
Abstract
We present a new computational approach, named Watermelon, designed for the development of pharmacophore models based on receptor structures. The methodology involves the sampling of potential hotspots for ligand interactions within a protein target’s binding site, utilising molecular fragments as probes. By employing docking and molecular dynamics (MD) simulations, the most significant interactions formed by these probes within distinct regions of the binding site are identified. These interactions are subsequently transformed into pharmacophore features that delineates key anchoring sites for potential ligands. The reliability of the approach was experimentally validated using the monoacylglycerol lipase (MAGL) enzyme. The generated pharmacophore model captured features representing ligand-MAGL interactions observed in various X-ray co-crystal structures and was employed to screen a database of commercially available compounds, in combination with consensus docking and MD simulations. The screening successfully identified two new MAGL inhibitors with micromolar potency, thus confirming the reliability of the Watermelon approach.
Introduction
The development of a receptor-based pharmacophore model is a valuable approach often employed in structure-based virtual screening (SBVS) to identify novel active compounds. The essential requirement for the application of this strategy is the spatial and chemical characterisation of favourable ligand-protein interaction sites: this enables the definition of the chemical features a molecule should possess to bind to the target protein and exhibit a specific biological activity. Classically, the aforementioned functionalities, known as pharmacophoric features, are inferred by analysing crystal structures of the protein complexed with known active ligands or chemically diverse probes, as in the case of complexes obtained via multiple solvent crystal structures (MSCS) experimentsCitation1. Nevertheless, the necessity of at least one solved holo complex is the principal limitation of this approach. It is also worth mentioning that a receptor-based pharmacophore model developed considering just one ligand-protein complex accounts for specific interactions identified in the specific structure, but inevitably excludes other potential key interactions not shown by the reference ligand. Therefore, different computational approaches have been proposed to overcome the prohibitive aspects that characterise this scenario, with many efforts being concentrated in setting up protocols for the identification of favourable ligand-protein interaction sites with the solely knowledge of the target protein’s apo structure, to enable the development of receptor-based pharmacophore models in situations of limited or absent ligands information availability. Although being united by their purpose of mapping the protein binding cavity searching for possible hotspots, these methods can be broadly divided into two groups with respect to how the protein structure is analysed. Some approaches consider the protein as a static rigid entity, while some others account for protein flexibility: the former group includes GRID-based mapping techniques as well as docking-based approaches, while the latter comprises molecular dynamics (MD) based methods.
GRID-based approaches, such as the recently reported T2F, identify energetically favoured interaction sites by means of a 3D grid positioned in the protein’s region of interestCitation2. By iteratively placing chemically heterogeneous molecular probes at each node of the grid they calculate the binding energies with different chemical groups along the whole analysed surface. The resulting 3D energy map can then be used to place pharmacophoric features at locations where the interactions that the protein establishes with specific functional groups appear to be energetically advantageous. In docking-based approaches, such as the FTmap algorithm, the binding cavity of the protein is mapped through the docking of multiple small molecular probesCitation3. In this specific protocol, the docking procedure is then followed by an energy minimisation step, at the end of which the minimised fragments are clustered and hotspots are defined as consensus sites where probes clusters resulted to overlap. In another recently developed workflow, namely apo2ph4, the docking procedure is performed using several drug-like fragmentsCitation4. In this method, each fragment-receptor complex is directly submitted to LigandScout pharmacophore modelling softwareCitation5 to retrieve pharmacophoric features; subsequently, the final pharmacophore model to be used for VS is obtained by clustering, scoring and filtering all the generated features. Considering the importance of accounting for receptor flexibility when searching for relevant ligand-protein interactions, many authors focused on identifying key ligand anchoring sites through the use of MD techniques. According to their setup, MD simulations can be performed in explicit solvent using a solution of water with one or more cosolvents. Carlson and co-workers prioritised the mixed-solvent MD (MixMD) approach in which water-miscible molecular probes are added in a specific percentage to the bulk solvent. Besides considering the protein flexibility, this method also allows probe molecules to dynamically compete with water to interact with the protein’s surface and is therefore thought to represent a valuable computational alternative to MSCS for the identification of interaction hotspots in receptor-based pharmacophore modellingCitation6. However, surrounding the protein with a non-natural environment as in mixed solvent MDs might result in biased protein conformations, especially when using probes more lipophilic than water as cosolvents. For this reason, and also to avoid probe aggregation, the fraction of cosolvent in water needs to be well calibratedCitation7.
Driven by the idea that integrating different techniques in a unique framework could leverage the strengths of each approach, enhancing the effectiveness of the entire protocol, we developed Watermelon, an in silico procedure that brings together principles from GRID-based approach, fragment docking calculations and MD simulations to identify favourable ligand-protein interaction sites with the solely knowledge of the target protein’s apo form. The Watermelon procedure consists of three steps: (a) firstly, multiple partially superimposed 3D docking grids are used to “slice” the protein’s binding site into equally sized portions; (b) multiple chemically diverse probes are then individually docked into each section of the binding site; (c) finally, all energetically favoured protein-probe complexes generated by the previous steps are subjected to MD simulations in explicit water. This way, multiple potential key anchoring sites for each fragment probe are identified through the docking step, and the corresponding fragment-protein complexes are then used as starting points for a more exhaustive MD-based hotspot sampling, which allows to simulate the dynamics of the system in its natural environment and to search for the most stable fragment-protein interactions. The potential pharmacophoric sites identified through the Watermelon protocol, which is named after the resemblance between seeds in the slices of the watermelon fruit and fragments bound to the different portions of the protein binding site, can then be used to develop a pharmacophore model for SBVS.
In this work, to validate the Watermelon protocol and demonstrate its utility in discovering new potential ligands of therapeutically interesting protein targets, we applied it to generate a pharmacophore model for the monoacylglycerol lipase (MAGL) enzyme. MAGL is primarily found in the endocannabinoid system where it regulates the levels of 2-arachidonoylglycerol (2-AG)Citation8,Citation9. By cleaving 2-AG, MAGL influences various physiological processes such as pain modulation, inflammation, and energy homeostasis, and has therefore gained attention as a potential therapeutic target for various conditions, including pain management, neuroinflammation, and metabolic disordersCitation10. The pharmacophore model obtained through the Watermelon procedure was then used to perform a virtual screening (VS) study to identify new potential MAGL inhibitors. By combining pharmacophore screening with consensus docking and MD simulations we discovered two new MAGL inhibitors with micromolar potency, thus confirming the soundness of the Watermelon protocol and identifying valuable starting points for hit-to-lead and future lead optimisation studies.
Material and methods
Fragment generation
Chemical probes were built by employing the last version of MolBook UNIPICitation11. The following fragments were considered and constructed: benzene and cyclopropane, for possible hydrophobic interactions; methylamine, formaldehyde and methanol for H-bond interactions; acetate and ammonium for possible H-bond and salt bridge interactions. For each fragment a low-energy conformer was generated.
