189.
3-Vinylphenyl piperidine-1-carboxylate (6g)
Pale yellow oil; yield: 52% (1.00 g); Rf: 0.33 (petroleum ether/ethyl acetate, 10:1, v/v). 1H NMR (600 MHz, CDCl3) δ 7.31–7.29 (m, 1H), 7.24–7.21 (m, 1H), 7.17–7.15 (m, 1H), 7.01 − 6.99 (m, 1H), 6.68 (dd, J = 17.6, 10.9 Hz, 1H), 5.76–5.72 (m, 1H), 5.27–5.25 (m, 1H), 3.61 (s, 2H), 3.51 (s, 2H), 1.65–1.63 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 152.76, 151.61, 138.43, 135.87, 129.28, 122.98, 121.35, 119.31, 115.11, 45.01, 44.52, 25.44, 25.10, 23.66. HRMS (ESI) (m/z): calculated for C14H17NO2 [M + H]+ 232.1338, found 232.1340.
3-Vinylphenyl morpholine-4-carboxylate (6h)
White solid; yield: 88% (1.70 g); Rf: 0.42 (petroleum ether/ethyl acetate, 3:1, v/v). 1H NMR (600 MHz, CDCl3) δ 7.33–7.30 (m, 1H), 7.26–7.24 (m, 1H), 7.17–7.15 (m, 1H), 7.02–7.00 (m, 1H), 6.68 (dd, J = 17.6, 10.9 Hz, 1H), 5.77–5.73 (m, 1H), 5.29–5.26 (m, 1H), 3.77–3.74 (m, 4H), 3.68 (s, 2H), 3.57 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 152.93, 151.42, 138.50, 135.82, 129.36, 123.22, 121.32, 119.26, 115.21, 65.74, 44.52, 43.81. HRMS (ESI) (m/z): calculated for C13H15NO3 [M + Na]+ 256.0950, found 256.0952.
3-Vinylphenyl 4-benzylpiperidine-1-carboxylate (6i)
Yellow oil; yield: 33% (0.67 g); Rf: 0.29 (petroleum ether/ethyl acetate, 10:1, v/v). 1H NMR (600 MHz, DMSO-d6) δ 7.44–7.26 (m, 5H), 7.24–7.13 (m, 4H), 7.01–6.99 (m, 0.5H), 6.72 (dd, J = 17.7, 10.9 Hz, 0.5H), 5.86 (d, J = 17.6 Hz, 0.5H), 5.29 (d, J = 11.0 Hz, 0.5H), 4.16–3.94 (m, 2H), 3.00–2.75 (m, 2H), 2.56–2.54 (m, 2H), 1.79–1.72 (m, 1H), 1.62 (d, J = 13.3 Hz, 2H), 1.24–1.13 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 152.74, 151.58, 138.43, 135.85, 129.29, 128.97, 128.14, 125.81, 123.01, 121.35, 119.30, 115.14, 44.28, 43.90, 41.99, 36.99, 31.43, 31.11. HRMS (ESI) (m/z): calculated for C21H23NO2 [M + Na]+ 344.1626, found 344.1634.
3-Vinylphenyl 4-benzylpiperazine-1-carboxylate (6j)
White solid; yield: 27% (0.55 g); Rf: 0.24 (petroleum ether/ethyl acetate, 3:1, v/v). 1H NMR (600 MHz, DMSO-d6) δ 7.36–7.31 (m, 6H), 7.30–7.22 (m, 2H), 7.02 (d, J = 7.3 Hz, 1H), 6.72 (dd, J = 17.6, 11.0 Hz, 1H), 5.87 (d, J = 17.7 Hz, 1H), 5.29 (d, J = 11.0 Hz, 1H), 3.59 (s, 2H), 3.53 (s, 2H), 3.43 (s, 2H), 2.42 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 152.54, 148.25, 137.78, 129.98, 129.85, 128.86, 128.67, 128.19, 127.01, 125.97, 125.65, 123.20, 116.51, 61.86, 52.35, 52.14, 44.24, 43.69. HRMS (ESI) (m/z): calculated for C20H22N2O2 [M + H]+ 323.1760, found 323.1765.
1-(Piperidin-1-yl)-2–(3-vinylphenoxy)ethan-1-one (6k)
White oil; yield: 65% (0.80 g); Rf: 0.21 (petroleum ether/ethyl acetate, 3:1, v/v). 1H NMR (600 MHz, CDCl3) δ 7.25–7.22 (m, 1H), 7.05–7.02 (m, 1H), 7.01–6.98 (m, 1H), 6.87–6.84 (m, 1H), 6.68 (dd, J = 17.6, 10.9 Hz, 1H), 5.76–5.71 (m, 1H), 5.27–5.22 (m, 1H), 4.68 (s, 2H), 3.58–3.55 (m, 2H), 3.51–3.47 (m, 2H), 1.61–1.52 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 166.20, 158.39, 139.27, 136.63, 129.70, 124.73, 119.75, 113.99, 112.55, 67.86, 46.55, 43.32, 26.57, 25.62, 24.54. HRMS (ESI) (m/z): calculated for C15H19NO2 [M + H]+ 246.1494, found 246.1488.
General procedures for the synthesis of compounds 7a–7p
A mixture of compound 6a (0.40 g, 2.09 mmol), 3a (0.93 g, 3.14 mmol), triethylamine (0.42 g, 4.18 mmol), and palladium acetate (0.09 g, 0.42 mmol) in N,N-dimethylformamide (10 ml) was stirred at 100 °C under N2 atmosphere. Once compound 6a had disappeared (as detected by TLC), the mixture was filtered through a Celite pad. Water was then added, and the resulting mixture was extracted with ethyl acetate. The organic solvent phase was washed with brine, dried over anhydrous Na2SO4, and evaporated under a vacuum. The desired product 7a was obtained through purification using column chromatography (petroleum ether/ethyl acetate, 1:1, v/v). Compounds 7b–7p were synthesised using a similar procedure as compound 7a.
