
ABSTRACT
Cold-chain requirements, limited stockpiling potential and the lack of potent immune responses are major challenges of parenterally formulated influenza vaccines. Decreased cold chain dependence and stockpiling can be achieved if vaccines are formulated in a dry state using suitable excipients and drying technologies. Furthermore, having the vaccine in a dry state enables the development of non-parenteral patient friendly dosage forms: microneedles for transdermal administration, tablets for oral administration, and powders for epidermal, nasal or pulmonary administration. Moreover, these administration routes have the potential to elicit an improved immune response. This review highlights the rationale for the development of dried influenza vaccines, as well as processes used for the drying and stabilization of influenza vaccines; it also compares the immunogenicity of dried influenza vaccines administered via non-invasive routes with that of parenterally administered influenza vaccines. Finally, it discusses unmet needs, challenges and future developments in the field of dried influenza vaccines.
Introduction
Parenteral vaccination against influenza is the gold standard for controlling dissemination of the disease. Low costs per dosage unit and ease of formulation still renders it suitable for mass vaccination programs all over the world. However, current seasonal and pandemic influenza vaccines need to be stored and distributed at refrigerated temperatures (2–8°C); the so-called cold chain must be applied to ensure product stability and prevent antigen degradation. Stabilization of the antigen by drying with suitable excipients would greatly improve storage stability. The restricted molecular mobility in the dry state may preserve the conformational and structural integrity of the antigen which could make the cold chain superfluous [Citation1,Citation2]. In addition, improved storage stability can greatly enhance stockpile potential in case of a pandemic outbreak.
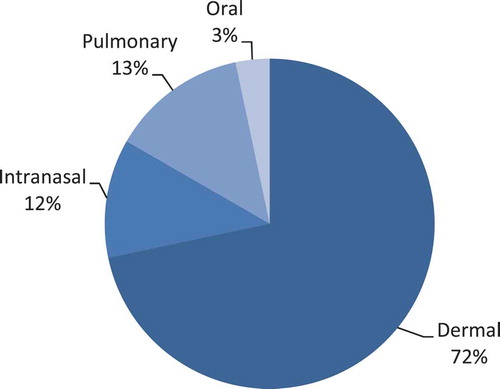
Drying techniques like spray drying [Citation3–Citation5], freeze drying [Citation6–Citation8], spray freeze drying [Citation9–Citation11] and air or vacuum drying [Citation12,Citation13] can be used in combination with suitable excipients not only to improve antigen stability but also to provide a solid carrier or vesicle to reach the desired target site, depending upon the route of administration. The currently preferred injection site for influenza vaccination is the deltoid muscle. Since muscle tissue has a low number of antigen presenting cells (APCs) and lacks major histocompatibility complex class II cells, its potential to induce potent humoral and cellular immune responses is limited [Citation14]. Further, passive drainage of the antigen to lymph nodes might require high antigen doses, thereby posing the risk of shortage in case of pandemics. Alternative routes that target areas rich in APCs like mucosal surfaces and the skin might be better target sites for influenza vaccination. Vaccines could, for example, be in the form of a dry powder with a certain particle size, shape, and density that targets a specific area in the lungs [Citation15], nasal cavity [Citation12], or dermis [Citation9] (pulmonary, nasal, and epidermal powder immunization [EPI]); or a film-coated [Citation16] or dissolvable matrix [Citation17] to target the skin (dermal vaccination); or a tablet to target the sublingual (s.l.) [Citation18], buccal [Citation19], and gut regions [Citation20] (oral vaccination). To be more specific, these alternative forms could target immune cells resident in these areas to provide a more potent humoral and cellular immune response than currently achieved by conventional parenteral vaccination. The use of new routes could lead to reduction of doses, improve the efficacy of the vaccine, and make possible simple and patient-friendly ways to execute influenza vaccination. presents an overview of the publications found on dry influenza vaccines per route of administration expressed as percentage. This review gives an overview of current developments in dry influenza vaccines, including their drying techniques and various alternative routes of administration. We will compare these formulations with standard parenteral influenza vaccines in terms of stability and efficacy. Finally, we will share a perspective on these dry influenza vaccines and their possibilities to improve current influenza vaccination practices.
Figure 1. Publications found on dry influenza vaccines per route of administration expressed as percentage. Numbers are based on literature found from 2000 to 2015 using Embase and Pubmed.
Commonly used drying techniques for influenza vaccines
Typical drying processes use convection, conduction, or radiation (infrared) as methods of heat transfer [Citation21]. Pharmaceuticals such as antigens in influenza vaccine formulations are often prone to degradation due to heat, cooling, or freezing, as well as shear and dehydration stresses caused by the drying process. To prevent degradation during the drying process and improve storage stability at room temperature, one can use stabilizing excipients like polysaccharides, such as inulin or dextran, and disaccharides, such as trehalose [Citation22–Citation24]. During drying hydroxyl groups of the saccharides replace the hydrogen bonds of water surrounding the antigen, thereby preserving the protein’s three-dimensional structure. Furthermore, upon drying the antigen is also stabilized by vitrification when the saccharide forms a glassy matrix around the antigen [Citation25–Citation27]. Therefore, one should select a saccharide with a high glass transition temperature (Tg) as the residual moisture can strongly decrease the Tg because of the plasticizing effects of water. Since antigens in influenza vaccines are usually given in very low doses (only a few micrograms), the saccharides can also be used as a bulking agent. The most frequently used methods for drying influenza vaccines are spray drying, (spray)freeze drying, and air drying or vacuum drying. We will therefore discuss these techniques in the sections below.
Spray drying
Spray drying is a well-established technique to produce dry powders. In general, a pumpable liquid or solution (also called the feed material) is atomized into small droplets by a nozzle. Atomization is the process by which liquid is broken up into fine droplets (usually in a micrometer range). The droplets containing the antigen are sprayed under atomization into the drying-chamber where they come in contact with a stream of dry hot air, resulting in the evaporation of liquid to form dry particles [Citation28,Citation29]. The atomization of the liquid, the subsequent drying of the droplet, and the effect of the outlet temperature will induce shear, dehydration, and heat stress, respectively, thereby possibly resulting in degradation and loss of potency of the antigen. For this reason, stabilizing excipients like (poly)saccharides are used during the spray drying process [Citation30]. In most cases, the spray-dried particles are separated from the stream of air by a cyclone and collected in the attached vial. However, collection by bag filters or electrostatic precipitation is also used [Citation31,Citation32]. Often spherically shaped hollow particles with a shell are obtained, which have a lower density than solid particles. A spray dried solid particle usually looks like a raisin due to its wrinkled appearance [Citation33]. Since the size, shape, and density of a dry particle play an important role in pulmonary, epidermal, and (to a lesser extent) intranasal (i.n.) dry powder immunization it is important to understand the factors that influence these characteristics. The relation between particle characteristics and administration routes will be discussed later in this review.
(Spray)freeze drying
While spray drying utilizes heat to dry the desired product, freeze drying utilizes a partial vacuum to dry the product while in the frozen state. In general, a liquid formulation containing the solute(s) (e.g. antigen, saccharide, salts, and buffer components) and a suitable solvent (usually water) is first completely solidified by freezing. During the freezing of an aqueous solution, water will start to crystallize into ice crystals that form a matrix among the remaining solution. Due to the presence of solutes the remaining liquid water will start to crystallize at lower temperature due to freezing point depression. Upon further cooling, more water will crystallize and the remaining solution will become more concentrated until the glass transition temperature of the maximally freeze-concentrated fraction (Tg’) will be reached and water will no longer crystallize. Instead, the remaining maximally freeze-concentrated solution will form a glass [Citation34]. To obtain a product in the glassy state, cooling should be fast enough to prevent crystallization of the saccharide. The liquid formulation is usually frozen on the shelf of the freeze dryer at a temperature of ‒20 to ‒100°C, but can also be snap frozen outside the freeze dryer, for example in liquid nitrogen. Snap freezing is also used during spray freeze drying where the solution is atomized (with a technology similar to that used in spray drying) after which the formed droplets are collected and frozen into liquid nitrogen [Citation35] or onto a cold surface [Citation36]. The drying process is initiated by lowering the pressure in the chamber of the freeze dryer to a partial vacuum (microbar range). The drying process consists of two stages, namely primary and secondary drying. During primary drying, ice crystals will sublimate from the frozen formulation. During this process, to prevent crystallization of the saccharide, the temperature of the frozen solution should not exceed the Tg’. Once all the ice crystals have been sublimated from the frozen formulation, the primary drying process is completed and secondary drying starts. During secondary drying, water evaporates from the maximally freeze-concentrated fraction, the pressure in the freeze drying chamber is further lowered (usually by a factor of 10) and the temperature is gradually increased. The secondary drying phase is completed when the desired residual moisture content is achieved to ensure product stability. With conventional freeze drying, the product consists of a porous cake, and with spray freeze drying, it consists of porous spherical particles [Citation37].