Fragment docking studies
The crystal structure of the human MAGL (PDB code: 3PE6Citation12) was taken from the Protein Data BankCitation13. Docking calculations were performed by employing AUTODOCK 4.0 softwareCitation14. The initial docking site used for calculations was defined by a box of 68, 30, 40 points in the x, y, and z directions, which was manually centred at the centre of the MAGL catalytic site. Subsequently, the binding site was “sliced” by dividing the initial docking site into five different sections. For each section, a distinct docking grid of 22, 30, and 40 points in the x, y, and z directions was generated. In practice, we maintained two of the three dimensions of the initial docking site, and we reduced the dimension related to the longer axis of the protein binding site, adapting it to the size of the probes. This way, the five grids were defined so that their centres only differed for a single spatial coordinate, and were thus laying on the same axis, corresponding to the longer axis of the protein binding site. The number of grids was determined to ensure both the complete site coverage and the partial overlap of adjacent grids, to ensure that all biding site residues were sampled at least once by each probe. A grid spacing of 0.375 Å and a distance-dependent function of the dielectric constant were employed for the energetic map calculations in each of the 5 different grids. The fragments were subjected to 100 runs of Autodock search, using the Lamarckian Genetic Algorithm with 2 500 000 steps of energy evaluation, while all other settings were left as their defaults. The best pose (showing the best binding energy) of each fragment into each of the five docking grids was chosen for the next steps of the analysis. This step made it possible to effectively sample the active site, identifying the best anchor points for each fragment into each of the five different portions of the enzyme binding site, to seek potential pharmacophoric hotspots. Each generated fragment-protein complex was then subjected to MD simulations.
MD simulations of fragment-protein and ligand-protein complexes
All MD simulations were performed using AMBER, version 20Citation15,Citation16. General amber force field (GAFF) parametersCitation17 were assigned to the ligands, while partial charges were determined using the AM1-BCC methodCitation18, as implemented in the Antechamber suiteCitation19. The fragment–protein complexes were placed in a rectangular parallelepiped water-box by using the TIP3P explicit solvent model and were solvated by employing a minimum distance of 5 Å of water between the protein and the sides of the box. Sodium ions were added as counterions in order to neutralise the systems. Before the MD simulations, a stage of minimisation was carried out, minimising the entire system through 5000 steps of steepest descent followed by the conjugate gradient (CG) until convergence of 0.05 kcal/Å•mol, applying a position restraint of 10 kcal/mol•Å2 only on the protein α carbons. A constant-volume MD simulation was performed for the first 1 ns, during which the temperature of the systems was raised from 0 to 300 K. The systems were then equilibrated through 4 ns of constant pressure simulation, using the Langevin thermostat in order to maintain the temperature of the systems constant. Then, 96 ns of constant pressure periodic boundary MD was carried out at 300 K by using the Langevin thermostat. Hence, a total of 101 ns of MD simulation was carried out for each protein–probe complex analysed in this study. For each fragment, a total trajectory including the results of the five different simulations performed was created, and all trajectory frames (generated every 100 ps of simulation) were extracted and saved to be further analysed in order to create the pharmacophore model. The MD simulations of the ligand-protein complexes were performed using the same settings employed for fragment-protein simulations, with only a few differences in the applied protocol. In particular, a water-box of 20 Å was employed, and chloride and sodium ions were added as counterions to neutralise the systems. After energy minimisation, each ligand-protein complex was subjected to a 20 ns MD simulation. Initially, a simulation at constant volume raised the temperature from 0 to 300 K in 0.5 ns, followed by 3 ns of equilibrium at constant pressure, applying a position restraint of 10 kcal/mol•Å2 only on the protein α carbons. Subsequently, 16.5 ns of MD was conducted at constant pressure at 300 K, without position restraints applied on the α carbons. All the obtained MD trajectories were analysed using the pytraj and cpptraj programsCitation20 implemented in Amber 20.
Clustering and interactions analysis
For each MD trajectory obtained from each fragment, the root-mean-square deviation (RMSD) matrix of the frames was calculated by means the function pytraj.pairwise_rmsd of the pytraj library, a Python front-end of the popular cpptraj package, belonging to AMBER suite. In particular, the different MD frames obtained for each probe were clustered based on the spatial coordinates of the probe within the solvated probe-protein system. The average-linkage method of the clustering function of the scipy library was employed as a hierarchical clustering algorithm, with an RMSD cut-off of 2.0 Å, thus generating clusters of frames with reciprocal distances in terms of RMSD below 2.0 Å. Only clusters containing at least 10% of the total frames obtained for each probe was considered for the interaction analyses. For each frame of the selected clusters, the probe-protein interactions were calculated with Binana softwareCitation21, summarising the main interactions between the fragment and the MAGL binding site.
Pharmacophore model generation and screening
The pharmacophore model was created using Ligandscout 4.4Citation5. The pharmacophore hypothesis was built by importing the X-ray structure of MAGL in complex with the representative fragments, which were selected after analysing the most representative clusters and interactions obtained from the analysis of the corresponding MD trajectories. An exhaustive pharmacophore model including all possible features was constructed. Such pharmacophore included two H-bond acceptors, two H-bond donors, two aromatic and two hydrophobic features, for a total of eight different features. Moreover, the excluded volume spheres defined based on the receptor structure, as implemented in the default LigandScout configuration, were added to the model. Approximately 4 million compounds belonging from the Vitas-M, ChemBridge, Enamine, and Pharmeks commercial databases were used as the screening database. The software iConCitation22 implemented in LigandScout was used to execute ligand conformational sampling and to set up the screening database. The previously created pharmacophore model, including the excluded volume spheres, was used to filter the generated screening database and to search for compounds with the desired properties. The screening was performed setting the two hydrophobic and the two H-bond acceptor features of the model as mandatory, whereas the two aromatic and the two H-bond donor features were set as optional. Only compounds that matched at least the four mandatory features were retrieved during the screening.
Consensus docking evaluation
All docking calculations were carried out using the same X-ray structure of MAGL (PDB code: 3PE6) already employed for pharmacophore modelling. Thirteen different docking procedures were used in this study: AutodockCitation14, Autodock VINACitation23, Glide 5.0 with the standard precision (SP) and extra precision (XP) methodCitation24, GOLD 5.1 with ChemScore, GoldScore, ASP and ChemPLP fitness functionsCitation25, GLAMDOCKCitation26, PLANTSCitation27, DOCKCitation28, FREDCitation29, and rDOCKCitation30, according to the procedures described in our previous studiesCitation31. Prior to the consensus docking evaluation a self-docking analysis for each docking program was performed by calculating the RMSD between the binding pose predicted by docking and the known experimental disposition of the ligand co-crystallized with MAGL in the reference X-ray structure, using the rms_analysis software of Gold suite. Each compound selected through the pharmacophore screening step was docked into the MAGL binding site using the 13 different docking procedures. For each compound, 13 different binding poses were thus obtained (considering the top-scored pose for each docking method). The RMSD of each docking pose relative to the remaining docking dispositions was calculated through the rms_analysis software from the Gold suite, and a 13 × 13 matrix was generated, reporting the RMSD results. With the application of an in-house python script, results were clustered so that similar docking poses were grouped together. The complete-linkage method was employed as a hierarchical clustering algorithm, using an RMSD cut-off of 2.0 Å, thus generating clusters of poses with mutual distances in terms of RMSD values up to 2.0 Å. For each ligand, the consensus level was defined as the number of docking poses that were clustered within the RMSD cut-off of 2.0 Å and, consequently, as the number of docking methods that generated similar binding poses.