(E)-3–(2-(3-(benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl dimethylcarbamate (7a)
Pale yellow solid; yield: 16% (0.14 g); Rf: 0.38 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 96.2–96.6 °C; purity: 99.60%. 1H NMR (600 MHz, DMSO-d6) δ 7.47–7.44 (m, 2H), 7.42–7.37 (m, 2H), 7.36–7.34 (m, 2H), 7.31–7.28 (m, 1H), 7.27 (t, J = 1.9 Hz, 1H), 7.24 (d, J = 16.3 Hz, 1H), 7.11–7.09 (m, 1H), 6.98 (d, J = 16.4 Hz, 1H), 6.27 (d, J = 0.7 Hz, 1H), 5.14 (s, 2H), 3.07 (s, 3H), 2.94 (s, 3H), 2.31 (d, J = 0.5 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 174.76, 164.30, 153.96, 153.86, 151.73, 141.17, 136.67, 136.55, 132.76, 129.68, 128.98, 128.27, 124.36, 122.68, 120.31, 115.95, 113.89, 73.36, 36.29, 36.09, 19.04. HRMS (ESI) (m/z): calculated for C24H23NO5 [M + H]+ 406.1654, found 406.1669.
(E)-3–(2-(3-(benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl ethyl(methyl)carbamate (7b)
Yellow solid; yield: 19% (0.30 g); Rf: 0.45 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 111.6–112.8 °C; purity: 96.82%. 1H NMR (600 MHz, DMSO-d6) δ 7.46 (d, J = 7.1 Hz, 2H), 7.42–7.33 (m, 4H), 7.32–7.22 (m, 3H), 7.10 (d, J = 6.6 Hz, 1H), 6.98 (d, J = 16.3 Hz, 1H), 6.27 (s, 1H), 5.14 (s, 2H), 3.40 (dq, J = 68.9, 7.9, 6.9 Hz, 2H), 2.99 (d, J = 78.4 Hz, 3H), 2.32 (s, 3H), 1.17 (dt, J = 54.3, 6.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 174.81, 164.38, 154.03, 151.73, 141.19, 136.69, 136.58, 132.83, 129.73, 129.03, 128.30, 124.36, 122.74, 120.37, 115.95, 113.92, 73.42, 43.53, 39.92, 33.95 (1/2 C), 33.63 (1/2 C), 19.09, 13.09 (1/2 C), 12.28 (1/2 C). HRMS (ESI) (m/z): calculated for C25H25NO5 [M + H]+ 420.1811, found 420.1818.
(E)-3–(2-(3-(benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl diethylcarbamate (7c)
Yellow solid; yield: 7% (0.12 g); Rf: 0.42 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 94.9–95.3 °C; purity: 98.94%. 1H NMR (400 MHz, CD3CN-d3) δ 7.46 (d, J = 7.5 Hz, 2H), 7.40–7.34 (m, 3H), 7.31 (d, J = 7.8 Hz, 2H), 7.25 (d, J = 16.4 Hz, 1H), 7.20 (s, 1H), 7.08 (d, J = 7.4 Hz, 1H), 7.00 (d, J = 16.3 Hz, 1H), 6.14 (s, 1H), 5.16 (s, 2H), 3.47 (q, J = 7.3 Hz, 2H), 3.37 (q, J = 7.2 Hz, 2H), 2.29 (s, 3H), 1.26 (t, J = 6.3 Hz, 3H), 1.18 (t, J = 6.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 176.06, 164.28, 155.62, 154.13, 152.10, 141.74, 137.03, 136.77, 133.87, 129.70, 129.29, 128.66, 124.34, 122.85, 120.78, 116.15, 114.41, 74.68, 42.49, 42.11, 19.94, 14.45, 13.57. HRMS (ESI) (m/z): calculated for C26H27NO5 [M + H]+ 434.1967, found 434.1949.
(E)-3–(2-(3-(benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl diisopropylcarbamate (7d)
White solid; yield: 47% (0.87 g); Rf: 0.31 (petroleum ether/ethyl acetate, 1:1, v/v); mp: 105.8–107.6 °C; purity: 96.09%. 1H NMR (600 MHz, DMSO-d6) δ 7.47–7.44 (m, 2H), 7.41–7.33 (m, 4H), 7.32–7.22 (m, 3H), 7.11–7.07 (m, 1H), 6.96 (d, J = 16.4 Hz, 1H), 6.28 (s, 1H), 5.14 (s, 2H), 4.02 (s, 2H), 2.32 (s, 3H), 1.31 (s, 6H), 1.24 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 174.75, 164.30, 153.99, 152.59, 151.53, 141.11, 136.65, 136.55, 132.80, 129.70, 129.00, 128.25, 124.05, 122.66, 120.33, 115.85, 113.89, 73.36, 46.21, 45.76, 21.25, 20.14, 19.03. HRMS (ESI) (m/z): calculated for C28H31NO5 [M + H]+ 462.2280, found 462.2300.
(E)-3–(2-(3-(benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl diphenylcarbamate (7e)
Orange solid; yield: 33% (0.56 g); Rf: 0.37 (petroleum ether/ethyl acetate, 1:1, v/v); mp: 137.6–138.5 °C; purity: 98.57%. 1H NMR (600 MHz, DMSO-d6) δ 7.52–7.43 (m, 10H), 7.42–7.39 (m, 3H), 7.35–7.29 (m, 4H), 7.28–7.19 (m, 3H), 7.05–6.99 (d, J = 16.4 Hz, 1H), 6.27 (s, 1H), 5.15 (s, 2H), 2.32 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 174.79, 164.34, 153.97, 152.37, 151.27, 142.16, 141.16, 136.71, 136.60, 132.59, 129.83, 129.16, 129.04, 128.27, 127.18, 126.74, 124.85, 122.58, 120.24, 116.14, 113.92, 73.36, 19.05. HRMS (ESI) (m/z): calculated for C34H27NO5 [M + H]+ 530.1967, found 530.1979.
(E)-3–(2-(3-(benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl pyrrolidine-1-carboxylate (7f)
White solid; yield: 40% (0.80 g); Rf: 0.40 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 140.0–140.5 °C; purity: 97.77%. 1H NMR (600 MHz, CD3CN-d3) δ 7.46 (d, J = 6.8 Hz, 2H), 7.39–7.35 (m, 3H), 7.33–7.29 (m, 2H), 7.28–7.19 (m, 2H), 7.09 (d, J = 7.9 Hz, 1H), 7.00 (d, J = 16.3 Hz, 1H), 6.14 (s, 1H), 5.16 (s, 2H), 3.56 (t, J = 6.4 Hz, 2H), 3.40 (t, J = 6.5 Hz, 2H), 2.29 (s, 3H), 1.97 (d, J = 5.7 Hz, 2H), 1.93–1.89 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 174.76, 164.31, 153.96, 152.05, 151.60, 141.16, 136.67, 136.54, 132.80, 129.68, 128.98, 128.27, 124.30, 122.70, 120.35, 115.92, 113.89, 73.35, 46.26, 46.05, 25.31, 24.46, 19.04. HRMS (ESI) (m/z): calculated for C26H25NO5 [M + H]+ 432.1811, found 432.1818.
