
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Novel series of nitric oxide-releasing thiazolidine-2,4-diones (NO-TZD-3a-d,5,6) and 3,4,5-trimethoxychalcone-based multifunctional 1,4-dihydropyrimidines (CDHPM-10a-g) have been designed and synthesised as potent broad-spectrum anticancer agents with potential VEGFR-2 inhibition. The designed analogs were evaluated for their anticancer activities towards a full panel of NCI-60 tumour cell lines and CDHPM-10a-g emerged mean %inhibitions ranging from 76.40 to 147.69%. Among them, CDHPM-10e and CDHPM-10f demonstrated the highest MGI% of 147.69 and 140.24%, respectively. Compounds CDHPM-10a,b,d-f showed higher mean %inhibitory activity than the reference drug sorafenib (MGI% = 105.46%). Superiorly, the hybrid CDHPM-10e displayed the highest potencies towards all the herein tested subpanels of nine types of cancer with MGI50 of 1.83 µM. Also, it revealed potent cytostatic single-digit micromolar activity towards the herein examined cancer cell lines. The designed compounds CDHPM-10a-g were exposed as potent non-selective broad-spectrum anticancer agents over all NCI subpanels with an SI range of 0.66–1.97. In addition, the target analog CDHPM-10e revealed potency towards VEGFR-2 kinase comparable to that of sorafenib with a sub-micromolar IC50 value of 0.11 µM. Also, CDHPM-10e could effectively induce Sub-G1-phase arrest and prompt apoptosis via caspase and p53-dependent mechanisms. Furthermore, CDHPM-10e revealed significant anti-metastatic activity as detected by wound healing assay. The modelling study implies that CDHPM-10e overlaid well with sorafenib and formed a strong H-bond in the DFG binding domain. The ADMET studies hinted out that CDHPM-10e met Pfizer’s drug-likeness criteria. The presented novel potent anticancer agent merits further devotion as a new lead product in developing more chalcone-based VEGFR-2 inhibitors.
GRAPHICAL ABSTRACT
Introduction
Cancer is an enormous burden for all worldwide. About 10 million people die annually, with more than $1.16 trillion costCitation1–3. Metastasis represents the major cause of death and is regulated mainly by angiogenesis, implying proliferation, metastasis, and endothelial differentiationCitation4. Tumour aggressiveness and angiogenesis are strongly associated with KDR (Flk-1/VEGFR-2) kinaseCitation5. KDR has arisen increasing attention due to overexpression in many cancer types such as renalCitation6,Citation7, liverCitation6,Citation7, non-small cell lung (NSCLC)Citation6,Citation8–10, cervicalCitation6,Citation8, breastCitation11,Citation12, colonCitation6,Citation12, and ovarianCitation13–15 cancers. In addition, vascular endothelial growth factor VEGF is overexpressed in solid tumours, initiating the activation of KDRCitation16 and hence triggering Raf/MEK/ERK downstream signalling cascade, leading ultimately too extreme acceleration of angiogenesis, tumour growth, and metastasisCitation7,Citation16–18. Sorafenib (SF) is a well-known potent KDR, c-Kit, PDGFR-ß, and VEGFR-3 kinases inhibitor targeting downstream MAPK cascade with potent activation of downstream apoptotic markersCitation19–21. Interestingly, sorafenib and the next-generation anti-angiogenic drugs sunitinib, cabozantinib, regorafenib, nintedanib, and Lenvatinib have attracted increasing attention due to their activity towards a vast array of cancer typesCitation22. Regrettably, the major leading causes of concern are resistance, dose-limiting adverse effects, and impact on patient’s health, such as thrombotic effects, cardiotoxicity, and arterial hypertensionCitation22,Citation23. Therefore, this study endeavours to develop a novel class of low-toxicity KDR TKIs and enrich the anti-angiogenic potential.
The straightforward pharmacophoric characteristics of sorafenib and the next-generation KDR inhibitors are summarised as follows. Firstly, the hydrophobic tail which occupies the allosteric binding site. Secondly, the H-bond acceptor and donor group interact with DFG domain residues (Glu:885 and Asp:1046). Thirdly, the central het/aryl moiety occupies the spacer domain. Fourthly, the heterocyclic scaffold interacts with the ATB binding domain, as seen in . Therefore, developing a novel generation with these pharmacophoric characteristics has increased attention.
Figure 1. Basic pharmacophoric properties of KDR inhibitor sorafenib.
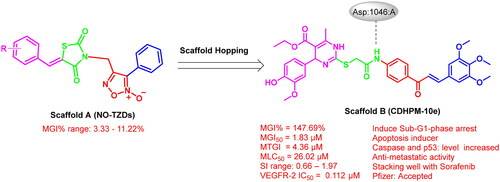
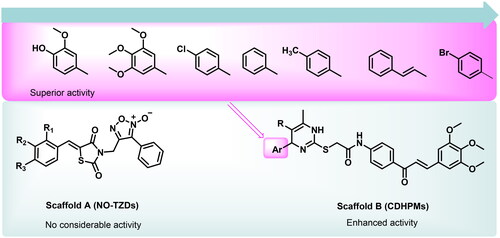
Based on the electrostatic potential similarities and physically realistic shapes utilising OpenEye’s EON moduleCitation24,Citation25, scaffolds “A” and “B” (), have been identified as promising motifs for designing a novel generation of angiogenesis inhibitors with enhanced bioavailability and improved biological activityCitation20. The pharmacophoric properties of scaffold “A” are characterised by furoxan moiety which occupies the linker region, the TZD group which inserts into the DFG motif, the aryl moiety which interacts with the hydrophobic region and the terminal phenyl group which occupies head hinge region. Interestingly, the chemical fragments, furoxan, and TZD, have been confounded in numerous hybrids as potential anticancer agentsCitation20,Citation26. Our group has recently conferred that furoxan-based pyrazolo[3,4-d]pyrimidines showed potential anti-VEGFR-2 activityCitation20. In addition, Taghour et al.Citation27 and Upadhyay et al.Citation28 have unveiled some TZDs hybrids as potential VEGFR-2 inhibitors. Therefore, scaffold “A” could serve as a promising target for angiogenesis inhibition.
Figure 2. Scaffold hopping of target compounds NO-TZDs and CDHPMs.
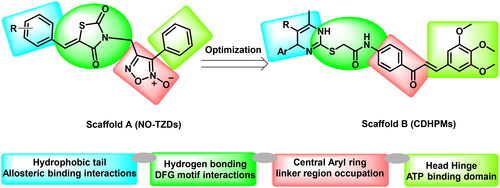
On the other hand, EON scaffold hopping offered scaffold “B” which is rationalised as follows. Firstly, the incorporation of 3,4,5-trimethoxyphenyledine base scaffold to fit tightly in the ATP-binding region. Secondly, the introduction of a phenyl ketone group to occupy the linker region. Thirdly, the open S-acetamide bridge allows hydrogen bonding formation with the DFG domain. Fourthly, endowing with 1,4-dihydropyrimidine scaffold as a hydrophobic tail to allow deep insertion into the allosteric active site. Finally, steric, and electronic effects, hydrophilicity, and hydrophobicity were also explored.
It is worth stressing that the isosteric 1,4-dihydropyrimidine scaffold has been reported in numerous anti-tumour agentsCitation29,Citation30. Particularly, monastrol is a well-known anticancer drug with a 1,4-dihydropyrimidine-2-thione coreCitation31–33. Also, Marzouk et al. described a potent KDR inhibitor (compound I) with a 1,6-dihydropyrimidine-2-thio scaffoldCitation33. Therefore, the privileged 1,4-dihydropyrimidine core has been deemed a promising motif for discovering a novel VEGFR-2 inhibitor generation.
Furthermore, the 3,4,5-trimethoxychalcone motif has been exposed to be beneficial for antitumor action in earlier SAR studiesCitation34–37. Remarkably, this flavonoid class plays a pivotal role in exhibiting promising biological effects towards tumour hallmarks such as proliferationCitation4, stemnessCitation4, inflammationCitation3,Citation4,Citation37 angiogenesisCitation4,Citation37, metastasisCitation4, invasionCitation4, regulation of tumour epigeneticsCitation35,Citation36, cell cycle arrest and apoptosisCitation37, inhibition of multidrug resistance proteinsCitation37,Citation38 as well as potent inhibitory activity towards VEGFR-2Citation39. Interestingly, besides methoxylated chalcones, other marine symmetric bromophenol natural products derived from macroalgae displayed potent anticancer activitiesCitation40. Of particular interest, a chalcone bromophenol analog BDDE with a three-atom bridging two phenyl groups has drawn considerable attention due to its potential antitumor activity. BDDE displayed broad-spectrum in vitro anticancer activities with potent induction of apoptosis through mitochondrial cascadeCitation40. BDDE has also been found to inhibit VEGF, VEGFR, and topoisomerase I, thus potentially repressing angiogenesisCitation41. Therefore, the 3,4,5-trimethoxychalcone scaffold spaced with enone moiety in the three-atom linker may significantly impact tumour growth and metastasisCitation33.
Enlightened by those above and in continuation to our effortsCitation20, we have been inspired to develop furoxan-based thiazolidine-2,4-diones and trimethoxychalcone-based dihydopyrimidines as a novel generation of VEGFR-2 TKIs based on isosterism and combination principles.
Results and discussion
Chemistry
Scheme 1 demonstrates the synthetic pathway of target compounds NO-TZD-3a-d and NO-TZD-5. Condensation of TZD-1 with different aldehydes in absolute ethanol containing 0.8 equivalent of piperazineCitation42–44 provided the key intermediates TZDs 2a-d and 4Citation43,Citation45,Citation46 which underwent N-alkylation with furoxan mesylate in DMF containing potassium carbonate at 65 °C to yield NO-TZD-3a-d and NO-TZD-5. On the other hand, O-allylation of NO-TZD-3c with allyl bromide in DMF containing potassium carbonate at 80 °C afforded the allyl derivative NO-TZD-6 (Scheme 2). Since the chemical shift of the exocyclic olefinic proton in the (Z)-isomer is relatively deshielded rather than the (E)-one, the Z-configuration of TZDs was verified by comparing with the incremented chemical shiftsCitation43,Citation45–47. Hence, the existence of singlet signal corresponding to the vinylic proton at 7.93–8.09 ppm demonstrates the Z-configuration of TZDs.
Scheme 1. Synthesis of target furoxan-based thiazolidine-2,4-diones NO-TZD-3a-d,5. (i) HCl 33%, reflux; (ii) Araldehyde, piperazine (0.8 eq.), EtOH, 1–2 h, reflux; (iii) Furoxan mesylate, K2CO3, DMF, 65 °C, 1 h; (iv) cinnamaldehyde, piperazine (0.8 eq.), EtOH, 2 h, reflux.
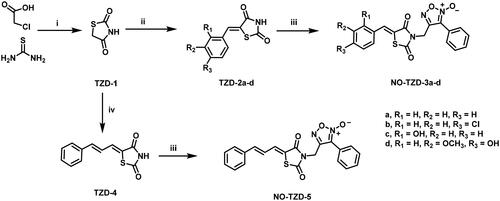
Scheme 2. Synthesis of allyl derivative NO-TZD-6. (i) allyl bromide, K2CO3, DMF, 80 °C, 1 h.
The 3,4,5-trimethoxychalcone-based 1,4-dihydropyrimidine-2-thio analogs CDHPM-10a-g were prepared as depicted in Scheme 3. The reaction route started with monastrol analog precursors (DHPM-7a-g) which were prepared according to Biginelli’s one-pot reaction of araldehyde, ethyl acetoacetate, and thiourea in 0.13 M ethanolic HClCitation48–53. On the other hand, the condensation of p-aminoacetophenone with 3,4,5-trimethoxybenzaldehyde upon stirring with potassium hydroxide in absolute ethanol afforded 3,4,5-trimethoxychalconamine C-1Citation54,Citation55, which underwent acetylation with chloroacetyl chloride to yield the chalcone chloride intermediate C-2Citation56–58. The target compounds 3,4,5-trimethoxychalcone-based 1,4-Dihydropyrimidine-2-thio analogs (CDHPM-10a-g) were then synthesised through S-alkylation of monastrol analogs and chalcone chloride C-2 in presence of K2CO3 and catalytic amount of sodium iodide. Regarding the precursor DHPM-7a (Figure S13, Supplementary Material), the R-configuration around C4 was assigned by 1H NMR signal at about 5.19 ppm corresponding to pyrimidine C4-H upon comparing to the R-isomers reported by Alvim et al.Citation59 at 5.10–514 ppm. In addition, the 1H NMR of methyl ester analog of DHPM-7a (Figure S14, Supplementary Material) characterised the C4-H at 5.19 ppm which augments the R-configuration assignments. On the other hand, Wang et al.Citation50 reported the S-isomer which characterised by down field shifting of C4-H at 5.41 ppm. Moreover, the E-configuration of CDHPMs was verified by the doublet signals of CHs with J-coupling ≈ 15.4 Hz corresponding to the exocyclic olefinic proton. Spectra are given in Figures S1–S29 (see Supplementary Material) and have been entirely consistent with the synthesised analogs.