MAGL inhibition assay
The compounds identified by the VS were purchased from Aldrich Market Select and their inhibitory activity against MAGL was experimentally evaluated as previously reportedCitation32. Human recombinant MAGL and 4-nitrophenylacetate (4-NPA) were commercially available from Cayman Chemical. IC50 values were generated in 96-well microtiter plates. The MAGL reaction was performed at room temperature, at a final volume of 200 μL in 10 mM Tris buffer, pH 7.2, containing 1 mM EDTA and 0.1 mg/mL bovine serum albumin (BSA). A total of 150 μL of 4-NPA 133.3 μM was added to 10 μL of DMSO containing the suitable amount of compound. The reaction was started by adding 40 μL of MAGL (11 ng/well) so that the assay was linear over 30 min. The final concentration of the tested compounds ranged for from 320 to 0.02 nM. After 30 min from the start of the reaction, the absorbance values were measured by using Victor X3 Microplates Reader (PerkinElmer®) at 405 nm. Two reactions were also performed: (1) a reaction containing no compound and (2) a reaction containing neither compound nor MAGL. IC50 values were derived from experimental data using the Sigmoidal dose − response fitting of GraphPad Prism software. Final values were obtained from duplicates of three independent experiments. To remove possible false-positive results, a blank analysis was performed for each compound concentration, and the final absorbance results were obtained by subtracting the absorbance produced by the presence of all the components except MAGL under the same conditions. MAGL activity was measured by recording the increase in absorbance of 4-nitrophenol using Victor X3 Microplates Reader (PerkinElmer®). In the enzyme kinetics experiments, compound VS5 was tested in the presence of scalar concentrations of 4-NPA. It was added in scalar amounts (at a concentration of 20 μM and 10 μM) to a reaction mixture containing scalar concentrations of 4-NPA (15–1400 μM). Finally, the MAGL solution was added (11 ng/well). MAGL activity was measured by recording the increase in absorbance of 4-NPA. The experimental data were analysed by non-linear regression analysis with GraphPad Prism software, using second order polynomial regression analysis and by applying the mixed model inhibition fit.
DTT interference assay
The inhibition assay was the same described above with the exception that, prior to the addition of 40 µL of MAGL (11 ng/well), the compound-substrate mixture was incubated for 15 min in the presence of DTT at a 10 µM concentration.
MAGL preincubation assay
The MAGL reaction was conducted under the same conditions reported above. A total of 150 µL of MAGL (11 ng/well) was added to 10 µL of DMSO containing the appropriate amount of compound. After 0 min, 30 min, and 60 min of incubation time, the reaction was started by adding 40 µL of 4-NPA 500 µM. The enzyme activity was then measured after 30 min from the start of the reaction according to the procedure described above. Final values were obtained from triplicates of two independent experiments.
NRF2 assays
Compounds VS2 and VS5 were evaluated for their ability to modulate the antioxidant response pathway by monitoring the luciferase activity, strictly correlated with NRF2 activation as reported by Ferrario et al.Citation33 with minor changes. Experiments were performed using NRF2/ARE Responsive Luciferase Reporter HEK293 stable cell line (Signosis, Santa Clara, CA, USA) in Dulbecco’s modified Eagle medium (DMEM; Lonza, Verviers, Belgium) supplemented with 10% foetal bovine serum (FBS; Gibco, Gaithersburg, MD, USA), 1% Penicillin/Streptomycin (Lonza), and 50 μg/mL of G418 sulphate solution (Promega Corporation, Madison, WI, USA). Constitutive MAGL protein expression in HEK293 cells was assessed by immunocytochemistry studies by Zygmunt et al. in 2013Citation34. HEK293 cells were treated with different concentrations of compounds VS2 and VS5 for 18 h after seeding in a white 96-well plate (BRANDplates®, cell grade) at 12 000 cells/well. Subsequently, to avoid any interference on the reading of luciferase activity, media were removed and 50 μL/well of PBS was added. ONE-GloTM Luciferase Assay Substrate (purchased from Promega Corporation, Madison, WI, USA) (50 μL/well) was directly added to the wells, followed by a luciferase measurement performed using a luminometer (Wallac Victor2 1420, Perkin-ElmerTM Life Science, Monza, Italy). Experiments were performed with biological and technical replicates, with results shown as mean ± SD compared to untreated control cells. Statistical analysis was performed using one-way ANOVA with Bonferroni’s multiple comparisons test (p < 0.05 was considered significant). The cell viability was assessed with MTT assay on HEK293 cells treated with all the concentrations of the two tested compounds for antioxidant activity.
Cell viability assays
The cell viability for all the concentrations of the tested compounds was verified by MTT assay on HEK293 cells in transparent 96-well plates seeded with 12 000 cells/well, in Dulbecco’s modified Eagle medium (DMEM; Lonza, Verviers, Belgium) supplemented with 10% foetal bovine serum (FBS; Gibco, Gaithersburg, MD, USA) and 1% Penicillin/Streptomycin (Lonza). After 18 h incubation with the compounds at the appropriate concentrations, media were removed. Subsequently, 100 μL/well of DMEM was added, followed by 11 μL/well MTT (3–(4,5-Dimethyl-2- thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide, purchased from Merck KGaA, Darmstadt, Germany) reagent (5 mg/mL). After 4 h incubation, DMEM was removed, cells were treated with lysis buffer (100 μL/well) (HCl 8 mM, 5% TWEEN20, DMSO), and the 96-well plate was shaken for 15 min in a plate shaker in the dark. Absorbance at 575 nm and 630 nm was measured using a plate reader (BioTek’s PowerWave HT, Winooski, VT, USA). Cells incubated with media were used as a control of 100% proliferation, while cells incubated with DMSO (4%) were used as a negative control.