Air drying, nitrogen purging, and vacuum drying
More simplistic approaches to achieve a dry vaccine product are by air drying, nitrogen purging, or vacuum drying. Usually the product is first air dried or dried in a stream of inert gas, for example nitrogen, and then, if the product needs further drying, it is often subjected to a partial vacuum. For this purpose an airtight container like a desiccator can be used. A stopcock attached to the desiccator can be connected to a vacuum pump to apply a partial vacuum. A highly hygroscopic material (like silica gel) on the bottom of the desiccator absorbs the evaporated water from the product. During these drying processes, the formulation remains for a substantial period of time (h) in the rubbery state before it is vitrified. This might be detrimental to the antigen for two reasons [Citation38,Citation39]. First, during drying in this state, the solution becomes more concentrated while the antigen is not immobilized. This may easily cause changes in the three-dimensional structure of the antigen, or aggregation with loss of potency as a result. Second, the saccharide may crystallize, thereby fully losing its stabilizing effects [Citation40]. These drying methods may therefore not be most suitable for obtaining a stable dry vaccine formulation.
Routes of administration
Transdermal dry influenza vaccine delivery
The barrier property of the outermost layer of the skin that is stratum corneum (10–20 µm) protects the body from the surrounding environment and prevents the entry of pathogens. On the one hand, the barrier function prevents antigen uptake in or through the skin. This explains the need to apply an administration technique that penetrates the stratum corneum to obtain adequate antigen delivery. On the other hand, the abundance of large numbers of diverse immune cells like epidermal Langerhans cells (LC) and dermal dendritic cells (DC) make the skin a highly suitable immunological organ for vaccination. Both LC and DC serve as immune responsive APCs, which are involved in the uptake and presentation of pathogen-derived antigens to naïve B and T cells, hence inducing an adaptive immune response [Citation14,Citation41].
EPI as a dry influenza vaccination method
Dry influenza vaccines can be used for dermal vaccination by ballistic powder delivery or EPI. Elongated tubular devices or ballistic injectors like the PowderJect use compressed sterile helium gas to fire the dry powder vaccine from a compartment or cassette through a nozzle into the skin. These dry powders can be produced by conventional drying methods like spray drying, or freeze drying or purging with nitrogen gas, followed by desiccation and grinding to achieve the desired particle size. The desired particle size depends on the particle density and can range from 0.1 to 250 µm [Citation42]. Key factors determining the depth of penetration into the (epi)dermis (about 100–500 µm) are particle size, density, shape, and velocity. Improved designs of epidermal injectors focus mainly on creating high uniform particle velocities, which are necessary to ensure deposition of the particles within the dermis. Dry powder influenza vaccines suitable for epidermal powder delivery are usually dried with polysaccharides or disaccharides to improve stability [Citation43,Citation44]. As a result of this drying process, dry powder vaccines most often have a low particle density. To compensate for this low density, the dry powder particles need to be relatively large in size (20–60 µm) and dispersed at high velocities (300–1000 m/s) in order to penetrate the skin at the desired depth [Citation42]. However, some studies have shown to improve particle density by increasing the solid content of the spray freeze dried feed material, and utilize the promotion of particle shrinkage by using different excipients [Citation44]. Over the past 15 years, EPI against influenza has been investigated in preclinical studies as well as in a clinical study and compared with conventional liquid injections.
Chen et al. showed that administration of 25, 5, and 1 µg of whole inactivated virus (WIV) by EPI to mice induced significantly higher antibody titers than did parenteral administration () [Citation45]. Furthermore, EPI conferred 100% protection against lethal virus for all three doses whereas administration by conventional subcutaneous (s.c.) injection elicited only partial protection: 75% and 62.5% survival at a dose of 25 µg and 5 µg, respectively, and no survival at a dose of 1 µg [Citation45]. Likewise, in another mice study, EPI induced higher antibody titers than did liquid vaccine administered intramuscularly [Citation46]. However, in monkeys (rhesus macaques), unadjuvanted formulations elicited similar antibody titers by both intramuscular and epidermal routes [Citation46]. Nonetheless, the co-administration of adjuvant quillaja saponins-21 (QS-21) with the same influenza vaccine to monkeys by EPI elicited higher antibody titers than did only antigen or intramascular (i.m.) administration [Citation46].
Table 1. An overview of stability and immunogenicity of dry influenza vaccines described in literature.
Several other adjuvants like CpG oligonucleotide (CpG ODN), cholera toxin, cholera toxin B subunit, and bacterial toxin mutants (LTR72 or LTR63) coadministered with trivalent influenza vaccine were found to enhance serum antibody titers after EPI in mice [Citation46–Citation50,Citation85].
The mechanisms behind the enhanced immune response elicited by EPI were investigated by depleting epidermal LC and transfer of the LC isolated from the EPI sites to naïve mice [Citation50]. It was found that the depletion of LC caused a significant reduction in antibody responses whereas transfer to naïve mice induced robust antigen specific antibody responses [Citation50]. These results provided direct evidence that LC function as APCs following EPI to evoke an immune response.
In a phase 1 clinical trial conducted in healthy adults, EPI with trivalent influenza vaccine was shown to elicit strong antibody responses and high seroconversion rates that were equivalent or higher than in the i.m. group [Citation9].
The clinical trial and preclinical studies have shown the potential of EPI vaccination against influenza as it produces immune responses similar to or better than vaccination via the parenteral route.
Microneedle-mediated dry influenza vaccination
In the last decade, based on the aforementioned immunological properties of the skin, dry influenza vaccine delivery by microneedles has been extensively investigated. Administration via microneedles is also an attractive alternative to conventional hypodermic needles because it is a painless delivery system due to the relatively short needle length (about 200–700 µm), which does not reach the nerve endings. Different approaches have been used to deliver dry solid influenza vaccines using microneedles. One approach is to coat a solution containing the vaccine onto the outer surface of non-dissolving microneedles. Non-dissolving microneedles for influenza vaccination are usually made of materials such as stainless steel, titanium, silicon, or glass and manufactured by a chemical etching process, a strong cutting laser, or electropolishing [Citation86]. The coating is applied by dipping a small array of microneedles into a coating solution and then drying by an air or vacuum drying. A coating solution usually consists of a stabilizing saccharide like trehalose to prevent the antigen from losing its activity during drying/storage, a viscosity enhancer like carboxymethylcellulose (CMC) to eventually obtain a coating of sufficient thickness, and a wetting agent or surfactant like poloxamer to reduce the surface tension of the coating solution and to ensure uniform coating efficiency ((a)).
Figure 2. (a) Formulation excipients required for the production of dry, stable influenza vaccines per route of administration. (b) A general overview of advantages and disadvantages associated with non-parenteral routes used for influenza vaccine immunization in preclinical and clinical studies.
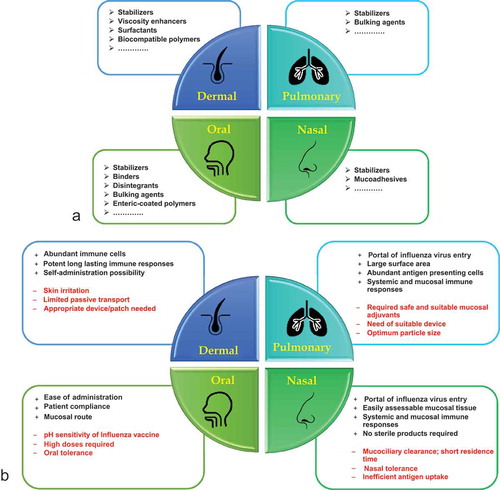
Each of these excipients has a unique potential to influence the stability of various influenza vaccines. For example, a high dose (10 µg) of WIV (A/PR8 H1N1) was coated onto solid microneedles in the absence of trehalose in the coating solution [Citation54]. The magnitude of immune response and protection against viral challenge elicited by coated microneedles was equivalent to i.m. immunization in mice [Citation54]. In addition, 3–10 µg of H3N2 WIV (A/Aichi H3N2) delivered to the skin induced a level of immune response similar to that after i.m. administration [Citation51]. The need for such high doses of influenza vaccine was hypothesized due to the stability issues arising from the sudden phase change of the vaccine from a liquid to a dry state [Citation52,Citation55]. Consequently, efforts were made to stabilize influenza vaccines coated on microneedles and the stabilization was speculated to play a role in dose sparing.
It was found that low dose (0.4 µg) of trehalose stabilized WIV (A/PR8 H1N1) administered to mice using microneedles, resulted in better viral protection and generation of rapid recall immune responses superior to i.m. formulations at the same dose () [Citation52,Citation53]. Similarly, a low dose (0.3–2 µg) of stabilized virus-like particles (VLPs) (A/PR8 H1N1, A/Vietnam H5N1) coated on microneedles and administered to mice showed superior Th1 responses [Citation55], potent recall responses and complete protection (100%) as compared to partial protection (≤40%) by intramuscularly immunized mice [Citation55–Citation57]. Notably, it has been shown that stabilized influenza VLPs could provide complete protection at a threefold lower dose than that of unstabilized influenza VLPs [Citation55]. Hence, the stabilization combined with skin vaccination using microneedles has potential to elicit strong antibody titers, superior Th1 responses and rapid recall immune responses with a low dose. This plays a vital role in providing microneedle-mediated superior protection. Dose sparing could also be achieved by the use of the Nanopatch (NP), a patch with densely packed (~21,000 microprojections/cm2 compared to ~≤321 microprojections/cm2 of microneedles) coated microprojections of shorter length (~110 µm compared to 700 µm microneedles) designed to target thousands of skin APCs [Citation60]. Chen et al. reported that the commercial trivalent split vaccine (Fluvax®) coated onto NP administered to mice is able to elicit comparable protective immune responses comparable to those found after administration via the i.m. route, but with a dose 30 times lower. Moreover, the NP was found to be stable for 6 months at room temperature, providing immunogenicity comparable to that of freshly prepared patches [Citation60]. Later, the co-delivery of trivalent split influenza vaccine and saponin Quil-A adjuvant (6.5 ng of vaccine and 1.4 µg of adjuvant) coated on NP applied to mice induced IgG antibody and hemagglutination inhibition (HAI) titers that were similar in magnitude to conventional i.m. injection (6000 ng of vaccine without adjuvant), but with a 900-fold lower dose [Citation62].
