
Abstract
Tyrosinase, a pivotal enzyme in melanin synthesis, is a primary target for the development of depigmenting agents. In this work, in vitro and in silico techniques were employed to identify novel tyrosinase inhibitors from a set of 12 anilino-1,4-naphthoquinone derivatives. Results from the mushroom tyrosinase activity assay indicated that, among the 12 derivatives, three compounds (1, 5, and 10) demonstrated the most significant inhibitory activity against mushroom tyrosinase, surpassing the effectiveness of the kojic acid. Molecular docking revealed that all studied derivatives interacted with copper ions and amino acid residues at the enzyme active site. Molecular dynamics simulations provided insights into the stability of enzyme–inhibitor complexes, in which compounds 1, 5, and particularly 10 displayed greater stability, atomic contacts, and structural compactness than kojic acid. Drug likeness prediction further strengthens the potential of anilino-1,4-naphthoquinones as promising candidates for the development of novel tyrosinase inhibitors for the treatment of hyperpigmentation disorders.
Graphical Abstract
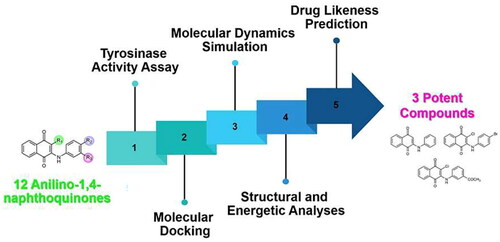
Introduction
The intricate process of melanin biosynthesis plays a predominant role in determining the colour of human skin, hair, and eyesCitation1. Tyrosinase () catalyses the rate-limiting step of melanin biosynthesis by converting tyrosine to DOPA and then to dopaquinoneCitation4, which serves as a precursor for the synthesis of eumelanin and pheomelaninCitation5 (). Although melanin serves essential photoprotective functions, its dysregulation often results in undesirable hyperpigmentation disordersCitation6, such as melasma and age spots. Consequently, the search for effective tyrosinase inhibitors has gained momentum, driven by both cosmetic and therapeutic interestsCitation3,Citation7. Currently, there are a few known tyrosinase inhibitors, but most inhibitors have side effects and lack clinical efficacyCitation8,Citation9. Moreover, safety and stability are significant concerns when using tyrosinase inhibitors, particularly over extended periods and at high dosesCitation10. For instance, hydroquinone can undergo catabolism leading to benzene metabolites, posing potential toxicity to bone marrow, carcinogenicityCitation3, skin irritation, and allergic reactionsCitation11. Arbutin has been reported to exhibit a counteractive effect on skin allergiesCitation12. The instability of kojic acid (KA, ), especially during storage, has been observedCitation3. Therefore, the search for novel potent tyrosinase inhibitors is needed.
Figure 1. (A) Crystal structure of tyrosinase (PDB ID: 2Y9XCitation2). The catalytic histidine residues and copper ions (CuA and CuB) were labelled in purple and orange, respectively. (B) Melanogenesis pathway produces eumelanin and pheomelaninCitation3. (C) Chemical structures of KA, lawsone, plumbagin, and 2-phenyl-1,4-naphthoquinones.
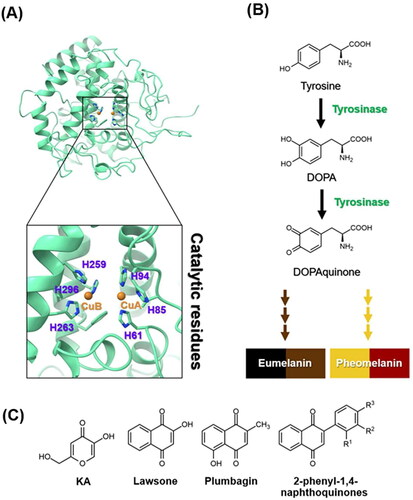
Many lines of evidence have shown that 1,4-naphthoquinone derivatives () can act as tyrosinase inhibitors. Lawsone can reduce melanin production by inhibiting tyrosinase activity and suppressing the expression of tyrosinase and microphthalmia-associated transcription factorCitation13. Plumbagin was found to inhibit α-MSH-induced melanin synthesis in B16F10 melanoma cells by suppressing tyrosinase activityCitation14. Additionally, 2-phenyl-1,4-naphthoquinones were reported to suppress both tyrosinase activity and melanin production in B16F10 murine melanoma cellsCitation15.
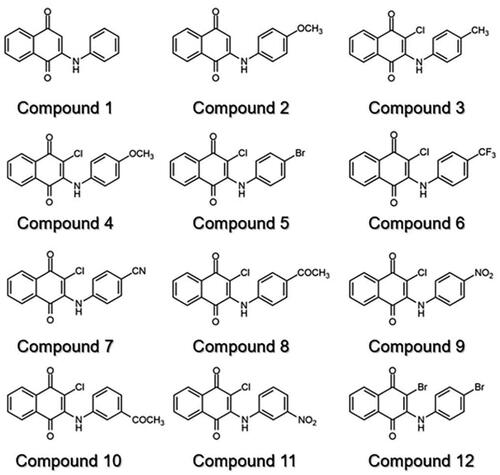
Previously, we found that anilino-1,4-naphthoquinone derivatives (), particularly compound 3, possess anti-cancer potential by inhibiting EGFR activity with a fourfold higher potency than erlotinibCitation16,Citation17. However, the anti-tyrosinase activity of these derivatives has not been reported yet. Herein, a series of 12 anilino-1,4-naphthoquinone derivatives were chosen to explore their inhibitory capabilities against tyrosinase through both in vitro and in silico studies. Mushroom tyrosinase activity assay was initially utilised to identify potent compounds from 12 derivatives. Subsequently, molecular docking, molecular dynamics (MD) simulations, and free energy calculation based on the molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) method were conducted to elucidate the binding mechanism of the screened anilino-1,4-naphthoquinones against mushroom tyrosinase. The results obtained from this study could provide valuable insights for further design and development of novel tyrosinase inhibitors based on anilino-1,4-naphthoquinone scaffolds.