Results and discussion
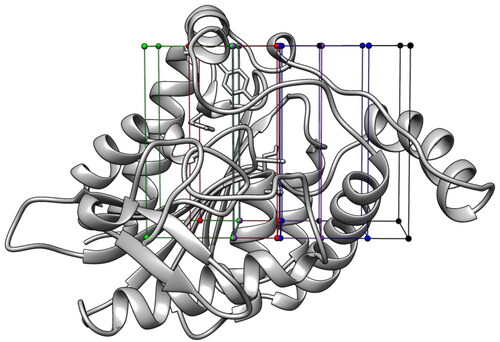
The strategy described here aims at the exploration of the binding cavity of a target protein by means of small chemical probes to identify interesting hotspots suitable for drug interactions, which can be exploited for the generation of purely receptor-based pharmacophore models and thus for the identification of novel potentially active ligands. The enzyme MAGL was used as a reference target for both developing and evaluating our in silico approach. Although various ligand-protein co-crystal structures of MAGL already exist, we focused on this target with the aim of using the experimentally observed ligand-MAGL interactions as a reference to evaluate the efficacy of our approach in identifying reliable ligand binding hotspots. As a first step in the development of the protocol we defined a library of fragments to be used as chemical probes able to mimic all potential ligand-protein interactions. In particular, we chose cyclopropane and benzene for identifying purely hydrophobic and hydrophobic/aromatic interactions, formaldehyde as an H-bond acceptor probe, methanol, and methylamine for their dual H-bond donor/acceptor properties, whereas acetate and ammonium were selected as probes accounting also for salt-bridge interactions. Once defined the probe fragments, we employed them for performing an initial docking-based sampling of the protein binding pocket using Autodock software. To ensure a thorough analysis of the most favourable anchoring points of each fragment, the catalytic site of MAGL was first defined by a single docking grid, and then “sliced” into five equally sized portions. Therefore, five equally sized grids (see ) were generated with the Autodock graphical interface (see Material and Methods for details).
Figure 1. Subdivision of MAGL catalytic site (PDB code: 3PE6) into 5 different grids.
The seven probes were then docked into each of the five portions of the protein binding site. For each fragment, a total of five docking calculations were thus performed, and five corresponding top-scored poses (one for each portion of the binding site) were selected. Considering the whole set of probes, 35 different fragment-protein complexes were generated, thus obtaining an initial overview of the potential ligand anchoring points present within the MAGL binding site. The following step of our in silico protocol was based on MD simulations. In particular, each fragment-protein complex generated by the docking-based analysis was used as a starting point for an MD simulation with the double purpose of both evaluating the stability of the fragment-protein interactions predicted by docking and, most importantly, allowing the probes to dynamically sample the protein binding site to identify additional binding hotspots. Each complex was analysed through 100 ns of MD simulation in explicit solvent (water) and, subsequently, the results obtained for the systems containing the same probe fragment were merged together, in order to obtain a comprehensive trajectory generated by each probe while sampling the protein binding site. This way, by gathering the results of the five different MD simulations performed for each fragment (for a total of 500 ns), we collected a total of 5000 snapshots of the dynamic binding site sampling produced by each probe, which were used for further analyses (see Materials and Methods for details). The different MD frames obtained for each probe were then clustered based on the spatial coordinates of the probe within the solvated probe-protein system, to identify the regions within the protein binding site where the probes were primarily placed during the MD and could thus form stable interactions with the binding site residues. The analysis was performed using an average-linkage hierarchical clustering algorithm, with an RMSD cut-off of 2.0 Å, thus generating clusters of frames in which a specific fragment presented mutual distances in terms of RMSD up to 2.0 Å. With the aim of focusing only on the most representative hotspots of each probe, which should correspond to the most stable fragment-protein interactions within the binding site, we considered and analysed only the most populated clusters, i.e. containing at least 10% of the total frames obtained for each probe. This threshold corresponds to 500 MD snapshots, accounting for a total of 50 ns of simulation. Considering the very small size of the fragments used as probes, and thus the very limited number and extent of interactions that each fragment may establish, we believed that an overall residence time of 50 ns for a specific probe in a single hotspot of the protein binding site could be enough to suggest the presence of considerably stable interactions between the probe and the binding site residues. Based on this criterion, two sufficiently populated clusters were identified for benzene, cyclopropane, and methylamine, whereas a single cluster was found for formaldehyde fragment. On the contrary, no sufficiently populated cluster could be identified for methanol, acetate, and ammonium probes, suggesting that no sufficiently strong interaction at a relevant hotspot within the protein binding site was established by these three probes (see ).
Figure 2. Population density of the clusters selected for the analysis.
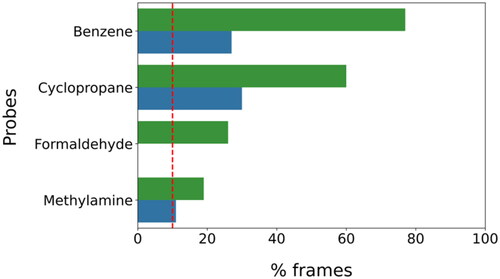
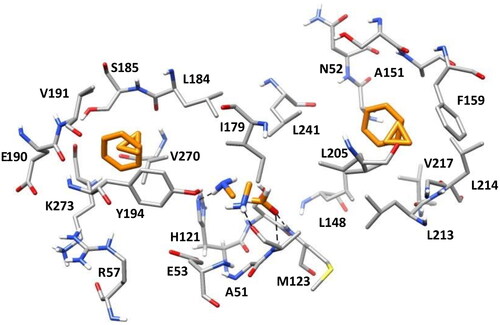
The selected clusters were inspected both from a qualitative and a quantitative point of view to identify the most relevant binding orientations of each probe and the corresponding protein-probe interactions, which were automatically detected with BINANA software. The most populated cluster identified for the benzene fragment (including 77% of the total MD frames) is concentrated in the hydrophobic pocket of the MAGL binding site defined by L148, A151, F159, L213, and L214 (see ). The other densely populated benzene cluster (23% of frames) was located in the lipophilic pocket delimited by L184, V191, Y194, V270, and K273, in which a relevant π–π stacking interaction with Y194 is observed in addition to multiple hydrophobic contacts with the surrounding residues (see ). Similar results were obtained analysing the two highly populated clusters identified for cyclopropane, accounting for about 60% and 30% of the total MD snapshots. In fact, the two clusters were placed in the same lipophilic pockets where benzene was localised. Moreover, the probe essentially showed the same hydrophobic interactions with the residues delimiting the two pockets (see ). Formaldehyde showed a single densely populated cluster, including 27% of the total MD simulation frames, in which the carbonyl oxygen of the fragment formed interactions with the backbone nitrogen of A51 and M123 (constituting MAGL oxyanion hole) as an H-bond acceptor (see ). Finally, methylamine formed two relevant clusters that were subjected to analysis, in which the probe interacted as an H-bond donor with two different protein residues. The most populated cluster contained 19% of the total MD frames and was characterised by a hydrogen bond between the probe and the backbone oxygen of A51, whereas the second one (including 10% of the MD snapshots), showed an H-bond with the side chain of H121 (see ).
Figure 3. The most populated clusters identified for each probe fragment studied are shown in orange: two for benzene, cyclopropane, and methylamine and one for formaldehyde. The protein residues surrounding the ligands are shown in gray. Probes-protein hydrogen bonds are highlighted with black dashed lines.