The long-term stability of coated microneedles is also a critical factor influencing the immunogenicity of the vaccine. Kim et al. investigated the influence of storage time on the immunogenicity of WIV coated on the microneedles [Citation67]. After 4 weeks of storage at room temperature, stabilized microneedles induced high antibody titers and protected mice from lethal viral challenge [Citation67]. Mechanistic studies on degradation during storage revealed a direct correlation between the degree of time-dependent phase transformation (crystallization and phase separation) of vaccine coating and hemagglutinin (HA) activity [Citation13]. Vaccine-coated microneedles stored at room temperature for 4 months were found to be phase transformed and poorly immunogenic as compared to fresh-coated formulations [Citation13]. Further, osmotic stresses during drying result in destabilization of WIV indicating the need for viscosity enhancers [Citation68,Citation74]. Viscosity enhancers like CMC also seem to play an important role in diminishing the surfactant-induced phase transformations of the vaccine coating, thus preserving antigen stability [Citation74]. Hence, the presence of viscosity enhancer augmented vaccine-specific systemic immune responses and provided better protection against viral challenge [Citation74].
The choice of excipients also depends on the nature and type of influenza vaccine. Microneedles were coated with H5 influenza HA encoding DNA vaccine using a viscosity enhancer and a surfactant. Mice vaccinated with coated microneedles elicited higher levels of antibody, HAI titers and a better viral protection than those vaccinated with conventional i.m. injection of the similar DNA vaccine, still the protection elicited by microneedles was only partial [Citation66]. The partial protection was attributed to the viscosity enhancer which diminished the expression efficiency of the DNA vaccine, thereby reducing its immunogenicity [Citation66]. Due to the inherent stability and viscosity of DNA vaccines, they have also been coated onto microneedles without additional excipients like stabilizers or viscosity enhancers. Microneedle-based skin delivery of HA encoding DNA vaccine (excipient-free coating) induced potent humoral, cellular, memory responses and better protection than did i.m. immunization [Citation65]. In another study, the co-delivery approach of WIV (A/PR8) and DNA vaccine encoding for HA (A/PR8) was investigated to achieve cross-protective immune responses against heterologous influenza strains (pandemic 2009 H1N1) [Citation69]. Due to the high viscosity of DNA vaccines, it played a dual role as an immunogen and a viscosity enhancer. Furthermore, sugar was incorporated in the formulation to stabilize WIV and its HA activity was fully maintained. The co-immunization of DNA vaccine together with WIV by microneedles generated both homologous (A/PR8) and heterologous (A/California/2009) immune responses in mice comparable to or better than i.m. vaccination [Citation69].
The long-term protective efficacy of different influenza vaccines coated on microneedles was investigated by viral challenge several weeks or months after vaccination. Microneedle-mediated or s.c. delivery of WIV in mice exhibited similar antibody titers and complete protection against viral challenge 6 weeks after vaccination [Citation61]. Six months post vaccination, the microneedle group had high antibody titers and complete viral protection whereas the s.c. group had a 60% decline in antibody titers and only partial protection was provided against a lethal viral challenge [Citation61]. Likewise, Koutsonanos et al. showed that subunit vaccine (A/Brisbane H1N1) generated peak antibody levels at week 8 when administered in mice by the i.m. route, as compared to week 12 in mice immunized by microneedles [Citation63]. Both vaccinated groups of mice were fully protected against lethal challenge at 4 or 12 weeks post immunization. However, at 36 weeks post immunization, 38% of the i.m. immunized animals had a significant decline in HAI titers below 40 (HAI < 40) whereas the microneedle group had HAI titers high enough to confer complete protection against lethal challenge (HAI > 40) [Citation63]. Similarly, the protective efficacy of stabilized VLPs (A/PR8 H1N1) coated on microneedles was investigated by viral challenge 14 months after a single vaccine dose in mice [Citation71]. Significant systemic, mucosal, and recall immune responses provided complete protection against viral challenge even after such a substantial period of time [Citation71]. These findings thus demonstrate that stabilized influenza vaccines delivered by microneedles can generate longer lasting immunity and better protection than conventional systemic routes.
To assess whether a vaccine formulation could induce a broad protective immunity and serve as a proof of concept for a universal flu vaccine, four repeats of a conserved part of the M2 protein linked to the Toll-like receptor-5 (TLR-5) ligand Salmonella typhimurium phase I flagellin (FliC) were coated on microneedles and administered to mice. A homo and heterosubtypic lethal viral challenge of mouse adapted A/Philippines (H3N2) and A/PR/8 (H1N1) showed that all the microneedle-immunized mice survived [Citation75]. Post-challenge lung viral titers of microneedle immunized mice were more effectively reduced than those of intramuscularly immunized mice [Citation75]. In another study, a patch of microneedles coated with VLPs containing heterologous M2e extracellular domains (M2e5x) of influenza virus stabilized with trehalose induced a broad heterosubtypic cross-protection in mice [Citation16]. Microneedle-immunized mice showed a strong induction of humoral and mucosal M2e antibody responses and were better cross-protected than i.m.-immunized mice against heterosubtypic (H1N1, H3N2, and H5N1) lethal viral challenge. Further, the antigenicity and immunogenicity of the M2e5x-VLP were maintained for at least 8 weeks at room temperature [Citation16]. Microneedle-mediated delivery of conserved epitopes show promising results and holds a great potential for further development of universal flu vaccination. Not only are broad and cross-reactive immune responses desired, but also an influenza vaccine that is safe and effective for all age groups would be preferable.
Children, elderly, and immunocompromised patients are more susceptible to influenza infection than other individuals. Therefore, the protective efficacy of skin-based delivery was also investigated in young mice [Citation73]. The microneedle-vaccinated group showed improved humoral responses, reduced lung viral titers and better viral protection after viral challenge than did the i.m. group. These potent humoral responses and better survival after challenge were attributed to higher numbers of antibody secreting cells and activated germinal center formation [Citation73]. Besides mice, guinea pigs were also inserted with microneedles coated with commercial trivalent vaccine. Comparable immune titers were observed for both the microneedle and i.m. group [Citation70].
Another approach uses (water) dissolving microneedles consisting of vaccines encapsulated in matrices of polymers like CMC, polyvinylpyrrolidone (PVP), polyethylene glycol, or polyvinylalcohol, polysaccharides like dextrin, dextran, or hyaluronic acid, disaccharides like maltose, and monosaccharides like galactose ((a)) [Citation86,Citation87]. These microneedles dissolve within minutes once inserted into the skin. Dissolving microneedles used for influenza vaccination are most often produced using a solvent casting method, whereby a solution containing the vaccine and a suitable matrix is poured (sometimes aided by centrifugation) into a mold where it solidifies to create the desired cast of a microneedle array. Solidification of the matrix can be the result of a photo-polymerization process or a drying step [Citation58]. Since a certain force, velocity, and sharpness are needed to penetrate the microneedles into the skin, the mechanical strength of the water dissolving matrix material used to encapsulate the vaccine is also important and should be well investigated when developing (dissolving) microneedles.
In-vivo studies have been carried out using dissolving microneedles and the immunogenicity was compared to either coated microneedles or conventional parenteral routes. Sullivan et al. used dissolving PVP based microneedles encapsulating lyophilized WIV and compared the immunogenicity and protective efficacy to that in i.m. vaccinated mice () [Citation58]. Humoral as well as cellular immune responses and improved serological memory, strong enough to protect against lethal challenge were found after dissolvable microneedle vaccination. In comparison to i.m. route, reduced lung viral titers and enhanced cellular recall responses were determined after microneedle immunization. Moreover, dissolvable microneedles were found to have comparable humoral and superior induction of cellular responses when compared with coated microneedles [Citation58]. Recently, Vassilieva et al. developed a gelatine-based microneedle patch encapsulating different strains of WIV and found the induction of neutralizing antibody titers better (all strains) than after conventional i.m. immunization [Citation72]. Further, antigen stability was retained after storage for 3 months at room temperature [Citation72]. Also, a clinical study investigated the safety and efficacy of dissolving microneedles containing sodium hyaluronate, dextran 70, povidone, and trivalent seasonal influenza subunit vaccine [Citation17]. No severe local and systemic adverse events were observed, however, at the site of application the skin displayed local temporary erythema. Although the efficacy of the vaccine against the B strain was stronger than after s.c. immunization, immune responses against A/H1N1 and A/H3N2 were equally induced [Citation17].