Scheme 3. Synthetic route of target compounds CDHPM-10a-g. (i) EtOH/HCl, reflux, 6 h; (ii) chloroacetyl chloride, TEA, ACN, RT, 1 h; (iii) K2CO3, NaI, DMF, RT, 2 h.
Biological evaluation
NCI-USA anticancer activities
The herein designed conjugates NO-TZD-3a-d,5,6 (NSCs: 838 404, 844 385, 844 376, 838 406, 838 405, and 845 060, respectively) and CDHPM-10a-g (NSCs: 842 638, 842 636, 842 637, 842 635, 842 634, 842 639, and 842 640, respectively) have been screened at NCI-DTP (MD, USA) for their in vitro antitumor activities towards a full panel of nine types of cancers comprising leukaemia, CNS, colon, ovarian, breast, melanoma, prostate, renal and NSCLC cancers using SRB assay following their standard protocolCitation60–62. The herein designed analogs were examined firstly at 10 µM (single-dose screen) and then at 0.01–100 µM (five-dose screen) for those that demonstrated high potency at 10 µM concentration.
Preliminary NCI-USA one-dose screen
The accomplished results () unveiled that the target compounds NO-TZD-3a-d exhibited very low activity towards the full NCI cell panel with mean growth inhibition percentage (MGI%) ranging from 3.33 to 11.22%. Also, the modified hydrophobic tail targets NO-TZD-5 and NO-TZD-6 did not improve the overall activity displaying an MGI% of 5.49%. On the other hand, the herein examined 3,4,5-trimethoxychalcone-based multifunctional 1,4-dihydropyrimidines, CDHPM-10a-g, demonstrated high potency with significant MGI% spanning in the range: 76.40–147.69%. Superiorly, CDHPM-10e, and CDHPM-10f exhibited mean %inhibitions of 147.69 and 140.24%, respectively, whereas compounds CDHPM-10a, CDHPM-10b, and CDHPM-10d displayed MGI% of 113.88, 116.97, and 111.87%, respectively. Compared to the reference drug sorafenib (MGI% = 105.46%), compounds CDHPM-10a, CDHPM-10b, and CDHPM-10d-f were more potent. Moreover, the hybrids CDHPM-10c and CDHPM-10g exhibited significant MGI% of 76.40 and 91.51%, respectively, but lower than sorafenib. In addition, the maximum %inhibition achieved against the Leukaemia subpanel was 100%, while the other types of cancer were more. It is worth underlining that all the herein designed CDHPM-10a-g analogs demonstrated variable lethal effects (>100% growth inhibition) at a 10 µM screen. Compounds CDHPM-10a-g were lethal over 31, 33, 9, 29, 46, 44, and 15 cells, respectively. The influence of substitution at 1,4-dihydropyrimidine C4 was also studied. The results hinted out that the 4-hydroxy-3-methoxy derivative CDHPM-10e was more active than the corresponding 3,4,5-trimethoxy derivative CDHPM-10f as the m-methoxy group of the head phenyl may form an additional hydrogen bond inside the receptor active pocket leading to optimal binding interaction, and therefore higher anticancer action. Furthermore, the chloro derivative CDHPM-10b showed higher activities than the corresponding unsubstituted analog CDHPM-10a, which is more active than the tolyl analog CDHPM-10d. This may be ascribed to the influence of hydrophobic interactions within the receptor’s active site. Moreover, the styryl hybrid CDHPM-10g exerted a significant inhibitory activity than the bromo derivative CDHPM-10c since the styryl moiety may geometrically enable tight fitting in the receptor’s active site. Therefore, it can be inferred that the steric and electronic effect(s) of hydrophilic motifs at C4 of the privileged 1,4-dihydropyrimidine scaffold could impact the affinities towards the receptor’s active site. Interestingly, the 3,4,5-trimethoxyphenyledine head base scaffold could help to fit tightly in the head hinge ATP-binding domain, the phenyl ketone moiety forms hydrophobic interactions with the linker region, the open S-acetamide bridge allows hydrogen bonding formation with the DFG motif and finally endowing with multifunctional 1,4-dihydropyrimidine scaffold could deserve deep insertion into the allosteric active site of the herein target enzyme. The herein reported analogs, CDHPM-10a-g, displayed high mean %inhibitions with broad-spectrum anticancer activity over the NCI-tested panel (); thus, they were selected for five-dose testing.
Figure 3. Mean %inhibitions of the designed analogs NO-TZD-3a-d,5,6, CDHPM-10a-g and reference drug sorafenib (SF) at 10 µM over the herein examined NCI cancer cells.
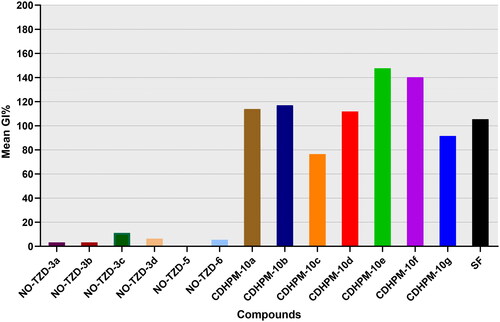
Table 1. Percentage inhibitions of hybrids CDHPM-10a-g and sorafenib (SF) towards the herein tested NCI tumour cells.
In vitro five-dose screening
The promising designed hybrids, CDHPM-10a-g, were further tested to determine their 50% cell growth inhibition (GI50), total cell growth inhibition (TGI), and median lethal concentration (LC50) against the herein examined nine cancer types using a five-dose assay according to NCI standard protocol; the data are presented in and (see Supplementary Material).
Table 2. GI50 (µM) of target analogs CDHPM-10a-g and sorafenib (SF).
GI50
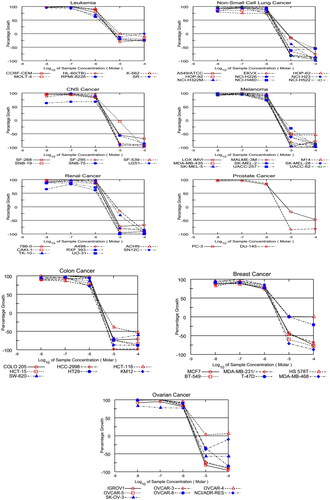
The GI50s of the herein examined hybrids in a five-dose screen against all nine subpanels are described in , using the reference drug sorafenib. The data demonstrated that Colon HCT-15 and Renal RXF 393 tumour cells were the most susceptible cell lines to all the examined conjugates with a 1-digit micromolar GI50 scale. In addition, the CNS SNB-75 tumour cell was the most sensitive cell line among the herein examined 60 cell lines with a sub-micromolar GI50 value of 0.83 µM. Among all the tested targets, CDHPM-10e with hydrophilic moiety showed the highest potencies over all the herein examined 60 tumour cells with single-digit micromolar scale. Compound CDHPM-10e was also found to be more potent than the reference drug sorafenib for the nine types of cancer. At the same time, the least potent hybrid was found to be the lipophilic-containing moiety CDHPM-10c. Notably, the more hydrophilic moiety endows with, the more potent antiproliferative activity will be. Interestingly, the hybrid drug candidate CDHPM-10e certainly has dose-dependent activity towards the herein examined cancer cell lines ().
Figure 4. Dose-response curves for compound CDHPM-10e.
TGI
In addition, most compounds showed variable cytostatic activity ranging from potent to no impact (Table S1, see Supplementary Material). Melanoma (MDA-MB-435 and LOX IMVI), Colon (SW-620, HT29, HCT-15, and HCT-116), Ovarian (OVCAR-4 and IGROV1), Renal (RXF 393) and Breast (MDA-MB-468) tumour cells were the most susceptible to the target analogs with potent cytostatic effect at a 1-digit micromolar scale. Notably, the hybrid CDHPM-10e demonstrated potent cytostatic action at a 1-digit micromolar range with TGI spanning in the interval: 2.43–9.81 µM towards 57 cells belonging to all herein tested tumour panels (Table S1). While compound CDHPM-10e, whose TGI > 100 µM had no cytostatic effect towards Ovarian OVCAR-4 cancer cell lines, exhibited good activity towards Breast HS578T cells with a TGI of 30.79 µM. In addition, compound CDHPM-10e emerged with two-fold more significant cytostatic activity (mean TGI = 4.36 µM) than sorafenib (NSC: 747 971Citation63; mean TGI = 9.11 µM). In contrast, compound CDHPM-10c had the least cytostatic activity towards the herein explored cell lines.
LC50
Furthermore, most target compounds emerged as non-lethal agents towards most of the herein examined tumour cells. All the target analogs were found to be non-lethal towards Leukaemia (RPMI-8226, MOLT-4, K-562, HL-60(TB), CCRF-CEM and SR), Ovarian (OVCAR-4 and NCI/ADR-RES), Breast (T-47D and HS 578 T) and Prostate (PC-3) tumour cells with LC50 > 100 µM. Among the target compounds, CDHPM-10e displayed LC50 values spanning in the interval: 2.04–70.60 µM (Table S2, see Supplementary Material). Also, the mean LC50 value of CDHPM-10e was 26.02 µM while sorafenib (NSC: 747 971Citation63) had 43.10 µM.
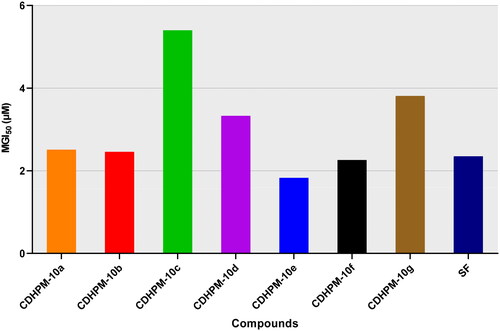
Moreover, all the herein tested compounds revealed high potency with a single-digit micromolar mean GI50 scale against all the nine cancer types (). All the MGI50 values were spanning in the range from 1.83 to 5.40 µM. Of particular interest, the hybrid CDHPM-10e with vanillin-like moiety displayed the highest potencies towards all the herein tested subpanels with an MGI50 of 1.83 µM. In comparison, the bromo derivative CDHPM-10c was the least active hybrid among the herein examined compounds with MGI50 of 5.40 µM. In addition, derivative CDHPM-10e was more potent than sorafenib, while compounds CDHPM-10a, CDHPM-10b, and CDHPM-10f were approximately equipotent.
Figure 5. Full panel MGI50 of target hybrids CDHPM-10a-g and SF.
Selectivity index (SI)
Again, in , the SI was calculated by division of MGI50 (full panel) by MGI50 (subpanel). Non-selectivity is denoted by a value <3, while moderate and high selectivity are denoted by a range from 3 to 6 and a value >6, respectivelyCitation64. The herein reported analogs CDHPM-10a-g have been exposed as potent non-selective broad-spectrum anticancer agents over all NCI subpanels with an SI range of 0.66–1.97, while sorafenib fell in the average of 0.99. Thus, the promising anti-proliferative profile of phenolic derivative CDHPM-10e over all the herein examined 60 tumour cells implied to explore its mechanism of action further.
Table 3. SI of target hybrids CDHPM-10a-g and the reference drug sorafenib towards nine cancer types.
Structure-activity relationships (SAR)
Initially, the NO-TZDs (scaffold A) did not show considerable activity, therefore, the rational design has been directed to scaffold hopping yielding trimethoxychalcone-based dihydropyrimidines (scaffold B). This optimisation allowed better interaction within the receptor’s active site. Since the investigated analogs exhibited variable anticancer activities towards tumour cells, the SAR of scaffold “B” could be summarised as follows. Firstly, substituting the terminal phenyl group with 4-hydroxy-3-methoxyphenyl moiety (afforded compound CDHPM-10e) would significantly increase its anti-proliferative activity by 34.01%, while substitution with 3,4,5-trimethoxyphenyl motif as in CDHPM-10f, the %inhibition was increased by 26.39%. Secondly, substituting terminal phenyl moiety with chlorine atom at the para position (afforded compound CDHPM-10b) would increase the inhibitory activity by 3.09%. Thirdly, introducing the p-methyl group at the terminal phenyl group (afforded compound CDHPM-10d) would decrease the inhibitory activity by 2.01%. Fourthly, inserting an ethylene linker between the terminal phenyl group and the pyrimidine core, as in CDHPM-10g, led to a significant decrease in its anti-proliferative activity by 22.37%. In comparison, substituting the terminal phenyl moiety with a more lipophilic bromine atom at the para position, as in CDHPM-10c, led to a marked activity loss of 37.48%. Finally, CDHPM-10e established the most potent anticancer agent among the herein examined series towards the full cell panel. In brief, we could conclude that the installation of a more hydrophilic group at the terminal phenyl moiety of the pyrimidine core, which is endowed with 3,4,5-trimethoxychalcone through the S-acetamide bridge, could significantly improve the anti-proliferative activity as shown in .
Figure 6. SAR study of designed hybrids CDHPM-10a-g.