After analysing the clusters and the most representative interactions established by the corresponding fragments, the centroid frame of each selected cluster was chosen to build a pharmacophore model. A multifragment-protein complex was thus generated by combining the reference X-ray structure of MAGL with the structures of the probes in the coordinates assumed in the centroid frame of each selected cluster, and a receptor-based pharmacophore model was then built by using the Ligandscout program. Based on the disposition and interactions with the binding site residues shown by the different probe fragments, a total of eight pharmacophore features were included in the pharmacophore model (see ).
Figure 4. Receptor-based pharmacophore model derived from the most relevant fragment-probe interactions observed.
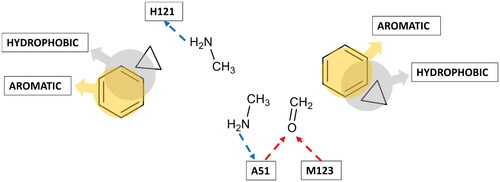
The accuracy of the pharmacophore features identified using the watermelon approach and their consistency with known protein-ligand interactions observed in the available MAGL-ligand X-ray structures was assessed through a retrospective evaluation. The formation of H-bonds with the backbone amide groups of A51 and M123, within the oxyanion hole of the MAGL catalytic site, appears to be crucial for the inhibitory activity of small molecules. Indeed, as can be observed in the available MAGL-inhibitor co-crystal structures, A51 and M123 constitute key anchoring sites shared by almost all non-covalent ligands. In fact, the inhibitor ZYH, co-crystallized with MAGL in the X-ray structure employed as a reference in this work (PDB code 3PE6), establishes H-bond interactions with the backbone nitrogen of these two residues through its central carbonyl group. Our fragment-based approach properly identified these two pharmacophoric interactions through the formaldehyde probe, recognising A51 and M123 as key anchor points for H-bond acceptors within MAGL active site. Another hotspot for H-bonds identified by our approach involved the side chain of residue H121. Although no interaction with this residue can be observed within our reference X-ray structure (PDB code 3PE6), H121 acts as a ligand anchoring point in the four crystallographic complexes of MAGL bound to a 3-Oxo-3,4-dihydro-2H-benzo[b][1,4]oxazin-6-yl inhibitor (PDB code 74LT) and its isosteric derivatives (PDB codes 7L4U, 7L4W and 7L50)Citation35. These crystallographic inhibitors, besides forming the two H-bonds with A51 and M123, through their central carbonyl group, also show an H-bond with the side chain of H121 via the NH group of their lactam and cyclic carbamate moieties. The watermelon approach correctly identified H121 as a hotspot for ligand H-bond donor moieties based on the interactions analysed for the methylamine probe, thus demonstrating its ability to spot key pharmacohporic interactions within MAGL binding site that could not be observed in the reference co-crystal structure used for the study. Finally, our approach identified a further pharmacophore feature corresponding to the H-bond interaction of a ligand donor moiety (again represented by the methylamine probe) with the carbonyl oxygen of A51. Interestingly, such direct interaction has never been experimentally observed. Nevertheless, in three crystallographic structures (PDB code 3PE6, 5ZUN, and 7PRM), it is possible to observe a water bridged interaction between the charged piperazine nitrogen of the inhibitors and the backbone carbonyl of A51, mediated by a structural water molecule that also interacts with Y194. Moreover, the interaction with the carbonyl oxygen of A51 was predicted by our previous molecular modelling studies for the sulphonamide moiety of a promising inhibitor identified during a virtual screeningCitation36.
The reliability of the pharmacophore features accounting for non-polar interactions was also confirmed by our retrospective evaluation. In three MAGL co-crystal structures (PDB codes 3PE6Citation12, 7PRMCitation37, and 5ZUNCitation38) it is possible to observe a face-to-face π-π stacking between an aromatic moiety of the bound inhibitor (the pyrimidine ring of ZYH, the furan ring of 81I and the thiazole ring of 3I, respectively) and the side chain of Y194. Moreover, the same aromatic groups of these inhibitors are inserted into a lipophilic pocket mainly delimited by L184, V191, and V270, thus forming hydrophobic interactions with these residues. These evidences confirm the reliability of both the aromatic and the hydrophobic features identified by our approach in this portion of the MAGL binding site. Finally, the inhibitors bound with MAGL in both the reference (PDB code 3PE6) and two recent (PDB codes 7PRM and 7L50) X-ray structures respectively present an alicyclic and aromatic group placed into a second lipophilic pocket represented by the residues L148, A156, F159, L213 and L214, with which they form hydrophobic interactions, in agreement with the hydrophobic feature identified by our approach, with the cyclopropyl probe, in the same binding site pocket. Based on this retrospective analysis, the watermelon approach appears to be able to identify reasonable pharmacophoric hotspots for ligand interactions, thus representing a profitable strategy for receptor-based pharmacophore modelling in case of targets for which scarce experimental information about ligand-protein interactions is available. The pharmacophore model generated through the watermelon approach was thus refined to be used for a VS study aimed at identifying novel MAGL inhibitors. In particular, the two H-bond acceptor features corresponding to the key interactions with A51 and M123 were considered as mandatory, as well as the two hydrophobic features representing the lipophilic interactions with residues L148, A156, F159, L213, and L214, on one side of the protein binding cavity, and residues L184, V191, and V270 on the other one. The remaining four features of the pharmacophore were set as optional, which included the two aromatic features representing the π-π stacking interactions with Y194 and F159, as well as the two H-bond donor features representing the interactions with the side chain of H121 and with the backbone oxygen of A51. The developed pharmacophore model was screened against a combination of four commercial databases (i.e. Enamine, ChemBridge, Pharmeks, and Vitas-M) that comprise around 4 million compounds. Through the application of the pharmacophore filter, a set of 67 528 compounds endowed with structural moieties satisfying at least the 4 mandatory features of the model were initially selected. Since many of the identified compounds were also found to match additional features that were set as optional during the pharmacophore screening (), the initial pool of pharmacophore hits was further filtered. In particular, only compounds satisfying at least a total of 6 different pharmacophore features were retained and subjected to a consensus docking evaluation, which represented the following stage of our receptor-based VS protocol.
Table 1. Pharmacophore screening results.
Prior to the consensus docking study, the reliability of 13 different docking procedures was tested through self-docking, using the reference MAGL-inhibitor complex (PDB code 3PE6) as a reference. In particular, the bound inhibitor ZYH was subjected to docking studies within the MAGL binding site using all procedures and the RMSD between each predicted binding mode and the experimental disposition of the ligand was calculated. As shown in Supplementary Table S1, all 13 docking methods employed showed RMSD values less than 2.0 Å and were thus selected for the consensus docking step.