Intranasal and pulmonary dry powder influenza vaccine delivery
Targeting influenza vaccines to the mucosal sites in the airways might be advantageous because of the enormous surface area and extensively developed innate and adaptive immune system [Citation88]. The presence of APCs such as DC, macrophages, and B cells enables the transport of antigens to the lymph nodes and the initiation of immune responses against the antigens [Citation88]. The mucosal surface is a well-developed system consisting of nasal-associated lymphoid tissue (NALT) in the upper respiratory tract, and inducible bronchus-associated lymphoid tissue in the lower respiratory tract [Citation89]. These lymphoid tissues play a major role in the stimulation and modulation of immune responses in the upper and lower respiratory tract [Citation89]. Hence, in cases of respiratory infectious diseases such as influenza, the delivery of antigen at the natural portal of virus entry might reduce antigen dose and induce local (mucosal) immune responses.
Nasal dry powder influenza vaccine delivery
When targeting the intranasal area using dry powders, one should take into account several factors like particle size, density, and air velocity during administration. Particles bigger than 50 µm usually show reproducible intranasal deposition and do not follow the streamline direction of inhaled air; they thereby prevent deposition in the lung [Citation90]. The high clearance rate of the nose might potentiate nasal tolerance against influenza vaccination. This would make the use of mucoadhesives like chitosan or hypromellose necessary to increase the residence time of the vaccine ((a)).
In a study conducted in rats by Huang et al., the nasal delivery of freeze dried and subsequently milled WIV (A/PR8 H1N1) formulations blended with mucoadhesive (chitosan), generated comparable systemic and better nasal antibody titers than were found after i.m. administration [Citation76]. Moreover, the dry powder formulation remained completely stable (as determined by the hemagglutination assay) when stored at 25°C/25% RH for 12 weeks, whereas the potency of the liquid formulation was reduced to 12.5% [Citation76]. Several other mucoadhesives like sodium alginate or cellulose derivatives like CMC and HPMC were used by Garmise et al. in a similar way () [Citation77]. It was found that i.n. WIV (A/PR8 H1N1) formulations, formulated with or without these mucoadhesives, elicited similar serum antibody titers and higher nasal IgA titers than formulations administered by i.m. route to rats. Also, the stability of the powder formulations was well preserved for 25°C/40% RH for 12 weeks whereas liquid vaccine lost 70% of its stability under similar conditions (measured by HA titer determination) [Citation77].
The potential of in situ gelling nasal inserts as a delivery system for influenza vaccine was investigated by Bertram et al. [Citation78]. The inserts were manufactured by freeze drying hydrophilic polymer solutions containing influenza split vaccine (H1N1) with or without several adjuvants. Upon contact with the nasal mucosa, the hydrophilic polymeric matrix takes up water leading to gel formation after which the vaccine is released in a controlled manner. In-vivo studies in rats revealed that freeze dried influenza vaccine incorporated in xanthan gum with or without cationic lipid (CL) adjuvant, elicited serum IgG titers similar to those of pure i.n. liquid solution [Citation78]. The authors hypothesized a probable interaction between oppositely charged xanthan gum and CL, which could inhibit antigen–adjuvant interaction to boost the immune response. The production of xanthan gum nasal inserts might be an interesting alternative to enhance the stability of influenza antigen while maintaining an immune response similar in magnitude to that of liquid formulations. Vacuum-dried chitosan nanospheres encapsulating WIV (A/New Caledonia H1N1) and adjuvants like CpG ODN or Quillaja saponins were also tested for their suitability as nasal particulate delivery system[Citation12,Citation79]. The structure of WIV was unaffected by encapsulation. The chitosan nanospheres encapsulated with influenza whole virus and CpG ODN generated both local and systemic humoral and cellular immune responses in rabbits, and induced higher levels of IgA than did liquid nasal and i.m. formulations [Citation79].
Therefore, it can be concluded from the aforementioned studies that the influenza vaccine in a dry state not only enhances the stability of the antigen but also generates immune response comparable to that of liquid i.m. or i.n. formulations.
Pulmonary dry powder influenza vaccine delivery
Spherically shaped influenza vaccine powder particles, suitable for pulmonary delivery have been prepared by spray or spray freeze drying using saccharides like inulin and trehalose as stabilizers and bulking agents ((a)). To reach the central and peripheral airways particle size, particle density and particle shape play an important role. To do so effectively, the aerodynamic particle size should be in the range of 1–5 µm. The aerodynamic size or diameter of a particle takes into account the particle’s density and the shape of the particle (dynamic shape factor), and is defined as the diameter of a perfect spherical particle with a density of 1 g/cm3 having the same terminal settling velocity in still air as the particle in consideration. Particles with an aerodynamic size greater than 5 µm will show high deposition in the throat, upper and central airways while a large fraction of particles with an aerodynamic diameter smaller than 1 µm will be exhaled [Citation91]. It is still not completely known which part of the lungs should be targeted for optimal pulmonary immunization against influenza. Studies conducted by Waldman et al. in the late 1960s did show protective antibodies against influenza in children and adults without severe adverse reactions after pulmonary administration of liquid aerosolized inactivated influenza vaccine by nebulization [Citation92,Citation93]. Various in-vivo studies have shown that pulmonary immunization against influenza by dry powder delivery is a promising approach.
Amorij et al. demonstrated with mice that pulmonary delivery of influenza subunit vaccine spray freeze dried in the presence of inulin (A/Panama H3N2) resulted in a better immune response than did i.m. liquid immunization () [Citation80]. Pulmonary powder delivery was able to elicit increased systemic humoral (IgG), mucosal (IgA and IgG), and cell-mediated immune responses (IFN-γ and IL-4) as compared to i.m. vaccination. Moreover, pulmonary powder immunization induced a balanced superior Th1/Th2 immune response as compared to the Th2 dominant response after i.m. injection. A Th1 or a balanced Th1/Th2 response is considered to be superior because it plays a key role in virus neutralization and provides a certain degree of cross-reactive immunity and thus results in better protection against infection [Citation94,Citation95]. The occurrence of high levels of IgG and IgA antibodies in the lungs and minor antibody titers in the nose was attributed to the migration of immune effector cells from the primary mucosal induction site (lungs) to the secondary distant mucosal site (nose) [Citation80]. In a follow-up study, Saluja et al. showed that the integrity of the antigen was best conserved after spray drying and spray freeze drying when formulated in phosphate buffer saline and hepes buffer saline, respectively [Citation24]. The stability of the dried antigen as determined by single radial immunodiffusion assay was preserved for 3 years at room temperature whereas the potency of the liquid vaccine was below detection limits after 3 years of storage at 4°C [Citation24]. Long-term immunogenic and physical stability of WIV at elevated storage temperatures was achieved after spray freeze drying them in the presence of suitable stabilizers [Citation11].
Audouy et al. compared the virus protecting potential of two doses of WIV (A/PR8) spray freeze dried powders administered via the pulmonary route with a single dose of subunit vaccine (A/PR8) administered by i.m. injection in mice [Citation81]. After a viral challenge, the weight loss of the animals vaccinated with dry powders via the pulmonary route was significantly lower than the weight loss of the animals which were administered with liquid vaccine via the i.m. route. Moreover, powder-treated animals had the largest reduction in lung virus titer [Citation81].
Peeters et al. assessed the protective efficacy of unadjuvanted pulmonary delivered dry powder influenza vaccines against viral challenge in chickens [Citation82]. It was found that the antibody levels induced by unadjuvanted dry powder WIV (H5N1) formulations were sufficient to fully protect chickens from morbidity and mortality after highly pathogenic avian influenza virus challenge (H5N1) [Citation82]. In contrast to the results of Amorij et al. [Citation80], other studies with dry unadjuvanted WIV or subunit formulations showed that the latter elicited a Th2 dominated response, evidenced by the high IgG1 antibody titers, as compared to IgG2a antibody titers after pulmonary administration [Citation11,Citation81]. Besides a Th2 dominated response, low nasal IgA titers by pulmonary delivered dry powders also indicated the need for using suitable mucosal adjuvants. Hence, influenza vaccine formulations were adjuvanted with saponin adjuvant GP-0100 and the TLR ligands, monophosphoryl lipid A (MPLA), palmitoyl-3-cysteine-lysine-serine-4 (Pam3CSK4), and CpG ODN to increase the magnitude of the mucosal immune response and to direct the immune response toward the Th1 phenotype [Citation83,Citation84]. These adjuvants were selected because they have been shown to enhance systemic humoral responses after parenteral administration of influenza vaccines [Citation96,Citation97]. Besides this, they have also been used as mucosal adjuvants for influenza and tuberculosis antigens [Citation98–Citation100]. Dry powder pulmonary immunization of WIV (A/Hiroshima H3N2) adjuvanted with MPLA induced a higher production of superior Th1 type serum antibody titers (IgG2a) than did unadjuvanted formulations. Moreover, the dry MPLA adjuvanted influenza vaccine powders induced higher mucosal IgA and IgG antibody titers in both nose and lungs before and after challenge. Pulmonary immunization with WIV adjuvanted formulations was found to be as effective as standard subunit i.m. formulation in reducing lung virus titers after challenge (A/PR8). Hence, the co-administration of WIV with MPLA adjuvant could steer to a Th1 type immune response and be able to elicit potent mucosal antibody titers in the lungs [Citation83]. Likewise, other TLR- and saponin-based adjuvants were also found to direct the immune response of pulmonary delivered dry powder WIV (A/California H1N1) formulations to either a dominant Th1 type or a balanced Th1/Th2 type [Citation84]. GP-0100 enhanced mucosal (IgA) antibody titers in lungs and nose and was able to provide partial protection against heterologous virus challenge (A/PR8 H1N1) [Citation84].