(E)-3–(2-(3-(benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl piperidine-1-carboxylate (7g)
White solid; yield: 34% (0.66 g); Rf: 0.50 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 112.4–112.9 °C; purity: 99.54%. 1H NMR (600 MHz, DMSO-d6) δ 7.48–7.44 (m, 2H), 7.42–7.38 (m, 2H), 7.37–7.34 (m, 2H), 7.31–7.28 (m, 1H), 7.27–7.23 (m, 2H), 7.11–7.09 (m, 1H), 6.98 (d, J = 16.4 Hz, 1H), 6.27 (s, 1H), 5.14 (s, 2H), 3.58 (s, 2H), 3.43 (s, 2H), 2.32 (s, 3H), 1.65–1.52 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 174.77, 164.33, 153.98, 152.67, 151.71, 141.17, 136.68, 136.55, 132.79, 129.69, 129.01, 128.29, 128.27, 124.32, 122.69, 120.34, 115.95, 113.89, 73.38, 45.07, 44.59, 25.50, 25.10, 23.68, 19.05. HRMS (ESI) (m/z): calculated for C27H27NO5 [M + H]+ 446.1967, found 446.1991.
(E)-3–(2-(3-(benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl morpholine-4-carboxylate (7h)
Pale yellow solid; yield: 52% (1.00 g); Rf: 0.30 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 162.9–164.3 °C; purity: 97.85%. 1H NMR (600 MHz, DMSO-d6) δ 7.46 (d, J = 6.8 Hz, 2H), 7.43–7.39 (m, 2H), 7.36 (t, J = 7.3 Hz, 2H), 7.32–7.28 (m, 2H), 7.25 (d, J = 16.3 Hz, 1H), 7.16–7.11 (m, 1H), 7.00 (d, J = 16.3 Hz, 1H), 6.27 (s, 1H), 5.14 (s, 2H), 3.67 (s, 4H), 3.62 (s, 2H), 3.45 (s, 2H), 2.32 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 174.80, 164.34, 153.97, 152.86, 151.55, 141.21, 136.70, 136.63, 132.74, 129.76, 129.00, 128.31, 128.30, 124.53, 122.65, 120.33, 116.02, 113.92, 73.38, 65.79, 44.58, 43.84, 19.05. HRMS (ESI) (m/z): calculated for C26H25NO6 [M + H]+ 448.1760, found 448.1736.
(E)-3–(2-(3-(benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl 4-benzylpiperidine-1-carboxylate (7i)
White solid; yield: 18% (0.19 g); Rf: 0.37 (petroleum ether/ethyl acetate, 1:1, v/v); mp: 113.1–113.3 °C; purity: 95.46%. 1H NMR (600 MHz, DMSO-d6) δ 7.46 (d, J = 7.5 Hz, 2H), 7.42–7.33 (m, 4H), 7.29 (t, J = 6.7 Hz, 3H), 7.25 (d, J = 15.6 Hz, 2H), 7.20 (d, J = 7.2 Hz, 3H), 7.10 (d, J = 7.1 Hz, 1H), 6.99 (d, J = 16.3 Hz, 1H), 6.27 (s, 1H), 5.14 (s, 2H), 4.09 (dd, J = 80.4, 11.9 Hz, 2H), 3.05–2.79 (m, 2H), 2.57 (d, J = 6.8 Hz, 2H), 2.32 (s, 3H), 1.84–1.74 (m, 1H), 1.66 (d, J = 12.2 Hz, 2H), 1.30–1.12 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 174.76, 164.30, 153.96, 152.63, 151.68, 141.18, 139.93, 136.68, 136.55, 132.77, 129.67, 128.98, 128.26, 128.15, 125.83, 124.31, 122.67, 120.33, 115.94, 113.89, 73.37, 44.27, 43.88, 42.04, 37.03, 31.53, 31.19, 19.04. HRMS (ESI) (m/z): calculated for C34H33NO5 [M + H]+ 536.2437, found 536.2452.
(E)-3–(2-(3-(benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl 4-benzylpiperazine-1-carboxylate (7j)
Orange solid; yield: 13% (0.12 g); Rf: 0.50 (ethyl acetate); mp: 98.4–100.5 °C; purity: 98.32%. 1H NMR (600 MHz, DMSO-d6) δ 7.46 (d, J = 7.0 Hz, 2H), 7.42–7.39 (m, 2H), 7.37–7.34 (m, 6H), 7.31–7.22 (m, 4H), 7.11 (d, J = 6.9 Hz, 1H), 6.98 (d, J = 16.4 Hz, 1H), 6.27 (s, 1H), 5.14 (s, 2H), 3.63 (s, 2H), 3.54 (s, 2H), 3.47 (s, 2H), 2.45 (d, J = 7.9 Hz, 4H), 2.32 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 174.77, 164.31, 153.96, 152.68, 151.56, 141.19, 137.77, 136.68, 136.60, 132.74, 129.73, 129.00, 128.87, 128.28, 128.21, 127.03, 124.47, 122.66, 120.31, 115.99, 113.91, 73.38, 61.88, 52.24, 52.10, 44.21, 43.65, 19.05. HRMS (ESI) (m/z): calculated for C33H32N2O5 [M + H]+ 537.2389, found 537.2407.
(E)-3-(benzyloxy)-6-methyl-2–(3–(2-oxo-2-(piperidin-1-yl)ethoxy)styryl)-4H-pyran-4-one (7k)
Pale yellow solid; yield: 11% (0.16 g); Rf: 0.47 (ethyl acetate); mp: 117.9–118.1 °C; purity: 99.35%. 1H NMR (600 MHz, DMSO-d6) δ 7.47–7.44 (m, 2H), 7.37 − 7.34 (m, 2H), 7.33–7.28 (m, 2H), 7.20 (d, J = 16.3 Hz, 1H), 7.13–7.10 (m, 2H), 7.01 (d, J = 16.3 Hz, 1H), 6.93–6.90 (m, 1H), 6.26 (d, J = 0.6 Hz, 1H), 5.15 (s, 2H), 4.85 (s, 2H), 3.45–3.39 (m, 4H), 2.32 (s, 3H), 1.61–1.57 (m, 2H), 1.56–1.51 (m, 2H), 1.46 − 1.41 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 174.77, 165.31, 164.29, 158.47, 154.11, 140.92, 136.59, 136.54, 133.49, 129.85, 128.97, 128.31, 128.28, 120.07, 115.78, 115.45, 113.88, 113.10, 73.22, 66.08, 45.22, 42.16, 25.96, 25.28, 23.92, 19.07. HRMS (ESI) (m/z): calculated for C28H29NO5 [M + Na]+ 482.1943, found 482.1935.