The 1080 compounds retrieved from the pharmacophore screening were docked in the X-ray structure of MAGL using the thirteen selected docking approaches, thus obtaining thirteen different binding poses for each compound. A consensus docking analysis was then performed as reported in the Materials and Methods section and the docked ligands were then classified according to the resulting consensus level (i.e. the number of docking results sharing the same binding mode). The outcomes derived from this analysis, as presented in , indicated that only one ligand attained complete agreement across all docking methods. These findings were consistent with our previous evaluations of the consensus docking approach and prospective VS studies employing consensus dockingCitation39, in which we observed that only a very small percentage of the analysed compounds achieves a high consensus among numerous docking proceduresCitation40,Citation41, and often no compound obtains the maximum consensus level achievableCitation42–44. Based on the results obtained through the described consensus docking protocol, 31 molecules showing a consensus level of at least 10 were selected for further investigations.
Table 2. Consensus docking results.
The 31 selected molecules were subjected to MD simulations aimed at assessing the stability of their predicted binding modes and key interactions with the residues of the enzyme binding site. Specifically, the 31 ligand–protein complexes were studied through 20 ns of MD simulation, and the results were then analysed in terms of RMSD of ligand disposition during the simulation with respect to the starting coordinates, as well as stability of key H-bonds with A51 and M123 represented in the pharmacophore model. Based on this analysis, only 8 ligands out of the 31 analysed compounds, showing an average RMSD below 2.0 Å and maintaining at least the two fundamental H-bonds with A51 and M123 for more than 60% of the MD, were ultimately selected to be purchased and subjected to biological evaluations together with the reference MAGL inhibitor 6, which was used as a positive controlCitation45. The enzyme inhibition assays revealed appreciable MAGL inhibitory activity for two out of seven compounds, corresponding to a hit rate of about 29%. Compounds VS5 and VS2 compounds showed a promising inhibitory activity, with IC50 values of 14.6 and 31.4 µM, respectively ().
Table 3. In vitro inhibitory activity on MAGL (IC50, µM) of compounds VS1 − 7 and the reference compound 6.
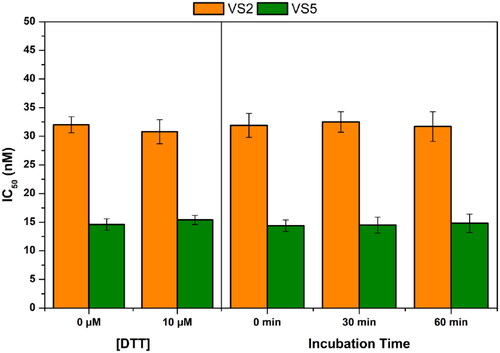
To exclude the possibility that the inhibitory effect of these compounds was due to a covalent interaction with cysteine residues of MAGL, we evaluated the activity of VS2 and VS5 in the presence of the thiol-containing agent 1,4-dithio-dl-threitol (DTT). Moreover, the inhibitory potency of the two ligands was also tested after different pre-incubation time with the protein, with the aim of confirming their reversible mechanism of action. As displayed in , the activity of compounds VS2 and VS5 was found to be altered by neither the presence of DDT nor any of the different pre-incubation times tested. These results confirmed the absence of any appreciable interaction between the two ligands and MAGL cysteine residues, as well as the reversibility of their binding mode. The mode of inhibition of compound VS5 was then evaluated by measuring the Michaelis–Menten kinetics at various inhibitor concentrations. The datasets were plotted as substrate concentration versus enzyme activity and analysed by applying the mixed model inhibition fit. Kinetic studies indicate for compound VS5 an α value greater than 10 000, thus supporting the competitive behaviour for this compound and a measured Ki value of 10.4 ± 0.4 µM (see Figure S1).
Figure 5. Analysis of the mechanism of MAGL inhibition of compounds VS2 and VS5. (A) Effect of DTT on MAGL inhibition activity. (B) IC50 (nM) values at different preincubation times with MAGL (0 min, 30 min and 60 min).
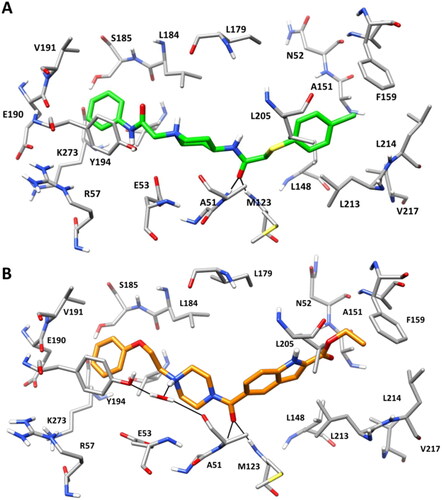
shows the binding modes refined through MD simulations predicted for compounds VS2 and VS5. Both compounds were predicted to form H-bond interactions with A51 and M123, which were maintained for 80% of the whole MD, through their carbonyl groups. A face-to-face π-π stacking is observed between the phenyl rings of the compounds and Y194, as observed for the reference ligand ZYH in the corresponding X-ray structure. Moreover, the chlorophenzyl group of VS2 and the ethyloxycarbonyl group of VS5 fit inside the hydrophobic pocket defined by A151, F159, L148, L213, L214 in agreement with the pharmacophore model (). In addition, a H-bond water-bridge interaction was observed between the piperazine nitrogen of VS5 and the backbone carbonyl of A51, promoted by a structural water molecule that also interacts with Y194 (), thus justifying the higher activity of VS5 with respect to VS2. Finally, VS5-MAGL MD simulation was subjected to a trajectory analysis including the evaluation of the RMSD and the root-mean-square fluctuation (RMSF) of the protein α carbons (Figure S2), as well as of secondary structure analyses (Table S2). The model showed an average RMSD during the whole MD around 1.0 Å, thus confirming the stability of the system during the simulation. The RMSF analysis confirmed this stability since, with the exception of the residues 145–175, corresponding to a part of the flexible lid domain of the protein, the highest fluctuation was found to be lower than 1.5 Å. Regarding the secondary structure analysis, more than 95% of residues maintained the initial secondary structure, thus further supporting the stability of the system during the MD simulation.
Figure 6. Minimised average structure of compound VS2 (A) and VS5 (B) within MAGL catalytic site. The protein residues surrounding the ligands are shown. Ligand-protein hydrogen bonds are highlighted with black dashed lines.