Besides being stable and immunogenic, a vaccine and an adjuvant should not induce undesirable side effects. Audouy et al. investigated the safety of pulmonary influenza vaccination [Citation81]. Administration to mice of spray freeze dried WIV in the presence of inulin did induce mild transient cell influx but the inflammatory reactions subsided with time [Citation81]. Also, pulmonary administration of the GP-100 formulation did not induce gross lung inflammation [Citation84].
In conclusion, the dry influenza powders (adjuvanted/unadjuvanted) were stable and their delivery by pulmonary route was safe, effective, and capable of protection against viral challenge.
Sublingual, buccal, and gastrointestinal dry influenza vaccine delivery
Immunization via the oral route (mucosal areas of mouth and gastrointestinal tract) is a potentially attractive site to target for influenza vaccination. The mucosa of s.l. and buccal regions in the mouth contains DC and LC which are involved in the uptake, processing and presentation of the antigen to T-cells to induce an adaptive immune response [Citation101]. Liquid influenza vaccination by the s.l. route has already been shown in various studies to induce systemic, mucosal, and cellular immune responses in mice [Citation102,Citation103]. However, dry influenza vaccine delivery by s.l. or buccal routes has not yet been investigated in detail. Recently, Murugappan et al. incorporated lyophilized WIV into a tablet with a good crushing strength and a low disintegration time making it suitable for s.l. administration [Citation18]. Moreover, the hemagglutination capacity of the antigen was well preserved after freeze drying and tablet manufacture. However, the lack of dissolution of these tablets under the tongue in appropriate animal models like mice led to reconstitution of the powders to show the potential of s.l. administration [Citation18]. McNeilly et al. targeted murine buccal mucosa with dried split influenza vaccine (Fluvax) coated on a Nanopatch and then compared the elicited immunogenicity to liquid formulations given i.m. or orally (gastrointestinal tract) [Citation19]. The systemic IgG titers induced by Nanopatch buccally applied were comparable to those after i.m. administration and significantly higher than after gastrointestinal immunization. Furthermore, four out of five mice immunized with Nanopatch were found to exceed the protective HAI threshold (HAI ≤ 40), whereas only one out of five animals immunized i.m. reached the minimum value of 40 and no HAI activity was detected in gastrointestinal immunized mice. Hence, the buccal Nanopatch delivery of dry split influenza vaccine generated comparable and better immune responses than did the i.m. and oral routes, respectively [Citation19]. The better immune responses generated by the buccal route were attributed to the uptake of antigen by oral mucosa rather than oral ingestion of the vaccine [Citation19].
The ease of administration, patient compliance and the presence of gut-associated lymphoid tissue (GALT) make oral vaccine for intestinal delivery an attractive administration route. However, the potential of the oral route for influenza vaccine delivery in the intestines is yet to be proven. Enormous challenges like acidic gastric pH, suitable oral adjuvants, oral tolerance, choice of which intestinal region to target, a suitable delivery system, delivery mechanism, and the vaccine’s immunogenicity need to be dealt with so as to design a successful oral influenza vaccine formulation. Some progress has however, been made. Recently, an adenovirus-based oral influenza vaccine enteric coated tablet (H1N1) was manufactured to investigate its safety and immunogenicity in healthy adults () [Citation20]. It was well tolerated and the majority of the adults reached HAI titers ≥40, whereas no seroconversion was found in the placebo group. In addition, a fourfold increase in the micro-neutralization titers for the vaccine group as compared to the control group was recorded. Hence, more than 90% of the individuals had high humoral responses elicited by oral influenza vaccine delivery [Citation20].
Dry influenza vaccine formulations for oral delivery are still at a very early stage of development. However, the stability and immunogenicity of influenza vaccines formulated in tablets or coated on Nanopatch and administered by oral, s.l. or buccal routes seem very promising and hold great potential for future developments in this field.
Expert commentary
Parenteral influenza vaccines have, for more than 80 years now, been considered to be the benchmark in influenza vaccination. However, the need for a cold chain and often insufficient immune responses are the major disadvantages of currently available injectable influenza vaccines. The development of stable dry influenza vaccines would alleviate the necessity of a cold chain, thereby making stockpiling possible and improving availability to the public. Drying an influenza vaccine under appropriate conditions and using suitable stabilizers would not only provide better stability of the antigen but also improve its efficacy when administered by a suitable route of interest like the skin or mucosal areas [Citation56,Citation79,Citation80,Citation19]. (b) gives a general overview of the advantages and disadvantages of dry influenza vaccination per route of administration. As shown by various preclinical and clinical studies, vaccinating the skin against influenza by microneedles or epidermal powder delivery results in a direct activation of the residing immune cells providing immune responses stronger or equal to those evoked by parenteral influenza vaccination [Citation9,Citation17,Citation61,Citation63]. However, the high costs of epidermal powder delivery by a device like the PowderJect limit its relevance for mass vaccination [Citation104]. Other disadvantages of EPI are that it causes erythema, edema, petechiae, skin discoloration, and flaking of the skin [Citation9] which could lead to reduced patient compliance. Therefore, application of coated non-dissolving microneedles is an attractive alternative as it is cheap, it can be self-administered and it does not cause the side effects related to ballistic administration. Despite the numerous advantages of the use of microneedles, antigen integrity during drying/storage remains a troublesome issue. It has been found that the presence of surfactant causes crystallization of stabilizing excipients and thus phase separation of vaccine coatings during drying or during long-term storage by which the stabilizing effects of the excipients are lost [Citation13,Citation68]. Another disadvantage is the risk of needles being broken off and left in the skin during administration. The use of dissolving microneedles would be an attractive alternative to circumvent this problem. However, long-term storage stability of the antigen in these microneedles has not been addressed thus far. In addition, the production and formulation of microneedles is labor intensive and challenging at a laboratory scale. New processes should be developed to efficiently produce these microneedles on an industrial scale. Furthermore, clinically relevant models are necessary to investigate the efficacy of microneedles for vaccination against influenza in humans. Recently, the group of Prausnitz has started a phase 1 clinical trial to investigate the immunogenicity, safety, reactogenicity, and acceptability of an inactivated influenza vaccine delivered using a microneedle patch [Citation105]. This could lead to new insights in the field of transtradermal influenza vaccination.
While transdermal vaccination is a promising route, the initial transmission of influenza virus takes place via the airways through inhalation of small droplets from the air or by contaminated surfaces. Curbing the infection at the portal of entry would therefore seem to be a more logical approach. Intranasal and pulmonary influenza vaccination might be a more direct way to neutralize the virus by evoking a local immune response once it has entered the airways [Citation106]. Intranasal immunization has been shown to provide broad immune responses and considerable protection [Citation107,Citation108]. However, dry powder intranasal influenza vaccines are still in a process of preclinical development. Low residence time, inefficient antigen uptake, and the need for adjuvanted formulation with the subsequent risk of ciliotoxicity are the major hurdles to overcome when developing liquid and dry powder intranasal vaccines [Citation109]. Some adjuvants have been shown to redirect an antigen to the olfactory neuroepithelium, thereby inducing inflammatory responses in the nasal cavity [Citation110]. Also, subsequent retrograde transport of the adjuvant into the olfactory bulb of the central nervous system has been indicated [Citation110]. Mucoadhesives, particulate systems, as well as safe, effective, and tolerable adjuvanted formulations are being investigated to overcome these hurdles, thereby increasing the complexity of such formulations in terms of stability and immunogenicity [Citation88,Citation109]. Another factor to consider is which specific site to target within the nasal cavity. Anterior nasal mucosa does not include cilia; this reduces the risk of quick clearance of the administered vaccine, a risk that is greater in the highly ciliated posterior part of the nasal cavity [Citation111]. However, targeting of the antigen to the posterior part might be desirable because it is close to the NALT. Nonetheless, the targeting areas within the upper and lower airways most likely to induce the best protective immune responses are still not yet completely known. Spray or spray freeze dried influenza vaccines administered by the pulmonary route have been shown to elicit local humoral and cellular immune responses, providing comparable or better protection than parenteral administration [Citation24,Citation80,Citation83]. Since the particle size and the delivery device play an important role in the deposition within the airways, it is essential to characterize these components. The currently used method of administering a spray dried or spray freeze dried influenza vaccine to animals is by insufflation, using a device such as the Penn-Century dry powder insufflator. Insufflation by this device is based upon intubating the trachea of an animal like a rodent and then active dispersion of a dry powder using a certain volume of air (200–1000 µl). The limited volume of air that can be used for the dispersion of dry powder into rodents and high particle velocity generated during dispersion of the dry powder from the insufflator cause a high deposition within the upper part of the lungs [Citation15,Citation112]. This makes it complicated to investigate the relationship between the site of deposition and the protective immunity against influenza. New ways of pulmonary administration of dry powder influenza vaccines in a preclinical setting should therefore be investigated. A novel aerosol generator did show improved lung deposition in mice as compared to the dry powder insufflator from Penn-Century. This aerosol generator would seem to be a step toward improved devices for preclinical pulmonary animal studies [Citation15]. The successful approach by Waldman et al. in the 1960s and 1970s already showed the effectiveness of liquid influenza vaccination after pulmonary administration via a nebulizer in humans [Citation92,Citation93]. However, these studies were probably stopped because a suitable inhalation system and formulation were not available at that time. To deliver a dry powder influenza vaccine via the pulmonary route a device should be disposable, cheap, and easy to operate in case of pandemics without the need of health-care personnel. Nowadays, such devices are widely available [Citation113], as for example the single-use disposable inhaler the Twincer®, with inulin stabilized solid influenza powder [Citation114]. In-vitro experiments using a cascade impactor have shown that the dispersion of spray dried and spray freeze dried subunit influenza vaccine from the Twincer® is suitable for the administration to the human respiratory tract [Citation24]. Although a single-use disposable inhaler like the Twincer® could be used in mass vaccination against influenza, it is not suitable for children up to 4 years old since they are not able to make the necessary inhalation maneuver [Citation115]. It is also necessary to conduct clinical trials to investigate the safety, tolerability, and efficacy of pulmonary delivered dry powder influenza vaccines.