VEGFR-2 inhibitory activity
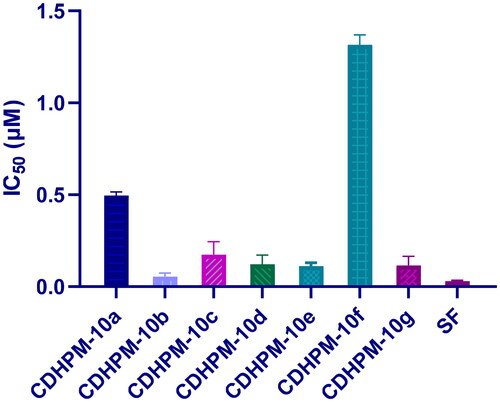
The most active analogs, CDHPM-10a-g, were examined for their inhibitory action towards VEGFR-2 utilising VEGFR-2 Kinase assay following the manufacturer’s instruction and sorafenib as a reference. The data showed that the hybrids CDHPM-10b-e and CDHPM-10g revealed potency comparable to that of sorafenib with sub-micromolar IC50 values of 0.054–0.174 µM (), demonstrating the anti-angiogenic action of them.
Figure 7. VEGFR-2 blockade of CDHPM-10e and the reference drug sorafenib.
In vitro anti-proliferative activities (SRB assay)
The most promising analog, CDHPM-10e, was evaluated for its growth inhibitory activity towards the MCF-7 cells, a highly expressing VEGFR-2 kinaseCitation12. Serial dilutions of hybrid CDHPM-10e were incubated with MCF-7 cells for 72 h. SRB assay was conducted to evaluate the cell viability utilising the reference drug SFCitation65. The target compound CDHPM-10e registered two-fold more potent anti-proliferative activity than sorafenib since the IC50 values were 2.52 and 5.10 µM, respectively, towards the MCF-7 cell line with a selectivity index of 0.91 ().
Table 4. IC50 values for target analog CDHPM-10e and sorafenib towards breast cancer cells.
MTT assay
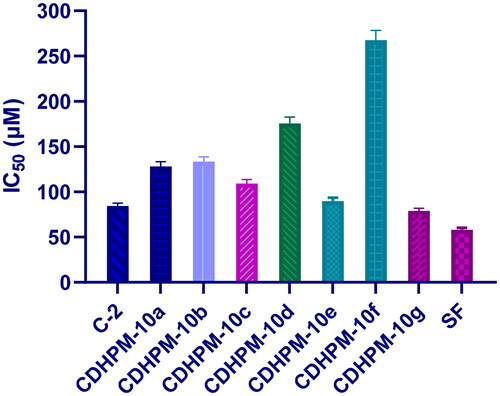
The chalcone chloride intermediate C-2 and the most promising analogs CDHPM-10a-g were evaluated for their growth inhibitory activity towards the human umbilical vein endothelial cells (HUVECs) as they are widely used as a source of primary endothelial cells for in vitro studies of the vasculature and angiogenesis using MTT assay along with sorafenib as a reference drug (). Serial dilutions of chalcone chloride intermediate C-2 and hybrids CDHPM-10a-g were incubated with HUVECs cells for 48 h using sorafenib as a reference. Among the target compounds, CDHPM-10e and CDHPM-10g exhibited antiproliferative activity comparable to that of the reference drug with IC50s equal 89.91, 78.79, and 58.08 µM, respectively, towards the HUVECs cell line. In addition, chalcone chloride intermediate C-2 was tested to examine the hybridisation of trimethoxy chalcone motif with dihydrpyrimidine scaffold. The results proved the importance of trimethoxy chalcone scaffold for biological activity with an IC50 of 84.29 µM. Furthermore, coupling the styryl derivative of DHPM with trimethoxy chalcone scaffold C-2, as in CDHPM-10g, improved the antiproliferative activity. Therefore, the results thoroughly delineate the hybrid candidates CDHPM-10e and CDHPM-10g as good lead compounds and anti-angiogenic agents for further study.
Figure 8. Cytotoxicity activity for chalcone chloride intermediate C-2, target analogs CDHPM-10a-g and reference drug sorafenib towards human umbilical vein endothelial cells (HUVECs).
Cell cycle study
The activity of CDHPM-10e on the distribution of the MCF-7 cell cycle was explored to verify its mechanism of action with propidium iodide (PI) using flow cytometryCitation66,Citation67. Cells were treated with 2.52 µM of CDHPM-10e and DMSO (NC) for 48 h. The data confounded that CDHPM-10e notably enhanced the sub-G1 phase cells compared to DMSO (negative control; NC). The %cells of the subG1/apoptosis phase increased to 45.64% for CDHPM-10e from 0.82% for NC (). Furthermore, the results revealed a decrease of the G2/M, G1, and S peaks, indicating the loss of DNA during apoptosis, thus leading to a lower PI intensity. Therefore, compound CDHPM-10e has the potential to induce Sub-G1-phase arrest.
Figure 9. Flow cytometric studies: cell cycle analysis; (A) DMSO, (B) CDHPM-10e, (C) %Cells in cycle stages (G1, S, G2/M, Sub-G1); apoptosis induction; (D) histogram for induction of apoptosis, (E) NC, (F) CDHPM-10e.
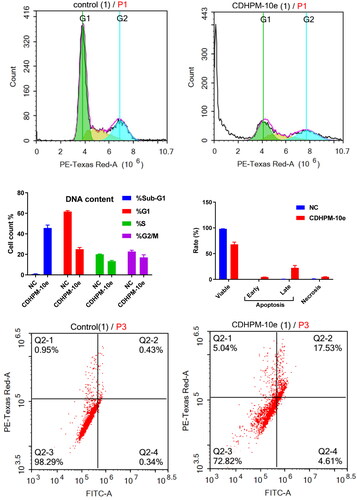
Apoptosis analysis
The double staining assay annexin-V-FITC/propidium iodide (PI) was conducted to verify the mode of cell death induction in the MCF-7 cells at the sub-G1 area. At a concentration of 2.52 μM, the cells were treated with the examined analog CDHPM-10e along with NC for 24 h. The study disclosed that the total induced apoptosis percentage was 31.99%. There was an increase in the apoptotic cell rate in the early phase from 0.33% for NC up to 4.52%. In addition, there was a significant increase in the apoptotic rate in the late phase, to attain 22.50% compared to 0.49% in NC cells. These results exposed that analog CDHPM-10e induced the pre-G1 apoptotic stage and caused cell cycle arrest at the sub-G1 area (). Nonetheless, this ratio implied that the anticancer activity of derivative CDHPM-10e has relied on apoptosis. After 48 h, ACEA measurements employing MCF-7 revealed that CDHPM-10e mainly triggers apoptosis in which 22.50% of the cells were late apoptosis and 4.52% early apoptosis whereas only 4.97% were necrotic, indicating high sensitivity towards many types of cancers which augmented by its antiproliferative activity.
Effect of CDHPM-10e on Caspase-3 level
The human active caspase-3 protein (a marker of apoptosis) level was examined utilising a western blot assay following the manufacturer’s protocol. The stimulation of Cas-3 plays a vital role in apoptosis induction. DNA fragmentation is a result of its activation through an initiator caspasesCitation68. It’s well known that VEGFR-2 TKIs increase the Cas-3 enzymatic activity, thus potentiating the intrinsic apoptotic cascadeCitation68,Citation69. Therefore, to explore the effect of analog CDHPM-10e on the apoptotic Cas-3 level, the MCF-7 tumour cell line was treated with the herein examined analog at 2.52 µM for 24 h using DMSO (Negative Control; NC). The data indicated that hybrid CDHPM-10e exhibited a significant increase in the active Cas-3 level compared to NC ().
Figure 10. Western blot analysis of compound CDHPM-10e along with DMSO control.
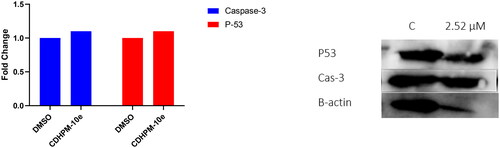
Effect of CDHPM-10e on P53 level
P53 protein was proven to have a vital role in the induction of apoptosis, cell cycle arrest, DNA repair, and angiogenesis inhibitionCitation70. Further evaluation of CDHPM-10e at 2.52 µM towards MCF-7 cells clearly boosted the tumour suppressor protein p53 expression level (). Therefore, the obtained data from western blot analysis exposed that compound CDHPM-10e might prompt apoptosis and cell cycle arrest via caspase and p53-dependent mechanisms.
Cell migration assay
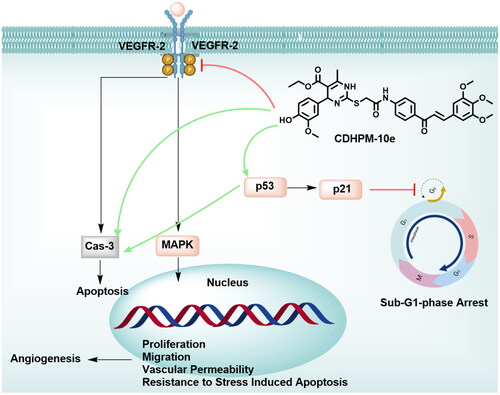
The anti-metastatic action of CDHPM-10e was also investigated using a simple and economical wound-healing assay. MCF-7 cells were incubated with derivative CDHPM-10e in serum-free medium (SFM) for 48 h at a concentration of 2.52 µM. shows that hybrid CDHPM-10e markedly decreased the cell migration rate in SFM compared to NC cells. Therefore, the results hinted out that hybrid CDHPM-10e is a potent anticancer agent with anti-metastatic activity. Overall, the anticancer mechanisms of CDHPM-10e associated with VEGFR-2 TK inhibition are illustrated in .
Figure 11. Wound healing inhibition in CDHPM-10e-treated MCF-7 cell line.
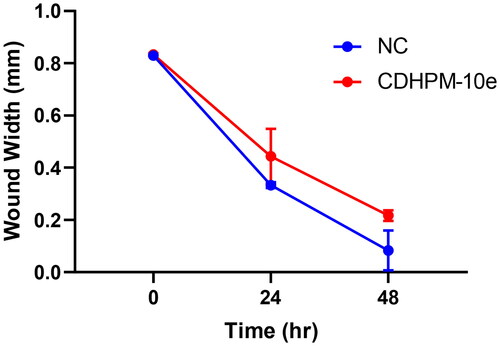
Figure 12. Potential anticancer mechanisms of CDHPM-10e.
In-silico molecular modelling studies
EON scaffold hopping
Concerning sorafenib as an existing anti-angiogenic drug, OpenEye’s EON module utilises electrostatic potential similarities and physically realistic shapes to discover new molecular targets utilising full Poisson–Boltzmann (PB) electrostatics to generate electrostatic Tanimoto (ET) gridsCitation24,Citation25. The formal charges and pKa state majorly affect the electrostatics, so the database and query drug are examined in a neutral pH model. EON module bestows substantially to lead discovery and design. Results were visualised and analysed by the EON view tool of OpenEye’s VIDA visualisation moduleCitation71. In addition, the query drug sorafenib was comprised in the library for results validation. EON ranks the designed compounds based on their electrostatic similarity and shape to the query drug. The top-scoring analogs, NO-TZD-3a-d,5,6 and CDHPM-10g, offered the best alignment with the query are cited in .
Table 5. EON electrostatic potential similarity and shape of the herein examined analogs concerning sorafenib as a query drug.
Docking studies
Modelling studies utilised OpenEye® Scientific Software (2023) to investigate the interacting poses of the most potent herein designed analogs, CDHPM-10a-g, with the KDR binding pocket whose co-crystallised drug sorafenib (PDB ID: 3WZE). The sum of hydrogen bonding, desolvation, and shape energies (FRED Chemgauss4 scores) were utilised to examine compounds in which the lowest scores indicate the best binding poses, as shown in . Sorafenib was re-docked utilising the standard method to rationalise the protein–ligand poses of the herein examined analogs in the VEGFR-2 active site and validate the results. The data demonstrated that the docking interaction of sorafenib was the same as that of the co-crystallised ligand ().
Figure 13. 3D binding interactions of sorafenib (green) in the VEGFR-2 pocket. Green dash lines denote H-bonds.
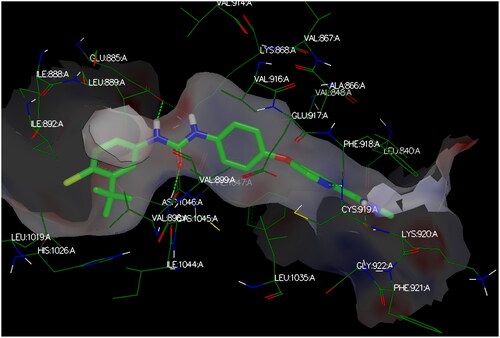
Table 6. Chemgauss4 scores of the designed analogs CDHPM-10a-g and sorafenib.