Figure 2. Chemical structure of 12 anilino-1,4-naphthoquinone derivatives.
Materials and methods
Mushroom tyrosinase activity assay
Enzymatic conversion of l-DOPA into dopachrome by mushroom tyrosinase (Sigma Aldrich, St. Louis, MO) was detected using spectrophotometric evaluation. The mushroom tyrosinase activity assay was performed according to a previous reportCitation18. In this study, the enzyme assay was conducted at 37 °C, which is in the optimal temperature range of tyrosinaseCitation19, as per previous studiesCitation20,Citation21. The enzyme, substrate, and inhibitor(s) were dissolved in potassium phosphate buffer at a pH of 6.8, which is in the optimal pH range of tyrosinaseCitation22. Note that, the solvent for preparing the stock solution of inhibitors was dimethyl sulphoxide. The 40 μL of mushroom tyrosinase (5 U/μL) was added to each well of a 96-well plate, followed by adding 50 μL of a prepared solution of 12 compounds and KA (Sigma Aldrich, St. Louis, MO) at various concentrations. Then, the substrate (10 μL of 50 mM of l-DOPA) was introduced to the samples, followed by incubation at 37 °C for 15 min in darkness. The absorbance was later measured at 450 nm. The experiment was conducted in triplicate. IC50 values were determined through dose–response curve analysis using GraphPad Prism 10.2.2 software (La Jolla, CA).
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM) from three independent experiments (n = 3). Statistical comparisons between two groups were conducted using the t-test, with a p value of <0.05 considered to be statistically significant.
System preparation and molecular docking
The crystal structure of tyrosinase from Agaricus bisporus (PDB ID: 2Y9XCitation2) was retrieved from the Protein Data Bank. The protonation state of the protein was assessed at a pH of 7.4 using the PDB2PQR web serverCitation23. The covalent thioether bond between the carbon atom of H85 and the sulphur atom of C83 was constructed according to the previous reportCitation2. The 3D structures of 12 anilino-1,4-naphthoquinones and KA were constructed using the GaussView 6.1 programCitation24. The protonation state of all investigated ligands was assessed at a pH of 7.4 using the Marvinsketch program softwareCitation25. According to standard protocolsCitation16,Citation26–28, the electrostatic potential (ESP) charges of each ligand were calculated with the HF/6-31G(d) level of theory using the Gaussian 09 programCitation29. Molecular docking was conducted using the CB-DOCK2 serverCitation30. The 2D interaction profile was generated using the Discovery Studio visualiser. The docked complexes with the lowest docking energy were selected for MD simulations and free energy calculations.
Molecular dynamics (MD) simulation
The all-atom MD simulation with a time step of 2 fs of each docked complex was performed using the AMBER16 software packageCitation31. The parameters for both bonded and non-bonded interactions of all inhibitors were handled using the general Amber force field (GAFF)Citation32. The protein parameters were defined using the AMBER ff14SB force fieldCitation33. The missing hydrogen atoms were added using the LEaP module. Each system was solvated using TIP3P water moleculesCitation34. To maintain neutrality, Na+ counterions were included. In the isobaric–isothermal (NPT) ensemble, a boundary condition was set with a constant pressure of 1 atm and a temperature of 310 K. The SHAKE algorithmCitation35 was employed to constrain all bonds involving hydrogen. Non-bonded interactions were computed with a residue-based cut-off of 12 Å. The particle mesh Ewald methodCitation36 was utilised for handling long-range electrostatic interactions. To eliminate unfavourable interactions, we conducted 1000 iterations of the steepest descent (SD) method and 2000 iterations of conjugated gradient (CG) energy minimisation on the complex structure. Subsequently, the entire system was gradually heated to 310 K over a 100 ps. The systems underwent restrained MD simulations for a total of 5.0 ns, with progressively decreasing restraints of 50, 30, 20, 10, 5, and 1 kcal/mol·Å2. Afterward, the unrestrained MD simulations were conducted for 500 ps. Finally, MD simulations in the NPT ensemble (1 atm and 310 K) without any restraints were performed until reaching 10 ns.
Structural and energetic analyses
The CPPTRAJ moduleCitation37 of AMBER16 was used to compute structural parameters, including root mean square displacement (RMSD), the number of atom contacts (#Contacts), radius of gyration (Rg), and solvent accessible surface area (SASA) from the last 5 ns simulation. The obtained structural and energetic data were plotted using the OriginPro 2023 program. Additionally, hotspot residues involved in the binding process of ligand–protein complexes were determined using decomposition free energy calculation (ΔGbind, res) based on the MM/PBSA methodCitation38,Citation39 with an interior dielectric constant of 5.0.
Drug likeness prediction
Drug-likeness of all studied compounds was predicted using the SwissADME web tool (www.swissadme.ch)Citation40.