Oral administration of influenza vaccines could be suitable for all age groups due to its ease of acceptance. APCs in the mucosal areas of the s.l., buccal, and gastrointestinal region can be targeted by tablets [Citation20] or coated micro-projections like the Nanopatch [Citation19], which show promising results. Viral vectors in tablets for gastrointestinal targeting can prevent gastric acid degradation. Coating strategies like the ColoPulse that utilize a delayed pulsatile release profile based on intestinal pH, could also be a potential way to deliver an antigen to the ileocolonic segment [Citation116]. Still like the delivery of influenza vaccines into the airways, the optimal antigen deposition site within the gastrointestinal tract is still to be investigated.
Dry influenza vaccines show potential in terms of stability, efficacy, and their administration via non-parenteral routes is a convenient alternative to traditional parenteral influenza vaccination, thus encouraging further developments.
Five-year view
Dry influenza vaccines administered by non-parenteral routes are expected to draw more attention. Better preclinical models will be used to mimic human anatomy and physiology. More clinical trials will be conducted to examine the efficacy, safety, and tolerability of newly developed dry influenza vaccines. These vaccines, delivered by different routes of administration, will result in great advancements in the field of dry influenza vaccines.
Key issues
Parenteral vaccines are the cornerstone for mass influenza vaccination, yet the induction of broad humoral and cellular responses is limited.
Liquid influenza vaccines require cold chain conditions.
Properly dried influenza vaccines can be stored at room temperature with improved efficacy and enhanced patient acceptance by utilizing different routes of administration.
The numerous APCs in the skin and mucosal areas of the airways, mouth and gastrointestinal tract make these areas excellent targets for influenza vaccination.
Dry influenza vaccines should induce strong, and preferably cross-reactive immune responses.
Dry influenza vaccines should be suitable for all age groups.
Dry influenza vaccination will result in immune responses equal to or better than those with conventional parenteral vaccination.
The number of clinical studies on dry influenza vaccination by different routes of administration is limited; the subject deserves more attention.
Declaration of interests
This work was supported by the European Union's Seventh Framework Programme for research, technological development and demonstration [grant number 602012]. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
- Wilschut J, de Jonge J, Huckriede A, et al. Preservation of influenza virosome structure and function during freeze-drying and storage. J Liposome Res. 2007;17(3–4):173–182.
- Geeraedts F, Saluja V, ter Veer W, et al. Preservation of the immunogenicity of dry-powder influenza H5N1 whole inactivated virus vaccine at elevated storage temperatures. AAPS J. 2010;12(2):215–222.
- Muttil P, Prego C, Garcia-Contreras L, et al. Immunization of guinea pigs with novel hepatitis B antigen as nanoparticle aggregate powders administered by the pulmonary route. AAPS J. 2010;12(3):330–337.
- Garcia-Contreras L, Wong Y-L, Muttil P, et al. Immunization by a bacterial aerosol. Proc Natl Acad Sci U S A. 2008;105(12):4656–4660.
- Muttil P, Pulliam B, Garcia-Contreras L, et al. Pulmonary immunization of guinea pigs with diphtheria CRM-197 antigen as nanoparticle aggregate dry powders enhance local and systemic immune responses. AAPS J. 2010;12(4):699–707.
- de Jonge J, Amorij J-P, Hinrichs WLJ, et al. Inulin sugar glasses preserve the structural integrity and biological activity of influenza virosomes during freeze-drying and storage. Eur J Pharm Sci. 2007;32(1):33–44.
- LiCalsi C, Maniaci MJ, Christensen T, et al. A powder formulation of measles vaccine for aerosol delivery. Vaccine. 2001;19(17–19):2629–2636.
- Amorij J-P, Meulenaar J, Hinrichs WLJ, et al. Rational design of an influenza subunit vaccine powder with sugar glass technology: preventing conformational changes of haemagglutinin during freezing and freeze-drying. Vaccine. 2007;25(35):6447–6457.
- Dean HJ, Chen D. Epidermal powder immunization against influenza. Vaccine. 2004;23(5):681–686.
- Maa Y-F, Ameri M, Shu C, et al. Influenza vaccine powder formulation development: spray-freeze-drying and stability evaluation. J Pharm Sci. 2004;93(7):1912–1923.
- Murugappan S, Patil HP, Kanojia G, et al. Physical and immunogenic stability of spray freeze-dried influenza vaccine powder for pulmonary delivery: comparison of inulin, dextran, or a mixture of dextran and trehalose as protectants. Eur J Pharm Biopharm. 2013;85(3):716–725.
- Dehghan S, Tavassoti Kheiri M, Tabatabaiean M, et al. Dry-powder form of chitosan nanospheres containing influenza virus and adjuvants for nasal immunization. Arch Pharm Res. 2013;36(8):981–992.
- Choi H-J, Yoo D-G, Bondy BJ, et al. Stability of influenza vaccine coated onto microneedles. Biomaterials. 2012;33(14):3756–3769.
- Gill HS, Kang S-M, Quan F-S, et al. Cutaneous immunization: an evolving paradigm in influenza vaccines. Expert Opin Drug Deliv. 2014;11(4):615–627.
- Tonnis WF, Bagerman M, Weij M, et al. A novel aerosol generator for homogenous distribution of powder over the lungs after pulmonary administration to small laboratory animals. Eur J Pharm Biopharm. 2014;88(3):1056–1063.
- Kim M, Lee J, Choi H, et al. Microneedle patch delivery to the skin of virus-like particles containing heterologous M2e extracellular domains of influenza virus induces broad heterosubtypic cross-protection. J Control Release. 2015;210:208–216.
- Hirobe S, Azukizawa H, Hanafusa T, et al. Clinical study and stability assessment of a novel transcutaneous influenza vaccination using a dissolving microneedle patch. Biomaterials. 2015;57:50–58.
- Murugappan S, Patil HP, Frijlink HW, et al. Simplifying influenza vaccination during pandemics: sublingual priming and intramuscular boosting of immune responses with heterologous whole inactivated influenza vaccine. AAPS J. 2014;16(2):342–349.
- McNeilly CL, Crichton ML, Primiero CA, et al. Microprojection arrays to immunise at mucosal surfaces. J Control Release. 2014;196:252–260.
- Liebowitz D, Lindbloom JD, Brandl JR, et al. High titre neutralising antibodies to influenza after oral tablet immunisation: a phase 1, randomised, placebo-controlled trial. Lancet Infect Dis. 2015;15(9):1041–1048.
- Guerrero M, Albet C, Palomer A, et al. Drying in pharmaceutical and biotechnological industries. Food Sci Technol Int. 2003;9(3):237–243.
- Pizzuto MS, De Benedictis P, Maniero S, et al. Improving heat stability of haemagglutinating antigens for avian influenza. Biologicals. 2011;39(3):149–151.
- Tonnis WF, Mensink M, de Jager A, et al. Size and molecular flexibility of sugars determine the storage stability of freeze-dried proteins. Mol Pharm. 2015;12(3):684–694.
- Saluja V, Amorij J-P, Kapteyn JC, et al. A comparison between spray drying and spray freeze drying to produce an influenza subunit vaccine powder for inhalation. J Control Release. 2010;144(2):127–133.
- Chang L, Pikal MJ. Mechanisms of protein stabilization in the solid state. J Pharm Sci. 2009;98(9):2886–2908.
- Chang L, Shepherd D, Sun J, et al. Mechanism of protein stabilization by sugars during freeze-drying and storage: native structure preservation, specific interaction, and/or immobilization in a glassy matrix? J Pharm Sci. 2005;94(7):1427–1444.