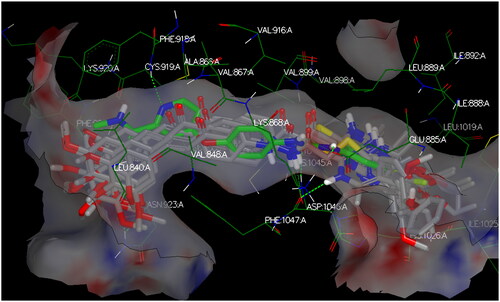
The target compounds spatially stacked well together, showing an overly with sorafenib (). Compounds CDHPM-10d-f displayed hydrogen bonds coming from acetamide NH and Asp:1046 residue in the DFG binding domain with lengths of 1.85, 2.09, and 2.41 Å, correspondingly, while compounds CDHPM-10a, CDHPM-10b, CDHPM-10c, and CDHPM-10g revealed mainly electrostatic and hydrophobic interactions within the active site. In addition, compounds CDHPM-10d and CDHPM-10e revealed an additional hydrogen bond coming from m-methoxy group of the head phenyl and NH of Asn:923:A with lengths of 2.22 and 2.12 Å, respectively. The results displayed an excellent fitting of the designed analogs to the binding pocket of KDR with FRED Chemgauss4 score interval from −10.1780 to −12.6031 compared to −18.408 for sorafenib.
Figure 14. 3D binding interactions show an overlay between target compounds (grey) and sorafenib (green) in the VEGFR-2 pocket. Green dash lines denote H-bonds.
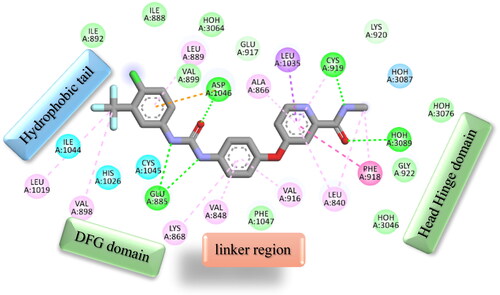
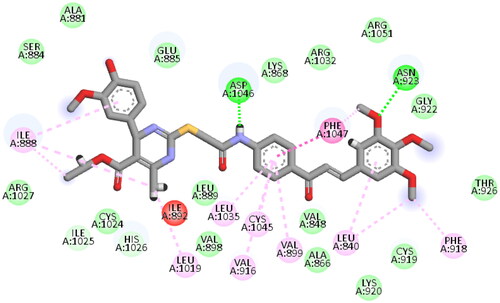
Again, the most promising hybrid towards NCI-60 tumour cells with broad-spectrum antiproliferative activity, CDHPM-10e, was chosen as the most plausible interacting conformation, so we studied its interacting modes within the active site. The analog CDHPM-10e presented a unique interacting pose that could inspire its broad-spectrum antitumor activity. As illustrated in , compound CDHPM-10e presented a strong H-bond within the DFG binding region and ATB binding domain overlaid well with the reference drug, and fit tightly inside the VEGFR-2 binding region. In addition, the 2D interacting pose of the KDR with CDHPM-10e shown in was generated by Discovery Studio VisualiserCitation72,Citation73. Compound CDHPM-10e showed hydrophobic and electrostatic interactions with the contacted amino acids. The pyrimidine C-4 phenyl group formed Pi-alkyl hydrophobic interaction with Ile:888:A, while the linker’s phenyl group formed pi–pi T-shaped hydrophobic interactions with Phe:1047:A. Furthermore, the ethyl and m-methoxy groups formed alkyl interactions with Ile:888:A and Leu:840:A, respectively. Also, the linker’s phenyl group formed pi-alkyl hydrophobic interactions with Leu:1035:A, Val:899:A, Val:916, and Cys:1045:A. In addition, the two m-methoxy groups of the head phenyl and pyrimidine C6 methyl formed pi-alkyl hydrophobic interactions with Phe:1047:A, Phe:918:A, and Leu:1019:A. Additionally, CDHPM-10e developed other mixed hydrophobic and Van der Waals interactions.
Figure 15. 3D interactions; (A) docking pose of CDHPM-10e (grey) inside the binding region of VEGFR-2 demonstrating hydrogen bonding (green dash line) with Asp:1046:A and hydrophobic interactions; (B) overlay of CDHPM-10e with the reference drug sorafenib (green).
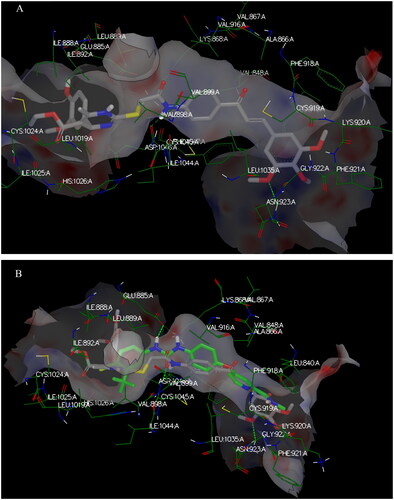
Figure 16. 2D depiction of CDHPM-10e showing hydrophilic and hydrophobic interactions inside VEGFR-2 active site.
Furthermore, the dock report tool provided an additional property for CDHPM-10e optimisation at vanillin-like moiety, pyrimidine C-5 ester group, pyrimidine N1-H, and central aryl moiety, demonstrating protein-ligand desolvation, hydrogen bonding, and shape energies, recognised by Chemgauss4 score (Figure S49, see Supplementary Material). The analog CDHPM-10e showed a FRED chemgauss4 score of −11.1431 comparable to that of sorafenib (-18.408). Furthermore, the modelling study implies that CDHPM-10e was a potent VEGFR-2 inhibitor but was less potent than sorafenib, which is aligned with the enzyme assay inhibitory data.
Drug-likeness characteristics and ADMET prediction
Drug-likeness and pharmacokinetics ADMET features for the most promising hybrid, CDHPM-10e, were predicted using the ADMETlab2.0 web-based toolCitation74,Citation75. shows that CDHPM-10e disclosed 2 violations from Lipinski’s rule (MW > 500, N or O > 10) while Pfizer accepted its drug-likeness properties. The ADMET prediction data exposed that CDHPM-10e has a good synthetic accessibility score, a high GIT absorption rate, and moderate solubility in water. Besides, it showed >20% bioavailability and >90% plasma protein binding. Additionally, it could be CYP2D6, CYP2C19, and CYP1A2 enzymes non-inhibitor, implying a low possibility of producing drug-drug interaction. Moreover, acute toxicity and carcinogenicity tests demonstrated that hybrid CDHPM-10e is neither toxic nor carcinogenic, proving a good profile of ADMET properties ().
Table 7. Lipinski’s rule of five characteristics for hybrid CDHPM-10e.
Table 8. ADMET parameters for hybrid CDHPM-10e.
Materials and methods
Chemistry
1H and 13C NMR spectra were acquired on a Bruker AVANCE and Varian Unity INOVA-400 MHz NMR spectrometer. TLC was applied on pre-coated silica gel 60 F254 (Merck) sheets and visualised by UV (254 nm). Column chromatography was used to purify the synthesised compounds using silica gel 150–250 μm. All melting points were measured on digital melting point device (FALC Instruments s.r.l., Model MPD-03, Italy) and were uncorrected. Mass spectra were carried out on Direct Inlet part to mass analyser in Thermo Scientific GC-MS model ISQ (Waltham, MA, USA). Vario elemental analyser III (Vario, Germany) was used for C, H, and N analysis. The results agreed with the designed compounds within ±0.4% of the theoretical values. All reagents and solvents were acquired from commercially available suppliers. The intermediates TZD-1Citation76,Citation77, TZD-2a-dCitation43,Citation45,Citation46, TZD-4Citation43, DHPM-7a-gCitation48–53, C-8Citation48–53, C-9Citation56–58 were prepared as previously described.
General procedure for synthesis of NO-TZD-3a-d and NO-TZD-5
To a stirred solution of TZDs 2a-d and 4 (1 mmol) in dry DMF (2 ml), furoxan mesylate (1.1 mmol, 0.297 g) and K2CO3 (1 mmol, 0.138 g) were added. The reaction mixture was heated at 65 °C for 1 h as indicated by TLC. The crude mixture was poured onto ice-cold water (50 ml) and then decanted. The resulting crude product was boiled in 70% ethanol and then filtered, dried, and crystallised from absolute ethanol to afford the target compounds NO-TZD-3a-d and NO-TZD-5 in good yields.
(Z)-4-((5-benzylidene-2,4-dioxothiazolidin-3-yl)methyl)-3-phenyl-1,2,5-oxadiazole 2-oxide (NO-TZD-3a)
Yield (75%); yellow powder; Melting point: 169.6–170.4 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.93 (s, 1H, CH-Olefinic), 7.81 (dd, J = 7.4, 1.9 Hz, 2H, Ar-H), 7.69–7.59 (m, 5H, Ar-H), 7.54 (q, J = 7.5, 6.9 Hz, 3H, Ar-H), 5.04 (s, 2H, CH2); 13C NMR (101 MHz, DMSO) δ 167.10, 165.27, 157.74, 134.60, 133.13, 131.82, 131.44, 130.72, 129.92, 129.76, 128.55, 126.06, 120.51, 112.03, 34.92; GC-MS m/z calcd for [M]+ C19H13N3O4S: 379.39, found: 379.57.
(Z)-4-((5–(4-chlorobenzylidene)-2,4-dioxothiazolidin-3-yl)methyl)-3-phenyl-1,2,5-oxadiazole 2-oxide (NO-TZD-3b)
Yield (86%); yellowish white powder; Melting point: 162.6–163 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.93 (s, 1H, CH-Olefinic), 7.81 (d, J = 7.0 Hz, 2H, Ar-H), 7.62 (d, J = 5.2 Hz, 7H, Ar-H), 5.03 (s, 2H, CH2); 13C NMR (101 MHz, DMSO) δ 166.83, 165.15, 157.71, 136.05, 133.27, 132.31, 132.03, 131.80, 129.97, 129.74, 128.54, 126.07, 121.29, 111.94, 34.96; GC-MS m/z calcd for [M]+ C19H12ClN3O4S: 413.83, found: 413.75.
(Z)-4-((5–(2-hydroxybenzylidene)-2,4-dioxothiazolidin-3-yl)methyl)-3-phenyl-1,2,5-oxadiazole 2-oxide (NO-TZD-3c)
Yield (51%); yellow powder; Melting point: 201.1–201.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H, OH, D2O exchangeable), 8.09 (s, 1H, CH-Olefinic), 7.84–7.80 (m, 2H, Ar-H), 7.63 (dd, J = 9.2, 6.4 Hz, 3H, Ar-H), 7.55 (dd, J = 8.1, 6.4 Hz, 1H, Ar-H), 7.37–7.31 (m, 1H, Ar-H), 6.99–6.94 (m, 1H, Ar-H), 6.77 (t, J = 5.6 Hz, 1H, Ar-H), 5.02 (s, 2H, CH2); 13C NMR (101 MHz, DMSO) δ 167.44, 165.51, 157.90, 133.39, 131.81, 131.54, 129.76, 129.54, 128.55, 128.37, 120.30, 120.02, 118.97, 116.72, 114.70, 112.08, 34.84; GC-MS m/z calcd for [M]+ C19H13N3O5S: 395.39, found: 395.48.
(Z)-4-((5–(4-hydroxy-3-methoxybenzylidene)-2,4-dioxothiazolidin-3-yl)methyl)-3-phenyl-1,2,5-oxadiazole 2-oxide (NO-TZD-3d)
Yield (55%); yellow powder; Melting point: 164–164.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.97 (s, 1H, OH, D2O exchangeable), 7.96 (s, 1H, CH-Olefinic), 7.80 (d, J = 7.2 Hz, 2H, Ar-H), 7.61 (q, J = 7.5, 6.9 Hz, 4H, Ar-H), 7.14 (s, 1H, Ar-H), 7.07 (d, J = 8.2 Hz, 1H, Ar-H), 6.89 (d, J = 8.3 Hz, 1H, Ar-H), 5.02 (s, 2H, CH2), 3.81 (s, 3H, OCH3); 13C NMR (101 MHz, DMSO) δ 167.30, 165.37, 157.73, 148.71, 135.45, 131.81, 129.74, 129.66, 128.53, 128.41, 126.05, 125.33, 116.90, 114.71, 112.12, 56.06, 34.81; GC-MS m/z calcd for [M]+ C20H15N3O6S: 425.42, found: 425.62.