Results and discussion
Anti-tyrosinase activity of anilino-1,4-naphthoquinones
The initial screening of the 12 synthesised anilino-1,4-naphthoquinone derivatives was conducted against tyrosinase at a concentration of 160 μM to identify compounds with potent inhibitory activity. illustrates that compounds 1 (49.58 ± 1.39%), 5 (46.05 ± 2.02%), and 10 (37.87 ± 4.96%) demonstrated higher anti-tyrosinase activity compared to the remaining derivatives (>50%). Therefore, these three potent compounds were selected to determine their half-maximal inhibitory concentration (IC50) value. As shown in and , all screened compounds inhibited tyrosinase activity in a concentration-dependent manner, similar to KA. Compound 1, lacking functional groups on both the naphthoquinone and aniline moieties, inhibited tyrosinase activity with an IC50 value of 151.13 ± 4.39 μM. Introduction of bromine (–Br) on the aniline moiety and chlorine (–Cl) on the naphthoquinone ring (compound 5) led to enhanced anti-tyrosinase activity (IC50 = 138 ± 6.07 μM). Notably, compound 10, containing a –Cl substituent on the quinone ring and an acetyl group (–COCH3) on the aniline moiety, exhibited the highest tyrosinase inhibitory activity (IC50 = 111.95 ± 9.18 μM). The IC50 of KA was 168.90 ± 3.30 μM, which is similar to the previously reported IC50 values (235–250 μMCitation41,Citation42).
Figure 3. Mushroom tyrosinase inhibitory activity of 12 anilino-1,4-naphthoquinone derivatives at a concentration of 160 μM. Data are shown as the mean ± standard error of the mean (SEM) (n = 3). **p < 0.01, ***p < 0.001, and ****p < 0.0001 vs. control.
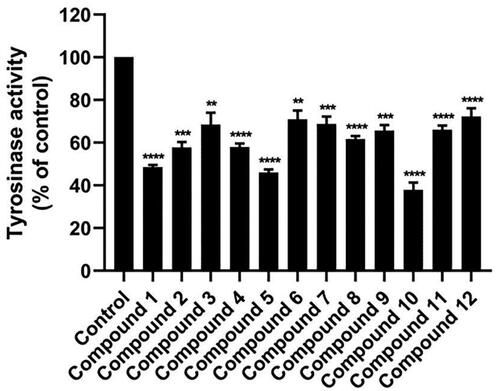
Figure 4. Tyrosinase inhibitory activity of (A) compound 1, (B) compound 5, (C) compound 10, and (D) KA using l-DOPA as a substrate. Data are shown as the mean ± SEM (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 vs. control.
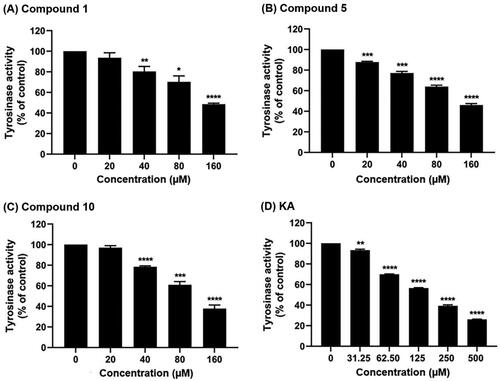
Table 1. IC50 values of compounds 1, 5, 10, and KA against tyrosinase.
It is worth noting that these three anilino-1,4-naphthoquinones showed greater inhibition than KA, as well as than the previously reported tyrosinase inhibitors, including morin (IC50 = 2320 μM)Citation43, amphotericin B (IC50 = 263.36 ± 11.76 μM)Citation44, glabrene (IC50 = 7600 μM)Citation45, and methimazole derivatives (IC50 = 4100 μM) Citation46, indicating the potential for further development of anilino-1,4-naphthoquinones as novel tyrosinase inhibitors.
The highest anti-tyrosinase inhibitory effect of compound 10 was in good agreement with high structural stability and the compactness of the ligand–protein complex, as discussed later. Altogether, the three potent compounds (1, 5, and 10) were subjected to further analyses, including (i) molecular docking, (ii) MD simulation, (iii) key binding residues analysis, and (iv) drug likeness prediction.
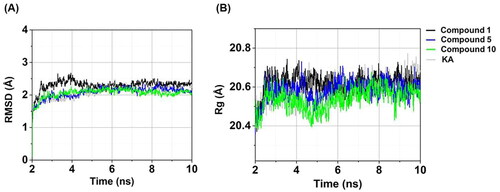
Figure 6. Time evolution of (A) RMSD and (B) Rg of compounds 1, 5, 10, and KA in complexes with tyrosinase.
Molecular docking
The binding mode between the screened compounds (1, 5, and 10) and KA in complexes with tyrosinase was studied using the CB-DOCK2 server. As shown in , the interaction energy of compounds 1 (−6.8 kcal/mol), 10 (−6.6 kcal/mol), and 5 (−6.1 kcal/mol) was lower than that of the KA–tyrosinase system (−5.3 kcal/mol).
Table 2. CB-DOCK2 interaction energy of compounds 1, 5, 10, and KA against tyrosinase.