- Grasmeijer N, Stankovic M, de Waard H, et al. Unraveling protein stabilization mechanisms: vitrification and water replacement in a glass transition temperature controlled system. Biochim Biophys Acta. 2013;1834(4):763–769.
- Sadek C, Schuck P, Fallourd Y, et al. Drying of a single droplet to investigate process-structure-function relationships: a review. Dairy Sci Technol. 2015;95(6):771–794.
- Sander A, Penović T. Droplet size distribution obtained by atomization with two-fluid nozzles in a spray dryer. Chem Eng Technol. 2014;37(12):2073–2084.
- Sou T, Morton DAV, Williamson M, et al. Spray-dried influenza antigen with trehalose and leucine produces an aerosolizable powder vaccine formulation that induces strong systemic and mucosal immunity after pulmonary administration. J Aerosol Med Pulm Drug Deliv. 2015;28(5):361–371.
- Huang L, Mujumdar AS. Numerical study of two-stage horizontal spray dryers using computational fluid dynamics. Dry Technol. 2006;24(6):727–733.
- Gu B, Linehan B, Tseng Y-C. Optimization of the Büchi B-90 spray drying process using central composite design for preparation of solid dispersions. Int J Pharm. 2015;491(1–2):208–217.
- Littringer EM, Paus R, Mescher A, et al. The morphology of spray dried mannitol particles – the vital importance of droplet size. Powder Technol. 2013;239:162–174.
- Jennings T. Lyophilization intro and basics. New York (NY): CRC Press; 1999.
- Tonnis WF, Amorij J-P, Vreeman MA, et al. Improved storage stability and immunogenicity of hepatitis B vaccine after spray-freeze drying in presence of sugars. Eur J Pharm Sci. 2014;55(1):36–45.
- Li X, Thakkar SG, Ruwona TB, et al. A method of lyophilizing vaccines containing aluminum salts into a dry powder without causing particle aggregation or decreasing the immunogenicity following reconstitution. J Control Release. 2015;204:38–50.
- Wanning S, Süverkrüp R, Lamprecht A. Pharmaceutical spray freeze drying. Int J Pharm. 2015;488(1–2):136–153.
- Roos Y, Karel M. Plasticizing effect of water on thermal behavior and crystallization of amorphous food models. J Food Sci. 1991;56(1):38–43.
- Saleki-Gerhardt A, Zografi G. Non-isothermal and isothermal crystallization of sucrose from the amorphous state. Pharm Res Off J Am Assoc Pharm Sci. 1994;11(8):1166–1173.
- Bhandari BR, Howes T. Implication of glass transition for the drying and stability of dried foods. J Food Eng. 1999;40(1–2):71–79.
- Ioanna S, Compans RW. Skin immunization with influenza vaccines. Curr Top Microbiol Immunol. 2013;358(Jan):3–32.
- Bellhouse BJ, Sarphie DF, Greenford JC Method of delivering powder transdermally with needless injector. US005630796A. 1997.
- Anamur C, Winter G, Engert J. Stability of collapse lyophilized influenza vaccine formulations. Int J Pharm. 2015;483(1–2):131–141.
- Maa Y, Ameri M, Shu C, et al. Influenza vaccine powder formulation development: spray-freeze-drying and stability evaluation. J Pharm Sci. 2004;93(7):1912–1923.
- Chen D, Endres RL, Erickson CA, et al. Epidermal immunization by a needle-free powder delivery technology: immunogenicity of influenza vaccine and protection in mice. Nat. Med. 2000;6(10):1187–1190.
- Chen D, Endres R, Maa Y-F, et al. Epidermal powder immunization of mice and monkeys with an influenza vaccine. Vaccine. 2003;21(21–22):2830–2836.
- Chen D, Endres RL, Erickson CA, et al. Epidermal powder immunization using non-toxic bacterial enterotoxin adjuvants with influenza vaccine augments protective immunity. Vaccine. 2002;20:2671–2679.
- Chen D, Weis KF, Chu Q, et al. Epidermal powder immunization induces both cytotoxic T-lymphocyte and antibody responses to protein antigens of influenza and hepatitis B viruses. J Virol. 2001;75(23):11630–11640.
- Chen D, Periwal SB, Larrivee K, et al. Serum and mucosal immune responses to an inactivated influenza virus vaccine induced by epidermal powder immunization serum and mucosal immune responses to an inactivated influenza virus vaccine induced by epidermal powder immunization. J Virol. 2001;75(17):7956–7965.
- Chen D, Burger M, Chu Q, et al. Epidermal powder immunization: cellular and molecular mechanisms for enhancing vaccine immunogenicity. Virus Res. 2004;103:147–153.
- Koutsonanos DG, del Pilar Martin M, Zarnitsyn VG, et al. Transdermal influenza immunization with vaccine-coated microneedle arrays. PLoS One. 2009;4(3):e4773.
- Quan F-S, Kim Y-C, Yoo D-G, et al. Stabilization of influenza vaccine enhances protection by microneedle delivery in the mouse skin. PLoS One. 2009;4(9):e7152.
- Kim Y-C, Quan F-S, Yoo D-G, et al. Improved influenza vaccination in the skin using vaccine coated microneedles. Vaccine. 2009;27(49):6932–6938.
- Zhu Q, Zarnitsyn VG, Ye L, et al. Immunization by vaccine-coated microneedle arrays protects against lethal influenza virus challenge. Proc Natl Acad Sci U S A. 2009;106:7968–7973.
- Quan F-S, Kim Y-C, Vunnava A, et al. Intradermal vaccination with influenza virus-like particles by using microneedles induces protection superior to that with intramuscular immunization. J Virol. 2010;84(15):7760–7769.
- Quan F-S, Kim Y-C, Compans RW, et al. Dose sparing enabled by skin immunization with influenza virus-like particle vaccine using microneedles. J Control Release. 2010;147(3):326–332.
- Song J-M, Kim Y-C, Barlow PG, et al. Improved protection against avian influenza H5N1 virus by a single vaccination with virus-like particles in skin using microneedles. Antiviral Res. 2010;88(2):244–247.
- Sullivan SP, Koutsonanos DG, Martin P, et al. Dissolving polymer microneedle patches for influenza vaccination. Nat Med. 2010;16(8):915–920.
- Kim Y-C, Quan F-S, Compans RW, et al. Formulation of microneedles coated with influenza virus-like particle vaccine. AAPS PharmSciTech. 2010;11(3):1193–1201.
- Chen X, Fernando GJP, Crichton ML, et al. Improving the reach of vaccines to low-resource regions, with a needle-free vaccine delivery device and long-term thermostabilization. J Control Release. 2011;152(3):349–355.
- Koutsonanos DG, del Pilar Martin M, Zarnitsyn VG, et al. Serological memory and long-term protection to novel H1N1 influenza virus after skin vaccination. J Infect Dis. 2011;204(4):582–591.
- Fernando GJP, Chen X, Primiero CA, et al. Nanopatch targeted delivery of both antigen and adjuvant to skin synergistically drives enhanced antibody responses. J Control Release. 2012;159(2):215–221.
- Koutsonanos DG, Vassilieva EV, Stavropoulou A, et al. Delivery of subunit influenza vaccine to skin with microneedles improves immunogenicity and long-lived protection. Sci Rep. 2012;2:1–10.
- Chen X, Fernando GJP, Raphael AP, et al. Rapid kinetics to peak serum antibodies is achieved following influenza vaccination by dry-coated densely packed microprojections to skin. J Control Release. 2012;158(1):78–84.
- Song J-M, Kim Y-C, O E, et al. DNA vaccination in the skin using microneedles improves protection against influenza. Mol Ther. 2012;20(7):1472–1480.
- Kim Y-C, Song J-M, Lipatov AS, et al. Increased immunogenicity of avian influenza DNA vaccine delivered to the skin using a microneedle patch. Eur J Pharm Biopharm. 2012;81(2):239–247.
- Kim Y-C, Quan F-S, Compans RW, et al. Stability kinetics of influenza vaccine coated onto microneedles during drying and storage. Pharm Res. 2011;28(1):135–144.
- Choi H-J, Bondy BJ, Yoo D-G, et al. Stability of whole inactivated influenza virus vaccine during coating onto metal microneedles. J Control Release. 2013;166(2):159–171.
- Kim Y-C, Yoo D-G, Compans RW, et al. Cross-protection by co-immunization with influenza hemagglutinin DNA and inactivated virus vaccine using coated microneedles. J Control Release. 2013;172(2):579–588.
- Kommareddy S, Baudner BC, Bonificio A, et al. Influenza subunit vaccine coated microneedle patches elicit comparable immune responses to intramuscular injection in guinea pigs. Vaccine. 2013;31(34):3435–3441.
- Quan F-S, Kim Y-C, Song J-M, et al. Long-term protective immunity from an influenza virus-like particle vaccine administered with a microneedle patch. Clin Vaccine Immunol. 2013;20(9):1433–1439.
- Vassilieva EV, Kalluri H, McAllister D, et al. Improved immunogenicity of individual influenza vaccine components delivered with a novel dissolving microneedle patch stable at room temperature. Drug Deliv Transl Res. 2015;5:360–371.