(E)-3–(2-(3-(benzyloxy)-4-oxo-4H-pyran-2-yl)vinyl)phenyl piperidine-1-carboxylate (7l)
White solid; yield: 26% (0.12 g); Rf: 0.53 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 126.6–127.3 °C; purity: 97.46%. 1H NMR (600 MHz, DMSO-d6) δ 8.12 (d, J = 5.7 Hz, 1H), 7.48–7.46 (m, 2H), 7.42–7.34 (m, 4H), 7.32–7.29 (m, 1H), 7.28–7.27 (m, 1H), 7.24 (d, J = 16.4 Hz, 1H), 7.13–7.09 (m, 1H), 7.02 (d, J = 16.4 Hz, 1H), 6.42 (d, J = 5.6 Hz, 1H), 5.16 (s, 2H), 3.59 (s, 2H), 3.43 (s, 2H), 1.65–1.52 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 174.42, 154.77, 154.75, 152.70, 151.73, 142.37, 136.60, 136.49, 133.23, 129.71, 129.04, 128.35, 128.30, 124.55, 122.91, 120.31, 116.32, 115.97, 73.46, 45.09, 44.61, 25.50, 25.13, 23.69. HRMS (ESI) (m/z): calculated for C26H25NO5 [M + Na]+ 454.1630, found 454.1624.
(E)-3–(2-(3-(benzyloxy)-6-(hydroxymethyl)-4-oxo-4H-pyran-2-yl)vinyl)phenyl piperidine-1-carboxylate (7m)
White solid; yield: 13% (0.16 g); Rf: 0.56 (ethyl acetate); mp: 128.6–129.1 °C; purity: 96.00%. 1H NMR (600 MHz, DMSO-d6) δ 7.48–7.46 (m, 2H), 7.41 (t, J = 7.8 Hz, 1H), 7.39–7.34 (m, 3H), 7.32–7.25 (m, 3H), 7.12–7.09 (m, 1H), 7.01 (d, J = 16.3 Hz, 1H), 6.34 (s, 1H), 5.74 (t, J = 6.3 Hz, 1H), 5.16 (s, 2H), 4.38 (d, J = 6.2 Hz, 2H), 3.59 (s, 2H), 3.43 (s, 2H), 1.67–1.50 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 174.86, 167.06, 153.92, 152.68, 151.75, 141.42, 136.66, 136.54, 133.16, 129.75, 129.05, 128.34, 128.31, 124.34, 122.75, 120.37, 115.92, 111.54, 73.44, 59.37, 45.10, 44.58, 25.52, 25.13, 23.70. HRMS (ESI) (m/z): calculated for C27H27NO6 [M + Na]+ 484.1736, found 484.1729.
(E)-3–(2-(6-methyl-3-(naphthalen-2-ylmethoxy)-4-oxo-4H-pyran-2-yl)vinyl)phenyl piperidine-1-carboxylate (7n)
Yellow solid; yield: 54% (0.29 g); Rf: 0.48 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 126.3–127.1 °C; purity: 98.59%. 1H NMR (600 MHz, DMSO-d6) δ 7.96 (s, 1H), 7.93–7.89 (m, 2H), 7.88–7.85 (m, 1H), 7.67 (dd, J = 8.4, 1.7 Hz, 1H), 7.51–7.48 (m, 2H), 7.37–7.29 (m, 3H), 7.23 (d, J = 16.4 Hz, 1H), 7.10–7.06 (m, 2H), 6.28 (d, J = 0.7 Hz, 1H), 5.33 (s, 2H), 3.58 (s, 2H), 3.43 (s, 2H), 2.32–2.28 (m, 3H), 1.65–1.50 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 174.82, 164.35, 153.96, 152.68, 151.71, 141.18, 136.51, 134.31, 132.92, 132.68, 132.65, 129.62, 127.86, 127.76, 127.50, 126.79, 126.28, 126.24, 124.24, 122.72, 120.40, 115.92, 113.92, 73.38, 45.06, 44.61, 25.49, 25.11, 23.69, 19.05. HRMS (ESI) (m/z): calculated for C31H29NO5 [M + Na]+ 518.1943, found 518.1937.
(E)-3–(2-(3-([1,1′-biphenyl]-4-ylmethoxy)-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl piperidine-1-carboxylate (7o)
White solid; yield: 41% (0.28 g); Rf: 0.50 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 131.3–131.8 °C; purity: 99.38%. 1H NMR (600 MHz, DMSO-d6) δ 7.63–7.59 (m, 2H), 7.55–7.50 (m, 4H), 7.41–7.37 (m, 2H), 7.36–7.33 (m, 2H), 7.32–7.28 (m, 2H), 7.22 (d, J = 16.3 Hz, 1H), 7.08–7.05 (m, 1H), 6.99 (d, J = 16.3 Hz, 1H), 6.25 (d, J = 0.8 Hz, 1H), 5.15 (s, 2H), 3.50 (s, 2H), 3.36 (s, 2H), 2.29 (d, J = 0.6 Hz, 3H), 1.58–1.42 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 174.79, 164.37, 154.06, 152.65, 151.72, 141.18, 140.13, 139.67, 136.55, 135.81, 132.86, 129.69, 129.61, 128.85, 127.49, 126.61, 124.42, 122.67, 120.21, 115.95, 113.94, 73.10, 44.99, 44.52, 25.51, 25.16, 23.66, 19.08. HRMS (ESI) (m/z): calculated for C33H31NO5 [M + Na]+ 544.2100, found 544.2093.