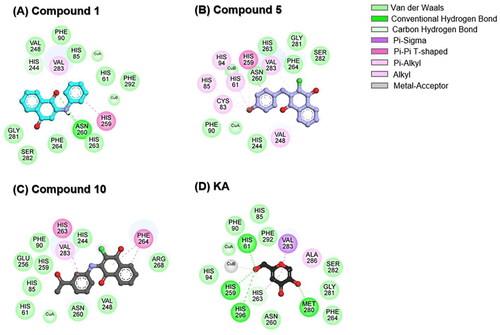
To delve into further detail, the two-dimensional (2D) interaction profile of each ligand within the active site of tyrosinase was analysed, and the results are depicted in . Results revealed that three anilino-1,4-naphthoquinone derivatives could interact with the catalytic copper centre of tyrosinase. The −NH and C═O groups of compound 1 can form hydrogen bonds with N260 (), in good agreement with the previous study showing that Thiamidol™ had the capability to form hydrogen bond with N260 residueCitation47. As compared to compound 1, the substituted –Br atom of compound 5 can form halogen interactions with H61, C83, H85, H94, H259, and V283 residues (). Previous study by Paudel et al. indicated that an increase in the number of –Br moieties in the chemical structure correlates with the enhanced halogen interactions, contributing to their potent anti-tyrosinase activityCitation48. For compound 10, its –COCH3 substitution resulted in increased van der Waals (vdW) forces with catalytic residues H85 and H259 (). These structural findings support the increased tyrosinase inhibitory activity of compounds 5 and 10 when compared to compound 1 ().
Figure 5. 2D interaction profile of (A) compound 1, (B) compound 5, (C) compound 10, and (D) KA in complexes with tyrosinase.
The aromatic ring of three anilino-1,4-naphthoquinones engaged in Pi interactions with V248, H259, H263, F264, and V283 residues located in the active site of tyrosinase. The vdW interactions were found to mainly stabilise the binding between ligands and coppers/tyrosinase at the catalytic site, which was similar to previously reported luteolin and luteolin 5-O-β-d-glucopyranoside in complexes with tyrosinaseCitation49. In the case of KA–tyrosinase system, hydrogen bonds were found between the hydroxyl (−OH) groups of KA and the H61, H259, H296, and M280 residues (). The –OH group attached to the methylene (–CH2) group in KA was positioned close to the copper atoms, resembling the molecular docking simulation observed in previous reports between KA and tyrosinaseCitation49,Citation50. It should be noted that the H61, H85, F90, H259, N260, H263, F264, and V283 residues found in KA–tyrosinase system were matched with those found in anilino-1,4-naphthoquinone–tyrosinase complexes. The binding of three potent anilino-1,4-naphthoquinones to the active site of tyrosinase is similar to the binding of 2-phenyl-1,4-naphthoquinonesCitation15, epicatechinCitation51, catechinCitation52, carvacrolsCitation53, benzimidazothiazolone analogsCitation54, 2,3-dihydro-1,5-benzothiazepine derivativesCitation55, kuwanon G, mulberrofuran G, and albanol BCitation56 to the tyrosinase.
Although compounds 1, 5, and 10 lack the –OH functional group, their chemical structure contains other moieties (i.e. –Br, –COCH3, −NH, and C═O) that can form potential interactions with the tyrosinase as mentioned above. Supportively, previous reports have shown that several compounds lacking the –OH group (e.g. aryl pyrazoles, thiosemicarbazones, 5-benzylidene(thio)barbiturate-β-d-glycosides, and 3-/4-aminoacetophenones) exhibited higher tyrosinase inhibition than the well-known tyrosinase inhibitors, kojic acid and arbutinCitation57–62.
Taken together, compounds 1, 5, and 10 can bind to the catalytic histidine residues (H61, H85, H94, H259, H263, and H296) and amino acid residues located in the active site of the tyrosinase. Additionally, they can interact with Cu ions (CuA and CuB). This finding suggested that our studied compounds could function as competitive inhibitors.
Structural stability and compactness
The RMSD aids in the assessment of whether the simulated systems attain a state of stability. As shown in , the RMSD values of the three anilino-1,4-naphthoquinones–tyrosinase complexes exhibited a gradual increase until reaching a stabilised level of approximately 2.11–2.32 Å after 5 ns. Similarly, the RMSD value for the KA–tyrosinase complex ultimately achieved a state of equilibrium following 5 ns, registering at 2.23 ± 0.09 Å. These outcomes implied that all studied systems achieved a state of equilibrium at 5 ns and maintained this stability until the conclusion of the MD simulations. Notably, the compound 10–tyrosinase complex displayed the highest stability within the last 5 ns timeframe (RMSD of ∼2.11 ± 0.07 Å) compared to the other systems (RMSD of ∼2.17–2.32 Å).
We further computed the Rg values for all investigated systems to evaluate the compactness of the complexCitation63. As illustrated in , the Rg values for all systems were stable within a range of 20.46–20.62 Å after the last 5 ns, similar to the RMSD results (). Remarkably, compound 10 in complex with tyrosinase exhibited the highest compactness (Rg of ∼20.46 ± 0.04 Å within the last 5 ns) compared to the other systems (Rg of ∼20.54–20.62 Å).
Taken together, these three anilino-1,4-naphthoquinone–tyrosinase complexes, particularly compound 10–tyrosinase complex, exhibited greater stability and compactness than the KA–tyrosinase system, which was in good agreement with the results of tyrosinase activity ().
Atomic contacts and water accessibility
Atomic contacts play a role in the protein–ligand binding affinityCitation64. In this work, the #Contacts within a 4 Å radius of ligand was calculated for all systems. As shown in , three anilino-1,4-naphthoquinones in complexes with tyrosinase exhibited the higher #Contacts (55.63 ± 8.67 to 67.76 ± 12.32, calculated from the last 5 ns) than the KA–tyrosinase complex (12.43 ± 1.63). Notably, compound 10–tyrosinase system demonstrated the highest #Contacts (67.76 ± 12.32). These findings are strongly supported by the structural stability () and the compactness in protein–ligand complex () results as mentioned above.