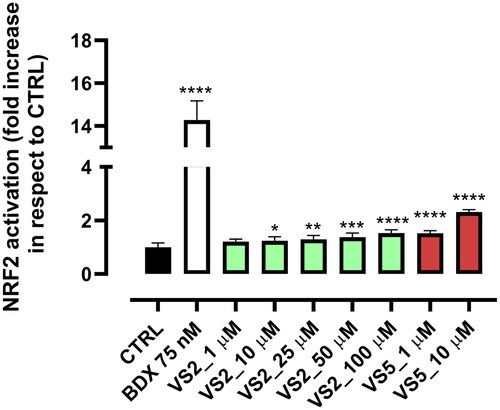
The two most promising MAGL inhibitors, compound VS2 and VS5, were further selected to determine their ability to modulate the Nrf2 pathway. Nrf2 is a transcription factor involved in the transcription of antioxidant and detoxifying genes. Under basal conditions, Nrf2 is bound to Keap1, a cytoskeleton-binding protein, and targeted for ubiquitination and proteasomal degradation by Cul3-E3-ligaseCitation46. However, this equilibrium can be altered by oxidant species, electrophilic oxidation byproducts or electrophilic xenobiotics which induce Nrf2 interacting, via Michael Addition, with the cysteine residues present in the thiol-rich domain of Keap1 by covalent binding, by oxidation or alkylation. This causes a conformational change which prevents Nrf2 from ubiquitination and leads to its translocation into the nucleus, binding to the antioxidant-responsive element (ARE) and thus activating a subset of cytoprotective genesCitation33,Citation47. In the case of MAGL inhibition, the activation of Nrf2 occurs indirectly. More specifically, MAGL-induced 2-AG accumulation is able to significantly enhance extracellular signal-regulated kinase 1/2 (ERK1/2) phosphorylation by activating the CB2 receptor. Under oxidative stress conditions, ERK1/2 phosphorylation upregulates Nrf2 expression by binding Nrf2 to ARECitation48. In this work, the antioxidant potential of compounds VS2 and VS5 was evaluated in HEK293 cell lines, in comparison with Bardoxolone (BDX), a synthetic Nrf2 activator, which was used as a positive control. Data reported in show a significant increase of Nrf2 activation compared to the control. The activity is significant from a concentration of 10 μM for compound VS2, and already at 1 μM for compound VS5, in agreement with its higher enzymatic MAGL inhibitory activity. Cell viability was tested through MTT assay and was found not toxic for both compounds for all the tested concentrations (data not shown).
Figure 7. Dose-dependent effect of compounds VS2 and VS5 on NRF2 nuclear translocation. Statistical analysis was calculated by one-way ANOVA with Bonferroni’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Conclusions
In conclusion, we developed and applied a computational approach able to create pharmacophore models based on receptor structures only, without the need of any bound ligand. The proposed in silico approach, named watermelon, consists in sampling the potential hotsposts for ligand interactions within the binding site of a protein target employing small molecular fragments as probes. Docking and MD simulations are used to identify the most relevant interactions formed by the chemical probes within different portions of the binding site. Such interactions are then converted into pharmacophore features, thus generating a purely receptor-based pharmacophore model representing the main anchoring sites for potential ligands of the target protein. In this work, the X-ray structure of MAGL protein was used as a reference to validate the reliability of the protocol. The generated pharmacophore model, which showed to include features representing ligand-MAGL interactions experimentally observed in different X-ray co-crystal structures, was employed to screen a database of commercially available compounds and the results were further filtered by means of docking and MD simulations. The VS approach allowed us to identify two new MAGL inhibitors with micromolar potency, which confirmed the reliability of our pharmacophore modelling and screening protocol. The antioxidant potential of the two compounds was also evaluated by testing their effects on the Nrf2 pathway and, without exerting cytotoxic effects on the tested cell line, they were able to activate Nrf2 at a micromolar concentrations. Furthermore, the predicted binding dispositions of the two compounds in the active site of MAGL provided a valuable starting point for structure-based hit optimisation studies. Overall, these results validated the reliability of the watermelon protocol, which can be profitably applied to all proteins for which scarce information related to ligand-protein interactions is available.
Authors contributions
Conceptualisation, M.M., T.T., and G.P.; methodology, M.D.S. and S.G.; validation, M.D.S., S.G., F.G., and L.P.; formal analysis, M.D.S., S.G., C.G., F.G., M.M., and A.G.; resources, T.T. and M.M.; data curation, M.D.S., S.G., C.G. and A.G.; writing-original draft preparation, M.D.S., S.G., T.T., and G.P.; supervision, T.T.; funding acquisition, T.T., and M.M. All authors have read and agreed to the published version of the manuscript. All authors agree to be accountable for all aspects of the work.
Supplemental Material
Download PDF (769.2 KB)Disclosure statement
No potential competing interest was reported by all authors except TT. Tiziano Tuccinardi is a member of the Editorial Board of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Data availability statement
The data that support the findings of this study are available from the corresponding author, [TT], upon reasonable request.
Additional information
Funding
References
- Mattos C, Bellamacina CR, Peisach E, Pereira A, Vitkup D, Petsko GA, Ringe D. Multiple solvent crystal structures: probing binding sites, plasticity and hydration. J Mol Biol. 2006;357(5):1471–1482.
- Mortier J, Dhakal P, Volkamer A. Truly target-focused pharmacophore modeling: a novel tool for mapping intermolecular surfaces. Molecules. 2018;23(8):1959.
- Brenke R, Kozakov D, Chuang GY, Beglov D, Hall D, Landon MR, Mattos C, Vajda S. Fragment-based identification of druggable “hot spots” of proteins using Fourier domain correlation techniques. Bioinformatics. 2009;25(5):621–627.
- Heider J, Kilian J, Garifulina A, Hering S, Langer T, Seidel T. Apo2ph4: a versatile workflow for the generation of receptor-based pharmacophore models for virtual screening. J Chem Inf Model. 2023;63(1):101–110.
- Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45(1):160–169.
- Ung PMU, Ghanakota P, Graham SE, Lexa KW, Carlson HA. Identifying binding hot spots on protein surfaces by mixed-solvent molecular dynamics: HIV-1 protease as a test case. Biopolymers. 2016;105(1):21–34.
- Graham SE, Smith RD, Carlson HA. Predicting displaceable water sites using mixed-solvent molecular dynamics. J Chem Inf Model. 2018;58(2):305–314.
- Manera C, Tuccinardi T, Martinelli A. Indoles and related compounds as cannabinoid ligands. Mini Rev Med Chem. 2008;8(4):370–387.
- Deng H, Li W. Monoacylglycerol lipase inhibitors: modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm Sin B. 2020;10(4):582–602.
- Tuccinardi T, Granchi C, Rizzolio F, Caligiuri I, Battistello V, Toffoli G, Minutolo F, Macchia M, Martinelli A. Identification and characterization of a new reversible MAGL inhibitor. Bioorg Med Chem. 2014;22(13):3285–3291.
- Galati S, Di Stefano M, Macchia M, Poli G, Tuccinardi T. MolBook UNIPI─create, manage, analyze, and share your chemical data for free. J Chem Inf Model. 2023;63(13):3977–3982.
- Schalk-Hihi C, Schubert C, Alexander R, Bayoumy S, Clemente JC, Deckman I, DesJarlais RL, Dzordzorme KC, Flores CM, Grasberger B, et al. Crystal structure of a soluble form of human monoglyceride lipase in complex with an inhibitor at 1.35 Å resolution. Protein Sci. 2011;20(4):670–683.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791.
- Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688.
- Case DA, Aktulga HM, Belfon K, Cerutti DS, Cisneros GA, Cruzeiro VWD, Forouzesh N, Giese TJ, Götz AW, Gohlke H, et al. AmberTools. J Chem Inf Model. 2023;63(20):6183–6191.