(E)-3-(2-(3-methoxy-6-methyl-4-oxo-4H-pyran-2-yl)vinyl)phenyl piperidine-1-carboxylate (7p)
Pale yellow solid; yield: 44% (0.35 g); Rf: 0.32 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 106.7–106.9 °C; purity: 97.43%. 1H NMR (600 MHz, DMSO-d6) δ 7.54–7.50 (m, 2H), 7.42 (t, J = 7.9 Hz, 1H), 7.38 (d, J = 16.3 Hz, 1H), 7.26 (d, J = 16.3 Hz, 1H), 7.13–7.10 (m, 1H), 6.24 (d, J = 0.7 Hz, 1H), 3.83 (s, 3H), 3.57 (s, 2H), 3.41 (s, 2H), 2.34 (d, J = 0.6 Hz, 3H), 1.64–1.50 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 174.61, 164.29, 153.43, 152.73, 151.79, 143.00, 136.63, 133.28, 129.69, 124.72, 122.86, 120.52, 115.74, 113.94, 60.21, 45.07, 44.59, 25.45, 25.09, 23.68, 19.08. HRMS (ESI) (m/z): calculated for C21H23NO5 [M + Na]+ 392.1474, found 392.1468.
General procedures for the synthesis of compounds 6l and 6m
Compound 3a (1.00 g, 3.39 mmol), 7-(Bpin)naphthalen-2-ol (1.10 g, 4.07 mmol), anhydrous potassium carbonate (0.70 g, 5.08 mmol), and [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloride (0.19 g, 0.25 mmol) were dissolved in a mixture of tetrahydrofuran and water (20 ml, 9:1, v/v). The resulting solution was stirred overnight at 75 °C under N2 atmosphere. Subsequently, the mixture was extracted with ethyl acetate after the addition of water. The organic layers collected were then washed with brine, dried over anhydrous Na2SO4, and concentrated using rotational evaporation. The desired product 6l was obtained through purification via column chromatography (petroleum ether/ethyl acetate, 3:1, v/v). Compound 6m was synthesised using a similar procedure as compound 6l.
3-(Benzyloxy)-2–(7-hydroxynaphthalen-2-yl)-6-methyl-4H-pyran-4-one (6l)
Brown solid; yield: 49% (0.59 g); Rf: 0.30 (petroleum ether/ethyl acetate, 1:1, v/v). 1H NMR (600 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.20 (d, J = 1.1 Hz, 1H), 7.85 (d, J = 8.7 Hz, 1H), 7.81 (d, J = 8.8 Hz, 1H), 7.67 (dd, J = 8.6, 1.7 Hz, 1H), 7.34–7.31 (m, 2H), 7.29 − 7.27 (m, 3H), 7.17 (dd, J = 8.8, 2.4 Hz, 1H), 7.12 (d, J = 2.3 Hz, 1H), 6.37 (d, J = 0.5 Hz, 1H), 5.12 (s, 2H), 2.38 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 175.45, 165.07, 155.92, 155.18, 142.45, 136.68, 133.83, 129.17, 128.32, 128.20, 128.04, 127.66, 127.63, 126.47, 121.32, 120.35, 113.91, 109.42, 72.72, 19.16. HRMS (ESI) (m/z): calculated for C23H18O4 [M + H]+ 359.1283, found 359.1278.
3-(Benzyloxy)-2–(3′-hydroxy-[1,1′-biphenyl]-4-yl)-6-methyl-4H-pyran-4-one (6m)
White solid; yield: 46% (0.18 g); Rf: 0.35 (petroleum ether/ethyl acetate, 1:1, v/v). 1H NMR (600 MHz, DMSO-d6) δ 9.61 (s, 1H), 7.94–7.90 (m, 2H), 7.73–7.69 (m, 2H), 7.34–7.26 (m, 6H), 7.15–7.12 (m, 1H), 7.09 (t, J = 2.0 Hz, 1H), 6.84–6.79 (m, 1H), 6.36 (d, J = 0.7 Hz, 1H), 5.11 (s, 2H), 2.36 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 175.35, 164.99, 157.87, 154.55, 142.26, 141.90, 140.39, 136.58, 130.06, 129.02, 128.41, 128.38, 128.17, 128.04, 126.43, 117.52, 115.08, 113.88, 113.48, 72.67, 19.10. HRMS (ESI) (m/z): calculated for C25H20O4 [M + H]+ 385.1440, found 385.1434.
General procedures for the synthesis of compounds 7q and 7r
A mixture of piperidine-1-carbonyl chloride (0.12 g, 0.84 mmol), anhydrous potassium carbonate (0.15 g, 1.12 mmol), and 4-dimethylaminopyridine (0.01 g, 0.11 mmol) in anhydrous acetonitrile (10 ml) was prepared. Then, compound 6l (0.20 g, 0.59 mmol) was added at 0 °C. The resulting mixture was stirred overnight at 75 °C under N2 atmosphere. After the reaction was completed, the solvent was evaporated under reduced pressure. The residue was extracted with ethyl acetate after adding water. The combined organic solvents were washed with brine, dried over Na2SO4, and evaporated under vacuum. Finally, the residue was purified by column chromatography (petroleum ether/ethyl acetate, 2:1, v/v) to obtain product 7q. Compound 7r was synthesised using a similar procedure as compound 7q.
7–(3-(Benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)naphthalen-2-yl piperidine-1-carboxylate (7q)
White solid; yield: 53% (0.15 g); Rf: 0.47 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 105.9–106.7 °C; purity: 99.65%. 1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 7.99 (t, J = 8.6 Hz, 2H), 7.88 (dd, J = 8.7, 1.7 Hz, 1H), 7.67 (d, J = 2.1 Hz, 1H), 7.40 (dd, J = 8.9, 2.3 Hz, 1H), 7.34–7.28 (m, 2H), 7.28–7.21 (m, 3H), 6.38 (s, 1H), 5.15 (s, 2H), 3.61 (s, 2H), 3.45 (s, 2H), 2.38 (s, 3H), 1.66–1.52 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 175.43, 165.12, 154.66, 152.80, 149.57, 142.59, 136.60, 132.62, 130.92, 128.80, 128.41, 128.15, 128.07, 128.03, 127.75, 127.70, 124.10, 123.68, 119.14, 113.97, 72.81, 45.18, 44.65, 40.13, 25.55, 25.13, 23.71, 19.14. HRMS (ESI) (m/z): calculated for C29H27NO5 [M + Na]+ 492.1787, found 492.1781.