4-(((Z)-2,4-dioxo-5-((E)-3-phenylallylidene)thiazolidin-3-yl)methyl)-3-phenyl-1,2,5-oxadiazole 2-oxide (NO-TZD-5)
Yield (62%); yellow powder; Melting point: 137.6–138.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.81–7.78 (m, 1H, CH-Olefinic), 7.71–7.68 (m, 1H, Ar-H), 7.62 (ddd, J = 9.1, 6.0, 3.5 Hz, 4H, Ar-H), 7.42 (d, J = 7.1 Hz, 3H, Ar-H), 7.36 (d, J = 15.1 Hz, 2H, Ar-H), 6.99 (dd, J = 15.2, 11.5 Hz, 2H, 2 x CH-Olefinic), 5.00 (s, 2H, CH2); 13C NMR (101 MHz, DMSO) δ 167.00, 164.71, 157.69, 145.40, 135.88, 134.94, 131.82, 130.52, 129.74, 129.43, 128.52, 126.01, 123.55, 121.52, 112.06, 34.82; GC-MS m/z calcd for [M]+ C21H15N3O4S: 405.43, found: 405.46.
Synthesis of (Z)-4-((5–(4-(allyloxy)-3-methoxybenzylidene)-2,4-dioxothiazolidin-3-yl)methyl)-3-phenyl-1,2,5-oxadiazole 2-oxide (NO-TZD-6)
To a stirred solution of NO-TZD-3c (1 mmol, 0.425 g) in DMF (2 ml), allyl bromide (5 mmol, 0.42 ml), K2CO3 (1 mmol, 0.138 g) were added. The reaction mixture was heated at 80 °C for 1 h. The crude mixture was poured onto ice-cold water, filtered, dried, and crystallised from ethanol absolute to afford O-allyl derivative NO-TZD-6 in good yield.
Yield (53%); yellow powder; Melting point: 154.4–155 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.96 (s, 1H, CH-Olefinic), 7.83 (d, J = 22.2 Hz, 2H, Ar-H), 7.68–7.56 (m, 3H, Ar-H), 7.32–7.03 (d, J = 23.0 Hz, 3H, Ar-H), 6.16–5.94 (m, 1H, CH-Olefinic), 5.42 (d, J = 17.2 Hz, 1H, CH-Olefinic), 5.34–5.19 (m, 1H, CH-Olefinic), 5.03 (s, 2H, NCH2), 4.75–4.54 (m, 2H, OCH2), 3.82 (s, 3H, OCH3); 13C NMR (101 MHz, DMSO) δ 167.16, 165.30, 157.72, 150.55, 149.63, 134.91, 133.66, 131.81, 129.74, 128.54, 126.04, 125.96, 124.41, 118.60, 117.42, 114.22, 113.94, 112.05, 69.40, 56.12, 34.89; GC-MS m/z calcd for [M]+ C23H19N3O6S: 465.10, found: 465.03.
General procedure for the synthesis of the designed analogs CDHPM-10a-g
To a mixture of monastrol analogs DHPM-7a-g (1 mmol) in DMF (3 ml), K2CO3 (1 mmol, 0.138 g), NaI (0.050 g), and trimethoxychalcone chloride C-9 (1 mmol, 0.389 g) were added. The chemical mixture was stirred at RT for 2 h as monitored by TLC and then added portion-wise to ice-cold water. The formed precipitate was filtered off and dried. Column chromatography (EtAc: Hex 1:1) afforded the target analogs CDHPM-10a-g in good yields.
Ethyl (R,E)-6-methyl-2-((2-oxo-2-((4–(3-(3,4,5-trimethoxyphenyl)acryloyl)phenyl)amino)ethyl)thio)-4-phenyl-1,4-dihydropyrimidine-5-carboxylate (CDHPM-10a)
Yield (85%); Light yellow powder; Melting point: 99–100 °C; 1H NMR (DMSO-d6, δ = ppm, 400 MHz) δ 10.59 (s, 1H, NH amide), 9.82 (s, 1H, Py-NH), 8.15 (d, J = 7.9 Hz, 2H, Ar-H), 7.91 (d, J = 15.2 Hz, 1H, COCH = CH), 7.72–7.66 (m, 3H, Ar-H and COCH=CH), 7.25–7.22 (m, 7H, Ar-H), 5.55 (s, 1H, Py-CH), 4.07–3.96 (m, 4H, SCH2 and CH3CH2), 3.89 (s, 6H, two m-OCH3), 3.74 (s, 3H, p-OCH3), 2.25 (s, 3H, Py-CH3), 1.14 (t, J = 7.1 Hz, 3H, CH3CH2); 13C NMR (DMSO-d6, δ = ppm, 101 MHz) δ 187.98, 167.47, 166.67, 153.61, 150.58, 146.23, 145.71, 144.38, 143.65, 140.23, 132.98, 130.81, 130.33, 128.98, 128.61, 127.18, 127.05, 126.67, 121.65, 118.90, 107.00, 99.20, 60.63, 59.61, 59.35, 56.64, 35.48, 17.98, 14.60; GC-MS m/z calcd for [M]+ C34H35N3O7S: 629.73, found: 629.46; Anal. Calcd.: C, 64.85; H, 5.60; N, 6.67; Found: C, 65.07; H, 5.74; N, 6.89.
Ethyl (R,E)-4–(4-chlorophenyl)-6-methyl-2-((2-oxo-2-((4–(3-(3,4,5-trimethoxyphenyl)acryloyl)phenyl)amino)ethyl)thio)-1,4-dihydropyrimidine-5-carboxylate (CDHPM-10b)
Yield (83%); Light yellow powder; Melting point: 163.2–164 °C; 1H NMR (DMSO-d6, δ = ppm, 400 MHz) δ 10.51 (s, 1H, NH amide), 9.86 (s, 1H, Py-NH), 8.14 (d, J = 8.3 Hz, 2H, Ar-H), 7.90 (d, J = 15.2 Hz, 1H, COCH = CH), 7.69 (d, J = 15.3 Hz, 1H, COCH=CH), 7.63 (d, J = 8.4 Hz, 2H, Ar-H), 7.24 (s, 2H, Ar-H), 7.21 (s, 4H, Ar-H), 5.51 (s, 1H, Py-CH), 4.01 (q, J = 15.4, 11.9 Hz, 4H, SCH2 and CH3CH2), 3.88 (s, 6H, two m-OCH3), 3.73 (s, 3H, p-OCH3), 2.25 (s, 3H, Py-CH3), 1.12 (t, J = 7.0 Hz, 3H, CH3CH2); 13C NMR (DMSO-d6, δ = ppm, 101 MHz) δ 187.92, 167.40, 166.50, 153.60, 150.69, 146.58, 144.61, 144.31, 143.62, 140.16, 132.94, 131.73, 130.82, 130.29, 128.94, 128.60, 128.55, 121.61, 118.85, 106.93, 98.67, 60.62, 59.67, 58.86, 56.61, 35.44, 18.00, 14.59; GC-MS m/z calcd for [M]+ C34H34ClN3O7S: 663.18, found: 663.82; Anal. Calcd.: C, 61.49; H, 5.16; N, 6.33; Found: C, 61.65; H, 5.37; N, 6.59.
Ethyl (R,E)-4–(4-bromophenyl)-6-methyl-2-((2-oxo-2-((4–(3-(3,4,5-trimethoxyphenyl)acryloyl)phenyl)amino)ethyl)thio)-1,4-dihydropyrimidine-5-carboxylate (CDHPM-10c)
Yield (81%); Yellow powder; Melting point: 110–111 °C; 1H NMR (DMSO-d6, δ = ppm, 400 MHz) δ 10.53 (s, 1H, NH amide), 9.87 (s, 1H, Py-NH), 8.15 (d, J = 8.4 Hz, 2H, Ar-H), 7.90 (d, J = 15.5 Hz, 1H, COCH = CH), 7.69 (d, J = 15.4 Hz, 1H, COCH=CH), 7.63 (d, J = 8.3 Hz, 2H, Ar-H), 7.35 (d, J = 8.0 Hz, 2H, Ar-H), 7.24 (s, 2H, Ar-H), 7.15 (d, J = 8.1 Hz, 2H, Ar-H), 5.50 (s, 1H, Py-CH), 4.06–3.97 (m, 4H, SCH2 and CH3CH2), 3.88 (s, 6H, two m-OCH3), 3.73 (s, 3H, p-OCH3), 2.25 (s, 3H, Py-CH3), 1.12 (t, J = 7.1 Hz, 3H, CH3CH2); 13C NMR (DMSO-d6, δ = ppm, 101 MHz) δ 187.91, 167.40, 166.49, 153.59, 150.73, 146.61, 145.00, 144.30, 143.63, 140.13, 132.93, 131.47, 130.82, 130.30, 129.32, 121.61, 120.26, 118.85, 106.91, 98.57, 60.62, 59.69, 58.90, 56.61, 35.43, 18.00, 14.59; GC-MS m/z calcd for [M]+ C34H34BrN3O7S: 707.13, found: 707.06; Anal. Calcd.: C, 57.63; H, 4.84; N, 5.93; Found: C, 57.90; H, 5.01; N, 6.12.
Ethyl (R,E)-6-methyl-2-((2-oxo-2-((4–(3-(3,4,5-trimethoxyphenyl)acryloyl)phenyl)amino)ethyl)thio)-4-(p-tolyl)-1,4-dihydropyrimidine-5-carboxylate (CDHPM-10d)
Yield (83%); Light yellow powder; Melting point: 114.7–115.6 °C; 1H NMR (DMSO-d6, δ = ppm, 400 MHz) δ 10.53 (s, 1H, NH amide), 9.77 (s, 1H, Py-NH), 8.14 (d, J = 8.4 Hz, 2H, Ar-H), 7.90 (d, J = 15.6 Hz, 1H, COCH = CH), 7.70 (d, J = 15.4 Hz, 1H, COCH=CH), 7.62 (d, J = 8.3 Hz, 2H, Ar-H), 7.25 (s, 2H, Ar-H), 7.09 (d, J = 7.6 Hz, 2H, Ar-H), 7.00 (d, J = 7.8 Hz, 2H, Ar-H), 5.49 (s, 1H, Py-CH), 4.04–3.96 (m, 4H, SCH2 and CH2CH3), 3.89 (s, 6H, two m-OCH3), 3.74 (s, 3H, p-OCH3), 2.24 (s, 3H, Py-CH3), 2.21 (s, 3H, p-CH3), 1.14 (t, J = 7.0 Hz, 3H, CH3CH2); 13C NMR (DMSO-d6, δ = ppm, 101 MHz) δ 187.93, 167.52, 166.67, 153.60, 150.32, 145.97, 144.35, 143.62, 142.88, 140.18, 136.22, 132.92, 130.81, 130.26, 129.49, 129.17, 127.01, 126.61, 121.60, 118.89, 106.96, 99.35, 60.62, 59.56, 59.17, 56.62, 35.40, 21.09, 17.96, 14.61, 14.58; GC-MS m/z calcd for [M]+ C35H37N3O7S: 643.76, found: 643.51; Anal. Calcd.: C, 65.30; H, 5.79; N, 6.53; Found: C, 65.24; H, 5.98; N, 6.71.
Ethyl (R,E)-4–(4-hydroxy-3-methoxyphenyl)-6-methyl-2-((2-oxo-2-((4–(3-(3,4,5-trimethoxyphenyl)acryloyl)phenyl)amino)ethyl)thio)-1,4-dihydropyrimidine-5-carboxylate (CDHPM-10e)
Yield (61%); Light yellow powder; Melting point: 111.6–112 °C; 1H NMR (DMSO-d6, δ = ppm, 400 MHz) δ10.56 (s, 1H, CONH), 9.76 (s, 1H, Py-NH), 8.84 (s, 1H, OH), 8.13 (d, J = 8.4 Hz, 2H, Ar-H), 7.89 (d, J = 15.5 Hz, 1H, COCH = CH), 7.69 (d, J = 15.5 Hz, 1H, COCH=CH), 7.59 (d, J = 8.4 Hz, 2H, Ar-H), 7.25 (s, 2H, Ar-H), 6.78 (d, J = 2.1 Hz, 1H, Ar-H), 6.62 (dd, J = 8.9, 7.0 Hz, 2H, Ar-H), 5.45 (s, 1H, Py-CH), 4.06–3.99 (m, 4H, SCH2 and CH3CH2), 3.89 (s, 6H, two m-OCH3), 3.73 (s, 3H, p-OCH3), 3.69 (s, 3H, m-OCH3), 2.23 (s, 3H, Py-CH3), 1.15 (t, J = 7.3 Hz, 3H, CH3CH2); 13C NMR (DMSO-d6, δ = ppm, 101 MHz) δ 187.96, 167.57, 166.77, 153.59, 150.27, 147.66, 146.00, 145.59, 144.37, 143.57, 140.15, 137.12, 132.97, 130.81, 130.31, 121.63, 119.43, 118.86, 115.69, 111.60, 106.94, 99.80, 60.62, 59.56, 59.01, 56.61, 55.95, 40.61, 40.40, 40.19, 39.98, 39.78, 39.57, 39.36, 35.32, 17.95, 14.66; GC-MS m/z calcd for [M]+ C35H37N3O9S: 675.75, found: 675.91; Anal. Calcd.: C, 62.21; H, 5.52; N, 6.22; Found: C, 62.39; H, 5.64; N, 6.48.