Figure 7. Time evolution of (A) #Contacts and (B) SASA of compounds 1, 5, 10, and KA in complexes with tyrosinase.
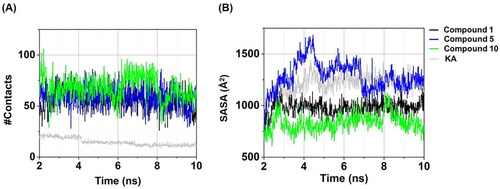
SASA yields valuable insights into the exposed surface area of a ligand–protein complex, with a particular focus on elucidating the interactions with surrounding water moleculesCitation64,Citation65. In this work, the amino acid residues of tyrosinase surrounding the ligand within a radius of 7.5 Å were taken into account, and the obtained results are presented in . Among three anilino-1,4-naphthoquinones in complexes with tyrosinase, compound 10–tyrosinase complex presented the lowest SASA (858.30 ± 79.40 Å2, calculated from the last 5 ns) compared to compound 1 (981.35 ± 58.51 Å2), compound 5 (1269.22 ± 98.84 Å2), and KA (1242.78 ± 61.11 Å2). These results were consistent with the aforementioned RMSD, Rg, #Contacts, and particularly with the experimental data on tyrosinase inhibitory activity.
Key binding residues
To explore the pivotal amino acid residues involved in the binding of three anilino-1,4-naphthoquinones to tyrosinase, the ΔGbind, res calculation based on the MM/PBSA methodCitation38,Citation39 was performed. This analysis was carried out using a selection of 100 snapshots derived from the last 5 ns of MD simulations, and the obtained results are shown in . The ΔGbind, res values of ≤ −1.0 kcal/mol were highlighted in this study.
Figure 8. (Left) ΔGbind, res of compounds 1, 5, 10, and KA in complexes with tyrosinase. (Right) Representative structures showing the ligand orientation in the catalytic site drawn from the last 5 ns MD snapshots. The copper ions were hidden. The residues involved in the ligand binding (energy stabilisation of ≤ −1.0 kcal/mol) were coloured according to their ΔGbind, res values, where the highest to lowest ΔGbind, res values were shaded from yellow to red, respectively.
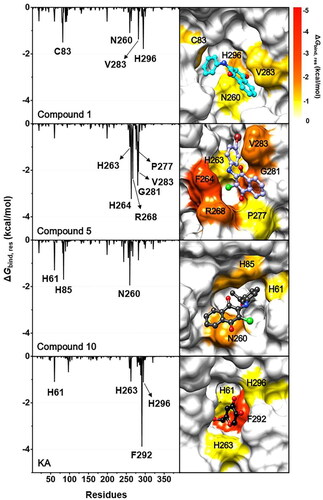
Results suggested that amino acid residues that made substantial contributions to KA’s binding were H61, H263, F292, and H296, which align well with the binding patterns identified in a previous study on the interaction between KA and tyrosinaseCitation50. The compound 5–tyrosinase complex exhibited the highest number of stabilising residues, including H263, F264, R268, P277, G281, and V283. The key binding residues for compound 1 and compound 10 were (i) C83, N260, V283, and H296 and (ii) H61, H85, and N260, respectively. To note that the H61, H263, and H296 residues found in KA–tyrosinase system were matched with those found in anilino-1,4-naphthoquinone–tyrosinase complexes. These aforementioned hotspot residues were also detected in the binding of 2-phenyl-1,4-naphthoquinonesCitation15, carvacrol derivativesCitation53, benzimidazothiazolone derivativesCitation54, 2,3-dihydro-1,5-benzothiazepine derivativesCitation55, kuwanon G, mulberrofuran G, and albanol BCitation56 to the active site of the tyrosinase. It should be noted that, all studied anilino-1,4-naphthoquinones can interact with the catalytic histidine residues in the enzyme active site, especially compound 10, which could bind to the two catalytic H61 and H85 residues.
Drug likeness prediction
Predicting the drug-likeness of compounds is a crucial element in drug developmentCitation66. In this study, we employed the SwissADME web toolCitation40 to predict the drug-likeness of three selected anilino-1,4-naphthoquinones together with KA. The parameters under investigation included molecular weight (MW), the number of hydrogen bond donors and acceptors (HBD and HBA), the number of rotatable bonds (RB), topological polar surface area (TPSA), and lipophilicity (log P). The results revealed that three compounds conformed to Lipinski’s rule of five criteria: (i) MW ≤500 Da, (ii) HBD ≤5 and HBA ≤10, (iii) RB ≤10, (iv) TPSA ≤140 Å2, and (v) log P ≤5Citation67,Citation68 as shown in . These findings suggested that our three compounds possess drug-likeness properties, laying the foundation for their potential advancement as novel drugs targeting the tyrosinase enzyme.
Table 3. Predicted values of drug-likeness parameters according to Lipinski’s rule of five criteria for compounds 1, 5, 10, and KA.