4′-(3-(Benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)-[1,1′-biphenyl]-3-yl piperidine-1-carboxylate (7r)
White solid; yield: 47% (0.10 g); Rf: 0.50 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 101.7–102.9 °C; purity: 95.01%. 1H NMR (400 MHz, DMSO-d6) δ 7.94 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 7.8 Hz, 1H), 7.52–7.46 (m, 2H), 7.36–7.26 (m, 5H), 7.18–7.13 (m, 1H), 6.36 (s, 1H), 5.11 (s, 2H), 3.58 (s, 2H), 3.42 (s, 2H), 2.36 (s, 3H), 1.67–1.49 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 175.34, 165.01, 154.40, 152.79, 151.92, 142.32, 140.71, 140.15, 136.54, 129.83, 129.39, 128.44, 128.37, 128.18, 128.04, 126.58, 123.46, 121.61, 120.16, 113.89, 72.63, 45.07, 44.62, 25.44, 25.06, 23.69, 19.10. HRMS (ESI) (m/z): calculated for C31H29NO5 [M + H]+ 496.2124, found 496.2117.
Preparation of compound 7s
Compound 7g (0.45 g, 1.01 mmol) and 10% Pt/C (0.05 g, 0.03 mmol) were added to tetrahydrofuran (15 ml) and stirred at 50 °C under H2 atmosphere. The reaction was thoroughly monitored using TLC. Subsequently, the mixture was filtered through a Celite pad and extracted with ethyl acetate, followed by the addition of water. The organic solvent phase was washed with brine, dried over anhydrous Na2SO4, and evaporated under a vacuum. The crude product was purified by silica gel chromatography (petroleum ether/ethyl acetate, 2:1, v/v) to give product 7s.
3–(2-(3-(Benzyloxy)-6-methyl-4-oxo-4H-pyran-2-yl)ethyl)phenyl piperidine-1-carboxylate (7s)
White solid; yield: 82% (0.37 g); Rf: 0.45 (petroleum ether/ethyl acetate, 1:2, v/v); mp: 98.6–99.1 °C; purity: 97.68%. 1H NMR (400 MHz, DMSO-d6) δ 7.40–7.32 (m, 5H), 7.25 (t, J = 7.8 Hz, 1H), 6.97–6.91 (m, 2H), 6.90–6.87 (m, 1H), 6.24–6.18 (m, 1H), 4.85 (s, 2H), 3.52 (s, 2H), 3.39 (s, 2H), 2.81–2.65 (m, 4H), 2.21 (s, 3H), 1.65–1.46 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 174.50, 164.57, 159.88, 152.74, 151.32, 141.91, 141.60, 137.01, 129.01, 128.52, 128.29, 128.06, 124.95, 121.62, 119.70, 113.86, 72.61, 45.01, 44.52, 31.84, 29.35, 25.48, 25.12, 23.68, 18.88. HRMS (ESI) (m/z): calculated for C27H29NO5 [M + Na]+ 470.1943, found 470.1938.
Biological evaluation
AChE and BuChE assays
The cholinesterase inhibitory activity was determined using a modified Ellman’s method. The assay was performed in PBS buffer (100 mM, pH 8.0) with a final reaction mixture of 100 μL. Concisely, the tested compounds (10 μL), DTNB (20 µL, 10 mM), AChE or BuChE (10 µL, 2.5 U/mL for eeAChE and eqBuChE, 0.75 U/mL for huBuChE), and 40 µL PBS were incubated at 37 °C for 10 min, and then ATC or BTC (20 µL, 75 mM) was added and incubated for another 10 min (for eeAChE and eqBuChE) or 30 min (for huBuChE) at 37 °C. Finally, the absorbance value was measured at 412 nm using a microplate reader (Epoch, BioTek). The IC50 values were calculated using the GraphPad Prism 8.0 software.
Anti-neuroinflammation
Cell viability assay
BV-2 cells (1 × 104 cells/well) were seeded in 96-well plates and incubated for 24 h. Subsequently, the medium containing compounds was added and the cells were further cultured for an additional 24 h. After that, 20 μL of MTT solution (5 mg/mL) was added to each well and incubated for another 4 h. The cell culture medium was then removed, and 150 μL of dimethyl sulfoxide (DMSO) was added to dissolve the formazan. The absorbance was measured at 490 nm using a microplate reader.
Nitric oxide measurement
BV-2 cells (5 × 104 cells/well) were inoculated into 96-well plates and cultured for 24 h. The tested compounds were added for 1 h, and then LPS (1 μg/mL, final concentration) was added to all wells except for the control. Following 24 h of treatment, the supernatant medium was harvested, and the level of nitric oxide was measured using the nitric oxide assay kit (Beyotime) according to the manufacturer’s instructions.
Kinetic studies of BuChE inhibition
The enzyme kinetics of compound 7p were investigated using a modified Ellman’s method. A mixture containing different concentrations of compound 7p (10 μL), DTNB (20 µL, 10 mM), eqBuChE (10 µL, 2.5 U/mL), and 40 µL PBS was incubated at 37 °C for 10 min. Following that, BTC (20 µL, 1.25–10.0 mM) was added, and the absorbance value was immediately measured on a multi-mode microplate reader (SpectarMax pardigm, Molecular Devices) at 412 nm every minute for 10 min, with automatic shaking of the well plates before each reading. Dixon and Cornish-Bowden plots were constructed using linear regression in GraphPad Prism 8.0 software to determine the type and constants of inhibitionCitation41.
Docking studies
The docking studies were conducted using AutoDock 4.2 software. The crystal structures of BuChE (PDB: 1P0I)Citation42 and AChE (PDB: 4EY7)Citation43 were retrieved from the Protein Data Bank (https://rcsb.org). Pymol software was utilised to modify both proteins by removing the ligand, water molecules, and unbonded co-crystallized compounds. Using Autodock software, proteins and ligands were further prepared by adding polar hydrogen atoms and Gasteiger charges to enzymes, as well as charge calculations and setting up rotatable bonds for ligands. The Lamarckian Genetic Algorithm was used with a random individual operation of 100, while the remaining parameters were kept at their default values. The binding mode was selected based on the minimal-energy docked conformation obtained from AutoDock software. The graphic manipulation and visualisation were further performed using Pymol software.
SwissADME calculations
The physicochemical properties, drug-likeness, and BBB permeation of compounds 7a–7s were predicted using the online software tool SwissADME (http://www.swissadme.ch/).