Ethyl (R,E)-6-methyl-2-((2-oxo-2-((4–(3-(3,4,5-trimethoxyphenyl)acryloyl)phenyl)amino)ethyl)thio)-4–(3,4,5-trimethoxyphenyl)-1,4-dihydropyrimidine-5-carboxylate (CDHPM-10f)
Yield (55%); Light yellow powder; Melting point: 114–115 °C; 1H NMR (DMSO-d6, δ = ppm, 400 MHz) δ 10.59 (s, 1H, NH amide), 9.86 (s, 1H, Py-NH), 8.12 (d, J = 8.2 Hz, 2H, Ar-H), 7.88 (d, J = 15.0 Hz, 1H, COCH = CH), 7.70–7.64 (m, 3H, Ar-H and COCH=CH), 7.23 (s, 2H, Ar-H), 6.49 (s, 2H, Ar-H), 5.51 (s, 1H, Py-CH), 4.15–4.02 (m, 4H, SCH2 and CH3CH2), 3.88 (s, 6H, two m-OCH3), 3.73 (s, 3H, p-OCH3), 3.70 (s, 6H, two m-OCH3), 3.58 (s, 3H, p-OCH3), 2.23 (s, 3H, Py-CH3), 1.18 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (DMSO-d6, δ = ppm, 101 MHz) δ 187.96, 167.48, 166.75, 153.60, 153.05, 145.95, 144.37, 143.66, 141.45, 140.15, 136.89, 132.95, 130.81, 130.32, 121.61, 118.81, 106.95, 104.22, 99.52, 60.62, 60.38, 59.69, 59.24, 56.62, 56.13, 35.34, 17.98, 14.71; GC-MS m/z calcd for [M]+ C37H41N3O10S: 719.81, found: 719.80; Anal. Calcd.: C, 61.74; H, 5.74; N, 5.84; Found: C, 61.82; H, 5.91; N, 6.03.
Ethyl (R)-6-methyl-2-((2-oxo-2-((4-((E)-3–(3,4,5-trimethoxyphenyl)acryloyl)phenyl)amino)ethyl)thio)-4-((E)-styryl)-1,4-dihydropyrimidine-5-carboxylate (CDHPM-10g)
Yield (58%); Yellow powder; Melting point: 98.2–98.5 °C; 1H NMR (DMSO-d6, δ = ppm, 400 MHz) δ 10.67 (s, 1H, CONH), 9.80 (s, 1H, Py-NH), 8.12 (d, J = 8.4 Hz, 2H, Ar-H), 7.87 (d, J = 15.3 Hz, 1H, COCH = CH), 7.78 (d, J = 8.3 Hz, 2H, Ar-H), 7.69 (d, J = 15.3 Hz, 1H COCH=CH), 7.35–7.12 (m, 7H, Ar-H), 6.27 (d, J = 15.8 Hz, 1H, PhCH=CH), 6.11 (dt, J = 16.3, 5.7 Hz, 1H, PhCH = CH), 5.13 (d, J = 5.5 Hz, 1H, Py-CH), 4.15–4.04 (m, 4H, SCH2 and CH3CH2), 3.88 (s, 6H, two m-OCH3), 3.73 (s, 3H, p-OCH3), 2.22 (s, 3H, Py-CH3), 1.21 (t, J = 7.2 Hz, 3H, CH3CH2); 13C NMR (DMSO-d6, δ = ppm, 101 MHz) δ 187.91, 167.61, 166.37, 153.60, 153.57, 151.02, 146.73, 144.34, 143.72, 140.16, 137.14, 132.97, 130.81, 130.62, 130.37, 128.91, 128.32, 126.64, 121.59, 118.94, 106.94, 97.79, 60.62, 59.65, 57.09, 56.61, 56.58, 35.55, 17.97, 14.75; GC-MS m/z calcd for [M]+ C36H37N3O7S: 655.24, found: 655.38; Anal. Calcd.: C, 65.94; H, 5.69; N, 6.41; Found: C, 66.13; H, 5.85; N, 6.59.
Biological activity
NCI-60 cells screening
The herein designed compounds CDHPM-10a-g were screened at NCI-DTP (MD, USA) on a full panel of 60 tumour cell lines comprising 9 different tumour types: Breast, Prostate, Renal, Ovarian, Melanoma, CNS, Colon, Leukaemia, and NSCL cancers. All the target analogs were initially tested at 10 µM (single dose). All compounds represented by scaffold “B” expressed sufficient potency in one-dose assay so that they were examined further at five-dose mode (0.01–100 µM) following the SRB standard procedure of the Drug Evaluation BranchCitation62,Citation78. In the Supplementary Material, the experimental methods were specified.
SRB cytotoxicity assay
The assay was applied following the procedure described elsewhereCitation65. MCF-7/HSF cell lines were purchased from Nawah Scientific Company, Egypt. Briefly, in a 96-well plate, MCF-7/HSF cells (100 μL of 5 × 103 cells/well) were seeded and incubated for 24 h. Various concentrations of the examined analogs (100 μL) were added and incubated for 72 h. Plates were then fixed with 150 μL of 10% TCA and incubated at 4 °C for 1 h. Cells were washed five times with deionised water. After that, 70 μL of SRB solution (0.4% w/v) was added and incubated in the dark for 10 min at RT. Cells were washed thrice with acetic acid (1%) and air-dried overnight. Finally, the protein-bound SRB stain was dissolved by adding 150 μL TRIS (10 mM), and BMG LABTECH®-FLUOstar Omega microplate reader (Ortenberg, Germany) was used to measure the absorbance at 540 nm. The selectivity index (SI) was calculated from the following equation. The test compound and NC were examined in triplicate, and the results were denoted as ±SD.
MTT cytotoxicity assay
The assay was performed as described earlierCitation20,Citation79–81. Human umbilical vein endothelial cell line (HUVECs) was purchased from Vacsera, Egypt, of ATCC origin. Briefly, in a 96-well plate, HUVECs were seeded (106 cells/cm2) and incubated overnight at 37 °C and CO2 (5%). The cells were then incubated for 48 h with serial dilutions of C-2, CDHPM-10a-g utilising SF as a reference drug. After 4 h incubation with MTT (Cat. No. M-5655) 5 mg/mL, anhydrous isopropanol containing 10% Triton X-100 plus 0.1 N HCl was added. The absorbance was read using a Bioline plate reader at λ570- 630 nm.
Cell cycle analysis
The assay was conducted following the manufacturer’s instructionsCitation20,Citation66,Citation67,Citation79,Citation82. Briefly, MCF-7 cells (105 cells/mL) were treated with either test compound CDHPM-10e (IC50) or DMSO (NC) for 48 h. Then, MCF-7 cells were trypsinized, washed with PBS (ice-cold; pH 7.4), fixed with ice-cold ethanol (60%; 2 ml), and incubated for 1 h at 4 °C. The fixed cells were rewashed with PBS, stained with 50 µg/mL RNAase A and 10 µg/mL propidium iodide (PI), and incubated in the dark for 20 min at 37 °C. They were analysed utilising FL2 (λex/em 535/617 nm; 12 000 events) signal detector (ACEA Novocyte™ flow cytometer, ACEA Biosciences Inc., San Diego, CA, USA) for DNA contents. Cell cycle analysis was calculated utilising ACEA NovoExpress™ software (ACEA Biosciences Inc., San Diego, CA, USA). The test compound and NC were examined in triplicate, and the data were denoted as ±SD.
Apoptosis analysis
The MCF-7 cells (105 cells/mL) were incubated with either 2.52 µM of CDHPM-10e or NC for 48 h. After that, cells were trypsinized, washed two times with PBS (ice-cold; pH 7.4), and treated with 0.5 ml Annexin V-FITC/PI solution in the dark at RT for 0.5 h. Cells were then injected via ACEA Novocyte™ flow cytometer (ACEA Biosciences Inc., San. Diego, CA, USA) and analysed fluorescent signals for FITC and PI utilising FL1 and FL2. Signal detector, correspondingly (λex/em 488/530 nm for FITC and λex/em 535/617 nm for PI). Twelve thousand events are acquired, and positive FITC and PI cells are quantified by quadrant analysis and calculated utilising ACEA NovoExpress™ software (ACEA Biosciences Inc., San Diego, CA, USA). The test compound and NC were examined in triplicate, and the data were denoted as ±SD.
VEGFR-2 inhibitory activity
The assay was conducted following the BPS Bioscience® procedureCitation17,Citation83–86. Briefly, Kinase Buffer 1 (5×), ATP, and PTK substrate (50×) of KDR (Cat. No. 40325) were thawed, and 25 µl/well master mixture was prepared, added, and then the test compound CDHPM-10e was added using sorafenib a reference drug. To the Blank wells, Kinase Buffer 1 (1×) was added. VEGFR-2 enzyme in Kinase Buffer 1 (1×) was added to sorafenib and test compound wells after incubation for 45 min. At 30 °C, ADP-Glo reagent/Kinase-Glo Max reagent was added, and the plate was incubated at RT for 0.25 h. in a dark place. Tecan–spark® reader was used to measure the luminescence immediately. From all readings, the “Blank” was subtracted. The test compound and sorafenib were examined in duplicate, and IC50 values were denoted as ±SD.
Western blotting
MCF-7 cell line (2.5*105 cells/mL) was seeded and treated as described above. Cells were then lysed using RIPA buffer containing 1x protease and phosphatase inhibitor cocktail (Thermo Scientific™) after 48 h of incubation with hybrid CDHPM-10e. After centrifugation at 4 °C, the supernatant was collected. The Pierce BCA Protein Assay Kit (Thermo Scientific™, 23225) was utilised for protein concentration detection. Cas-3 and p53 were separated using SDS-PAGE for 45 min at 100 V. Then, they transferred to a nitrocellulose membrane, which was incubated with an appropriate primary antibody for 12 h. After that, an HRP-conjugated secondary antibody was added. The loading control B-actin was used. The membrane was then developed utilising WesternBright® ECL (K-12045-D20), and the signals were recorded by ChemiDoc XRS+ (1708265, Bio-Rad). A CCD camera-based imager was used to capture the chemiluminescent signals. The band intensities were analysed using the image analysis softwareCitation20,Citation87. The antibodies were purchased from Cell Signalling Technology and Invitrogen, as depicted in .
Table 9. Antibodies used in the assay.
Cell migration assay
MCF-7 cell line (2 × 105/well) was seeded in a coated 12-well plate for scratch cell migration assay and incubated in 5% FBS-DMEM for 12 h at 5% CO2 and 37 °C. Then, horizontal scratches were applied to the confluent monolayer. PBS was used to wash the plate thoroughly. Test wells were treated with fresh medium containing CDHPM-10e, while the control wells were replenished with fresh media. Images were captured utilising an inverted microscope at specified time intervals. The plate was then incubated at various intervals at 5% CO2 and 37 °C. MII ImageView software version 3.7 was used to analyse the captured images.
In silico molecular modelling studies
The state-of-the-art OpenEye® scientific software (2023) was utilised for in silico molecular modelling evaluation. The software comprises EONCitation24, OMEGACitation88, SpruceCitation89, FREDCitation90, Docking-ReportCitation91, and VIDACitation71 modules. Initially, a library of the designed hybrids CDHPM-10a-g and SOR was energy minimised using a force field of MMFF94. SOR was used as a query ligand for the designed library in EON scaffold hoping. On the other hand, the docking scheme began with multi-conformers using the OMEGA® application. Then, the Spruce® application was used for VEGFR-2 (PDB ID: 3WZE) receptor preparation. After that, the FRED Chemgauss4 was generated utilising the FRED® application. The Docking-Report was obtained from OpenEye’s Docking-Report application. Finally, visualisation of the examined compounds’ binding interaction towards the VEGFR-2 binding pocket was accomplished by the Vida® module. Moreover, the 2D diagram of the compound’s pose was carried out by the Discovery Studio Visualisation application (DS).
ADMET prediction and drug-likeness properties
The efficient ADMETlab 2.0, a web-based tool, was utilised to analyse ADMET and drug-likeness parameters for the most potent analog, CDHPM-10eCitation74,Citation75.