Conclusions
This study employed in vitro and in silico approaches to evaluate the inhibitory potential of 12 synthesised anilino-1,4-naphthoquinone derivatives against mushroom tyrosinase. Compounds 1, 5, and 10 exhibited superior anti-tyrosinase activity, surpassing the inhibitory activity of KA. Notably, compound 10 exhibited the highest anti-tyrosinase inhibitory activity, which was supported by the structural stability, the compactness of the ligand–protein complex, atomic contacts, and the water accessibility results. H61, C83, H85, N260, H263, F264, R268, P277, G281, V283, F292, and H296 were found to be hotspot residues for the binding of compounds 1, 5, 10, and KA. All the compounds studied can interact with the catalytic histidine residues. The drug likeness prediction further strengthens the potential of compounds 1, 5, and especially 10 as promising candidates for the development of novel tyrosinase inhibitors. These findings pave the way for further development and potential application of anilino-1,4-naphthoquinones in dermatological and cosmetic products.
Author contributions
P.M.: conceptualisation. S.S., S.N., N.K., and P.M.: investigation, formal analysis, and methodology. S.S., S.N., P.M., and R.P.: data curation, validation, and visualisation. S.S. and P.M.: writing – original draft, writing – review and editing. P.M.: funding acquisition and project administration. P.M. and A.S.: supervision. P.M., A.S., R.P., and T.R.: resources. R.P.: chemical synthesis. T.R.: software. All authors have read and agreed to the published version of the manuscript.
Disclosure statement
The authors report no conflicts of interest.
Data availability statement
The datasets presented in the current study are available upon reasonable request.
Additional information
Funding
References
- Kanteev M, Goldfeder M, Fishman A. Structure–function correlations in tyrosinases. Protein Sci. 2015;24(9):1–11.
- Ismaya WT, Rozeboom HJ, Weijn A, Mes JJ, Fusetti F, Wichers HJ, Dijkstra BW. Crystal structure of Agaricus bisporus mushroom tyrosinase: identity of the tetramer subunits and interaction with tropolone. Biochemistry. 2011;50(24):5477–5486.
- Pillaiyar T, Manickam M, Namasivayam V. Skin whitening agents: medicinal chemistry perspective of tyrosinase inhibitors. J Enzyme Inhib Med Chem. 2017;32(1):403–425.
- Lu Y, Tonissen KF, Di Trapani G. Modulating skin colour: role of the thioredoxin and glutathione systems in regulating melanogenesis. Biosci Rep. 2021;41(5):BSR20210427.
- Sansinenea E, Ortiz A. Melanin: a photoprotection for Bacillus thuringiensis based biopesticides. Biotechnol Lett. 2015;37(3):483–490.
- Solano F. Photoprotection and skin pigmentation: melanin-related molecules and some other new agents obtained from natural sources. Molecules. 2020;25(7):1537.
- Zolghadri S, Bahrami A, Hassan Khan MT, Munoz-Munoz J, Garcia-Molina F, Garcia-Canovas F, Saboury AA. A comprehensive review on tyrosinase inhibitors. J Enzyme Inhib Med Chem. 2019;34(1):279–309.
- Kumari S, Tien Guan Thng S, Kumar Verma N, Gautam HK. Melanogenesis inhibitors. Acta Derm Venereol. 2018;98(10):924–931.
- Mann T, Gerwat W, Batzer J, Eggers K, Scherner C, Wenck H, Stäb F, Hearing VJ, Röhm K-H, Kolbe L. Inhibition of human tyrosinase requires molecular motifs distinctively different from mushroom tyrosinase. J Invest Dermatol. 2018;138(7):1601–1608.
- Zolghadri S, Beygi M, Mohammad TF, Alijanianzadeh M, Pillaiyar T, Garcia-Molina P, Garcia-Canovas F, Munoz-Munoz J, Saboury AA. Targeting tyrosinase in hyperpigmentation: current status, limitations and future promises. Biochem Pharmacol. 2023;212:115574.
- Hu ZM, Zhou Q, Lei TC, Ding SF, Xu SZ. Effects of hydroquinone and its glucoside derivatives on melanogenesis and antioxidation: biosafety as skin whitening agents. J Dermatol Sci. 2009;55(3):179–184.
- Numata T, Tobita R, Tsuboi R, Okubo Y. Contact dermatitis caused by arbutin contained in skin-whitening cosmetics. Contact Dermatitis. 2016;75(3):187–188.
- Lee SM, Chen YS, Lin CC, Chen KH. Hair dyes resorcinol and lawsone reduce production of melanin in melanoma cells by tyrosinase activity inhibition and decreasing tyrosinase and microphthalmia-associated transcription factor (MITF) expression. Int J Mol Sci. 2015;16(1):1495–1508.
- Oh TI, Yun JM, Park EJ, Kim YS, Lee YM, Lim JH. Plumbagin suppresses alpha-MSH-induced melanogenesis in B16F10 mouse melanoma cells by inhibiting tyrosinase activity. Int J Mol Sci. 2017;18(2):320.
- Ullah S, Akter J, Kim SJ, Yang J, Park Y, Chun P, Moon HR. The tyrosinase-inhibitory effects of 2-phenyl-1,4-naphthoquinone analogs: importance of the (E)-β-phenyl-α,β-unsaturated carbonyl scaffold of an endomethylene type. Med Chem Res. 2018;28(1):95–103.
- Mahalapbutr P, Leechaisit R, Thongnum A, Todsaporn D, Prachayasittikul V, Rungrotmongkol T, Prachayasittikul S, Ruchirawat S, Prachayasittikul V, Pingaew R. Discovery of anilino-1,4-naphthoquinones as potent EGFR tyrosine kinase inhibitors: synthesis, biological evaluation, and comprehensive molecular modeling. ACS Omega. 2022;7(21):17881–17893.