In vitro blood-brain barrier permeation assay
The BBB penetration of compound 7p was assessed using the parallel artificial membrane permeation assay. Commercial drugs and test compound were dissolved in DMSO at a concentration of 5 mg/mL, and then diluted in PBS (pH = 7.4) to a final concentration of 25 μg/mL. The filter membrane in the donor microplate was impregnated with porcine brain lipid (PBL, 4 μL, 20 mg/mL) in dodecane. Subsequently, the donor and acceptor microplates were filled with a test solution (150 μL) and PBS buffer (300 μL), respectively. The donor filter microplate was carefully placed on top of the acceptor microplate to form a “sandwich” assembly. The assembly was then incubated at 25 °C for 10 h. After incubation, the donor microplate was carefully removed, and the solution from the acceptor well was transferred to a UV-96 microplate. The absorbance was measured at the maximum absorption wavelength using a multi-mode microplate reader, and the concentration was calculated using a standard curve (Table S2). Pe value was calculated using the following formula: Pe = {−VdVa/[(Vd+Va)At]}ln(1-drugacceptor/drugequilibrium). Six commercial drugs with known permeability were used to validate the model, and a linear correlation was established using the values of reference [Pe(ref.)] and experimental [Pe(exp.)] permeability.
Acute toxicity test
The experimental protocols were evaluated and approved by the Ethics Committee of Fujian Medical University (IACUC FJMU 2023–0288). The acute toxicity test was conducted on ICR mice (25–30 g, half male and half female). Sixteen mice were randomly divided into two groups (n = 8). Compound 7p was administered intragastrically at a single dose of 1000 mg/kg, while the control group was given a solvent (0.5% CMC-Na). Following administration, the status and behaviour of mice were continuously observed for 14 d, including their diet, water intake, body weight, and survival. At the end of the observation period, mice were sacrificed, dissected, and macroscopically inspected for possible heart, liver, spleen, and brain damage.
Behaviour studies
The experimental protocols were evaluated and approved by the Ethics Committee of Fujian Medical University (IACUC FJMU 2023–0288). Behaviour studies were performed using C57BL/6J mice (6–8 weeks old, half male and half female) through the Morris water maze test. The mice were divided into the following groups (n = 8): control, scopolamine, Rivastigmine (1 mg/kg), and compound 7p (1 and 5 mg/kg). Except for the control group, the other four groups were given the intraperitoneal injection of scopolamine (5 mg/kg) throughout the entire period (days 1–26). After ten days of injections, treatment with rivastigmine and compound 7p began for the remaining days (days 11–26), with a once-daily oral dose. The behaviour studies were conducted on days 21–26.
The MWM is a circular pool filled with water that was made opaque by adding non-toxic white TiO2. A camera was used to record the tracks of the animal. The pool was divided into four equal quadrants with visual cues, and an escape platform was placed in the centre of one quadrant. The behavioural studies consisted of five consecutive days of learning and memory training, during which the mice were trained to find the platform in the four quadrants. If a mouse failed to find the platform within 60 s, it was gently placed on the hidden platform for 15 s. On the last day, the platform was removed and a probe trial was performed. Each animal was released from a starting point and allowed to swim for 60 s to assess its spatial memory. The tracking system recorded the observed data used for the analysis of animal performance, which was performed using SMART 3.0.0.3.
Authors contributions
Chuanyu Yu designed and synthesised compounds, conducted in vitro and in vivo activity evaluation, and wrote the manuscript; Xueyan Liu conducted in vivo activity evaluation and molecular docking; Bingxiang Ma conducted in vitro activity evaluation and ADME calculations; Jiexin Xu assisted in the synthesis of compounds; Yiquan Chen and Chaoxian Dai assisted in animal experiments; Huaping Peng guided the design and implementation of the project; Daijun Zha designed and guided the entire project and edited the manuscript.
Supplemental Material
Download PDF (4.4 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, Cummings J, van der Flier WM. Alzheimer’s disease. Lancet. 2021;397(10284):1–16.
- Meyers EA, Sexton C, Snyder HM, Carrillo MC. Impact of Alzheimer’s association support and engagement in the AD/ADRD research community through the COVID-19 pandemic and beyond. Alzheimers Dement. 2023;19(7):3222–3225.
- Giacobini E, Cuello AC, Fisher A. Reimagining cholinergic therapy for Alzheimer’s disease. Brain. 2022;145(7):2250–2275.
- Srivastava S, Ahmad R, Khare SK. Alzheimer’s disease and its treatment by different approaches: a review. Eur J Med Chem. 2021;216:113320.
- Wang Y, Xiong B, Lin H, Li Q, Yang H, Qiao Y, Li Q, Xu Z, Lyu W, Qu W, et al. Design, synthesis and evaluation of fused hybrids with acetylcholinesterase inhibiting and Nrf2 activating functions for Alzheimer’s disease. Eur J Med Chem. 2022;244:114806.
- Padhi D, Govindaraju T. Mechanistic insights for drug repurposing and the design of hybrid drugs for Alzheimer’s disease. J Med Chem. 2022;65(10):7088–7105.
- Jing L, Wu G, Kang D, Zhou Z, Song Y, Liu X, Zhan P. Contemporary medicinal-chemistry strategies for the discovery of selective butyrylcholinesterase inhibitors. Drug Discov Today. 2019;24(2):629–635.
- Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018;25(1):59–70.
- Ha ZY, Mathew S, Yeong KY. Butyrylcholinesterase: A multifaceted pharmacological target and tool. Curr Protein Pept Sci. 2020;21(1):99–109.
- Xing S, Li Q, Xiong B, Chen Y, Feng F, Liu W, Sun H. Structure and therapeutic uses of butyrylcholinesterase: application in detoxification, Alzheimer’s disease, and fat metabolism. Med Res Rev. 2021;41(2):858–901.
- Košak U, Brus B, Knez D, Šink R, Žakelj S, Trontelj J, Pišlar A, Šlenc J, Gobec M, Živin M, et al. Development of an in-vivo active reversible butyrylcholinesterase inhibitor. Sci Rep. 2016;6(1):39495.
- Košak U, Brus B, Knez D, Žakelj S, Trontelj J, Pišlar A, Šink R, Jukič M, Živin M, Podkowa A, et al. The magic of crystal structure-based inhibitor optimization: Development of a butyrylcholinesterase inhibitor with picomolar affinity and in vivo activity. J Med Chem. 2018;61(1):119–139.
- Panek D, Pasieka A, Latacz G, Zaręba P, Szczęch M, Godyń J, Chantegreil F, Nachon F, Brazzolotto X, Skrzypczak-Wiercioch A, et al. Discovery of new, highly potent and selective inhibitors of BuChE - design, synthesis, in vitro and in vivo evaluation and crystallography studies. Eur J Med Chem. 2023;249:115135.