Conclusions
In this study, a novel series of furoxan-based thiazolidine-2,4-diones and 1,4-dihydropyrimidines tethered to 3,4,5-trimethoxyxhalcone through S-acetamide bridge have been designed and synthesised. The target analogs were evaluated for their anticancer activity towards a full panel of 60 tumour cells. Among the tested hybrids, CDHPM-10a-g revealed mean %inhibitions range from 76.40 to 147.69%. Superiorly, compounds CDHPM-10e and CDHPM-10f exhibited the highest MGI% of 147.69 and 140.24%, respectively. Analogs CDHPM-10a, CDHPM-10b, and CDHPM-10d-f displayed higher mean %inhibitory activity than the reference drug sorafenib (MGI% = 105.46%). Five-dose testing also exposed that compound CDHPM-10e with hydrophilic moiety showed the highest potencies over all the herein examined 60 tumour cells with single-digit micromolar GI50 scale and mean GI50 value of 1.83 µM. Notably, the hybrid CDHPM-10e exhibited potent cytostatic effects at a single-digit micromolar range (TGI spanning in the interval: 2.43–9.81 µM) towards 58 tumour cells. All the target analogs were found to be non-lethal towards Leukaemia (CCRF-CEM, HL-60(TB), K-562, MOLT-4, RPMI-8226 and SR), Ovarian (NCI/ADR-RES and OVCAR-4), Breast (HS 578 T and T-47D) and Prostate (PC-3) cancer cells with LC50 > 100 µM. Among the target compounds, CDHPM-10e displayed LC50 values spanning in the interval: 2.04–70.60 µM. The designed compounds CDHPM-10a-g were exposed as potent non-selective broad-spectrum anticancer agents over all NCI subpanels with an SI range of 0.66–1.97. Also, compound CDHPM-10e revealed potency towards KDR comparable to that of sorafenib with a sub-micromolar IC50 of 0.112 µM. Furthermore, CDHPM-10e was more potent than sorafenib, with IC50s of 2.52 and 5.10 µM towards the MCF-7 cell line. The data showed that CDHPM-10e could effectively enhance Sub-G1-stage arrest and prompt apoptosis via caspase and p53-dependent mechanisms. Compound CDHPM-10e revealed significant anti-metastatic activity as detected by wound healing assay. The modelling study implies that CDHPM-10e overlaid well with sorafenib and formed a strong H-bond in the DFG binding domain. The ADMET and Drug-likeness study exposed that compound CDHPM-10e met the criteria of Pfizer. Therefore, the presented novel potent anti-cancer agent merits further devotion in developing VEGFR-2 inhibitors.
Author contributions
Conceptualisation, MAA and MM; methodology, MAA, MM, and SME; compounds synthesis, MAA; software, MAA, MHN, AHA, SME, and HHA; validation, MM, MHN, HHA, AHA, and MAA; formal analysis, MM, MHN, SME, AHA, and MAA; investigation, MAA, HHA, MHN, and AHA; resources, MM, MHN, AHA, and HHA; data curation, MAA, HHA, SME, and MHN; writing—original draft preparation, MM and MAA; writing—review and editing, MM, MAA, and SME; visualisation, MAA, HHA, SME, AHA, and MHN; supervision, MAA; project administration, MM and MAA; funding acquisition, MM and MAA. All authors have read and agreed to the published version of the manuscript.
Supplemental Material
Download PDF (8.4 MB)Acknowledgements
The authors are thankful to the Deanship of Graduate Studies and Scientific Research at Najran University for funding this work under the Elite Funding Program grant code NU/EP/MRC/13/541. The authors would also like to acknowledge OpenEye Scientific Cadence Molecular Sciences, Santa Fe, NM, USA for providing the academic license.
Disclosure statement
The authors report no conflicts of interest.
Data availability statement
Additional data may be requested from the authors.
Additional information
Funding
References
- WHO. Cancer. [cited 2024 Mar 18]. Available from: https://www.who.int/
- WHO. Global cancer burden growing, amidst mounting need for services. [cited 2024 Mar 18]. Available from: https://www.who.int/
- Kirby A, Drummond FJ, Lawlor A, Murphy A. Counting the social, psychological, and economic costs of COVID-19 for cancer patients. Support Care Cancer. 2022;30(11):1–24.
- NCI. Angiogenesis inhibitors. [cited 2024 Mar 18]. Available from: https://www.cancer.gov/
- Shaldam MA, Almahli H, Angeli A, Badi RM, Khaleel EF, Zain-Alabdeen AI, Elsayed ZM, Elkaeed EB, Salem R, Supuran CT, et al. Discovery of sulfonamide-tethered isatin derivatives as novel anticancer agents and VEGFR-2 inhibitors. J Enzym Inhib Med Chem. 2023;38(1):2203389–2203400.
- Haffez H, Elsayed NA, Ahmed MF, Fatahala SS, Khaleel EF, Badi RM, Elkaeed EB, El Hassab MA, Hammad SF, Eldehna WM, et al. Novel N-arylmethyl-aniline/chalcone hybrids as potential VEGFR inhibitors: synthesis, biological evaluations, and molecular dynamic simulations. J Enzym Inhib Med Chem. 2023;38(1):2278022–2278042.
- Modi SJ, Kulkarni VM. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med Drug Discov. 2019;2:100009.
- Guo S, Colbert LS, McGlothen TZ, Gonzalez-Perez RR. Regulation of angiogenesis in human cancer via vascular endothelial growth factor receptor-2 (VEGFR-2). In: Ran S, editor. Tumor angiogenesis. Rijeka: IntechOpen; 2012. p. 27–66.
- Hilbe W, Dirnhofer S, Oberwasserlechner F, Schmid T, Gunsilius E, Hilbe G, Wöll E, Kähler CM. CD133 positive endothelial progenitor cells contribute to the tumour vasculature in non-small cell lung cancer. J Clin Pathol. 2004;57(9):965–969.
- Donnem T, Al-Shibli K, Andersen S, Al-Saad S, Busund L-T, Bremnes RM. Combination of low vascular endothelial growth factor A (VEGF-A)/VEGF receptor 2 expression and high lymphocyte infiltration is a strong and independent favorable prognostic factor in patients with nonsmall cell lung cancer. Cancer. 2010;116(18):4318–4325.
- Parameshwaraiah SM, Shivakumar R, Xi Z, Siddappa TP, Ravish A, Mohan A, Poonacha LK, Uppar PM, Basappa S, Dukanya D, et al. Development of novel indazolyl-acyl hydrazones as antioxidant and anticancer agents that target VEGFR-2 in human breast cancer cells. Chem Biodivers. 2024;21(3):e202301950.
- Guo S, Colbert LS, Fuller M, Zhang Y, Gonzalez-Perez RR. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim Biophys Acta. 2010;1806(1):108–121.
- Li W, Zhang K, Wang W, Liu Y, Huang J, Zheng M, Li L, Zhang X, Xu M, Chen G, et al. Combined inhibition of HER2 and VEGFR synergistically improves therapeutic efficacy via PI3K-AKT pathway in advanced ovarian cancer. J Exp Clin Canc Res. 2024;43(1):56–56.
- Adham SA, Sher I, Coomber BL. Molecular blockade of VEGFR2 in human epithelial ovarian carcinoma cells. Lab Invest. 2010;90(5):709–723.
- He Z, Li B, Rankin GO, Rojanasakul Y, Chen YC. Selecting bioactive phenolic compounds as potential agents to inhibit proliferation and VEGF expression in human ovarian cancer cells. Oncol Lett. 2015;9(3):1444–1450.
- Wang D, Liu K, Li X, Lu G, Xue W ,Qian X, Mohamed O K, Meng F. Design, synthesis, and in vitro and in vivo anti-angiogenesis study of a novel vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitor based on 1,2,3-triazole scaffold. Eur J Med Chem. 2021;211:113083.
- Fontanella C, Ongaro E, Bolzonello S, Guardascione M, Fasola G, Aprile G. Clinical advances in the development of novel VEGFR2 inhibitors. Ann Transl Med. 2014;2(12):123.
- Abdel-Mohsen HT, Omar MA, El Kerdawy AM, Mahmoud AEE, Ali MM, El Diwani HI. Novel potent substituted 4-amino-2-thiopyrimidines as dual VEGFR-2 and BRAF kinase inhibitors. Eur J Med Chem. 2019;179:707–722.
- Rodríguez-Hernández A, Navarro-Villarán E, González R, Pereira S, Soriano-De Castro LB, Sarrias-Giménez A, Barrera-Pulido L, Álamo-Martínez JM, Serrablo-Requejo A, Blanco-Fernández G, et al. Regulation of cell death receptor S-nitrosylation and apoptotic signaling by sorafenib in hepatoblastoma cells. Redox Biol. 2015;6:174–182.
- Mahnashi MH, El-Senduny FF, Alshahrani MA, Abou-Salim MA. Design, synthesis, and biological evaluation of a novel VEGFR-2 inhibitor based on a 1,2,5-oxadiazole-2-oxide scaffold with MAPK signaling pathway inhibition. Pharmaceuticals. 2022;15(2):246–281.
- Hao S-H, Ma X-D, Xu L, Xie J-D, Feng Z-H, Chen J-W, Chen R-X, Wang F-W, Tang Y-H, Xie D, et al. Dual specific phosphatase 4 suppresses ferroptosis and enhances sorafenib resistance in hepatocellular carcinoma. Drug Resist Update. 2024;73:101052.
- El-Metwally SA, Abou-El-Regal MM, Eissa IH, Mehany ABM, Mahdy HA, Elkady H, Elwan A, Elkaeed EB. Discovery of thieno[2,3-d]pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg Chem. 2021;112:104947.
- Yousef EH, Abo El-Magd NF, El Gayar AM. Carvacrol enhances anti-tumor activity and mitigates cardiotoxicity of sorafenib in thioacetamide-induced hepatocellular carcinoma model through inhibiting TRPM7. Life Sci. 2023;324:121735.
- EON 2.4.1.1: OpenEye. Santa Fe (NM): Cadence Molecular Sciences. [cited 2023 Jan 11]. Available from: http://www.eyesopen.com
- OpenEye: Lead Optimization EON. [cited 2023 Jan 10]. Available from: https://www.eyesopen.com/
- El-Kashef H, Badr G, Abo El-Maali N, Sayed D, Melnyk P, Lebegue N, Abd El-Khalek R. Synthesis of a novel series of (Z)-3,5-disubstituted thiazolidine-2,4-diones as promising anti-breast cancer agents. Bioorg Chem. 2020;96:103569.
- Taghour MS, Elkady H, Eldehna WM, El-Deeb NM, Kenawy AM, Elkaeed EB, Alsfouk AA, Alesawy MS, Metwaly AM, Eissa IH. Design and synthesis of thiazolidine-2,4-diones hybrids with 1,2-dihydroquinolones and 2-oxindoles as potential VEGFR-2 inhibitors: in-vitro anticancer evaluation and in-silico studies. J Enzym Inhib Med Chem. 2022;37(1):1903–1917.
- Upadhyay N, Tilekar K, Safuan S, Kumar AP, Schweipert M, Meyer-Almes FJ, Ramaa CS. Multi-target weapons: diaryl-pyrazoline thiazolidinediones simultaneously targeting VEGFR-2 and HDAC cancer hallmarks. RSC Med Chem. 2021;12(9):1540–1554.
- Talath S, Wali AF, Nisar B, Wani HA, Mushtaq S, Rehman MU. Chapter 8 – novel dihydropyrimidine as potential cytotoxic agents in hepatic and blood cancers. In: Ahmad Bhat M, Rehman MU, Verma A, editors. Dihydropyrimidinones as potent anticancer agents. MA: Elsevier; 2023. p. 199–222.
- Naishima NL, Faizan S, Raju RM, Sruthi ASVL, Ng V, Sharma GK, Vasanth KS, Shivaraju VK, Ramu R, Kumar BRP. Design, synthesis, analysis, evaluation of cytotoxicity against MCF-7 breast cancer cells, 3D QSAR studies and EGFR, HER2 inhibition studies on novel Biginelli 1,4-dihydropyrimidines. J Mol Struct. 2023;1277:134848.
- Wazalwar SS, Banpurkar AR, Perdih F. Synthesis, crystal structure and molecular docking study of new monastrol analogues as inhibitors of epidermal growth factor receptor tyrosine kinase. J Mol Struct. 2023;1274:134508.
- Nikam D, Jain A. Advances in the discovery of DHPMs as Eg5 inhibitors for the management of breast cancer and glioblastoma: a review. Results Chem. 2023;5:100718.
- Marzouk AA, Abdel-Aziz SA, Abdelrahman KS, Wanas AS, Gouda AM, Youssif BGM, Abdel-Aziz M. Design and synthesis of new 1,6-dihydropyrimidin-2-thio derivatives targeting VEGFR-2: molecular docking and antiproliferative evaluation. Bioorg Chem. 2020;102:104090.
- Li T, Li W, Yang X, Chen G, Jin X, Chen W, Ye L. Design, synthesis, anticancer evaluation and in silico studies of 2,4,6-trimethoxychalcone derivatives. Saudi Pharm J. 2023;31(1):65–84.
- Rao YK, Fang S-H, Tzeng Y-M. Synthesis and biological evaluation of 3′,4′,5′-trimethoxychalcone analogues as inhibitors of nitric oxide production and tumor cell proliferation. Bioorg Med Chem. 2009;17(23):7909–7914.
- WalyEldeen AA, Sabet S, El-Shorbagy HM, Abdelhamid IA, Ibrahim SA. Chalcones: promising therapeutic agents targeting key players and signaling pathways regulating the hallmarks of cancer. Chem Biol Interact. 2023;369:110297.
- Michalkova R, Kello M, Cizmarikova M, Bardelcikova A, Mirossay L, Mojzis J. Chalcones and gastrointestinal cancers: experimental evidence. Int J Mol Sci. 2023;24(6):5964.