- Prachayasittikul V, Pingaew R, Worachartcheewan A, Nantasenamat C, Prachayasittikul S, Ruchirawat S, Prachayasittikul V. Synthesis, anticancer activity and QSAR study of 1,4-naphthoquinone derivatives. Eur J Med Chem. 2014;84:247–263.
- Teeravirote K, Sutthanut K, Thonsri U, Mahalapbutr P, Seubwai W, Luang S, Tippayawat P, Kanthawong S, Pipattanaboon C, Duangjinda M, et al. Anserine/carnosine-rich extract from Thai native chicken suppresses melanogenesis via activation of ERK signaling pathway. Molecules. 2022;27(21):7440.
- Zaidi KU, Ali AS, Ali SA. Purification and characterization of melanogenic enzyme tyrosinase from button mushroom. Enzyme Res. 2014;2014:120739.
- Wang Y, Hao MM, Sun Y, Wang LF, Wang H, Zhang YJ, Li HY, Zhuang PW, Yang Z. Synergistic promotion on tyrosinase inhibition by antioxidants. Molecules. 2018;23(1):106.
- Kim J-K, Park K-T, Lee H-S, Kim M, Lim Y-H. Evaluation of the inhibition of mushroom tyrosinase and cellular tyrosinase activities of oxyresveratrol: comparison with mulberroside A. J Enzyme Inhib Med Chem. 2012;27(4):495–503.
- Inamdar S, Joshi S, Bapat V, Jadhav J. Purification and characterization of RNA allied extracellular tyrosinase from Aspergillus species. Appl Biochem Biotechnol. 2013;172(3):1183–1193.
- Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K, et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018;27(1):112–128.
- Roy Dennington TAK, Millam JM. Gaussview. Shawnee Mission (KS): Semichem Inc.; 2016.
- Salvador GA, Oteiza PI. Iron overload triggers redox-sensitive signals in human IMR-32 neuroblastoma cells. NeuroToxicology. 2011;32(1):75–82.
- Mahalapbutr P, Nutho B, Wolschann P, Chavasiri W, Kungwan N, Rungrotmongkol T. Molecular insights into inclusion complexes of mansonone E and H enantiomers with various β-cyclodextrins. J Mol Graph Model. 2018;79:72–80.
- Sanachai K, Mahalapbutr P, Sanghiran Lee V, Rungrotmongkol T, Hannongbua S. In silico elucidation of potent inhibitors and rational drug design against SARS-CoV-2 papain-like protease. J Phys Chem B. 2021;125(50):13644–13656.
- Oo A, Kerdpol K, Mahalapbutr P, Rungrotmongkol T. Molecular encapsulation of emodin with various β-cyclodextrin derivatives: a computational study. J Mol Liq. 2022;347:118002.
- Frisch MJ, Trucks G, Schlegel HB, Scuseria GE, Robb MA, Cheeseman J, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. Gaussian 09 revision A.1. Wallingford: Gaussian Inc.; 2009.
- Liu Y, Yang X, Gan J, Chen S, Xiao Z-X, Cao Y. CB-Dock2: improved protein–ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022;50(W1):W159–W164.
- Salomon-Ferrer R, Case DA, Walker RC. An overview of the Amber biomolecular simulation package. WIREs Comput Mol Sci. 2012;3(2):198–210.
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general Amber force field. J Comput Chem. 2004;25(9):1157–1174.
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11(8):3696–3713.
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935.
- Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. Comput Phys. 1977;23(3):327–341.
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092.
- Roe DR, Cheatham TE.III. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9(7):3084–3095.
- Miller BR, III, McGee TD, Jr., Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput. 2012;8(9):3314–3321.
- Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449–461.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717.
- Bernard P, Berthon JY. Resveratrol: an original mechanism on tyrosinase inhibition. Int J Cosmet Sci. 2000;22(3):219–226.
- Li CY, Wu TS. Constituents of the stigmas of Crocus sativus and their tyrosinase inhibitory activity. J Nat Prod. 2002;65(10):1452–1456.
- Kubo I, Kinst-Hori I, Chaudhuri SK, Kubo Y, Sánchez Y, Ogura T. Flavonols from Heterotheca inuloides: tyrosinase inhibitory activity and structural criteria. Bioorg Med Chem. 2000;8(7):1749–1755.
- Mahalapbutr P, Sabuakham S, Nasoontorn S, Rungrotmongkol T, Silsirivanit A, Suriya U. Discovery of amphotericin B, an antifungal drug as tyrosinase inhibitor with potent anti-melanogenic activity. Int J Biol Macromol. 2023;246:125587.
- Nerya O, Vaya J, Musa R, Izrael S, Ben-Arie R, Tamir S. Glabrene and isoliquiritigenin as tyrosinase inhibitors from licorice roots. J Agric Food Chem. 2003;51(5):1201–1207.
- Chan C-F, Lai S-T, Guo Y-C, Chen M-J. Inhibitory effects of novel synthetic methimazole derivatives on mushroom tyrosinase and melanogenesis. Bioorg Med Chem. 2014;22(9):2809–2815.
- Mahalapbutr P, Nuramrum N, Rungrotmongkol T, Kongtaworn N, Sabuakham S. Structural dynamics and susceptibility of isobutylamido thiazolyl resorcinol (Thiamidol™) against human and mushroom tyrosinases. J Biomol Struct Dyn. 2023;41(21):11810–11817.
- Paudel P, Wagle A, Seong SH, Park HJ, Jung HA, Choi JS. A new tyrosinase inhibitor from the red alga Symphyocladia latiuscula (Harvey) Yamada (Rhodomelaceae). Mar Drugs. 2019;17(5):295.