- Pereira CF, Santos AE, Moreira PI, Pereira AC, Sousa FJ, Cardoso SM, Cruz MT. Is Alzheimer’s disease an inflammasomopathy? Ageing Res Rev. 2019;56:100966.
- Wang C, Zong S, Cui X, Wang X, Wu S, Wang L, Liu Y, Lu Z. The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Front Immunol. 2023;14:1117172.
- Ozben T, Ozben S. Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clin Biochem. 2019;72:87–89.
- Twarowski B, Herbet M. Inflammatory processes in Alzheimer’s disease – pathomechanism, diagnosis and treatment: A review. Int J Mol Sci. 2023;24(7):6518.
- Kumari S, Dhapola R, Sharma P, Singh SK, Reddy DH. Implicative role of cytokines in neuroinflammation mediated AD and associated signaling pathways: Current progress in molecular signaling and therapeutics. Ageing Res Rev. 2023;92:102098.
- Choi S-B, Kwon S, Kim J-H, Ahn N-H, Lee J-H, Yang S-H. The molecular mechanisms of neuroinflammation in Alzheimer’s disease, the consequence of neural cell death. Int J Mol Sci. 2023;24(14):11757.
- Savelieff MG, Nam G, Kang J, Lee HJ, Lee M, Lim MH. Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis in the last decade. Chem Rev. 2019;119(2):1221–1322.
- Chen H, Mi J, Li S, Liu Z, Yang J, Chen R, Wang Y, Ban Y, Zhou Y, Dong W, et al. Design, synthesis and evaluation of quinoline-O-carbamate derivatives as multifunctional agents for the treatment of Alzheimer’s disease. J Enzyme Inhib Med Chem. 2023;38(1):2169682.
- Zaręba P, Łątka K, Mazur G, Gryzło B, Pasieka A, Godyń J, Panek D, Skrzypczak-Wiercioch A, Höfner GC, Latacz G, et al. Discovery of novel multifunctional ligands targeting GABA transporters, butyrylcholinesterase, β-secretase, and amyloid β aggregation as potential treatment of Alzheimer’s disease. Eur J Med Chem. 2023;261:115832.
- He M, Fan M, Peng Z, Wang G. An overview of hydroxypyranone and hydroxypyridinone as privileged scaffolds for novel drug discovery. Eur J Med Chem. 2021;221:113546.
- Liu X, Yu C, Yao Y, Lai H, Ye X, Xu J, Guo J, Xiao X, Lin C, Huang Z, et al. Novel neuroprotective pyromeconic acid derivatives with concurrent anti-Aβ deposition, anti-inflammatory, and anti-oxidation properties for treatment of Alzheimer’s disease. Eur J Med Chem. 2023;248:115120.
- Ghosh AK, Brindisi M. Organic carbamates in drug design and medicinal chemistry. J Med Chem. 2015;58(7):2895–2940.
- Zhang H, Wang Y, Wang Y, Li X, Wang S, Wang Z. Recent advance on carbamate-based cholinesterase inhibitors as potential multifunctional agents against Alzheimer’s disease. Eur J Med Chem. 2022;240:114606.
- Bar-On P, Millard CB, Harel M, Dvir H, Enz A, Sussman JL, Silman I. Kinetic and structural studies on the interaction of cholinesterases with the anti-Alzheimer drug rivastigmine. Biochemistry. 2002;41(11):3555–3564.
- Ellman GL, Courtney KD, Andres V, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7(2):88–95.
- Ruan BF, Ge WW, Cheng HJ, Xu HJ, Li QS, Liu XH. Resveratrol-based cinnamic ester hybrids: Synthesis, characterization, and anti-inflammatory activity. J Enzyme Inhib Med Chem. 2017;32(1):1282–1290.
- Fu WY, Wang X, Ip NY. Targeting neuroinflammation as a therapeutic strategy for Alzheimer’s disease: mechanisms, drug candidates, and new opportunities. ACS Chem Neurosci. 2019;10(2):872–879.
- Kulkarni B, Cruz-Martins N, Kumar D. Microglia in Alzheimer’s disease: An unprecedented opportunity as prospective drug target. Mol Neurobiol. 2022;59(5):2678–2693.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. Autodock4 and autodocktools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1-3):3–25.
- Xiong B, Wang Y, Chen Y, Xing S, Liao Q, Chen Y, Li Q, Li W, Sun H. Strategies for structural modification of small molecules to improve blood-brain barrier penetration: a recent perspective. J Med Chem. 2021;64(18):13152–13173.
- Di L, Kerns EH, Fan K, McConnell OJ, Carter GT. High throughput artificial membrane permeability assay for blood–brain barrier. Eur J Med Chem. 2003;38(3):223–232.
- Guo J, Zhang Y, Zhang C, Yao C, Zhang J, Jiang X, Zhong Z, Ge J, Zhou T, Bai R, et al. N-propargylamine-hydroxypyridinone hybrids as multitarget agents for the treatment of Alzheimer’s disease. Bioorg Chem. 2021;113:105013.
- Lazarova M, Tancheva L, Alexandrova A, Tsvetanova E, Georgieva A, Stefanova M, Tsekova D, Vezenkov L, Kalfin R, Uzunova D, et al. Effects of new galantamine derivatives in a scopolamine model of dementia in mice. J Alzheimers Dis. 2021;84(2):671–690.
- Du C, Wang L, Guan Q, Yang H, Chen T, Liu Y, Li Q, Lyu W, Lu X, Chen Y, et al. N-benzyl benzamide derivatives as selective sub-nanomolar butyrylcholinesterase inhibitors for possible treatment in advanced Alzheimer’s disease. J Med Chem. 2022;65(16):11365–11387.
- Tang KS. The cellular and molecular processes associated with scopolamine-induced memory deficit: a model of Alzheimer’s biomarkers. Life Sci. 2019;233:116695.
- Liu X, Yu C, Su B, Zha D. Synthesis and properties of the Kojic acid dimer and its potential for the treatment of Alzheimer’s disease. RSC Med Chem. 2023;14(2):268–276.
- Cornish-Bowden A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem J. 1974;137(1):143–144.
- Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J Biol Chem. 2003;278(42):41141–41147.
- Cheung J, Rudolph MJ, Burshteyn F, Cassidy MS, Gary EN, Love J, Franklin MC, Height JJ. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J Med Chem. 2012;55(22):10282–10286.