- Jesus A, Durães F, Szemerédi N, Freitas-Silva J, da Costa PM, Pinto E, Pinto M, Spengler G, Sousa E, Cidade H. BDDE-inspired chalcone derivatives to fight bacterial and fungal infections. Mar Drugs. 2022;20(5):315.
- Ahmed MF, Santali EY, El-Haggar R. Novel piperazine–chalcone hybrids and related pyrazoline analogues targeting VEGFR-2 kinase; design, synthesis, molecular docking studies, and anticancer evaluation. J Enzym Inhib Med Chem. 2021;36(1):308–319.
- Liu M, Zhang W, Wei J, Qiu L, Lin X. Marine bromophenol bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, induces mitochondrial apoptosis in K562 cells and inhibits topoisomerase I in vitro. Toxicol Lett. 2012;211(2):126–134.
- Qi X, Liu G, Qiu L, Lin X, Liu M. Marine bromophenol bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, represses angiogenesis in HUVEC cells and in zebrafish embryos via inhibiting the VEGF signal systems. Biomed Pharmacother. 2015;75:58–66.
- Šlachtová V, Šebela M, Torfs E, Oorts L, Cappoen D, Berka K, Bazgier V, Brulíková L. Novel thiazolidinedione-hydroxamates as inhibitors of Mycobacterium tuberculosis virulence factor Zmp1. Eur J Med Chem. 2020;185:111812.
- Meirelles LV, de Castro PP, Passos STA, Carvalho BBPP, Franco CHJ, Correa JR, Neto BAD, Amarante GW. Diverse 3-methylthio-4-substituted maleimides through a novel rearrangement reaction: synthesis and selective cell imaging. J Org Chem. 2022;87(5):2809–2820.
- Thari FZ, Tachallait H, El Alaoui N-E, Talha A, Arshad S, Álvarez E, Karrouchi K, Bougrin K. Ultrasound-assisted one-pot green synthesis of new N-substituted-5-arylidene-thiazolidine-2,4-dione-isoxazoline derivatives using NaCl/Oxone/Na3PO4 in aqueous media. Ultrason Sonochem. 2020;68:105222.
- Huan CL, Pham-The H, Le-Thi-Thu H, Thao PT, Que ND, Trang N-T, Phuong Dung TP, Pyo M, Han S-B, Thuan TN, et al. Exploration of some thiazolidine-2,4-dione and 2-oxoindoline derivatives incorporating 3,4,5-trimethoxybenzyl moiety as novel anticancer agents. Lett Drug Des Discov. 2018;15(4):375–387.
- Ha YM, Park YJ, Kim J-A, Park D, Park JY, Lee HJ, Lee JY, Moon HR, Chung HY. Design and synthesis of 5-(substituted benzylidene)thiazolidine-2,4-dione derivatives as novel tyrosinase inhibitors. Eur J Med Chem. 2012;49:245–252.
- Omar MT, Fouli A-E, El-Garhi MZ. Studies on 4-thiazolidinones. IX, the conversion of 5-substituted 2-thioxo-4-thiazolidinones into corresponding 2,4-thiazolidinediones. B Chem Soc Jpn. 2006;64(2):750–752.
- Kappe CO. The Biginelli reaction. In: Zhu J, Bienaymé H, editors. Multicomponent reactions. Weinheim: Wiley-VCH; 2005. p. 95–120.
- Kappe CO. Recent advances in the Biginelli dihydropyrimidine synthesis. New tricks from an old dog. Acc Chem Res. 2000;33(12):879–888.
- Wang Y, Yang H, Yu J, Miao Z, Chen R. Highly enantioselective Biginelli reaction promoted by chiral bifunctional primary amine-thiourea catalysts: asymmetric synthesis of dihydropyrimidines. Adv Synth Catal. 2009;351(18):3057–3062.
- Oliver Kappe C. 100 Years of the Biginelli dihydropyrimidine synthesis. Tetrahedron. 1993;49(32):6937–6963.
- Kappe CO, Roschger P. Synthesis and reactions of “Biginelli-compounds”. Part I. J Heterocycl Chem. 1989;26(1):55–64.
- Sherif SM, Youssef MM, Mobarak KM, Abdel-Fattah A-SM. A convenient synthesis of thiazolopyrimidines, thiazolodipyrimidines and heterocyclothiazolopyrimidines. Tetrahedron. 1993;49(42):9561–9572.
- Patel S, Challagundla N, Rajput RA, Mishra S. Design, synthesis, characterization and anticancer activity evaluation of deoxycholic acid-chalcone conjugates. Bioorg Chem. 2022;127:106036–106044.
- Dizdaroglu Y, Albay C, Arslan T, Ece A, Turkoglu EA, Efe A, Senturk M, Supuran CT, Ekinci D. Design, synthesis and molecular modelling studies of some pyrazole derivatives as carbonic anhydrase inhibitors. J Enzym Inhib Med Chem. 2020;35(1):289–297.
- Fu D-J, Li J-H, Yang J-J, Li P, Zhang Y-B, Liu S, Li Z-R, Zhang S-Y. Discovery of novel chalcone-dithiocarbamates as ROS-mediated apoptosis inducers by inhibiting catalase. Bioorg Chem. 2019;86:375–385.
- Uniyal A, Thakur BK, Singh S, Gairolla J, Anthwal A. Synthesis, in-silico antiviral screening and in-vitro antibacterial screening of a novel chalcone derivatives. J Indian Chem Soc. 2023;100(7):101038.
- Lu C-F, Wang S-H, Pang X-J, Zhu T, Li H-L, Li Q-R, Li Q-Y, Gu Y-F, Mu Z-Y, Jin M-J, et al. Synthesis and biological evaluation of amino chalcone derivatives as antiproliferative agents. Molecules. 2020;25(23):5530.
- Alvim HGO, Pinheiro DLJ, Carvalho-Silva VH, Fioramonte M, Gozzo FC, da Silva WA, Amarante GW, Neto BAD. Combined role of the asymmetric counteranion-directed catalysis (ACDC) and ionic liquid effect for the enantioselective Biginelli multicomponent reaction. J Org Chem. 2018;83(19):12143–12153.
- Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigro-Wolff A. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer I. 1991;83(11):757–766.
- Boyd MR, Paull KD. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev Res. 1995;34(2):91–109.
- NCI-60 screening methodology. [cited 2024 Mar 15]. Available from: https://dtp.cancer.gov/
- NCI-DTP: Search for all data by compound identifiers. [cited 2023 Oct 7]. Available from: https://dtp.cancer.gov/
- Al-Warhi T, Abo-Ashour MF, Almahli H, Alotaibi OJ, Al-Sanea MM, Al-Ansary GH, Ahmed HY, Elaasser MM, Eldehna WM, Abdel-Aziz HA. Novel [(N-alkyl-3-indolylmethylene)hydrazono]oxindoles arrest cell cycle and induce cell apoptosis by inhibiting CDK2 and Bcl-2: synthesis, biological evaluation and in silico studies. J Enzym Inhib Med Chem. 2020;35(1):1300–1309.
- Allam RM, Al-Abd AM, Khedr A, Sharaf OA, Nofal SM, Khalifa AE, Mosli HA, Abdel-Naim AB. Fingolimod interrupts the cross talk between estrogen metabolism and sphingolipid metabolism within prostate cancer cells. Toxicol Lett. 2018;291:77–85.
- Henriksen M, Miller B, Newmark J, Al-Kofahi Y, Holden E. Chapter 7 – laser scanning cytometry and its applications: a pioneering technology in the field of quantitative imaging cytometry. In: Darzynkiewicz Z, Holden E, Orfao A, Telford W, Wlodkowic D, editors. Methods in cell biology. Vol. 102. USA: Academic Press; 2011. p. 159–205.
- Gray JW, Dolbeare F, Pallavicini MG, Beisker W, Waldman F. Cell cycle analysis using flow cytometry. Int J Radiat Biol. 1986;49(2):237–255.
- El-Dash Y, Elzayat E, Abdou AM, Hassan RA. Novel thienopyrimidine-aminothiazole hybrids: design, synthesis, antimicrobial screening, anticancer activity, effects on cell cycle profile, caspase-3 mediated apoptosis and VEGFR-2 inhibition. Bioorg Chem. 2021;114:105137.
- Huang R, Chen XQ, Huang Y, Chen N, Zeng H. The multikinase inhibitor sorafenib induces caspase-dependent apoptosis in PC-3 prostate cancer cells. Asian J Androl. 2010;12(4):527–534.
- Abdallah AE, Mabrouk RR, Elnagar MR, Farrag AM, Kalaba MH, Sharaf MH, El-Fakharany EM, Bakhotmah DA, Elkaeed EB, Al Ward MMS. New series of VEGFR-2 inhibitors and apoptosis enhancers: design, synthesis and biological evaluation. Drug Des Dev Ther. 2022;16:587–606.
- VIDA 5.0.3.1: OpenEye. Santa Fe (NM): Cadence Molecular Sciences. [cited 2023 Jan 10]. Available from: http://www.eyesopen.com
- Patil SM, Maruthi KR, Bajpe SN, Vyshali VM, Sushmitha S, Akhila C, Ramu R. Comparative molecular docking and simulation analysis of molnupiravir and remdesivir with SARS-CoV-2 RNA dependent RNA polymerase (RdRp). Bioinformation. 2021;17(11):932–939.
- BIOVIA Discovery Studio Visualizer. [cited 2023 Dec 2]. Available from: https://discover.3ds.com/
- Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, Yin M, Zeng X, Wu C, Lu A, et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49(W1):W5–W14.
- Computational Biology & Drug Design Group: ADMETLAB2.0. [cited 2023 Nov 6]. Available from: https://admetmesh.scbdd.com/
- Manikala VK, Rao VM. Synthesis and anticancer activity of (E)-5-[(1-aryl-1H-1,2,3-triazol-4-yl)methylene]thiazolidine-2,4-diones. Russ J Org Chem. 2020;56(5):863–868.
- Asati V, Bharti SK. Design, synthesis and molecular modeling studies of novel thiazolidine-2,4-dione derivatives as potential anti-cancer agents. J Mol Struct. 2018;1154:406–417.
- Elbadawi MM, Eldehna WM, Wang W, Agama KK, Pommier Y, Abe M. Discovery of 4-alkoxy-2-aryl-6,7-dimethoxyquinolines as a new class of topoisomerase I inhibitors endowed with potent in vitro anticancer activity. Eur J Med Chem. 2021;215:113261–113281.
- Mahnashi M, Alshahrani MM, Al Ali A, Asiri A, Abou-Salim MA. Novel Glu-based pyrazolo[3,4-d]pyrimidine analogues: design, synthesis and biological evaluation as DHFR and TS dual inhibitors. J Enzym Inhib Med Chem. 2023;38(1):2203879.
- Abou-Salim MA, Shaaban MA, Abd El Hameid MK, Elshaier YAMM, Halaweish F. Design, synthesis and biological study of hybrid drug candidates of nitric oxide releasing cucurbitacin-inspired estrone analogs for treatment of hepatocellular carcinoma. Bioorg Chem. 2019;85:515–533.
- Elshaier YAMM, Shaaban MA, Abd El Hamid MK, Abdelrahman MH, Abou-Salim MA, Elgazwi SM, Halaweish F. Design and synthesis of pyrazolo[3,4-d]pyrimidines: nitric oxide releasing compounds targeting hepatocellular carcinoma. Bioorg Med Chem. 2017;25(12):2956–2970.
- Fekry MI, Ezzat SM, Salama MM, Alshehri OY, Al-Abd AM. Bioactive glycoalkaloides isolated from Solanum melongena fruit peels with potential anticancer properties against hepatocellular carcinoma cells. Sci Rep. 2019;9(1):1746.
- Sharma K, Suresh P, Mullangi R, Srinivas N. Quantitation of VEGFR2 (vascular endothelial growth factor receptor) inhibitors–review of assay methodologies and perspectives. Biomed Chromatogr. 2015;29(6):803–834.
- Bioscience: VEGFR2 (KDR) Kinase Assay Kit. [cited 2021 Dec 5]. Available from: https://bpsbioscience.com/
- Bioscience: FLT1 Kinase Assay Kit. [cited 2021 Dec 5]. Available from: https://bpsbioscience.com/
- Bioscience: FLT3 Kinase Assay Kit. [cited 2021 Dec 5]. Available from: https://bpsbioscience.com/
- Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290.
- OMEGA 4.2.1.1: OpenEye. Santa Fe (NM): Cadence Molecular Sciences. [cited 2023 Jan 14]. Available from: http://www.eyesopen.com
- Spruce-1.5.2.1: OpenEye. Santa Fe (NM): Cadence Molecular Sciences. [cited 2023 Jan 10]. Available from: http://www.eyesopen.com
- FRED 4.2.1.1: OpenEye. Santa Fe (NM): Cadence Molecular Sciences. [cited 2023 Jan 15]. Available from: http://www.eyesopen.com
- Dock report utility: OpenEye. Santa Fe (NM): Cadence Molecular Sciences. [cited 2023 Jan 15]. Available from: http://www.eyesopen.com