- Wagle A, Seong SH, Joung E-J, Kim H-R, Jung HA, Choi JS. Discovery of a highly potent tyrosinase inhibitor, luteolin 5-O-β-d-glucopyranoside, isolated from Cirsium japonicum var. maackii (Maxim.) Matsum., Korean Thistle: kinetics and computational molecular docking simulation. ACS Omega. 2018;3(12):17236–17245.
- Choi H, Young Ryu I, Choi I, Ullah S, Jin Jung H, Park Y, Hwang Y, Jeong Y, Hong S, Chun P, et al. Identification of (Z)-2-benzylidene-dihydroimidazothiazolone derivatives as tyrosinase inhibitors: anti-melanogenic effects and in silico studies. Comput Struct Biotechnol J. 2022;20:899–912.
- Mechqoq H, Hourfane S, El Yaagoubi M, El Hamdaoui A, da Silva Almeida JR, Rocha JM, El Aouad N. Molecular docking, tyrosinase, collagenase, and elastase inhibition activities of argan by-products. Cosmetics. 2022;9(1):24.
- Tang H, Cui F, Li H, Huang Q, Li Y. Understanding the inhibitory mechanism of tea polyphenols against tyrosinase using fluorescence spectroscopy, cyclic voltammetry, oximetry, and molecular simulations. RSC Adv. 2018;8(15):8310–8318.
- Ashraf Z, Rafiq M, Nadeem H, Hassan M, Afzal S, Waseem M, Afzal K, Latip J. Carvacrol derivatives as mushroom tyrosinase inhibitors; synthesis, kinetics mechanism and molecular docking studies. PLOS One. 2017;12(5):e0178069.
- Jung HJ, Choi DC, Noh SG, Choi H, Choi I, Ryu IY, Chung HY, Moon HR. New benzimidazothiazolone derivatives as tyrosinase inhibitors with potential anti-melanogenesis and reactive oxygen species scavenging activities. Antioxidants. 2021;10(7):1078.
- Al-Rooqi MM, Sadiq A, Obaid RJ, Ashraf Z, Nazir Y, Jassas RS, Naeem N, Alsharif MA, Shah SWA, Moussa Z, et al. Evaluation of 2,3-dihydro-1,5-benzothiazepine derivatives as potential tyrosinase inhibitors: in vitro and in silico studies. ACS Omega. 2023;8(19):17195–17208.
- Koirala P, Seong SH, Zhou Y, Shrestha S, Jung HA, Choi JS. Structure–activity relationship of the tyrosinase inhibitors kuwanon G, mulberrofuran G, and albanol B from Morus species: a kinetics and molecular docking study. Molecules. 2018;23(6):1413.
- Channar PA, Saeed A, Larik FA, Batool B, Kalsoom S, Hasan MM, Erben MF, El-Seedi HR, Ali M, Ashraf Z. Synthesis of aryl pyrazole via Suzuki coupling reaction, in vitro mushroom tyrosinase enzyme inhibition assay and in silico comparative molecular docking analysis with kojic acid. Bioorg Chem. 2018;79:293–300.
- Liu J, Yi W, Wan Y, Ma L, Song H. 1-(1-Arylethylidene)thiosemicarbazide derivatives: a new class of tyrosinase inhibitors. Bioorg Med Chem. 2008;16(3):1096–1102.
- Liu J, Li M, Yu Y, Cao S. Novel inhibitors of tyrosinase produced by the 4-substitution of TCT (capital PE, Cyrillic). Int J Biol Macromol. 2017;103:1096–1106.
- Dong H, Liu J, Liu X, Yu Y, Cao S. Molecular docking and QSAR analyses of aromatic heterocycle thiosemicarbazone analogues for finding novel tyrosinase inhibitors. Bioorg Chem. 2017;75:106–117.
- Yan Q, Cao R, Yi W, Yu L, Chen Z, Ma L, Song H. Synthesis and evaluation of 5-benzylidene(thio)barbiturate-beta-d-glycosides as mushroom tyrosinase inhibitors. Bioorg Med Chem Lett. 2009;19(15):4055–4058.
- You A, Zhou J, Song S, Zhu G, Song H, Yi W. Structure-based modification of 3-/4-aminoacetophenones giving a profound change of activity on tyrosinase: from potent activators to highly efficient inhibitors. Eur J Med Chem. 2015;93:255–262.
- Zhan F, Ding S, Xie W, Zhu X, Hu J, Gao J, Li B, Chen Y. Towards understanding the interaction of β-lactoglobulin with capsaicin: multi-spectroscopic, thermodynamic, molecular docking and molecular dynamics simulation approaches. Food Hydrocolloids. 2020;105:105767.
- Wang DD, Ou-Yang L, Xie H, Zhu M, Yan H. Predicting the impacts of mutations on protein–ligand binding affinity based on molecular dynamics simulations and machine learning methods. Comput Struct Biotechnol J. 2020;18:439–454.
- Cao Y, Li L. Improved protein–ligand binding affinity prediction by using a curvature-dependent surface-area model. Bioinformatics. 2014;30(12):1674–1680.
- Hu Q, Feng M, Lai L, Pei J. Prediction of drug-likeness using deep autoencoder neural networks. Front Genet. 2018;9:585.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26.
- Benet LZ, Hosey CM, Ursu O, Oprea TI. BDDCS, the rule of 5 and drugability. Adv Drug Deliv Rev. 2016;101:89–98.