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174.
- Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23(16):1623–1641.
- Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25(2):247–260.
- Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9(7):3084–3095.
- Durrant JD, McCammon JA. BINANA: a novel algorithm for ligand-binding characterization. J Mol Graph Model. 2011;29(6):888–893.
- Poli G, Seidel T, Langer T. Conformational sampling of small molecules with iCon: performance assessment in comparison with OMEGA. Front Chem. 2018;6:229.
- Tanchuk VY, Tanin VO, Vovk AI, Poda G. A new, improved hybrid scoring function for molecular docking and scoring based on AutoDock and AutoDock Vina. Chem Biol Drug Des. 2016;87(4):618–625.
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47(7):1750–1759.
- Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52(4):609–623.
- Tietze S, Apostolakis J. GlamDock: development and validation of a new docking tool on several thousand protein-ligand complexes. J Chem Inf Model. 2007;47(4):1657–1672.
- Korb O, Stützle T, Exner TE. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J Chem Inf Model. 2009;49(1):84–96.
- Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, Case DA, Kuntz ID, Rizzo RC. DOCK 6: Impact of new features and current docking performance. J Comput Chem. 2015;36(15):1132–1156.
- OEDOCKING 4.3.0.3. OpenEye. Santa Fe (NM): Cadence Molecular Sciences, Inc., [accessed 2024 Apr 8]. http://www.eyesopen.com.
- Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, Garmendia-Doval AB, Juhos S, Schmidtke P, Barril X, Hubbard RE, Morley SD. rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLOS Comput Biol. 2014;10(4):e1003571.
- Tuccinardi T, Poli G, Romboli V, Giordano A, Martinelli A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J Chem Inf Model. 2014;54(10):2980–2986.
- Granchi C, Lapillo M, Glasmacher S, Bononi G, Licari C, Poli G, El Boustani M, Caligiuri I, Rizzolio F, Gertsch J, et al. Optimization of a benzoylpiperidine class identifies a highly potent and selective reversible monoacylglycerol lipase (MAGL) inhibitor. J Med Chem. 2019;62(4):1932–1958.
- Ferrario G, Baron G, Gado F, Della Vedova L, Bombardelli E, Carini M, D’Amato A, Aldini G, Altomare A. Polyphenols from thinned young apples: HPLC-HRMS profile and evaluation of their anti-oxidant and anti-inflammatory activities by proteomic studies. Antioxidants. 2022;11(8):1577.
- Zygmunt PM, Ermund A, Movahed P, Andersson DA, Simonsen C, Jönsson BA, Blomgren A, Birnir B, Bevan S, Eschalier A, et al. Monoacylglycerols activate TRPV1–a link between phospholipase C and TRPV1. PLOS One. 2013;8(12):e81618.
- Ikeda S, Sugiyama H, Tokuhara H, Murakami M, Nakamura M, Oguro Y, Aida J, Morishita N, Sogabe S, Dougan DR, et al. Design and synthesis of novel spiro derivatives as potent and reversible monoacylglycerol lipase (MAGL) inhibitors: bioisosteric transformation from 3-Oxo-3,4-dihydro-2 H -benzo [b] [1,4]oxazin-6-yl moiety. J Med Chem. 2021;64(15):11014–11044.
- Jha V, Biagi M, Spinelli V, Di Stefano M, Macchia M, Minutolo F, Granchi C, Poli G, Tuccinardi T. Discovery of monoacylglycerol lipase (MAGL) inhibitors based on a pharmacophore-guided virtual screening study. Molecules. 2021;26(1):78.
- He Y, Schild M, Grether U, Benz J, Leibrock L, Heer D, Topp A, Collin L, Kuhn B, Wittwer M, et al. Development of high brain-penetrant and reversible monoacylglycerol lipase PET tracers for neuroimaging. J Med Chem. 2022;65(3):2191–2207.
- Aida J, Fushimi M, Kusumoto T, Sugiyama H, Arimura N, Ikeda S, Sasaki M, Sogabe S, Aoyama K, Koike T. Design, synthesis, and evaluation of piperazinyl pyrrolidin-2-ones as a novel series of reversible monoacylglycerol lipase inhibitors. J Med Chem. 2018;61(20):9205–9217.
- Poli G, Martinelli A, Tuccinardi T. Reliability analysis and optimization of the consensus docking approach for the development of virtual screening studies. J Enzyme Inhib Med Chem. 2016;31(suppl 2):167–173.
- Russo Spena C, De Stefano L, Poli G, Granchi C, El Boustani M, Ecca F, Grassi G, Grassi M, Canzonieri V, Giordano A, et al. Virtual screening identifies a PIN1 inhibitor with possible antiovarian cancer effects. J Cell Physiol. 2019;234(9):15708–15716.
- Chiarelli LR, Mori M, Barlocco D, Beretta G, Gelain A, Pini E, Porcino M, Mori G, Stelitano G, Costantino L, et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. Eur J Med Chem. 2018;155:754–763.
- Poli G, Giuntini N, Martinelli A, Tuccinardi T. Application of a FLAP-consensus docking mixed strategy for the identification of new fatty acid amide hydrolase inhibitors. J Chem Inf Model. 2015;55(3):667–675.
- Lapillo M, Salis B, Palazzolo S, Poli G, Granchi C, Minutolo F, Rotondo R, Caligiuri I, Canzonieri V, Tuccinardi T, et al. First-of-its-kind STARD 3 inhibitor: in silico identification and biological evaluation as anticancer agent. ACS Med Chem Lett. 2019;10(4):475–480.
- Galati S, Sainas S, Giorgis M, Boschi D, Lolli ML, Ortore G, Poli G, Tuccinardi T. Identification of human dihydroorotate dehydrogenase inhibitor by a pharmacophore-based virtual screening study. Molecules. 2022;27(12):3660.
- Granchi C, Rizzolio F, Palazzolo S, Carmignani S, Macchia M, Saccomanni G, Manera C, Martinelli A, Minutolo F, Tuccinardi T. Structural optimization of 4-chlorobenzoylpiperidine derivatives for the development of potent, reversible, and selective monoacylglycerol lipase (MAGL) inhibitors. J Med Chem. 2016;59(22):10299–10314.
- Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev. 2018;98(3):1169–1203.
- Della Vedova L, Ferrario G, Gado F, Altomare A, Carini M, Morazzoni P, Aldini G, Baron G. Liquid chromatography–high-resolution mass spectrometry (LC-HRMS) profiling of commercial enocianina and evaluation of their antioxidant and anti-inflammatory activity. Antioxidants. 2022;11(6):1187.
- Zhao Q, He Z, Chen N, Cho YY, Zhu F, Lu C, Ma WY, Bode AM, Dong Z. 2-Arachidonoylglycerol stimulates activator protein-1-dependent transcriptional activity and enhances epidermal growth factor-induced cell transformation in JB6 P + cells. J Biol Chem. 2005;280(29):26735–26742.