- Koutsonanos DG, Esser ES, McMaster SR, et al. Enhanced immune responses by skin vaccination with influenza subunit vaccine in young hosts. Vaccine. 2015;33(37):4675–4682.
- Choi H-J, Song J-M, Bondy BJ, et al. Effect of osmotic pressure on the stability of whole inactivated influenza vaccine for coating on microneedles. PLoS One. 2015;10(7):e0134431.
- Wang B-Z, Gill HS, He C, et al. Microneedle delivery of an M2e-TLR5 ligand fusion protein to skin confers broadly cross-protective influenza immunity. J Control Release. 2014;178:1–7.
- Huang J, Garmise RJ, Crowder TM, et al. A novel dry powder influenza vaccine and intranasal delivery technology: induction of systemic and mucosal immune responses in rats. Vaccine. 2004;23(6):794–801.
- Garmise RJ, Staats HF, Hickey AJ. Novel dry powder preparations of whole inactivated influenza virus for nasal vaccination. AAPS PharmSciTech. 2007;8(4):2–10.
- Bertram U, Bernard M-C, Haensler J, et al. In situ gelling nasal inserts for influenza vaccine delivery. Drug Dev Ind Pharm. 2010;36(5):581–593.
- Dehghan S, Tafaghodi M, Bolourieh T, et al. Rabbit nasal immunization against influenza by dry-powder form of chitosan nanospheres encapsulated with influenza whole virus and adjuvants. Int J Pharm. 2014;475(1–2):1–8.
- Amorij J-P, Saluja V, Petersen AH, et al. Pulmonary delivery of an inulin-stabilized influenza subunit vaccine prepared by spray-freeze drying induces systemic, mucosal humoral as well as cell-mediated immune responses in BALB/c mice. Vaccine. 2007;25(52):8707–8717.
- Audouy SAL, van der Schaaf G, Hinrichs WLJ, et al. Development of a dried influenza whole inactivated virus vaccine for pulmonary immunization. Vaccine. 2011;29(26):4345–4352.
- Peeters B, Tonnis WF, Murugappan S, et al. Pulmonary immunization of chickens using non-adjuvanted spray-freeze dried whole inactivated virus vaccine completely protects against highly pathogenic H5N1 avian influenza virus. Vaccine. 2014;32(48):6445–6450.
- Patil HP, Murugappan S, ter Veer W, et al. Evaluation of monophosphoryl lipid A as adjuvant for pulmonary delivered influenza vaccine. J Control Release. 2014;174:51–62.
- Patil HP, Murugappan S, de Vries-Idema J, et al. Comparison of adjuvants for a spray freeze-dried whole inactivated virus influenza vaccine for pulmonary administration. Eur J Pharm Biopharm. 2015;93:231–241.
- Chen D, Erickson CA, Endres RL, et al. Adjuvantation of epidermal powder immunization. Vaccine. 2001;19(20–22):2908–2917.
- Kis EE, Winter G, Myschik J. Devices for intradermal vaccination. Vaccine. 2012;30(3):523–538.
- Kim YC, Jarrahian C, Zehrung D, et al. Delivery systems for intradermal vaccination. Curr Top Microbiol Immunol. 2012;351(Jan):77–112.
- Jia Y, Krishnan L, Omri A. Nasal and pulmonary vaccine delivery using particulate carriers. Expert Opin Drug Deliv. 2015;12(6):993–1008.
- Rose MA, Zielen S, Baumann U. Mucosal immunity and nasal influenza vaccination. Expert Rev Vaccines. 2012;11(5):595–607.
- Newman SP, Pitcairn GR, Dalby RN. Drug delivery to the nasal cavity: in vitro and in vivo assessment. Crit Rev Ther Drug Carrier Syst. 2004;21(1):46–66.
- Hinds WC. Aerosol technology: properties, behavior, and measurement of airborne particles. New York (NY): Wiley; 1999.
- Waldman RH, Wood SH, Torres EJ, et al. Influenza antibody response following aerosal administration of inactivated virus. Am J Epidemiol. 1970;91(6):574–585.
- Waldman RH, Coggins WJ. Influenza immunization: field trial on a university campus. J Infect Dis. 1972;126(3):242–248.
- Moran TM, Park H, Fernandez-Sesma A, et al. Th2 responses to inactivated influenza virus can be converted to Th1 responses and facilitate recovery from heterosubtypic virus infection. J Infect Dis. 1999;180(3):579–585.
- Rimmelzwaan GF, Fouchier RA, Osterhaus AD. Influenza virus-specific cytotoxic T lymphocytes: a correlate of protection and a basis for vaccine development. Curr Opin Biotechnol. 2007;18(6):529–536.
- Caproni E, Tritto E, Cortese M, et al. MF59 and Pam3CSK4 boost adaptive responses to influenza subunit vaccine through an IFN type I-independent mechanism of action. J Immunol. 2012;188(7):3088–3098.
- Moldoveanu Z, Love-Homan L, Huang WQ, et al. CpG DNA, a novel immune enhancer for systemic and mucosal immunization with influenza virus. Vaccine. 1998;16(11–12):1216–1224.
- Shafique M, Meijerhof T, Wilschut J, et al. Evaluation of an intranasal virosomal vaccine against respiratory syncytial virus in mice: effect of TLR2 and NOD2 ligands on induction of systemic and mucosal immune responses. PLoS One. 2013;8(4):e61287.
- Todoroff J, Lemaire MM, Fillee C, et al. Mucosal and systemic immune responses to Mycobacterium tuberculosis antigen 85A following its co-delivery with CpG, MPLA or LTB to the lungs in mice. PLoS One. 2013;8(5):e63344.
- Liu H, Patil HP, de Vries-Idema J, et al. Enhancement of the immunogenicity and protective efficacy of a mucosal influenza subunit vaccine by the saponin adjuvant GPI-0100. PLoS One. 2012;7(12):e52135.
- Kraan H, Vrieling H, Czerkinsky C, et al. Buccal and sublingual vaccine delivery. J Control Release. 2014;190:580–592.
- Çuburu N, Kweon M-N, Song J-H, et al. Sublingual immunization induces broad-based systemic and mucosal immune responses in mice. Vaccine. 2007;25:8598–8610.
- Song J-H, Nguyen HH, Cuburu N, et al. Sublingual vaccination with influenza virus protects mice against lethal viral infection. Proc Natl Acad Sci U S A. 2008;105(5):1644–1649.
- Mitragotri S. Immunization without needles. Nat Rev Immunol. 2005;5(12):905–916.
- U.S. National Institutes of Health. Inactivated influenza vaccine delivered by microneedle patch or by hypodermic needle [Internet]. 2016 [cited 2016 Jan 21]. Available from: https://clinicaltrials.gov/ct2/show/NCT02438423?term=prausnitz&rank=1
- Dean HJ. Alternative routes of influenza vaccine delivery. Expert Opin Drug Deliv. 2006;3(5):557–561.
- Greenbaum E, Furst A, Kiderman A, et al. Mucosal [SIgA] and serum [IgG] immunologic responses in the community after a single intra-nasal immunization with a new inactivated trivalent influenza vaccine. Vaccine. 2002;20(7–8):1232–1239.
- Glezen WP, Gaglani MJ, Kozinetz CA, et al. Direct and indirect effectiveness of influenza vaccination delivered to children at school preceding an epidemic caused by 3 new influenza virus variants. J Infect Dis. 2010;202(11):1626–1633.
- Amorij J-P, Kersten GFA, Saluja V, et al. Towards tailored vaccine delivery: needs, challenges and perspectives. J Control Release. 2012;161(2):363–367.
- van Ginkel FW, Jackson RJ, Yoshino N, et al. Enterotoxin-based mucosal adjuvants alter antigen trafficking and induce inflammatory responses in the nasal tract. Infect Immun. 2005;73(10):6892–6902.
- Tafaghodi M, Abolghasem Sajadi Tabassi S, Jaafari M-R, et al. Evaluation of the clearance characteristics of various microspheres in the human nose by gamma-scintigraphy. Int J Pharm. 2004;280(1–2):125–135.
- Hoppentocht M, Hoste C, Hagedoorn P, et al. In vitro evaluation of the DP-4M PennCentury™ insufflator. Eur J Pharm Biopharm. 2014;88(1):153–159.
- Friebel C, Steckel H. Single-use disposable dry powder inhalers for pulmonary drug delivery. Expert Opin Drug Deliv. 2010;7(12):1359–1372.
- de Boer AH, Hagedoorn P, Westerman EM, et al. Design and in vitro performance testing of multiple air classifier technology in a new disposable inhaler concept (Twincer) for high powder doses. Eur J Pharm Sci. 2006;28(3):171–178.
- Tonnis WF, Lexmond AJ, Frijlink HW, et al. Devices and formulations for pulmonary vaccination. Expert Opin Drug Deliv. 2013;10(10):1383–1397.
- Maurer JM, Schellekens RCA, van Rieke HM, et al. Gastrointestinal pH and transit time profiling in healthy volunteers using the intellicap system confirms ileo-colonic release of ColoPulse tablets. PLoS One. 2015;10(7):e0129076.